
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med. , 23 May 2022
Sec. Cardiovascular Genetics and Systems Medicine
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.857952
 Anzhu Wang1,2
Anzhu Wang1,2 Zhendong Li3
Zhendong Li3 Sun Zhuo3
Sun Zhuo3 Feng Gao1,2
Feng Gao1,2 Hongwei Zhang1,2
Hongwei Zhang1,2 Zhibo Zhang1,4Gaocan Ren1,2
Zhibo Zhang1,4Gaocan Ren1,2 Xiaochang Ma1,5*
Xiaochang Ma1,5*Purpose: Sodium-glucose cotransporter 2 (SGLT2) inhibitors have cardiorenal protective effects regardless of whether they are combined with type 2 diabetes mellitus, but their specific pharmacological mechanisms remain undetermined.
Materials and Methods: We used databases to obtain information on the disease targets of “Chronic Kidney Disease,” “Heart Failure,” and “Type 2 Diabetes Mellitus” as well as the targets of SGLT2 inhibitors. After screening the common targets, we used Cytoscape 3.8.2 software to construct SGLT2 inhibitors' regulatory network and protein-protein interaction network. The clusterProfiler R package was used to perform gene ontology functional analysis and Kyoto encyclopedia of genes and genomes pathway enrichment analyses on the target genes. Molecular docking was utilized to verify the relationship between SGLT2 inhibitors and core targets.
Results: Seven different SGLT2 inhibitors were found to have cardiorenal protective effects on 146 targets. The main mechanisms of action may be associated with lipid and atherosclerosis, MAPK signaling pathway, Rap1 signaling pathway, endocrine resistance, fluid shear stress, atherosclerosis, TNF signaling pathway, relaxin signaling pathway, neurotrophin signaling pathway, and AGEs-RAGE signaling pathway in diabetic complications were related. Docking of SGLT2 inhibitors with key targets such as GAPDH, MAPK3, MMP9, MAPK1, and NRAS revealed that these compounds bind to proteins spontaneously.
Conclusion: Based on pharmacological networks, this study elucidates the potential mechanisms of action of SGLT2 inhibitors from a systemic and holistic perspective. These key targets and pathways will provide new ideas for future studies on the pharmacological mechanisms of cardiorenal protection by SGLT2 inhibitors.
Diabetes mellitus (DM) is one of the world's most common public health issues, with the 10th edition of the International Diabetes Federation estimating a prevalence of 537 million people in 2021, and ~783.2 million people are expected to have DM by 2045 (1). Type 2 Diabetes Mellitus (T2DM) is the most common type of diabetes, accounting for more than 90% of all diabetic patients and being one of the primary causes of chronic kidney disease (CKD) and cardiovascular disease (CVD) (2). For instance, in clinical trials for patients with T2DM, the prevalence of heart failure (HF) at baseline ranged from ~10 to 30% (3–5). In clinical trials of patients with chronic HF, the prevalence of T2DM was nearly 30% regardless of HF phenotype (6–8). T2DM is found in ~40–45% of patients hospitalized with HF in North America and Europe (9, 10).10% of deaths in patients with T2DM are attributed to renal failure (11). According to the China Kidney Disease Network 2015 Annual Data Report, the prevalence of CKD is 10.8%, and diabetic nephropathy accounting for 26.9% of CKD hospitalizations (12). Diabetic macrovascular and microangiopathy result in cardiovascular and renal endpoint events, which are the leading cause of death in diabetic patients. In contrast, the improved mortality outcomes obtained with specific classes of hypoglycemic agents are largely independent of their glycemic effects (13).
Sodium-glucose cotransporter 2 (SGLT2) is a key glucose transporter protein in the kidney that accounts for nearly 90% of glucose reabsorption from primary urine, and SGLT2 inhibitors are novel oral drugs for the treatment of T2DM. The United States Food and Drug Administration and the European Union have currently approved four SGLT2 inhibitors (Canagliflozin, Empagliflozin, Dapagliflozin, and Ertugliflozin) (14). Ipragliflozin, Tofogliflozin, and Luseogliflozin are some of the other medications in the family that have been authorized in Japan (15–17). Recent evidence suggests that SGLT2 inhibitors can help individuals with or without T2DM improve their renal and cardiovascular outcomes (Table 1).

Table 1. Large clinical trials of SGLT2 inhibitors.
Network pharmacology is a discipline based on systems biology and multidirectional pharmacology that uses specific nodes and adopts biomolecular network analysis methods to conduct drug molecular design and target analysis (29). When compared to traditional experimental pharmacology approaches, network pharmacology is based on a complete system and can more effectively investigate the target and pathway relationships between medications and disorders more effectively. Therefore, we utilized the network pharmacology analysis system to determine the targets of SGLT2 inhibitors on the cardiorenal protection of T2DM and identify the biological pathways involved to provide directions for in-depth investigation of SGLT2 inhibitors.
The structural formulae of Ertugliflozin, Sotagliflozin, Canagliflozin, Dapagliflozin, Empagliflozin, Tofogliflozin, Luseogliflozin, and Ipragliflozin were obtained from PubChem (https://pubchem.ncbi.nlm.nih.gov/) (30). The structural formulae were added to the SwissTargetPrediction database (https://www.drugbank.com/) with probability>0 screening (31). To predict the targets, the DrugBank (https://www.drugbank.com/) was also utilized to forecast the targets (32). The intersection of the results was used.
In order to obtain potential targets, the keywords “Chronic Kidney Disease,” “Heart Failure” and “Type 2 Diabetes Mellitus” were retrieved from Genecards (http://www.genecards.org/), Disgenet (https://www.disgenet.org/), Online Mendelian Inheritance in Man (OMIM, https://omim.org/), Pharmacogenomics Knowledgebase (PharmGKB, https://www.pharmgkb.org/) and Therapeutic Target Database (TTD, http://db.idrblab.net/ttd/).
Venn diagrams were created using R packages (“VennDiagram”) from the intersection of medication targets acquired in item 2.1 and illness targets obtained in item 2.2. The regulatory network was constructed using the Cytoscape3.8.2 software, with SGLT2 inhibitors and intersection targets serving as nodes.
The common interaction targets were imported into the Search Tool for the Retrieval of Interacting Genes database (STRING, Version 11.5, https://www.string-db.org/), screened with an interaction score ≥0.4, unlinked nodes in the network were hidden, the remaining parameters were left unchanged, and the obtained results were exported in text form into the Cytoscape 3.8.2 software for visualization (33). The Cytohubba and Molecular Complex Detection (MCODE) plug-ins in Cytoscape 3.8.2 were used to filter the modules of the PPI network with high significance. The cytohubba plug-in filtering criterion was to compute the node scores and rank the top 10 significant genes based on the maximal clique centrality (MCC) (34); the MCODE plug-in filtering criterion was degree cutoff = 2, node score cutoff = 0.2, K-score = 2, Max depth = 100, with P < 0.05 indicating a statistically significant difference (35). The previously mentioned two intersection targets were chosen after screening the targets separately.
Gene ontology (GO) categorizes genes into three groups: biological process (BP), cellular component (CC), and molecular function (MF). The Kyoto encyclopedia of genes and genomes (KEGG) was used to investigate the signaling pathways involved in genes. GO and KEGG enrichment analysis visualization is completed using R packages “org.hs.eg. db,” “DOSE,” “clusterProfiler,” “enrichPlot,” “Colorspace,” “stringi” and “GGploT2,” and output the enriched bubble plots in accordance the number and significance of enrichment (p-value cutoff <0.05, q-value cutoff <0.05). The obtained KEGG pathway was brought into Cytoscape 3.8.2, and the “pathway-gene” network was built based on the degree value.
Cytohubba and MCODE screened intersecting targets, and the first three core targets in the “pathway-gene” network were molecularly docked to SGLT2 inhibitors. The protein 3D format of the core targets was downloaded from the Protein Data Bank (PDB, https://www.rcsb.org/) (36), and operations such as dehydration, hydrogenation, and ligand extraction were carried out using Pymol 2.5 software (37). The Autodock Vina 1.1.2 program was used for molecular docking (38).
SwissTargetPrediction was utilized to obtain 71 Ertugliflozin targets, 103 Sotagliflozin targets, 53 Canagliflozin targets, 60 Dapagliflozin targets, 17 Empagliflozin targets, 57 Tofogliflozin targets, and 59 Ipragliflozin targets. Eight Canagliflozin targets, 11 Dapagliflozin targets, and 10 Empagliflozin targets were found in Drugbank. After de-duplicating and combining, 164 SGLT2 inhibitor targets were found.
After de-duplication and merging, a total of 12,132 HF-related targets, 12,091 CKD-related targets, and 9,910 T2DM-related targets were obtained by searching in Genecards, DisGeNet, OMIM, PharmGKB, and TTD databases. A Venn diagram was used to identify the 146 targets of SGLT2 inhibitors that intersected with disease (Figure 1A). The 146 targets were loaded into Cytoscape 3.8.2 to generate a drugs-targets regulatory network with 153 nodes and 409 edges, indicating the interaction between the SGLT2 inhibitors and the putative targets (Figure 1B).
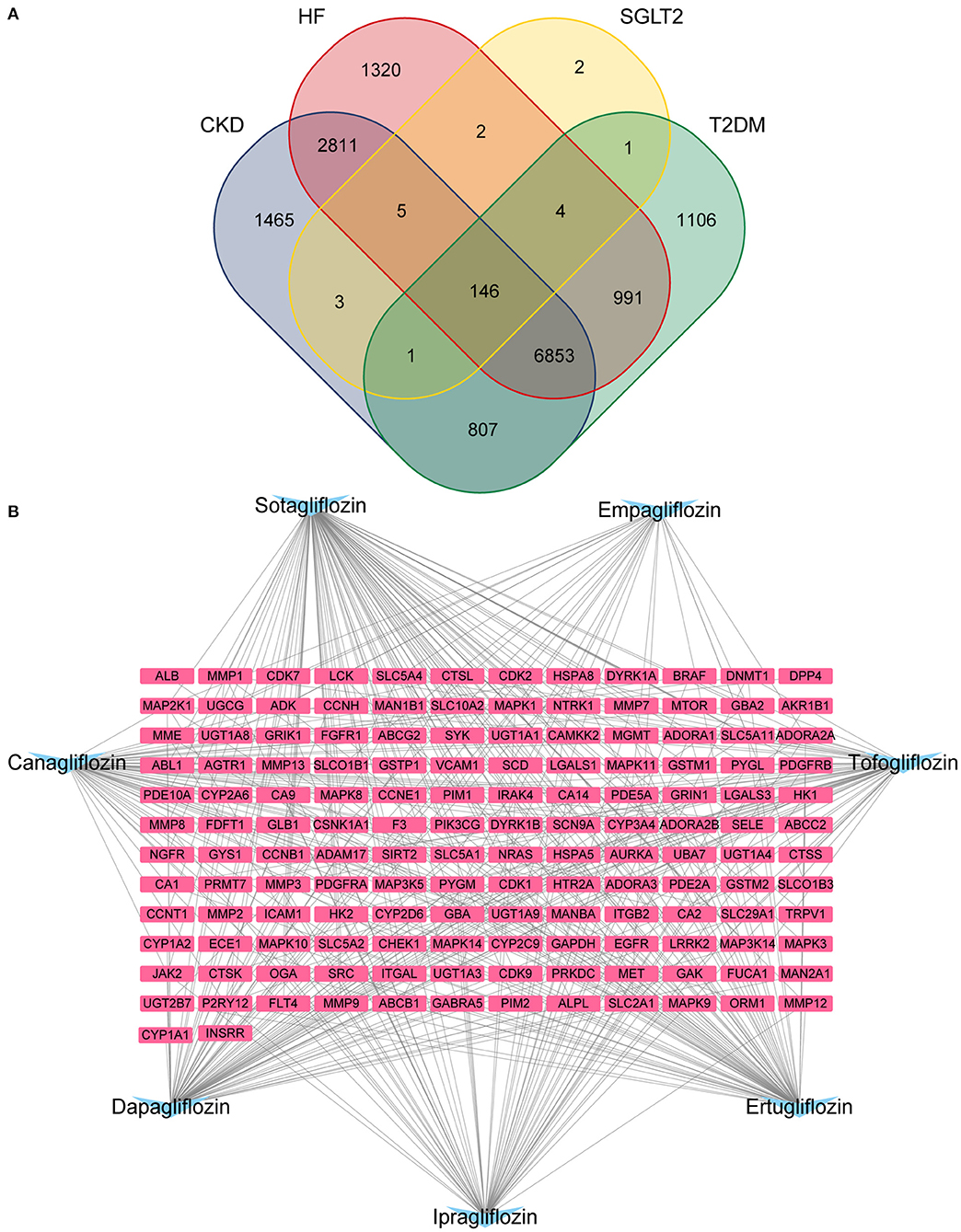
Figure 1. Potential SGLT2 inhibitors-related targets. (A) Venn diagram showing that 146 targets were common to CKD, HF, SGLT2, and T2DM; (B) Interaction network to indicate drugs-targets composited of SGLT2 inhibitors (blue) and 146 targets (pink).
The PPI network had 1,049 pairs of interactions, and the top 10 key targets in the PPI network obtained by the Cytohubba plug-in were: SRC, EGFR, MAPK3, GAPDH, ALB, MMP9, MMP2, ICAM1, VCAM1, and MMP1 (Figure 2A); the detailed results of the MCODE plug-in clustering analysis are depicted in Table 2, and Figures 2B–G; the key targets of their intersection are GAPDH, MAPK3, and MMP9.
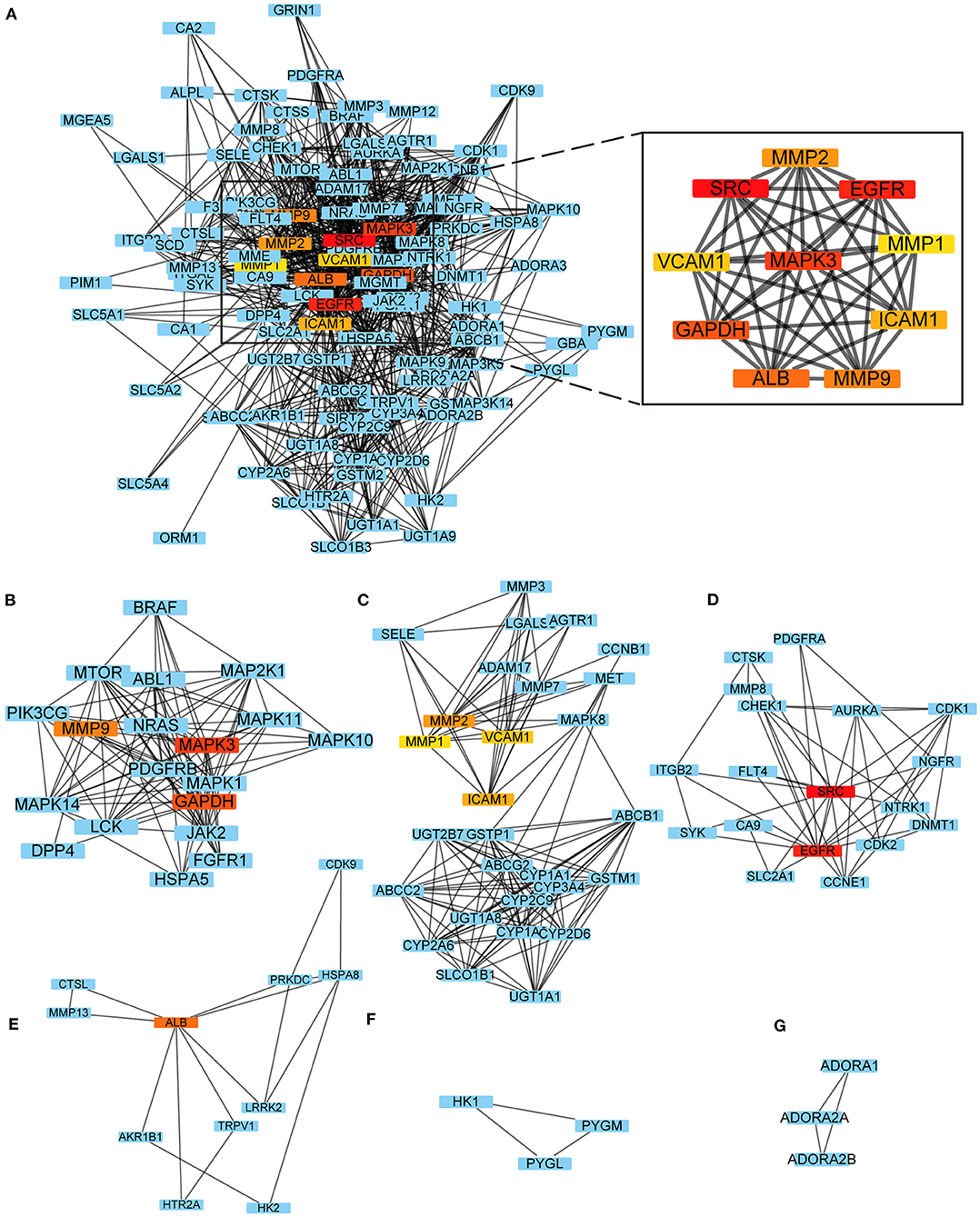
Figure 2. PPI network. (A) Top 10 key targets in the PPI network obtained by the Cytohubba; (B–G) The results of the MCODE plug-in clustering analysis.

Table 2. Cluster details based on MCODE plug-in.
GO functional analysis was performed on 146 intersecting targets, and the results were screened at P < 0.05. The enrichment results revealed a total of 1,089 BP, which mainly involved regulating protein serine/threonine kinase activity, peptidyl-tyrosine phosphorylation, and peptidyl-tyrosine modification. The overall number of CC was 66, with most of them relating to membrane raft and membrane microdomains. The total number of MF included 145, with protein serine/threonine kinase activity and protein tyrosine kinase activity. Figure 3 shows the top 10 items in the BP, CC, and MF categories.
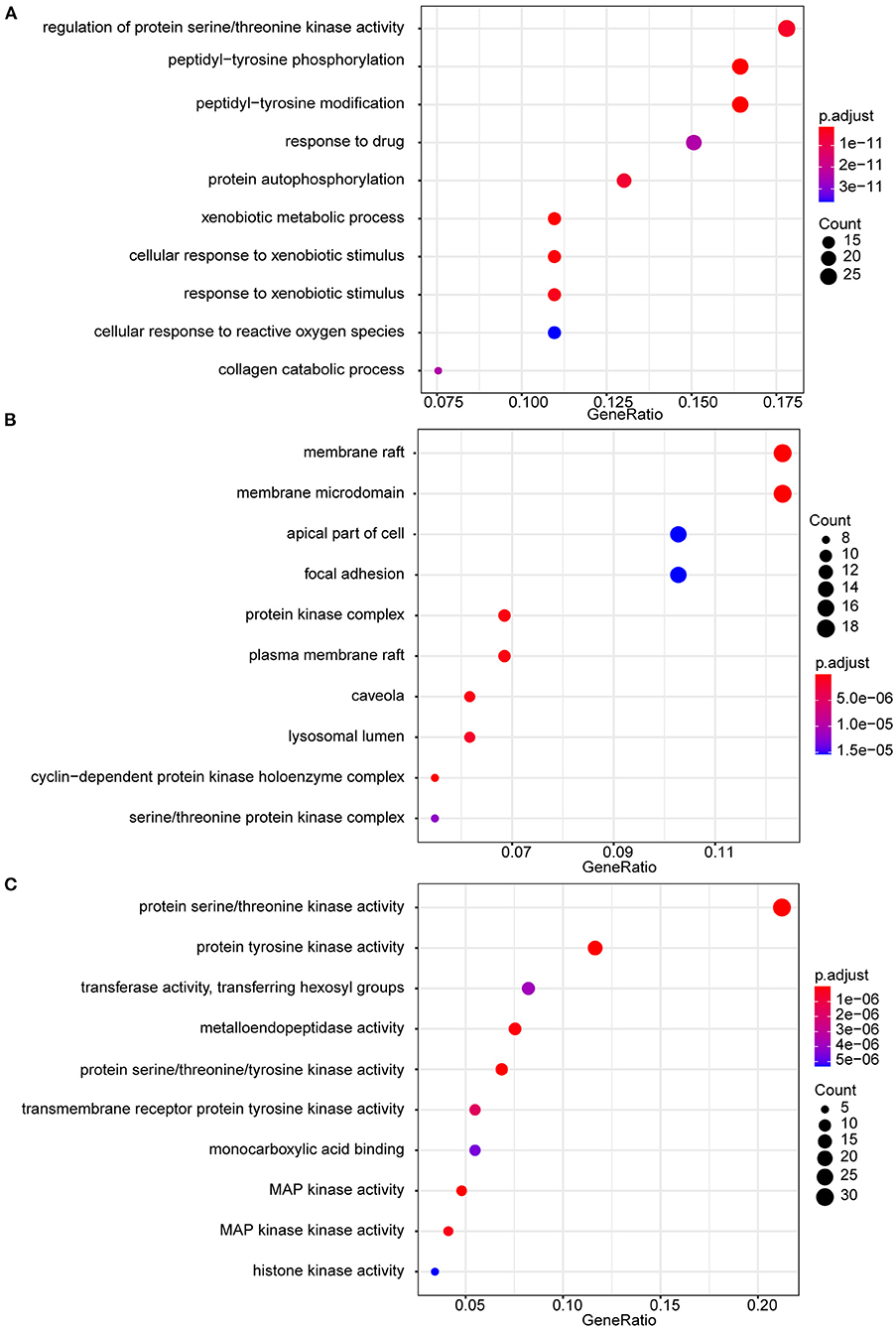
Figure 3. GO enrichment analysis. (A) GO biological processes of 146 core genes; (B) GO cellular component of 146 core genes; (C) GO molecular function f 146 core genes.
The cardiorenal protective effect of SGLT2 inhibitors may be related to 147 KEGG signaling pathways in diabetic complications, including lipid and atherosclerosis, MAPK signaling pathway, Rap1 signaling pathway, endocrine resistance, fluid shear stress, and atherosclerosis, TNF signaling pathway, relaxin signaling pathway, neurotrophin signaling pathway, and AGEs-RAGE signaling pathway (Figure 4A). The top 20 enriched pathways were imported into Cytoscape 3.8.2 to build a pathways-targets network, and the results revealed that the top 10 targets associated with the enriched pathway are MAPK1, MAPK3, NRAS, MAP2K1, MAPK11, MAPK14, MAPK10, MAPK8, and MAPK9 (Figure 4B).
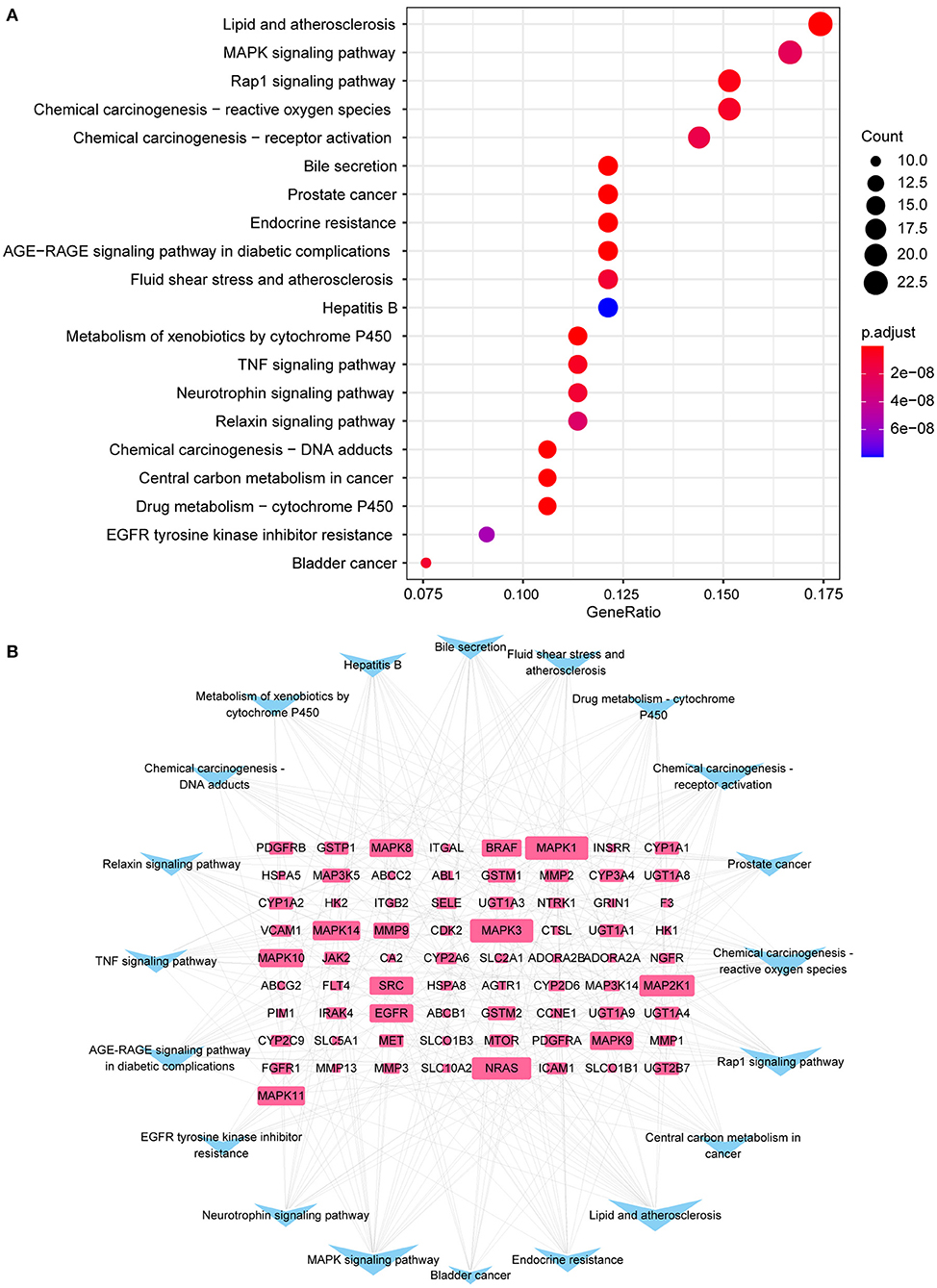
Figure 4. KEGG enrichment analysis. (A) The top 20 enriched pathways; (B) Interaction network of 73 targets (blue) and top 20 pathways (pink) to indicate pathways-targets network.
The intersection targets of PPI network and pathways-targets network GAPDH, MAPK3, MMP9, MAPK1, and NRAS were docked with SGLT2 inhibitors. Binding energy <0 indicates that the ligand and receptor are spontaneous bindings, and the lower the affinity value in the docking result, the more stable the interaction between the target and the active ingredient. Molecular docking revealed that the five important targets mentioned above have good binding activity to SGLT2 inhibitors (Table 3). The top 10 docking results were selected for visual analysis (Figure 5). The dashed lines in the graph are hydrogen bonds and the values are bond lengths. The names represent residues in the binding sites.
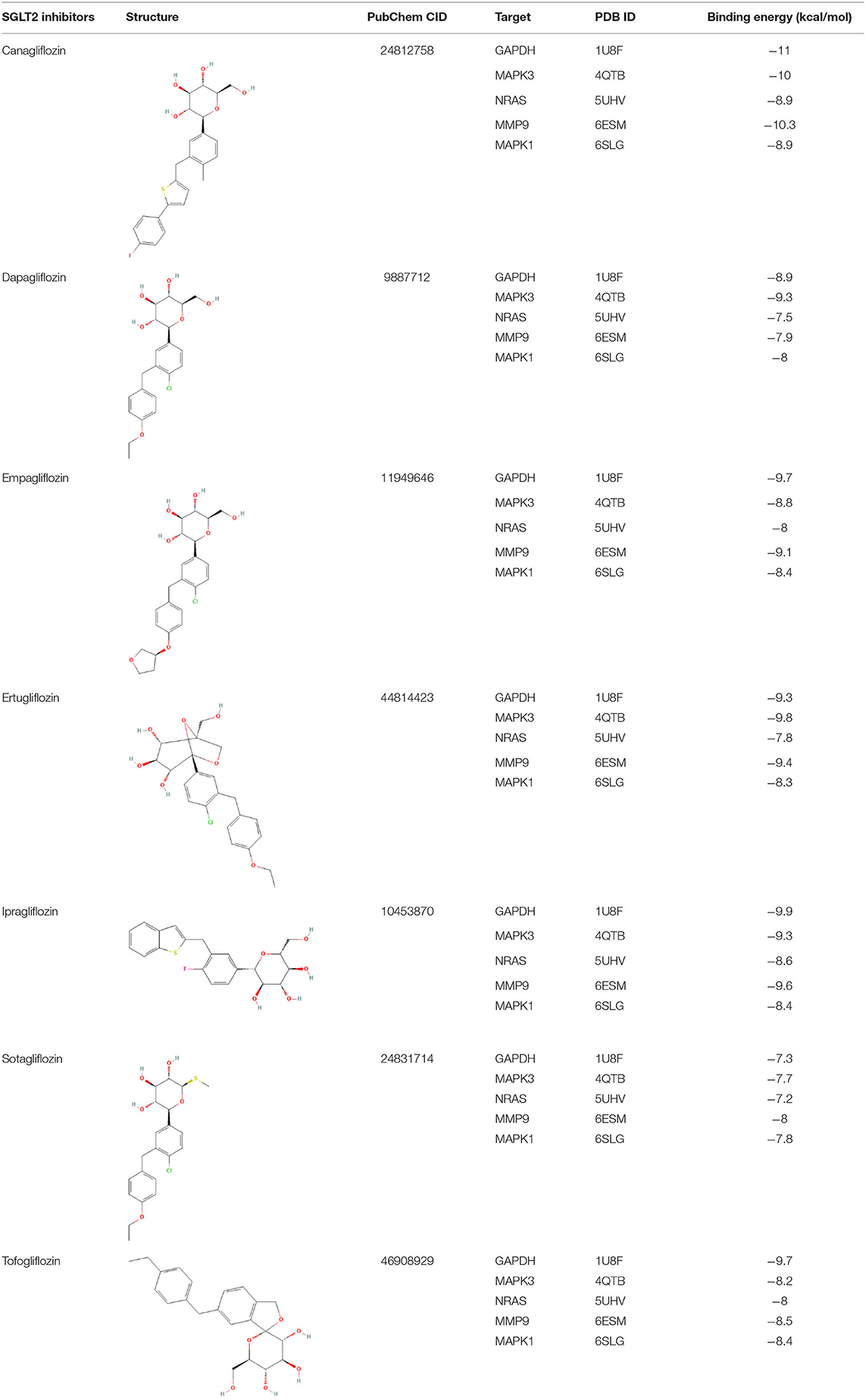
Table 3. Molecular docking results of SGLT2 inhibitors.
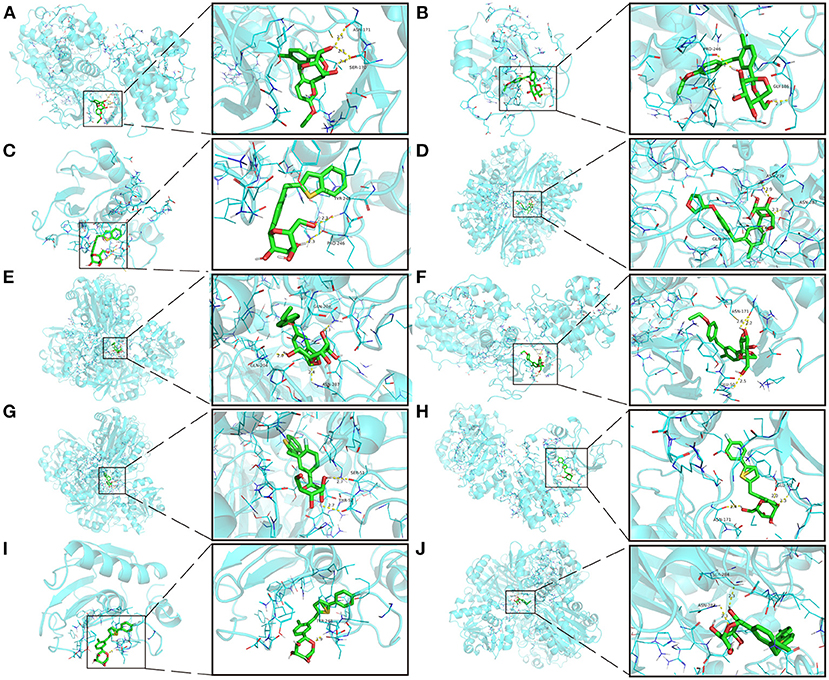
Figure 5. The top 10 docking results. (A) Dapagliflozin-MAPK3; (B) Ertugliflozin-MMP9; (C) Ipragliflozin-MMP9; (D) Empagliflozin-GAPDH; (E) Tofogliflozin-GAPDH; (F) Ertugliflozin-MAPK3; (G) Ipragliflozin-GAPDH; (H) Canagliflozin-MAPK3; (I) Canagliflozin-MMP9; (J) Canagliflozin-GAPDH. The dashed lines in the graph are hydrogen bonds and the values are bond lengths. The names represent residues in the binding sites.
The main complications of T2DM are HF and CKD, and the three can interact. Patients with T2DM develop a variety of cardiac abnormalities, a phenomenon known as “diabetic cardiomyopathy” (39). Furthermore, the accumulation of AGEs due to hyperglycaemia can lead to microvascular remodeling and cardiac fibrosis (40). The primary mechanism for the progression of HF stems from the stimulation of the renin-angiotensin-aldosterone system (RAAS) (41), and excessive activation of the RAAS can interfere with insulin production, secretion, and metabolic pathways, leading to reduced insulin sensitivity and disturbed glucose metabolism (42). These metabolic changes may not only impair myocardial energetics and reduce the efficiency of mechanical work but also promote systemic insulin resistance, creating a vicious cycle in which HF causes metabolic modifications and thus inducing HF (43). The multiple metabolic and haemodynamic alterations caused by T2DM promote structural changes in the kidney, primarily by affecting microcirculation. These changes further activate harmful pathways (e.g., inflammation), leading to glomerular fibrosis and tubular atrophy and eventually to end-stage renal disease (44). Clinically and pathophysiologically, HF and CKD are significantly correlated, and this association is especially pronounced in patients with coexisting T2DM (45). Haemodynamic changes owing to decreased cardiac output and/or altered venous return, sympathetic activation, and/or stimulation of the RAAS controlling the (neuro)hormonal axis are all pathophysiological processes that contribute to the genesis and progression of cardio-renal and renal-heart interactions. In addition to other factors that accelerate the progression of HF and CKD (local and systemic inflammation, metabolic changes, anemia, etc.) (46). With the progression of clinical studies with SGLT2 inhibitors, the results suggest a cardiorenal protective effect in patients with or without DM. Although several hypotheses have been proposed, the mechanisms are unknown; hence, we used a network pharmacology method to investigate the mechanisms of cardiorenal protection.
In the regulatory network, there are 7 SGLT2 inhibitors, namely Ertugliflozin, Sotagliflozin, Canagliflozin, Dapagliflozin, Empagliflozin, Tofogliflozin, and Ipragliflozin. Sotagliflozin is an SGLT2 inhibitor with minor SGLT1 inhibitory activity (47). Several recent studies have revealed potential mechanisms of SGLT2 inhibitors. DM induces reactive oxygen species (ROS), apoptosis, and endoplasmic reticulum (ER) stress, which could be reversed with Dapagliflozin therapy (48). The renoprotective effects of dapagliflozin in prediabetes by alleviating obesity-induced renal inflammation, fibrosis, ER stress, apoptosis, and lipid accumulation (49). Dapagliflozin also slowed the progression of diabetic kidney disease by decreasing cellular senescence and oxidative stress via ketone-induced nuclear erythroid-related factor 2 (NRF2) activation (50). The vasoprotective effect of dapagliflozin may be due to improved transport of bone marrow-derived haematopoietic cells to the site of vascular injury (51). Empagliflozin-associated renoprotection in injured proximal tubules of mice with apolipoprotein E (APOE) deficiency given a high-fat diet is mediated by increased ketone body, which corrects mechanistic target of rapamycin complex 1 (mTORC1) hyperactivation detected in non-proteinuric and proteinuric diabetic kidney disease (52). The etiology of HF is linked to the activation of the late component of the cardiac sodium channel current (late-INa), which is a molecular target for Empagliflozin (53). Empagliflozin's prevention of HF progression is linked to reduced proximal tubule sodium-hydrogen exchanger 3 (NHE3) activity, euvolemia restoration, and renal mass maintenance (54). A study on human cardiac microvascular endothelial cells in a co-culture system with adult rat ventricular cardiomyocytes to measure diastolic and systolic function revealed that serum from CKD patients impaired the beneficial effects of cardiac microvascular endothelial cells on cardiomyocyte function and that Empagliflozin restored this effect by reducing mitochondrial oxidative damage (55). Reversion-inducing-cysteine-rich protein with Kazal motifs (RECK), a membrane-anchored matrix metalloproteinase (MMP) regulator, is linked to fibrosis. Empagliflozin reduces systemic and renal arterial stiffness and reverses RECK expression in T2DM female mice, alleviating kidney damage (56).
The key targets obtained by the PPI network are 3-phosphoglyceraldehyde dehydrogenase (GAPDH), mitogen-activated protein kinase 3 (MAPK3), MAPK1, NRAS and MMP9 combined with KEGG enrichment results. GAPDH is a simple “housekeeping” protein that plays a role in a variety of physiological and pathological processes and has a variety of non-glycolytic functions in addition to its part in glycolysis (57). These multifunctional properties of GAPDH may depend on its posttranslational modification, oligomerization and significant subcellular localizations (including cytoplasm, nucleus, mitochondria and vesicles) (58). The significant expression of GAPDH in the cytoplasm may enable it to operate as an intracellular sensor capable of directly transmitting signals to numerous organelles, such as the mitochondria. GAPDH translocation to mitochondria has been proven to cause the inner transmembrane potential to be lost, the matrix to swell, and the inner mitochondrial membrane to permeabilize (59). These changes lead to apoptosis, which is closely associated with CKD and HF (60, 61). Hyperglycemia, on the other hand, causes an excess of acetoacetyl coenzyme A, which feeds into the tricarboxylic acid cycle, resulting in an excess of nicotinamide adenine dinucleotide (NADH), putting the mitochondrial electron transport chain under a lot of stress (62). Thus, mitochondrial oxidation of overproduced NADH will eventually result in the creation of more superoxide and hence more ROS, which can target and inactivate GAPDH (63). This would cause a buildup of glycolytic metabolites upstream of glyceraldehyde 3-phosphate as well as the activation of alternate glucose disposal pathways, all of which are associated with ROS generation and hence enhance oxidative stress (9). Dapagliflozin has been proven in studies to alleviate glucotoxicity by lowering excessive glucose influx into renal tubular epithelial cells under high glucose circumstances through activating GAPDH (64). Furthermore, dapagliflozin alleviates pressure overload-induced myocardial remodeling in mice via inhibiting ER stress-mediated apoptosis (65). Empagliflozin improved renal ischemia/reperfusion injury in non-diabetic rats by promoting mitochondrial biogenesis and inhibiting oxidative stress and apoptosis (66). In the murine model of left ventricular pressure overload, empagliflozin aids the suppression of endothelial apoptosis, the maintenance of capillarization, and the improvement of cardiac systolic dysfunction (67). MAPK3/MAPK1, also known as extracellular signal-regulated kinase 1/2 (ERK1/2), is a member of the MAPK protein family (68). MAPKs are serine/threonine protein kinases that phosphorylate a varied array of downstream targets in distinct compartments in order to carry out highly specific physiological responses in response to activation (69). MAPK signaling has four major branching routes, including ERK1/2, which is often mitogen sensitive but also the most widely responsive to a large range of stimuli; ERK5, which is functionally similar to ERK1/2; and the c-Jun N-terminal kinases (JNK) as well as p38 families, which are activated by environmental stress changes and inflammatory factors (70). ERK1/2 is the most critical effector in cardiomyocytes, particularly cardiac hypertrophy, and has also been identified as a key component of the cardioprotective reperfusion damage salvage kinase pathway (71, 72). In adult organs, ERK1/2 mediates transcription and post-transcriptional regulation of numerous nephron channels and transporters, resulting in acid-base and electrolyte balance stabilization (73). ERK1/2 s also closely related to the pathogenesis of hypoxia-induced renal fibrosis (74). Pretreatment with empagliflozin protects the heart from I/R injury-induced severe fatal ventricular arrhythmia, which might be the result of glucose-independent stimulation of the ERK1/2 (75). Canagliflozin, not empagliflozin or dapagliflozin, alleviates inflammation and lowers Hexokinase II expression by inhibiting ERK1/2 phosphorylation in lipopolysaccharide-stimulated human coronary artery endothelial cells (76). Neuroblastoma RAS Viral Gene Homolog (NRAS) is a RAS family member that regulates the MAPK signaling pathway. Because it has been examined extensively in tumor illnesses, it is rarely discussed (77, 78). Both CKD and HF are associated with changes in the extracellular matrix (ECM), and MMPs are primarily responsible for ECM degradation (79, 80). MMP9, also known as gelatinase B, can degrade elastin and collagen and is involved in inflammatory responses, and is one of the most extensively studied MMPs in cardiovascular and renal research (81, 82). Low-dose empagliflozin can improve systolic heart function after MI in rats by regulating MMP9 (83). Luseogliflozin regulates the expression of multiple mRNA, including MMP9, and inhibits the progression of atherosclerosis in diabetic APOE-deficient mice (84).
The results of GO functional enrichment are primarily related to protein serine/threonine kinase activity. Protein kinases and protein phosphatases maintain the balance of protein phosphorylation regulation as two major classes of proteins, phosphorylation, and dephosphorylation, respectively. Phosphorylation of proteins occurs most commonly at serines and threonines, where protein kinases transfer phosphate groups to these amino acid residues, usually using adenosine triphosphate as a substrate, and protein phosphatases hydrolyze phosphate from the amino acid residues of the protein. They collaborate to regulate protein function, stability, and signaling, as well as carrying out physiopathological tasks (85). Protein kinase C (PKC) isozymes are a group of serine/threonine kinases whose activity and levels are dispensed in several pathological heart conditions, including atherosclerosis (AS), myocardial infarction (MI), acute ischaemic, cardiac hypertrophy, cardiac arrhythmia, HF, and cardiac fibrosis (86, 87). The AKT (also known as protein kinase B, PKB) family also belongs to serine/threonine protein kinases, and its activation is critical in a variety of signaling pathways linked to kidney injury (88). Some serine/threonine kinases, such as Rho-associated, coiled-coil containing kinases (ROCK), have been implicated in cardiovascular–renal remodeling via oxidative stress and oxidative stress-related signaling via nicotinamide adenine dinucleotide phosphate (NADPH) oxidase induction (89).
According to the KEGG enrichment results, SGLT2 inhibitors' cardiorenal protection may be associated with Lipid and AS, MAPK signaling pathway, Ras-related Protein 1 (Rap1) signaling pathway, Endocrine resistance, Fluid shear stress, and atherosclerosis, tumor necrosis factor (TNF) signaling pathway, Relaxin signaling pathway, Neurotrophin signaling pathway, Advanced glycation end-products (AGEs)- receptor for advanced glycation end-products (RAGE) signaling pathway in diabetic complications. AS is a slow-progressing inflammatory illness that is a primary underlying pathology for CVD, including MI, HF, stroke, and peripheral artery disease, which are the top causes of mortality in DM and CKD patients (90). DM is a risk factor for AS, and CKD can also accelerate AS through increased inflammation, lipid metabolism disorders, and other mechanisms (91). According to the findings of a systematic review and meta-analysis, glucose-lowering medications, such as SGLT2 inhibitors, can greatly reduce the risk events of AS (92). A wide range of multinational observational studies in individuals with T2DM and cardiovascular risk has also demonstrated that the beneficial effects of SGLT2 inhibitors are associated with MI and stroke, with AS and its clinical consequences being the most closely linked (93, 94). The results of network pharmacology suggest a mechanism of SGLT2 associated with AS, which is supported by experimental evidence. Canagliflozin, for example, in the APOE knockout mice model, could slow the progression of AS by decreasing the expression of inflammatory molecules such as monocyte chemoattractant protein-1 (MCP-1) and vascular cell adhesion molecule-1 (VCAM-1). Additionally, it could increase the stability of atherosclerotic plaques by increasing the tissue inhibitor of metalloproteinase 1 (TIMP1)/MMP2 ratio (95). Empagliflozin has been shown to reduce lipid profiles and sympathetic activity in AS (96). ERK1/2 in the MAPK signaling pathway have been mentioned above, and ERK5 has a similar function with relatively fewer studies, but it also plays an important role. ERK5 is a protein that is predominantly located in the glomerular mesangium and is expressed in the kidney. It is involved in the contraction, proliferation, and ECM accumulation of disease-induced mesangial cells (97, 98). The use of lentivirus to overexpress full-length ERK5 in the kidneys of mice protected them from renal I/R injury (99). Targeted ERK5 deletion reduces hypertrophy and increases pressure overload-induced apoptosis in the heart (100). In female mice, ERK5 regulates body weight and systemic energy balance, most likely through its expression in hypothalamus neurons, making it a target for metabolic illnesses such as T2DM (101). The involvement that JNK plays in cardiac hypertrophy and I/R is less clear because JNK activation is most likely a dynamic signal transduction event that can be modified by the nature of the stimuli and that different JNK (JNK1-3) isoforms may play separate roles in the process (102–104). In addition, reduced myocardial connexin 43 expression and gap junction integrity, caused by the activation of the JNK signaling pathway, have been associated with conduction abnormalities and abrupt HF in mice (105). The JNK pathway can be triggered by a variety of stimuli associated with acute and chronic kidney injury, including pro-inflammatory cytokines, pro-fibrotic factors, risk-associated molecular pattern ligands, oxidative stress and nephrotoxins. The activated JNK pathway may either promote renal fibrosis through its pro-apoptotic and pro-inflammatory effects or directly enhance the fibrotic response (106). JNK also interacts with other pro-fibrotic pathways, including the transforming growth factor-β (TGF-β)/small mothers against decapentaplegic SMAD pathway (107). JNK is also involved in the regulation of insulin resistance and β-cell dysfunction (108). The quantity, length, mode, and timing of induction involving different isoforms (α, β, γ, and δ) and upstream/downstream pathways determine the specific role of p38 in myocardial ischemia damage and protection (109). Acute p38 signaling activation may serve as an adaptive response to extracellular stresses in the early phases of hypertrophy, but prolonged p38 signaling activation appears to have deleterious implications, including suboptimal cardiac remodeling and HF (110). The majority of evidence suggests that p38 activation has a deleterious influence on the onset of HF. The mechanisms at work include the formation of cardiac fibrosis, alterations in Ca2+ handling proteins, and gap junction regulation in cardiomyocytes (111). Activation of P38 in intrinsic renal cells (endothelial cells, podocytes and tubular cells) and infiltrating leukocytes is associated with renal dysfunction and histopathology, and plays an important pathogenic role in human glomerulonephritis, contributing to the development of CKD (112). Another interesting finding was the occurrence of renal dysfunction and fibrosis and the activation of P38 in both cerebral infarction and MI mouse models, suggesting that P38 plays a key role in the fibrotic response induced by signals from different injury modes (113, 114). In RAW 264.7 macrophages stimulated by lipopolysaccharide, empagliflozin had no effect on phosphorylation of p38 and ERK, but it did reduce phosphorylation of JNK (115). Empagliflozin reduces high glucose-induced RECK suppression, oxidative stress and epithelial-to-mesenchymal transition in cultured kidney proximal tubule cells and these beneficial effects are partially associated with p38 (116). Empagliflozin also attenuates the aging of cardiac stromal cells and improves heart function in a diabetic mouse model by targeting P38 (117). Canagliflozin protects endothelium function in APOE -deficient mice via inhibiting P38 activation (118). MAPK signaling plays a critical and complicated role in cardiorenal protection, which can only be briefly summarized here. The protective mechanism of SGLT2 seems to be more related to the ERK1/2 and P38 pathways. Rap1 is a small molecular weight GTPase that belongs to the Ras family. On the one hand, it maintains telomere function in the nucleus, and on the other hand, the Rap1 signaling pathway is primarily involved in cell adhesion, cell connection formation, and endothelial barrier protection (119). Rap1 is required to form a podocyte slit diaphragm, a specialized junction universally injured in proteinuric diseases and as an intracellular signaling hub (120). In rats with streptozotocin-induced diabetes, Rap1 ameliorates tubular damage in diabetic nephropathy by modulating primarily the mitochondrial-derived oxidative stress (121). In the cardiovascular system, Rap1 signaling promotes angiogenesis and maintenance of vascular stability by promoting vascular endothelial growth factor receptor 2 (VEGFR2) activation or acting downstream from vascular endothelial growth factor (VEGF), fibroblast growth factor 2 (FGF2), and sphingosine 1-phosphate (S1P) receptors in signaling pathways. Rap1 also regulates cell-cell junctions and heart contractility, but there is little evidence that Rap1 directly promotes hypertrophy (122). Rap1 has been linked to pathologies like metabolism, inflammation and oxidative stress, and regulation of telomeric length, and it may play an important role in diabetic cardiomyopathy (123). Rap1 can also mediate the MAPK pathway through ERK.
Tumor necrosis factor (TNF) is a tiny molecular protein secreted by monocyte-macrophages, including both TNF-α and TNF-β, that binds to two TNF receptors (TNFR) on the cell surface. Besides, it is a cytokine with inflammatory mediator effects (124). Either type of HF is associated with activation of inflammatory signaling (125), and TNF-α-mediated unfavorable remodeling and development of HF may entail impacts on cardiomyocytes, macrophages, and the ECM (126). TNF induces cardiomyocyte apoptosis by activating multiple cell death pathways (127), and RNA sequencing of HF cardiac fibroblasts indicated that TNF-signaling pathways were connected to highly modulated genes involved in ECM structure (128). There is growing evidence that TNF contributes significantly to the pathogenesis of CKD, promoting inflammation, apoptosis, and ECM accumulation, reducing glomerular blood flow, and destroying the glomerular permeability barrier as proteinuria progresses (129). Soluble TNFR-1, the circulating version of the membrane-bound receptor, is correlated with a long-term decline in kidney function for a decade in a multiethnic population (130). SGLT2 inhibitors have been shown to decrease TNF-α, TNF-β or TNFR expression in patients and experimental models (131–133). AGEs are a kind of chemical created by the non-enzymatic interaction between proteins and sugar residues known as glycation or the Maillard reaction (134). AGEs accumulate in the body with age, and DM accelerates this process. The accumulation of AGEs may contribute to the development of HF and CKD by causing protein modifications and triggering numerous inflammatory responses, oxidative stress, and enhancing fibrosis via receptors for advanced glycation end-products (135, 136). Dapagliflozin protects podocytes from advanced AGEs via the adenosine monophosphate-activated protein kinase (AMPK)/mammalian target of rapamycin (mTOR)-mediated autophagy pathway (137). Empagliflozin inhibits the AGEs-receptor axis, which contributes to its anti-inflammatory and antifibrotic actions in experimental diabetic nephropathy (138). Relaxin is a naturally occurring peptide hormone that produces nitric oxide, inhibits endothelin and angiotensin II, produces VEGF and MMPs, and acts as a pleiotropic vasodilator with several pleiotropic effects for HF treatment (139). Several studies have identified the relaxin signaling pathway as a potential therapeutic target for heart failure with preserved ejection fraction (HFpEF) (140, 141). The relaxin signaling pathway also can attenuate tubular epithelial cell apoptosis and prevent renal interstitial fibrosis (142, 143). The neurotrophins family of dimeric polypeptides includes nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), neurotrophin 3, and neurotrophin 4/5 (144). Even though neurotrophins are well-recognized for their effects on neuronal survival, they also exert cardiovascular effects under physiological and pathological conditions, including pro-angiogenic effects, promoting cardiomyocyte survival, and affecting cardiac sympathetic and parasympathetic nerve activity (145, 146). BDNF effectively repairs podocyte damage by microRNA-134 and microRNA-132 mediated increase in actin polymerization (147). Reduced renal function usually leads to significant disturbances in hormone levels, while HF is strongly associated with insulin resistance, and the potential impact of SGLT2 inhibitors on the heart and kidneys may lie in improving insulin resistance (148, 149).
Network pharmacology is based on a variety of databases and analysis software that can predict the most probable protein targets of SGLT2 inhibitors, thus obtaining the key links in their mechanism of action. However, the main limitation of web-based pharmacology studies is the lack of experimental validation. Pharmacological research will thus be critical in further clarifying the cardiorenal protective effects of SGLT2 inhibitors.
Clinical research is focused on cardiac and renal protection, regardless of whether DM is comorbid or not. Therefore, we have constructed a network between SGLT2 inhibitors and targets related to CKD, T2DM, and HF and analyzed the potential mechanisms of SGLT2 inhibitors' cardioprotective effects through network pharmacology, which contributes to deepening the understanding of SGLT2 and provide a theoretical for future studies.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/supplementary material.
AW: conceptualization, methodology, validation, formal analysis, investigation, data curation, writing—original draft, and visualization. ZL and SZ: conceptualization, methodology, and writing—review and editing. FG: investigation, formal analysis, and data curation. HZ: investigation and data curation. ZZ: formal analysis and visualization. GR: writing–review and editing and supervision. XM: conceptualization, writing–review and editing, supervision, and project administration. All authors contributed to the article and approved the submitted version.
This work was supported by the National Key Research and Development Program of China (No. 2018YFC1707410-02). The funder had no role in the study design, data analysis, or decision to publish.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to acknowledge the Home for Researchers editorial team (www.home-for-researchers.com) for their language editing service.
AGEs, Advanced glycosylation end products; AKT/PKB, Protein kinase B; AS, Atherosclerosis; AMPK, Adenosine monophosphate-activated protein kinase; APOE, Apolipoprotein E; BDNF, Brain-derived neurotrophic factor; BP, Biological process; CC, Cellular component; CKD, Chronic kidney disease; CVD, Cardiovascular disease; DM, Diabetes mellitus; ECM, Extracellular matrix; ER, Endoplasmic reticulum; ERK1, Extracellular signal-regulated kinase 1; FGF2, Fibroblast growth factor 2; GAPDH, 3-phosphoglyceraldehyde dehydrogenase; GO, Gene ontology; H9C2, Rat cardiomyocytes; HF, Heart failure; HFpEF, Heart failure with preserved ejection fraction; JNK, c-Jun N-terminal kinases; KEGG, Kyoto encyclopedia of genes and genomes; MAPK, Mitogen-activated protein kinase; MCC, Maximal clique centrality; MCODE, Molecular Complex Detection; MCP-1, Monocyte chemoattractant protein-1; MF, Molecular function; MI, Myocardial infarction; MMP, Matrix metalloproteinase; mTOR, Mammalian target of rapamycin; mTORC1, Mechanistic target of rapamycin complex 1; NADH, Nicotinamide adenine dinucleotide; NADPH, Nicotinamide adenine dinucleotide phosphate; NRAS, Neuroblastoma RAS Viral Gene Homolog; NGF, Nerve growth factor; NHE3, Sodium-hydrogen exchanger 3; NRF2, Nuclear erythroid-related factor 2; OMIM, Online Mendelian Inheritance in Man; PDB, Protein Data Bank; PharmGKB, Pharmacogenomics Knowledgebase; PKC, Protein kinase C; PPI, Protein protein interaction; RAAS, Renin-angiotensin-aldosterone system; RAGE, Receptor for advanced glycation end-products; Rap1, Ras-related Protein 1; RECK, Reversion-inducing-cysteine-rich protein with Kazal motifs; ROCK, Rho-associated, coiled-coil containing kinases; ROS, reactive oxygen species; SAMD, small mothers against decapentaplegic; S1P, Sphingosine 1-phosphate; SGLT2, Sodium-glucose cotransporter 2; STRING, Search Tool for the Retrieval of Interacting Genes database; T2DM, Type 2 Diabetes Mellitus; TGF-β, Transforming growth factor-β; TIMP-1, Tissue inhibitor of metalloproteinase 1; TNF, Tumor necrosis factor; TNFR, Tumor necrosis factor receptors; TTD, Therapeutic Target Database; VCAM-1, Vascular cell adhesion molecule-1; VEGF, Vascular endothelial growth factor; VEGFR2, Vascular endothelial growth factor receptor 2.
1. Sun H, Saeedi P, Karuranga S, Pinkepank M, Ogurtsova K, Duncan BB, et al. IDF Diabetes Atlas: global, regional and country-level diabetes prevalence estimates for 2021 and projections for 2045. Diabetes Res Clin Pract. (2022) 183:109119. doi: 10.1016/j.diabres.2021.109119
2. Holman N, Young B, Gadsby R. Current prevalence of type 1 and type 2 diabetes in adults and children in the UK. Diabet Med. (2015) 32:1119–20. doi: 10.1111/dme.12791
3. Cornel JH, Bakris GL, Stevens SR, Alvarsson M, Bax WA, Chuang LM, et al. Effect of sitagliptin on kidney function and respective cardiovascular outcomes in type 2 diabetes: outcomes from TECOS. Diabetes Care. (2016) 39:2304–10. doi: 10.2337/dc16-1415
4. Holman RR, Bethel MA, Mentz RJ, Thompson VP, Lokhnygina Y, Buse JB, et al. Effects of once-weekly exenatide on cardiovascular outcomes in type 2 diabetes. N Engl J Med. (2017) 377:1228–39. doi: 10.1056/NEJMoa1612917
5. Scirica BM, Braunwald E, Raz I, Cavender MA, Morrow DA, Jarolim P, et al. Heart failure, saxagliptin, and diabetes mellitus: observations from the SAVOR-TIMI 53 randomized trial. Circulation. (2015) 132:e198. doi: 10.1161/CIRCULATIONAHA.115.015511
6. Banks AZ, Mentz RJ, Stebbins A, Mikus CR, Schulte PJ, Fleg JL, et al. Response to exercise training and outcomes in patients with heart failure and diabetes mellitus: insights from the HF-ACTION trial. J Card Fail. (2016) 22:485–91. doi: 10.1016/j.cardfail.2015.12.007
7. Packer M, O'Connor C, McMurray JJV, Wittes J, Abraham WT, Anker SD, et al. Effect of ularitide on cardiovascular mortality in acute heart failure. N Engl J Med. (2017) 376:1956–64. doi: 10.1056/NEJMoa1601895
8. Pitt B, Pfeffer MA, Assmann SF, Boineau R, Anand IS, Claggett B, et al. Spironolactone for heart failure with preserved ejection fraction. N Engl J Med. (2014) 370:1383–92. doi: 10.1056/NEJMoa1313731
9. Targher G, Dauriz M, Laroche C, Temporelli PL, Hassanein M, Seferovic PM, et al. In-hospital and 1-year mortality associated with diabetes in patients with acute heart failure: results from the ESC-HFA Heart Failure Long-Term Registry. Eur J Heart Fail. (2017) 19:54–65. doi: 10.1002/ejhf.679
10. Win TT, Davis HT, Laskey WK. Mortality among patients hospitalized with heart failure and diabetes mellitus: results from the national inpatient sample 2000 to 2010. Circ Heart Fail. (2016) 9:e003023. doi: 10.1161/CIRCHEARTFAILURE.115.003023
11. Zheng Y, Ley SH, Hu FB. Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol. (2018) 14:88–98. doi: 10.1038/nrendo.2017.151
12. Zhang L, Zhao MH, Zuo L, Wang Y, Yu F, Zhang H, et al. China Kidney Disease Network (CK-NET) 2015 annual data report. Kidney Int Suppl. (2019) 9:e1–81. doi: 10.1016/j.kisu.2018.11.001
13. Rodriguez-Gutierrez R, Gonzalez-Gonzalez JG, Zuniga-Hernandez JA, McCoy RG. Benefits and harms of intensive glycemic control in patients with type 2 diabetes. BMJ. (2019) 367:l5887. doi: 10.1136/bmj.l5887
14. Fediuk DJ, Nucci G, Dawra VK, Cutler DL, Amin NB, Terra SG, et al. Overview of the clinical pharmacology of ertugliflozin, a novel Sodium-Glucose Cotransporter 2 (SGLT2) inhibitor. Clin Pharmacokinet. (2020) 59:949–65. doi: 10.1007/s40262-020-00875-1
15. Markham A, Elkinson S. Luseogliflozin: first global approval. Drugs. (2014) 74:945–50. doi: 10.1007/s40265-014-0230-8
16. Poole RM, Dungo RT. Ipragliflozin: first global approval. Drugs. (2014) 74:611–17. doi: 10.1007/s40265-014-0204-x
17. Poole RM, Prossler JE. Tofogliflozin: first global approval. Drugs. (2014) 74:939–44. doi: 10.1007/s40265-014-0229-1
18. Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. (2015) 373:2117–28. doi: 10.1056/NEJMoa1504720
19. Packer M, Anker SD, Butler J, Filippatos G, Pocock SJ, Carson P, et al. Cardiovascular and renal outcomes with empagliflozin in heart failure. N Engl J Med. (2020) 383:1413–24. doi: 10.1056/NEJMoa2022190
20. Anker SD, Butler J, Filippatos G, Ferreira JP, Bocchi E, Bohm M, et al. Empagliflozin in heart failure with a preserved ejection fraction. N Engl J Med. (2021) 385:1451–61. doi: 10.1056/NEJMoa2107038
21. Perkovic V, Jardine MJ, Neal B, Bompoint S, Heerspink HJL, Charytan DM, et al. Canagliflozin and renal outcomes in type 2 diabetes and nephropathy. N Engl J Med. (2019) 380:2295–306. doi: 10.1056/NEJMoa1811744
22. Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. (2017) 377:644–57. doi: 10.1056/NEJMoa1611925
23. Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med. (2019) 380:347–57. doi: 10.1056/NEJMoa1812389
24. McMurray JJV, Solomon SD, Inzucchi SE, Kober L, Kosiborod MN, Martinez FA, et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med. (2019) 381:1995–2008. doi: 10.1056/NEJMoa1911303
25. Heerspink HJL, Stefansson BV, Correa-Rotter R, Chertow GM, Greene T, Hou FF, et al. Dapagliflozin in patients with chronic kidney disease. N Engl J Med. (2020) 383:1436–46. doi: 10.1056/NEJMoa2024816
26. Cannon CP, Pratley R, Dagogo-Jack S, Mancuso J, Huyck S, Masiukiewicz U, et al. Cardiovascular outcomes with ertugliflozin in type 2 diabetes. N Engl J Med. (2020) 383:1425–35. doi: 10.1056/NEJMoa2004967
27. Bhatt DL, Szarek M, Steg PG, Cannon CP, Leiter LA, McGuire DK, et al. Sotagliflozin in patients with diabetes and recent worsening heart failure. N Engl J Med. (2021) 384:117–28. doi: 10.1056/NEJMoa2030183
28. Bhatt DL, Szarek M, Pitt B, Cannon CP, Leiter LA, McGuire DK, et al. Sotagliflozin in patients with diabetes and chronic kidney disease. N Engl J Med. (2021) 384:129–39. doi: 10.1056/NEJMoa2030186
29. Gosak M, Markovic R, Dolensek J, Slak Rupnik M, Marhl M, Stozer A, et al. Network science of biological systems at different scales: a review. Phys Life Rev. (2018) 24:118–35. doi: 10.1016/j.plrev.2017.11.003
30. Kim S, Chen J, Cheng T, Gindulyte A, He J, He S, et al. PubChem in 2021: new data content and improved web interfaces. Nucleic Acids Res. (2021) 49:D1388–95. doi: 10.1093/nar/gkaa971
31. Daina A, Michielin O, Zoete V. SwissTargetPrediction: updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res. (2019) 47:W357–64. doi: 10.1093/nar/gkz382
32. Barneh F, Jafari M, Mirzaie M. Updates on drug-target network; facilitating polypharmacology and data integration by growth of DrugBank database. Brief Bioinform. (2016) 17:1070–80. doi: 10.1093/bib/bbv094
33. Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, et al. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. (2021) 49:D605–12. doi: 10.1093/nar/gkaa1074
34. Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol. (2014) 8 (Suppl. 4):S11. doi: 10.1186/1752-0509-8-S4-S11
35. Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. (2003) 4:2. doi: 10.1186/1471-2105-4-2
36. Burley SK, Bhikadiya C, Bi C, Bittrich S, Chen L, Crichlow GV, et al. RCSB Protein Data Bank: powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. (2021) 49:D437–51. doi: 10.1093/nar/gkaa1038
37. Mooers BHM. Shortcuts for faster image creation in PyMOL. Protein Sci. (2020) 29:268–76. doi: 10.1002/pro.3781
38. Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. (2010) 31:455–61. doi: 10.1002/jcc.21334
39. Aneja A, Tang WH, Bansilal S, Garcia MJ, Farkouh ME. Diabetic cardiomyopathy: insights into pathogenesis, diagnostic challenges, therapeutic options. Am J Med. (2008) 121:748–57. doi: 10.1016/j.amjmed.2008.03.046
40. Senatus L, MacLean M, Arivazhagan L, Egana-Gorrono L, Lopez-Diez R, Manigrasso MB, et al. Inflammation meets metabolism: roles for the receptor for advanced glycation end products axis in cardiovascular disease. Immunometabolism. (2021) 3:e210024. doi: 10.20900/immunometab20210024
41. Garcia-Garduno TC, Padilla-Gutierrez JR, Cambron-Mora D, Valle Y. RAAS: a convergent player in ischemic heart failure and cancer. Int J Mol Sci. (2021) 22:7106. doi: 10.3390/ijms22137106
42. Aroor AR, McKarns S, Demarco VG, Jia G, Sowers JR. Maladaptive immune and inflammatory pathways lead to cardiovascular insulin resistance. Metabolism. (2013) 62:1543–52. doi: 10.1016/j.metabol.2013.07.001
43. Kenny HC, Abel ED. Heart failure in type 2 diabetes mellitus. Circ Res. (2019) 124:121–41. doi: 10.1161/CIRCRESAHA.118.311371
44. Anders HJ, Huber TB, Isermann B, Schiffer M. CKD in diabetes: diabetic kidney disease versus nondiabetic kidney disease. Nat Rev Nephrol. (2018) 14:361–77. doi: 10.1038/s41581-018-0001-y
45. Verma S, Wanner C, Zwiener I, Ofstad AP, George JT, Fitchett D, et al. Influence of microvascular disease on cardiovascular events in type 2 diabetes. J Am Coll Cardiol. (2019) 73:2780–2. doi: 10.1016/j.jacc.2019.03.002
46. Schefold JC, Filippatos G, Hasenfuss G, Anker SD, von Haehling S. Heart failure and kidney dysfunction: epidemiology, mechanisms and management. Nat Rev Nephrol. (2016) 12:610–23. doi: 10.1038/nrneph.2016.113
47. Koufakis T, Mustafa OG, Tsimihodimos V, Ajjan RA, Kotsa K. Insights into the results of sotagliflozin cardiovascular outcome trials: is dual inhibition the cherry on the cake of cardiorenal protection? Drugs. (2021) 81:1365–71. doi: 10.1007/s40265-021-01559-1
48. Shih JY, Lin YW, Fisch S, Cheng JT, Kang NW, Hong CS, et al. Dapagliflozin suppresses ER stress and improves subclinical myocardial function in diabetes: from bedside to bench. Diabetes. (2021) 70:262–7. doi: 10.2337/db20-0840
49. Jaikumkao K, Pongchaidecha A, Chueakula N, Thongnak LO, Wanchai K, Chatsudthipong V, et al. Dapagliflozin, a sodium-glucose co-transporter-2 inhibitor, slows the progression of renal complications through the suppression of renal inflammation, endoplasmic reticulum stress and apoptosis in prediabetic rats. Diabetes Obes Metab. (2018) 20:2617–26. doi: 10.1111/dom.13441
50. Kim MN, Moon JH, Cho YM. Sodium-glucose cotransporter-2 inhibition reduces cellular senescence in the diabetic kidney by promoting ketone body-induced NRF2 activation. Diabetes Obes Metab. (2021) 23:2561–71. doi: 10.1111/dom.14503
51. Albiero M, Tedesco S, Amendolagine FI, D'Anna M, Migliozzi L, Zuccolotto G, et al. Inhibition of SGLT2 rescues bone marrow cell traffic for vascular repair: role of glucose control and ketogenesis. Diabetes. (2021) 70:1767–79. doi: 10.2337/db20-1045
52. Tomita I, Kume S, Sugahara S, Osawa N, Yamahara K, Yasuda-Yamahara M, et al. SGLT2 inhibition mediates protection from diabetic kidney disease by promoting ketone body-induced mTORC1 inhibition. Cell Metab. (2020) 32:404–19 e6. doi: 10.1016/j.cmet.2020.06.020
53. Philippaert K, Kalyaanamoorthy S, Fatehi M, Long W, Soni S, Byrne NJ, et al. Cardiac late sodium channel current is a molecular target for the sodium/glucose cotransporter 2 inhibitor empagliflozin. Circulation. (2021) 143:2188–204. doi: 10.1161/CIRCULATIONAHA.121.053350
54. Borges-Junior FA, Silva Dos Santos D, Benetti A, Polidoro JZ, Wisnivesky ACT, Crajoinas RO, et al. Empagliflozin Inhibits Proximal Tubule NHE3 Activity, Preserves GFR, and Restores Euvolemia in Nondiabetic Rats with Induced Heart Failure. J Am Soc Nephrol. (2021) 32:1616–29. doi: 10.1681/ASN.2020071029
55. Juni RP, Al-Shama R, Kuster DWD, van der Velden J, Hamer HM, Vervloet MG, et al. Empagliflozin restores chronic kidney disease-induced impairment of endothelial regulation of cardiomyocyte relaxation and contraction. Kidney Int. (2021) 99:1088–101. doi: 10.1016/j.kint.2020.12.013
56. Aroor AR, Das NA, Carpenter AJ, Habibi J, Jia G, Ramirez-Perez FI, et al. Glycemic control by the SGLT2 inhibitor empagliflozin decreases aortic stiffness, renal resistivity index and kidney injury. Cardiovasc Diabetol. (2018) 17:108. doi: 10.1186/s12933-018-0750-8
57. Krasnov GS, Dmitriev AA, Snezhkina AV, Kudryavtseva AV. Deregulation of glycolysis in cancer: glyceraldehyde-3-phosphate dehydrogenase as a therapeutic target. Expert Opin Ther Targets. (2013) 17:681–93. doi: 10.1517/14728222.2013.775253
58. Tristan C, Shahani N, Sedlak TW, Sawa A. The diverse functions of GAPDH: views from different subcellular compartments. Cell Signal. (2011) 23:317–23. doi: 10.1016/j.cellsig.2010.08.003
59. Tarze A, Deniaud A, Le Bras M, Maillier E, Molle D, Larochette N, et al. GAPDH, a novel regulator of the pro-apoptotic mitochondrial membrane permeabilization. Oncogene. (2007) 26:2606–20. doi: 10.1038/sj.onc.1210074
60. Del Re DP, Amgalan D, Linkermann A, Liu Q, Kitsis RN. Fundamental mechanisms of regulated cell death and implications for heart disease. Physiol Rev. (2019) 99:1765–817. doi: 10.1152/physrev.00022.2018
61. Garg JP, Vucic D. Targeting cell death pathways for therapeutic intervention in kidney diseases. Semin Nephrol. (2016) 36:153–61. doi: 10.1016/j.semnephrol.2016.03.003
62. Nyengaard JR, Ido Y, Kilo C, Williamson JR. Interactions between hyperglycemia and hypoxia: implications for diabetic retinopathy. Diabetes. (2004) 53:2931–8. doi: 10.2337/diabetes.53.11.2931
63. Rains JL, Jain SK. Oxidative stress, insulin signaling, and diabetes. Free Radic Biol Med. (2011) 50:567–75. doi: 10.1016/j.freeradbiomed.2010.12.006
64. Eleftheriadis T, Pissas G, Tsogka K, Nikolaou E, Liakopoulos V, Stefanidis I. A unifying model of glucotoxicity in human renal proximal tubular epithelial cells and the effect of the SGLT2 inhibitor dapagliflozin. Int Urol Nephrol. (2020) 52:1179–89. doi: 10.1007/s11255-020-02481-3
65. Ren FF, Xie ZY, Jiang YN, Guan X, Chen QY, Lai TF, et al. Dapagliflozin attenuates pressure overload-induced myocardial remodeling in mice via activating SIRT1 and inhibiting endoplasmic reticulum stress. Acta Pharmacol Sin. (2021). doi: 10.1038/s41401-021-00805-2. [Epub ahead of print].
66. Ala M, Khoshdel MRF, Dehpour AR. Empagliflozin enhances autophagy, mitochondrial biogenesis, and antioxidant defense and ameliorates renal ischemia/reperfusion in nondiabetic rats. Oxid Med Cell Longev. (2022) 2022:1197061. doi: 10.1155/2022/1197061
67. Nakao M, Shimizu I, Katsuumi G, Yoshida Y, Suda M, Hayashi Y, et al. Empagliflozin maintains capillarization and improves cardiac function in a murine model of left ventricular pressure overload. Sci Rep. (2021) 11:18384. doi: 10.1038/s41598-021-97787-2
68. McKay MM, Morrison DK. Integrating signals from RTKs to ERK/MAPK. Oncogene. (2007) 26:3113–21. doi: 10.1038/sj.onc.1210394
69. Qi M, Elion EA. MAP kinase pathways. J Cell Sci. (2005) 118 (Pt. 16):3569–72. doi: 10.1242/jcs.02470
70. Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. (2002) 298:1911–12. doi: 10.1126/science.1072682
71. Gilbert CJ, Longenecker JZ, Accornero F. ERK1/2: an integrator of signals that alters cardiac homeostasis and growth. Biology. (2021) 10:346. doi: 10.3390/biology10040346
72. Hausenloy DJ, Yellon DM. Reperfusion injury salvage kinase signalling: taking a RISK for cardioprotection. Heart Fail Rev. (2007) 12:217–34. doi: 10.1007/s10741-007-9026-1
73. Capolongo G, Suzumoto Y, D'Acierno M, Simeoni M, Capasso G, Zacchia M. ERK1,2 signalling pathway along the nephron and its role in acid-base and electrolytes balance. Int J Mol Sci. (2019) 20:4153. doi: 10.3390/ijms20174153
74. Liu M, Ning X, Li R, Yang Z, Yang X, Sun S, et al. Signalling pathways involved in hypoxia-induced renal fibrosis. J Cell Mol Med. (2017) 21:1248–59. doi: 10.1111/jcmm.13060
75. Hu Z, Ju F, Du L, Abbott GW. Empagliflozin protects the heart against ischemia/reperfusion-induced sudden cardiac death. Cardiovasc Diabetol. (2021) 20:199. doi: 10.1186/s12933-021-01392-6
76. Uthman L, Kuschma M, Romer G, Boomsma M, Kessler J, Hermanides J, et al. Novel anti-inflammatory effects of canagliflozin involving hexokinase II in lipopolysaccharide-stimulated human coronary artery endothelial cells. Cardiovasc Drugs Ther. (2021) 35:1083–94. doi: 10.1007/s10557-020-07083-w
77. Adam C, Fusi L, Weiss N, Goller SG, Meder K, Frings VG, et al. Efficient suppression of NRAS-driven melanoma by co-inhibition of ERK1/2 and ERK5 MAPK pathways. J Invest Dermatol. (2020) 140:2455–65 e10. doi: 10.1016/j.jid.2020.03.972
78. Nguyen MQ, Teh JLF, Purwin TJ, Chervoneva I, Davies MA, Nathanson KL, et al. Targeting PHGDH upregulation reduces glutathione levels and resensitizes resistant NRAS-mutant melanoma to MAPK kinase inhibition. J Invest Dermatol. (2020) 140:2242–52 e7. doi: 10.1016/j.jid.2020.02.047
79. Frangogiannis NG The extracellular matrix in ischemic and nonischemic heart failure. Circ Res. (2019) 125:117–46. doi: 10.1161/CIRCRESAHA.119.311148
80. Rayego-Mateos S, Campillo S, Rodrigues-Diez RR, Tejera-Munoz A, Marquez-Exposito L, Goldschmeding R, et al. Interplay between extracellular matrix components and cellular and molecular mechanisms in kidney fibrosis. Clin Sci. (2021) 135:1999–2029. doi: 10.1042/CS20201016
81. Nandi SS, Katsurada K, Sharma NM, Anderson DR, Mahata SK, Patel KP. MMP9 inhibition increases autophagic flux in chronic heart failure. Am J Physiol Heart Circ Physiol. (2020) 319:H1414–437. doi: 10.1152/ajpheart.00032.2020
82. Wang H, Gao M, Li J, Sun J, Wu R, Han D, et al. MMP-9-positive neutrophils are essential for establishing profibrotic microenvironment in the obstructed kidney of UUO mice. Acta Physiol. (2019) 227:e13317. doi: 10.1111/apha.13317
83. Goerg J, Sommerfeld M, Greiner B, Lauer D, Seckin Y, Kulikov A, et al. Low-dose empagliflozin improves systolic heart function after myocardial infarction in rats: regulation of MMP9, NHE1, and SERCA2a. Int J Mol Sci. (2021) 22:5437. doi: 10.3390/ijms22115437
84. Nakatsu Y, Kokubo H, Bumdelger B, Yoshizumi M, Yamamotoya T, Matsunaga Y, et al. The SGLT2 inhibitor luseogliflozin rapidly normalizes aortic mRNA levels of inflammation-related but not lipid-metabolism-related genes and suppresses atherosclerosis in diabetic ApoE KO mice. Int J Mol Sci. (2017) 18:1704. doi: 10.3390/ijms18081704
85. Zhang Q, Fan Z, Zhang L, You Q, Wang L. Strategies for targeting serine/threonine protein phosphatases with small molecules in cancer. J Med Chem. (2021) 64:8916–38. doi: 10.1021/acs.jmedchem.1c00631
86. Newton AC, Antal CE, Steinberg SF. Protein kinase C mechanisms that contribute to cardiac remodelling. Clin Sci. (2016) 130:1499–510. doi: 10.1042/CS20160036
87. Singh RM, Cummings E, Pantos C, Singh J. Protein kinase C and cardiac dysfunction: a review. Heart Fail Rev. (2017) 22:843–59. doi: 10.1007/s10741-017-9634-3
88. Lan A, Du J. Potential role of Akt signaling in chronic kidney disease. Nephrol Dial Transplant. (2015) 30:385–94. doi: 10.1093/ndt/gfu196
89. Seccia TM, Rigato M, Ravarotto V, Calo LA. ROCK (RhoA/Rho Kinase) in cardiovascular-renal pathophysiology: a review of new advancements. J Clin Med. (2020) 9:1328. doi: 10.3390/jcm9051328
90. Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, et al. Atherosclerosis. Nat Rev Dis Primers. (2019) 5:56. doi: 10.1038/s41572-019-0106-z
91. Gistera A, Hansson GK. The immunology of atherosclerosis. Nat Rev Nephrol. (2017) 13:368–80. doi: 10.1038/nrneph.2017.51
92. Ghosh-Swaby OR, Goodman SG, Leiter LA, Cheng A, Connelly KA, Fitchett D, et al. Glucose-lowering drugs or strategies, atherosclerotic cardiovascular events, and heart failure in people with or at risk of type 2 diabetes: an updated systematic review and meta-analysis of randomised cardiovascular outcome trials. Lancet Diabetes Endocrinol. (2020) 8:418–35. doi: 10.1016/S2213-8587 (20)30038-3
93. Kosiborod M, Birkeland KI, Cavender MA, Fu AZ, Wilding JP, Khunti K, et al. Rates of myocardial infarction and stroke in patients initiating treatment with SGLT2-inhibitors versus other glucose-lowering agents in real-world clinical practice: results from the CVD-REAL study. Diabetes Obes Metab. (2018) 20:1983–7. doi: 10.1111/dom.13299
94. Kosiborod M, Lam CSP, Kohsaka S, Kim DJ, Karasik A, Shaw J, et al. Cardiovascular events associated with SGLT-2 inhibitors versus other glucose-lowering drugs: the CVD-REAL 2 study. J Am Coll Cardiol. (2018) 71:2628–39. doi: 10.1016/j.jacc.2018.03.009
95. Nasiri-Ansari N, Dimitriadis GK, Agrogiannis G, Perrea D, Kostakis ID, Kaltsas G, et al. Canagliflozin attenuates the progression of atherosclerosis and inflammation process in APOE knockout mice. Cardiovasc Diabetol. (2018) 17:106. doi: 10.1186/s12933-018-0749-1
96. Liu Y, Xu J, Wu M, Xu B, Kang L. Empagliflozin protects against atherosclerosis progression by modulating lipid profiles and sympathetic activity. Lipids Health Dis. (2021) 20:5. doi: 10.1186/s12944-021-01430-y
97. Dorado F, Velasco S, Esparis-Ogando A, Pericacho M, Pandiella A, Silva J, et al. The mitogen-activated protein kinase Erk5 mediates human mesangial cell activation. Nephrol Dial Transplant. (2008) 23:3403–11. doi: 10.1093/ndt/gfn333
98. Urushihara M, Takamatsu M, Shimizu M, Kondo S, Kinoshita Y, Suga K, et al. ERK5 activation enhances mesangial cell viability and collagen matrix accumulation in rat progressive glomerulonephritis. Am J Physiol Renal Physiol. (2010) 298:F167–76. doi: 10.1152/ajprenal.00124.2009
99. Kawakami T, Park SW, Kaku R, Yang J. Extracellular-regulated-kinase 5-mediated renal protection against ischemia-reperfusion injury. Biochem Biophys Res Commun. (2012) 418:603–8. doi: 10.1016/j.bbrc.2012.01.043
100. Kimura TE, Jin J, Zi M, Prehar S, Liu W, Oceandy D, et al. Targeted deletion of the extracellular signal-regulated protein kinase 5 attenuates hypertrophic response and promotes pressure overload-induced apoptosis in the heart. Circ Res. (2010) 106:961–70. doi: 10.1161/CIRCRESAHA.109.209320
101. Horie T, Park G, Inaba Y, Hashiuchi E, Iezaki T, Tokumura K, et al. MAPK Erk5 in leptin receptorexpressing neurons controls body weight and systemic energy homeostasis in female mice. Endocrinology. (2019) 160:2837–48. doi: 10.1210/en.2019-00090
102. Boluyt MO, Loyd AM, Roth MH, Randall MJ, Song EY. Activation of JNK in rat heart by exercise: effect of training. Am J Physiol Heart Circ Physiol. (2003) 285:H2639–47. doi: 10.1152/ajpheart.00596.2003
103. Chambers JW, Pachori A, Howard S, Iqbal S, LoGrasso PV. Inhibition of JNK mitochondrial localization and signaling is protective against ischemia/reperfusion injury in rats. J Biol Chem. (2013) 288:4000–11. doi: 10.1074/jbc.M112.406777
104. Kumar A, Singh UK, Kini SG, Garg V, Agrawal S, Tomar PK, et al. JNK pathway signaling: a novel and smarter therapeutic targets for various biological diseases. Future Med Chem. (2015) 7:2065–86. doi: 10.4155/fmc.15.132
105. Ursitti JA, Petrich BG, Lee PC, Resneck WG, Ye X, Yang J, et al. Role of an alternatively spliced form of alphaII-spectrin in localization of connexin 43 in cardiomyocytes and regulation by stress-activated protein kinase. J Mol Cell Cardiol. (2007) 42:572–81. doi: 10.1016/j.yjmcc.2006.11.018
106. Grynberg K, Ma FY, Nikolic-Paterson DJ. The JNK signaling pathway in renal fibrosis. Front Physiol. (2017) 8:829. doi: 10.3389/fphys.2017.00829
107. Meng XM, Nikolic-Paterson DJ, Lan HY. TGF-beta: the master regulator of fibrosis. Nat Rev Nephrol. (2016) 12:325–38. doi: 10.1038/nrneph.2016.48
108. Yung JHM, Giacca A. Role of c-Jun N-terminal Kinase (JNK) in obesity and type 2 diabetes. Cells. (2020) 9:706. doi: 10.3390/cells9030706
109. Yokota T, Wang Y. p38 MAP kinases in the heart. Gene. (2016) 575 (Pt. 2):36976. doi: 10.1016/j.gene.2015.09.030
110. Marber MS, Rose B, Wang Y. The p38 mitogen-activated protein kinase pathway–a potential target for intervention in infarction, hypertrophy, heart failure. J Mol Cell Cardiol. (2011) 51:485–90. doi: 10.1016/j.yjmcc.2010.10.021
111. Romero-Becerra R, Santamans AM, Folgueira C, Sabio G. p38 MAPK pathway in the heart: new insights in health and disease. Int J Mol Sci. (2020) 21:7412. doi: 10.3390/ijms21197412
112. Stambe C, Nikolic-Paterson DJ, Hill PA, Dowling J, Atkins RC. p38 Mitogen-activated protein kinase activation and cell localization in human glomerulonephritis: correlation with renal injury. J Am Soc Nephrol. (2004) 15:326–36. doi: 10.1097/01.ASN.0000108520.63445.E0
113. Cai Y, Lu X, Cheng X, Lv Q, Xu G, Liu X. Increased renal dysfunction, apoptosis, and fibrogenesis through sympathetic hyperactivity after focal cerebral infarction. Transl Stroke Res. (2021). doi: 10.1007/s12975-021-00900-w. [Epub ahead of print].
114. Molkentin JD, Bugg D, Ghearing N, Dorn LE, Kim P, Sargent MA, et al. Fibroblast-specific genetic manipulation of p38 mitogen-activated protein kinase in vivo reveals its central regulatory role in fibrosis. Circulation. (2017) 136:549–61. doi: 10.1161/CIRCULATIONAHA.116.026238
115. Lee N, Heo YJ, Choi SE, Jeon JY, Han SJ, Kim DJ, et al. Anti-inflammatory effects of empagliflozin and gemigliptin on LPS-stimulated macrophage via the IKK/NF-kappaB, MKK7/JNK, and JAK2/STAT1 signalling pathways. J Immunol Res. (2021) 2021:9944880. doi: 10.1155/2021/9944880
116. Das NA, Carpenter AJ, Belenchia A, Aroor AR, Noda M, Siebenlist U, et al. Empagliflozin reduces high glucose-induced oxidative stress and miR-21-dependent TRAF3IP2 induction and RECK suppression, and inhibits human renal proximal tubular epithelial cell migration and epithelial-to-mesenchymal transition. Cell Signal. (2020) 68:109506. doi: 10.1016/j.cellsig.2019.109506
117. Madonna R, Doria V, Minnucci I, Pucci A, Pierdomenico DS, De Caterina R. Empagliflozin reduces the senescence of cardiac stromal cells and improves cardiac function in a murine model of diabetes. J Cell Mol Med. (2020) 24:12331–40. doi: 10.1111/jcmm.15699
118. Rahadian A, Fukuda D, Salim HM, Yagi S, Kusunose K, Yamada H, et al. Canagliflozin prevents diabetes-induced vascular dysfunction in ApoE-deficient mice. J Atheroscler Thromb. (2020) 27:1141–51. doi: 10.5551/jat.52100
119. Jaskiewicz A, Pajak B, Orzechowski A. The many faces of Rap1 GTPase. Int J Mol Sci. (2018) 19:2848. doi: 10.3390/ijms19102848
120. Ni J, Bao S, Johnson RI, Zhu B, Li J, Vadaparampil J, et al. MAGI-1 interacts with nephrin to maintain slit diaphragm structure through enhanced Rap1 activation in podocytes. J Biol Chem. (2016) 291:24406–17. doi: 10.1074/jbc.M116.745026
121. Xiao L, Zhu X, Yang S, Liu F, Zhou Z, Zhan M, et al. Rap1 ameliorates renal tubular injury in diabetic nephropathy. Diabetes. (2014) 63:1366–80. doi: 10.2337/db13-1412
122. Chrzanowska-Wodnicka M. Distinct functions for Rap1 signaling in vascular morphogenesis and dysfunction. Exp Cell Res. (2013) 319:2350–9. doi: 10.1016/j.yexcr.2013.07.022
123. Cai Y, Kandula V, Kosuru R, Ye X, Irwin MG, Xia Z. Decoding telomere protein Rap1: its telomeric and nontelomeric functions and potential implications in diabetic cardiomyopathy. Cell Cycle. (2017) 16:1765–73. doi: 10.1080/15384101.2017.1371886
124. Feldman AM, Kadokami T, Higuichi Y, Ramani R, McTiernan CF. The role of anticytokine therapy in heart failure: recent lessons from preclinical and clinical trials? Med Clin North Am. (2003) 87:419–40. doi: 10.1016/S0025-7125 (02)00189-X
125. Dick SA, Epelman S. Chronic heart failure and inflammation: what do we really know? Circ Res. (2016) 119:159–76. doi: 10.1161/CIRCRESAHA.116.308030
126. Hanna A, Frangogiannis NG. Inflammatory cytokines and chemokines as therapeutic targets in heart failure. Cardiovasc Drugs Ther. (2020) 34:849–63. doi: 10.1007/s10557-020-07071-0
127. Haudek SB, Taffet GE, Schneider MD, Mann DL. TNF provokes cardiomyocyte apoptosis and cardiac remodeling through activation of multiple cell death pathways. J Clin Invest. (2007) 117:2692–701. doi: 10.1172/JCI29134
128. Perestrelo AR, Silva AC, Oliver-De La Cruz J, Martino F, Horvath V, Caluori G, et al. Multiscale analysis of extracellular matrix remodeling in the failing heart. Circ Res. (2021) 128:24–38. doi: 10.1161/CIRCRESAHA.120.317685
129. Sanchez-Nino MD, Benito-Martin A, Goncalves S, Sanz AB, Ucero AC, Izquierdo MC, et al. TNF superfamily: a growing saga of kidney injury modulators. Mediat Inflamm. (2010) 2010:182958. doi: 10.1155/2010/182958
130. Bhatraju PK, Zelnick LR, Shlipak M, Katz R, Kestenbaum B. Association of soluble TNFR-1 concentrations with long-term decline in kidney function: the multi-ethnic study of atherosclerosis. J Am Soc Nephrol. (2018) 29:2713–21. doi: 10.1681/ASN.2018070719
131. Heerspink HJL, Perco P, Mulder S, Leierer J, Hansen MK, Heinzel A, et al. Canagliflozin reduces inflammation and fibrosis biomarkers: a potential mechanism of action for beneficial effects of SGLT2 inhibitors in diabetic kidney disease. Diabetologia. (2019) 62:1154–66. doi: 10.1007/s00125-019-4859-4
132. Hussein AM, Eid EA, Taha M, Elshazli RM, Bedir RF, Lashin LS. Comparative study of the effects of GLP1 analog and SGLT2 inhibitor against diabetic cardiomyopathy in type 2 diabetic rats: possible underlying mechanisms. Biomedicines. (2020) 8:43. doi: 10.3390/biomedicines8030043
133. Sardu C, Massetti M, Testa N, Martino LD, Castellano G, Turriziani F, et al. Effects of Sodium-Glucose Transporter 2 Inhibitors (SGLT2-I) in patients with Ischemic Heart Disease (IHD) treated by coronary artery bypass grafting via MiECC: inflammatory burden, and clinical outcomes at 5 years of follow-up. Front Pharmacol. (2021) 12:777083. doi: 10.3389/fphar.2021.777083
134. Frimat M, Daroux M, Litke R, Neviere R, Tessier FJ, Boulanger E. Kidney, heart and brain: three organs targeted by ageing and glycation. Clin Sci. (2017) 131:1069–92. doi: 10.1042/CS20160823
135. Hartog JW, Voors AA, Bakker SJ, Smit AJ, van Veldhuisen DJ. Advanced glycation end-products (AGEs) and heart failure: pathophysiology and clinical implications. Eur J Heart Fail. (2007) 9:1146–55. doi: 10.1016/j.ejheart.2007.09.009
136. Mallipattu SK, Uribarri J. Advanced glycation end product accumulation: a new enemy to target in chronic kidney disease? Curr Opin Nephrol Hypertens. (2014) 23:547–54. doi: 10.1097/MNH.0000000000000062
137. Yang L, Liang B, Li J, Zhang X, Chen H, Sun J, et al. Dapagliflozin alleviates advanced glycation end product induced podocyte injury through AMPK/mTOR mediated autophagy pathway. Cell Signal. (2022) 90:110206. doi: 10.1016/j.cellsig.2021.110206
138. Ojima A, Matsui T, Nishino Y, Nakamura N, Yamagishi S. Empagliflozin, an inhibitor of sodium-glucose cotransporter 2 exerts anti-inflammatory and antifibrotic effects on experimental diabetic nephropathy partly by suppressing AGEs-receptor axis. Horm Metab Res. (2015) 47:686–92. doi: 10.1055/s-0034-1395609
139. Teichman SL, Unemori E, Dschietzig T, Conrad K, Voors AA, Teerlink JR, et al. Relaxin, a pleiotropic vasodilator for the treatment of heart failure. Heart Fail Rev. (2009) 14:321–9. doi: 10.1007/s10741-008-9129-3
140. Dschietzig TB. Relaxin-2 for heart failure with preserved ejection fraction (HFpEF): rationale for future clinical trials. Mol Cell Endocrinol. (2019) 487:54–8. doi: 10.1016/j.mce.2019.01.013
141. Warbrick I, Rabkin SW. Effect of the peptides Relaxin, Neuregulin, Ghrelin and Glucagon-like peptide-1, on cardiomyocyte factors involved in the molecular mechanisms leading to diastolic dysfunction and/or heart failure with preserved ejection fraction. Peptides. (2019) 111:33–41. doi: 10.1016/j.peptides.2018.05.009
142. Sasser JM. New targets for renal interstitial fibrosis: relaxin family peptide receptor 1-angiotensin type 2 receptor heterodimers. Kidney Int. (2014) 86:9–10. doi: 10.1038/ki.2014.22
143. Xie XC, Zhao N, Xu QH, Yang X, Xia WK, Chen Q, et al. Relaxin attenuates aristolochic acid induced human tubular epithelial cell apoptosis in vitro by activation of the PI3K/Akt signaling pathway. Apoptosis. (2017) 22:769–76. doi: 10.1007/s10495-017-1369-z
144. Tsai SJ. Role of neurotrophic factors in attention deficit hyperactivity disorder. Cytokine Growth Factor Rev. (2017) 34:35–41. doi: 10.1016/j.cytogfr.2016.11.003
145. Geissler A, Ryzhov S, Sawyer DB. Neuregulins: protective and reparative growth factors in multiple forms of cardiovascular disease. Clin Sci. (2020) 134:2623–43. doi: 10.1042/CS20200230
146. Pius-Sadowska E, Machalinski B. Pleiotropic activity of nerve growth factor in regulating cardiac functions and counteracting pathogenesis. ESC Heart Fail. (2021) 8:974–87. doi: 10.1002/ehf2.13138
147. Li M, Armelloni S, Zennaro C, Wei C, Corbelli A, Ikehata M, et al. BDNF repairs podocyte damage by microRNA-mediated increase of actin polymerization. J Pathol. (2015) 235:731–44. doi: 10.1002/path.4484
148. Guo CA, Guo S. Insulin receptor substrate signaling controls cardiac energy metabolism and heart failure. J Endocrinol. (2017) 233:R131–43. doi: 10.1530/JOE-16-0679
Keywords: type 2 diabetes mellitus, chronic kidney disease, heart failure, sodium-glucose cotransporter 2 inhibitors, cardiorenal protection
Citation: Wang A, Li Z, Zhuo S, Gao F, Zhang H, Zhang Z, Ren G and Ma X (2022) Mechanisms of Cardiorenal Protection With SGLT2 Inhibitors in Patients With T2DM Based on Network Pharmacology. Front. Cardiovasc. Med. 9:857952. doi: 10.3389/fcvm.2022.857952
Received: 24 January 2022; Accepted: 04 May 2022;
Published: 23 May 2022.
Edited by:
Christoph Sinning, University Medical Center Hamburg-Eppendorf, GermanyReviewed by:
Ander Vergara Arana, Vall d'Hebron University Hospital, SpainCopyright © 2022 Wang, Li, Zhuo, Gao, Zhang, Zhang, Ren and Ma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaochang Ma, bWF4aWFvY2hhbmdAeDI2My5uZXQ=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.