 Steven P. Grover
Steven P. Grover Nigel Mackman
Nigel Mackman
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 03 May 2022
Sec. Thrombosis and Haemostasis
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.878199
This article is part of the Research Topic Unraveling Clot Composition and Structure in Vitro, in Vivo and in Silico View all 4 articles
Appropriate activation of coagulation requires a balance between procoagulant and anticoagulant proteins in blood. Loss in this balance leads to hemorrhage and thrombosis. A number of endogenous anticoagulant proteins, such as antithrombin and heparin cofactor II, are members of the serine protease inhibitor (SERPIN) family. These SERPIN anticoagulants function by forming irreversible inhibitory complexes with target coagulation proteases. Mutations in SERPIN family members, such as antithrombin, can cause hereditary thrombophilias. In addition, low plasma levels of SERPINs have been associated with an increased risk of thrombosis. Here, we review the biological activities of the different anticoagulant SERPINs. We further consider the clinical consequences of SERPIN deficiencies and insights gained from preclinical disease models. Finally, we discuss the potential utility of engineered SERPINs as novel therapies for the treatment of thrombotic pathologies.
Appropriate activation of coagulation is essential in limiting blood loss from a closed circulatory system. On vascular injury, exposure of sub-endothelial tissue factor (TF) results in activation of the extrinsic pathway of coagulation through exposure of the TF: factor (F) VIIa complex to substrate FX resulting in generation of FXa (Figure 1) (1). Coagulation can also be initiated by activation of the intrinsic pathway through autoactivation of FXII to FXIIa (2, 3). FXIIa generation is enhanced by reciprocal activation of FXII by plasma kallikrein and its cofactor high molecular weight kininogen. FXIIa activates FXI to FXIa that itself activates FIX to FIXa. FIXa in complex with cofactor FVIIIa catalyzes additional FXa generation (Figure 1). FXa, in complex with the essential cofactor FVa and in the presence of additional cofactors phospholipid and calcium, catalyzes the conversion of prothrombin zymogen to thrombin. As the terminal coagulation protease thrombin catalyzes cleavage of soluble fibrinogen to insoluble fibrin leading to the formation of a fibrin mesh that functions to limit blood loss at the site of injury. Additionally, thrombin facilitates the formation of a platelet rich plug at the site of vascular injury through direct activation of platelets.
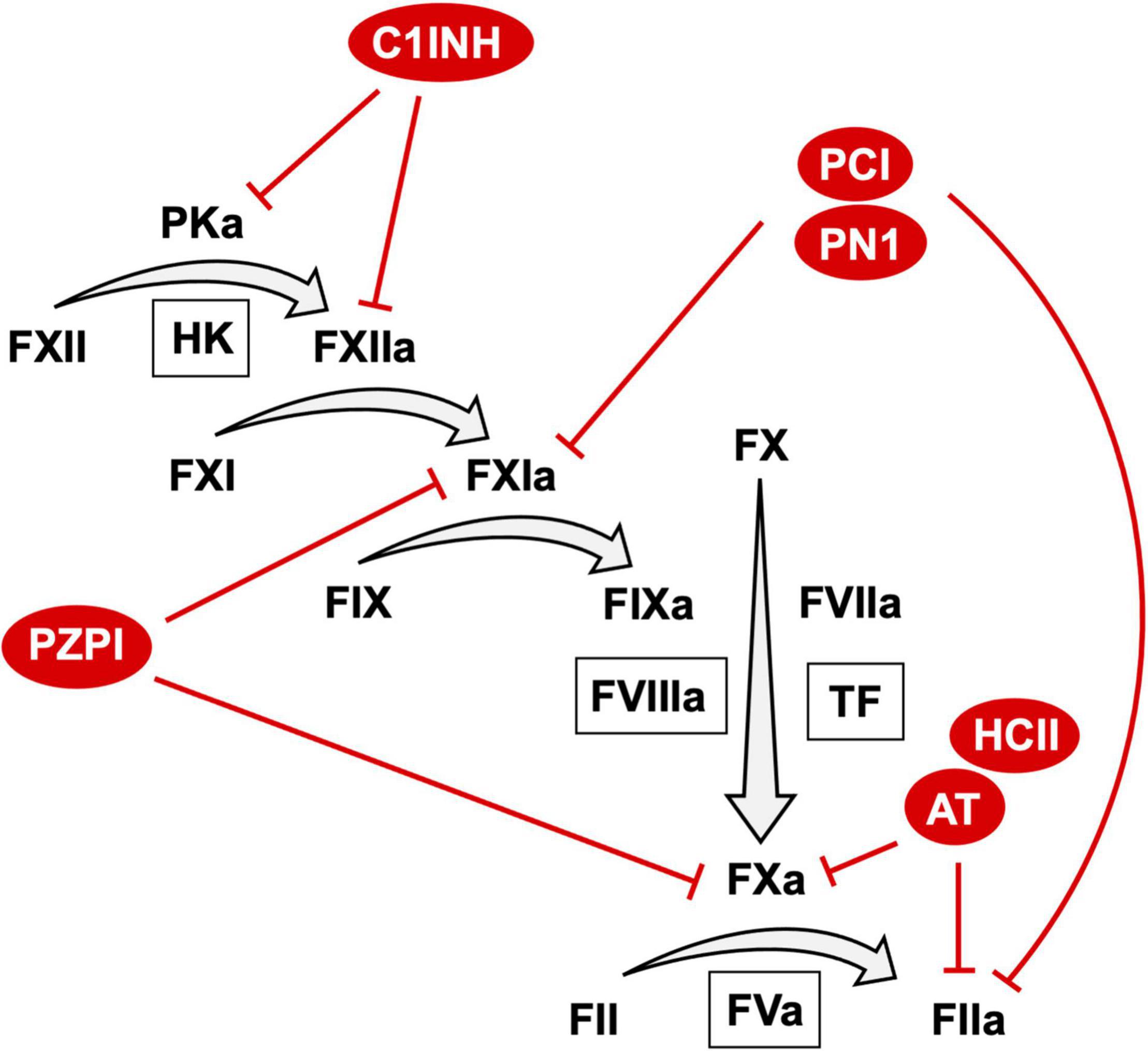
Figure 1. The coagulation cascade and anticoagulant SERPIN targets. A schematic representation of the coagulation cascade annotated with anticoagulant SEPRIN targets. For clarity only the top targets of each SERPIN with second order inhibitory rate constants > 1.0 × 105 are represented. A comprehensive list of targets and second order inhibitory rate constants is provided in Table 1.
Thrombin generation can be further enhanced through feedback activation of the intrinsic pathway of coagulation by the extrinsic pathway. The TF:FVIIa complex is an effective activator of FIX and the TF:FVIIa:FXa ternary complex can activate FVIII (4, 5). In addition, thrombin can directly activate FXI leading to further intrinsic pathway derived thrombin generation (6). Together these feedback pathways can generate significant quantities of thrombin and contribute to hemostasis in a tissue specific manner (7).
Inappropriate activation of coagulation underpins a number of common cardiovascular and hematological diseases. An impaired ability to mount an effective hemostatic response to vascular injury can lead to bleeding disorders, such as those observed in individuals with genetic deficiencies for FVIII and FIX that cause hemophilia A and B, respectively (8). Alternatively, excessive activation of coagulation can lead to thrombotic disorders such as venous thromboembolism (VTE) that includes both deep vein thrombosis (DVT) and pulmonary embolism (PE) (1, 2, 9). Excessive activation of coagulation can be caused by resistance of coagulation factors to inhibition, such as is observed in Factor V Leiden, (10).
For this reason, the coagulation system must be tightly regulated. Negative regulation of coagulation is achieved by anticoagulant proteins. These anticoagulant proteins take the form of protease inhibitors that together function to inactivate all proteases of the coagulation system. A number of these protease inhibitors are members of the serine protease inhibitor (SERPINs) superfamily (11). Anticoagulant SERPINs include antithrombin (AT), heparin co-factor II (HCII), protein Z dependent protease inhibitor (PZPI), protease nexin 1 (PN1) and C1-inhibitor (C1INH) (Figure 1). The inhibitory activity of SERPINs is complemented by a number of non-SERPIN anticoagulants. This includes tissue factor pathway inhibitor, which is the primary inhibitor of the TF:FVIIa complex (12). In addition, activated protein C (aPC) and its essential cofactor protein S function as an important physiological inhibitor of FVa and FVIIIa (13).
In this review, we introduce the properties of SERPIN family members that facilitate inhibition of target coagulation proteases. We further discuss the anticoagulant properties of specific SERPINs and consider how these properties enable regulation of hemostatic and thrombotic processes. Particular attention is paid to congenital SERPIN deficiencies, more broadly referred to as serpinopathies, that are associated with altered thrombotic risk (14, 15). Finally, we review advances in the development of variant SERPIN proteins with therapeutic potential.
Despite the identification of numerous non-inhibitory family members, the SERPIN superfamily name remains. Members are subdivided into clades based on phylogenetics. To date, sixteen clades, named A through P, have been identified with an additional number of SERPINs remaining as unclassified orphan members (16, 17). Based on the clade naming system, alpha 1 antitrypsin (A1AT) is the first member of clade A and is designated SERPINA1 encoded by the SERPINA1 gene (16, 17). Despite the unifying clade-based nomenclature for SERPINs the majority of family member proteins are referred to by the name given to them on first description. SERPINs with anticoagulant activities are dispersed throughout the clades (Table 1).
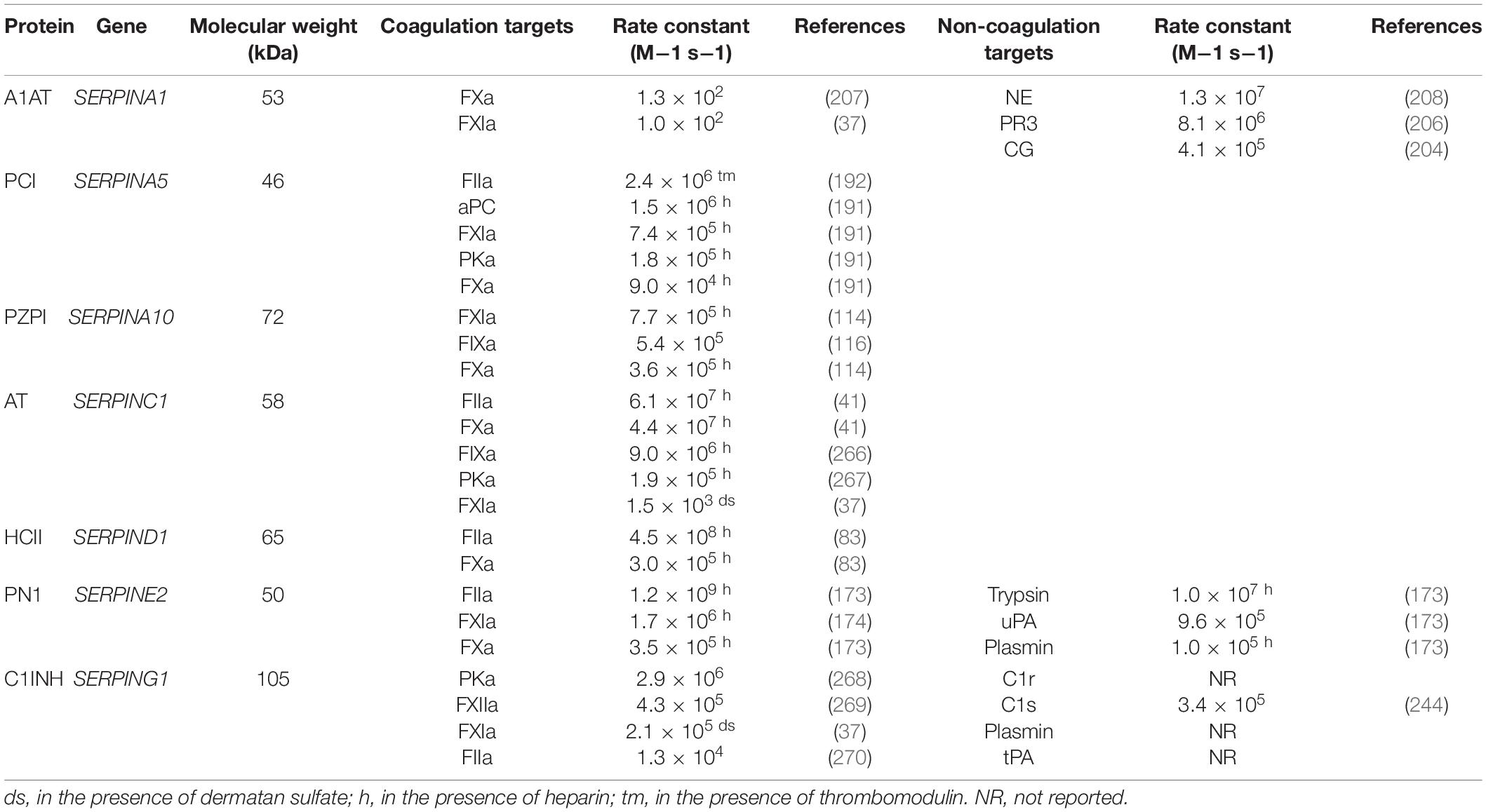
Table 1. Anticoagulant SERPINs and targets.
The initial identification of the SERPIN family of serine protease inhibitors was based on primary sequence similarities identified between AT and A1AT (18). Subsequent structural studies demonstrated that, despite relatively low sequence identity, relatively high levels of sequence homology enabled SERPINs to retain a well-preserved sequence of secondary structures consisting of three β-sheets and 8–9 α-helices that form a core structural domain (Figure 2) (16). In addition, SERPINs contain a reactive center loop (RCL) that functions as a critical determinant for protease specificity (19). As a result of these structural similarities the SERPIN family of proteins demonstrate a remarkable degree of structural homology (20). Outside of this core structural domain SERPINs possess variable N and C terminal regions that contribute to the wide range of observed molecular weights. For example, unlike the smaller SERPINs A1AT and PCI that are 45–55 kDa in weight, C1INH inhibitor possesses a heavily glycosylated N-terminal extension resulting in an observed molecular weight of 105 kDa.
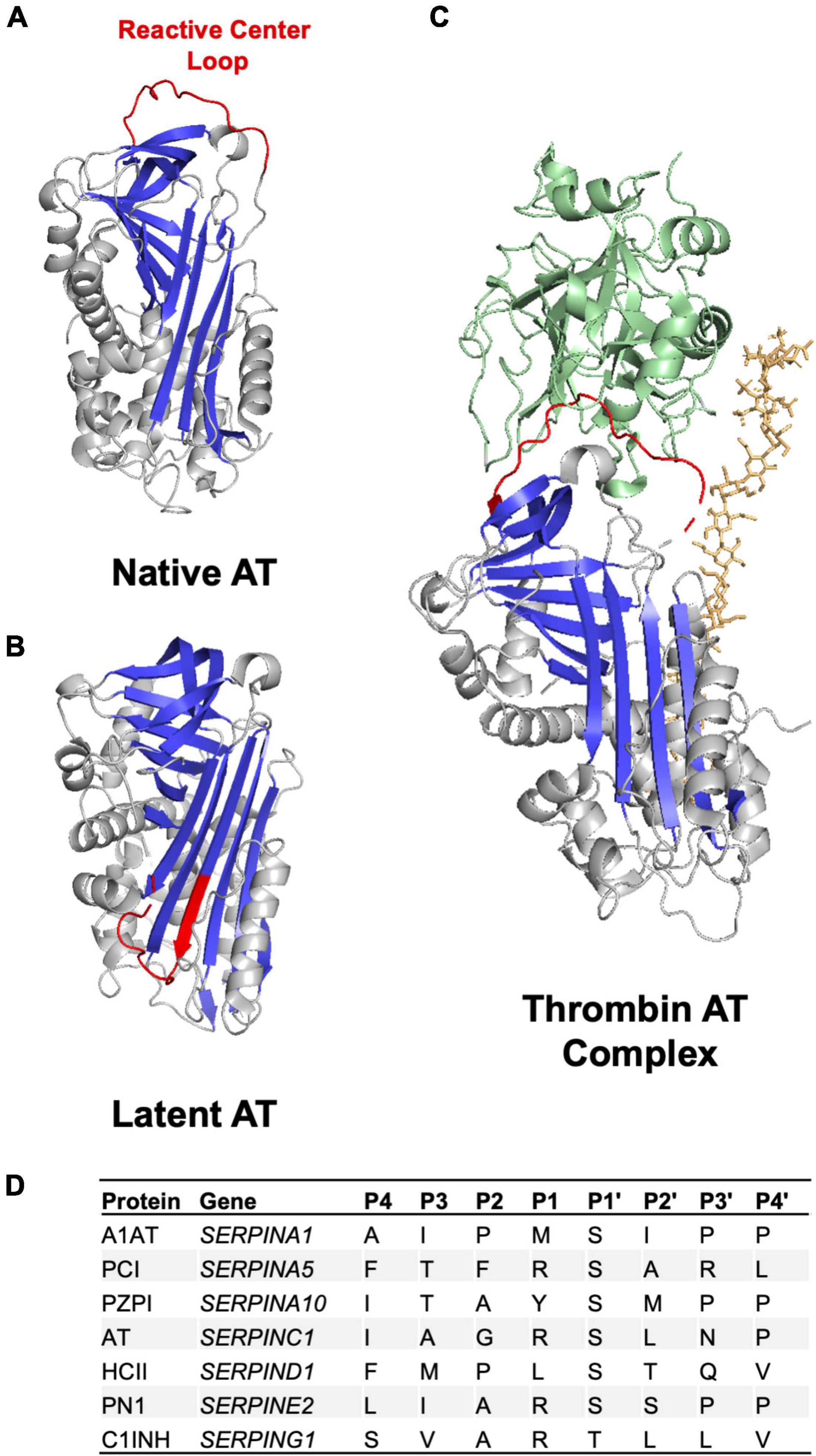
Figure 2. Anticoagulant SERPIN structure and RCL. (A) Crystal structure of the anticoagulant SERPIN AT in the native conformation with a protease accessible RCL (B) Crystal structure of AT in the latent conformation in which the RCL is buried in the protein body and inaccessible to protease. The AT RCL is colored red, β sheets are colored in blue and α helices in gray. (C) Crystal structure of AT in complex with thrombin (green) in the presence of heparin (orange). Images made in PyMol using PDB files 1T1F, 2BEH, and 1TB6. (D) Amino acid residues in anticoagulant SERPINs in the P4-P4’ region of the RCL.
The RCL is a 16–17 amino acid sequence found toward the SERPIN C terminus (Figure 2) (21). This sequence is critical to the inhibitory specificity of a given SERPIN. The RCL contains a protease recognition sequence that functions as molecular “bait” for target proteases (19). A target protease binds to the SERPIN through the protease recognition sequence and forms a reversible Michaelis-Menten complex. The protease recognition sequence contains a proteolytic cleavage site with RCL amino acid residues being annotated with P for N terminal residues to the cleavage site and P’ for C terminal residues to the cleavage site as per convention (Figure 2) (22). The specificity of a given SERPIN for a given protease appears to be particularly dependent on the amino acids present at the P1-P1’ positions but can also be influenced by the sequence of amino acids in the P4-P4’ positions (Figure 2) (19). Cleavage of the SERPIN by a target protease results in the formation of a covalent bond between the protease and the main chain carbonyl bond of the SERPIN P1 residue (21). This SERPIN cleavage event results in a marked conformational change as discussed below.
SERPINs can exist in either a native metastable or a non-native hyperstable conformational state (Figure 2). In the native metastable conformation, the RCL is externalized and free to interact with target proteases (21). Upon cleavage by a target protease the RCL inserts into β-sheet A forming the hyperstable conformation. Insertion of the RCL into the SERPIN body results in translocation of the target protease interrupting the final hydrolysis of the bond between SERPIN and protease. Trapping of the protease in this manner prevents release and results in the formation of an irreversible SERPIN-protease complex (21). The speed with which a SERPIN, when cleaved by a protease, transitions between the native metastable state and latent hyperstable state is important in determining inhibitory capacity (19, 21). A slow transition allows for hydrolysis of the bond between the SERPIN and protease regenerating functional protease and leaving hyperstable SERPIN. In addition to inhibiting the proteolytic function of the protease, SERPIN complex formation also significantly disrupts the structure of the protease making it more susceptible to proteolytic degradation (23).
It is also possible for SERPINs to be present in a latent conformational state. The latent conformational state bears some structural similarity to the hyperstable state in that the RCL inserts into β-sheet A but this process is not dependent on interaction with a target protease (21). Interestingly, AT has been found to be present as a conformational heterodimer with one monomer present in the latent state (24).
The inhibitory capacity of most anticoagulant SERPINs, including AT, C1INH, HCII PCI, and PN1, is enhanced by binding to negatively-charged glycosaminoglycans (GAGs), such as heparin (Figure 3) (25). The inhibitory activity of these SERPINs can be increased by as much as several thousand-fold by GAG binding. The ability of the GAG heparin to potentiate the activity of anticoagulant SERPINs has been leveraged clinically with unfractionated and low molecular weight formulations used as thromboprophylactic agents to prevent VTE (26).
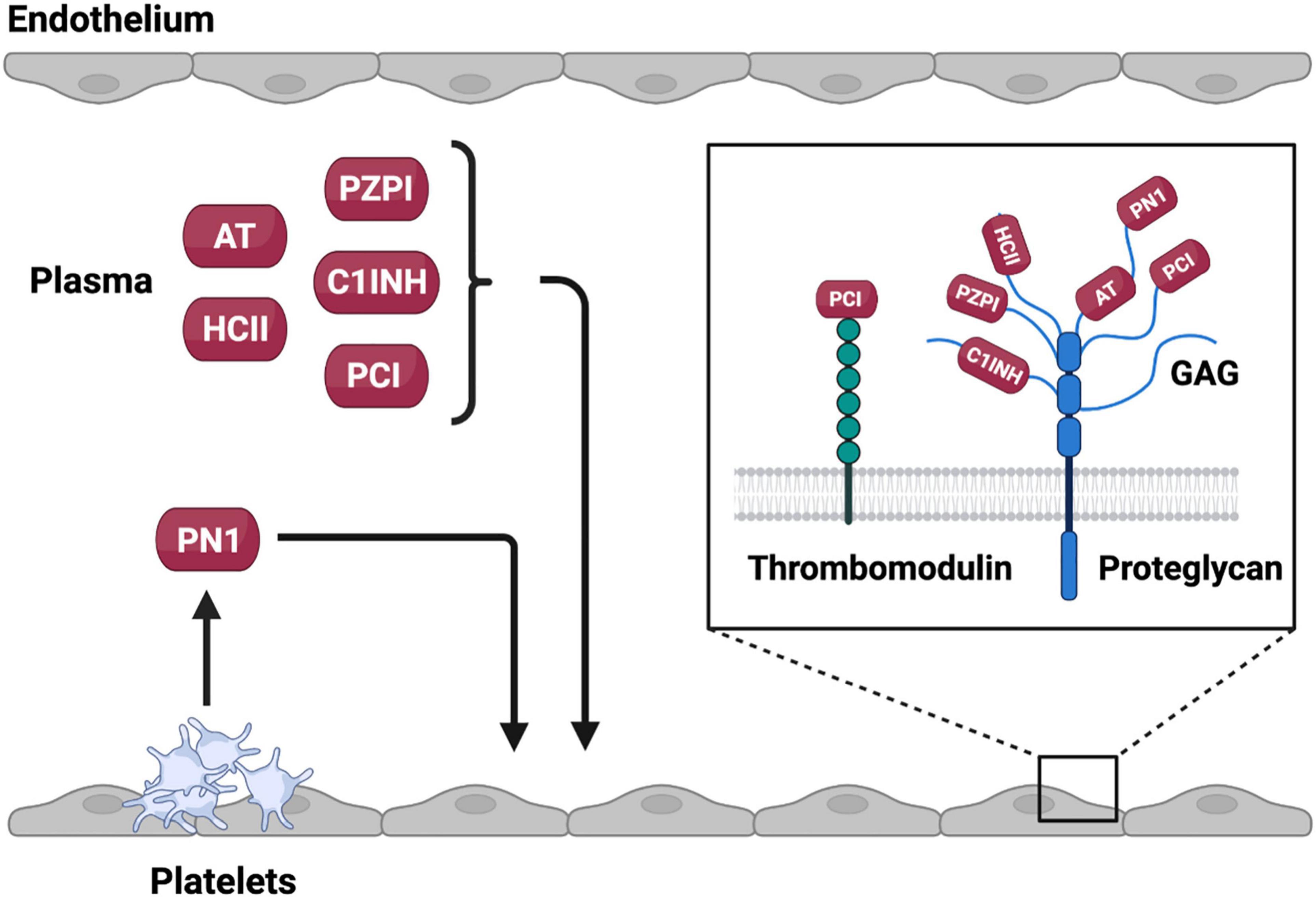
Figure 3. Source and site of activity of anticoagulant SERPINs. Anticoagulant SERPINs AT, HCII, PZPI, C1INH, and PCI are present in the plasma whereas PN1 is stored in platelet alpha granules where it can be released on platelet activation. Anticoagulant SERPINs bind to glycosaminoglycans (GAGs) or protein cofactors on the endothelial surface that potentiate inhibition of target proteases. Created with BioRender.com.
Two major mechanisms of GAG mediated rate enhancement have been proposed. First, GAGs can act as a bridge binding to both the SERPIN and the protease bringing them together in a confirmation preferable for the protease to interact with the RCL. This interaction has been described to occur between AT and thrombin in the presence of heparin (Figure 2) (27, 28). Second, binding of the GAGs to the SERPIN, which may occur at a distal site, can result in a conformational change in the SERPIN that makes it more reactive toward the target protease. GAGs have been found to facilitate this interaction between HCII and thrombin (29–31). It is likely that GAGs function as both bridging molecules and allosteric modulators for a given SERPIN (32). Indeed, the native forms of both AT and HCII have RCLs that are partially incorporated into the SERPIN body with GAG binding promoting RCL exposure (31, 33, 34). GAGs are present on the endothelial surface forming part of the glycocalyx. Binding of SERPINs to GAGs facilitates localization of these inhibitors to the vessel surface and may help prevent inappropriate intravascular coagulation. At sites of vessel injury where endothelial bound GAGs are not present activation of coagulation is allowed to continue to form a hemostatic plug. This property of SERPINs may be of particular interest when considering the use of these inhibitors as therapeutic agents.
AT is a 58 kDa serine protease inhibitor encoded by the SERPINC1 gene. AT is present in plasma at a concentration of approximately 120 μg/ml with a circulating half-life of roughly 3 days. AT functions as a major endogenous inhibitor of thrombin and FXa but also inhibits FIXa, FXIa, FXIIa, and plasma kallikrein (Table 1) (35–41). The ability of AT to inhibit target proteases is dramatically enhanced (2,000–4,000 times) in the presence of heparin (42–44). Interestingly, the GAG heparan sulfate appears to be a much more effective potentiator of AT activity than others, such as dermatan sulfate and chondroitin sulfate (45). This is likely due to the fact that heparin and heparan sulfate contain a specific sulfated pentasaccharide (46–48). The RCL of native AT was shown to be partially incorporated into the SERPIN body limiting interaction with target proteases (33, 34). Critically, binding of the specific heparin pentasaccharide induced a confirmational change in native AT expelling the RCL from the SERPIN body providing accessibility to target proteases (49, 50). The altered presentation of the AT reactive center loop may underpin the enhanced inhibitory activity observed in the presence of heparin. As discussed earlier native and latent AT can form heterodimers (24, 33). Formation of heterodimers was found to reduce the thrombin inhibitory capacity of AT (24). It has been proposed that some AT variants associated with VTE, that retain significant activity against thrombin, may have an increased tendency to be present in the latent conformation and may disrupt the inhibitory capacity of native AT through heterodimer formation (24, 51, 52).
Congenital deficiency for AT is relatively common in the general population with an estimated prevalence of between 1:500 and 1:2,000 (53, 54). To date more than 200 mutations in the SERPINC1 gene have been identified that cause AT deficiency (55). Mutations in the SERPINC1 gene can lead to reduced levels of AT, causing type I AT deficiency, or to expression of AT with reduced anticoagulant activity, causing type II AT deficiency (56). Type II AT deficiency can be further divided into additional subtypes: type IIa caused by mutations in the thrombin binding site, type IIb caused by mutations in the heparin binding site or type IIc in which mutations near the reactive center loop have pleiotropic effects on both heparin and thrombin interactions (56). Interestingly, a SERPINC1 variant causing type II AT deficiency has been shown to induce polymerization of AT in the plasma (57). This mechanism is distinct from other SERPIN family variants described that destabilize protein structure and cause intracellular aggregation and retention. Type II AT deficiencies are much more common in the general population than type I deficiencies (58).
Between 1 and 2% of all VTE events are associated with AT deficiency (59, 60). Strikingly, individuals with AT deficiency have a 50% chance of suffering a VTE by the age of 50 (61, 62). Consistent with this, individuals with AT deficiency have a markedly increased risk of a first VTE compared to controls (relative risk 8.1) (Table 2) (61). Further, a number of studies have reported an increase in the rate of VTE recurrence in individuals with overt (< 70% normal AT levels) deficiency (hazard ratios 1.9–5.9) (63–65). On presenting with a first proximal DVT event individuals with AT deficiency had an increased risk of symptomatic PE (relative risk 2.4) (60). Interestingly, individuals with mild AT deficiency (70–80% of normal) also have an increased risk of venous thromboembolism recurrence (hazard ratio 3.9) (64). The robustness of this observation is unclear, however, with a subsequent study finding a weaker not significant association (65). AT deficiency appears to be associated with a modestly increased risk of arterial thrombosis (66, 67). Further, low plasma levels of AT were found to be associated with a significantly increased risk of arterial thrombotic events (68). Additional studies are warranted to determine the effect of AT deficiency on risk of arterial thrombosis. Importantly, human purified plasma-derived AT products with elimination half-lives of 2–3 days are used for thromboprophylactic replacement therapy in patients with AT deficiency undergoing surgical procedures (69).

Table 2. Associations between SERPIN abnormalities and atherothrombotic diseases.
AT is a negative acute phase reactant, being depleted under inflammatory conditions, that is also thought to have important anti-inflammatory functions (70). In the inflammatory setting of sepsis patients that did not survive had significantly lower plasma levels of AT than those that survived (71). Consistent with the anti-inflammatory effects of AT, treatment of sepsis patients with exogenous AT significantly reduced plasma levels of interleukin 6 and c reactive protein (72). Given the proinflammatory activities of AT targets FXa and thrombin it is likely that the anti-inflammatory effects of AT are secondary to inhibition of FXa (70).
Attempts have been made to generate AT deficient mice (73). Complete deficiency for AT was found to result in embryonic lethality at mid-gestation (73). AT deficient embryos had evidence of intravascular fibrin deposition and extensive hemorrhage consistent with a consumptive coagulopathy (73). Interestingly, transgenic mice expressing an AT variant, R48C, with reduced ability to bind heparin cofactor have also been generated (74). Embryos homozygous for this variant AT were present at mid-gestation at the expected frequency (74). However, reduced peri and post-natal survival was observed with mice homozygous for the variant AT (74). In addition, adult mice homozygous for the variant AT had evidence of spontaneous thrombosis, particularly in the heart and liver (74). In a complementary approach short interfering RNA mediated silencing of the SERPINC1 gene in mice resulted in acute and extensive thrombus formation in the head, limbs and liver (75). Crossing of AT deficient mice with those expressing low levels of TF enabled deficient embryos to persist to late gestation but was not sufficient to generate viable mice (76). Disruption of AT in zebrafish also resulted in reduced survival of AT deficient offspring with evidence of spontaneous thrombosis (77). Surprisingly, AT deficient larvae demonstrated prolonged occlusion times in a caudal vein laser injury thrombosis model (77). This was explained by the presence of hypofibrinogenemia in AT deficient larvae likely caused by a consumptive coagulopathy (77). These studies demonstrate that the anticoagulant activity of AT is essential for appropriate regulation of coagulation in vivo. Further, loss of AT anticoagulant activity can lead to spontaneous thrombotic events.
In light of the critical anticoagulant activity of AT, inhibition of this protein has been explored as a potential therapy for hemophilia (78). FVIII deficient mice that were also heterozygous for AT had increased thrombin generation and demonstrated shortened bleeding times in a tail transection model compared to FVIII deficient mice (79). Short interfering RNA mediated gene silencing of the SERPINC1 gene was also found to reduce bleeding in FVIII deficient mice subject to a saphenous vein transection model (80). SERPINC1 gene silencing in FVIII deficient mice supported increased platelet and fibrin accumulation at sites of vascular injury in a cremaster arteriole laser injury model of thrombosis (80). A single-domain inhibitory anti-AT antibody has also been developed (81). FVIII or FIX deficient mice administered an adenoviral vector expressing the single-domain inhibitory anti-AT antibody had significantly reduced blood loss in a tail vein transection bleeding model compared to controls (81). Importantly, this anti-AT antibody was equally as effective in reducing blood loss in FIX deficient mice with inhibitory FIX autoantibodies compared to those without (81). Based on these studies, a SERPINC1 targeting short interfering RNA is currently being evaluated in patients with hemophilia A and B (82).
AT functions as a potent inhibitor of thrombin and FXa. A significant body of clinical evidence indicates that AT deficiency is associated with an increased risk of VTE. Consistent with the essential anticoagulant functions of AT mice deficient for this SERPIN are not viable. AT inhibitors have shown promise as prohemostatic therapies in preclinical models of hemophilia supporting enhanced thrombin generation.
HCII, encoded by the SERPIND1 gene, is a 65 kDa serine protease inhibitor that serves as a potent and selective inhibitor of thrombin and to a lesser extent FXa (Table 1) (83). HCII is present in plasma at a concentration of approximately 80μg/ml with a circulating half-life of 2–3 days (83, 84). Unlike AT, HCII is a stronger inhibitor of thrombin and has markedly weaker activity against FXa (83). As is the case with AT, native HCII is a poor inhibitor of thrombin owing to the lack of accessibility of the RCL in this conformation (31). Binding of HCII to GAGs drives the conformational transformation of native inactive HCII into the native active form expelling the partially incorporated RCL and markedly enhancing activity toward target proteases (31). Unlike AT, that is selectively activated by specific pentasaccharides present in a small percentage of GAGs, HCII is less selective and can be activated by a host of GAGs, including heparan sulfate, chondroitin sulfate, and dermatan sulfate (85–87).
HCII deficiency in humans has been described, with a number of individuals identified after episodes of arterial or venous thrombosis (88–92). In two studies patients with a history of arterial and venous thrombosis were found to have plasma HCII levels 50% of normal (88, 89). Further, an individual presenting with multiple episodes of DVT and PE was found to have a homozygous deficiency for HCII (91). This individual was also found to have a heterozygous AT deficiency that likely contributed to the thrombotic phenotype given that another family member with a homozygous HCII deficiency had no history of VTE (91). It is possible that additional thrombophilias are required to reveal the thrombotic phenotype of HCII deficiency. Indeed, compound heterozygous mutations in HCII and either factor V or protein C have been reported to result in thrombotic manifestations (93).
Anecdotal case series-based evidence has led to further systematic studies aimed at identifying a formal association between HCII deficiency and risk of thrombosis. In a study of 277 individuals with unexplained VTE 3 cases of HCII deficiency were identified (94). However, it was determined that HCII deficiency was equally prevalent in a healthy control population and thus unlikely to be a major driver of thrombotic risk (94). In a study of 583 individuals with anticoagulant protein deficiencies no association between HCII deficiency and VTE was observed (95). It is important to note, however, that this study included only 6 individuals with HCII deficiency, of which 4 had suffered a thrombotic event (95). Owing to the small number of HCII deficient individuals the study was likely not sufficiently powered to determine an association between HCII deficiency and VTE risk (95). Taken together, the available evidence does not support an association between HCII deficiency and thrombotic events. This is in contrast to the strong association between AT deficiency and VTE, suggesting that HCII may be a less important anticoagulant compared to AT. However, given the apparent rarity of HCII deficiency additional larger scale studies may provide further insights.
The presence of advanced atherosclerotic disease in several patients with HCII deficiency has led to the suggestion that HCII may have additional vascular protective effects (90, 96). Despite the lack of an association between plasma HCII and thrombosis, significant associations between plasma levels of HCII and atherosclerotic disease have been reported (Table 2) (97). In one study of 306 elderly individuals higher plasma levels of HCII were associated with reduced carotid artery plaque thickness (98). In a second study of 134 sequential patients undergoing percutaneous coronary intervention high plasma levels of HCII were associated with a reduced rate of in-stent restenosis (99). Complementary findings were made in a similar study of 63 patients undergoing percutaneous interventions for peripheral artery disease where high plasma levels of HCII were associated with reduced restenosis (100).
HCII deficient mice have been used to study the anticoagulant effect of endogenous HCII (101). On first description HCII deficient mice were generated at the expected frequency demonstrating normal development and survival (102). However, in a subsequent study strain dependent embryonic lethality of homozygous HCII deficient mice was observed (103). In a carotid artery rose bengal induced thrombosis model HCII deficient mice were found to have significantly shortened occlusion times. Importantly, administration of dermatan sulfate was found to prolong occlusion times in wildtype mice but not HCII deficient mice (104). Moreover, in contrast to wildtype HCII protein or HCII variant protein with reduced heparin binding (K173Q), variant HCII protein with reduced affinity for dermatan sulfate (R189H) was not able to normalize occlusion times (105). These findings suggest that dermatan sulfate is an important activator of HCII in vivo. Consistent with the anticoagulant activity of HCII, administration of exogenous purified HCII was found to significantly prolong occlusion times in a rat femoral artery rose bengal induced thrombosis model (106).
In addition to anticoagulant functions an important vascular protective function of HCII has been described (101). When crossed onto a ApoE deficient background mice with heterozygous or homozygous HCII deficiency demonstrated enhanced atherosclerotic lesion development (107, 108). A number of studies have also shown that heterozygous or homozygous HCII deficient mice had increased intimal hyperplasia in arteries subject to cuff or wire-based injury (103, 107, 108). The mechanism by which HCII deficiency promotes these pathologic processes has yet to be determined. Taken together these studies indicate that HCII has important anticoagulant and vascular protective activity in preclinical models of atherothrombotic disease (101). Further preclinical studies are warranted to evaluate if endogenous HCII plays a role in venous thrombosis.
HCII is a potent GAG-enhanced inhibitor of thrombin. Available clinical evidence is limited and does not support an association between HCII deficiency and VTE. However, in preclinical studies endogenous and exogenous HCII inhibited arterial thrombosis. Critically high plasma levels of HCII have been associated with reduced atherosclerosis, a finding that has been complemented by preclinical studies.
PZPI is a 72 kDa serine protease inhibitor encoded by the SERPINA10 gene. PZPI is present in plasma at a concentration of approximately 5 μg/ml (109). PZPI, in complex with the vitamin K-dependent cofactor protein Z (PZ), functions as a selective inhibitor of FXa, FIXa, and FXIa (Table 1) (110, 111). Lipid membrane surface and heparin have been identified as additional co-factors that enhance the inhibitory activity of PZPI toward FXa and FXIa (111–114). Although it was initially thought that FXa bound to FVa in the prothrombinase complex was protected from PZPI mediated inhibition, recent work has demonstrated that FXa is effectively targeted for inhibition by PZ bound PZPI (115). Whereas binding of PZ to PZPI was found to markedly enhance inhibitory activity toward FXa, PZPI efficiently inhibits FIXa in the absence of PZ (116). In fact, PZ binding has been found to reduce the efficiency with which PZPI inhibits FXIa (117).
Mutations in the PZPI gene SERPINA10 have been associated with an increased risk of VTE (Table 2). In one study, sequencing of the SERPINA10 gene revealed a significantly increased prevalence of loss of function PZPI variants (R67X, W303X) in 250 VTE patients compared to a cohort of 250 control individuals (118). A subsequent study of 1,018 VTE patients and 1,018 healthy controls confirmed that the presence of the R67X variant was associated with a significantly increased risk of VTE (119). Assessment of an additional set of SERPINA10 mutations resulting in nonsense and loss of function PZPI variants (R88X, W324X, Q384R, F145L) in a cohort of 550 VTE patients and 600 healthy controls found that these variants were significantly more prevalent in VTE patients (120). It is important to note that the association between PZPI loss of function mutations and VTE risk was not confirmed in other similarly sized independent studies (121–124). It is possible that the association only holds for specific populations. The conflicting reports of the association between PZPI and thrombosis mirror that reported for the cofactor PZ in the setting of ischemic stroke with both positive and negative studies reported (125–128). Further population-based studies are required to more conclusively determine if an association between PZPI and VTE exists.
Preclinical studies have demonstrated an important role for PZPI and the cofactor PZ as regulators of coagulation. Mice deficient for PZ were generated at the expected frequency. However, when PZ deficient mice were crossed onto a prothrombotic homozygous FV Leiden background complete postnatal lethality was observed (129). A high proportion of FV Leiden embryos deficient for PZ had evidence of hemorrhage, which was thought to be secondary to a consumptive coagulopathy, and intravascular deposition of fibrin (129). Similarly, PZPI deficient mice were generated at the expected frequency but when crossed onto a homozygous FV Leiden background demonstrated complete postnatal lethality (130). When crossed to generate FV Leiden heterozygotes, deficiency for PZPI, but not PZ, was found to result in embryonic lethality at mid to late gestation (130). The stronger phenotype associated with PZPI deficiency in this setting suggests that PZPI may be an important regulator of FIXa and FXIa during embryonic development (130). Interestingly, equivalent phenotypes have been observed in PZPI deficient and PZ deficient mice in the absence of an additional procoagulant phenotype. Both PZPI and PZ deficient mice demonstrated significantly reduced survival in a collagen epinephrine model of pulmonary embolism. Further, both PZPI and PZ deficient mice demonstrated increased occlusion in a carotid artery ferric chloride model (130). The equivalent phenotype associated with PZPI and PZ deficiency alone suggests that an additional procoagulant stimulus is required to reveal phenotypic differences between the SERPIN and cofactor (130). Modulating the anticoagulant activity of PZPI and PZ has also been evaluated as a potential therapy for hemophilia. Deficiency for either PZPI or PZ was found to significantly reduce bleeding times in FVIII deficient mice and supported enhanced plasma thrombin generation (131).
PZPI is a selective inhibitor of FXa, FIXa, and FXIa. Clinical studies provide evidence of a potential association between loss of function PZPI variants and VTE risk. Complementary preclinical studies have shown that loss of PZPI or its cofactor PZ enhances arterial thrombus formation in mouse models. PZPI is a potential target for novel hemophilia therapies given its function as an inhibitor of FIXa. Indeed, PZPI deficiency improved the hemostatic response in a mouse model of hemophilia A.
C1INH is a 105 kDa serine protease inhibitor encoded by the SERPING1 gene. C1INH is present in plasma at a concentration of 200 μg/ml with a circulating half-life of approximately 3 days (132). C1INH is also present in platelet alpha granules and is released upon platelet activation (133). C1INH serves as the major endogenous inhibitor of FXIIa and plasma kallikrein (Table 1) (134–137). C1INH is also an effective inhibitor of FXIa (138). As the name indicates, C1INH was first identified for the ability to inhibit the first component of complement (139). C1INH also inhibits a host of other proteases, including plasmin, tissue plasminogen activator, mannan-binding lectin serine protease 1 and mannan-binding lectin serine protease 2 (140–142). Importantly GAGs have been found to selectively enhance the activity of C1INH toward FXIa (37, 143). However, GAGs were found to decrease inhibitory activity of C1INH toward FXIIa (37, 144). More recently the polyanion polyphosphate has been found to enhance the inhibitory activity of C1INH toward the first component of complement (145). The effect of polyphosphate on inhibition of coagulation proteases by C1INH has yet to be determined. Unlike other anticoagulant SERPINs, C1INH undergoes extensive glycosylation resulting in a markedly increased apparent molecular weight. C1INH contains 3 C-terminal and 3 N-terminal N-linked glycosylation sites and up to 26 O-linked N-terminal glycosylation sites (146). The impact of glycosylation on C1INH function remains unclear with conflicting reports on the contribution of glycosylation to circulating half-life and inhibitory efficacy of C1INH (147–150).
C1INH deficiency in humans results in a condition called hereditary angioedema (HAE), a life-threatening syndrome triggered by episodes of bradykinin induced swelling that can lead to asphyxiation (151). Mutations in the SERPING1 gene can result in reduced expression of C1INH, leading to type I HAE, or expression of C1INH with reduced function, leading to type II HAE (151). Interestingly, the vast majority of patients presenting with HAE are heterozygous for these mutations indicating an autosomal dominant pattern of inheritance (152). HAE patients with heterozygous mutations in the C1INH gene typically present with plasma levels of C1INH antigen or activity markedly lower than the expected 50% (153). This may be due to the fact that variant C1INH proteins can form intracellular aggregates with wildtype protein leading to reduced secretion (154). To date over 500 disease-causing SERPING1 variants have been reported (152).
A number of treatments have been used to manage attacks in patients with HAE (151). C1INH products have been developed to serve as replacement therapies and include human purified plasma-derived and recombinant C1INH protein preparations (151). Plasma-derived C1INH products have elimination half-lives of 2–5 days depending on the route of administration (132). Additional therapies including the bradykinin receptor inhibitor icatibant and the kallikrein inhibitors ecallantide and berotralstat have also been developed (151). These therapies function by either inhibiting bradykinin generation or inhibiting activation of the bradykinin receptor that drives swelling in patients with HAE.
Given that the primary phenotype of patients with HAE is related to excess activation of FXIIa and kallikrein mediated bradykinin generation, efforts have been made to evaluate the effect of C1INH deficiency on the contact system (155). Levels of plasma kallikrein and FXIIa activity were markedly elevated in patients with HAE when compared to controls (156). Similarly, levels of FXIIa and cleaved kininogen were markedly higher in patients with HAE during acute attacks compared to when in remission (157).
HAE patients have been reported to have elevated plasma levels of markers of activation of coagulation and fibrin degradation, including prothrombin fragment F1+2, thrombin-antithrombin (TAT) complexes and D-Dimer compared to healthy controls (157–159). Further, these markers are elevated in HAE patients during acute attacks compared to when in remission (157–159). Activated partial thromboplastin times were significantly shorter in patients with HAE when compared to controls and significantly shorter in HAE patients during attacks than during remission (159). Plasma from patients with HAE was also found to support a modest but significant increase in TF-initiated thrombin generation when compared to controls (160). This study was conducted in the absence of a contact pathway inhibitor, such as corn trypsin inhibitor, and as such this phenotype could be attributed to residual activation of the contact pathway (160).
The relationship between C1INH deficiency and thrombosis has been less clear. Initial observations suggested that treatment of HAE patients with C1INH was associated with an increased risk of thrombosis (161). However, a large-scale registry study did not observe an association between C1INH treatment and thrombotic events (162). In a recent study HAE was associated with an increased risk of ATE (odds ratio 6.7) and VTE (odds ratio 4.2) (BRSK01,). Further, in a retrospective study treatment of HAE patients with C1INH resulted in a 10-fold reduction in the incidence of VTE on long-term follow-up compared to untreated patients (164). In a small plasma proteomics-based biomarker discovery study an association between plasma levels of C1INH and future risk of VTE was observed (Table 2) (165). In this study, low plasma levels of C1INH were found to negatively associate with future risk of VTE (165). This finding is consistent with the anticoagulant activity of C1INH and suggests that plasma levels of C1INH modulate VTE risk in the general population. Epidemiological studies have also demonstrated that elevated plasma levels of complement factor C3 and C5 are associated with an increased risk of VTE (166, 167). It is possible that the anti-complement activities of C1INH could contribute to the observed association with VTE. Further work is required to investigate this association and the contributing mechanisms.
It is interesting to note that in addition to possessing anti-complement and anti-coagulant properties, C1INH is also an acute phase reactant with anti-inflammatory properties (168). Indeed, C1INH administration reduced plasma levels of inflammatory cytokines in a clinical endotoxemia model (169). Further, C1INH administration reduced plasma levels of inflammatory cytokines and mortality in patients with sepsis (170).
Mice deficient for C1INH have been generated to model HAE (171). C1INH deficient mice developed normally and were present at the expected frequency (171). As observed in patients with HAE, mice with a heterozygous deficiency for C1INH had markedly lower plasma C1INH levels than the expected 50% (171). Consistent with the clinical phenotype, bradykinin-mediated vascular permeability was increased in C1INH deficient mice upon challenge and could be corrected by C1INH replacement or inhibition of bradykinin receptor signaling (171).
The anticoagulant activity of exogenous C1INH has been evaluated in the preclinical setting. Administration of human purified C1INH to rabbits significantly reduced intrinsic pathway-initiated thrombin generation and prolonged the activated partial thromboplastin time (172). In a rabbit femoral artery ferric chloride thrombosis model administration of human C1INH significantly reduced vessel occlusion (172). This study demonstrates that exogenous C1INH is effective in limiting activation of coagulation and arterial thrombosis. Additional studies are required to evaluate the role of C1INH in other thrombotic pathologies.
C1INH is a major endogenous inhibitor of intrinsic pathway factors PKa, FXIIa, and FXIa. A congenital deficiency in C1INH, causing HAE, has been shown to result in increased plasma levels of markers of activation of coagulation. Epidemiological evidence has indicated that low plasma levels of C1INH are associated with an increased risk of VTE. Preclinical studies indicated that exogenous C1INH is able to effectively suppress intrinsic pathway mediated activation of coagulation and arterial thrombosis.
PN1 is a 50 kDa protein encoded by the SERPINE2 gene and functions as a broad serine protease inhibitor. PN1 is a potent inhibitor of the coagulation proteases thrombin and FXIa, and to a lesser extent FXa (Table 1) (173, 174). PN1 also shows inhibitory activity toward trypsin, urokinase plasminogen activator and plasmin (173). As with other anticoagulant SERPINs the activity of PN1 toward target proteases is significantly enhanced by heparin (173). However, unlike the majority of other anticoagulant SERPINs, PN1 is not expressed by the liver and is not present at detectable levels in plasma (175). Instead, PN1 is expressed in a number of other tissues, including the brain, heart, spleen, kidney and lung (176). PN1 is also present in platelets and monocytes (177). PN1 is stored in platelet alpha granules that may serve as a labile pool of this SERPIN (178). It is likely that release of PN1 from activated platelets accumulating at sites of vascular injury results in high localized levels of this SERPIN. Such a regulated mechanism would be distinct from that of other anticoagulant SERPINs that are present at high levels in plasma.
The SERPINE2 gene was identified as a candidate susceptibility gene for chronic obstructive pulmonary disease (179). The association between SERPINE2 gene polymorphisms and chronic obstructive pulmonary disease was confirmed in a subsequent study (180). Additional studies indicate that SERPINE2 polymorphisms may be associated with a wider spectrum of lung diseases, including asthma and emphysema (181–183). To date, SERPINE2 polymorphisms have not been associated with thrombotic pathologies.
PN1 deficient mice have been developed as a tool to study the biological function of this SERPIN (184). PN1 deficient mice were viable and developed normally with no overt phenotype (184). Subsequent studies demonstrated that mice deficient for PN1 had markedly enhanced thrombus formation in both mesenteric venule and arteriole ferric chloride injury models (178, 185). Platelet PN1 was found to inhibit thrombin activity with platelet-rich plasma from PN1 deficient mice supporting increased thrombin generation (178). Interestingly, platelets from PN1 deficient mice also demonstrated enhanced P-selectin exposure and aggregation in response to thrombin (178). These findings suggest that the platelet is an important source of PN1 and confers anticoagulant activity through the ability to inhibit thrombin (185). Consistent with the clinically observed lung phenotype, PN1 deficient mice have reduced survival in a bleomycin induced lung injury model (186). Loss of PN1 led to increased inflammation and activation of coagulation in the lung (186). Bone marrow chimeras demonstrated that this phenotype was driven by loss of PN1 in hematopoietic cells and could be reversed by inhibition of thrombin or PAR4 activation (186).
The effect of PN1 on hemostatic processes has also been investigated. Plasma from FVIII deficient mice also deficient for PN1 or treated with an anti-PN1 antibody supported enhanced thrombin generation (187). FVIII deficient mice also deficient for PN1 demonstrated markedly reduced bleeding in a tail amputation model compared with controls (187). Further, translating these findings anti-PN1 single domain antibodies have been developed (188). The anti-PN1 antibodies restored thrombin activity in purified and plasma-based systems (188). The procoagulant effect of anti-PN1 antibodies could find utility as a novel hemostatic therapy for patients with hemophilia.
In addition to inhibiting procoagulants PN1 also inhibits fibrinolytic enzymes. Ex vivo, tPA initiated clot lysis was enhanced in plasma from PN1 deficient mice. In vivo, thrombi formed in PN1 deficient mice were found to be more susceptible to tPA induced lysis (189). This suggests that PN1 has important anticoagulant and antifibrinolytic functions. While PN1 may inhibit thrombus formation, thrombi formed in the presence of PN1 may be more resistant to lysis.
PN1 functions as an inhibitor of thrombin, FXa and FXIa. Although PN1 variants are associated with pulmonary pathologies, their effects on thrombotic and hemostatic disorders have not been reported. In preclinical studies, PN1 deficiency was found to enhance thrombus formation in mice. Additional studies indicate that PN1 may be a suitable target for novel hemophilia therapies with inhibition of PN1 found to normalize hemostasis in a mouse model.
PCI is a 46 kDa serine protease inhibitor encoded by the SERPINA5 gene and is present in plasma at a concentration of roughly 5 μg/ml (190). PCI is a potent inhibitor of the anticoagulant aPC conferring PCI with procoagulant activity, unlike the other SERPINs reviewed here (190). However, in addition to aPC, PCI also inhibits several coagulation proteases, including thrombin, FXa, FXIa, and plasma kallikrein providing PCI with anticoagulant activity (Table 1) (191). As with other SERPINs the activity of PCI against coagulation proteases is enhanced by heparin (191). In addition, the activity of PCI against thrombin is markedly enhanced by thrombomodulin (192).
To date no congenital deficiency for PCI has been described. Early studies postulated that PCI deficiency may underpin combined FV and FVIII deficiency as plasma PCI activity was undetectable in patients with this deficiency (193). Mechanistically, an inability to inhibit aPC could lead to a constitutive reduction in FVa and FVIIIa activity. Subsequent studies revealed that individuals with combined FV and FVIII deficiency had normal levels of functional PCI (194). Involvement of PCI in the combined deficiency was more conclusively excluded when the locus for the SERPINA5 gene was mapped on chromosome 14 with the genetic defect associated with FV and FVIII deficiency mapped to chromosome 18 (195, 196). Although there have been no reports on individuals with congenital PCI deficiency, available evidence from the whole exome sequencing database gnomAD indicates that predicted loss of function variants are present at the expected frequency (197).
Studies of PCI in mice are complicated by the fact that the pattern of tissue expression in this species differs compared to humans. Whereas PCI is expressed in the liver in humans expression is largely absent in the liver of mice (198). Given that the liver is a critical organ for release of proteins into the blood the lack of expression in this organ in mice likely explains the observed absence of PCI in mouse plasma (199). These species differences notwithstanding, PCI deficient mice have been generated (199). PCI deficient mice were generated at the expected frequency and did not demonstrate any gross hemostatic defects. However, male PCI deficient mice were found to be infertile (199). PCI was found to be present at very high levels in human seminal fluid suggesting an important role in male fertility (200). Further studies demonstrated impaired spermatogenesis in PCI deficiency mice (201). To study the role of plasma PCI in the mouse a transgenic approach was used in which human PCI was expressed in the mouse liver (198). Transgenic overexpression of PCI resulted in mouse plasma levels of human PCI roughly double that observed normal human plasma (198). As expected, human PCI transgenic mouse plasma was able to effectively inhibit exogenous human aPC (198). However, in an LPS endotoxemia model no difference in survival was observed between human PCI transgenic mice and controls (198). An additional transgenic mouse expressing human PCI under the control of the human promoter resulted in plasma levels of PCI approximately four times that found in human plasma (202). In these mice expression of human PCI resulted in shortened activated partial thromboplastin plasma clotting times (202). Interestingly, in an LPS endotoxemia model these human PCI transgenic mice demonstrated an enhanced prolongation of activated partial thromboplastin times and an enhanced reduction in plasma fibrinogen compared to controls (202). The high plasma levels of PCI in these mice likely inhibited endogenous mouse aPC leading to a consumptive coagulopathy. The findings with human PCI transgenic mice indicate that the predominant effect of PCI is as a procoagulant.
Despite being an inhibitor of multiple coagulation proteases the potent activity of PCI toward aPC confers this SERPIN with net procoagulant activity. No clinical association between PCI and bleeding or thrombosis has been established. Preclinical studies indicate that PCI potently enhances activation of coagulation induced by endotoxemia.
A1AT is a 53 kDA serine protease inhibitor encoded by the SERPINA1 gene. A1AT circulates in plasma at a concentration of approximately 1.2 mg/ml with a half-life of 4–5 days. Although A1AT is primarily expressed in the liver it is also produced in monocytes and macrophages owing to the presence of an alternative promoter (203). A1AT serves as the major endogenous inhibitor of neutrophil elastase (204). A1AT also functions as a potent inhibitor of other neutrophil related proteases, including proteinase 3 and cathepsin G (205, 206). A1AT demonstrates weak inhibitory activity against FXa and FXIa (Table 1) (37, 207). Although GAGs enhance the inhibitory activity of a number of SERPINs in the case of A1AT the presence of heparin has been found to markedly reduce inhibitory activity (37, 208).
Congenital A1AT deficiency is a common genetic disease (209). The vast majority of severe A1AT deficiencies result from homozygosity for a single variant that causes an amino acid substitution (Q342K) frequently referred to as the Z allele (209). The Z allele is particularly prevalent in individuals of European descent (heterozygous, 1:25: homozygous 1:2,000) (209). Variant Z allele protein polymerizes in the endoplasmic reticulum resulting in reduced secretion (210). Another single acid substitution (Q264V) referred to as the S allele is less common and results in more modest A1AT deficiency (211). Despite a preponderance for the Z and S alleles in individuals with A1AT deficiency to date more than 120 variants have been described causing some degree of A1AT deficiency (212). Individuals with A1AT deficiency are at increased risk of emphysema and chronic obstructive pulmonary disease (213, 214). Indeed, 5% of all chronic obstructive pulmonary disease diagnoses may be attributable to A1AT deficiency (215). Individuals with A1AT deficiency were also found to be at increased risk of liver disease (213, 216). Variant Z allele A1AT has been shown to fold incorrectly forming toxic aggregates in hepatocytes that function as a driver of liver disease in this patient population (217). Human purified plasma-derived A1AT augmentation therapies with elimination half-lives of 2–3 days have been developed as effective treatments for patients with A1AT deficiency associated chronic obstructive pulmonary disease (218).
More recently, it was found that individuals with A1AT deficiency are at increased risk of VTE (Table 2). In one study, individuals with severe A1AT deficiency, caused by homozygosity for the Z allele, had a significantly increased risk of VTE compared to controls (hazard ratio 4.2) (219). In a second population-based study individuals homozygous for the Z allele were also found to have a significantly increased risk of VTE (hazard ratio 2.2) (220). Interestingly, severe A1AT deficiency has been associated with a reduced risk of ischemic heart disease (221). The mechanism by which loss of endogenous A1AT results in an increased risk of VTE but reduced risk of ischemic heart disease remains to be determined.
A naturally occurring A1AT variant, M358R, termed A1AT-Pittsburgh was identified as the underlying genetic cause of recurrent bleeding events in a young patient (222). Subsequent studies revealed that the M358R single amino acid substitution transformed A1AT from an inhibitor of neutrophil proteases to a broad inhibitor of coagulation associated proteases, including thrombin, aPC, plasmin, FXIa, FXa, plasma kallikrein, and FXIIa (223–225). The A1AT-Pittsburgh variant is very rare with only 2 pedigrees reported to date and is associated with a variable bleeding phenotype (222, 226).
A1AT is an acute phase reactant reported to have important anti-inflammatory and immunomodulatory functions. A1AT deficiency is associated with the inflammatory condition rheumatoid arthritis (227, 228). The anti-inflammatory and anti-viral properties of A1AT products are currently being evaluated in the setting of COVID-19 (229).
A1AT deficiency has proven challenging to model in mice owing to the presence of fives SERPINA1 paralogs. Early efforts deleting a single SERPINA1 paralog resulted in unexpected embryonic lethality (230, 231). This is not consistent with the observed phenotype of humans with a severe A1AT deficiency and suggested that murine A1AT may have gained additional developmental functions in the mouse. More recently, however, a complete A1AT deficient mouse has been developed lacking all five of the murine SERPINA1 paralogs (232). These A1AT deficient mice demonstrated normal survival (232). Consistent with the reported role of A1AT deficiency in lung disease, aged deficient mice demonstrated spontaneous emphysema with evidence of increased neutrophil, monocyte and lymphocyte counts in bronchial alveolar lavage fluid (232). Further studies are required to determine if these deficient mice can model the prothrombotic phenotype observed in humans (232).
The primary activity of A1AT is as a potent inhibitor of neutrophil-derived proteases. A1AT possesses relatively weak anticoagulant activity. Despite this weak anticoagulant activity recent clinical studies have revealed that patients with A1AT deficiency are at increased risk of VTE. The mechanism by which endogenous A1AT prevents VTE remains to be elucidated.
Generation of SERPIN variant proteins was initially used as a powerful tool to decipher what components of a given SERPIN’s structure contributed to the observed biological activity. For example, generation of variants has facilitated the identification of GAG binding regions and provided mechanistic insights into how these molecules enhance SERPIN activity. However, based on the important contribution of anticoagulant SERPINs to hemostatic and thrombotic processes considerable efforts have also been made to engineer variants with altered biological activities and properties (19, 233, 234). Engineered variant SERPIN proteins are typically designed to either alter substrate selectivity or improve biological stability. The overarching goal of these efforts is to generate novel engineered SERPINs that possess attractive biochemical and biophysical properties for potential use as novel therapeutic agents.
Significant efforts have been made to alter and refine the selectivity of a number of anticoagulant SERPINs. This has primarily been achieved by substitution of residues in the RCL that serves as an important determinant of selectivity. Initial efforts focused on generating chimeric SERPINs that substituted partial RCL sequence of one SERPIN into another. Using this approach chimeric SERPINs have been made that swap portions of the RCL of AT into A1AT and plasminogen activator inhibitor 1 (235–237). While in both cases insertion of the AT RCL conferred thrombin inhibitory capacity the second order rate constants were markedly lower than that of wildtype AT. This reinforces that although the RCL sequence is an important determinant of substrate selectivity other regions play a role.
Further efforts have focused on single or multiple amino acid substitutions of the RCL in a given SERPIN. A1AT has been the focus of concerted efforts likely owing to the presence of the naturally occurring A1AT-Pittsburgh variant, M358R, that demonstrated the profound effects that a single amino acid substitution could have on substrate selectivity (223–225). An additional amino acid substitution at the preceding P2 residue (357AR358) further modified the activity of the A1AT-Pittsburgh variant conferring increased activity toward plasma kallikrein (238). This substitution was found to protect mice from bradykinin induced hypertension (238). Greater selectivity of A1AT toward plasma kallikrein was achieved by substituting the P3 and P2 residues in the A1AT-Pittsburgh variant (356PFR358) (239). Two additional A1AT-Pittsburgh based variants have been developed (355SLLRV359 and 355SMTRV359) that demonstrate improved activity against plasma kallikrein and reduced activity against both thrombin and aPC (240). These variants were effective in reducing bradykinin induced edema and arterial thrombosis in mouse models (240). To improve the selectivity of the A1AT-Pittsburgh variant for thrombin over aPC additional A1AT-Pittsburgh based variants have been evaluated (357AR358, 357GR358, and 358RT359) but no marked difference in selectivity was observed (237).
A1AT variant design has also been informed by the crystal structure of target proteases. In one approach the A1AT variant 357KRK359 was designed to selectively inhibit aPC over thrombin due to the steric constraints of the thrombin active site (241). As predicted, the A1AT variant 357KRK359 was highly selective for aPC and significantly enhanced thrombin generation in the plasma of hemophilia A patients (241). In FVIII deficient mice the A1AT variant 357KRK359 was also found to reduce bleeding in a tail amputation model and increase platelet and fibrin accumulation at sites of vascular injury (241).
The effect of RCL amino acid substitutions in other SERPINs has also been evaluated. Systematic substitution of the P1 residue of C1INH demonstrated the importance of this amino acid to inhibitor activity and selectivity (242). The majority of C1INH P1 variants demonstrated reduced or absent activity toward C1s, plasma kallikrein, FXIIa and plasmin (242). However, one variant, R442K, demonstrated preserved activity toward C1s with reduced activity toward plasma kallikrein and FXIIa (242). A naturally occurring C1INH P2 variant, A443V, has been identified and was found to have increased specificity toward plasma kallikrein and FXIIa (243). In a systematic substitution approach for the P2 residue of C1INH the naturally occurring A443V variant was found to have increased activity toward trypsin and thrombin (244). Similarly, the P1 variant R444L was found to have increased heparin dependent activity toward thrombin (245). One additional variant of particular interest, A443T, showed markedly enhanced specificity for plasma kallikrein and FXIIa over C1s, thrombin and plasmin (244).
Larger scale systematic approaches have also been evaluated. A phage display approach has been used to identify A1AT RCL sequences with enhanced specificity for FXIa (246). In a multistage approach, fragments of the A1AT-Pittsburgh RCL sequence were mutated and screened for the ability to inhibit FXIa in a process termed biopanning. A1AT containing the FXIa optimized RCL (346HASTGQFLEAIPR358) demonstrated strong selectivity for FXIa over thrombin (246). In another approach the P4 to P4’ residues of A1AT, covering a region particularly important for substrate selectivity, were systematically substituted (247). Recombinant variant A1AT proteins containing single amino acid substitutions for every amino acid at each of these positions were generated and their activity toward target proteases evaluated (247). A platform was developed to predict the effect of multiple amino acid substitutions on substrate specificity (247). Using this platform variants with predicted potency and specificity for aPC were selected for evaluation as novel hemophilia therapies (247). A number of these predicted A1AT variants were found to be selective inhibitors of aPC. One variant, 355KMPRRIPA362, restored hemostasis more effectively in FVIII deficient mice than the previously reported A1AT 357KRK359 (247).
Numerous efforts have been made to improve the circulating half-life of anticoagulant SERPINS. This is due to the fact that some natural SERPINs and other recombinantly produced SERPINs have relatively short circulating half-lives.
One approach to improve the half-life of circulating SERPINs is to modify protein glycosylation in a process termed glycoengineering (248). N-linked glycosylation of natural A1AT aids in correct folding of the protein into the metastable state and extends the circulating half-life (249, 250). While purified A1AT contains normally glycosylated protein, A1AT produced recombinantly often contains sub optimally glycosylated protein. In one approach the effect of adding additional glycosylation sites to A1AT on the circulating half-life was assessed. A recombinant A1AT variant containing one additional N-linked glycosylation site (G147N/K149T) had a significantly longer circulating half-life in mice (251). Similar findings were made with a further recombinant A1AT variant containing two additional N-linked glycosylation sites (Q9N and D12N/S14T) that resulted in a significantly prolonged circulating half-life in rats (252). As an alternative approach, efforts have also been undertaken to improve glycosylation of natural sites in recombinantly produced A1AT. An optimized Chinese ovarian hamster cell line that overexpresses a human glycosylation gene produced recombinant A1AT with a glycosylation profile similar to that of purified A1AT (253). However, it remains to be determined to what extent this improves the circulating half-life of recombinant A1AT.
It should be noted that SERPIN glycosylation has effects beyond simply prolonging the circulating half-life. Wildtype AT contains four sites for N-linked glycosylation with one site demonstrating poor glycosylation. Addition of N-linked glycosylation sites has had variable effects on secretion with modifications at some positions impairing secretion while at other sites modification improved secretion (254). These differing results are likely a result of the effect of N-linked glycosylation on protein folding. Indeed, N-linked glycosylation has been shown to contribute to the efficient folding and secretion of AT (255). Additional N-linked glycosylation could also alter SERPIN activity. The potential for this to occur has been highlighted by the varying activities of the two naturally occurring forms of AT, the α and β forms. The α form of AT, that is glycosylated at all four sites, demonstrates impaired activity against thrombin compared to the β form, that is glycosylated at three sites, with the additional glycosylation being found to disrupt heparin interactions (256). This indicates that although N-linked glycosylation may improve biological half-life this modification may have unpredictable, and possibly deleterious, effects on protein secretion and function.
In an additional approach the effect of conjugating A1AT with polyethylene glycol (PEG), in a process termed PEGylation, has been evaluated. PEGylation of recombinant A1AT markedly reduced renal clearance resulting in a prolonged circulating half-life (257). PEGylation of a single amino acid residue of recombinant A1AT (C232) increased the circulating half-life in mice with longer PEG polymers having a greater effect (258). A PEGylated version of the Myxomavirus derived SERPIN Serp-1 has also been developed demonstrating improved efficacy in a mouse model of diffuse alveolar hemorrhage (259). Serp-1 is a broad acting SERPIN with anti-inflammatory and anti-fibrinolytic activities (260). In addition, however, Serp-1 also functions as a heparin dependent inhibitor of thrombin (261, 262). Serp-1 is currently being evaluated as a novel therapy for treatment of acute coronary syndrome (263). It is interesting to consider if the thrombin inhibitory activity of Serp-1 may contribute to the therapeutic effect of this SERPIN.
The generation of A1AT fusion proteins has also been explored. A1AT has been conjugated to the Fc portion of immunoglobulin (264). Conjugation of intact A1AT with the Fc portion of immunoglobulin resulted in a fusion protein that retained inhibitory activity. Although not directly determined it was inferred that this fusion protein should have a prolonged circulating half-life through interaction with Fc receptors present on immune cells (264). Importantly, A1AT-Fc was found to be more effective in preserving lung function in murine emphysema models than purified A1AT (265).
Anticoagulant SERPINs serve as critical negative regulators of coagulation. Consistent with their essential anticoagulant function congenital deficiencies of specific SERPINs, such as AT and HCII, are associated with an increased risk of VTE in humans. Moreover, targeting of these anticoagulants is being explored as a novel approach to reduce bleeding in hemophilia patients. Concerted efforts have been made to develop novel therapeutic SERPINs with altered selectivity and specificity for target coagulation proteases. Such therapeutic SERPINs may find utility as novel therapies for thrombotic pathologies.
SG conducted the literature search and wrote the manuscript. NM critically reviewed and edited the manuscript. Both authors contributed to the article and approved the submitted version.
This work was supported by awards from the National Heart Blood and Lung Institute of the National Institutes of Health for SG (T32HL007149) and NM (R35HL155657). Additional support was provided by a CSL Behring Prof Heimburger Award to SG.
SG has been a consultant for CSL Behring and has received research support from CSL Behring.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Frank C. Church (UNC Chapel Hill) for helpful comments.
1. Grover SP, Mackman N. Tissue factor: an essential mediator of hemostasis and trigger of thrombosis. Arterioscler Thromb Vasc Biol. (2018) 38:709–25. doi: 10.1161/ATVBAHA.117.309846
2. Grover SP, Mackman N. Intrinsic pathway of coagulation and thrombosis. Arterioscler Thromb Vasc Biol. (2019) 39:331–8. doi: 10.1161/ATVBAHA.118.312130
3. Gailani D, Renne T. The intrinsic pathway of coagulation: a target for treating thromboembolic disease? J Thromb Haemost. (2007) 5:1106–12. doi: 10.1111/j.1538-7836.2007.02446.x
4. Josso F, Prou-Wartelle O. Interaction of tissue factor and factor VII at the earliest phase of coagulation. Thromb Diath Haemorrh Suppl. (1965) 17:35–44.
5. Kamikubo Y, Mendolicchio GL, Zampolli A, Marchese P, Rothmeier AS, Orje JN, et al. Selective factor VIII activation by the tissue factor-factor VIIa-factor Xa complex. Blood. (2017) 130:1661–70. doi: 10.1182/blood-2017-02-767079
6. Gailani D, Broze GJ Jr. Factor Xi activation in a revised model of blood coagulation. Science. (1991) 253:909–12. doi: 10.1126/science.1652157
7. Grover SP, Schmedes CM, Auriemma AC, Butler E, Parrish ML, Miszta A, et al. Differential roles of factors IX and XI in murine placenta and hemostasis under conditions of low tissue factor. Blood Adv. (2020) 4:207–16. doi: 10.1182/bloodadvances.2019000921
8. Bolton-Maggs PH, Pasi KJ. Haemophilias a and B. Lancet. (2003) 361:1801–9. doi: 10.1016/S0140-6736(03)13405-8
9. Mackman N. New insights into the mechanisms of venous thrombosis. J Clin Invest. (2012) 122:2331–6. doi: 10.1172/JCI60229
10. Esmon CT. Basic mechanisms and pathogenesis of venous thrombosis. Blood Rev. (2009) 23:225–9. doi: 10.1016/j.blre.2009.07.002
11. Rau JC, Beaulieu LM, Huntington JA, Church FC. Serpins in thrombosis, hemostasis and fibrinolysis. J Thromb Haemost. (2007) 5(Suppl. 1):102–15. doi: 10.1111/j.1538-7836.2007.02516.x
12. Mast AE. Tissue factor pathway inhibitor: multiple anticoagulant activities for a single protein. Arterioscler Thromb Vasc Biol. (2016) 36:9–14. doi: 10.1161/ATVBAHA.115.305996
13. Griffin JH, Zlokovic BV, Mosnier LO. Protein C anticoagulant and cytoprotective pathways. Int J Hematol. (2012) 95:333–45. doi: 10.1007/s12185-012-1059-0
14. Lomas DA, Carrell RW. Serpinopathies and the conformational dementias. Nat Rev Genet. (2002) 3:759–68. doi: 10.1038/nrg907
15. Gooptu B, Lomas DA. Polymers and inflammation: disease mechanisms of the serpinopathies. J Exp Med. (2008) 205:1529–34. doi: 10.1084/jem.20072080
16. Law RH, Zhang Q, McGowan S, Buckle AM, Silverman GA, Wong W, et al. An overview of the serpin superfamily. Genome Biol. (2006) 7:216. doi: 10.1186/gb-2006-7-5-216
17. Silverman GA, Bird PI, Carrell RW, Church FC, Coughlin PB, Gettins PG, et al. The serpins are an expanding superfamily of structurally similar but functionally diverse proteins. evolution, mechanism of inhibition, novel functions, and a revised nomenclature. J Biol Chem. (2001) 276:33293–6. doi: 10.1074/jbc.R100016200
18. Hunt LT, Dayhoff MO. A surprising new protein superfamily containing ovalbumin, antithrombin-iii, and alpha 1-proteinase inhibitor. Biochem Biophys Res Commun. (1980) 95:864–71. doi: 10.1016/0006-291x(80)90867-0
19. Sanrattana W, Maas C, de Maat S. Serpins-from trap to treatment. Front Med (Lausanne). (2019) 6:25. doi: 10.3389/fmed.2019.00025
20. Huntington JA. Serpin structure, function and dysfunction. J Thromb Haemost. (2011) 9(Suppl. 1):26–34. doi: 10.1111/j.1538-7836.2011.04360.x
21. Gettins PG. Serpin structure, mechanism, and function. Chem Rev. (2002) 102:4751–804. doi: 10.1021/cr010170
22. Schechter I, Berger A. On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun. (1967) 27:157–62. doi: 10.1016/s0006-291x(67)80055-x
23. Stavridi ES, O’Malley K, Lukacs CM, Moore WT, Lambris JD, Christianson DW, et al. Structural change in alpha-chymotrypsin induced by complexation with alpha 1-antichymotrypsin as seen by enhanced sensitivity to proteolysis. Biochemistry. (1996) 35:10608–15. doi: 10.1021/bi9605806
24. Zhou A, Huntington JA, Carrell RW. Formation of the antithrombin heterodimer in vivo and the onset of thrombosis. Blood. (1999) 94:3388–96. doi: 10.1182/blood.v94.10.3388.422k20_3388_3396
25. Huntington JA. Mechanisms of glycosaminoglycan activation of the serpins in hemostasis. J Thromb Haemost. (2003) 1:1535–49. doi: 10.1046/j.1538-7836.2003.00305.x
26. Hirsh J, Anand SS, Halperin JL, Fuster V, American Heart A. Guide to anticoagulant therapy: heparin : a statement for healthcare professionals from the American heart association. Circulation. (2001) 103:2994–3018. doi: 10.1161/01.cir.103.24.2994
27. Olson ST, Bjork I, Sheffer R, Craig PA, Shore JD, Choay J. Role of the antithrombin-binding pentasaccharide in heparin acceleration of antithrombin-proteinase reactions. Resolution of the antithrombin conformational change contribution to heparin rate enhancement. J Biol Chem. (1992) 267:12528–38. doi: 10.1016/s0021-9258(18)42309-5
28. Chuang YJ, Swanson R, Raja SM, Olson ST. Heparin enhances the specificity of antithrombin for thrombin and factor Xa independent of the reactive center loop sequence. Evidence for an exosite determinant of factor Xa specificity in heparin-activated antithrombin. J Biol Chem. (2001) 276:14961–71. doi: 10.1074/jbc.M011550200
29. Hortin GL, Trimpe BL. Allosteric changes in thrombin’s activity produced by peptides corresponding to segments of natural inhibitors and substrates. J Biol Chem. (1991) 266:6866–71. doi: 10.1016/s0021-9258(20)89581-7
30. Van Deerlin VM, Tollefsen DM. The N-terminal acidic domain of heparin cofactor II mediates the inhibition of alpha-thrombin in the presence of glycosaminoglycans. J Biol Chem. (1991) 266:20223–31. doi: 10.1016/s0021-9258(18)54913-9
31. Baglin TP, Carrell RW, Church FC, Esmon CT, Huntington JA. Crystal structures of native and thrombin-complexed heparin cofactor Ii reveal a multistep allosteric mechanism. Proc Natl Acad Sci USA. (2002) 99:11079–84. doi: 10.1073/pnas.162232399
32. Izaguirre G, Aguila S, Qi L, Swanson R, Roth R, Rezaie AR, et al. Conformational activation of antithrombin by heparin involves an altered exosite interaction with protease. J Biol Chem. (2014) 289:34049–64. doi: 10.1074/jbc.M114.611707
33. Carrell RW, Stein PE, Fermi G, Wardell MR. Biological implications of a 3 a structure of dimeric antithrombin. Structure. (1994) 2:257–70. doi: 10.1016/s0969-2126(00)00028-9
34. Schreuder HA, de Boer B, Dijkema R, Mulders J, Theunissen HJ, Grootenhuis PD, et al. The intact and cleaved human antithrombin III complex as a model for serpin-proteinase interactions. Nat Struct Biol. (1994) 1:48–54. doi: 10.1038/nsb0194-48
35. Pike RN, Buckle AM, le Bonniec BF, Church FC. Control of the coagulation system by serpins. Getting by with a little help from glycosaminoglycans. FEBS J. (2005) 272:4842–51. doi: 10.1111/j.1742-4658.2005.04880.x
37. Wuillemin WA, Eldering E, Citarella F, de Ruig CP, ten Cate H, Hack CE. Modulation of contact system proteases by glycosaminoglycans. Selective enhancement of the inhibition of factor Xia. J Biol Chem. (1996) 271:12913–8. doi: 10.1074/jbc.271.22.12913
38. Sanchez J, Elgue G, Riesenfeld J, Olsson P. Studies of adsorption, activation, and inhibition of factor XII on immobilized heparin. Thromb Res. (1998) 89:41–50. doi: 10.1016/s0049-3848(97)00310-1
39. Burrowes CE, Habal FM, Movat HZ. The inhibition of human plasma kallikrein by antithrombin Iii. Thromb Res. (1975) 7:175–83. doi: 10.1016/0049-3848(75)90134-6
40. Vennerod AM, Laake K. Inhibition of purified plasma kallikrein by antithrombin iii and heparin. Thromb Res. (1975) 7:223–6. doi: 10.1016/0049-3848(75)90138-3
41. Rezaie AR. Calcium enhances heparin catalysis of the antithrombin-factor Xa reaction by a template mechanism. evidence that calcium alleviates Gla domain antagonism of heparin binding to factor Xa. J Biol Chem. (1998) 273:16824–7. doi: 10.1074/jbc.273.27.16824
42. Jin L, Abrahams JP, Skinner R, Petitou M, Pike RN, Carrell RW. The anticoagulant activation of antithrombin by heparin. Proc Natl Acad Sci USA. (1997) 94:14683–8. doi: 10.1073/pnas.94.26.14683
43. Jordan RE, Oosta GM, Gardner WT, Rosenberg RD. The kinetics of hemostatic enzyme-antithrombin interactions in the presence of low molecular weight heparin. J Biol Chem. (1980) 255:10081–90. doi: 10.1016/s0021-9258(19)70431-1
44. Griffith MJ. Kinetics of the heparin-enhanced antithrombin III/thrombin reaction. Evidence for a template model for the mechanism of action of heparin. J Biol Chem. (1982) 257:7360–5. doi: 10.1016/s0021-9258(18)34385-0
45. Ofosu FA, Modi GJ, Smith LM, Cerskus AL, Hirsh J, Blajchman MA. Heparan sulfate and dermatan sulfate inhibit the generation of thrombin activity in plasma by complementary pathways. Blood. (1984) 64:742–7. doi: 10.1182/blood.v64.3.742.bloodjournal643742
46. Lindahl U, Backstrom G, Thunberg L, Leder IG. Evidence for a 3-O-sulfated D-glucosamine residue in the antithrombin-binding sequence of heparin. Proc Natl Acad Sci USA. (1980) 77:6551–5. doi: 10.1073/pnas.77.11.6551
47. Atha DH, Lormeau JC, Petitou M, Rosenberg RD, Choay J. Contribution of 3-O- and 6-O-sulfated glucosamine residues in the heparin-induced conformational change in antithrombin III. Biochemistry. (1987) 26:6454–61. doi: 10.1021/bi00394a024
48. Sobczak AIS, Pitt SJ, Stewart AJ. Glycosaminoglycan neutralization in coagulation control. Arterioscler Thromb Vasc Biol. (2018) 38:1258–70. doi: 10.1161/ATVBAHA.118.311102
49. Li W, Johnson DJ, Esmon CT, Huntington JA. Structure of the antithrombin-thrombin-heparin ternary complex reveals the antithrombotic mechanism of heparin. Nat Struct Mol Biol. (2004) 11:857–62. doi: 10.1038/nsmb811
50. Johnson DJ, Langdown J, Li W, Luis SA, Baglin TP, Huntington JA. Crystal structure of monomeric native antithrombin reveals a novel reactive center loop conformation. J Biol Chem. (2006) 281:35478–86. doi: 10.1074/jbc.M607204200
51. Bruce D, Perry DJ, Borg JY, Carrell RW, Wardell MR. Thromboembolic disease due to thermolabile conformational changes of antithrombin Rouen-VI (187 Asn–>Asp). J Clin Invest. (1994) 94:2265–74. doi: 10.1172/JCI117589
52. Beauchamp NJ, Pike RN, Daly M, Butler L, Makris M, Dafforn TR, et al. Antithrombins wibble and wobble (T85m/K): archetypal conformational diseases with in vivo latent-transition, thrombosis, and heparin activation. Blood. (1998) 92:2696–706. doi: 10.1182/blood.v92.8.2696
53. Rosenberg RD. Actions and interactions of antithrombin and heparin. N Engl J Med. (1975) 292:146–51. doi: 10.1056/NEJM197501162920307
54. Tait RC, Walker ID, Davidson JF, Islam SI, Mitchell R. Antithrombin III activity in healthy blood donors: age and sex related changes and prevalence of asymptomatic deficiency. Br J Haematol. (1990) 75:141–2. doi: 10.1111/j.1365-2141.1990.tb02635.x
55. Luxembourg B, Delev D, Geisen C, Spannagl M, Krause M, Miesbach W, et al. Molecular basis of antithrombin deficiency. Thromb Haemost. (2011) 105:635–46. doi: 10.1160/TH10-08-0538
56. Patnaik MM, Moll S. Inherited antithrombin deficiency: a review. Haemophilia. (2008) 14:1229–39. doi: 10.1111/j.1365-2516.2008.01830.x
57. Bhakuni T, Sharma A, Rashid Q, Kapil C, Saxena R, Mahapatra M, et al. Antithrombin III deficiency in Indian patients with deep vein thrombosis: identification of first India based at variants including a novel point mutation (T280a) that leads to aggregation. PLoS One. (2015) 10:e0121889. doi: 10.1371/journal.pone.0121889
58. Finazzi G, Caccia R, Barbui T. Different prevalence of thromboembolism in the subtypes of congenital antithrombin III deficiency: review of 404 cases. Thromb Haemost. (1987) 58:1094. doi: 10.1055/s-0038-1646063
59. Rosendaal FR. Risk factors for venous thrombotic disease. Thromb Haemost. (1999) 82:610–9. doi: 10.1055/s-0037-1615887
60. Rossi E, Za T, Ciminello A, Leone G, De Stefano V. The risk of symptomatic pulmonary embolism due to proximal deep venous thrombosis differs in patients with different types of inherited thrombophilia. Thromb Haemost. (2008) 99:1030–4. doi: 10.1160/TH08-02-0069
61. Martinelli I, Mannucci PM, De Stefano V, Taioli E, Rossi V, Crosti F, et al. Different risks of thrombosis in four coagulation defects associated with inherited thrombophilia: a study of 150 families. Blood. (1998) 92:2353–8. doi: 10.1182/blood.v92.7.2353.2353_2353_2358
62. Bucciarelli P, Rosendaal FR, Tripodi A, Mannucci PM, De Stefano V, Palareti G, et al. Risk of venous thromboembolism and clinical manifestations in carriers of antithrombin, protein C, protein S deficiency, or activated protein c resistance: a multicenter collaborative family Study. Arterioscler Thromb Vasc Biol. (1999) 19:1026–33. doi: 10.1161/01.atv.19.4.1026
63. De Stefano V, Simioni P, Rossi E, Tormene D, Za T, Pagnan A, et al. The risk of recurrent venous thromboembolism in patients with inherited deficiency of natural anticoagulants antithrombin, protein C and protein S. Haematologica. (2006) 91:695–8.
64. Di Minno MN, Dentali F, Lupoli R, Ageno W. Mild antithrombin deficiency and risk of recurrent venous thromboembolism: a prospective cohort study. Circulation. (2014) 129:497–503. doi: 10.1161/CIRCULATIONAHA.113.003756
65. Sokol J, Timp JF, le Cessie S, van Hylckama-Vlieg A, Rosendaal FR, Kubisz P, et al. Mild antithrombin deficiency and risk of recurrent venous thromboembolism: results from the mega follow-up study. J Thromb Haemost. (2018) 16:680–8. doi: 10.1111/jth.13960
66. Vossen CY, Rosendaal FR, Group ES. Risk of arterial thrombosis in carriers of familial thrombophilia. J Thromb Haemost. (2006) 4:916–8. doi: 10.1111/j.1538-7836.2006.01838.x
67. Mahmoodi BK, Veeger NJ, Middeldorp S, Lijfering WM, Brouwer JL, Ten Berg J, et al. Interaction of hereditary thrombophilia and traditional cardiovascular risk factors on the risk of arterial thromboembolism: pooled analysis of four family cohort studies. Circ Cardiovasc Genet. (2016) 9:79–85. doi: 10.1161/CIRCGENETICS.115.001211
68. Croles FN, Van Loon JE, Dippel DWJ, De Maat MPM, Leebeek FWG. Antithrombin levels are associated with the risk of first and recurrent arterial thromboembolism at a young age. Atherosclerosis. (2018) 269:144–50. doi: 10.1016/j.atherosclerosis.2018.01.014
69. Menache D, Grossman BJ, Jackson CM. Antithrombin III: physiology, deficiency, and replacement therapy. Transfusion. (1992) 32:580–8. doi: 10.1046/j.1537-2995.1992.32692367206.x
70. Levy JH, Sniecinski RM, Welsby IJ, Levi M. Antithrombin: anti-inflammatory properties and clinical applications. Thromb Haemost. (2016) 115:712–28. doi: 10.1160/TH15-08-0687
71. Wildhagen KC, Wiewel MA, Schultz MJ, Horn J, Schrijver R, Reutelingsperger CP, et al. Extracellular histone H3 levels are inversely correlated with antithrombin levels and platelet counts and are associated with mortality in sepsis patients. Thromb Res. (2015) 136:542–7. doi: 10.1016/j.thromres.2015.06.035
72. Inthorn D, Hoffmann JN, Hartl WH, Muhlbayer D, Jochum M. Effect of antithrombin III supplementation on inflammatory response in patients with severe sepsis. Shock. (1998) 10:90–6. doi: 10.1097/00024382-199808000-00002
73. Ishiguro K, Kojima T, Kadomatsu K, Nakayama Y, Takagi A, Suzuki M, et al. Complete antithrombin deficiency in mice results in embryonic lethality. J Clin Invest. (2000) 106:873–8. doi: 10.1172/JCI10489
74. Dewerchin M, Herault JP, Wallays G, Petitou M, Schaeffer P, Millet L, et al. Life-threatening thrombosis in mice with targeted Arg48-to-Cys mutation of the heparin-binding domain of antithrombin. Circ Res. (2003) 93:1120–6. doi: 10.1161/01.RES.0000103634.69868.4F
75. Safdar H, Cheung KL, Salvatori D, Versteeg HH, Laghmani el H, Wagenaar GT, et al. Acute and severe coagulopathy in adult mice following silencing of hepatic antithrombin and protein C production. Blood. (2013) 121:4413–6. doi: 10.1182/blood-2012-11-465674
76. Hayashi M, Matsushita T, Mackman N, Ito M, Adachi T, Katsumi A, et al. Fatal thrombosis of antithrombin-deficient mice is rescued differently in the heart and liver by intercrossing with low tissue factor mice. J Thromb Haemost. (2006) 4:177–85. doi: 10.1111/j.1538-7836.2005.01679.x
77. Liu Y, Kretz CA, Maeder ML, Richter CE, Tsao P, Vo AH, et al. Targeted mutagenesis of zebrafish antithrombin iii triggers disseminated intravascular coagulation and thrombosis, revealing insight into function. Blood. (2014) 124:142–50. doi: 10.1182/blood-2014-03-561027
78. Ragni MV. Targeting antithrombin to treat hemophilia. N Engl J Med. (2015) 373:389–91. doi: 10.1056/NEJMcibr1505657
79. Bolliger D, Szlam F, Suzuki N, Matsushita T, Tanaka KA. Heterozygous antithrombin deficiency improves in vivo haemostasis in factor VIII-deficient mice. Thromb Haemost. (2010) 103:1233–8. doi: 10.1160/TH09-10-0732
80. Sehgal A, Barros S, Ivanciu L, Cooley B, Qin J, Racie T, et al. An RNAi therapeutic targeting antithrombin to rebalance the coagulation system and promote hemostasis in hemophilia. Nat Med. (2015) 21:492–7. doi: 10.1038/nm.3847
81. Barbon E, Ayme G, Mohamadi A, Ottavi JF, Kawecki C, Casari C, et al. Single-domain antibodies targeting antithrombin reduce bleeding in hemophilic mice with or without inhibitors. EMBO Mol Med. (2020) 12:e11298. doi: 10.15252/emmm.201911298
82. Pasi KJ, Rangarajan S, Georgiev P, Mant T, Creagh MD, Lissitchkov T, et al. Targeting of antithrombin in hemophilia a or B with RNAi therapy. N Engl J Med. (2017) 377:819–28. doi: 10.1056/NEJMoa1616569
83. Tollefsen DM, Majerus DW, Blank MK. Heparin cofactor II. Purification and properties of a heparin-dependent inhibitor of thrombin in human plasma. J Biol Chem. (1982) 257:2162–9. doi: 10.1016/s0021-9258(18)34900-7
84. Sie P, Dupouy D, Pichon J, Boneu B. Turnover study of heparin cofactor II in healthy man. Thromb Haemost. (1985) 54:635–8. doi: 10.1055/s-0038-1660087
85. Liaw PC, Austin RC, Fredenburgh JC, Stafford AR, Weitz JI. Comparison of heparin- and dermatan sulfate-mediated catalysis of thrombin inactivation by heparin cofactor II. J Biol Chem. (1999) 274:27597–604. doi: 10.1074/jbc.274.39.27597
86. Whinna HC, Choi HU, Rosenberg LC, Church FC. Interaction of heparin cofactor II with biglycan and decorin. J Biol Chem. (1993) 268:3920–4. doi: 10.1016/s0021-9258(18)53560-2
87. Shirk RA, Parthasarathy N, San Antonio JD, Church FC, Wagner WD. Altered dermatan sulfate structure and reduced heparin cofactor II-stimulating activity of biglycan and decorin from human atherosclerotic plaque. J Biol Chem. (2000) 275:18085–92. doi: 10.1074/jbc.M001659200
88. Tran TH, Marbet GA, Duckert F. Association of hereditary heparin co-factor II deficiency with thrombosis. Lancet. (1985) 2:413–4. doi: 10.1016/s0140-6736(85)92736-9
89. Sie P, Dupouy D, Pichon J, Boneu B. Constitutional heparin co-factor II deficiency associated with recurrent thrombosis. Lancet. (1985) 2:414–6. doi: 10.1016/s0140-6736(85)92737-0
90. Matsuo T, Kario K, Sakamoto S, Yamada T, Miki T, Hirase T, et al. Hereditary heparin cofactor II deficiency and coronary artery disease. Thromb Res. (1992) 65:495–505. doi: 10.1016/0049-3848(92)90201-k
91. Villa P, Aznar J, Vaya A, Espana F, Ferrando F, Mira Y, et al. Hereditary homozygous heparin cofactor II deficiency and the risk of developing venous thrombosis. Thromb Haemost. (1999) 82:1011–4. doi: 10.1055/s-0037-1614320
92. Simioni P, Lazzaro AR, Coser E, Salmistraro G, Girolami A. Hereditary heparin cofactor II deficiency and thrombosis: report of six patients belonging to two separate kindreds. Blood Coagul Fibrinolysis. (1990) 1:351–6. doi: 10.1097/00001721-199010000-00001
93. Bernardi F, Legnani C, Micheletti F, Lunghi B, Ferraresi P, Palareti G, et al. A heparin cofactor II mutation (HCII Rimini) combined with factor V Leiden or type I protein C deficiency in two unrelated thrombophilic subjects. Thromb Haemost. (1996) 76:505–9. doi: 10.1055/s-0038-1650612
94. Bertina RM, van der Linden IK, Engesser L, Muller HP, Brommer EJ. Hereditary heparin cofactor II deficiency and the risk of development of thrombosis. Thromb Haemost. (1987) 57:196–200. doi: 10.1055/s-0038-1651093
95. Mateo J, Oliver A, Borrell M, Sala N, Fontcuberta J. Increased risk of venous thrombosis in carriers of natural anticoagulant deficiencies. Results of the family studies of the Spanish multicenter study on thrombophilia (Emet study). Blood Coagul Fibrinolysis. (1998) 9:71–8. doi: 10.1097/00001721-199801000-00009
96. Kanagawa Y, Shigekiyo T, Aihara K, Akaike M, Azuma H, Matsumoto T. Molecular mechanism of type I congenital heparin cofactor (HC) II deficiency caused by a missense mutation at reactive P2 site: HC II tokushima. Thromb Haemost. (2001) 85:101–7. doi: 10.1055/s-0037-1612911
97. Tollefsen DM. Does heparin cofactor II modulate atherosclerosis and restenosis? Circulation. (2004) 109:2682–4. doi: 10.1161/01.CIR.0000130436.14464.FC
98. Aihara K, Azuma H, Takamori N, Kanagawa Y, Akaike M, Fujimura M, et al. Heparin cofactor II is a novel protective factor against carotid atherosclerosis in elderly individuals. Circulation. (2004) 109:2761–5. doi: 10.1161/01.CIR.0000129968.46095.F3
99. Takamori N, Azuma H, Kato M, Hashizume S, Aihara K, Akaike M, et al. High plasma heparin cofactor II activity is associated with reduced incidence of in-stent restenosis after percutaneous coronary intervention. Circulation. (2004) 109:481–6. doi: 10.1161/01.CIR.0000109695.39671.37
100. Schillinger M, Exner M, Sabeti S, Mlekusch W, Amighi J, Handler S, et al. High plasma heparin cofactor II activity protects from restenosis after femoropopliteal stenting. Thromb Haemost. (2004) 92:1108–13. doi: 10.1160/TH04-05-0311
101. Tollefsen DM. Heparin cofactor II modulates the response to vascular injury. Arterioscler Thromb Vasc Biol. (2007) 27:454–60. doi: 10.1161/01.ATV.0000256471.22437.88
102. He L, Vicente CP, Westrick RJ, Eitzman DT, Tollefsen DM. Heparin cofactor II inhibits arterial thrombosis after endothelial injury. J Clin Invest. (2002) 109:213–9. doi: 10.1172/JCI13432
103. Aihara K, Azuma H, Akaike M, Ikeda Y, Sata M, Takamori N, et al. Strain-dependent embryonic lethality and exaggerated vascular remodeling in heparin cofactor II-deficient mice. J Clin Invest. (2007) 117:1514–26. doi: 10.1172/JCI27095
104. Vicente CP, He L, Pavao MS, Tollefsen DM. Antithrombotic activity of dermatan sulfate in heparin cofactor II-deficient mice. Blood. (2004) 104:3965–70. doi: 10.1182/blood-2004-02-0598
105. He L, Giri TK, Vicente CP, Tollefsen DM. Vascular dermatan sulfate regulates the antithrombotic activity of heparin cofactor II. Blood. (2008) 111:4118–25. doi: 10.1182/blood-2007-12-127928
106. Yamanaga K, Yuuki T, Tsukada M, Koshiba H, Nakajima T, Takechi K, et al. Heparin cofactor II inhibits thrombus formation in a rat thrombosis model. Thromb Res. (2000) 98:95–101. doi: 10.1016/s0049-3848(99)00201-7
107. Vicente CP, He L, Tollefsen DM. Accelerated atherogenesis and neointima formation in heparin cofactor II deficient mice. Blood. (2007) 110:4261–7. doi: 10.1182/blood-2007-04-086611
108. Aihara K, Azuma H, Akaike M, Sata M, Matsumoto T. Heparin cofactor II as a novel vascular protective factor against atherosclerosis. J Atheroscler Thromb. (2009) 16:523–31. doi: 10.5551/jat.1552
109. Bolkun L, Galar M, Piszcz J, Lemancewicz D, Kloczko J. Plasma concentration of protein Z and protein Z-dependent protease inhibitor in patients with haemophilia A. Thromb Res. (2013) 131:e110–3. doi: 10.1016/j.thromres.2012.11.031
110. Han X, Fiehler R, Broze GJ Jr. Isolation of a protein Z-dependent plasma protease inhibitor. Proc Natl Acad Sci USA. (1998) 95:9250–5. doi: 10.1073/pnas.95.16.9250
111. Han X, Fiehler R, Broze GJ Jr. Characterization of the protein Z-dependent protease inhibitor. Blood. (2000) 96:3049–55. doi: 10.1182/blood.v96.9.3049.h8003049_3049_3055
112. Huang X, Swanson R, Broze GJ Jr, Olson ST. Kinetic characterization of the protein Z-dependent protease inhibitor reaction with blood coagulation factor Xa. J Biol Chem. (2008) 283:29770–83. doi: 10.1074/jbc.M805214200
113. Huang X, Dementiev A, Olson ST, Gettins PG. Basis for the specificity and activation of the serpin protein Z-dependent proteinase inhibitor (ZPI) as an inhibitor of membrane-associated factor Xa. J Biol Chem. (2010) 285:20399–409. doi: 10.1074/jbc.M110.112748
114. Huang X, Rezaie AR, Broze GJ Jr, Olson ST. Heparin is a major activator of the anticoagulant serpin, protein Z-dependent protease inhibitor. J Biol Chem. (2011) 286:8740–51. doi: 10.1074/jbc.M110.188375
115. Huang X, Swanson R, Kroh HK, Bock PE. Protein Z-dependent protease inhibitor (ZPI) is a physiologically significant inhibitor of prothrombinase function. J Biol Chem. (2019) 294:7644–57. doi: 10.1074/jbc.RA118.006787
116. Heeb MJ, Cabral KM, Ruan L. Down-regulation of factor IXa in the factor Xase complex by protein Z-dependent protease inhibitor. J Biol Chem. (2005) 280:33819–25. doi: 10.1074/jbc.M506502200
117. Tabatabai A, Fiehler R, Broze GJ Jr. Protein Z circulates in plasma in a complex with protein Z-dependent protease inhibitor. Thromb Haemost. (2001) 85:655–60. doi: 10.1055/s-0037-1615649
118. Van de Water N, Tan T, Ashton F, O’Grady A, Day T, Browett P, et al. Mutations within the protein Z-dependent protease inhibitor gene are associated with venous thromboembolic disease: a new form of thrombophilia. Br J Haematol. (2004) 127:190–4. doi: 10.1111/j.1365-2141.2004.05189.x
119. Corral J, Gonzalez-Conejero R, Soria JM, Gonzalez-Porras JR, Perez-Ceballos E, Lecumberri R, et al. A nonsense polymorphism in the protein Z-dependent protease inhibitor increases the risk for venous thrombosis. Blood. (2006) 108:177–83. doi: 10.1182/blood-2005-08-3249
120. Young LK, Birch NP, Browett PJ, Coughlin PB, Horvath AJ, Van de Water NS, et al. Two missense mutations identified in venous thrombosis patients impair the inhibitory function of the protein Z dependent protease inhibitor. Thromb Haemost. (2012) 107:854–63. doi: 10.1160/TH11-10-0708
121. Martinelli I, Razzari C, Biguzzi E, Bucciarelli P, Mannucci PM. Low levels of protein Z and the risk of venous thromboembolism. J Thromb Haemost. (2005) 3:2817–9. doi: 10.1111/j.1538-7836.2005.01664.x
122. Al-Shanqeeti A, van Hylckama Vlieg A, Berntorp E, Rosendaal FR, Broze GJ Jr. Protein Z and protein Z-dependent protease inhibitor. Determinants of levels and risk of venous thrombosis. Thromb Haemost. (2005) 93:411–3. doi: 10.1160/TH04-11-0715
123. Razzari C, Martinelli I, Bucciarelli P, Viscardi Y, Biguzzi E. Polymorphisms of the protein Z-dependent protease inhibitor (ZPI) gene and the risk of venous thromboembolism. Thromb Haemost. (2006) 95:909–10. doi: 10.1160/th06-01-0060
124. Folsom AR, Cushman M, Rasmussen-Torvik LJ, Heckbert SR, Tsai MY. Prospective study of polymorphisms of the protein Z-dependent protease inhibitor and risk of venous thromboembolism. Thromb Haemost. (2007) 97:493–4. doi: 10.1160/th06-11-0636
125. Vasse M, Guegan-Massardier E, Borg JY, Woimant F, Soria C. Frequency of protein Z deficiency in patients with ischaemic stroke. Lancet. (2001) 357:933–4. doi: 10.1016/S0140-6736(00)04218-5
126. Heeb MJ, Paganini-Hill A, Griffin JH, Fisher M. Low protein Z levels and risk of ischemic stroke: differences by diabetic status and gender. Blood Cells Mol Dis. (2002) 29:139–44. doi: 10.1006/bcmd.2002.0549
127. Kobelt K, Biasiutti FD, Mattle HP, Lammle B, Wuillemin WA. Protein Z in ischaemic stroke. Br J Haematol. (2001) 114:169–73. doi: 10.1046/j.1365-2141.2001.02913.x
128. McQuillan AM, Eikelboom JW, Hankey GJ, Baker R, Thom J, Staton J, et al. Protein Z in ischemic stroke and its etiologic subtypes. Stroke. (2003) 34:2415–9. doi: 10.1161/01.STR.0000092124.52084.4B
129. Yin ZF, Huang ZF, Cui J, Fiehler R, Lasky N, Ginsburg D, et al. Prothrombotic phenotype of protein Z deficiency. Proc Natl Acad Sci USA. (2000) 97:6734–8. doi: 10.1073/pnas.120081897
130. Zhang J, Tu Y, Lu L, Lasky N, Broze GJ Jr. Protein Z-dependent protease inhibitor deficiency produces a more severe murine phenotype than protein Z deficiency. Blood. (2008) 111:4973–8. doi: 10.1182/blood-2007-12-126391
131. Girard TJ, Lasky NM, Grunz K, Broze GJ Jr. Suppressing protein Z-dependent inhibition of factor Xa improves coagulation in hemophilia A. J Thromb Haemost. (2019) 17:149–56. doi: 10.1111/jth.14337
132. Martinez-Saguer I, Cicardi M, Suffritti C, Rusicke E, Aygoren-Pursun E, Stoll H, et al. Pharmacokinetics of plasma-derived C1-esterase inhibitor after subcutaneous versus intravenous administration in subjects with mild or moderate hereditary angioedema: the passion study. Transfusion. (2014) 54:1552–61. doi: 10.1111/trf.12501
133. Schmaier AH, Smith PM, Colman RW. Platelet C1- inhibitor. A secreted alpha-granule protein. J Clin Invest. (1985) 75:242–50. doi: 10.1172/JCI111680
134. Schreiber AD, Kaplan AP, Austen KF. Inhibition by C1inh of hagemann factor fragment activation of coagulation, fibrinolysis, and kinin generation. J Clin Invest. (1973) 52:1402–9. doi: 10.1172/JCI107313
135. Pixley RA, Schapira M, Colman RW. The Regulation of Human Factor XIIa by plasma proteinase inhibitors. J Biol Chem. (1985) 260:1723–9. doi: 10.1016/s0021-9258(18)89653-3
136. McConnell DJ. Inhibitors of kallikrein in human plasma. J Clin Invest. (1972) 51:1611–23. doi: 10.1172/jci106962
137. van der Graaf F, Koedam JA, Bouma BN. Inactivation of kallikrein in human plasma. J Clin Invest. (1983) 71:149–58. doi: 10.1172/jci110743
138. Wuillemin WA, Minnema M, Meijers JC, Roem D, Eerenberg AJ, Nuijens JH, et al. Inactivation of factor XIa in human plasma assessed by measuring factor XIa-protease inhibitor complexes: major role for C1-inhibitor. Blood. (1995) 85:1517–26. doi: 10.1182/blood.v85.6.1517.bloodjournal8561517
139. Ratnoff OD, Lepow IH. Some properties of an esterase derived from preparations of the first component of complement. J Exp Med. (1957) 106:327–43. doi: 10.1084/jem.106.2.327
140. Harpel PC, Cooper NR. Studies on human plasma C1 inactivator-enzyme interactions. I. Mechanisms of interaction with C1s, plasmin, and trypsin. J Clin Invest. (1975) 55:593–604. doi: 10.1172/JCI107967
141. Huisman LG, van Griensven JM, Kluft C. On the role of C1-inhibitor as inhibitor of tissue-type plasminogen activator in human plasma. Thromb Haemost. (1995) 73:466–71. doi: 10.1055/s-0038-1653798
142. Presanis JS, Hajela K, Ambrus G, Gal P, Sim RB. Differential substrate and inhibitor profiles for human Masp-1 and Masp-2. Mol Immunol. (2004) 40:921–9. doi: 10.1016/j.molimm.2003.10.013
143. Mauron T, Lammle B, Wuillemin WA. Influence of low molecular weight heparin and low molecular weight dextran sulfate on the inhibition of coagulation factor XIa by serpins. Thromb Haemost. (1998) 80:82–6. doi: 10.1055/s-0037-1615143
144. Pixley RA, Schmaier A, Colman RW. Effect of negatively charged activating compounds on inactivation of factor XIIa by Cl inhibitor. Arch Biochem Biophys. (1987) 256:490–8. doi: 10.1016/0003-9861(87)90606-0
145. Wijeyewickrema LC, Lameignere E, Hor L, Duncan RC, Shiba T, Travers RJ, et al. Polyphosphate is a novel cofactor for regulation of complement by a serpin C1 inhibitor. Blood. (2016) 128:1766–76. doi: 10.1182/blood-2016-02-699561
146. Stavenhagen K, Kayili HM, Holst S, Koeleman CAM, Engel R, Wouters D, et al. N- and O-glycosylation analysis of human C1-inhibitor reveals extensive mucin-type O-glycosylation. Mol Cell Proteomics. (2018) 17:1225–38. doi: 10.1074/mcp.RA117.000240
147. Minta JO. The role of sialic acid in the functional activity and the hepatic clearance of C1-INH. J Immunol. (1981) 126:245–9.
148. Reboul A, Prandini MH, Colomb MG. Proteolysis and deglycosylation of human C1 inhibitor. Effect on functional properties. Biochem J. (1987) 244:117–21. doi: 10.1042/bj2440117
149. Bos IG, Lubbers YT, Roem D, Abrahams JP, Hack CE, Eldering E. The functional integrity of the serpin domain of C1-inhibitor depends on the unique N-terminal domain, as revealed by a pathological mutant. J Biol Chem. (2003) 278:29463–70. doi: 10.1074/jbc.M302977200
150. Ghannam A, Sellier P, Fain O, Martin L, Ponard D, Drouet C. C1 Inhibitor as a glycoprotein: the influence of polysaccharides on its function and autoantibody target. Mol Immunol. (2016) 71:161–5. doi: 10.1016/j.molimm.2016.02.007
151. Busse PJ, Christiansen SC. Hereditary angioedema. N Engl J Med. (2020) 382:1136–48. doi: 10.1056/NEJMra1808012
152. Ponard D, Gaboriaud C, Charignon D, Ghannam A, Wagenaar-Bos IGA, Roem D, et al. Serping1 mutation update: mutation spectrum and C1 inhibitor phenotypes. Hum Mutat. (2020) 41:38–57. doi: 10.1002/humu.23917
153. Cicardi M, Igarashi T, Rosen FS, Davis AE III. Molecular basis for the deficiency of complement 1 Inhibitor in type I hereditary angioneurotic edema. J Clin Invest. (1987) 79:698–702. doi: 10.1172/JCI112873
154. Haslund D, Ryo LB, Seidelin Majidi S, Rose I, Skipper KA, Fryland T, et al. Dominant-negative serping1 variants cause intracellular retention of C1 inhibitor in hereditary angioedema. J Clin Invest. (2019) 129:388–405. doi: 10.1172/JCI98869
155. De Maat S, Hofman ZLM, Maas C. Hereditary angioedema: the plasma contact system out of control. J Thromb Haemost. (2018) 16:1674–85. doi: 10.1111/jth.14209
156. Joseph K, Tholanikunnel TE, Kaplan AP. Treatment of episodes of hereditary angioedema with C1 inhibitor: serial assessment of observed abnormalities of the plasma bradykinin-forming pathway and fibrinolysis. Ann Allergy Asthma Immunol. (2010) 104:50–4. doi: 10.1016/j.anai.2009.11.014
157. Cugno M, Cicardi M, Bottasso B, Coppola R, Paonessa R, Mannucci PM, et al. Activation of the coagulation cascade in C1-inhibitor deficiencies. Blood. (1997) 89:3213–8. doi: 10.1182/blood.v89.9.3213
158. Cugno M, Zanichelli A, Bellatorre AG, Griffini S, Cicardi M. Plasma biomarkers of acute attacks in patients with angioedema due to C1-inhibitor deficiency. Allergy. (2009) 64:254–7. doi: 10.1111/j.1398-9995.2008.01859.x
159. Csuka D, Veszeli N, Imreh E, Zotter Z, Skopal J, Prohaszka Z, et al. Comprehensive study into the activation of the plasma enzyme systems during attacks of hereditary angioedema due to C1-inhibitor deficiency. Orphanet J Rare Dis. (2015) 10:132. doi: 10.1186/s13023-015-0351-5
160. van Geffen M, Cugno M, Lap P, Loof A, Cicardi M, van Heerde W. Alterations of coagulation and fibrinolysis in patients with angioedema due to C1-inhibitor deficiency. Clin Exp Immunol. (2012) 167:472–8. doi: 10.1111/j.1365-2249.2011.04541.x
161. Gandhi PK, Gentry WM, Bottorff MB. Thrombotic events associated with C1 esterase inhibitor products in patients with hereditary angioedema: investigation from the United States food and drug administration adverse event reporting system database. Pharmacotherapy. (2012) 32:902–9. doi: 10.1002/j.1875-9114.2012.01126
162. Riedl MA, Bygum A, Lumry W, Magerl M, Bernstein JA, Busse P, et al. Safety and usage of C1-inhibitor in hereditary angioedema: berinert registry data. J Allergy Clin Immunol Pract. (2016) 4:963–71. doi: 10.1016/j.jaip.2016.04.018
163. Sundler BL, Persson B, Aronsson D, Skattum L, Nordenfelt P, Egesten A Comorbidities in hereditary angioedema-a population-based cohort study. Clin Transl Allergy. (2022) 12:e12135. doi: 10.1002/clt2.12135
164. Farkas H, Kohalmi KV, Veszeli N, Zotter Z, Varnai K, Varga L. Risk of thromboembolism in patients with hereditary angioedema treated with plasma-derived C1-inhibitor. Allergy Asthma Proc. (2016) 37:164–70. doi: 10.2500/aap.2016.37.3933
165. Jensen SB, Hindberg K, Solomon T, Smith EN, Lapek JD Jr, Gonzalez DJ, et al. Discovery of novel plasma biomarkers for future incident venous thromboembolism by untargeted synchronous precursor selection mass spectrometry proteomics. J Thromb Haemost. (2018) 16:1763–74. doi: 10.1111/jth.14220
166. Norgaard I, Nielsen SF, Nordestgaard BG. Complement C3 and high risk of venous thromboembolism: 80517 individuals from the Copenhagen general population study. Clin Chem. (2016) 62:525–34. doi: 10.1373/clinchem.2015.251314
167. Skjeflo EW, Braekkan SK, Ludviksen JK, Snir O, Hindberg K, Mollnes TE, et al. Elevated plasma concentration of complement factor C5 is associated with risk of future venous thromboembolism. Blood. (2021) 138:2129–37. doi: 10.1182/blood.2021010822
168. Caliezi C, Wuillemin WA, Zeerleder S, Redondo M, Eisele B, Hack CE. C1-esterase inhibitor: an anti-inflammatory agent and its potential use in the treatment of diseases other than hereditary angioedema. Pharmacol Rev. (2000) 52:91–112.
169. Dorresteijn MJ, Visser T, Cox LA, Bouw MP, Pillay J, Koenderman AH, et al. C1-esterase inhibitor attenuates the inflammatory response during human endotoxemia. Crit Care Med. (2010) 38:2139–45. doi: 10.1097/CCM.0b013e3181f17be4
170. Igonin AA, Protsenko DN, Galstyan GM, Vlasenko AV, Khachatryan NN, Nekhaev IV, et al. C1-esterase inhibitor infusion increases survival rates for patients with sepsis*. Crit Care Med. (2012) 40:770–7. doi: 10.1097/CCM.0b013e318236edb8
171. Han ED, MacFarlane RC, Mulligan AN, Scafidi J, Davis AE III. Increased vascular permeability in C1 inhibitor-deficient mice mediated by the bradykinin type 2 receptor. J Clin Invest. (2002) 109:1057–63. doi: 10.1172/JCI14211
172. Schurmann D, Herzog E, Raquet E, Nolte MW, May F, Muller-Cohrs J, et al. C1-esterase inhibitor treatment: preclinical safety aspects on the potential prothrombotic risk. Thromb Haemost. (2014) 112:960–71. doi: 10.1160/TH13-06-0469
173. Evans DL, McGrogan M, Scott RW, Carrell RW. Protease specificity and heparin binding and activation of recombinant protease nexin I. J Biol Chem. (1991) 266:22307–12. doi: 10.1016/s0021-9258(18)54571-3
174. Knauer DJ, Majumdar D, Fong PC, Knauer MF. Serpin regulation of factor XII. The novel observation that protease nexin 1 in the presence of heparin is a more potent inhibitor of factor XIa than C1 inhibitor. J Biol Chem. (2000) 275:37340–6. doi: 10.1074/jbc.M003909200
175. Baker JB, Gronke RS. Protease nexins and cellular regulation. Semin Thromb Hemost. (1986) 12:216–20. doi: 10.1055/s-2007-1003554
176. Mansuy IM, van der Putten H, Schmid P, Meins M, Botteri FM, Monard D. Variable and multiple expression of protease nexin-1 during mouse organogenesis and nervous system development. Development. (1993) 119:1119–34. doi: 10.1242/dev.119.4.1119
177. Mansilla S, Boulaftali Y, Venisse L, Arocas V, Meilhac O, Michel JB, et al. Macrophages and platelets are the major source of protease nexin-1 in human atherosclerotic plaque. Arterioscler Thromb Vasc Biol. (2008) 28:1844–50. doi: 10.1161/ATVBAHA.108.171389
178. Boulaftali Y, Adam F, Venisse L, Ollivier V, Richard B, Taieb S, et al. Anticoagulant and antithrombotic properties of platelet protease nexin-1. Blood. (2010) 115:97–106. doi: 10.1182/blood-2009-04-217240
179. Demeo DL, Mariani TJ, Lange C, Srisuma S, Litonjua AA, Celedon JC, et al. The serpine2 gene is associated with chronic obstructive pulmonary disease. Am J Hum Genet. (2006) 78:253–64. doi: 10.1086/499828
180. Zhu G, Warren L, Aponte J, Gulsvik A, Bakke P, Anderson WH, et al. The serpine2 gene is associated with chronic obstructive pulmonary disease in two large populations. Am J Respir Crit Care Med. (2007) 176:167–73. doi: 10.1164/rccm.200611-1723OC
181. Fujimoto K, Ikeda S, Arai T, Tanaka N, Kumasaka T, Ishii T, et al. Polymorphism of SERPINE2 gene is associated with pulmonary emphysema in consecutive autopsy cases. BMC Med Genet. (2010) 11:159. doi: 10.1186/1471-2350-11-159
182. Himes BE, Klanderman B, Ziniti J, Senter-Sylvia J, Soto-Quiros ME, Avila L, et al. Association of SERPINE2 with asthma. Chest. (2011) 140:667–74. doi: 10.1378/chest.10-2973
183. Kukkonen MK, Tiili E, Hamalainen S, Vehmas T, Oksa P, Piirila P, et al. SERPINE2 haplotype as a risk factor for panlobular type of emphysema. BMC Med Genet. (2011) 12:157. doi: 10.1186/1471-2350-12-157
184. Luthi A, Van der Putten H, Botteri FM, Mansuy IM, Meins M, Frey U, et al. Endogenous serine protease inhibitor modulates epileptic activity and hippocampal long-term potentiation. J Neurosci. (1997) 17:4688–99. doi: 10.1523/JNEUROSCI.17-12-04688.1997
185. Bouton MC, Boulaftali Y, Richard B, Arocas V, Michel JB, Jandrot-Perrus M. Emerging role of SERPINE2/protease nexin-1 in hemostasis and vascular biology. Blood (2012) 119:2452–7. doi: 10.1182/blood-2011-10-387464
186. Francois D, Arocas V, Venisse L, Aymonnier K, Idir L, Martos R, et al. Hematopoietic protease nexin-1 protects against lung injury by preventing thrombin signaling in mice. Blood Adv. (2018) 2:2389–99. doi: 10.1182/bloodadvances.2018018283
187. Aymonnier K, Kawecki C, Venisse L, Boulaftali Y, Christophe OD, Lenting PJ, et al. Targeting protease nexin-1, a natural anticoagulant serpin, to control bleeding and improve hemostasis in hemophilia. Blood. (2019) 134:1632–44. doi: 10.1182/blood.2019000281
188. Kawecki C, Aymonnier K, Ferriere S, Venisse L, Arocas V, Boulaftali Y, et al. Development and characterization of single-domain antibodies neutralizing protease nexin-1 as tools to increase thrombin generation. J Thromb Haemost. (2020) 18:2155–68. doi: 10.1111/jth.14940
189. Boulaftali Y, Ho-Tin-Noe B, Pena A, Loyau S, Venisse L, Francois D, et al. Platelet protease nexin-1, a serpin that strongly influences fibrinolysis and thrombolysis. Circulation. (2011) 123:1326–34. doi: 10.1161/CIRCULATIONAHA.110.000885
190. Meijers JC, Herwald H. Protein C inhibitor. Semin Thromb Hemost. (2011) 37:349–54. doi: 10.1055/s-0031-1276583
191. Espana F, Berrettini M, Griffin JH. Purification and characterization of plasma protein C inhibitor. Thromb Res. (1989) 55:369–84. doi: 10.1016/0049-3848(89)90069-8
192. Rezaie AR, Cooper ST, Church FC, Esmon CT. Protein C inhibitor is a potent inhibitor of the thrombin-thrombomodulin complex. J Biol Chem. (1995) 270:25336–9. doi: 10.1074/jbc.270.43.25336
193. Marlar RA, Griffin JH. Deficiency of protein C inhibitor in combined factor V/VIII deficiency disease. J Clin Invest. (1980) 66:1186–9. doi: 10.1172/JCI109952
194. Canfield WM, Kisiel W. Evidence of normal functional levels of activated protein C inhibitor in combined factor V/VIII deficiency disease. J Clin Invest. (1982) 70:1260–72. doi: 10.1172/jci110725
195. Billingsley GD, Walter MA, Hammond GL, Cox DW. Physical mapping of four serpin genes: alpha 1-antitrypsin, alpha 1-antichymotrypsin, corticosteroid-binding globulin, and protein C inhibitor, within a 280-Kb region on chromosome I4q32.1. Am J Hum Genet. (1993) 52:343–53.
196. Nichols WC, Seligsohn U, Zivelin A, Terry VH, Arnold ND, Siemieniak DR, et al. Linkage of combined factors V and VIII deficiency to chromosome 18q by homozygosity mapping. J Clin Invest. (1997) 99:596–601. doi: 10.1172/JCI119201
197. Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. (2020) 581:434–43. doi: 10.1038/s41586-020-2308-7
198. Wagenaar GT, van Vuuren AJ, Girma M, Tiekstra MJ, Kwast L, Koster JG, et al. Characterization of transgenic mice that secrete functional human protein C inhibitor into the circulation. Thromb Haemost. (2000) 83:93–101. doi: 10.1055/s-0037-1613763
199. Uhrin P, Dewerchin M, Hilpert M, Chrenek P, Schofer C, Zechmeister-Machhart M, et al. Disruption of the protein c inhibitor gene results in impaired spermatogenesis and male infertility. J Clin Invest. (2000) 106:1531–9. doi: 10.1172/JCI10768
200. Laurell M, Christensson A, Abrahamsson PA, Stenflo J, Lilja H. Protein C inhibitor in human body fluids. seminal plasma is rich in inhibitor antigen deriving from cells throughout the male reproductive system. J Clin Invest. (1992) 89:1094–101. doi: 10.1172/JCI115689
201. Uhrin P, Schofer C, Zaujec J, Ryban L, Hilpert M, Weipoltshammer K, et al. Male fertility and protein C inhibitor/plasminogen activator inhibitor-3 (PCI): localization of PCI in mouse testis and failure of single plasminogen activator knockout to restore spermatogenesis in PCI-deficient mice. Fertil Steril. (2007) 88(4 Suppl):1049–57. doi: 10.1016/j.fertnstert.2006.11.193
202. Hayashi T, Nishioka J, Kamada H, Asanuma K, Kondo H, Gabazza EC, et al. Characterization of a novel human protein C inhibitor (PCI) gene transgenic mouse useful for studying the role of PCI in physiological and pathological conditions. J Thromb Haemost. (2004) 2:949–61. doi: 10.1111/j.1538-7836.2004.00733.x
203. Kalsheker N, Morley S, Morgan K. Gene regulation of the serine proteinase inhibitors alpha1-antitrypsin and alpha1-antichymotrypsin. Biochem Soc Trans. (2002) 30:93–8.
204. Beatty K, Bieth J, Travis J. Kinetics of association of serine proteinases with native and oxidized alpha-1-proteinase inhibitor and alpha-1-antichymotrypsin. J Biol Chem. (1980) 255:3931–4. doi: 10.1016/s0021-9258(19)85615-6
205. Janciauskiene S, Wrenger S, Immenschuh S, Olejnicka B, Greulich T, Welte T, et al. The multifaceted effects of alpha1-antitrypsin on neutrophil functions. Front Pharmacol. (2018) 9:341. doi: 10.3389/fphar.2018.00341
206. Rao NV, Wehner NG, Marshall BC, Gray WR, Gray BH, Hoidal JR. Characterization of proteinase-3 (Pr-3), a neutrophil serine proteinase. structural and functional properties. J Biol Chem. (1991) 266:9540–8. doi: 10.1016/s0021-9258(18)92854-1
207. Ellis V, Scully M, MacGregor I, Kakkar V. Inhibition of human factor Xa by various plasma protease inhibitors. Biochim Biophys Acta. (1982) 701:24–31.
208. Frommherz KJ, Faller B, Bieth JG. Heparin strongly decreases the rate of inhibition of neutrophil elastase by alpha 1-proteinase inhibitor. J Biol Chem. (1991) 266:15356–62. doi: 10.1016/s0021-9258(18)98623-0
209. Strnad P, McElvaney NG, Lomas DA. Alpha1-antitrypsin deficiency. N Engl J Med. (2020) 382:1443–55. doi: 10.1056/NEJMra1910234
210. Le A, Ferrell GA, Dishon DS, Le QQ, Sifers RN. Soluble aggregates of the human Piz alpha 1-antitrypsin variant are degraded within the endoplasmic reticulum by a mechanism sensitive to inhibitors of protein synthesis. J Biol Chem. (1992) 267:1072–80. doi: 10.1016/s0021-9258(18)48397-4
211. McElvaney NG, Stoller JK, Buist AS, Prakash UB, Brantly ML, Schluchter MD, et al. Baseline characteristics of enrollees in the national heart, lung and blood institute registry of alpha 1-antitrypsin deficiency. alpha 1-antitrypsin deficiency registry study group. Chest. (1997) 111:394–403. doi: 10.1378/chest.111.2.394
212. Seixas S, Marques PI. Known mutations at the cause of alpha-1 antitrypsin deficiency an updated overview of serpina1 variation spectrum. Appl Clin Genet. (2021) 14:173–94. doi: 10.2147/TACG.S257511
213. Kelly E, Greene CM, Carroll TP, McElvaney NG, O’Neill SJ. Alpha-1 antitrypsin deficiency. Respir Med. (2010) 104:763–72. doi: 10.1016/j.rmed.2010.01.016
214. Foreman MG, Wilson C, DeMeo DL, Hersh CP, Beaty TH, Cho MH, et al. Alpha-1 Antitrypsin PiMZ genotype is associated with chronic obstructive pulmonary disease in two racial groups. Ann Am Thorac Soc. (2017) 14:1280–7. doi: 10.1513/AnnalsATS.201611-838OC
215. Lieberman J, Winter B, Sastre A. Alpha 1-antitrypsin Pi-types in 965 COPD patients. Chest. (1986) 89:370–3. doi: 10.1378/chest.89.3.370
216. Sveger T. Liver disease in alpha1-antitrypsin deficiency detected by screening of 200,000 infants. N Engl J Med. (1976) 294:1316–21. doi: 10.1056/NEJM197606102942404
217. Mitchell EL, Khan Z. Liver disease in alpha-1 antitrypsin deficiency: current approaches and future directions. Curr Pathobiol Rep. (2017) 5:243–52. doi: 10.1007/s40139-017-0147-5
218. Wewers MD, Crystal RG. Alpha-1 antitrypsin augmentation therapy. COPD. (2013) 10(Suppl. 1):64–7. doi: 10.3109/15412555.2013.764402
219. Basil N, Ekstrom M, Piitulainen E, Lindberg A, Ronmark E, Jehpsson L, et al. Severe alpha-1-antitrypsin deficiency increases the risk of venous thromboembolism. J Thromb Haemost. (2021) 19:1519–25. doi: 10.1111/jth.15302
220. Riis J, Nordestgaard BG, Afzal S. Alpha1 –antitrypsin Z allele and risk of venous thromboembolism in the general population. J Thromb Haemost. (2022) 20:115–25. doi: 10.1111/jth.15556
221. Tanash H, Ekstrom M, Basil N, Ronmark E, Lindberg A, Piitulainen E. Decreased risk of ischemic heart disease in individuals with severe alpha 1-antitrypsin deficiency (PiZZ) in comparison with the general population. Int J Chron Obstruct Pulmon Dis. (2020) 15:1245–52. doi: 10.2147/COPD.S247377
222. Lewis JH, Iammarino RM, Spero JA, Hasiba U. Antithrombin Pittsburgh: an alpha1-antitrypsin variant causing hemorrhagic disease. Blood. (1978) 51:129–37. doi: 10.1182/blood.v51.1.129.bloodjournal511129
223. Owen MC, Brennan SO, Lewis JH, Carrell RW. Mutation of antitrypsin to antithrombin. Alpha 1-antitrypsin pittsburgh (358 met leads to Arg), a fatal bleeding disorder. N Engl J Med. (1983) 309:694–8. doi: 10.1056/NEJM198309223091203
224. Scott CF, Carrell RW, Glaser CB, Kueppers F, Lewis JH, Colman RW. Alpha-1-antitrypsin-Pittsburgh. A potent inhibitor of human plasma factor XIa, kallikrein, and factor XIIf. J Clin Invest. (1986) 77:631–4. doi: 10.1172/JCI112346
225. Heeb MJ, Bischoff R, Courtney M, Griffin JH. Inhibition of activated protein C by recombinant alpha 1-antitrypsin variants with substitution of arginine or leucine for methionine358. J Biol Chem. (1990) 265:2365–9. doi: 10.1016/s0021-9258(19)39985-5
226. Hua B, Fan L, Liang Y, Zhao Y, Tuddenham EG. Alpha1-antitrypsin Pittsburgh in a family with bleeding tendency. Haematologica. (2009) 94:881–4. doi: 10.3324/haematol.2008.004739
227. Cox DW, Huber O. Rheumatoid arthritis and alpha-1-antitrypsin. Lancet. (1976) 1:1216–7. doi: 10.1016/s0140-6736(76)92163-2
228. Papiha SS, Pal B, Walker D, Mangion P, Hossain MA. Alpha 1 antitrypsin (Pi) phenotypes in two rheumatic diseases: a reappraisal of the association of Pi subtypes in rheumatoid arthritis. Ann Rheum Dis. (1989) 48:48–52. doi: 10.1136/ard.48.1.48
229. Yang C, Keshavjee S, Liu M. Alpha-1 antitrypsin for covid-19 treatment: dual role in antiviral infection and anti-inflammation. Front Pharmacol. (2020) 11:615398. doi: 10.3389/fphar.2020.615398
230. Kushi A, Akiyama K, Noguchi M, Edamura K, Yoshida T, Sasai H. Disruption of the murine alpha1-antitrypsin/Pi2 gene. Exp Anim. (2004) 53:437–43. doi: 10.1538/expanim.53.437
231. Wang D, Wang W, Dawkins P, Paterson T, Kalsheker N, Sallenave JM, et al. Deletion of serpina1a, a murine alpha1-antitrypsin ortholog, results in embryonic lethality. Exp Lung Res. (2011) 37:291–300. doi: 10.3109/01902148.2011.554599
232. Borel F, Sun H, Zieger M, Cox A, Cardozo B, Li W, et al. Editing out five serpina1 paralogs to create a mouse model of genetic emphysema. Proc Natl Acad Sci USA. (2018) 115:2788–93. doi: 10.1073/pnas.1713689115
233. Maas C, de Maat S. Therapeutic serpins: improving on nature. Front Cardiovasc Med. (2021) 8:648349. doi: 10.3389/fcvm.2021.648349
234. Bianchini EP, Auditeau C, Razanakolona M, Vasse M, Borgel D. Serpins in hemostasis as therapeutic targets for bleeding or thrombotic disorders. Front Cardiovasc Med. (2020) 7:622778. doi: 10.3389/fcvm.2020.622778
235. Ehrlich HJ, Gebbink RK, Keijer J, Linders M, Preissner KT, Pannekoek H. Alteration of serpin specificity by a protein cofactor. vitronectin endows plasminogen activator inhibitor 1 with thrombin inhibitory properties. J Biol Chem. (1990) 265:13029–35. doi: 10.1016/s0021-9258(19)38262-6
236. Lawrence DA, Strandberg L, Ericson J, Ny T. Structure-function studies of the serpin plasminogen activator inhibitor type 1. Analysis of chimeric strained loop mutants. J Biol Chem. (1990) 265:20293–301. doi: 10.1016/s0021-9258(17)30503-3
237. Hopkins PC, Crowther DC, Carrell RW, Stone SR. Development of a novel recombinant serpin with potential antithrombotic properties. J Biol Chem. (1995) 270:11866–71. doi: 10.1074/jbc.270.20.11866
238. Schapira M, Ramus MA, Waeber B, Brunner HR, Jallat S, Carvallo D, et al. Protection by recombinant alpha 1-antitrypsin Ala357 Arg358 against arterial hypotension induced by factor XII fragment. J Clin Invest. (1987) 80:582–5. doi: 10.1172/JCI113108
239. Sulikowski T, Bauer BA, Patston PA. Alpha(1)-proteinase inhibitor mutants with specificity for plasma kallikrein and C1s but not C1. Protein Sci. (2002) 11:2230–6. doi: 10.1110/ps.0207302
240. de Maat S, Sanrattana W, Mailer RK, Parr NMJ, Hessing M, Koetsier RM, et al. Design and characterization of alpha1-antitrypsin variants for treatment of contact system-driven thromboinflammation. Blood. (2019) 134:1658–69. doi: 10.1182/blood.2019000481
241. Polderdijk SG, Adams TE, Ivanciu L, Camire RM, Baglin TP, Huntington JA. Design and characterization of an apc-specific serpin for the treatment of hemophilia. Blood. (2017) 129:105–13. doi: 10.1182/blood-2016-05-718635
242. Eldering E, Huijbregts CC, Lubbers YT, Longstaff C, Hack CE. Characterization of recombinant C1 inhibitor P1 variants. J Biol Chem. (1992) 267:7013–20. doi: 10.1016/s0021-9258(19)50529-4
243. Zahedi R, Wisnieski J, Davis AE III. Role of the P2 residue of complement 1 inhibitor (Ala443) in determination of target protease specificity: inhibition of complement and contact system proteases. J Immunol. (1997) 159:983–8.
244. Zahedi R, MacFarlane RC, Wisnieski JJ, Davis AE III. C1 Inhibitor: analysis of the role of amino acid residues within the reactive center loop in target protease recognition. J Immunol. (2001) 167:1500–6. doi: 10.4049/jimmunol.167.3.1500
245. Derechin VM, Blinder MA, Tollefsen DM. Substitution of arginine for Leu444 in the reactive site of heparin cofactor II enhances the rate of thrombin inhibition. J Biol Chem. (1990) 265:5623–8. doi: 10.1016/s0021-9258(19)39407-4
246. Bhakta V, Hamada M, Nouanesengsy A, Lapierre J, Perruzza DL, Sheffield WP. Identification of an alpha-1 antitrypsin variant with enhanced specificity for factor XIa by phage display, bacterial expression, and combinatorial mutagenesis. Sci Rep. (2021) 11:5565. doi: 10.1038/s41598-021-84618-7
247. Sanrattana W, Sefiane T, Smits S, van Kleef ND, Fens MH, Lenting PJ, et al. A reactive center loop-based prediction platform to enhance the design of therapeutic serpins. Proc Natl Acad Sci USA. (2021) 118:e2108458118. doi: 10.1073/pnas.2108458118
248. Ma B, Guan X, Li Y, Shang S, Li J, Tan Z. Protein glycoengineering: an approach for improving protein properties. Front Chem. (2020) 8:622. doi: 10.3389/fchem.2020.00622
249. Sarkar A, Wintrode PL. Effects of glycosylation on the stability and flexibility of a metastable protein: the human serpin alpha(1)-antitrypsin. Int J Mass Spectrom. (2011) 302:69–75. doi: 10.1016/j.ijms.2010.08.003
250. Casolaro MA, Fells G, Wewers M, Pierce JE, Ogushi F, Hubbard R, et al. Augmentation of lung antineutrophil elastase capacity with recombinant human alpha-1-antitrypsin. J Appl Physiol. (1987) 63:2015–23. doi: 10.1152/jappl.1987.63.5.2015
251. Lusch A, Kaup M, Marx U, Tauber R, Blanchard V, Berger M. Development and analysis of alpha 1-antitrypsin neoglycoproteins: the impact of additional N-glycosylation sites on serum half-life. Mol Pharm. (2013) 10:2616–29. doi: 10.1021/mp400043r
252. Chung HS, Kim JS, Lee SM, Park SJ. Additional N-glycosylation in the N-terminal region of recombinant human alpha-1 antitrypsin enhances the circulatory half-Life in Sprague-Dawley rats. Glycoconj J. (2016) 33:201–8. doi: 10.1007/s10719-016-9657-3
253. Amann T, Hansen AH, Kol S, Hansen HG, Arnsdorf J, Nallapareddy S, et al. Glyco-engineered cho cell lines producing alpha-1-antitrypsin and c1 esterase inhibitor with fully humanized N-glycosylation profiles. Metab Eng. (2019) 52:143–52. doi: 10.1016/j.ymben.2018.11.014
254. Aguila S, Noto R, Luengo-Gil G, Espin S, Bohdan N, de la Morena-Barrio ME, et al. N-glycosylation as a tool to study antithrombin secretion, conformation, and function. Int J Mol Sci. (2021) 22:516. doi: 10.3390/ijms22020516
255. Chandrasekhar K, Ke H, Wang N, Goodwin T, Gierasch LM, Gershenson A, et al. Cellular folding pathway of a metastable serpin. Proc Natl Acad Sci USA. (2016) 113:6484–9. doi: 10.1073/pnas.1603386113
256. McCoy AJ, Pei XY, Skinner R, Abrahams JP, Carrell RW. Structure of beta-antithrombin and the effect of glycosylation on antithrombin’s heparin affinity and activity. J Mol Biol. (2003) 326:823–33. doi: 10.1016/s0022-2836(02)01382-7
257. Mast AE, Salvesen G, Schnebli HP, Pizzo SV. Evaluation of the rapid plasma elimination of recombinant alpha 1-proteinase inhibitor: synthesis of polyethylene glycol conjugates with improved therapeutic potential. J Lab Clin Med. (1990) 116:58–65.
258. Cantin AM, Woods DE, Cloutier D, Dufour EK, Leduc R. Polyethylene glycol conjugation at Cys232 prolongs the half-life of alpha1 proteinase inhibitor. Am J Respir Cell Mol Biol. (2002) 27:659–65. doi: 10.1165/rcmb.4866
259. Guo Q, Yaron JR, Wallen JW III, Browder KF, Boyd R, Olson TL, et al. Pegylated serp-1 markedly reduces pristane-induced experimental diffuse alveolar hemorrhage, altering upar distribution, and macrophage invasion. Front Cardiovasc Med. (2021) 8:633212. doi: 10.3389/fcvm.2021.633212
260. Yaron JR, Zhang L, Guo Q, Haydel SE, Lucas AR. Fibrinolytic serine proteases, therapeutic serpins and inflammation: fire dancers and firestorms. Front Cardiovasc Med. (2021) 8:648947. doi: 10.3389/fcvm.2021.648947
261. Nash P, Whitty A, Handwerker J, Macen J, McFadden G. Inhibitory specificity of the anti-inflammatory myxoma virus serpin SERP-1. J Biol Chem. (1998) 273:20982–91. doi: 10.1074/jbc.273.33.20982
262. Mahon BP, Ambadapadi S, Yaron JR, Lomelino CL, Pinard MA, Keinan S, et al. Crystal structure of cleaved SERP-1, a myxomavirus-derived immune modulating serpin: structural design of serpin reactive center loop peptides with improved therapeutic function. Biochemistry. (2018) 57:1096–107. doi: 10.1021/acs.biochem.7b01171
263. Tardif JC, L’Allier PL, Gregoire J, Ibrahim R, McFadden G, Kostuk W, et al. A randomized controlled, phase 2 trial of the viral serpin SERP-1 in patients with acute coronary syndromes undergoing percutaneous coronary intervention. Circ Cardiovasc Interv. (2010) 3:543–8. doi: 10.1161/CIRCINTERVENTIONS.110.953885
264. Lee S, Lee Y, Hong K, Hong J, Bae S, Choi J, et al. Effect of recombinant alpha1-antitrypsin Fc-Fused (AAT-Fc)protein on the inhibition of inflammatory cytokine production and streptozotocin-induced diabetes. Mol Med. (2013) 19:65–71. doi: 10.2119/molmed.2012.00308
265. Takeda K, Kim SH, Joetham A, Petrache I, Gelfand EW. Therapeutic benefits of recombinant alpha1-antitrypsin IgG1 Fc-fusion protein in experimental emphysema. Respir Res. (2021) 22:207. doi: 10.1186/s12931-021-01784-y
266. Izaguirre G, Zhang W, Swanson R, Bedsted T, Olson ST. Localization of an antithrombin exosite that promotes rapid inhibition of factors Xa and IXa dependent on heparin activation of the serpin. J Biol Chem. (2003) 278:51433–40. doi: 10.1074/jbc.M309266200
267. Olson ST, Sheffer R, Francis AM. High molecular weight kininogen potentiates the heparin-accelerated inhibition of plasma kallikrein by antithrombin: role for antithrombin in the regulation of kallikrein. Biochemistry. (1993) 32:12136–47. doi: 10.1021/bi00096a026
268. Silverberg M, Longo J, Kaplan AP. Study of the effect of high molecular weight kininogen upon the fluid-phase inactivation of kallikrein by C1 inhibitor. J Biol Chem. (1986) 261:14965–8. doi: 10.1016/s0021-9258(18)66813-9
269. Schousboe I. Binding of activated factor XII to endothelial cells affects its inactivation by the C1-esterase inhibitor. Eur J Biochem. (2003) 270:111–8. doi: 10.1046/j.1432-1033.2003.03367.x
Keywords: anticoagulants, coagulation factors, hemostasis, serine protease inhibitors, thrombosis
Citation: Grover SP and Mackman N (2022) Anticoagulant SERPINs: Endogenous Regulators of Hemostasis and Thrombosis. Front. Cardiovasc. Med. 9:878199. doi: 10.3389/fcvm.2022.878199
Received: 17 February 2022; Accepted: 29 March 2022;
Published: 03 May 2022.
Edited by:
Marina Panova-Noeva, Johannes Gutenberg University Mainz, GermanyReviewed by:
Fionnuala Ni Ainle, University College Dublin, IrelandCopyright © 2022 Grover and Mackman. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Steven P. Grover, c3RldmVuX2dyb3ZlckBtZWQudW5jLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.