 Maria Sancho
Maria Sancho Jacob Fletcher3
Jacob Fletcher3 Donald G. Welsh
Donald G. Welsh- 1Department of Pharmacology, University of Vermont, Burlington, VT, United States
- 2Department of Physiology, Faculty of Medicine, Universidad Complutense de Madrid, Madrid, Spain
- 3Department of Physiology and Pharmacology, Robarts Research Institute, University of Western Ontario, London, ON, Canada
Cerebral arteries contain two primary and interacting cell types, smooth muscle (SMCs) and endothelial cells (ECs), which are each capable of sensing particular hemodynamic forces to set basal tone and brain perfusion. These biomechanical stimuli help confer tone within arterial networks upon which local neurovascular stimuli function. Tone development is intimately tied to arterial membrane potential (VM) and changes in intracellular [Ca2+] driven by voltage-gated Ca2+ channels (VGCCs). Arterial VM is in turn set by the dynamic interplay among ion channel species, the strongly inward rectifying K+ (Kir) channel being of special interest. Kir2 channels possess a unique biophysical signature in that they strongly rectify, display negative slope conductance, respond to elevated extracellular K+ and are blocked by micromolar Ba2+. While functional Kir2 channels are expressed in both smooth muscle and endothelium, they lack classic regulatory control, thus are often viewed as a simple background conductance. Recent literature has provided new insight, with two membrane lipids, phosphatidylinositol 4,5-bisphosphate (PIP2) and cholesterol, noted to (1) stabilize Kir2 channels in a preferred open or closed state, respectively, and (2) confer, in association with the cytoskeleton, caveolin-1 (Cav1) and syntrophin, hemodynamic sensitivity. It is these aspects of vascular Kir2 channels that will be the primary focus of this review.
Introduction
Cerebral blood flow (CBF) is exquisitely controlled by a complex network of surface/penetrating resistance arteries and a dense capillary network that match local blood cell delivery with tissue metabolic demands (1). This vascular network is primarily composed of smooth muscle (SMC) and endothelial (EC) cells; however, a variety of other cell types including pacemakers, fibroblasts, and pericytes are also known to be present (2). The plasma membrane of vascular smooth muscle (SMC) and endothelial (EC) cells is capable of sensing electrical, neuronal, and/or physical stimuli (i.e., transmural flow and pressure) and transducing them into arterial diameter changes, a fundamental phenomenon for the precise blood flow delivery needed for optimal brain function (3–5). Smooth muscle cell contractility is tightly coupled to intracellular [Ca2+], a fundamental second messenger driving myosin light chain phosphorylation and the subsequent extent of cross-bridge cycling (6). The electrical driving force for extracellular Ca2+ entry is set by voltage-gated Ca2+ channels (VGCCs) which in turn, are critically regulated by the resting membrane potential (VM) (7). This absolute parameter is defined by the balance of inward depolarizing and outward hyperpolarizing currents in vascular cells. The latter is largely delivered via membrane-embedded K+ channels, including voltage-gated (Kv), ATP-sensitive (KATP), Ca2+-activated (BKCa, IKCa, SKCa), and inwardly rectifying (Kir) channels (8).
In the brain vasculature, Kir2 channels are prominently expressed in SMCs and ECs and exhibit key properties and functions. While Kir2 channels are often viewed as a resting conductance, their unique biophysical signature—inward rectification at VM values negative to EK, activation by extracellular K+, and rapid blockade by micromolar Ba2+ (9)—has posed them as significant contributors of arterial myogenic tone development and vascular pathology (10–16). Interestingly, numerous studies have reported direct interactions between Kir channels and their surrounding membrane lipid environment which places the channel in an active or silent state (17–23). At a functional level, their distinct properties enable Kir2 channels to contribute to K+-induced hyperpolarization, vessel dilation, and force-sensing (4, 21, 22). Particularly, Kir2 channels are noted to respond to specific hemodynamic stimuli, such as pressure and flow, thus contributing to basal tone and CBF control (22, 24).
In this review, we will summarize the major lipid constituents of the cellular plasma membrane, the structural and biophysical features of Kir2 channels, and the link between membrane lipid content and the activity of such ion channels. Thereafter, we will focus on the distinct molecular and mechanical attributes of the vascular plasma membrane, specifically within smooth muscle and the endothelium. This section will include a comprehensive description of mechanosensitive ion channels, cellular routes of force transmission and signaling membrane microdomains. Finally, we will provide the audience with a detailed picture of the unique lipid-protein interactions that confer vascular Kir2 channels the ability to sense mechanical forces (stretch and/or laminar flow), with a primary focus on cerebral vascular tone and perfusion control.
The (Vascular) Plasma Membrane: A Lipid Bilayer Containing Key Proteins
Every cell in a living organism contains a thick ∼4–10 nm hydrophobic layer which serves as a physical and selectively permeable barrier between two aqueous and heterogeneous environments: the cytoplasm and the outside of the cell. The plasma membrane represents a mosaic of multiple components—including lipids, embedded proteins, and carbohydrates—that are constantly subjected to spontaneous reorganization by rotational or lateral motion (25). Interestingly, the fluidity of the plasma membrane is determined by the ratio of rigid vs. fluid lipid components, which in turn depends primarily on the temperature, and the length/degree of unsaturation of phospholipid fatty acids.
Major Lipid Constituents of Vascular Plasma Membranes
More than 1,000 different species of lipids coexist within the plasma membrane, heterogeneously separated between the two leaflets and giving rise to complex lipid environments at the cell surface (26). These lipids are amphipathic or dual-loving molecules, containing a hydrophilic (“water loving”) and a hydrophobic (“water-fearing”) constituent. The lipid integral components—including phospholipids, cholesterol, and sphingolipids—in addition to being key structural components of the cell membrane, also interact with other non-lipid membrane constituents such as proteins. Specifically, a wide variety of ion channels, including the strongly inward rectifier potassium (Kir2) channel, require a specific local lipid microenvironment to properly function (22).
Phospholipids—built on a glycerol backbone, two fatty acyl chains and a polar head group—account for the most part of the membrane, accordingly termed phospholipid bilayer. Five types of phospholipids are found asymmetrically distributed across the membrane, including phosphatidylcholine, phosphatidylethanolamine, phosphatidylserine, sphingomyelin, and phosphatidylinositol. The latter group, while making up a minor fraction of cell phospholipids in comparison to the other families, represent key molecules involved in crucial cellular processes due to their ability to interact with a variety of integral proteins in the plasma membrane (27). Phosphatidylinositol and related phosphoinositides (PtdIns) contain negatively charged head groups and are non-uniformly distributed in the inner (cytosolic) leaflet of the membrane. Phosphatidylinositol 4,5-biphosphate (PIP2), which has emerged as a highly versatile player in cell signaling, constitutes only ∼1–3% of the phospholipids in eukaryotic cell membranes (28) and represents the most abundant of the doubly phosphorylated PtdIns species (>99%) (29). Measurements of local PIP2 concentrations in a cell are still imprecise, however, studies employing fluorescently tagged pleckstrin homology (PH) domains—which bind specifically to membrane PtdIns—have more accurately estimated its effective concentration to range from 2 to 30 μM (30).
Membrane PIP2 levels seem to be controlled by a concomitant interplay between breakdown and synthesis. PIP2 is synthetized by its precursor phosphatidylinositol-4-phosphate (PI4P) in two successive phosphorylation steps by the enzymes PI4 and PI5 kinases (PIK4 and PIK5). Moreover, the efficient hydrolysis of PIP2 provides the source of two important secondary messengers in the cell, inositol trisphosphate (IP3) and diacylglycerol (DAG). Structurally, PIP2 contains an inositol head group with an average orientation of 45° with respect to the bilayer surface (31), a phosphoglycerol backbone, and two acyl chains. The net charge of this key signaling phospholipid is believed to be -4, however, it may range from -3 to -5 depending on multiple factors, including the local pH and its interactivity with proteins (32, 33). Despite being a minority phospholipid of the membrane, a large list of ion channels and transporters are reported to require certain levels of PIP2 in the membrane to function correctly, thus acting as a local switch with the ability to control regional cellular activity (34–42).
Cholesterol (C27H46O) is a central lipid in mammalian cells involved in multiple cellular processes at the vascular plasma membrane. It accounts for up to 50% of total membrane lipid molecules, as more than 90% of total cellular cholesterol is confined to the plasma membrane (43). The cholesterol molecule contains four hydrophobic hydrocarbon rings and a weakly hydrophilic hydroxyl (OH) group attached to one end, conferring the molecule an amphipathic character. The OH group facilitates the orientation of the molecule—with the hydrophobic regions pointed toward the hydrophobic core of the membrane—and the interaction with water molecules or adjacent phospholipids, restricting their motion and modifying the intrinsic properties of the lipid bilayer such as fluidity, stiffness and permeability (44–46). Like PIP2, cholesterol binds to multiple transmembrane proteins including different types of ion channels (K+, Ca2+, Na+, and Cl– channels), altering their conformational states and regulating their activity (47–51). Cholesterol is additionally involved in determining the physical integrity and functionality of lipid rafts—specific low-fluidity nanoscale domains which function as anchors for residing membrane proteins, representing “signaling hotspots” for key cellular signaling and trafficking events and host-pathogen interactions (52–54).
The cholesterol content in the membrane is defined by the dynamic balance between de novo biosynthesis from acetyl coenzyme A in the membrane of the endoplasmic reticulum, receptor-mediated uptake via plasma lipoproteins, transport to the membrane by caveolins, export and storage. These processes are tightly controlled by specific feedback mechanisms to ensure appropriate cholesterol homeostasis and maintain its levels within a narrow range. Therefore, disturbed cholesterol balance may profoundly alter cell/tissue function, lead to cell death, and further contribute to not only severe cardiovascular pathologies—including acute thrombosis and vascular occlusion preceded by hypercholesterolemia and atherosclerosis (55)—but also multiple types of cancer. Interestingly, cholesterol accounts for a large portion of the adult brain (∼35 g), which contains approximately 25% of the total amount of the cholesterol present in the body, making it the most cholesterol-rich organ (56). This fact obviously implies that a dysregulation of brain cholesterol levels or its turnover could contribute to the development or progression of several neurodegenerative disorders (i.e., Alzheimer’s disease or multiple sclerosis) and may represent a target for potential therapeutic avenues.
Despite being less abundant, sphingolipids represent another class of bioactive membrane lipids. The backbone of sphingolipids is ceramide, a molecule composed of a sphingoid head, generally sphingosine—an amino alcohol that contains a long hydrocarbon chain—with a long-chain fatty acid linked via an amine bond. Sphingolipids are a particularly diverse lipid family which regulate structural characteristics of the membrane such as curvature and thickness (54), participate in important intracellular signaling pathways and, mediate dynamic interactions between cells and their surrounding extracellular microenvironment (57, 58). Furthermore, sphingolipids can interact with cholesterol constituting highly dynamic raft-like membrane domains which facilitate the interplay with membrane proteins (54). Particularly, the association of these cholesterol-sphingolipids enriched domains with the protein caveolin constitutes caveola, small (50–100 nm) flask-shaped invaginations of the plasma membrane considered as local hotspots for lipid- and protein-mediated signaling cascades with a key role in mechanosensation in vascular endothelial and smooth muscle cells (59).
Inward Rectifier K+ Channels: Key Membrane Proteins Modulated by Specific Lipid Interactions
Classical strong Kir channels are integral membrane proteins highly expressed in vascular cells from the cerebral (12, 24, 60, 61), coronary (61), mesenteric (62, 63), and renal (64) circulation. Structurally, Kir2 channels constitute tetramers of α-subunits (Kir2.1-2.4) sharing ∼50–70% of amino acid identity (65–67), and each containing two transmembrane domains (M1 and M2), an intracellular N- and C-terminus, and a highly conserved extracellular loop that serves as a K+ selectivity filter (Figure 1A), essential for channel regulation and the mechanism of inward rectification (68, 69). This fundamental property (originally defined in skeletal muscle as “anomalous rectification”) arises due to the intrinsic ability of inward rectifiers to conduct large K+ currents inward (at VM negative to the EK) while passing small outward currents at more depolarized voltages (24, 61). The inward rectification occurs because of the robust voltage-dependent pore blockade by impermeant intracellular cations mainly polyamines and Mg2+ at depolarized membrane voltages, whereas at hyperpolarizing VM the blocking cations are absent from the pore, enabling K+ to flow (65, 69). Despite being inward rectification the biophysical fingermark of Kir channels, the small (a few picoamperes of amplitude) outward current is largely important for controlling arterial VM, basal tone and blood flow. This tiny outward current displays a negative slope-conductance region at physiological voltages (66, 70, 71) that increases other outward K+ currents to boost the original hyperpolarizing/vasodilatory response (61, 62, 72), playing thus a fundamental role in the regulation of regional tissue blood flow and flow distribution (Figures 1B,C). This unique electrophysiological property is predominant in channels containing Kir2.1 and Kir2.2 subunits—elements robustly expressed in vascular SMCs and ECs (22, 24). Moreover, Kir channel activity relies on extracellular [K+] levels. Particularly, increases in [K+]o leads to an augmentation of Kir inward current and shifts the channel activation curve to more depolarized potentials and, a subsequent rightward shift in the EK (24, 73) (Figure 1D). In a physiological scenario, vascular cells display membrane potentials positive to the EK, this implies that a rightward shift in the channel activation curve may also enhance the amplitude of the outward current. Due to this particularity, Kir channels are poised to contribute to K+-evoked functional hyperemia in excitable tissues (i.e., brain or skeletal muscle), a fundamental process that ensures regional increases in blood flow in to maintain tissue function and health (12, 73, 74). Micromolar concentrations of extracellular BaCl2—a potent and selective voltage-dependent Kir channel blocker—represent an ideal tool to characterize the molecular basis of Kir2 currents in vascular tissues (4). Barium sensitivity substantially differs depending on the subunits that conform the Kir2 channel. For instance, heteromers of Kir2.1-Kir2.2 subunits (robustly expressed in the vasculature) exhibit IC50 values ranging from 0.7 to 5 μM (at -100 to -120 mV), different from that of homomeric Kir2.1 or Kir2.2 channels (IC50 = 16 μM and 2.3 μM for Kir2.1 and Kir2.2 at -120 mV, respectively) (21). The selectivity of BaCl2 to block Kir channels is well-validated by two important facts: (1) Ba2+ exclusively blocks the inward rectification at voltages more hyperpolarized than EK and, (2) tissues expressing Kir channels exclusively exhibit [K+]-induced vasodilation sensitive to Ba2+ (24, 61). Consistent with this, Ba2+ superfusion caused a pronounced depolarization and subsequent vasoconstriction of isolated intact arteries, with this effect being greater at low intravascular pressures when resistance vessels exhibit hyperpolarized VM values and limited myogenic tone (23, 60). Accordingly, in vivo studies in rodents have shown a potent BaCl2-induced vasoconstriction of brain surface (14) and cremaster arteries (75).
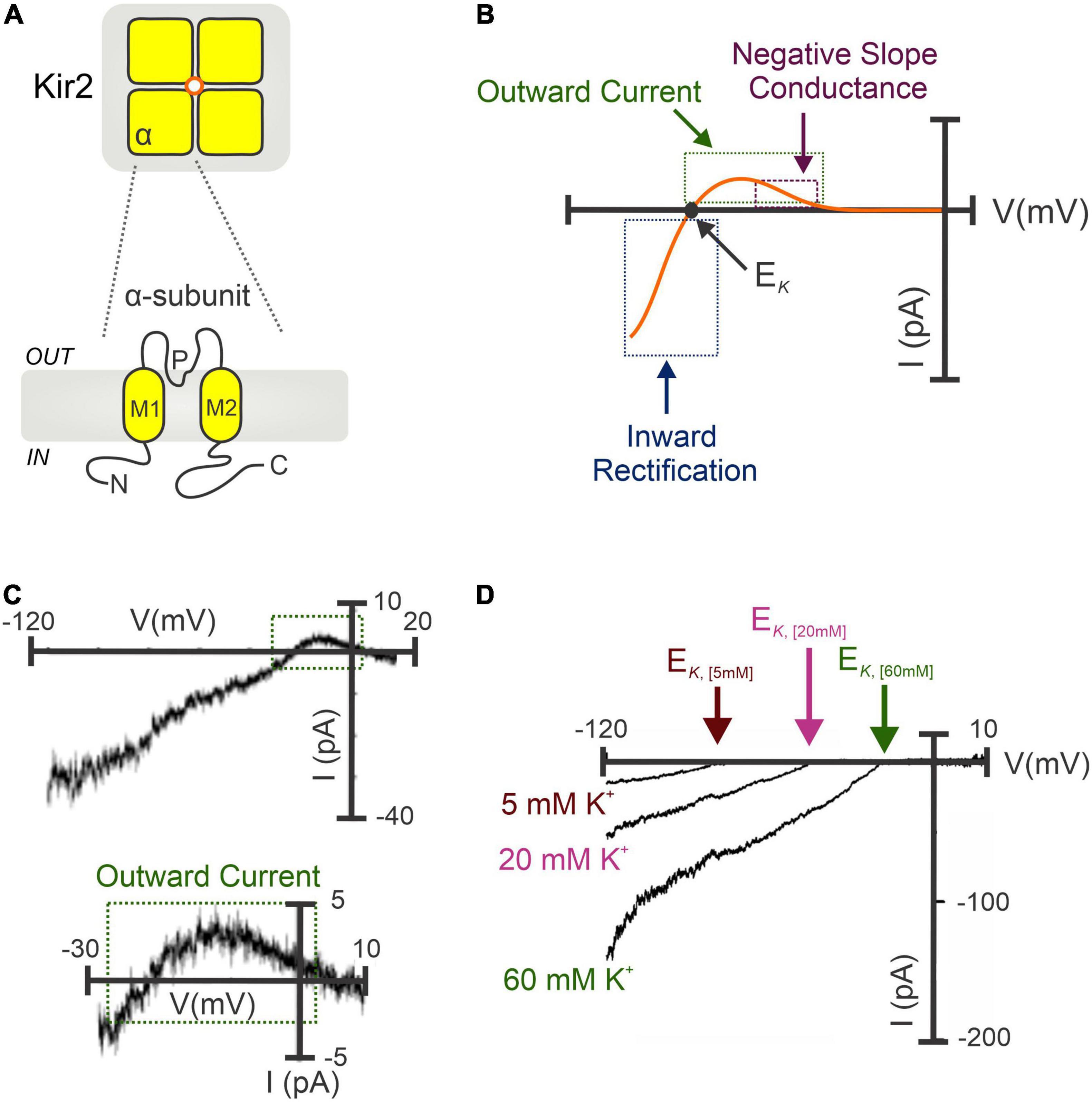
Figure 1. Molecular architecture and biophysical signature of strongly inward rectifying K+ (Kir2) channels. (A) Kir2 channels constitute tetramers of α-subunits (top). Each subunit contains two transmembrane domains (M1 and M2), a K+ selectivity pore (P) region, and C- and N-terminal domains (bottom). (B) Illustrative I-V relation of the Kir2 conductance involving Kir2.1 and/or Kir2.2 subunits. Notably, Kir2 whole-cell currents exhibit the characteristic inward rectification at hyperpolarized voltages (negative to the EK), and a small outward current with a negative slope conductance region at physiological VM. (C) Barium-sensitive Kir2 currents recorded in isolated cerebral SMCs exposed to high extracellular [K+] (60 mM) (top). A tiny (<5 pA) outward current with a negative slope conductance was successfully recoded (bottom, magnified from top). (D) Potentiation of Kir2 currents when raising the extracellular K+ concentrations, which increases Kir2 inward current density and shifts the channel activation curve to more positive VM, with a subsequent rightward shift in the EK. (C,D) Modified from Wu et al. (24) and Smith et al. (61).
Inward rectifiers were classically considered poor regulatory targets (62, 76), however, recent collecting evidence has shown that Kir2 channel activity is modulated by the surrounding membrane lipid environment. This includes two signaling lipid regulators with opposing effects, PIP2 and cholesterol, which stimulates and suppresses the Kir2 channel activity, respectively. Specifically, PIP2 is required at certain levels in the plasma membrane to stabilize Kir2 channels in a preferred open (active) state, and transient depletion of PIP2 by PLC activation in vivo suppresses these functions (17, 39). In this sense, sophisticated crystallographic and functional analysis have provided a detailed structural depiction of the binding sites by which negatively charged phosphates of PIP2 electrostatically interact with positively charged (basic) amino acid residues (arginines, lysines, and possibly histidines) of Kir2 channels to directly modulate their activity. In particular, the majority of these basic amino acid residues are positioned at the interface between the transmembrane and the cytosolic domains of the channel allowing PIP2 to interact with both regions (Figure 2A). Specifically, the inositol polar group of the phosphoinositide makes interactions with a specific phosphatidylinositol region in the cytoplasmic domain of the channel whereas a conserved non-specific phospholipid binding site (RWR; containing the amino acid residues ARG43, ARG45, and TRP44) located in the transmembrane domain (N-terminus of the outer helix) provides specificity for the acyl chains and phosphopglycerol backbone of PIP2 (39, 77) (Figure 2B). Nonetheless, the only crucial element of PIP2 for Kir channel regulation seems to be the number of phosphates in the polar head as other PtdIns species (PtdIns(4)P and/or PtdIns) exhibited diminished activity or failed to stimulate Kir2 channels (39). In this sense, various polyvalent cations including polyamines, trivalent metals, neomycin and polylysine are capable of blocking Kir channel activity by screening the negatively charged polar groups of PIP2. This membrane lipid binds to the channel and provokes a conformational change to open the gate, thus resulting in increased channel activity. Particularly, this conformational change includes the contraction of a peptide flexible linker into a compact helical structure which induces a reorientation and rotation of the cytoplasmic region, becoming tethered to the transmembrane domain which in turn, provides a tangential force that automatically opens the gate of the channel and promotes the conductance of K+ ions across the pore (77) (Figures 3A,B). A Previously published electrophysiology study employing Kir2.1 (monomeric, dimeric, or tetrameric) constructs expressed in Xenopus oocytes showed that only one subunit of the tetrameric arrangement of the Kir channel is sufficient for channel activation to its fully open state. Additionally, further interaction with additional subunits induces multilevel positive cooperativity, increasing channel availability to open (reducing dwell time) and facilitating channel conductance (78). Among the diverse Kir subunits, Kir2.1 channels possess high affinity for PIP2, and are not influenced by its depletion. In contrast, other subunits including Kir2.2 and Kir2.3 display relatively low PIP2 affinity and therefore are robustly influenced by PIP2 breakdown (79). Moreover, the strength of PIP2-Kir direct interactions not only enhances Kir channel activity but also controls the sensitivity of the channel to other regulatory factors such as pH, protein kinase C, Mg2+ or phosphorylation (79).
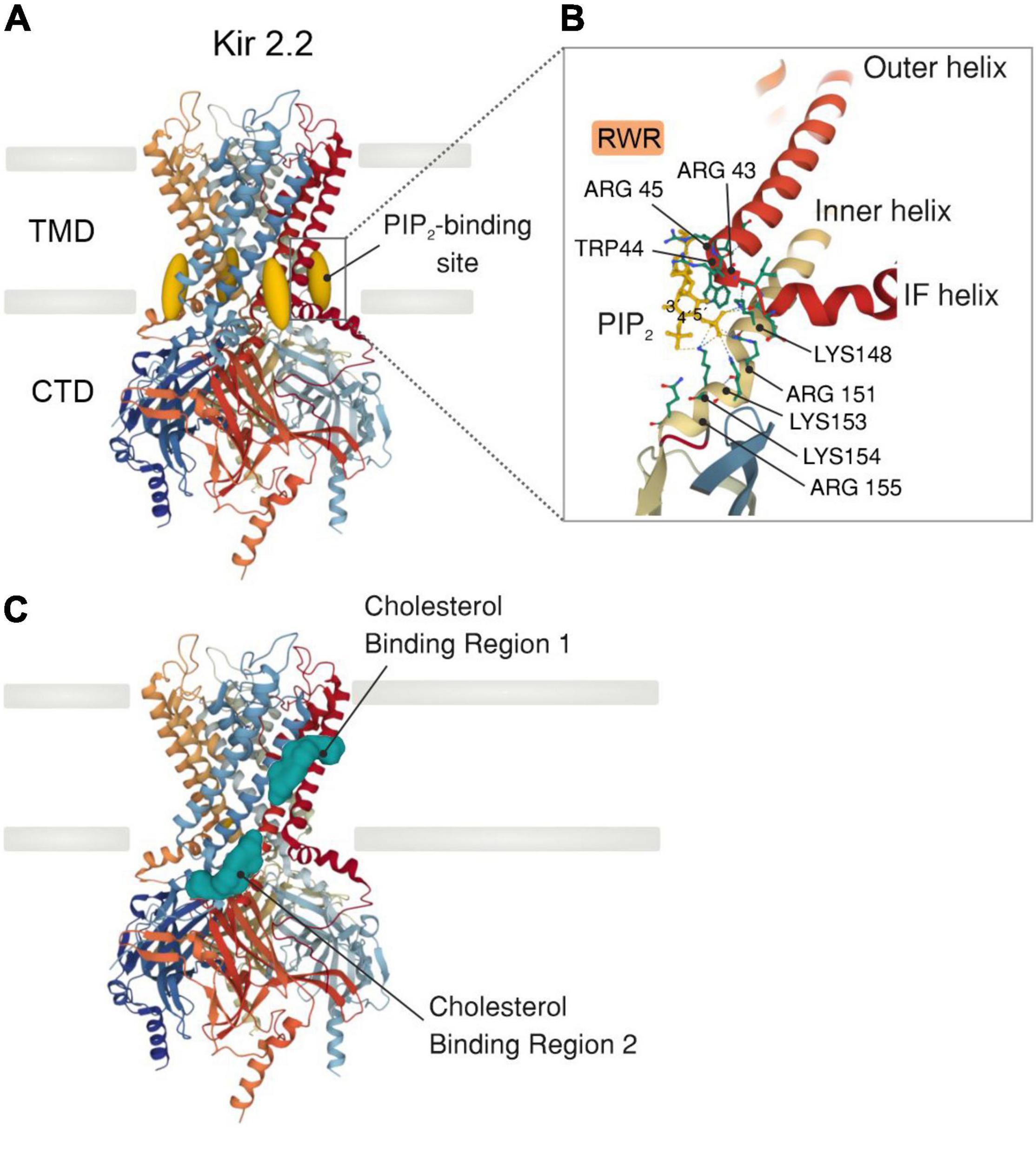
Figure 2. X-ray crystal structure of a Kir2.2 channel with PIP2 or cholesterol bound. (A) Side view of a model of a Kir2.2 tetramer consisting of a transmembrane domain (TMD), which comprises the potassium-selective pore, and a large cytosolic (CTD) domain (PDB ID: 3SPI). PIP2 molecules (dark yellow) bind at the interface between the TMD and the CTD. (B) Detailed illustration showing the PIP2-binding site in a similar orientation as outlined in A. The primary amino acid residues bound (dashed lines) to PIP2 are highlighted. All side chains are shown as solid lines. (C) Side view of a Kir2.2 channel model illustrating the two distinct cholesterol binding regions (green): one residing in the center of the TMD and the other at the interface between the TMD and the CTD.
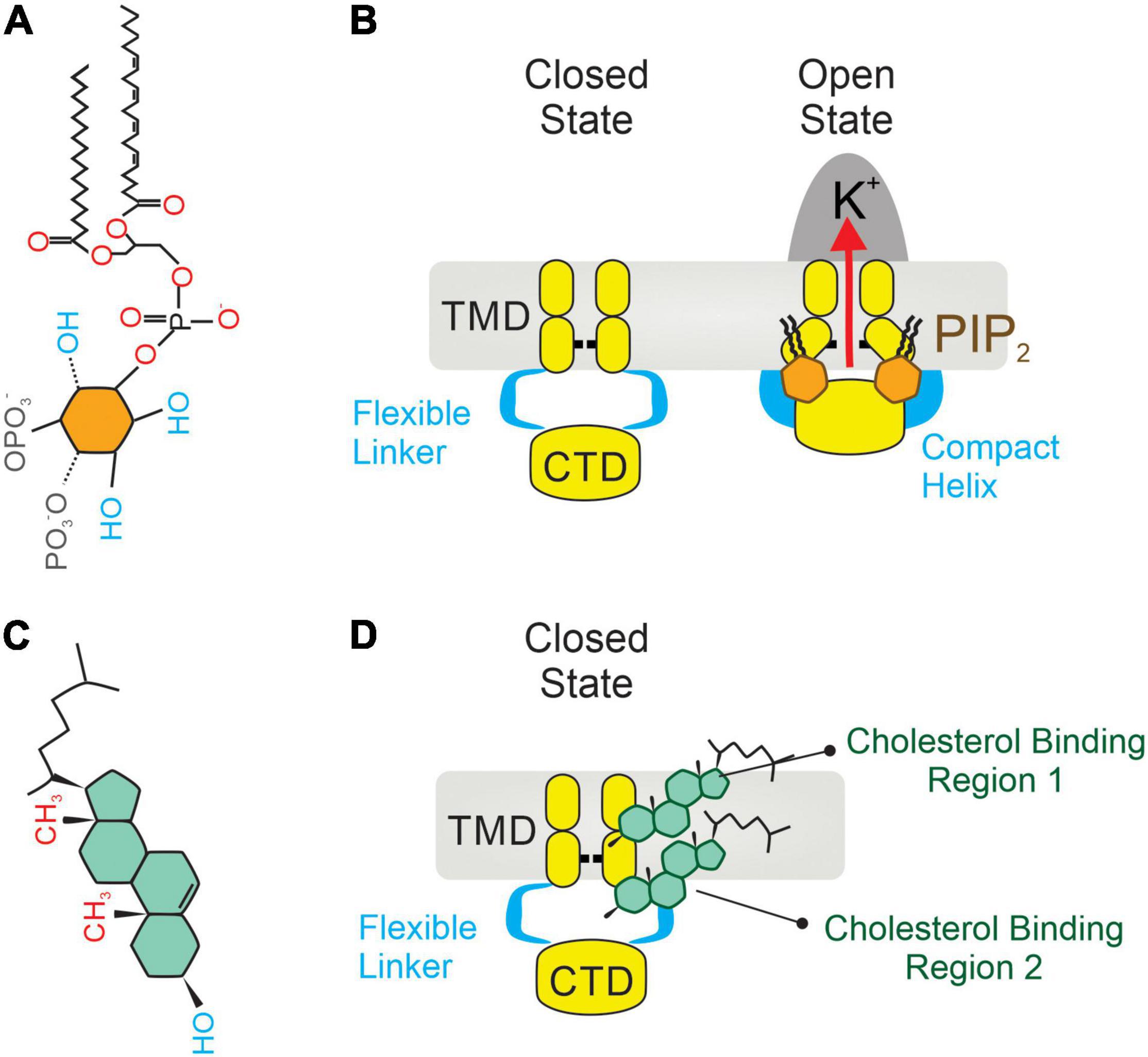
Figure 3. Two integral membrane lipids, PIP2 and cholesterol, bind to and regulate the activity of Kir2 channels. (A) Molecular architecture of a PIP2 molecule, constituted by an inositol polar group, a phosphoglycerol backbone and two acyl chains. (B) Mechanism of Kir2 stimulation by membrane PIP2 binding, which causes a change in the architecture of the flexible linker into a compact helix. Then the cytoplasmic domain (CTD) translates toward and becomes tethered to the transmembrane domain (TMD), which in turn mechanically opens the channel. (C) A cholesterol molecule contains three main parts: (1) tetracyclic carbon ring, (2) a polar 3β-hydroxyl (OH) group, and (3) a short non-polar carbon chain. (D) Location of two non-annular hydrophobic cholesterol binding domains in Kir2 channels: one located in the center of the TMD (1), and the second at the interface between the TMD and the CTD (2). Upon direct cholesterol interaction, the hinging motion of the transmembrane pore helix changes, causing channel stabilization in a preferred closed (silent) state.
In the brain circulation, our recent study (22) revealed that ECs, freshly isolated from third-order middle and posterior cerebral arteries, possess Kir2.1-Kir2.2 heterotetramers which are tightly controlled by membrane PIP2 levels. In this context, EC Kir2 conductance was impacted in a timely manner by dialyzing (conventional whole-cell patch-clamp configuration) the cells with diverse pharmacological drugs that modify membrane PIP2 concentrations. Consistent with PIP2 facilitating Kir channel activity, depletion of this signaling lipid by dropping the intracellular ATP concentration to zero, blocking PIK, or adding neomycin—an aminoglycoside that impairs the electrostatic interaction between PIP2 and the Kir channel—to the patch pipette induced an evident steady decrease of the Kir current. This whole-cell Kir current rundown was further prevented by incorporating the water-soluble short-chain analog of PIP2 (dioctanoyl-PIP2, diC8-PIP2) to the intracellular solution, a finding that poises membrane PIP2 as an intrinsic regulator of endothelial Kir2 channels by promoting channel stabilization in a preferred open (active) state. Surprisingly, Kir2 channels residing in SMCs were insensitive to these pharmacological manipulations over time, suggesting that membrane PIP2 is a minor modulator of this channel pool (22). These findings are consistent with the coexistence of two cellular populations of Kir2 channels (ECs vs. SMCs) in cerebral resistance arteries which are distinctly regulated by their neighboring lipid microenvironment.
As the brain vascular tree ramifies, pial (surface) arteries give rise to penetrating and parenchymal arterioles, and eventually to a capillary network containing hundreds of capillaries which vastly extend the arteriolar territory of perfusion. Capillaries, the narrowest blood vessels in the body, are tubes made up of a single layer of ECs which act as a neural activity-sensing network being thus much more than simple conduits for blood (12). At the capillary level, and consistent with our work, PIP2 has been reported as a requisite molecule to maintain EC Kir2.1 channels in a preferred open state. This property makes it essential for the generation of retrograde electrical signals from capillaries which dilate upstream arterioles and contribute to the fundamental process by which CBF increases to satisfy the metabolic demands of active neurons (functional hyperemia) (23). Since PIP2 is also known to exert opposite effects on capillary EC TRPV4 channel activity, this minor lipid has been suggested to serve as a regulatory switch that actively governs the balance between electrical (Kir2.1) and Ca2+ signaling (TRPV4) (80). In this context, Dabertrand and colleagues have recently revealed that exogenous application of PIP2 corrected CBF deficits by recovering capillary EC Kir2.1 activity in a monogenic model of Small Vessel Disease (SVD)—the major cause of stroke and dementia (15).
Contrarily to PIP2, membrane cholesterol strongly suppresses the activity of Kir channels by placing the channel in a preferred closed (silent or inactivated) state (81). This notion originated from electrophysiology studies that described a reduction in whole-cell Kir2 currents—being that the channel open probability, unitary conductance and channel protein expression were slightly impacted—when membrane cholesterol levels were increased (46). The molecular foundation of cholesterol regulation of Kir channels was effectively elucidated by the Levitan group which identified several cytosolic residues in the G-loop, the N-terminus and the peptide linker between the C-terminus and the inner transmembrane helix of the Kir2.1 channel. These residues form a belt that embraces the cytosolic pore and controls the cholesterol sensitivity of the channel. The cytoplasmic belt is different from the cholesterol binding site, but it allows cholesterol binding to a different part of the channel which is critical for the functionality of the gating system (82). Paradoxically, this cytosolic arrangement also contains some essential residues for PIP2 sensitivity, a fact consistent with both membrane lipids (PIP2 and cholesterol) sharing a common regulatory hub that controls the Kir2.1 channel state (open vs. silent). Interestingly, this attribute is absent in Kir2.2 channels as they lack the aforementioned overlap in PIP2- and cholesterol-binding residues (164). While cholesterol mediates Kir2 activity by binding to these specific residues, it has been also proposed to indirectly impact ion channel function during changes in the physical properties of the membrane (i.e., membrane stiffness). In this sense, the substitution of native cholesterol with its optical isomer (epicholesterol) not only abrogated the inhibitory effect, but also exerted opposing effects on endothelial Kir2 channel activity, revealing that the sensitivity of Kir2 to cholesterol is stereo-selective to cholesterol optical analogs (83). These findings effectively support the hypothesis that cholesterol suppresses Kir channel activity directly rather than by altering plasma membrane structure (84). In this context, Kir2 channels contain two non-annular cholesterol-binding motifs, one of which resides in the center of the transmembrane region and the second at the interface between transmembrane and cytoplasmic domains of the channel (Figure 2C). Upon favorable cholesterol-channel binding, the hinging motion of the transmembrane pore helix is altered, promoting thus channel stabilization in a preferred closed state (20) (Figures 3C,D).
Further studies using endothelial culture models revealed that Kir2.1 and Kir2.2, the major structural components of the endothelial Kir channel, are suppressed or activated following enriching or depleting membrane cholesterol, respectively, whereas Kir2.3 and Kir2.4 were less sensitive to cholesterol content manipulations (51). Moreover, the degree of Kir2 suppression matches with the amount of membrane cholesterol loading, being completely reversible following subtraction of cholesterol excess. The negative effect of cholesterol on EC Kir activity was also demonstrated in vivo, where Kir2 currents recorded in ECs isolated from the aorta of Yorkshire pigs fed atherogenic high-cholesterol diet were significantly diminished comparing to those recorded from healthy animals (85). In an attempt to translate these key findings into the brain vasculature, investigators have dialyzed native SMCs and ECs with methyl-β-cyclodextrin (MβCD), a compound widely used in cell biology to deplete cells of cholesterol (22). This pharmacological manipulation impacted Kir2 channel activity in a time-dependent manner, an effect that was subsequently prevented when the cells were exposed to cholesterol-saturated MβCD. Intriguingly, these responses were particularly evident in SMC Kir currents comparing to the endothelium, identifying cholesterol as a major modulator of the smooth muscle Kir channel population by promoting its stabilization in a preferred closed state.
The Plasma Membrane of Vascular Cells: An Efficient Mechanical Sensor
Resistance arteries are small blood vessels (<400 μm in diameter) that contribute to peripheral vascular resistance. The vascular wall of these blood vessels displays structural and intrinsic (local) responses to ever-dynamic forces to eventually regulate vascular tone and match blood flow to tissue metabolic demands. This ability is accomplished by the plasma membranes of two of the principal constituents of the vascular wall—smooth muscle and endothelium—which are constantly exposed to mechanical stimuli, mainly originated by a pulsatile blood flow—stretch (tensile stress) and/or shear stress (86). In this context, specific mechanosensitive molecules residing within the plasma membrane—ion channels, caveolae, and/or surface receptors—can modify their conformational state and electrical/chemical properties in response to mechanical disturbances and transduce them into a physiological response (87, 88).
Stretch and Myogenic Tone
The vascular smooth muscle layer of a resistance artery (or of downstream penetrating arterioles) possesses an inherent ability (independently of the endothelium and nerves) to contract—and reduce the luminal diameter—in response to an abrupt increase of transmural pressure (i.e., radial stretch) (89). In the brain, this phenomenon known as “myogenic tone” elevates resistance to blood flow (90, 91), and is essential for setting basal vascular tone, maintaining a constant perfusion over a range of intraluminal pressures, and fine-tuning local CBF while protecting downstream capillary networks from damage (6, 92). Interestingly, myogenic tone responsiveness becomes more robust as vessel size decreases (93, 94), and this could be explained by an increase in vessel wall distensibility as the vascular tree ramifies (95). Moreover, the myogenic tone of resistance arteries can be influenced by hemodynamic forces (i.e., flow), metabolic factors and vasoactive mediators released from other cell types, including the endothelium (96–98).
The underlying mechanisms of this phenomenon have been extensively studied over previous decades (90, 99–103). An increase of intraluminal pressure modulates the activity of stretch-sensitive ion channels expressed in SMCs resulting in membrane depolarization and the activation of voltage-gated calcium channels (VGCCs), which in turn elevates the intracellular levels of contractile Ca2+ (7, 104). As a result, mechanosensitive ion channels residing in SMCs are often investigated due to their ability to serve as pressure sensors by depolarizing arterial membrane potential (VM) and initiating the myogenic response (90, 105). In the brain, the myogenic response is imperative for precise vascular SMC function and accordingly, myogenic tone disruption has been associated with many vascular diseases including stroke, hypertension and dementia (106–108).
Shear Stress-Induced Vasodilation
The endothelial layer that lines the vasculature is experiencing a continuous frictional drag force of the blood flow and by rolling blood cells over its luminal surface. This type of internal stress known as (fluid) shear stress is opposed by tension and deformation in the endothelium and initiates a complex signaling cascade—involving mechanosensory complexes containing multiple surface molecules including ion channels and integrins—that in short timescales causes vessel dilation to preserve the functionality of the vasculature (109). ECs are exposed to diverse shear stress patterns in vivo that may vary in magnitude, direction and temporal characteristics in relation to their location within the vascular bed.
Although the exact signaling pathways that facilitate endothelial responses to flow are incompletely determined, mechanosensitive channels have been suggested as major sensors of laminar flow, being able to respond to changes in mechanical loading, integrate the signal and elaborate a response that impacts the ion channel permeability of the plasma membrane. A large number of mechanically activated calcium and potassium channels including P2RX4 (110), transient receptor potential (TRP) vanilloid subtype (TPV4) (111), Piezo 1 (112), Kir2.1 (113), and small-conductance Ca2+-activated K+ channels (SK) (114) have been suggested to play a role in endothelial responses to blood flow in various vascular beds (109). In the brain, endothelial cells express a complement of K+ channels including Kir, KATP, intermediate-conductance Ca2+-activated K+ channels (IK), and SK (11, 21, 61, 108), and other ion channel families such as TPV4 (80, 115) or Piezo1 (116). Notably, the expression of these ion channel species displays some differences depending on the anatomical location of the ECs along the vascular tree. Arterial/arteriolar ECs possess IK and SK channels that play a critical role in transducing rises in intracellular Ca2+ levels into hyperpolarizing/dilatory signals, whereas this ion channel family is absent in the capillary endothelium. Notably, ECs do not express voltage-gated channels; however, these cells are electrically coupled to SMCs and thus able to directly govern vascular basal tone (117).
Mechanosensitive Ion Channels, Cellular Routes for Force Transmission, and Signaling Microdomains
Mechanosensitive ion channels represent a firmly established set of biological molecules which possess an extraordinary capacity to sense and process external mechanical stimuli by initiating an electrical or chemical signal, eventually integrated into a coherent cellular response (118). Among them, several ion channels, including TREK-1 (119) and TRPC1 ion channels (120), are directly activated upon force transmission through the lipid bilayer without the involvement of any additional associated cellular components (i.e., cytoskeleton), a type of gating model typically observed in bacterial ion channels known as the “bilayer mechanism.” In contrast, the vast majority of mechanosensitive ion channels residing in animal cell membranes—including many TRP channels (121–125)—are governed by the “tethered mechanism” by which force is indirectly applied to them via cytoskeletal, extracellular matrix (ECM) molecules (e.g., fibronectin) and, accessory scaffolding components (39, 70). As a wide range of physical signals impact the plasma membrane, they are transferred across the ECM to the cytoskeleton (“dual-tether model”) through modulating spanning integrin receptors at focal adhesion sites (126). Therefore, cytoskeletal actin and additional structural components may represent key routes of cellular force transmission with clear impact on ion channel mechanosensitivity.
Actin is the most abundant cytoskeletal protein in vascular smooth muscle (∼20% of total protein content) with a critical role in the maintenance of cellular integrity (127), whereas contractile actin filaments associate with myosin to form the contractile machinery (128). The architecture of the actin cytoskeleton in SMCs is a dynamic process, switching from polymerized to depolymerized states and contributing to muscle contraction or relaxation, respectively. This process is mainly defined by the filamentous- (F) to globular- (G) actin ratio which under resting conditions—containing a significant pool (∼30%) of G-actin monomers—is approximately 2:1 (129). An external mechanical stimulus (pressure/stretch) provokes the transition of G- into F-actin leading to smooth muscle contraction and an increase in force generation. Moreover, studies using isolated and pressurized cerebral arteries demonstrated the importance of this ever-dynamic process for myogenic tone development as actin cytoskeletal disruption blunted pressure-induced constriction of cerebral arteries (129). In the context of ion channel function, pressurization of cerebral arteries treated with actin polymerization blockers (i.e., cytochalasin B) elevated Ca2+ influx and membrane depolarization. However, the precise identification of the ion channel species and scaffolding protein intermediates involved in this process remain undefined (130). In this context, members of the dystrophin-associated protein complex (DAPC) have been studied, as they mainly operate as connectors of the actin cytoskeleton to the membrane and ECM (131).
Dystrophin is a large (427 kDa; 110 nm) sub-membrane cytoskeleton protein that interacts with syntrophin and various protein elements to constitute the DAPC in vascular smooth muscle (131). Loss of function in the DMD gene encoding Dystrophin causes Duchenne muscular dystrophy, a rare but distinctive disease characterized by progressive muscle weakness and wasting primarily in males. The severe pathological effects of this rare genetic disease have stimulated a solid focus of research exploring the function of dystrophin and associated proteins (132, 133). Dystrophin spans actin filaments laterally, binding alongside to them through the N-terminal domain and forming a solid physical bridge with the cytoskeleton (134). The C-terminus of the dystrophin molecule enables the assembly of the DAPC by providing multiple binding sites for a group of signaling and channel proteins including syntrophins, sarcoglycans, dystroglycans, and dystrobrevins (131, 135). Collectively, the DAPC anchors the actin cytoskeleton to the ECM to physically protect the sarcolemma from the membrane perturbations developed during muscle contraction (136). Consequently, the lack of dystrophin increases the chance of sarcolemmal rupture following skeletal muscle contraction, a characteristic feature of Duchenne muscular dystrophy (136). In the vasculature, the loss of dystrophin in SMCs leads to decreased vessel contractility and impaired stretch-induced gene transcription, resulting in overall smooth muscle function and mechanosensitivity decline (137).
Syntrophins (from the Greek, syntrophos, thought to mean companion) are 58 kDa membrane-associated adaptor proteins involved in DAPC formation and the recruitment and regulation of surrounding signaling proteins—including ion channels—into a regulatory macromolecular assembly (138). The syntrophins are a multigene family involving five homologous isoforms (α1, β1, β2, γ1, and γ2) that display tissue-specific expression profiles. Among them, α1 is predominantly abundant at the sarcolemma of muscle fibers and directly binds to the C-terminal domain of dystrophin (139–141). Syntrophins lack intrinsic enzyme activity, however, they contain unique domains than enable them to bind other proteins and arrange signaling microdomains at the cell membrane. Structurally, all isoforms possess two pleckstrin-homology (PH) domains, one PDZ domain that facilitates protein-to-protein interactions, and a syntrophin unique (SU) domain located in the C-terminus which directly binds to dystrophin (140). Particularly, the PDZ domain recognizes and attaches to protein targets containing the C-terminal motif Ser/Thr-X-Φ-COOH, where X and Φ represent interchangeable and hydrophobic residues, respectively (142). Among the protein binding partners, syntrophins haven reported to recruit Kir2 channel subunits via their C-terminal PDZ binding motif (Ser-Glu-Ile) (143). This interaction enables syntrophins to connect Kir2 channels directly to the DAPC and form a structural link with the cytoskeleton. It has also been observed that this interaction is more strongly associated with the Kir2.2 subunit, with weaker interactions reported with Kir2.1 (143). While our understanding of the physiological relevance is still in its infancy, proteomic analyses have identified Kir2-synthrophin interactions in heart, skeletal muscle and brain (143, 144).
The clustering of signaling components with their protein targets—which facilitate rapid cellular responses—is mostly achieved by caveolae (or “little caves”) (145). Caveolae are unique cholesterol-enriched, flask-shaped and small (50–100 nm in diameter) membrane invaginations with a major role in many cellular processes including the regulation of cellular signaling cascades and adaptation to the constant tensional changes that cell membranes undergo (146). The curved structure of caveolae is shaped and stabilized by the insertion of caveolin proteins into the membrane. Among them, caveolin-1 (Cav1) and caveolin-2 (Cav2) are widely expressed across multiple tissues and cell types, whereas caveolin-3 (Cav3) is expressed primarily in smooth, skeletal and cardiac muscles (147, 148). Despite the three isoforms that have been identified in vascular smooth muscle, Cav1 is the only component that is required for caveolae formation and its deletion has been shown to weaken contractile responses (147, 149). In fact, the lack of this isoform leads to impaired endothelium-dependent relaxation, contractility and myogenic tone (149). Functionally, Cav1 and caveolae enhance signaling cascades by engaging key molecular components such as ion channels, adaptor proteins, and receptors within microdomains of the membrane (145). Moreover, Cav1 is known to regulate the activity of numerous ion channels and enzymes. As an example, swelling activated Cl– currents are suppressed in Cav1-deficient cell lines but restored by transient expression of Cav1 (150).
Actin filaments are known to associate with and stabilize caveolae, as depolymerization of the cytoskeleton leads to migration of Cav1 throughout the cell (145). This association creates a highly efficient mechanosensitive territory, providing a local area where mechanical stimuli can be focused, sensed, and processed. This mechanism has been proposed for stretch-activated Ca2+ channels as they reside in caveolae and their activity is tightly regulated by the actin cytoskeleton (145, 151, 152). Cav1 has been previously reported to directly bind Kir2 channels reducing current density without impairing the single channel properties or membrane protein expression. In this context, Han and colleagues previously demonstrated that Cav1 acts as a negative regulator of Kir channel activity and that Cav1 and cholesterol stabilized the channel in a closed silent state by a shared regulatory mechanism (153) although Cav1 is not required to confer cholesterol sensitivity to the channel. With the aid of crystallography this study effectively identified two putative Cav1 binding domains, the first one at the interface between the outer transmembrane helix and the N-terminus, and the second in the outer transmembrane helix close to the extracellular region of the channel. Moving to the brain circulation, we previously reported the close association (< 40 nm) of Kir2 channel subunits and Cav1 within caveolae structures in the membrane of brain vascular smooth muscle cells, a tandem arrangement that may enhance Kir2 channel mechanosensitivity (22).
Intriguingly, we recently demonstrated that simulated pressure using a hypoosmotic challenge, suppressed cerebral arterial smooth muscle Kir2 currents in whole-cell recordings, an effect that was entirely recovered by pre-incubation with actin-disrupting agents (i.e., latrunculin A and/or cytochalasin D). Disruption of caveolae-forming proteins similarly prevented suppression of smooth muscle Kir2 currents during cell-swelling. Moreover, immunofluorescence approaches revealed the expression of both scaffolding proteins, syntrophin and Cav1, in vascular smooth muscle and a proximity ligation assay highlighted the intimate structural association with Kir2.2 subunits (within 40 nm of one another), suggesting their participation in channel mechanosensing. These findings provided compelling evidence that Kir2 channel mechanosensitivity involves unique interactions with the cytoskeleton which are likely driven by scaffolding proteins.
Cellular Pools of Kir Channels, Biomechanical Stimulus, and PIP2- vs. Cholesterol-Kir2 Interactions
Numerous studies have proposed vascular Kir2 channels as mechanosensitive components, primarily involved in the initial response of both pressure-induced vasoconstriction and flow-induced vasodilation of resistance arteries (21, 23, 143). Wu et at. (24) originally demonstrated that smooth muscle Kir2 channels serve as molecular mechanosensors, playing a critical role in the development of pressure-induced constriction (myogenic tone) of brain resistance arteries. Specifically, Kir2 currents recorded from native SMCs were potently suppressed during hypoosmotic challenge (swelling), a stimulus known to activate mechanically sensitive inward currents, leading to myogenic-like responses in the resistance vasculature (103, 154, 155). We recently proved and further expanded these findings by applying negative pressure through the patch pipette (conventional whole-cell configuration) via pneumatic transducer (-15 to -45 mmHg). This complementary approach to reduce cell stretching caused an evident increase in the smooth muscle Kir2 conductance (22), reinforcing the view of Kir2 channels as exquisite mechosensitive elements of the vascular cell membrane. These impressive observations may imply a dynamic interplay between Kir2 and pressure-sensitive transient receptor potential (TRP) channels to facilitate myogenic tone development (105, 156). Accordingly, greater Ba2+-induced constrictions (employed as an index of Kir activity) were observed when the arteries were exposed to low intravascular pressures, where the channel is most active. We further demonstrated that the lipid microenvironment surrounding Kir2 can impact the ability of the channel to respond to the pressure stimuli. In this framework, membrane cholesterol depletion prevented pressure-induced Kir2 inhibition in vitro and significantly augmented Ba2+-induced vasoconstriction in response to high intraluminal pressures. Together, these provocative data suggest that cholesterol-smooth muscle Kir interactions are crucial for hemodynamic sensing of pressure in the cerebral vasculature (22).
The strategic location of the vascular endothelium—at the interface between tissue and blood—functionally implies the involvement of the endothelial Kir2 channel population in flow-sensing. Supporting this hypothesis, a great body of studies employing a combination of ex vivo functional and in vitro electrophysiology approaches identified the endothelial shear-sensitive Kir2.1 channel as a major contributor of flow-induced dilation of resistance arteries (63, 157). Briefly, elevations in blood flow-associated shear stress initiates endothelial cell membrane hyperpolarization (∼ 2–6 mV in response to shear stresses of 1–5 dyn/cm2) by increasing the open probability of K+ channels—a response entirely blocked by micromolar concentrations of extracellular Ba2+ (a specific blocker of Kir2 channels) or Cs+ and/or completely abolished in mesenteric ECs freshly isolated from genetically modified mice lacking the Kir2.1 gene (heterozygous deletion of Kir2.1) (63, 113, 158). Moving to the brain vasculature, we demonstrated that fluid shear stress enhanced the density of native endothelial Kir currents, a response that was functionally translated into flow-induced dilation of cerebral resistance arteries as this response was prevented by intraluminal perfusion BaCl2 perfusion. We further showed a decrease of shear stress potentiation of endothelial Kir2 channel activity and a consequent abolition of flow-induced vasodilation of endothelium-intact cerebral resistance arteries following neomycin-induced membrane PIP2 depletion (22). These impressive findings identify PIP2 as a crucial driver in conferring shear stress sensitivity to the endothelial Kir2 channel population (22). Paradoxically, recent work from Levitan’s group revealed a marked reduction in the flow-induced activation of endothelial Kir2.1 channel activity during hypercholesterolemic conditions (159). These findings were reinforced using isolated arteries from an established mouse model of dyslipidemia-induced endothelial dysfunction (Apolipoprotein E-deficient mouse; apoE–/–) (160), in which depletion of membrane cholesterol content rescued flow-induced vasodilation in a Kir2.1-dependent manner (159). In line with these findings, the authors propose that endothelium dysfunction exhibited during hypercholesterolemia (and potential arteriosclerotic vascular disease) may be attributable to cholesterol-induced inhibition of Kir2.1 channels. Brain vascular endothelial Kir2 channels display less sensitivity to cholesterol (22), however, enhanced levels of this lipid in plasma during dyslipidemia may induce a dynamic reorganization of lipid content at the membrane level with an increase in cholesterol and possibly a drop in PIP2. This resulting lipid membrane profile would push adjacent endothelial Kir2 channels to reside in a preferred silent state, therefore rendering them insensitive to shear stress stimulation. Consistent with this notion, a previous study from Levitan’s group (159) and preliminary findings from our group (161) revealed a marked reduction in endothelial Kir2 channel activity and a consequent impairment of laminar flow-induced vasodilation in cerebral arteries isolated from two distinct models of dyslipidemia in mice. These observations have also been observed in humans, manifesting as impaired flow-induced vasodilation in hypercholesterolemic patients, thus suggesting cholesterol-induced suppression of Kir2.1 as a mechanism underlying endothelial dysfunction in this pathology (162).
Concluding Remarks and Future Perspectives
Cerebral resistance arteries contain various cellular types including smooth muscle and endothelial cells—two intimately associated components that work in concert to set basal tone and control brain tissue perfusion. The plasma membranes of these vascular cells are constantly exposed to a range of biomechanical forces including membrane stretch (pressure) and shear stress (flow). These vascular cell types express functional Kir2 channels, and each cellular population is distinctly modulated by key lipid signaling molecules which can sense diverse hemodynamic forces. Particularly, Kir2 channels are uniquely modulated by membrane PIP2 and cholesterol depending on their cellular location (endothelium vs. smooth muscle); being these specific lipid-Kir2 channel interactions crucial to confer shear stress- and pressure sensitivity to the endothelial and smooth muscle Kir channel pools, respectively. These two cellular Kir2 populations have been proposed to actively interact to drive an integrated vascular Kir2 conductance designed to ultimately govern VM and basal tone in the brain circulation (Figure 4) (22). The role of cytoskeleton and focal adhesion proteins is likely to be a major player in these hemodynamic responses. In this context, we recently proposed a model in which smooth muscle Kir2 mechanosensitivity is in part, conferred by a multiprotein complex comprising Kir2.2, syntrophin, actin, and Cav1 (Figure 5). It is also intriguing to consider the involvement of the cytoskeleton and diverse scaffolding proteins in the context of shear stress responsive endothelial mechanisms. In the short-term future, genetic manipulations at the level of stretch- and shear-sensitive ion channels will represent valuable tools to further study vascular mechanosensation, a fascinating area of research.
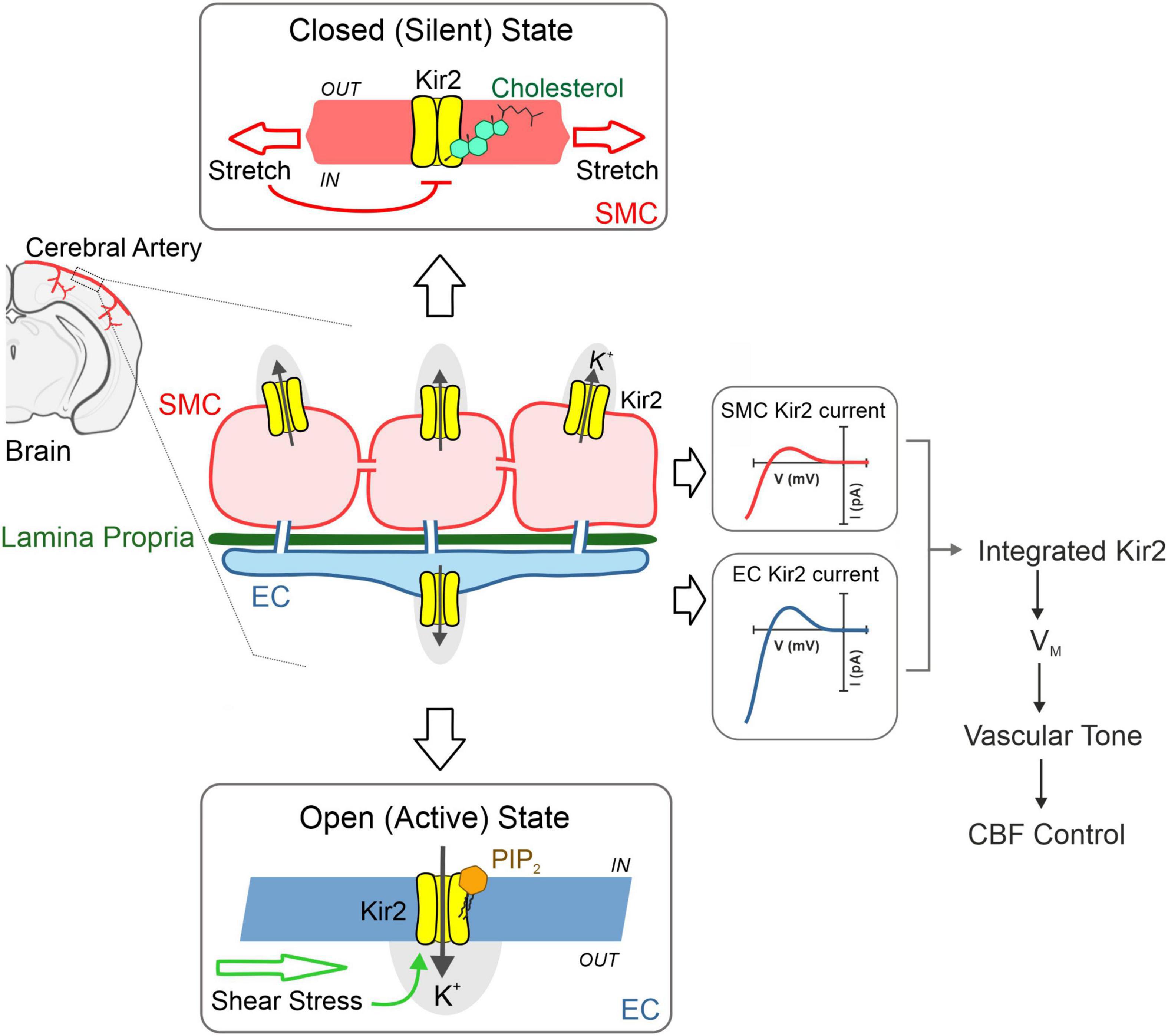
Figure 4. Cerebrovascular Kir2 channels and hemodynamic control. Schematic depiction explaining the modulation of two cellular populations of Kir2 channels, functionally expressed in smooth muscle and endothelium. Briefly, structural membrane lipids including PIP2, and cholesterol represent primary regulators of Kir2 channels, with the former robustly stimulating endothelial Kir2, whereas the latter preferentially suppresses smooth muscle Kir2 activity. These lipid-Kir interactions help confer pressure and shear stress sensing, hemodynamic forces that diminish and enhance smooth muscle and endothelial Kir2 channel activity, respectively. These cellular channel populations dynamically interact to drive an integrated Kir2 current through which hemodynamic stimuli contribute to set VM, arterial tone and cerebral blood flow (CFB) control. Modified from Sancho et al. (22).
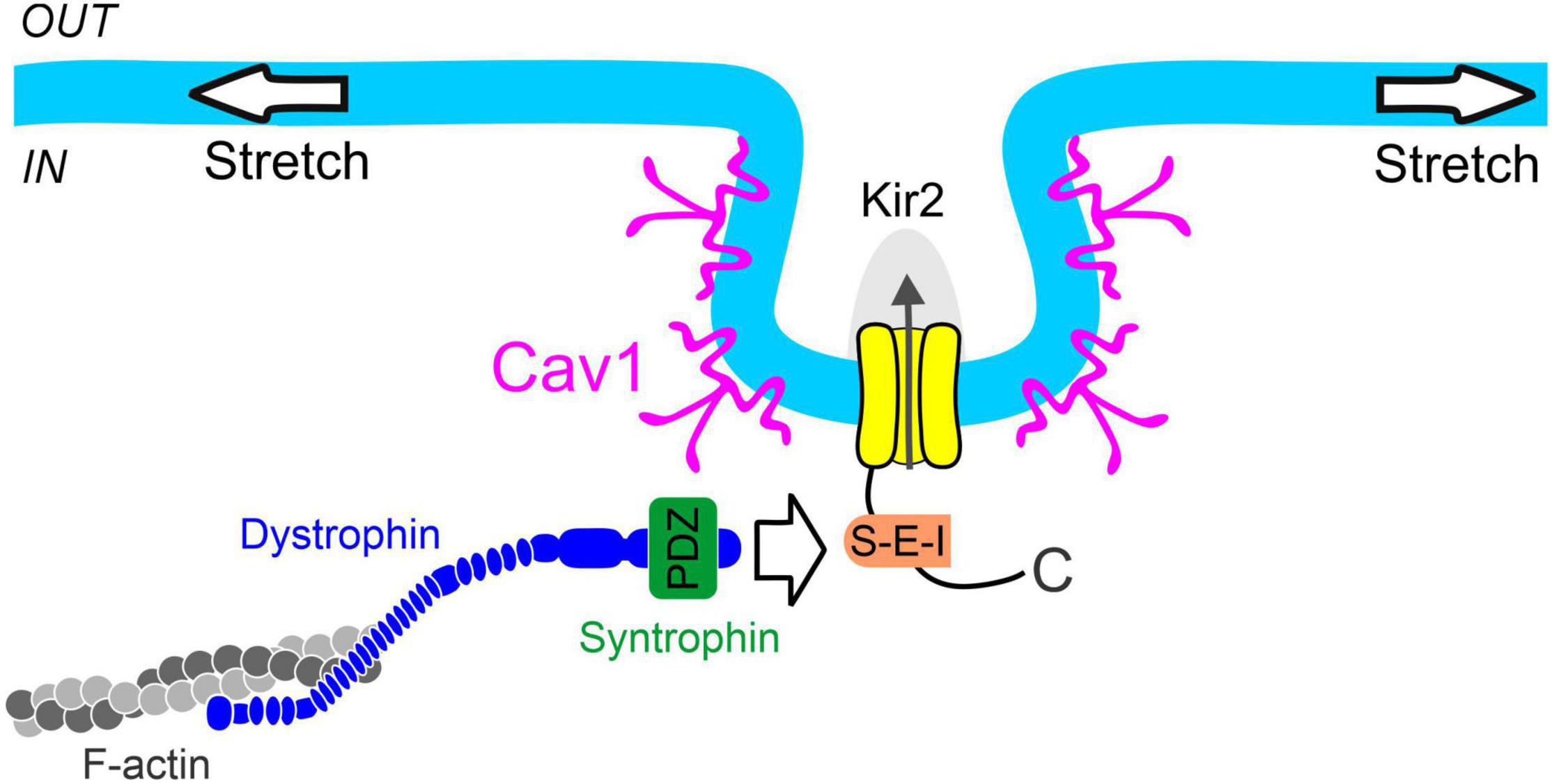
Figure 5. Proposed mechanism of smooth muscle Kir2 channel mechanosensitivity. Syntrophin binds to the C-terminal PDZ binding motif (S-E-I) of Kir2 channels residing in caveolae-like structures formed through the expression of caveolin-1 (Cav1). Dystrophin interacts with both syntrophin and filamentous actin (F-actin), providing a physical link between the Kir2 channel and cytoskeleton for force transmission. These structural components constitute a multiprotein signaling complex which confers pressure sensitivity to smooth muscle Kir2 channels.
These mechanistic insights may provide important translational implications in the framework of dyslipidemia, a lipid disorder characterized by elevated plasma LDL-cholesterol levels. Given the endothelium anatomical location, ECs would be constantly exposed to higher levels of LDL-cholesterol, which in turn may impact lipid-Kir interactions and cause Kir2 channel stabilization to a preferred closed state. Therefore, under dyslipidemia a regulatory switch (PIP2 to cholesterol) would suppress endothelial Kir2 activity, impair shear stress-induced activation, and ultimately impact brain blood flow. This future area of research is intended to analyze the possible role of endothelial dysfunction as an early predictor of atherosclerosis. Undoubtedly, the translation of these observations to humans will be the basis for the development of accurate therapeutic avenues to treat patients at risk of early cardiovascular disease (163).
Author Contributions
MS and JF wrote, designed the figures, and edited the manuscript. DGW edited the manuscript. All authors approved the submitted version.
Funding
This work was supported by operating grants to DGW from the Canadian Institute of Heath Research (FRN-159667). DGW was the Rorabeck Chair in Neuroscience and Vascular Biology at the University of Western Ontario.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Segal SS, Duling BR. Flow control among microvessels coordinated by intercellular conduction. Science. (1986) 234:868–70. doi: 10.1126/SCIENCE.3775368
2. Lee RMKW. Morphology of cerebral arteries. Pharmacol Ther. (1995) 66:149–73. doi: 10.1016/0163-7258(94)00071-A
3. Zhang R, Zuckerman JH, Iwasaki K, Wilson TE, Crandall CG, Levine BD. Autonomic neural control of dynamic cerebral autoregulation in humans. Circulation. (2002) 106:1814–20. doi: 10.1161/01.CIR.0000031798.07790.FE
4. Knot HJ, Zimmermann PA, Nelson MT. Extracellular K(+)-induced hyperpolarizations and dilatations of rat coronary and cerebral arteries involve inward rectifier K(+) channels. J Physiol. (1996) 492(Pt 2):419–30. doi: 10.1113/JPHYSIOL.1996.SP021318
5. Knot HJ, Nelson MT. Regulation of membrane potential and diameter by voltage-dependent K+ channels in rabbit myogenic cerebral arteries. Am J Physiol. (1995) 269:H348–55. doi: 10.1152/ajpheart.1995.269.1.h348
6. Cole WC, Welsh DG. Role of myosin light chain kinase and myosin light chain phosphatase in the resistance arterial myogenic response to intravascular pressure. Arch Biochem Biophys. (2011) 510:160–73. doi: 10.1016/J.ABB.2011.02.024
7. Thorneloe KS, Nelson MT. Ion channels in smooth muscle: regulators of intracellular calcium and contractility. Can J Physiol Pharmacol. (2005) 83:215–42. doi: 10.1139/Y05-016
8. Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol. (1995) 268:C799–822. doi: 10.1152/ajpcell.1995.268.4.c799
9. Quayle JM, McCarron JG, Brayden JE, Nelson MT. Inward rectifier K+ currents in smooth muscle cells from rat resistance-sized cerebral arteries. Am J Physiol. (1993) 265:C1363–70. doi: 10.1152/AJPCELL.1993.265.5.C1363
10. Goto K, Rummery NM, Grayson TH, Hill CE. Attenuation of conducted vasodilatation in rat mesenteric arteries during hypertension: role of inwardly rectifying potassium channels. J Physiol. (2004) 561:215–31. doi: 10.1113/JPHYSIOL.2004.070458
11. Marrelli SP, Johnson TD, Khorovets A, Childres WF, Bryan RM. Altered function of inward rectifier potassium channels in cerebrovascular smooth muscle after ischemia/reperfusion. Stroke. (1998) 29:1469–74. doi: 10.1161/01.STR.29.7.1469
12. Longden TA, Dabertrand F, Koide M, Gonzales AL, Tykocki NR, Brayden JE, et al. Capillary K +-sensing initiates retrograde hyperpolarization to increase local cerebral blood flow. Nat Neurosci. (2017) 20:717–26. doi: 10.1038/NN.4533
13. Park WS, Ko JH, Kim N, Son YK, Kang SH, Warda M, et al. Increased inhibition of inward rectifier K+ channels by angiotensin II in small-diameter coronary artery of isoproterenol-induced hypertrophied model. Arterioscler Thromb Vasc Biol. (2007) 27:1768–75. doi: 10.1161/ATVBAHA.107.143339
14. Mayhan WG, Mayhan JF, Sun H, Patel KP. In vivo properties of potassium channels in cerebral blood vessels during diabetes mellitus. Microcirculation. (2004) 11:605–13. doi: 10.1080/10739680490503410
15. Dabertrand F, Harraz OF, Koide M, Longden TA, Rosehart AC, Hill-Eubanks DC, et al. PIP 2 corrects cerebral blood flow deficits in small vessel disease by rescuing capillary Kir2.1 activity. Proc Natl Acad Sci USA. (2021) 118:e2025998118. doi: 10.1073/PNAS.2025998118
16. Koide M, Harraz OF, Dabertrand F, Longden TA, Ferris HR, Wellman GC, et al. Differential restoration of functional hyperemia by antihypertensive drug classes in hypertension-related cerebral small vessel disease. J Clin Invest. (2021) 131:e149029. doi: 10.1172/JCI149029
17. Enkvetchakul D, Jeliazkova I, Nichols CG. Direct modulation of Kir channel gating by membrane phosphatidylinositol 4,5-bisphosphate. J Biol Chem. (2005) 280:35785–8. doi: 10.1074/JBC.C500355200
18. Rohács T, Lopes CMB, Jin T, Ramdya PP, Molnár Z, Logothetis DE. Specificity of activation by phosphoinositides determines lipid regulation of Kir channels. Proc Natl Acad Sci USA. (2003) 100:745–50. doi: 10.1073/PNAS.0236364100
19. Rosenhouse-Dantsker A, Noskov S, Durdagi S, Logothetis DE, Levitan I. Identification of novel cholesterol-binding regions in Kir2 channels. J Biol Chem. (2013) 288:31154–64. doi: 10.1074/JBC.M113.496117
20. Rosenhouse-Dantsker A, Noskov S, Logothetis DE, Levitan I. Cholesterol sensitivity of KIR2.1 depends on functional inter-links between the N and C termini. Channels. (2013) 7:303–12. doi: 10.4161/CHAN.25437
21. Sancho M, Welsh DG. KIR channels in the microvasculature: regulatory properties and the lipid-hemodynamic environment. Curr Top Membr. (2020) 85:227–59. doi: 10.1016/BS.CTM.2020.01.006
22. Sancho M, Fabris S, Hald BO, Brett SE, Sandow SL, Poepping TL, et al. Membrane lipid-KIR2.x channel interactions enable hemodynamic sensing in cerebral arteries. Arterioscler Thromb Vasc Biol. (2019) 39:1072–87. doi: 10.1161/ATVBAHA.119.312493
23. Harraz OF, Longden TA, Dabertrand F, Hill-Eubanks D, Nelson MT. Endothelial GqPCR activity controls capillary electrical signaling and brain blood flow through PIP 2 depletion. Proc Natl Acad Sci USA. (2018) 115:E3569–77. doi: 10.1073/PNAS.1800201115
24. Wu BN, Luykenaar KD, Brayden JE, Giles WR, Corteling RL, Wiehler WB, et al. Hyposmotic challenge inhibits inward rectifying K+ channels in cerebral arterial smooth muscle cells. Am J Physiol Heart Circ Physiol. (2007) 292:H1085–94. doi: 10.1152/AJPHEART.00926.2006
25. Singer SJ, Nicolson GL. The fluid mosaic model of the structure of cell membranes. Science. (1972) 175:720–31. doi: 10.1126/SCIENCE.175.4023.720
26. van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Biol. (2008) 9:112–24. doi: 10.1038/nrm2330
27. Falkenburger BH, Jensen JB, Dickson EJ, Suh B-C, Hille B. Phosphoinositides: lipid regulators of membrane proteins. J Physiol. (2010) 588:3179. doi: 10.1113/JPHYSIOL.2010.192153
28. McLaughlin S, Wang J, Gambhir A, Murray D. PIP2 and proteins: interactions, organization, and information flow. Annu Rev Biophys Biomol Struct. (2003) 31:151–75. doi: 10.1146/ANNUREV.BIOPHYS.31.082901.134259
29. Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, et al. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem. (2003) 70:535–602. doi: 10.1146/ANNUREV.BIOCHEM.70.1.535
30. Lemmon MA, Ferguson KM. Signal-dependent membrane targeting by pleckstrin homology (PH) domains. Biochem J. (2000) 350(Pt 1):1–18. doi: 10.1042/bj3500001
31. Lupyan D, Mezei M, Logothetis DE, Osman R. A molecular dynamics investigation of lipid bilayer perturbation by PIP2. Biophys J. (2010) 98:240. doi: 10.1016/J.BPJ.2009.09.063
32. Toner M, Vaio G, McLaughlin A, McLaughlin S. Adsorption of cations to phosphatidylinositol 4,5-bisphosphate. Biochemistry. (1988) 27:7435–43. doi: 10.1021/BI00419A039
33. Wang J, Arbuzova A, Hangyas-Mihalyne G, McLaughlin S. The effector domain of myristoylated alanine-rich C kinase substrate binds strongly to phosphatidylinositol 4,5-bisphosphate. J Biol Chem. (2001) 276:5012–9. doi: 10.1074/JBC.M008355200
34. Hilgemann DW, Feng S, Nasuhoglu C. The complex and intriguing lives of PIP2 with ion channels and transporters. Sci STKE. (2001) 2001:re19. doi: 10.1126/stke.2001.111.re19
35. Suh BC, Hille B. Regulation of ion channels by phosphatidylinositol 4,5-bisphosphate. Curr Opin Neurobiol. (2005) 15:370–8. doi: 10.1016/J.CONB.2005.05.005
36. MacGregor GG, Dong K, Vanoye CG, Tang L, Giebisch G, Hebert SC. Nucleotides and phospholipids compete for binding to the C terminus of KATP channels. Proc Natl Acad Sci USA. (2002) 99:2726–31. doi: 10.1073/PNAS.042688899
37. Huang CL, Feng S, Hilgemann DW. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gβγ. Nature. (1998) 391:803–6. doi: 10.1038/35882
38. Shyng SL, Nichols CG. Membrane phospholipid control of nucleotide sensitivity of K(ATP) channels. Science. (1998) 282:1138–41. doi: 10.1126/SCIENCE.282.5391.1138
39. Rohács T, Chen J, Prestwich GD, Logothetis DE. Distinct specificities of inwardly rectifying K(+) channels for phosphoinositides. J Biol Chem. (1999) 274:36065–72. doi: 10.1074/JBC.274.51.36065
40. Loussouarn G, Park KH, Bellocq C, Baró I, Charpentier F, Escande D. Phosphatidylinositol-4,5-bisphosphate, PIP2, controls KCNQ1/KCNE1 voltage-gated potassium channels: a functional homology between voltage-gated and inward rectifier K+ channels. EMBO J. (2003) 22:5412–21. doi: 10.1093/EMBOJ/CDG526
41. Wu L, Bauer CS, Zhen XG, Xie C, Yang J. Dual regulation of voltage-gated calcium channels by PtdIns(4,5)P2. Nature. (2002) 419:947–52. doi: 10.1038/nature01118
42. Chemin J, Patel AJ, Duprat F, Lauritzen I, Lazdunski M, Honoré E. A phospholipid sensor controls mechanogating of thE K+ cHANNEL TREK-1. EMBO J. (2005) 24:44–53. doi: 10.1038/SJ.EMBOJ.7600494
43. Lange Y, Swaisgood MH, Ramos BV, Steck TL. Plasma membranes contain half the phospholipid and 90% of the cholesterol and sphingomyelin in cultured human fibroblasts. J Biol Chem. (1989) 264:3786–93. doi: 10.1016/S0021-9258(19)84918-9
44. Demel RA, Bruckdorfer KR, van Deenen LLM. The effect of sterol structure on the permeability of lipomes to glucose, glycerol and Rb+. Biochim Biophys Acta. (1972) 255:321–30. doi: 10.1016/0005-2736(72)90031-4
45. Xu X, London E. The effect of sterol structure on membrane lipid domains reveals how cholesterol can induce lipid domain formation. Biochemistry. (2000) 39:843–9. doi: 10.1021/bi992543v
46. Needham D, Nunn RS. Elastic deformation and failure of lipid bilayer membranes containing cholesterol. Biophys J. (1990) 58:997–1009. doi: 10.1016/S0006-3495(90)82444-9
47. Romanenko VG, Rothblat GH, Levitan I. Modulation of endothelial inward-rectifier K+ current by optical isomers of cholesterol. Biophys J. (2002) 83:3211–22. doi: 10.1016/S0006-3495(02)75323-X
48. Lundbæk JA, Birn P, Girshman J, Hansen AJ, Andersen OS. Membrane stiffness and channel function. Biochemistry. (1996) 35:3825–30. doi: 10.1021/bi952250b
49. Romanenko VG, Rothblat GH, Levitan I. Sensitivity of volume-regulated anion current to cholesterol structural analogues. J Gen Physiol. (2004) 123:77–87. doi: 10.1085/jgp.200308882
50. Mason RP, Jacob RF. Membrane microdomains and vascular biology: emerging role in atherogenesis. Circulation. (2003) 107:2270–3. doi: 10.1161/01.CIR.0000062607.02451.B6
51. Romanenko VG, Fang Y, Byfield F, Travis AJ, Vandenberg CA, Rothblat GH, et al. Cholesterol sensitivity and lipid raft targeting of Kir2.1 channels. Biophys J. (2004) 87:3850–61. doi: 10.1529/BIOPHYSJ.104.043273
52. Egawa J, Zemljic-Harpf A, Mandyam CD, Niesman IR, Lysenko LV, Kleschevnikov AM, et al. Neuron-targeted caveolin-1 promotes ultrastructural and functional hippocampal synaptic plasticity. Cereb Cortex. (2018) 28:3255. doi: 10.1093/CERCOR/BHX196
53. Sezgin E, Levental I, Mayor S, Eggeling C. The mystery of membrane organization: composition, regulation and roles of lipid rafts. Nat Rev Mol Cell Biol. (2017) 18:361–74. doi: 10.1038/nrm.2017.16
54. Lingwood D, Simons K. Lipid rafts as a membrane-organizing principle. Science. (2010) 327:46–50. doi: 10.1126/SCIENCE.1174621
55. Goldstein JL, Brown MS. The cholesterol quartet. Science. (2001) 292:1310–2. doi: 10.1126/SCIENCE.1061815
56. Björkhem I, Meaney S, Fogelman AM. Brain cholesterol: long secret life behind a barrier. Arterioscler Thromb Vasc Biol. (2004) 24:806–15. doi: 10.1161/01.ATV.0000120374.59826.1b
57. Lopez PH, Schnaar RL. Gangliosides in cell recognition and membrane protein regulation. Curr Opin Struct Biol. (2009) 19:549–57. doi: 10.1016/J.SBI.2009.06.001
58. Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. (2008) 9:139–50. doi: 10.1038/nrm2329
59. Parton RG, Simons K. The multiple faces of caveolae. Nat Rev Mol Cell Biol. (2007) 8:185–94. doi: 10.1038/nrm2122
60. Sancho M, Samson NC, Hald BO, Hashad AM, Marrelli SP, Brett SE, et al. KIR channels tune electrical communication in cerebral arteries. J Cerebr Blood Flow Metab. (2017) 37:2171–84. doi: 10.1177/0271678X16662041
61. Smith PD, Brett SE, Luykenaar KD, Sandow SL, Marrelli SP, Vigmond EJ, et al. KIR channels function as electrical amplifiers in rat vascular smooth muscle. J Physiol. (2008) 586:1147–60. doi: 10.1113/JPHYSIOL.2007.145474
62. Sonkusare SK, Dalsgaard T, Bonev AD, Nelson MT. Inward rectifier potassium (Kir2.1) channels as end-stage boosters of endothelium-dependent vasodilators. J Physiol. (2016) 594:3271–85. doi: 10.1113/JP271652
63. Ahn SJ, Fancher IS, Bian JT, Zhang CX, Schwab S, Gaffin R, et al. Inwardly rectifying K + channels are major contributors to flow-induced vasodilatation in resistance arteries. J Physiol. (2017) 595:2339–64. doi: 10.1113/JP273255
64. Chilton L, Loutzenhiser K, Morales E, Breaks J, Kargacin GJ, Loutzenhiser RD. Inward rectifier K(+) currents and Kir2.1 expression in renal afferent and efferent arterioles. J Am Soc Nephrol. (2008) 19:69–76. doi: 10.1681/ASN.2007010039
65. Raab-Graham KF, Vandenberg CA. Tetrameric subunit structure of the native brain inwardly rectifying potassium channel Kir 2.2. J Biol Chem. (1998) 273:19699–707. doi: 10.1074/JBC.273.31.19699
66. Schram G, Pourrier M, Wang Z, White M, Nattel S. Barium block of Kir2 and human cardiac inward rectifier currents: evidence for subunit-heteromeric contribution to native currents. Cardiovasc Res. (2003) 59:328–38. doi: 10.1016/S0008-6363(03)00366-3
67. Liu GX, Derst C, Schlichthörl G, Heinen S, Seebohm G, Brüggemann A, et al. Comparison of cloned Kir2 channels with native inward rectifier K+ channels from guinea-pig cardiomyocytes. J Physiol. (2001) 532:115–26. doi: 10.1111/J.1469-7793.2001.0115G.X
68. Bichet D, Haass FA, Jan LY. Merging functional studies with structures of inward-rectifier K(+) channels. Nat Rev Neurosci. (2003) 4:957–67. doi: 10.1038/NRN1244
69. Lu Z. Mechanism of rectification in inward-rectifier K+ channels. Annu Rev Physiol. (2004) 66:103–29. doi: 10.1146/ANNUREV.PHYSIOL.66.032102.150822
70. Matsuda H, Oishi K, Omori K. Voltage-dependent gating and block by internal spermine of the murine inwardly rectifying K+ channel, Kir2.1. J Physiol. (2003) 548:361–71. doi: 10.1113/JPHYSIOL.2003.038844
71. Dhamoon AS, Pandit SV, Sarmast F, Parisian KR, Guha P, Li Y, et al. Unique Kir2.x properties determine regional and species differences in the cardiac inward rectifier K+ current. Circ Res. (2004) 94:1332–9. doi: 10.1161/01.RES.0000128408.66946.67
72. Jantzi MC, Brett SE, Jackson WF, Corteling R, Vigmond EJ, Welsh DG. Inward rectifying potassium channels facilitate cell-to-cell communication in hamster retractor muscle feed arteries. Am J Physiol Heart Circ Physiol. (2006) 291:H1319–28. doi: 10.1152/AJPHEART.00217.2006
73. Filosa JA, Bonev AD, Straub SV, Meredith AL, Wilkerson MK, Aldrich RW, et al. Local potassium signaling couples neuronal activity to vasodilation in the brain. Nat Neurosci. (2006) 9:1397–403. doi: 10.1038/NN1779
74. Armstrong ML, Dua AK, Murrant CL. Potassium initiates vasodilatation induced by a single skeletal muscle contraction in hamster cremaster muscle. J Physiol. (2007) 581:841–52. doi: 10.1113/JPHYSIOL.2007.130013
75. Loeb AL, Godény I, Longnecker DE. Functional evidence for inward-rectifier potassium channels in rat cremaster muscle arterioles. Microvasc Res. (2000) 59:1–6. doi: 10.1006/MVRE.1999.2187
76. Quayle JM, Nelson MT, Standen NB. ATP-sensitive and inwardly rectifying potassium channels in smooth muscle. Physiol Rev. (1997) 77:1165–232. doi: 10.1152/PHYSREV.1997.77.4.1165
77. Hansen SB, Tao X, MacKinnon R. Structural basis of PIP2 activation of the classical inward rectifier K+ channel Kir2.2. Nature. (2011) 477:495. doi: 10.1038/NATURE10370
78. Xie LH, John Scott ASA, Ribalet B, Weiss JN. Phosphatidylinositol-4,5-bisphosphate (PIP2) regulation of strong inward rectifier Kir2.1 channels: multilevel positive cooperativity. J Physiol. (2008) 586:1833–48. doi: 10.1113/JPHYSIOL.2007.147868
79. Du X, Zhang H, Lopes C, Mirshahi T, Rohacs T, Logothetis DE. Characteristic interactions with phosphatidylinositol 4,5-bisphosphate determine regulation of kir channels by diverse modulators. J Biol Chem. (2004) 279:37271–81. doi: 10.1074/JBC.M403413200
80. Harraz OF, Longden TA, Eubanks DH, Nelson MT. PIP 2 depletion promotes TRPV4 channel activity in mouse brain capillary endothelial cells. Elife. (2018) 7:e38689. doi: 10.7554/ELIFE.38689
82. Rosenhouse-Dantsker A, Logothetis DE, Levitan I. Cholesterol sensitivity of Kir2.1 is controlled by a belt of residues around the cytosolic pore. Biophys J. (2011) 100:381–9. doi: 10.1016/J.BPJ.2010.11.086
83. Barbera N, Levitan I. Chiral specificity of cholesterol orientation within cholesterol binding sites in inwardly rectifying K + channels. Adv Exp Med Biol. (2019) 1115:77–95. doi: 10.1007/978-3-030-04278-3_4
84. Singh DK, Rosenhouse-Dantsker A, Nichols CG, Enkvetchakul D, Levitan I. Direct regulation of prokaryotic Kir channel by cholesterol. J Biol Chem. (2009) 284:30727–36. doi: 10.1074/JBC.M109.011221
85. Fang Y, Mohler ER, Hsieh E, Osman H, Hashemi SM, Davies PF, et al. Hypercholesterolemia suppresses inwardly rectifying K+ channels in aortic endothelium in vitro and in vivo. Circ Res. (2006) 98:1064–71. doi: 10.1161/01.RES.0000218776.87842.43
86. Lehoux S, Tedgui A. Signal transduction of mechanical stresses in the vascular wall. Hypertension. (1998) 32:338–45. doi: 10.1161/01.HYP.32.2.338
87. Ingber DE. Cellular mechanotransduction: putting all the pieces together again. FASEB J. (2006) 20:811–27. doi: 10.1096/FJ.05-5424REV
88. Martinac B. The ion channels to cytoskeleton connection as potential mechanism of mechanosensitivity. Biochim Biophys Acta. (2014) 1838:682–91. doi: 10.1016/J.BBAMEM.2013.07.015
89. Bayliss WM. On the local reactions of the arterial wall to changes of internal pressure. J Physiol. (1902) 28:220–31. doi: 10.1113/JPHYSIOL.1902.SP000911
90. Davis MJ, Hill MA. Signaling mechanisms underlying the vascular myogenic response. Physiol Rev. (1999) 79:387–423. doi: 10.1152/PHYSREV.1999.79.2.387
91. Kirby BS, Carlson RE, Markwald RR, Voyles WF, Dinenno FA. Mechanical influences on skeletal muscle vascular tone in humans: insight into contraction-induced rapid vasodilatation. J Physiol. (2007) 583:861–74. doi: 10.1113/JPHYSIOL.2007.131250
92. Hill MA, Davis MJ. Coupling a change in intraluminal pressure to vascular smooth muscle depolarization: still stretching for an explanation. Am J Physiol Heart Circ Physiol. (2007) 292:H2570–2. doi: 10.1152/AJPHEART.00331.2007
93. Davis MJ. Myogenic response gradient in an arteriolar network. Am J Physiol. (1993) 264:H2168–79. doi: 10.1152/AJPHEART.1993.264.6.H2168
94. Russell RWR, Simcock JP, Wilkinson IMS, Frears CC. The effect of blood pressure changes on the leptomeningeal circulation of the rabbit. Brain. (1970) 93:491–504. doi: 10.1093/BRAIN/93.3.491
95. Thorin-Trescases N, Bartolotta T, Hyman N, Penar PL, Walters CL, Bevan RD, et al. Diameter dependence of myogenic tone of human pial arteries. Stroke. (1997) 28:2486–92. doi: 10.1161/01.STR.28.12.2486
96. Garcia-Roldan JL, Bevan JA. Flow-induced constriction and dilation of cerebral resistance arteries. Circ Res. (1990) 66:1445–8. doi: 10.1161/01.RES.66.5.1445
97. Ngai AC, Winn HR. Modulation of cerebral arteriolar diameter by intraluminal flow and pressure. Circ Res. (1995) 77:832–40. doi: 10.1161/01.RES.77.4.832
98. Bevan JA, Joyce EH. Flow-induced resistance artery tone: balance between constrictor and dilator mechanisms. Am J Physiol. (1990) 258:H663–8. doi: 10.1152/AJPHEART.1990.258.3.H663
99. Brayden JE, Earley S, Nelson MT, Reading S. Transient receptor potential (TRP) channels, vascular tone and autoregulation of cerebral blood flow. Clin Exp Pharmacol Physiol. (2008) 35:1116–20. doi: 10.1111/J.1440-1681.2007.04855.X
100. Hill MA, Zou H, Potocnik SJ, Meininger GA, Davis MJ. Invited review: arteriolar smooth muscle mechanotransduction: Ca2+ signaling pathways underlying myogenic reactivity. J Appl Physiol. (2001) 91:973–85. doi: 10.1152/JAPPL.2001.91.2.973
101. Osol G, Brekke JF, McElroy-Yaggy K, Gokina NI. Myogenic tone, reactivity, and forced dilatation: a three-phase model of in vitro arterial myogenic behavior. Am J Physiol Heart Circ Physiol. (2002) 283:2260–7. doi: 10.1152/AJPHEART.00634.2002
102. Sharif-Naeini R, Folgering JHA, Bichet D, Duprat F, Delmas P, Patel A, et al. Sensing pressure in the cardiovascular system: Gq-coupled mechanoreceptors and TRP channels. J Mol Cell Cardiol. (2010) 48:83–9. doi: 10.1016/J.YJMCC.2009.03.020
103. Welsh DG, Nelson MT, Eckman DM, Brayden JE. Swelling-activated cation channels mediate depolarization of rat cerebrovascular smooth muscle by hyposmolarity and intravascular pressure. J Physiol. (2000) 527(Pt 1):139–48. doi: 10.1111/J.1469-7793.2000.T01-1-00139.X
104. Knot HJ, Nelson MT. Regulation of arterial diameter and wall [Ca2+] in cerebral arteries of rat by membrane potential and intravascular pressure. J Physiol. (1998) 508(Pt 1):199–209. doi: 10.1111/J.1469-7793.1998.199BR.X
105. Welsh DG, Morielli AD, Nelson MT, Brayden JE. Transient receptor potential channels regulate myogenic tone of resistance arteries. Circ Res. (2002) 90:248–50. doi: 10.1161/HH0302.105662
106. Izzard AS, Graham D, Burnham MP, Heerkens EH, Dominiczak AF, Heagerty AM. Myogenic and structural properties of cerebral arteries from the stroke-prone spontaneously hypertensive rat. Am J Physiol Heart Circ Physiol. (2003) 285:1489–94. doi: 10.1152/AJPHEART.00352.2003
107. Toth P, Tarantini S, Csiszar A, Ungvari Z. Functional vascular contributions to cognitive impairment and dementia: mechanisms and consequences of cerebral autoregulatory dysfunction, endothelial impairment, and neurovascular uncoupling in aging. Am J Physiol Heart Circ Physiol. (2017) 312:H1–20. doi: 10.1152/AJPHEART.00581.2016
108. Toth P, Tucsek Z, Sosnowska D, Gautam T, Mitschelen M, Tarantini S, et al. Age-related autoregulatory dysfunction and cerebromicrovascular injury in mice with angiotensin II-induced hypertension. J Cereb Blood Flow Metab. (2013) 33:1732–42. doi: 10.1038/JCBFM.2013.143
109. Gerhold KA, Schwartz MA. Ion channels in endothelial responses to fluid shear stress. Physiology. (2016) 31:359. doi: 10.1152/PHYSIOL.00007.2016
110. Yamamoto K, Furuya K, Nakamura M, Kobatake E, Sokabe M, Ando J. Visualization of flow-induced ATP release and triggering of Ca2+ waves at caveolae in vascular endothelial cells. J Cell Sci. (2011) 124:3477–83. doi: 10.1242/JCS.087221
111. Baratchi S, Almazi JG, Darby W, Tovar-Lopez FJ, Mitchell A, McIntyre P. Shear stress mediates exocytosis of functional TRPV4 channels in endothelial cells. Cell Mol Life Sci. (2016) 73:649–66. doi: 10.1007/S00018-015-2018-8
112. Coste B, Mathur J, Schmidt M, Earley TJ, Ranade S, Petrus MJ, et al. Piezo1 and piezo2 are essential components of distinct mechanically activated cation channels. Science. (2010) 330:55–60. doi: 10.1126/SCIENCE.1193270
113. Hoger JH, Ilyin VI, Forsyth S, Hoger A. Shear stress regulates the endothelial Kir2.1 ion channel. Proc Natl Acad Sci USA. (2002) 99:7780–5. doi: 10.1073/PNAS.102184999
114. Brähler S, Kaistha A, Schmidt VJ, Wölfle SE, Busch C, Kaistha BP, et al. Genetic deficit of SK3 and IK1 channels disrupts the endothelium-derived hyperpolarizing factor vasodilator pathway and causes hypertension. Circulation. (2009) 119:2323–32. doi: 10.1161/CIRCULATIONAHA.108.846634
115. Sonkusare SK, Bonev AD, Ledoux J, Liedtke W, Kotlikoff MI, Heppner TJ, et al. Elementary Ca2+ signals through endothelial TRPV4 channels regulate vascular function. Science. (2012) 336:597–601. doi: 10.1126/SCIENCE.1216283
116. Harraz OF, Klug NR, Senatore A, Koide M, Nelson MT. Piezo1 is a mechanosensor channel in CNS capillaries. J Gen Physiol. (2022) 154:e2021ecc12. doi: 10.1085/JGP.2021ECC12
117. Emerson GG, Segal SS. Electrical coupling between endothelial cells and smooth muscle cells in hamster feed arteries: role in vasomotor control. Circ Res. (2000) 87:474–9. doi: 10.1161/01.RES.87.6.474
118. Brohawn SG. How ion channels sense mechanical force: insights from mechanosensitive K2P channels TRAAK, TREK1, and TREK2. Ann N Y Acad Sci. (2015) 1352:20–32. doi: 10.1111/NYAS.12874
119. Honoré E. The neuronal background K2P channels: focus on TREK1. Nat Rev Neurosci. (2007) 8:251–61. doi: 10.1038/nrn2117
120. Maroto R, Raso A, Wood TG, Kurosky A, Martinac B, Hamill OP. TRPC1 forms the stretch-activated cation channel in vertebrate cells. Nat Cell Biol. (2005) 7:179–85. doi: 10.1038/NCB1218
121. Pedersen SF, Nilius B. Transient receptor potential channels in mechanosensing and cell volume regulation. Methods Enzymol. (2007) 428:183–207. doi: 10.1016/S0076-6879(07)28010-3
122. Kanzaki M, Zhang YQ, Mashima H, Li L, Shibata H, Kojima I. Translocation of a calcium-permeable cation channel induced by insulin-like growth factor-I. Nat Cell Biol. (1999) 1:165–70. doi: 10.1038/11086
123. Nilius B, Owsianik G, Voets T, Peters JA. Transient receptor potential cation channels in disease. Physiol Rev. (2007) 87:165–217. doi: 10.1152/PHYSREV.00021.2006
124. Zhu J, Yu Y, Ulbrich MH, Li MH, Isacoff EY, Honig B, et al. Structural model of the TRPP2/PKD1 C-terminal coiled-coil complex produced by a combined computational and experimental approach. Proc Natl Acad Sci USA. (2011) 108:10133–8. doi: 10.1073/PNAS.1017669108/-/DCSUPPLEMENTAL
125. Liman ER. The Ca 2 + -activated trp channels: TRPM4 and TRPM5. In: Liedtke W, Heller S, editors. TRP Ion Channel Function in Sensory Transduction and Cellular Signaling. Boca Raton, FL: CRC Press/Taylor & Francis (2007). p. 203–12.
126. Geiger B, Spatz JP, Bershadsky AD. Environmental sensing through focal adhesions. Nat Rev Mol Cell Biol. (2009) 10:21–33. doi: 10.1038/nrm2593
127. Hak RK, Gallant C, Leavis PC, Gunst SJ, Morgan KG. Cytoskeletal remodeling in differentiated vascular smooth muscle is actin isoform dependent and stimulus dependent. Am J Physiol Cell Physiol. (2008) 295:C768–78. doi: 10.1152/ajpcell.00174.2008
128. Tang DD, Anfinogenova Y. Physiologic properties and regulation of the actin cytoskeleton in vascular smooth muscle. J Cardiovasc Pharmacol Ther. (2008) 13:130–40. doi: 10.1177/1074248407313737
129. Cipolla MJ, Gokina NI, Osol G. Pressure-induced actin polymerization in vascular smooth muscle as a mechanism underlying myogenic behavior. FASEB J. (2002) 16:72–6. doi: 10.1096/CJ.01-0104HYP
130. Gokina NI, Osol G. Actin cytoskeletal modulation of pressure-induced depolarization and Ca(2+) influx in cerebral arteries. Am J Physiol Heart Circ Physiol. (2002) 282:H1410–20. doi: 10.1152/AJPHEART.00441.2001
131. Ehmsen J, Poon E, Davies K. The dystrophin-associated protein complex. J Cell Sci. (2002) 115:2801–3. doi: 10.1242/jcs.115.14.2801
132. Ohlendieck K, Matsumura K, Ionasescu VV, Towbin JA, Bosch EP, Weinstein SL, et al. Duchenne muscular dystrophy: deficiency of dystrophin-associated proteins in the sarcolemma. Neurology. (1993) 43:795–800. doi: 10.1212/WNL.43.4.795
133. Sadoulet-Puccio HM, Kunkel LM. Dystrophin and its isoforms. Brain Pathol. (1996) 6:25–35. doi: 10.1111/J.1750-3639.1996.TB00780.X
134. Rybakova IN, Ervasti JM. Dystrophin-glycoprotein complex is monomeric and stabilizes actin filaments in vitro through a lateral association. J Biol Chem. (1997) 272:28771–8. doi: 10.1074/JBC.272.45.28771
135. Suzuki A, Yoshida M, Yamamoto H, Ozawa E. Glycoprotein-binding site of dystrophin is confined to the cysteine-rich domain and the first half of the carboxy-terminal domain. FEBS Lett. (1992) 308:154–60. doi: 10.1016/0014-5793(92)81265-N
136. Petrof BJ, Shrager JB, Stedman HH, Kelly AM, Sweeney HL. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc Natl Acad Sci USA. (1993) 90:3710–4. doi: 10.1073/PNAS.90.8.3710
137. Turczyńska KM, Swärd K, Hien TT, Wohlfahrt J, Mattisson IY, Ekman M, et al. Regulation of smooth muscle dystrophin and synaptopodin 2 expression by actin polymerization and vascular injury. Arterioscler Thromb Vasc Biol. (2015) 35:1489–97. doi: 10.1161/ATVBAHA.114.305065
138. Constantin B. Dystrophin complex functions as a scaffold for signalling proteins. Biochim Biophys Acta. (2014) 1838:635–42. doi: 10.1016/J.BBAMEM.2013.08.023
139. Adams ME, Butler MH, Dwyer TM, Peters MF, Murnane AA, Froehner SC. Two forms of mouse syntrophin, a 58 kd dystrophin-associated protein, differ in primary structure and tissue distribution. Neuron. (1993) 11:531–40. doi: 10.1016/0896-6273(93)90157-M
140. Ahnt AH, Freener CA, Gussoni E, Yoshida M, Ozawa E, Kunkel LM. The three human syntrophin genes are expressed in diverse tissues, have distinct chromosomal locations, and each bind to dystrophin and its relatives. J Biol Chem. (1996) 271:2724–30. doi: 10.1074/JBC.271.5.2724
141. Piluso G, Mirabella M, Ricci E, Belsito A, Abbondanza C, Servidei S, et al. Gamma1- and gamma2-syntrophins, two novel dystrophin-binding proteins localized in neuronal cells. J Biol Chem. (2000) 275:15851–60. doi: 10.1074/JBC.M000439200
142. Schultz J, Hoffmuller U, Krause G, Ashurst J, Macias MJ, Schmieder P, et al. Specific interactions between the syntrophin PDZ domain and voltage-gated sodium channels. Nat Struct Biol. (1998) 5:19–24. doi: 10.1038/NSB0198-19
143. Leonoudakis D, Conti LR, Anderson S, Radeke CM, McGuire LMM, Adams ME, et al. Protein trafficking and anchoring complexes revealed by proteomic analysis of inward rectifier potassium channel (Kir2.x)-associated proteins. J Biol Chem. (2004) 279:22331–46. doi: 10.1074/JBC.M400285200
144. Willis BC, Ponce-Balbuena D, Jalife J. Protein assemblies of sodium and inward rectifier potassium channels control cardiac excitability and arrhythmogenesis. Am J Physiol Heart Circ Physiol. (2015) 308:H1463–73. doi: 10.1152/AJPHEART.00176.2015
145. Echarri A, del Pozo MA. Caveolae – mechanosensitive membrane invaginations linked to actin filaments. J Cell Sci. (2015) 128:2747–58. doi: 10.1242/JCS.153940
146. Harvey RD, Calaghan SC. Caveolae create local signalling domains through their distinct protein content, lipid profile and morphology. J Mol Cell Cardiol. (2012) 52:366–75. doi: 10.1016/J.YJMCC.2011.07.007
147. Hardin CD, Vallejo J. Caveolins in vascular smooth muscle: form organizing function. Cardiovasc Res. (2006) 69:808–15. doi: 10.1016/J.CARDIORES.2005.11.024
148. Williams TM, Lisanti MP. The caveolin proteins. Genome Biol. (2004) 5:214. doi: 10.1186/GB-2004-5-3-214
149. Drab M, Verkade P, Elger M, Kasper M, Lohn M, Lauterbach B, et al. Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science. (2001) 293:2449–52. doi: 10.1126/SCIENCE.1062688
150. Trouet D, Nilius B, Jacobs A, Remacle C, Droogmans G, Eggermont J. Caveolin-1 modulates the activity of the volume-regulated chloride channel. J Physiol. (1999) 520(Pt 1):113–9. doi: 10.1111/J.1469-7793.1999.T01-1-00113.X
151. Ito S, Suki B, Kume H, Numaguchi Y, Ishii M, Iwaki M, et al. Actin cytoskeleton regulates stretch-activated Ca2+ influx in human pulmonary microvascular endothelial cells. Am J Respir Cell Mol Biol. (2010) 43:26–34. doi: 10.1165/RCMB.2009-0073OC
152. Gervásio OL, Whitehead NR, Yeung EW, Phillips WD, Allen DG. TRPC1 binds to caveolin-3 and is regulated by Src kinase – role in duchenne muscular dystrophy. J Cell Sci. (2008) 121:2246–55. doi: 10.1242/JCS.032003
153. Han H, Rosenhouse-Dantsker A, Gnanasambandam R, Epshtein Y, Chen Z, Sachs F, et al. Silencing of Kir2 channels by caveolin-1: cross-talk with cholesterol. J Physiol. (2014) 592:4025–38. doi: 10.1113/JPHYSIOL.2014.273177
154. Ellershaw DC, Greenwood IA, Large WA. Modulation of volume-sensitive chloride current by noradrenaline in rabbit portal vein myocytes. J Physiol. (2002) 542:537–47. doi: 10.1113/JPHYSIOL.2002.018770
155. Yamazaki J, Duan D, Janiak R, Kuenzli K, Horowitz B, Hume JR. Functional and molecular expression of volume-regulated chloride channels in canine vascular smooth muscle cells. J Physiol. (1998) 507(Pt 3):729–36. doi: 10.1111/J.1469-7793.1998.729BS.X
156. Earley S, Waldron BJ, Brayden JE. Critical role for transient receptor potential channel TRPM4 in myogenic constriction of cerebral arteries. Circ Res. (2004) 95:922–9. doi: 10.1161/01.RES.0000147311.54833.03
157. Olesen SP, Claphamt DE, Davies PF. Haemodynamic shear stress activates a K+ current in vascular endothelial cells. Nature. (1988) 331:168–70. doi: 10.1038/331168A0
158. Lieu DK, Pappone PA, Barakat AI. Differential membrane potential and ion current responses to different types of shear stress in vascular endothelial cells. Am J Physiol Cell Physiol. (2004) 286:C1367–75. doi: 10.1152/AJPCELL.00243.2003
159. Fancher IS, Ahn SJ, Adamos C, Osborn C, Oh MJ, Fang Y, et al. Hypercholesterolemia-induced loss of flow-induced vasodilation and lesion formation in apolipoprotein E-deficient mice critically depend on inwardly rectifying K + channels. J Am Heart Assoc. (2018) 7:e007430. doi: 10.1161/JAHA.117.007430
160. Meyrelles SS, Peotta VA, Pereira TMC, Vasquez EC. Endothelial dysfunction in the apolipoprotein E-deficient mouse: insights into the influence of diet, gender and aging. Lipids Health Dis. (2011) 10:211. doi: 10.1186/1476-511X-10-211
161. Kowalewska P, Sancho M, Fabris S, Huff M, Gros R, Welsh D. Activity of inwardly rectifying K+ channels in cerebral arteries is diminished in dyslipidemia without overt effects on cerebral blood flow. FASEB J. (2021) 35. doi: 10.1096/FASEBJ.2021.35.S1.03299
162. Ahn SJ, Fancher IS, Granados ST, do Couto NF, Hwang CL, Phillips SA, et al. Cholesterol-induced suppression of endothelial Kir channels is a driver of impairment of arteriolar flow-induced vasodilation in humans. Hypertension. (2022) 79:126–38. doi: 10.1161/HYPERTENSIONAHA.121.17672
163. Anderson TJ, Charbonneau F, Title LM, Buithieu J, Rose MS, Conradson H, et al. Microvascular function predicts cardiovascular events in primary prevention: long-term results from the firefighters and their endothelium (FATE) study. Circulation. (2011) 123:163–9. doi: 10.1161/CIRCULATIONAHA.110.953653
Keywords: endothelium, smooth muscle, PIP2, cholesterol, Kir2 channels, cerebral blood flow, hemodynamic forces
Citation: Sancho M, Fletcher J and Welsh DG (2022) Inward Rectifier Potassium Channels: Membrane Lipid-Dependent Mechanosensitive Gates in Brain Vascular Cells. Front. Cardiovasc. Med. 9:869481. doi: 10.3389/fcvm.2022.869481
Received: 04 February 2022; Accepted: 28 February 2022;
Published: 28 March 2022.
Edited by:
Ibra S. Fancher, University of Delaware, United StatesReviewed by:
Anna Bukiya, University of Tennessee Health Science Center (UTHSC), United StatesIrena Levitan, University of Illinois at Chicago, United States
Copyright © 2022 Sancho, Fletcher and Welsh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria Sancho, bWFzYW5jNzVAdWNtLmVz; Donald G. Welsh, ZHdlbHNoQHJvYmFydHMuY2E=