 Diede Smeets
Diede Smeets Anton Gisterå
Anton Gisterå Stephen G. Malin
Stephen G. Malin Dimitrios Tsiantoulas
Dimitrios Tsiantoulas- 1Department of Laboratory Medicine, Medical University of Vienna, Vienna, Austria
- 2Center for Molecular Medicine, Karolinska University Hospital, Stockholm, Sweden
- 3Department of Medicine Solna, Karolinska Institutet, Stockholm, Sweden
B cells are a core element of the pathophysiology of atherosclerotic cardiovascular disease (ASCVD). Multiple experimental and epidemiological studies have revealed both protective and deleterious functions of B cells in atherosclerotic plaque formation. The spearhead property of B cells that influences the development of atherosclerosis is their unique ability to produce and secrete high amounts of antigen-specific antibodies that can act at distant sites. Exposure to an atherogenic milieu impacts B cell homeostasis, cell differentiation and antibody production. However, it is not clear whether B cell responses in atherosclerosis are instructed by atherosclerosis-specific antigens (ASA). Dissecting the full spectrum of the B cell properties in atherosclerosis will pave the way for designing innovative therapies against the devastating consequences of ASCVD.
Introduction
Heart attacks and strokes are the leading causes of mortality and morbidity worldwide (1–3). The main underlying pathology of these clinical manifestations is ASCVD, which leads to the formation of plaques in large and medium-sized arteries. Rupture or erosion of atherosclerotic plaques triggers thrombus formation thereby causing myocardial infarction (MI) or stroke (4, 5). Atherosclerosis is a lipid-driven chronic inflammatory disease characterized by progressive retention of cholesterol-carrying low-density lipoprotein (LDL) particles in the subendothelial space of arteries (6, 7) followed by a chronic maladaptive immune response (4, 8–12) and remodeling of the artery wall (13), fueled by genetic (14) and lifestyle risk factors (13). Local enzymes act on retained lipoproteins, which leads to LDL aggregation and oxidation (OxLDL) characterized by the formation of lipid peroxidation-derived products called oxidation-specific epitopes (OSE) (15, 16). The accumulating modified LDL particles stimulate endothelial cells to produce adhesion molecules and chemokines (17), which attract circulating leukocytes such as T lymphocytes (18) and monocytes (19) to the vessel wall.
The controlled double-blind clinical trials CANTOS (20), COLCOT (21), and LoDoCo2 (22) have demonstrated the therapeutic value of immunomodulation in secondary prevention of ASCVD. While these studies have shown that inflammation is crucially involved in human ASCVD, they also revealed the need for the development of precise immunotherapies that would limit side effects, such as the risk for fatal infections (23).
B Cells Are Key Pieces of the CVD “Immune-Mosaic”
Patients with autoimmune rheumatic diseases, who display dysregulated responses of adaptive immunity (B and T lymphocytes), are at high risk for premature ischemic heart disease due to accelerated development of atherosclerosis that cannot be fully explained by the traditional Framingham risk factors such as cholesterol levels, smoking and systolic blood pressure (24). Furthermore, mice lacking adaptive immunity display reduced atherosclerosis (25). These findings have highlighted the crucial role of adaptive immunity in modulating atherosclerosis. Several studies have revealed a broad spectrum of T cell [reviewed elsewhere; (18)] and B cell properties that affect atherosclerosis (26–28).
B cells have the unique ability to generate immunoglobulins that can be displayed on the cell surface in the form of the B cell receptor (BCR) or secreted as antibodies. In mouse B lymphopoiesis, B-cell-biased lymphoid progenitors (BLPs) differentiate via the pre-pro-B cell stage to committed pro-B cells, with commitment regulated by the transcription factor Pax5 (29). The successful display of a recombined heavy chain together with surrogate light chains on the cell surface provides proliferative signals to large pre-B cells and this cell division is followed by rearrangement of the light chain genes at the small pre-B cell stage, hence completing V(D)J recombination and resulting in an immature B cell that displays IgM on the cell surface. Upon completion of recombination events, the B cells can leave the bone marrow to further mature in secondary lymphoid organs. Although the marrow of long bones is often considered the predominant site of B lymphopoiesis, other locations are noticeable for B cell development, including the fetal liver, the calvaria of the skull (30), and also the mouse intestinal lamina propria (31).
Mature B cells consist of two main subsets, the conventional B-2 cells, and the less frequent B-1 cell subset (26). B-1 and B-2 cells display differences in their activation requirements, anatomical localization, and surface markers. B-1 cells are subdivided into B-1a and B-1b cells. B-1a cells are long-lived and self-renewing innate-like B cells that are derived from the fetal liver hematopoiesis, and are enriched within the peritoneal and pleural cavities, although a substantial population also can be found in the spleen (32). Notably, CD20+CD27+CD43+CD70– B cells were proposed to be the equivalent of mouse B-1 cells in humans (33). However, this remains unsettled considering the similarities of CD20+CD27+CD43+CD70– B cells with preplasmablasts (34, 35). On the other hand, B-2 cells display many similarities between mice and humans concerning their localization and function (36). B-2 cells include the follicular (FO) B cells and the marginal zone (MZ) B cells. Both subsets are generated through the maturation of splenic immature B cells, which have successfully escaped the bone marrow selection, via pertinent BCR signaling (37). In contrast to MZ B cells, FO B cells display circulating properties, which allow them to home to distant sites (37).
Early evidence supporting a role for B cells in human atherosclerosis is derived from studies more than 40 years ago that demonstrated the presence of immunoglobulins in atherosclerotic arteries (38, 39). Based on histological analyses, B cells are commonly detected in adventitia surrounding atherosclerotic regions with the ability to recirculate to draining lymph nodes (40), while they are an infrequent cell type in atherosclerotic plaques (41). However, although a technical contamination of circulating B cells cannot be excluded, a mass-cytometry analysis of human carotid atherosclerotic plaques revealed a substantial portion of plaque B cells (42). Besides being present in atherosclerotic arteries, a systems biology investigation of whole blood gene expression data from Framingham Heart Study participants and genome-wide association studies coupled to the construction of co-expression networks, identified coronary heart disease-specific causative modules enriched in genes regulating B-cell activation (43), thereby providing indications for a functional role of B cells in human atherosclerosis. In line with this, numbers of activated CD19+CD86+ B cells or IgM+ unswitched memory B cells display a positive and negative association, respectively, with increased risk for stroke in humans (44, 45), suggesting that B cell activation may be involved in the progression of atherosclerosis.
The first experimental evidence that B cells impact atherosclerosis was provided by Caligiuri et al., who showed that splenectomy-induced acceleration of atherosclerosis in Apolipoprotein E deficient (Apoe–/–) mice could be rescued upon transfer of splenic B cells that were isolated either from wild type or Apoe–/– donors (46). Next, Major et al. reported that lethally irradiated LDL receptor-deficient (Ldlr–/–) mice that were injected with bone marrow from B cell-deficient (μMT) donor mice developed increased atherosclerosis compared to controls (47). However, in a recent study Tay et al., reported that Apoe–/– μMT mice developed decreased atherosclerosis compared to control Apoe–/–mice (48). Apoe–/– mice accumulated predominately VLDL remnants in their circulation whereas in Ldlr–/– mice the main accumulating lipoprotein in plasma is the LDL (49). Apart from the obvious reasons, such as different experimental settings, the differences in lipoprotein profile and metabolism may be, at least in part, responsible for the differential effect in atherosclerosis upon B cell deficiency between Ldlr–/– and Apoe–/–mice.
B cell subsets exhibit distinct effects in atherosclerosis, which further emphasizes the sophisticated involvement of B cells in this disease (Figure 1). B-1 cells confer protection in atherosclerosis (50, 51). On the other hand, treatment of Apoe–/– or Ldlr–/– mice with a B cell depleting anti-CD20 antibody, which preferentially depletes B-2 cells, reduced atherosclerosis, and prevented the MI-induced acceleration of atherosclerosis (52–54). In addition, genetic deletion of the B cell transcription factor Pax5 in CD23-expressing cells (primarily mature B2 cells) (55), or treatment with an agonistic antibody specific for B- and T-lymphocyte attenuator (56) that reduced mature B-2 cells, also resulted in decreased atherosclerosis. Disruption of the B cell-activating factor receptor (BAFFR) pathway, which is essential for B-2 (but not B-1) cell survival (57), also conferred an atheroprotective effect (58–61). However, selective ablation of MZ B cells increases atherosclerosis (62), which indicates that therapeutic strategies targeting the entire B-2 cell compartment may not be optimal, and thus, dissecting the functions of B cell responses is essential for the designing of precise therapies in atherosclerosis.
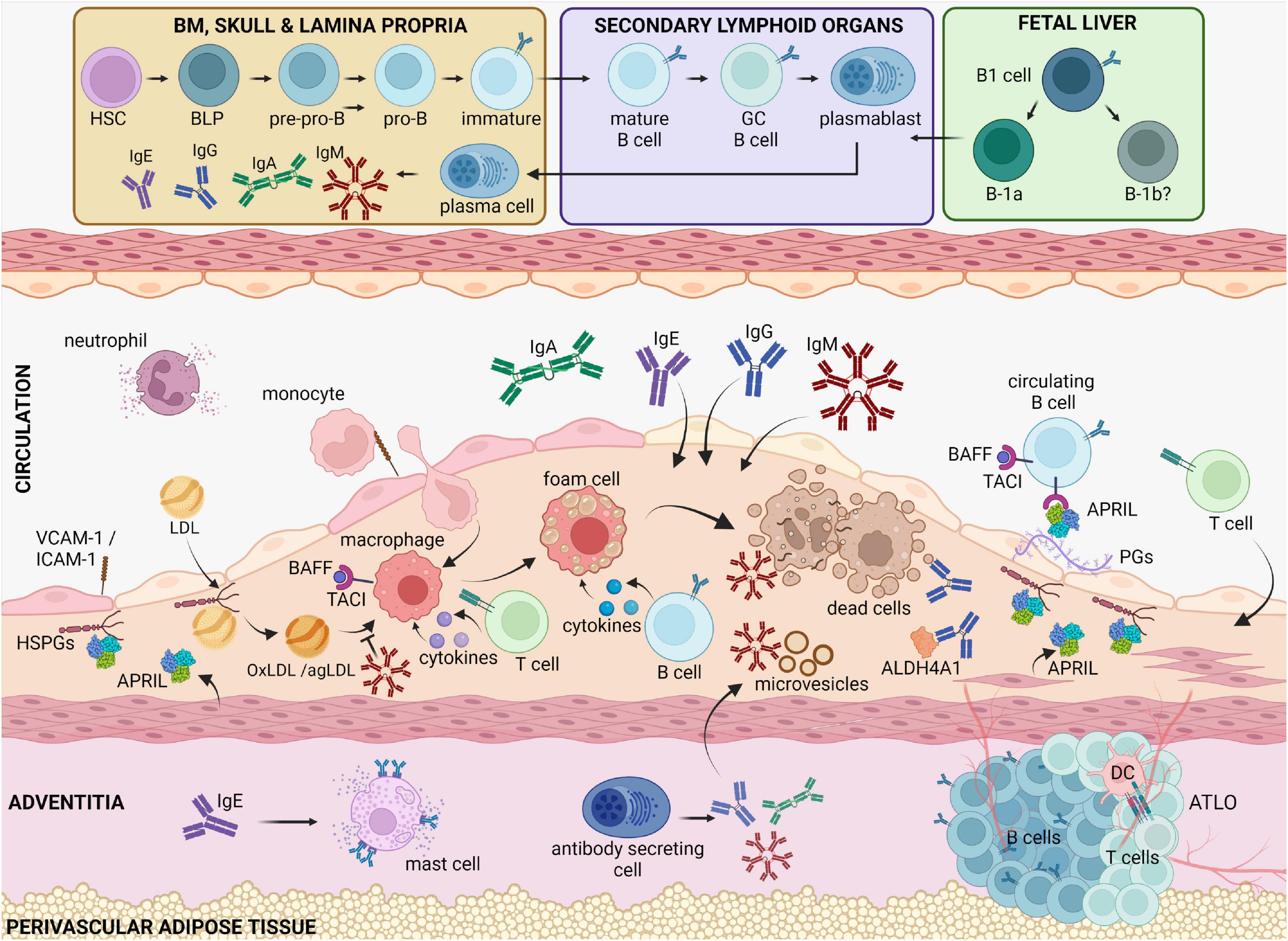
Figure 1. B cell functions in atherosclerotic disease. Circulating cholesterol-containing LDL particles are progressively retained in the subendothelial space of arteries. Oxidized or aggregated LDL particles are taken up by arterial macrophages, which turn to foam cells because of uncontrolled lipid uptake and thereby undergo apoptosis or necrosis. Antibody-producing cells that reside in the bone marrow, the spleen and in the adventitia, produce high amounts of antibodies that are deposited in the plaque. IgM is known to bind and block the proinflammatory effects of oxidized LDL, apoptotic cells and microvesicles. IgE antibodies by binding to FcεRI receptors activate powerful proinflammatory responses by mast cells and macrophages. IgG antibodies can also bind OxLDL as well as self-proteins, such as ALDH4A1, and modulate macrophage activation. BAFF and APRIL, which both bind the TACI receptor in B cells, dampen the proinflammatory responses by macrophages, and limit the LDL retention in the intima, respectively, thereby revealing an indirect property of B cells to regulate plaque inflammation. BLPs, B-cell-biased lymphoid progenitors; PGs, proteoglycans, TACI, Transmembrane activator and CAML interactor; BAFF, B cell activating factor; APRIL, A Proliferation Inducing Ligand, HSPGs, heparan sulfate proteoglycans; BM, bone marrow; ATLO, artery tertiary lymphoid organ; OxLDL, oxidized LDL; agLDL, aggregated LDL.
Antibody-Mediated Functions of B Cells in Atherosclerosis
The main property of B cells that plays a crucial role in atherosclerosis is antibody production. B-1 cells secrete high amounts of natural IgM antibodies, which are produced in absence of a foreign microbial threat (63). On the other hand, FO B cells can enter the germinal center (GC) reaction, which is notable for producing high-affinity antibodies through the process of somatic hypermutation, although this process is not exclusive to the GC. GCs can be found in the secondary lymphoid organs of Apoe–/– and Ldlr–/– mice (27, 55, 62, 64). Upon exit from the GC reaction, B cells can differentiate into short- and long-lived memory B cells as well as short- and long-lived antibody-producing cells (65).
Immunoglobulin M, a Trustworthy Groundkeeper
There is a consensus that IgM exhibits atheroprotective properties. For instance, transfer of B-1a cells into splenectomized mice, which exhibit a severe reduction in peritoneal B-1a cells and circulating IgM antibodies, reversed splenectomy-accelerated atherosclerosis (50). However, the protective effect of B-1a cell transfer in this setting was absent when B-1a cells deficient in secreted IgM (sIgM) were injected (50). Consistent with this, mice lacking sIgM develop aggravated atherosclerosis (66–68). Moreover, B cell-specific CXCR4 (C-X-C chemokine receptor type 4) deficiency, which resulted in reduced IgM levels in plasma, led to increased atherosclerosis in female mice (69). Thus, dissecting the molecular pathways that regulate the production of atheroprotective IgM antibodies may reveal new therapeutic strategies for atherosclerosis. Along this line, apoptotic cell injection (64), infusion of liposomes decorated with phosphatidylserine moieties (70), and genetically induced inhibition of antibody class-switching (71) led to increased total IgM levels in plasma and reduced atherosclerosis. In addition, the transfer of B-1b cells into lymphocyte-deficient atherosclerotic mice also led to increased plasma IgM and reduced plaque size (51). Thus, strategies directly promoting the expansion of B-1a or B-1b cells could be of interest. For instance, reduced atherosclerosis along with increased B-1a cell numbers and circulating IgM levels were reported in atherosclerosis-prone mice that were deficient in sialic acid-binding immunoglobulin-like lectin G (72) or had been treated with an antibody against the phosphatidylserine receptor T-cell immunoglobulin and mucin domain-1 (73). However, therapeutic strategies for the expansion of B-1a cells have to be considered with caution as they may be accompanied by an increase of the proatherogenic B-1a cell-derived subset, the innate response activator (IRA) B cells (74), which via producing granulocyte-macrophage colony-stimulating factor instruct a dendritic cell-mediated promotion of proatherogenic Th1 immunity (75).
The identification of atherosclerosis-specific antigens (ASA) will allow the designing of precise therapeutic strategies in atherosclerosis. Clinical studies have shown an inverse correlation of OSE-specific IgM against malondialdehyde and phosphorylcholine (PC), which are present on oxidized LDL (16, 76) and apoptotic cellular debris (77, 78), with atherosclerotic burden and cardiovascular outcomes (28, 79–82). The implication of OSE-specific IgM antibodies in atherosclerosis was originally shown by using the E06 IgM antibody that binds oxidized phospholipids (OxPLs) and has an identical CDR3 region to the germline-encoded B-1 cell-derived T15 clone (83). Immunization with heat-killed pneumococcal extracts led to a strong increase of the PC-specific T15/E06 IgM clonotype and decreased lesion formation (84). Furthermore, passive infusion of T15/E06 IgM antibodies reduced vein graft atherosclerosis in atherosclerotic Apoe–/– mice thereby providing direct evidence that the E06 IgM confers an atheroprotective effect (85). In a seminal study by Prof. Witztum’s lab, it has been shown that transgenic overexpression of the single-chain variable fragment of E06 strongly decreased atherosclerosis in Ldlr–/– mice (86). These data suggest that E06 acts as a blocking antibody limiting the proinflammatory effect of OxPLs in atherosclerosis in vivo. This is supported by the capacity of E06 to block OxLDL uptake by macrophages (87) and proinflammatory cytokine production by OxPL-stimulated macrophages (88) in vitro. While the expansion of OSE-specific IgM could be considered therapeutically in atherosclerosis, it is important to identify its right “therapeutic window.” This is essential as endogenous OSE-specific IgMs are present at high levels in both mice and humans (89) and increase over time in hypercholesterolemia (90). In fact, infusion of purified T15/E06 preparations (91) or the OxPL-neutralizing 10C12 IgM clone (68) had no effect in advanced atherosclerosis. Furthermore, it appears likely that long exposure to an atherogenic milieu might induce the expansion of IgM with shared antigen specificities. For instance, genetic deficiency of the VHS107.1.42 locus, which is essential for the successful production of T15 antibodies, did not affect experimental atherosclerosis in mice with marked dyslipidemia (92). IgMs also recognize other self-antigens and thereby regulate the maturation of B-2 cells (93–96) and circulating levels of other immunoglobulins (67, 97, 98). Notably, mice lacking sIgM, display high levels of IgE antibodies, which are responsible for the accelerated atherosclerosis in this setting (67). Taken together, the properties of IgM in atherosclerosis demonstrate the important role of (neo)-self-antigens in this disease.
Immunoglobulin E, a Powerful Assailant
The most well-documented properties of IgE antibodies are their role in triggering an allergic reaction and fighting microbial infections (99, 100). These properties of IgE are mediated via binding to high-affinity IgE receptor FcεRI, which is mainly present on mast cells, basophils, and eosinophils (101). Mice lacking the FcεRI receptor display reduced atherosclerotic plaque size, thereby suggesting that IgE antibodies play a role in atherosclerosis (102). The proatherogenic role of IgE antibodies was directly shown using a neutralizing anti-IgE antibody specific for free IgE that, as mentioned above, completely reversed the accelerated atherosclerosis in atherosclerotic sIgM deficient mice, which display high plasma IgE antibodies (67). In agreement with this, mice deficient in IgE antibodies developed decreased atherosclerosis (103). Furthermore, a systemic IgE-mediated mast cell activation in atherosclerotic mice lacking B cells resulted in increased lesion size (104). Mechanistically, IgEs promote mast cell and neutrophil activation, and the production of proinflammatory cytokines by macrophages and smooth muscle cells (67, 102, 103), which could be responsible for their effect in atherosclerosis in vivo. The detrimental role of IgE antibodies in atherosclerosis is also supported by several epidemiological studies (105, 106) that also implicate the mammalian oligosaccharide galactose-α-1,3-galactose as a candidate antigen (107). Future studies are required to identify the spectrum of proatherogenic IgE-specific antigens.
Immunoglobulin G, a Vault for Atherosclerosis-Specific Antigens?
IgG antibodies are produced in different subclasses: IgG1, IgG2, IgG3, and IgG4 in humans and IgG1, IgG2a/c, IgG2b, and IgG3 in mice (108). Tay et al., provided the first direct evidence on the role of IgG antibodies in atherosclerosis, by showing that mice lacking most endogenous immunoglobulins developed increased plaque size upon injection of purified total IgG from atherosclerotic mice compared to IgG from non-atherosclerotic donors (109). This study also suggests that exposure to an atherosclerotic milieu alters the antigen specificities of the IgG repertoire by inducing the expansion or even the de novo generation of B cell clonotypes that are likely to include specificities against ASA. In line with this, a protein array analysis revealed an altered repertoire of IgG1 protein targets in the serum of Apoe–/– vs. C57BL/6 mice fed an atherogenic diet (71). It is not clear whether the altered IgG repertoire is triggered upon chronic exposure to atherogenic pressure or already emerges at the initiation of the disease. In this regard, abrupt loss of APOE, which results in acute onset of dyslipidemia, triggered a rapid increase in IgG antibodies levels enriched in specificities against common autoantigens (55). These data suggest that the GC reaction is involved in atherosclerosis. In agreement with this, elimination of GC B cells achieved upon deletion of the key B cell transcription factor Pax5 in AID-expressing B cells, reduced atherosclerosis (55, 110). Furthermore, B cell-specific overexpression of the FcγRIIB receptor limited GC B cell responses and reduced atherosclerosis in male mice (111). Moreover, deletion of Prdm1 encoding the key transcription factor BLIMP1, in all B cells (110) or selectively in mature B cells (109), caused impaired plasma cell differentiation and a dramatic reduction in all immunoglobulin isotypes (particularly in IgG) and led to reduced atherosclerotic plaque size (109, 110). While these data show that the IgG antibodies confer an overall proatherogenic effect, B cell-specific deletion of the transcription factor x-box binding protein-1, which similarly to BLIMP-1 deficiency resulted in reduced levels of all immunoglobulins, increased early atherosclerosis (112). Thus, it is conceivable that the IgG repertoire also includes atheroprotective clones. Indeed, Lorenzo et al. have recently shown that GC-derived antibodies from hypercholesterolemic mice against mitochondrial dehydrogenase ALDH4A1 protect from atherosclerosis (27), thereby demonstrating that the repertoire of the antigen specificities of GC B cells includes also protective responses in atherosclerosis. This conclusion is also supported by Centa et al. showing that IgG antibodies could promote plaque stability (110). It appears promising that the identification of ASA recognized by IgG would reveal new mechanistic layers for the role of B cell responses in atherosclerosis.
Antibody-Independent Functions of B Cells in Atherosclerosis
B cells are an important source of cytokines (113). Transfer of CD21hiCD23hiCD24hi IL-10 secreting B cells isolated from renal lymph nodes into syngeneic mice increased plaque size in a perivascular collar injury model of the carotid artery (114). In contrast, B cell-specific IL-10 deficiency did not affect atherosclerosis in the aortic root (115). These data suggest that B cells may exhibit distinct effects in different atherosclerosis-prone sites. B-2 cell functions that affect atherosclerosis, such as antigen presentation via MHCII complexes (48, 109), CD40 (48, 109), and GITRL (116) signaling, involve interaction with T cells. In addition, MZ B cells mediate their protective effect in atherosclerosis via suppressing the proatherogenic responses of T follicular helper cells (62). However, lymphocyte-deficient mice that were injected with splenic B-2 cells developed increased atherosclerosis (53), which shows that B-2 cells can affect plaque formation in absence of T cells, for example via the production of TNF (117). Furthermore, BAFFR deficiency or blockage (that leads to dramatically reduced B cell numbers) limits atherosclerosis (58–60), whereas soluble BAFF neutralization aggravates atherosclerotic plaque size (118). Both B cell deficiency (48) as well BAFFR blockage (60) result in increased soluble BAFF levels that could be responsible for the atheroprotective effect in these settings. Similarly, A Proliferation Inducing Ligand (APRIL), which is recognized by B cells through shared receptors with BAFF, confers atheroprotection via binding to heparan-sulfate proteoglycans (HSPGs) in the artery wall (119). Therefore, it is likely that B cell depletion in the vessel wall (120, 121) would increase the availability of APRIL for binding to HSPGs.
Summary and Future Perspectives
B cells have the capacity to sense and respond to atherosclerosis. The revolutionary development of high-throughput technologies and methods for gene editing will allow the dissection of the mechanisms by which B cells impact atherosclerotic plaque formation in an unprecedented depth. A key point in this effort would be to identify if and how the B cell response in atherosclerosis is driven by ASA. This will set the ground for the development of precise therapies that will target selectively culprit or atheroprotective B cell clones.
Author Contributions
DS, AG, SM, and DT researched the data, wrote, and reviewed the manuscript. DS designed the figure (created with BioRender.com) with input from the other authors. DT supervised the preparation of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
DT was supported by the European Research Area Network on Cardiovascular Diseases (I4647), the Austrian Science Fund (I4963 and P35233) and the Austrian Heart Foundation. SM was funded by the Swedish Research Council (2020-01593), the Swedish Heart-Lung Foundation (20210520 and 20210532) and the Leducq Foundation Networks of Excellence Program grant B cells in Cardiovascular Disease. AG was funded by the Swedish Research Council (2020-01789), the Swedish Heart-Lung Foundation (20200333 and 20210469), Loo and Hans Osterman Foundation for Medical Research, Foundation for Geriatric Diseases at Karolinska Institutet, and Stiftelsen för Gamla Tjänarinnor.
Conflict of Interest
DT was a named inventor on a patent application (EP20217536.0) to exploit APRIL for diagnostic and therapeutic purposes in cardiovascular diseases that has been filed by the Medical University of Vienna (Austria) and CeMM Research Center for Molecular Medicine of the Austrian Academy of Sciences (Austria).
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Dai H, Much AA, Maor E, Asher E, Younis A, Xu Y, et al. Global, regional, and national burden of ischaemic heart disease and its attributable risk factors, 1990-2017: results from the Global Burden of Disease Study 2017. Eur Heart J Qual Care Clin Outc. (2022) 8:50–60. doi: 10.1093/ehjqcco/qcaa076
2. GBD 2017 Causes of Death Collaborators. Global, regional, and national age-sex-specific mortality for 282 causes of death in 195 countries and territories, 1980-2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet. (2018) 392:1736–88. doi: 10.1016/S0140-6736(18)32203-7
3. Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM, et al. Global burden of cardiovascular diseases and risk factors, 1990-2019: update from the GBD 2019 study. J Am Coll Cardiol. (2020) 76:2982–3021. doi: 10.1016/j.jacc.2020.11.010
4. Libby P, Lichtman AH, Hansson GK. Immune effector mechanisms implicated in atherosclerosis: from mice to humans. Immunity. (2013) 38:1092–104. doi: 10.1016/j.immuni.2013.06.009
5. Fahed AC, Jang IK. Plaque erosion and acute coronary syndromes: phenotype, molecular characteristics and future directions. Nat Rev Cardiol. (2021) 18:724–34. doi: 10.1038/s41569-021-00542-3
6. Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol. (1995) 15:551–61. doi: 10.1161/01.atv.15.5.551
7. Skalen K, Gustafsson M, Rydberg EK, Hulten LM, Wiklund O, Innerarity TL, et al. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature. (2002) 417:750–4. doi: 10.1038/nature00804
9. Everett BM, Cornel JH, Lainscak M, Anker SD, Abbate A, Thuren T, et al. Anti-inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation. (2019) 139:1289–99. doi: 10.1161/CIRCULATIONAHA.118.038010
10. Watkins H, Farrall M. Genetic susceptibility to coronary artery disease: from promise to progress. Nat Rev Genet. (2006) 7:163–73. doi: 10.1038/nrg1805
11. Tyrrell DJ, Goldstein DR. Ageing and atherosclerosis: vascular intrinsic and extrinsic factors and potential role of IL-6. Nat Rev Cardiol. (2021) 18:58–68. doi: 10.1038/s41569-020-0431-7
12. Zhao TX, Mallat Z. Targeting the Immune System in Atherosclerosis: JACC State-of-the-Art Review. J Am Coll Cardiol. (2019) 73:1691–706. doi: 10.1016/j.jacc.2018.12.083
13. Libby P. The changing landscape of atherosclerosis. Nature. (2021) 592:524–33. doi: 10.1038/s41586-021-03392-8
14. Klarin D, Natarajan P. Clinical utility of polygenic risk scores for coronary artery disease. Nat Rev Cardiol. (2021). [Online ahead of print]. doi: 10.1038/s41569-021-00638-w
15. Ruuth M, Nguyen SD, Vihervaara T, Hilvo M, Laajala TD, Kondadi PK, et al. Susceptibility of low-density lipoprotein particles to aggregate depends on particle lipidome, is modifiable, and associates with future cardiovascular deaths. Eur Heart J. (2018) 39:2562–73. doi: 10.1093/eurheartj/ehy319
16. Binder CJ, Papac-Milicevic N, Witztum JL. Innate sensing of oxidation-specific epitopes in health and disease. Nat Rev Immunol. (2016) 16:485–97. doi: 10.1038/nri.2016.63
17. Gimbrone MA Jr., Garcia-Cardena G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. (2016) 118:620–36. doi: 10.1161/CIRCRESAHA.115.306301
18. Saigusa R, Winkels H, Ley KT. Cell subsets and functions in atherosclerosis. Nat Rev Cardiol. (2020) 17:387–401. doi: 10.1038/s41569-020-0352-5
19. Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. (2011) 145:341–55. doi: 10.1016/j.cell.2011.04.005
20. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31.
21. Tardif JC, Kouz S, Waters DD, Bertrand OF, Diaz R, Maggioni AP, et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med. (2019) 381:2497–505. doi: 10.1056/NEJMoa1912388
22. Nidorf SM, Fiolet ATL, Mosterd A, Eikelboom JW, Schut A, Opstal TSJ, et al. Colchicine in patients with chronic coronary disease. N Engl J Med. (2020) 383:1838–47.
23. Lutgens E, Atzler D, Doring Y, Duchene J, Steffens S, Weber C. Immunotherapy for cardiovascular disease. Eur Heart J. (2019) 40:3937–46.
24. Skaggs BJ, Hahn BH, McMahon M. Accelerated atherosclerosis in patients with SLE–mechanisms and management. Nat Rev Rheumatol. (2012) 8:214–23. doi: 10.1038/nrrheum.2012.14
25. Song L, Leung C, Schindler C. Lymphocytes are important in early atherosclerosis. J Clin Invest. (2001) 108:251–9. doi: 10.1172/JCI11380
26. Tsiantoulas D, Diehl CJ, Witztum JL, Binder CJ. B cells and humoral immunity in atherosclerosis. Circ Res. (2014) 114:1743–56. doi: 10.1161/circresaha.113.301145
27. Lorenzo C, Delgado P, Busse CE, Sanz-Bravo A, Martos-Folgado I, Bonzon-Kulichenko E, et al. ALDH4A1 is an atherosclerosis auto-antigen targeted by protective antibodies. Nature. (2021) 589:287–92. doi: 10.1038/s41586-020-2993-2
28. Sage AP, Tsiantoulas D, Binder CJ, Mallat Z. The role of B cells in atherosclerosis. Nat Rev Cardiol. (2019) 16:180–96.
29. Medvedovic J, Ebert A, Tagoh H, Busslinger M. Pax5: a master regulator of B cell development and leukemogenesis. Adv Immunol. (2011) 111:179–206. doi: 10.1016/B978-0-12-385991-4.00005-2
30. Brioschi S, Wang WL, Peng V, Wang M, Shchukina I, Greenberg ZJ, et al. Heterogeneity of meningeal B cells reveals a lymphopoietic niche at the CNS borders. Science. (2021) 373:eabf9277. doi: 10.1126/science.abf9277
31. Wesemann DR, Portuguese AJ, Meyers RM, Gallagher MP, Cluff-Jones K, Magee JM, et al. Microbial colonization influences early B-lineage development in the gut lamina propria. Nature. (2013) 501:112–5. doi: 10.1038/nature12496
32. Baumgarth N. The double life of a B-1 cell: self-reactivity selects for protective effector functions. Nat Rev Immunol. (2011) 11:34–46. doi: 10.1038/nri2901
33. Griffin DO, Holodick NE, Rothstein TL. Human B1 cells in umbilical cord and adult peripheral blood express the novel phenotype CD20+ CD27+ CD43+ CD70. J Exp Med. (2011) 208:67–80. doi: 10.1084/jem.20101499
34. Warnatz K, Salzer U, Rizzi M, Fischer B, Gutenberger S, Bohm J, et al. B-cell activating factor receptor deficiency is associated with an adult-onset antibody deficiency syndrome in humans. Proc Natl Acad Sci USA. (2009) 106:13945–50. doi: 10.1073/pnas.0903543106
35. Covens K, Verbinnen B, Geukens N, Meyts I, Schuit F, Van Lommel L, et al. Characterization of proposed human B-1 cells reveals pre-plasmablast phenotype. Blood. (2013) 121:5176–83. doi: 10.1182/blood-2012-12-471953
36. Garraud O, Borhis G, Badr G, Degrelle S, Pozzetto B, Cognasse F, et al. Revisiting the B-cell compartment in mouse and humans: more than one B-cell subset exists in the marginal zone and beyond. BMC Immunol. (2012) 13:63. doi: 10.1186/1471-2172-13-63
37. Pillai S, Cariappa A. The follicular versus marginal zone B lymphocyte cell fate decision. Nat Rev Immunol. (2009) 9:767–77. doi: 10.1038/nri2656
38. Hollander W, Colombo MA, Kirkpatrick B, Paddock J. Soluble proteins in the human atheroschlerotic plaque With Spectral Reference to Immunoglobulins, C3-Complement Component, α1-Antitrypsin and α2-Macroglobulin. Atherosclerosis. (1979) 34:391–405. doi: 10.1016/0021-9150(79)90064-9
39. Parums D, Mitchinson MJ. Demonstration of immunoglobulin in the neighbourhood of advanced atherosclerotic plaques. Atherosclerosis. (1981) 38:211–6. doi: 10.1016/0021-9150(81)90118-0
40. Hamze M, Desmetz C, Berthe ML, Roger P, Boulle N, Brancherau P, et al. Characterization of resident B cells of vascular walls in human atherosclerotic patients. J Immunol. (2013) 191:3006–16. doi: 10.4049/jimmunol.1202870
41. Jonasson L, Holm J, Skalli O, Bondjers G, Hansson GK. Regional accumulations of T cells, macrophages, and smooth muscle cells in the human atherosclerotic plaque. Arteriosclerosis. (1986) 6:131–8. doi: 10.1161/01.atv.6.2.131
42. Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir ED, Amadori L, et al. Single-cell immune landscape of human atherosclerotic plaques. Nat Med. (2019) 25:1576–88. doi: 10.1038/s41591-019-0590-4
43. Huan T, Zhang B, Wang Z, Joehanes R, Zhu J, Johnson AD, et al. A systems biology framework identifies molecular underpinnings of coronary heart disease. Arterioscler Thromb Vasc Biol. (2013) 33:1427–34. doi: 10.1161/ATVBAHA.112.300112
44. Mantani PT, Ljungcrantz I, Andersson L, Alm R, Hedblad B, Björkbacka H, et al. Circulating CD40+ and CD86+ B cell subsets demonstrate opposing associations with risk of stroke. Arterioscler Thromb Vasc Biol. (2014) 34:211–8. doi: 10.1161/ATVBAHA.113.302667
45. Meeuwsen JAL, van Duijvenvoorde A, Gohar A, Kozma MO, van de Weg SM, Gijsberts CM, et al. High levels of (Un)Switched Memory B cells are associated with better outcome in patients with advanced atherosclerotic disease. J Am Heart Assoc. (2017) 6:e005747. doi: 10.1161/JAHA.117.005747
46. Caligiuri G, Nicoletti A, Poirier B, Hansson GK. Protective immunity against atherosclerosis carried by B cells of hypercholesterolemic mice. J Clin Investig. (2002) 109:745–53. doi: 10.1172/JCI7272
47. Major AS, Fazio S, Linton MF. B-Lymphocyte deficiency increases atherosclerosis in LDL receptor–null mice. Arterioscler Thromb Vasc Biol. (2002) 22:1892–8. doi: 10.1161/01.atv.0000039169.47943.ee
48. Tay C, Kanellakis P, Hosseini H, Cao A, Toh BH, Bobik A, et al. B Cell and CD4 T Cell interactions promote development of atherosclerosis. Front Immunol. (2019) 10:3046. doi: 10.3389/fimmu.2019.03046
49. Getz GS, Reardon CA. Do the Apoe-/- and Ldlr-/- Mice yield the same insight on atherogenesis? Arterioscler Thromb Vasc Biol. (2016) 36:1734–41. doi: 10.1161/ATVBAHA.116.306874
50. Kyaw T, Tay C, Krishnamurthi S, Kanellakis P, Agrotis A, Tipping P, et al. B1a B lymphocytes are atheroprotective by secreting natural IgM that increases IgM deposits and reduces necrotic cores in atherosclerotic lesions. Circ Res. (2011) 109:830–40. doi: 10.1161/CIRCRESAHA.111.248542
51. Rosenfeld SM, Perry HM, Gonen A, Prohaska TA, Srikakulapu P, Grewal S, et al. B-1b Cells Secrete Atheroprotective IgM and Attenuate Atherosclerosis. Circ Res. (2015) 117:e28–39. doi: 10.1161/CIRCRESAHA.117.306044
52. Ait-Oufella H, Herbin O, Bouaziz JD, Binder CJ, Uyttenhove C, Laurans L, et al. B cell depletion reduces the development of atherosclerosis in mice. J Exp Med. (2010) 207:1579–87. doi: 10.1084/jem.20100155
53. Kyaw T, Tay C, Khan A, Dumouchel V, Cao A, To K, et al. Conventional B2 B cell depletion ameliorates whereas its adoptive transfer aggravates atherosclerosis. J Immunol. (2010) 185:4410–9. doi: 10.4049/jimmunol.1000033
54. Kyaw T, Loveland P, Kanellakis P, Cao A, Kallies A, Huang AL, et al. Alarmin-activated B cells accelerate murine atherosclerosis after myocardial infarction via plasma cell-immunoglobulin-dependent mechanisms. Eur Heart J. (2021) 42:938–47. doi: 10.1093/eurheartj/ehaa995
55. Centa M, Prokopec KE, Garimella MG, Habir K, Hofste L, Stark JM, et al. Acute loss of apolipoprotein E triggers an autoimmune response that accelerates atherosclerosis. Arterioscler Thromb Vasc Biol. (2018) 38:e145–58. doi: 10.1161/ATVBAHA.118.310802
56. Douna H, Amersfoort J, Schaftenaar FH, Kroner MJ, Kiss MG, Slutter B, et al. B- and T-lymphocyte attenuator stimulation protects against atherosclerosis by regulating follicular B cells. Cardiovasc Res. (2020) 116:295–305. doi: 10.1093/cvr/cvz129
57. Mackay F, Schneider P. Cracking the BAFF code. Nat Rev Immunol. (2009) 9:491–502. doi: 10.1038/nri2572
58. Kyaw T, Tay C, Hosseini H, Kanellakis P, Gadowski T, MacKay F, et al. Depletion of B2 but not B1a B cells in BAFF receptor-deficient ApoE mice attenuates atherosclerosis by potently ameliorating arterial inflammation. PLoS One. (2012) 7:e29371. doi: 10.1371/journal.pone.0029371
59. Sage AP, Tsiantoulas D, Baker L, Harrison J, Masters L, Murphy D, et al. BAFF receptor deficiency reduces the development of atherosclerosis in mice–brief report. Arterioscler Thromb Vasc Biol. (2012) 32:1573–6. doi: 10.1161/ATVBAHA.111.244731
60. Kyaw T, Cui P, Tay C, Kanellakis P, Hosseini H, Liu E, et al. BAFF Receptor mAb Treatment Ameliorates Development and Progression of Atherosclerosis in Hyperlipidemic ApoE(-/-) Mice. PLoS One. (2013) 8:e60430. doi: 10.1371/journal.pone.0060430
61. Ponnuswamy P, Joffre J, Herbin O, Esposito B, Laurans L, Binder CJ, et al. Angiotensin II synergizes with BAFF to promote atheroprotective regulatory B cells. Sci Rep. (2017) 7:4111. doi: 10.1038/s41598-017-04438-6
62. Nus M, Sage AP, Lu Y, Masters L, Lam BYH, Newland S, et al. Marginal zone B cells control the response of follicular helper T cells to a high-cholesterol diet. Nat Med. (2017) 23:601–10. doi: 10.1038/nm.4315
63. Ehrenstein MR, Notley CA. The importance of natural IgM: scavenger, protector and regulator. Nat Rev Immunol. (2010) 10:778–86. doi: 10.1038/nri2849
64. Grasset EK, Duhlin A, Agardh HE, Ovchinnikova O, Hagglof T, Forsell MN, et al. Sterile inflammation in the spleen during atherosclerosis provides oxidation-specific epitopes that induce a protective B-cell response. Proc Natl Acad Sci USA. (2015) 112:E2030–8. doi: 10.1073/pnas.1421227112
65. De Silva NS, Klein U. Dynamics of B cells in germinal centres. Nat Rev Immunol. (2015) 15:137–48. doi: 10.1038/nri3804
66. Lewis MJ, Malik TH, Ehrenstein MR, Boyle JJ, Botto M, Haskard DO. Immunoglobulin M is required for protection against atherosclerosis in low-density lipoprotein receptor-deficient mice. Circulation. (2009) 120:417–26. doi: 10.1161/CIRCULATIONAHA.109.868158
67. Tsiantoulas D, Bot I, Ozsvar-Kozma M, Goderle L, Perkmann T, Hartvigsen K, et al. Increased Plasma IgE Accelerate Atherosclerosis in Secreted IgM Deficiency. Circ Res. (2017) 120:78–84.
68. Cherepanova OA, Srikakulapu P, Greene ES, Chaklader M, Haskins RM, McCanna ME, et al. Novel autoimmune IgM antibody attenuates atherosclerosis in IgM deficient low-fat diet-fed, but not western diet-fed Apoe(-/-) Mice. Arterioscler Thromb Vasc Biol. (2020) 40:206–19. doi: 10.1161/ATVBAHA.119.312771
69. Doring Y, Jansen Y, Cimen I, Aslani M, Gencer S, Peters LJF, et al. B-cell-specific CXCR4 protects against atherosclerosis development and increases plasma IgM levels. Circ Res. (2020) 126:787–8. doi: 10.1161/CIRCRESAHA.119.316142
70. Hosseini H, Li Y, Kanellakis P, Tay C, Cao A, Tipping P, et al. Phosphatidylserine liposomes mimic apoptotic cells to attenuate atherosclerosis by expanding polyreactive IgM producing B1a lymphocytes. Cardiovasc Res. (2015) 106:443–52. doi: 10.1093/cvr/cvv037
71. Hutchinson MA, Park HS, Zanotti KJ, Alvarez-Gonzalez J, Zhang J, Zhang L, et al. Auto-antibody production during experimental atherosclerosis in ApoE(-/-) Mice. Front Immunol. (2021) 12:695220. doi: 10.3389/fimmu.2021.695220
72. Gruber S, Hendrikx T, Tsiantoulas D, Ozsvar-Kozma M, Goderle L, Mallat Z, et al. Sialic acid-binding immunoglobulin-like lectin g promotes atherosclerosis and liver inflammation by suppressing the protective functions of B-1 cells. Cell Rep. (2016) 14:2348–61. doi: 10.1016/j.celrep.2016.02.027
73. Hosseini H, Yi L, Kanellakis P, Cao A, Tay C, Peter K, et al. Anti-TIM-1 monoclonal antibody (RMT1-10) attenuates atherosclerosis by expanding IgM-producing B1a cells. J Am Heart Assoc. (2018) 7:e008447. doi: 10.1161/JAHA.117.008447
74. Rauch PJ, Chudnovskiy A, Robbins CS, Weber GF, Etzrodt M, Hilgendorf I, et al. Innate response activator B cells protect against microbial sepsis. Science. (2012) 335:597–601. doi: 10.1126/science.1215173
75. Hilgendorf I, Theurl I, Gerhardt L, Robbins CS, Weber GF, Gonen A, et al. Innate response activator b cells aggravate atherosclerosis by stimulating T helper-1 adaptive immunity. Circulation. (2014) 129:1677–87. doi: 10.1161/CIRCULATIONAHA.113.006381
76. Palinski W, Hörkkö S, Miller E, Steinbrecher UP, Powell HC, Curtiss LK, et al. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E-deficient mice. Demonstration of epitopes of oxidized low density lipoprotein in human plasma. J Clin Investig. (1996) 98:800–14. doi: 10.1172/JCI118853
77. Chang MK, Binder CJ, Miller YI, Subbanagounder G, Silverman GJ, Berliner JA, et al. Apoptotic cells with oxidation-specific epitopes are immunogenic and proinflammatory. J Exp Med. (2004) 200:1359–70. doi: 10.1084/jem.20031763
78. Tsiantoulas D, Perkmann T, Afonyushkin T, Mangold A, Prohaska TA, Papac-Milicevic N, et al. Circulating microparticles carry oxidation-specific epitopes and are recognized by natural IgM antibodies. J Lipid Res. (2015) 56:440–8. doi: 10.1194/jlr.P054569
79. Tsimikas S, Willeit P, Willeit J, Santer P, Mayr M, Xu Q, et al. Oxidation-specific biomarkers, prospective 15-year cardiovascular and stroke outcomes, and net reclassification of cardiovascular events. J Am Coll Cardiol. (2012) 60:2218–29. doi: 10.1016/j.jacc.2012.08.979
80. Tsimikas S, Brilakis ES, Lennon RJ, Miller ER, Witztum JL, McConnell JP, et al. Relationship of IgG and IgM autoantibodies to oxidized low density lipoprotein with coronary artery disease and cardiovascular events. J Lipid Res. (2007) 48:425–33. doi: 10.1194/jlr.M600361-JLR200
81. van den Berg VJ, Haskard DO, Fedorowski A, Hartley A, Kardys I, Caga-Anan M, et al. IgM anti-malondialdehyde low density lipoprotein antibody levels indicate coronary heart disease and necrotic core characteristics in the Nordic Diltiazem (NORDIL) study and the Integrated Imaging and Biomarker Study 3 (IBIS-3). EBioMedicine. (2018) 36:63–72. doi: 10.1016/j.ebiom.2018.08.023
82. Boyle JJ, Christou I, Iqbal MB, Nguyen AT, Leung VW, Evans PC, et al. Solid-phase immunoglobulins IgG and IgM activate macrophages with solid-phase IgM acting via a novel scavenger receptor a pathway. Am J Pathol. (2012) 181:347–61. doi: 10.1016/j.ajpath.2012.03.040
83. Shaw PX, Horkko S, Chang MK, Curtiss LK, Palinski W, Silverman GJ, et al. Natural antibodies with the T15 idiotype may act in atherosclerosis, apoptotic clearance, and protective immunity. J Clin Invest. (2000) 105:1731–40. doi: 10.1172/JCI8472
84. Binder CJ, Hörkkö S, Dewan A, Chang M-K, Kieu EP, Goodyear CS, et al. Pneumococcal vaccination decreases atherosclerotic lesion formation: molecular mimicry between Streptococcus pneumoniae and oxidized LDL. Nat Med. (2003) 9:736. doi: 10.1038/nm876
85. Faria-Neto JR, Chyu K-Y, Li X, Dimayuga PC, Ferreira C, Yano J, et al. Passive immunization with monoclonal IgM antibodies against phosphorylcholine reduces accelerated vein graft atherosclerosis in apolipoprotein E-null mice. Atherosclerosis. (2006) 189:83–90. doi: 10.1016/j.atherosclerosis.2005.11.033
86. Que X, Hung MY, Yeang C, Gonen A, Prohaska TA, Sun X, et al. Oxidized phospholipids are proinflammatory and proatherogenic in hypercholesterolaemic mice. Nature. (2018) 558:301–6. doi: 10.1038/s41586-018-0198-8
87. Boullier A, Gillotte KL, Hörkkö S, Green SR, Friedman P, Dennis EA, et al. The binding of oxidized low density lipoprotein to mouse CD36 is mediated in part by oxidized phospholipids that are associated with both the lipid and protein moieties of the lipoprotein. J Biol Chem. (2000) 275:9163–9. doi: 10.1074/jbc.275.13.9163
88. Imai Y, Kuba K, Neely GG, Yaghubian-Malhami R, Perkmann T, van Loo G, et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell. (2008) 133:235–49. doi: 10.1016/j.cell.2008.02.043
89. Chou MY, Fogelstrand L, Hartvigsen K, Hansen LF, Woelkers D, Shaw PX, et al. Oxidation-specific epitopes are dominant targets of innate natural antibodies in mice and humans. J Clin Invest. (2009) 119:1335–49. doi: 10.1172/JCI36800
90. Khoo LHB, Thiam CH, Soh SY, Angeli V. Splenic extrafollicular reactions and BM plasma cells sustain IgM response associated with hypercholesterolemia. Eur J Immunol. (2015) 45:1300–12. doi: 10.1002/eji.201344347
91. Cesena FH, Dimayuga PC, Yano J, Zhao X, Kirzner J, Zhou J, et al. Immune-modulation by polyclonal IgM treatment reduces atherosclerosis in hypercholesterolemic apoE-/- mice. Atherosclerosis. (2012) 220:59–65. doi: 10.1016/j.atherosclerosis.2011.10.002
92. Centa M, Gruber S, Nilsson D, Polyzos KA, Johansson DK, Hansson GK, et al.. Atherosclerosis susceptibility in mice is independent of the V1 immunoglobulin heavy chain gene. Arterioscler Thromb Vasc Biol. (2016) 36:25–36. doi: 10.1161/ATVBAHA.115.305990
93. Tsiantoulas D, Kiss M, Bartolini-Gritti B, Bergthaler A, Mallat Z, Jumaa H, et al. Secreted IgM deficiency leads to increased BCR signaling that results in abnormal splenic B cell development. Sci Rep. (2017) 7:3540. doi: 10.1038/s41598-017-03688-8
94. Baker N, Ehrenstein MR. Cutting edge: selection of B lymphocyte subsets is regulated by natural IgM. J Immunol. (2002) 169:6686–90. doi: 10.4049/jimmunol.169.12.6686
95. Notley CA, Baker N, Ehrenstein MR. Secreted IgM enhances B cell receptor signaling and promotes splenic but impairs peritoneal B cell survival. J Immunol. (2010) 184:3386–93. doi: 10.4049/jimmunol.0902640
96. Nguyen TT, Elsner RA, Baumgarth N. Natural IgM Prevents Autoimmunity by Enforcing B Cell Central Tolerance Induction. J Immunol. (2015) 194:1489–502. doi: 10.4049/jimmunol.1401880
97. Boes M, Schmidt T, Linkemann K, Beaudette BC, Marshak-Rothstein A, Chen J. Accelerated development of IgG autoantibodies and autoimmune disease in the absence of secreted IgM. Proc Natl Acad Sci USA. (2000) 97:1184–9. doi: 10.1073/pnas.97.3.1184
98. Boes M, Esau C, Fischer MB, Schmidt T, Carroll M, Chen J. Enhanced B-1 cell development, but impaired IgG antibody responses in mice deficient in secreted IgM. J Immunol. (1998) 160:4776–87.
99. Wu LC, Zarrin AA. The production and regulation of IgE by the immune system. Nat Rev Immunol. (2014) 14:247–59. doi: 10.1038/nri3632
100. Starkl P, Watzenboeck ML, Popov LM, Zahalka S, Hladik A, Lakovits K, et al. IgE effector mechanisms, in concert with mast cells, contribute to acquired host defense against Staphylococcus aureus. Immunity. (2020) 53:1333. doi: 10.1016/j.immuni.2020.11.012
101. Gould HJ, Sutton BJ. IgE in allergy and asthma today. Nat Rev Immunol. (2008) 8:205–17. doi: 10.1038/nri2273
102. Wang J, Cheng X, Xiang M-X, Alanne-Kinnunen M, Wang J-A, Chen H, et al. IgE stimulates human and mouse arterial cell apoptosis and cytokine expression and promotes atherogenesis in Apoe–/– mice. J Clin Investig. (2011) 121:3564–77. doi: 10.1172/jci46028
103. Zhang X, Li J, Luo S, Wang M, Huang Q, Deng Z, et al. IgE contributes to atherosclerosis and obesity by affecting macrophage polarization, macrophage protein network, and foam cell formation. Arterioscler Thromb Vasc Biol. (2020) 40:597–610. doi: 10.1161/ATVBAHA.119.313744
104. Wezel A, Lagraauw MH, van der Velden D, de Jager S, Quax P, Kuiper J, et al. Mast cells mediate neutrophil recruitment during atherosclerotic plaque progression. Atherosclerosis. (2015) 241:289–96. doi: 10.1016/j.atherosclerosis.2015.05.028
105. Lippi G, Cervellin G, Sanchis-Gomar F. Immunoglobulin E (IgE) and ischemic heart disease. Which came first, the chicken or the egg? Ann Med. (2014) 46:456–63. doi: 10.3109/07853890.2014.927714
106. Kounis NG, Hahalis G. Serum IgE levels in coronary artery disease. Atherosclerosis. (2016) 251:498–500. doi: 10.1016/j.atherosclerosis.2016.05.045
107. Wilson JM, Nguyen AT, Schuyler AJ, Commins SP, Taylor AM, Platts-Mills TAE, et al. IgE to the Mammalian oligosaccharide galactose-alpha-1,3-galactose is associated with increased atheroma volume and plaques with unstable characteristics-brief report. Arterioscler Thromb Vasc Biol. (2018) 38:1665–9. doi: 10.1161/ATVBAHA.118.311222
108. Nimmerjahn F, Ravetch JV. Divergent immunoglobulin G subclass activity through selective fc receptor binding. Science. (2005) 310:1510–2. doi: 10.1126/science.1118948
109. Tay C, Liu YH, Kanellakis P, Kallies A, Li Y, Cao A, et al. Follicular B cells promote atherosclerosis via t cell-mediated differentiation into plasma cells and secreting pathogenic immunoglobulin G. Arterioscler Thromb Vasc Biol. (2018) 38:e71–84. doi: 10.1161/ATVBAHA.117.310678
110. Centa M, Jin H, Hofste L, Hellberg S, Busch A, Baumgartner R, et al. Germinal center-derived antibodies promote atherosclerosis plaque size and stability. Circulation. (2019) 139:2466–82. doi: 10.1161/CIRCULATIONAHA.118.038534
111. Bagchi-Chakraborty J, Francis A, Bray T, Masters L, Tsiantoulas D, Nus M, et al. B cell fcgamma receptor IIb modulates atherosclerosis in male and female mice by controlling adaptive germinal center and innate B-1-cell responses. Arterioscler Thromb Vasc Biol. (2019) 39:1379–89. doi: 10.1161/ATVBAHA.118.312272
112. Sage AP, Nus M, Bagchi Chakraborty J, Tsiantoulas D, Newland SA, Finigan AJ, et al. X-box binding protein-1 dependent plasma cell responses limit the development of atherosclerosis. Circ Res. (2017) 121:270–81. doi: 10.1161/CIRCRESAHA.117.310884
113. Shen P, Fillatreau S. Antibody-independent functions of B cells: a focus on cytokines. Nat Rev Immunol. (2015) 15:441–51. doi: 10.1038/nri3857
114. Strom AC, Cross AJ, Cole JE, Blair PA, Leib C, Goddard ME, et al. B regulatory cells are increased in hypercholesterolaemic mice and protect from lesion development via IL-10. Thromb Haemostasis. (2015) 114:835–47. doi: 10.1160/TH14-12-1084
115. Sage AP, Nus M, Baker LL, Finigan AJ, Masters LM, Mallat Z. Regulatory B cell-specific interleukin-10 is dispensable for atherosclerosis development in mice. Arterioscler Thromb Vasc Biol. (2015) 35:1770–3. doi: 10.1161/ATVBAHA.115.305568
116. Meiler S, Smeets E, Winkels H, Shami A, Pascutti MF, Nolte MA, et al. Constitutive GITR activation reduces atherosclerosis by promoting regulatory CD4+ T-cell responses-brief report. Arterioscler Thromb Vasc Biol. (2016) 36:1748–52. doi: 10.1161/ATVBAHA.116.307354
117. Tay C, Liu YH, Hosseini H, Kanellakis P, Cao A, Peter K, et al. B-cell-specific depletion of tumour necrosis factor alpha inhibits atherosclerosis development and plaque vulnerability to rupture by reducing cell death and inflammation. Cardiovasc Res. (2016) 111:385–97. doi: 10.1093/cvr/cvw186
118. Tsiantoulas D, Sage AP, Goderle L, Ozsvar-Kozma M, Murphy D, Porsch F, et al. B cell-activating factor neutralization aggravates atherosclerosis. Circulation. (2018) 138:2263–73. doi: 10.1161/CIRCULATIONAHA.117.032790
119. Tsiantoulas D, Eslami M, Obermayer G, Clement M, Smeets D, Mayer FJ, et al. APRIL limits atherosclerosis by binding to heparan sulfate proteoglycans. Nature. (2021) 597:92–6. doi: 10.1038/s41586-021-03818-3
120. Gjurich BN, Taghavie-Moghadam PL, Ley K, Galkina EV. L-selectin deficiency decreases aortic B1a and Breg subsets and promotes atherosclerosis. Thromb Haemost. (2014) 112:803–11. doi: 10.1160/TH13-10-0865
Keywords: B cells, atherosclerosis, antibodies, lipids, inflammation, cardiovascular disease
Citation: Smeets D, Gisterå A, Malin SG and Tsiantoulas D (2022) The Spectrum of B Cell Functions in Atherosclerotic Cardiovascular Disease. Front. Cardiovasc. Med. 9:864602. doi: 10.3389/fcvm.2022.864602
Received: 28 January 2022; Accepted: 02 March 2022;
Published: 15 April 2022.
Edited by:
Norbert Gerdes, Heinrich Heine University Düsseldorf, GermanyReviewed by:
Jürgen Bernhagen, Ludwig Maximilian University of Munich, GermanyKosuke Kataoka, Tokushima University, Japan
Copyright © 2022 Smeets, Gisterå, Malin and Tsiantoulas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dimitrios Tsiantoulas, ZGltaXRyaXMudHNpYW50b3VsYXNAbWVkdW5pd2llbi5hYy5hdA==