 Cristina Mas-Bargues
Cristina Mas-Bargues Consuelo Borrás
Consuelo Borrás Matilde Alique
Matilde Alique
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cardiovasc. Med., 15 April 2022
Sec. Atherosclerosis and Vascular Medicine
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.854726
This article is part of the Research TopicExtracellular Vesicles in Cardiovascular Inflammation and CalcificationView all 7 articles
Vascular calcification is an irreversible pathological process associated with a loss of vascular wall function. This process occurs as a result of aging and age-related diseases, such as cardiovascular and chronic kidney diseases, and leads to comorbidities. During these age-related diseases, the endothelium accumulates senescent cells, which stimulate calcification in vascular smooth muscle cells. Currently, vascular calcification is a silent pathology, and there are no early diagnostic tools. Therefore, by the time vascular calcification is diagnosed, it is usually untreatable. Some mediators, such as oxidative stress, inflammation, and extracellular vesicles, are inducers and promoters of vascular calcification. They play a crucial role during vascular generation and the progression of vascular calcification. Extracellular vesicles, mainly derived from injured endothelial cells that have acquired a senescent phenotype, contribute to calcification in a manner mostly dependent on two factors: (1) the number of extracellular vesicles released, and (2) their cargo. In this review, we present state-of-the-art knowledge on the composition and functions of extracellular vesicles involved in the generation and progression of vascular calcification.
Vascular calcification (VC) is a well-established multifactorial disorder characterized by calcium deposits along the vascular wall (1, 2). However, the calcification of vascular structures is unclear. Internal structures such as smooth muscle cells of the vascular wall undergo calcification that confers them osteoblast-like characteristics (3, 4). This prevents valvular or vascular adaptability and favors the development of pathologies. In general, there are three types of VC: medial arterial calcification, intimal calcification, and infantile calcification (2). This review mainly focuses on medial arterial calcification, which is often related to old age, diabetes mellitus, and cardiovascular diseases (CVD) associated with chronic kidney diseases (CKD) (5). In contrast, intimal calcification is only observed in patients with CVD such as atherosclerosis and hypertension (6). Infantile calcification refers to general arterial calcification in infants that is characterized mainly by medial calcification (7). Moreover, hydroxyapatite crystals could appear in cardiac valves (heart valve calcification) (6). Medial arterial calcification consists of hydroxyapatite crystals, which are calcium and phosphate mineral deposits found in bones (8–11). This type of calcification is a characteristic of CKD patients with CVD (6, 12–15). The pathology of medial arterial calcification is multifactorial (12). It is a gradual process resulting from disruptions in calcium, phosphate, and vitamin D homeostasis (8, 16). The pathophysiological process of vascular wall calcification involves some features of bone morphogenesis. Different cellular and molecular mediators are also involved such as pro-inflammatory molecules and molecules contributing to oxidative stress (8). Recently, extracellular vesicles (EVs) have been shown to play a role (17–21).
Chronic inflammatory diseases, including CKD, are considered synergistic pathologies due to the high risk of comorbidities, including CVD (12, 22–24). Furthermore, aging precipitates the appearance of age-related pathologies such as CKD and/or CVD, and vice versa; thus, these chronic inflammatory diseases trigger premature aging (19, 23). In general, the role of calcification in aging and age-related pathologies remains unclear. Calcification appears early and progresses rapidly, constituting a severe complication of kidney disease. Thus, VC is considered the primary cause of cardiovascular morbidity and mortality in patients with CKD. Due to its complex development mechanism, there are few tools for CKD treatment (22, 25). In this context, according to what we know so far, it would be essential to study the role of new mediators such as EVs, which are involved in aging, age-related diseases, and vascular calcification (18, 26, 27).
Vascular calcification generates complications in numerous cardiovascular pathologies. Coronary artery calcification is highly prevalent in CKD and coronary atherosclerotic plaques (5, 6, 28). Such calcification is classified as an intimal and medial calcification depending on the specific risk factor and is associated with major cardiovascular events (28). In the case of patients with acute myocardial infarction, they develop coronary calcification, which is associated with a high mortality rate (29). Furthermore, calcification is the most prevalent valvular disease in western societies, and calcific aortic stenosis is associated with a prevalence of 2% older (from 65 years) (30). Vascular calcification is also involved in aortic diseases, especially the ascending aorta called the porcelain aorta (31, 32).
Evidence suggests that chronic inflammation is a central factor in calcification. In the vasculature, chronic inflammation triggers atherosclerotic calcification (33). Subsequently, VC generates abnormalities in arteries, such as changes in blood flow due to the decreased wall elasticity and increased arterial stiffness, which decrease end-organ perfusion and cause injury (1, 8, 12). Consequently, VC may cause vascular alterations, cardiac arrest, and heart failure in patients with CKD (34).
Cellular senescence is a process where cells gain the maximum capacity of division and lose their division potential in somatic cells (35, 36). Some inflammatory chronic diseases are considered age-related diseases such as CKD and CVD, which are associated with kidney failure and vascular and valvular heart disease (36, 37) due to imbalance of oxidative stress, pro-inflammatory factors, and DNA damage that facilitate the accumulation of senescent cells (21, 37–39). Vascular aging is a consequence of premature aging in CKD that mediates medial VC that is a hallmark of senescence (37). Moreover, vascular and valvular heart diseases are associated with accelerated aging, accumulation of senescence, and increased inflammation that feedback these age-associated diseases and promote aortic calcification and calcific aortic valve diseases (CAVD) (36). CVD is the cause of death in 40% of the elderly (40).
Nowadays, few data describe the mechanisms/pathways by which EVs from vascular senescent cells mediate the development and progression of vascular calcification. This study compiles the role of senescent EVs from damage vasculature and the pathways and mechanisms described to date. However, more research is needed in this field. We know so far, the crossover between senescent EVs and VC has been less studied; therefore, the study of the EVs cargo generated by senescent cells and the signaling pathways in VC generation and progression could be critical in the VC prevention and treatment. In this way, senescent EV characterization and quantification could be a useful prognostic marker and therapeutic tool. This review explored the senescence-associated changes in EVs contributing to VC disease. However, due to the limited research, the work highlighted the background about this field described to date. Further studies should elucidate the role of senescent EVs in the mechanism implied in VC generation and progression.
In the last two decades, VC was identified as a manifestation of atherosclerosis associated with diabetes, hypertension, and dyslipidemia (41–45). Various therapeutic measures have been developed to treat the associated risk factors. However, results have been unsuccessful because VC pathophysiology remains poorly understood (8). Current knowledge of VC mainly indicates that it is caused by the progression of chronic silent inflammatory diseases (without clinical symptoms at the beginning) such as atherosclerosis, CKD, and CVD, among others (46, 47) and becomes manifest when calcification is advanced and irreversible (48). When finally diagnosed, there is no treatment; therefore, arterial calcification is considered a silent disease with no clinical symptoms or signs (18, 27, 46).
The strong correlation between VC and systemic inflammation has been described (49, 50). VC develops due to disturbances in the complex and subtle balance between inhibitors and promoters, acting at both systemic and local levels (5). Calcification involves proteins and mineralization mediators similar to those that regulate the ossification process (18). Inflammatory mediators, oxidative stress, and EVs induce the dedifferentiation of vascular smooth muscle cells (VSMCs) and cause endothelial damage (51), promoting the appearance of senescent cells in the vascular wall (18, 27, 52). Therefore, patients at a high risk of developing VC may experience cardiovascular events.
Recent evidence also suggests that an imbalance in the gut microbiota generates an accumulation of bioactive metabolites in the blood and activates cellular and molecular signaling, thereby disrupting homeostasis, and promoting diseases such as CKD. Moreover, CKD progression is often accompanied by VC that is linked to the dysregulation of gut microbiota and production of harmful metabolites, such as uremic blood toxins (gut microbiota-derived metabolites in CKD patients), that promote CKD development and therefore, they are implied in the calcification process (53). As a result, endothelial damage caused by uremic toxins is associated with the progression of CKD. In addition, uremic toxins promote vascular senescence and, finally, VC. Supporting this observation, in CKD, medial arterial calcification switches the VSMC phenotype and sometimes occurs in conjunction with calcium and phosphate accumulation within atherosclerotic lesions. Thus, vascular damage leads to VC, which is triggered by uremic toxins resulting from CKD. The imbalance of the bacterial metabolism in the gut microbiota promotes the production of uremic toxins and increases the possibility of VC development (54). Therefore, the generation of uremic toxins caused by microbiota imbalance that ultimately produces vascular senescence and VC in CKD was a novel way to develop VC.
The pathophysiology of VC involves multiple, complex signaling pathways that lead to mineralization (27). As already described, the loss of homeostasis leads to an accumulation of calcium and phosphate ions in the blood, causing spontaneous ion precipitation in the arteries, thereby inducing changes in the vasculature (12). VC caused by age-related pathologies, such as CKD and/or CVD, progresses in the same manner as that caused by physiological aging, but to a different extent (Figure 1).
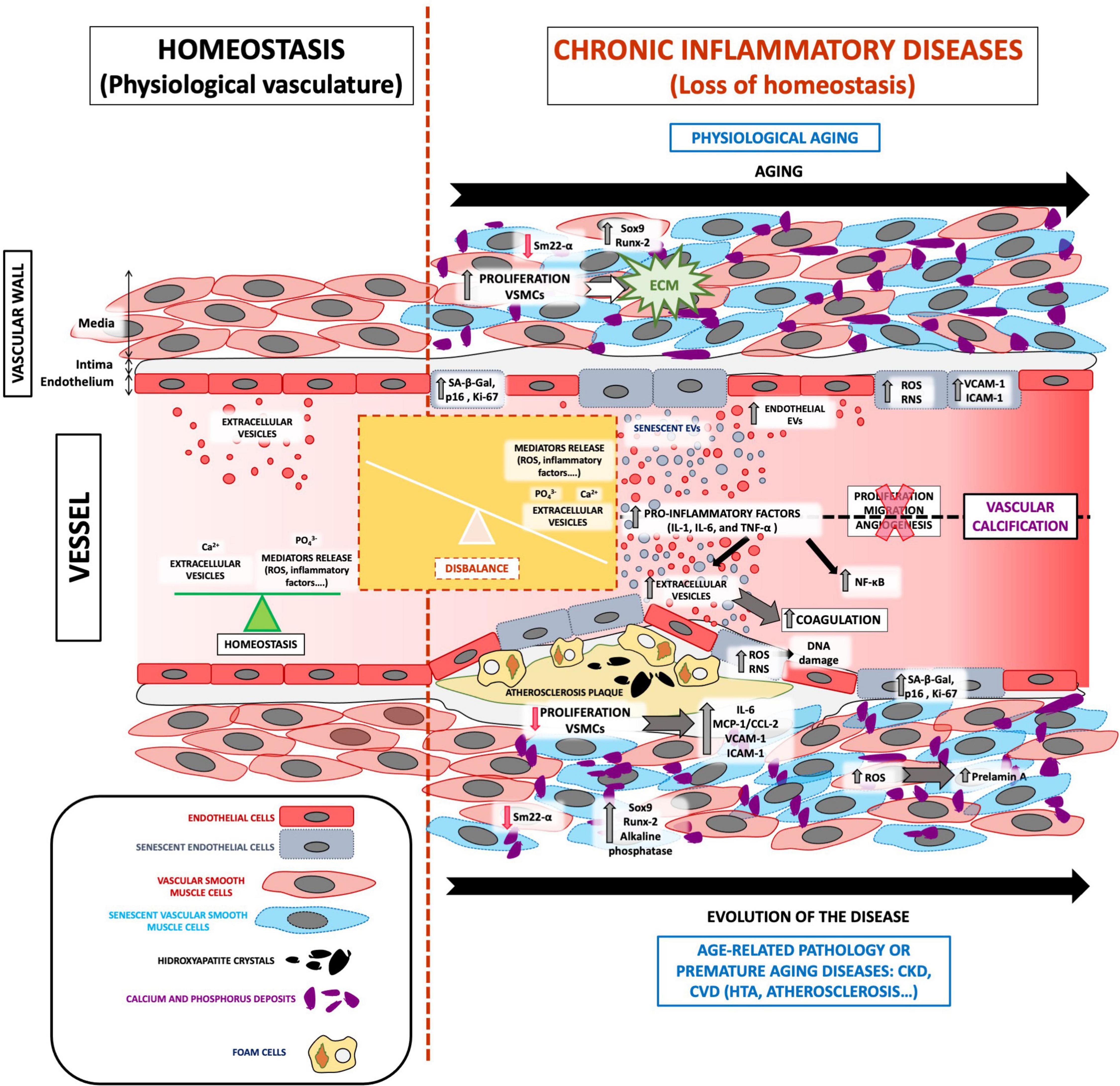
Figure 1. The role of EVs in VC development associated with vascular senescence. Under physiological conditions, the vasculature presents a balance between oxidative stress, inflammatory factors, and calcium and phosphate release and highlights the regular release of extracellular vesicles. All these mediators are focused on maintaining vessel homeostasis. The loss of homeostasis could appear with physiological aging and age-related pathologies such as CKD and/or CVD, which implies the imbalance of several processes. Both physiological and premature aging illnesses are characterized by an increase in ROS, calcium, and phosphate release, inflammatory mediators, adhesion molecules, coagulation process, the proliferation of extracellular matrix proliferation in the blood, and especially increased vasculature senescence. In addition, cells from a senescent vessel, especially endothelial cells and vascular smooth muscle cells, release extracellular vesicles that contain calcification factors. Complex signaling pathways in senescent extracellular vesicles cause spontaneous calcium ion precipitation in the arteries and thus contribute to VC development and pathophysiology.
Endothelial cells are continuously in contact with the blood in the vascular wall, and the imbalance of calcium/phosphate hampers endothelial cell function (55). Moreover, damage to endothelial cells in the vasculature generates oxidative stress through reactive oxygen species (ROS) production, which is a well-known cause of oxidative DNA damage and pro-inflammatory cytokine release (56, 57). These factors contribute actively to the appearance of extracellular matrix (ECM) deposits and increase calcium/phosphate accumulation in VSMCs, accelerating VC in CKD patients (58). It is noteworthy that damaged endothelial cells in this environment may adopt a senescent phenotype, thus contributing to VC development (18, 27).
Vascular smooth muscle cells are present alongside endothelial cells in the vasculature and are the primary cell type constituting the medial layer of the vascular wall (12). During the development of VC, VSMCs undergo dedifferentiation into osteoblast-like cells, promoting mineralization (an osteogenic transition) of the vessels. This process is associated with the upregulation of cellular and molecular targets of ROS, inflammatory factors, and EVs released from endothelial cells and VSMCs. This association highlights the multifactorial origin of VC (59, 60) occurring as a consequence of CKD (61).
Premature and physiological aging share many cellular phenotypes, including abnormal nuclear shape, loss of epigenetic markers, higher reactive oxygen and nitrogen species (RONS) levels, which increase lipid and protein production, cause DNA injury, increase calcium metabolism, and promote mitochondrial dysfunction (62). Cellular senescence is characterized by a stable cell cycle arrest (35) that causes inflammation and modifications of the microenvironment through the senescence-associated secretory phenotype (SASP). The SASP is a combination of molecules such as cytokines, ECM proteins, proteases, and other factors that alter the behavior of neighboring cells. Senescence is associated with phenotypic alterations that include morphological changes, such as a decrease in cellular proliferation and DNA replication. This phenotype is characterized by an increase in senescence marker levels (senescence-associated-β-galactosidase [SA-β-gal], p16INK4a, and Ki-67), lysosomal biogenesis, DNA repair protein levels, DNA injury, loss of cellular functionality, secretion of pro-inflammatory factors (IL-1, IL-6, and TNF-α), modulation of cell cycle arrest proteins, and reorganization of chromatin into discrete foci (21, 38).
In addition, senescence is exacerbated by reduced levels of renoprotective factors such as Klotho, vitamin D, and bone morphogenetic proteins (BMPs), and downregulated renoprotective mechanisms such as mitophagy (63). In this sense, senescence can be considered an adaptive cellular response to the external microenvironment. Accordingly, the interaction between endothelial cells and VSMCs in VC has been recently identified as an important factor; blocking the senescence process can attenuate osteogenic transformation (64).
Arterial aging in elderly patients leads to endothelial dysfunction, which eventually triggers a phenotypic change that results in cellular senescence (46). Endothelial cells damaged as a result of CKD may achieve a senescent phenotype characterized by a larger and flatter morphology and a polyploid nucleus, thus contributing to the evolution of VC (18, 27). Moreover, these cells exhibit changes in cytoskeleton integrity, proliferation rate, angiogenesis, and migration (21, 38). Furthermore, senescent endothelial cells demonstrate increased production of adhesion molecules (VCAM-1 and ICAM-1) and increased nuclear translocation of NF-κB. Therefore, the senescent endothelium is more susceptible to apoptosis, due to the presence of adhesion and inflammatory molecules (33, 65). Moreover, endothelial cell senescence is associated with an increase in EV release, which contributes actively to the generation and progression of VC in VSMCs (18, 21, 27). Overall, EVs from senescent endothelial cells play a critical role in VC and are considered a novel mediator.
Some reports have demonstrated that classical and novel mediators involved in VC may cause the VSMC phenotype to switch to an osteoblast-like phenotype in aortic vessels (12) and that senescent VSMCs generate a synergistic effect on the surrounding environment during the phenotypic transition. During this transition, the expression of some osteogenic/chondrogenic markers is modified. An early event in the VC signaling pathway is the downregulation of Sm22-α (a VSMC-specific marker) expression, while the expression of osteogenic/chondrogenic genes such as Sox9 and Runx-2 is upregulated (12, 18, 27). In this regard, VC in patients with CKD is not fully understood. It has been hypothesized that mediators such as oxidative stress, inflammation, and EVs are associated with a chronic inflammatory environment that regulates mineral metabolism in CKD patients (51, 66, 67).
Leopold et al. (68) reported that prelamin A levels increase in senescent cells through a mechanism involving ROS and pro-inflammatory cytokines, which are directly implicated in VC pathogenesis. Furthermore, it has been reported that atherosclerotic plaques increase morbidity and mortality rates in patients with advanced CKD and foster the development of atherosclerosis-related VC. This finding is associated with the development of VC in patients with CVD-associated chronic renal failure. Benz et al. (51) sought effective treatment to inhibit calcification mechanisms under CKD conditions. This finding focuses on atherosclerosis therapies to avoid calcification associated with CKD and these authors also highlighted the role of EVs in promoting inflammation and CV in CKD. Other studies that investigated VC in a model of accelerated aging reported that older subjects and younger CKD patients both presented phenotypically aged vascular walls, which was associated with upregulated prelamin A expression in calcified VSMCs (69–71). Moreover, senescent VSMCs present upregulated expression of Runx-2 and alkaline phosphatase, which are osteoblast transcription factors that enhance the transition of senescent VSMCs to the osteoblast-like phenotype and, therefore, promote calcification in vascular vessels (3, 72).
Furthermore, senescent cells in the vascular wall generate an imbalance of phosphate in VSMCs, thus promoting the pro-calcification phenotype of senescent vascular cells. Similarly, atherosclerosis is associated with premature cellular senescence; existing evidence demonstrates the presence of senescent VSMCs and atherosclerotic plaques in young patients (73). Senescent VSMCs exhibit decreased proliferation and a reduced capacity to repair plaques, with increased production of pro-inflammatory chemokines and adhesion molecule mediators (IL-6, MCP-1/CCL-2, VCAM-1, and ICAM-1) (74). Thus, inflammatory cells proliferate in this particular landscape.
Extracellular vesicles can be found in many body fluids, including plasma and urine. EVs are involved in physiological and pathophysiological processes through their involvement in the intercellular communication system. Under physiological conditions, EVs are involved in paracrine and/or endocrine communication mechanisms because they transmit biological signals to neighboring cells (75). In addition to delivering their cargo to nearby targets, EVs facilitate long-distance communication, thus regulating different biological and pathophysiological functions (76). Under pathological conditions, EVs participate in crucial processes such as inflammation, cell proliferation, and the immune response (38, 76–78). In addition, EV delivery is considered a biomarker per se and can be utilized to develop therapeutic strategies (76).
Indeed, EVs biogenesis, shedding and uptake, as well as their cargo content is known to be redox sensitive (79). In particular, senescence and oxidative stress promote EVs release (80–82), and recently, EVs are being included as part of the SASP (83). However, their specific content can include either antioxidants or ROS-generating enzymes; thus, oxidative-stress released EVs can trigger both antioxidant and pro-oxidant responses, thereby modulating the redox status of recipient cells (84). These EVs can also carry waste products, such as oxidized molecules, which would induce autophagy in target cells (85). Environmental factors influence the number and content of EVs in the development and progression of diseases (18, 27, 86). Levels of EVs are elevated in patients with vascular, metabolic, pulmonary, autoimmune, and neurodegenerative diseases, chronic inflammation, and cancer (87). Furthermore, an increase in EV levels is generated in vascular endothelial cells during atherosclerosis, due to stress (88). Hence, EVs are attracting increasing attention as markers for predicting, diagnosing, and monitoring complex diseases, with the potential to contribute to the identification of new therapeutic targets (88).
Some studies demonstrated that cells that arise SASP release different amounts of EVs compared with non-senescent cells and have also been shown that EVs from senescent cells present different EV cargo (18, 27, 82, 89). Moreover, these changes in EVs released by senescent cells are associated with an increase in the EV size and ultrastructural changes observed by electronic microscopy (18, 27). Furthermore, some evidence demonstrated that proteins expressed on the surface of EVs released by damaged cells is different from non-damaged cells such as cancer (90, 91) CVD (92), among others.
Extracellular vesicles general content includes calcification-promoting factors such as annexins, BMPs, and calcium. A recent study showed that senescent-cell-derived EV cargo is high in calcium, annexin A2, annexin A6, and BMP2 (18, 93). When VSMCs are cultured in the presence of EVs derived from senescent endothelial cells (93, 94), they undergo dedifferentiation (18), with a decrease in Sm22-α protein levels (27, 95). Recent evidence also suggests that EVs from the plasma of elderly subjects promote calcification in vascular muscle cells in vitro. Accordingly, EVs from senescent endothelial cells are involved in VC (18).
Chronic kidney disease is characterized by the accumulation of uremic toxins in the blood. Senescent endothelial cells stimulated by uremic toxins produce more EVs per cell (27). EVs produced by senescent endothelial cells, generated by treatment with primary plasma uremic toxin, indoxyl sulfate, and uremic serum from rats, cause calcification of the vasculature in vitro (27, 95). In the present study, senescent endothelial EV cargo was increased in calcium levels (27). The mechanism that senescent endothelial EVs generated in VSMCs mediated VC is remarkable. Recently, Alique et al. reported that senescent EVs generated an increase in Runx2 and BMP2 expression in VSMCs during VC progression (27). Moreover, these VSMCs change the phenotype to the procalcifying vascular phenotype indicated by a decrease in Sm22-α levels. During the development of this phenotype, vascular cells expressing different inflammatory cytokines such as TNF-α, TWEAK, MCP-1/CCL-2, CCL5, and IL-6 are implicated in VC (27). Furthermore, EVs play an essential role in angiogenesis, and together with their role in the development of VSMC senescence and VC generation, they mediate CKD progression and associated cardiovascular complications (96). Moreover, EVs in CKD patients have been proposed as therapeutic targets (97). Thus, the effect on the initiation and progression of VC depends on the number and cargo of EVs generated in patients with chronic inflammatory pathologies.
Recent evidence showed that EVs from senescent endothelial cells are implied in the VC (18, 27). Furthermore, some microRNAs are mediated in the endothelial senescence, highlighting the downregulation of miR126-3p, miR126-5p, miR21-3p, miR155, and miR210, critical keeping endothelial homeostasis. Therefore, finally, cellular damage carries endothelial cells to achieve the senescent phenotype (27). Moreover, as a consequence, senescent endothelial EVs release an increase of calcium and magnesium compared with “young” endothelial EVs (27). Moreover, it has been demonstrated that senescent cells suffer DNA damage that increases EV release in cancer (98). Finally, Wallis et al. (83) showed changes in EV cargo in senescent cells depending on their content, and senescent EVs have a different role, harmful or beneficial.
Vascular smooth muscle cells undergoing calcification may release calcifying EVs themselves (22), which contain metalloproteinase-2 (MMP-2); annexin A2, A5, and A6; and phosphatidylserine (PS) on the surface. The enrichment of these matrix EVs enhances calcium-binding, coagulation, hydroxyapatite formation, and subsequently, calcification of the vascular walls in aging-related calcification (55). Accumulating evidence suggests that senescent endothelial EVs act as promoters of VC, initiating a cascade of events that cause vascular injury and finally, CVD development (38, 99). The production of EVs by senescent endothelial cells is considered a pathogenic mechanism of endothelial dysfunction (21). Endothelial EVs promote damage to VSMCs and the vascular endothelium (38, 39). In this context, both CVD in the elderly and chronic diseases in younger patients cause vascular senescence and VC (1, 63).
Notably, the relationship between EVs, VC, and senescence is unclear. To date, a search in PubMed using the keywords “extracellular vesicles,” “vascular calcification,” and “senescence” gave 24 results (Table 1). Interestingly, 15 of the 24 results are reviews and only 8 are original research articles. The first study was published in 2014. More research is needed to continue unraveling the complex underlying mechanism that correlates all three factors.
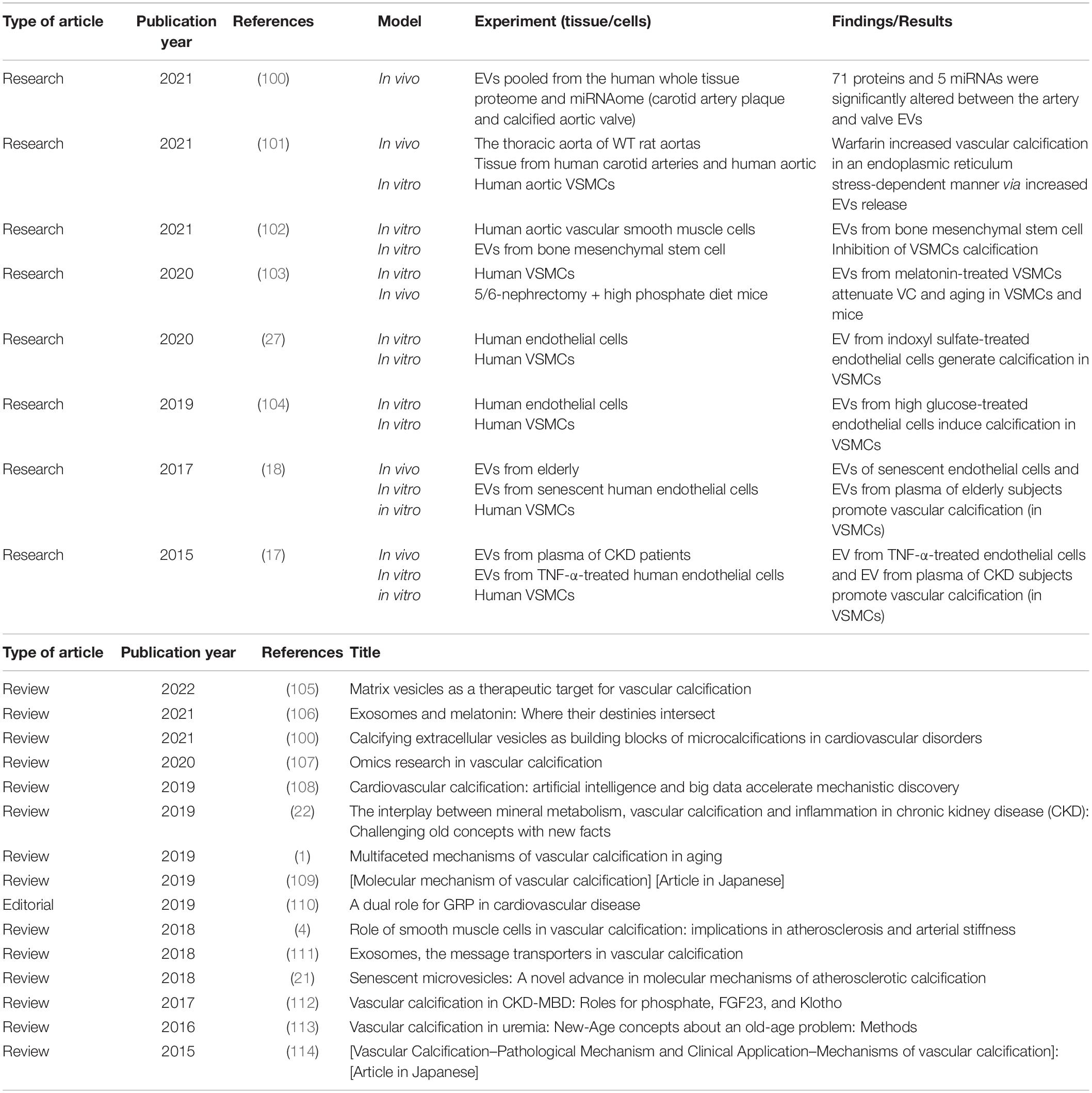
Table 1. Original research articles and reviews obtained after a search in PubMed with the following keywords: “extracellular vesicles” and “vascular calcification” and “senescence.”
The incidence of VC is increasing in developed countries, and VC can significantly increase cardiovascular risk. The processes and mechanisms involved in VC are unclear, and new therapeutic strategies are needed to prevent or reverse calcification. EVs have been outlined as a mediator in VC development, especially those released by senescent vasculature cells. Moreover, EVs are mediators that regulate aortic valve calcification evolution. Patients with CKD have a high prevalence of vascular calcification. To develop early diagnostic methods, evaluating EVs’ role in aging and age-related diseases such as CKD and VC is necessary. Consequently, studying EVs from damaged vasculature in physiological aging and age-related diseases to prevent the progression of VC is essential. It highlights EVs’ implication in the development of VC to develop early diagnostic methods to treat elderly and premature aging diseases (such as CVD associated with CKD) that will be achieved through VC.
MA: conceptualization, methodology, investigation, resources, and writing–original draft preparation. CM-B, CB, and MA: review and editing. CB and MA: funding acquisition. All authors have read and agreed to the published version of the manuscript.
This study was funded by Instituto de Salud Carlos III through the project “PI19/00240” (co-funded by the European Regional Development Fund “A Way to Make Europe”), Instituto de Salud Carlos III (ISCIII) FEDER funds RICORS program to RICORS2040 (RD21/0005/0002), Sociedad Española de Nefrología to MA, grant PID2020-113839RB-I00 funded by MCIN/AEI/10.13039/501100011033, PCIN-2017-117 of the Ministry of Economy and Competitiveness, and the EU Joint Programming Initiative “A Healthy Diet for a Healthy Life” (JPI HDHL INTIMIC-085) to CB.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Pescatore LA, Gamarra LF, Liberman M. Multifaceted mechanisms of vascular calcification in aging. Arterioscler Thromb Vasc Biol. (2019) 39:1307–16. doi: 10.1161/ATVBAHA.118.311576
2. Shi X, Gao J, Lv Q, Cai H, Wang F, Ye R, et al. Calcification in atherosclerotic plaque vulnerability: friend or foe? Front Physiol. (2020) 11:56. doi: 10.3389/fphys.2020.00056
3. Leopold JA. Vascular calcification: mechanisms of vascular smooth muscle cell calcification. Trends Cardiovasc Med. (2015) 25:267–74. doi: 10.1016/j.tcm.2014.10.021
4. Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM. Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc Res. (2018) 114:590–600. doi: 10.1093/cvr/cvy010
5. Otsuka F, Sakakura K, Yahagi K, Joner M, Virmani R. Has our understanding of calcification in human coronary atherosclerosis progressed? Arterioscler Thromb Vasc Biol. (2014) 34:724–36. doi: 10.1161/ATVBAHA.113.302642
6. Villa-Bellosta R. Vascular calcification: key roles of phosphate and pyrophosphate. Int J Mol Sci. (2021) 22:13536. doi: 10.3390/ijms222413536
7. Lanzer P, Boehm M, Sorribas V, Thiriet M, Janzen J, Zeller T, et al. Medial vascular calcification revisited: review and perspectives. Eur Heart J. (2014) 35:1515–25. doi: 10.1093/eurheartj/ehu163
8. Wang J, Zhou JJ, Robertson GR, Lee VW. Vitamin D in vascular calcification: a double-edged sword? Nutrients. (2018) 10:652. doi: 10.3390/nu10050652
9. Villa-Bellosta R. Synthesis of extracellular pyrophosphate increases in vascular smooth muscle cells during phosphate-induced calcification. Arterioscler Thromb Vasc Biol. (2018) 38:2137–47. doi: 10.1161/ATVBAHA.118.311444
10. Villa-Bellosta R, Sorribas V. Calcium phosphate deposition with normal phosphate concentration. Role of pyrophosphate. Circ J. (2011) 75:2705–10. doi: 10.1253/circj.cj-11-0477
11. St Hilaire C, Ziegler SG, Markello TC, Brusco A, Groden C, Gill F, et al. NT5E mutations and arterial calcifications. N Engl J Med. (2011) 364:432–42.
12. Opdebeeck B, Neven E, Millán JL, Pinkerton AB, D’Haese PC, Verhulst A. Chronic kidney disease-induced arterial media calcification in rats prevented by tissue non-specific alkaline phosphatase substrate supplementation rather than inhibition of the enzyme. Pharmaceutics. (2021) 13:1138. doi: 10.3390/pharmaceutics13081138
13. Goodman WG, London G, Amann K, Block GA, Giachelli C, Hruska KA, et al. Vascular calcification in chronic kidney disease. Am J Kidney Dis. (2004) 43:572–9.
14. Sharaf El, Din UA, Salem MM, Abdulazim DO. Vascular calcification: when should we interfere in chronic kidney disease patients and how? World J Nephrol. (2016) 5:398–417. doi: 10.5527/wjn.v5.i5.398
15. Jono S, Shioi A, Ikari Y, Nishizawa Y. Vascular calcification in chronic kidney disease. J Bone Miner Metab. (2006) 24:176–81.
16. Abedin M, Tintut Y, Demer LL. Vascular calcification: mechanisms and clinical ramifications. Arterioscler Thromb Vasc Biol. (2004) 24:1161–70. doi: 10.1161/01.ATV.0000133194.94939.42
17. Buendía P, Montes de Oca A, Madueño JA, Merino A, Martín-Malo A, Aljama P, et al. Endothelial microparticles mediate inflammation-induced vascular calcification. FASEB J. (2015) 29:173–81. doi: 10.1096/fj.14-249706
18. Alique M, Ruíz-Torres MP, Bodega G, Noci MV, Troyano N, Bohórquez L, et al. Microvesicles from the plasma of elderly subjects and from senescent endothelial cells promote vascular calcification. Aging (Albany NY). (2017) 9:778–89. doi: 10.18632/aging.101191
19. Alique M, Sánchez-López E, Bodega G, Giannarelli C, Carracedo J, Ramírez R. Hypoxia-inducible factor-1α: the master regulator of endothelial cell senescence in vascular aging. Cells. (2020) 9:195. doi: 10.3390/cells9010195
20. Luna C, Carmona A, Alique M, Carracedo J, Ramirez RTNF. α-damaged-HUVECs microparticles modify endothelial progenitor cell functional activity. Front Physiol. (2015) 6:395. doi: 10.3389/fphys.2015.00395
21. Alique M, Ramírez-Carracedo R, Bodega G, Carracedo J, Ramírez R. Senescent microvesicles: a novel advance in molecular mechanisms of atherosclerotic calcification. Int J Mol Sci. (2018) 19:2003. doi: 10.3390/ijms19072003
22. Viegas C, Araújo N, Marreiros C, Simes D. The interplay between mineral metabolism, vascular calcification and inflammation in chronic kidney disease (CKD): challenging old concepts with new facts. Aging (Albany NY). (2019) 11:4274–99. doi: 10.18632/aging.102046
23. Carracedo J, Alique M, Vida C, Bodega G, Ceprián N, Morales E, et al. Mechanisms of cardiovascular disorders in patients with chronic kidney disease: a process related to accelerated senescence. Front Cell Dev Biol. (2020) 8:185. doi: 10.3389/fcell.2020.00185
24. Mansouri L, Lundwall K, Moshfegh A, Jacobson SH, Lundahl J, Spaak J. Vitamin D receptor activation reduces inflammatory cytokines and plasma MicroRNAs in moderate chronic kidney disease – a randomized trial. BMC Nephrol. (2017) 18:161. doi: 10.1186/s12882-017-0576-8
25. Palit S, Kendrick J. Vascular calcification in chronic kidney disease: role of disordered mineral metabolism. Curr Pharm Des. (2014) 20:5829–33. doi: 10.2174/1381612820666140212194926
26. Mas-Bargues C, Alique M, Barrús-Ortiz MT, Borrás C, Rodrigues-Díez R. Exploring new kingdoms: the role of extracellular vesicles in oxi-inflamm-aging related to cardiorenal syndrome. Antioxidants. (2021) 11:78. doi: 10.3390/antiox11010078
27. Alique M, Bodega G, Corchete E, García-Menéndez E, de Sequera P, Luque R, et al. Microvesicles from indoxyl sulfate-treated endothelial cells induce vascular calcification. Comput Struct Biotechnol J. (2020) 18:953–66. doi: 10.1016/j.csbj.2020.04.006
28. Liu W, Zhang Y, Yu CM, Ji QW, Cai M, Zhao YX, et al. Current understanding of coronary artery calcification. J Geriatr Cardiol. (2015) 12668–75. doi: 10.11909/j.issn.1671-5411.2015.06.012
29. Yokoyama H, Higuma T, Endo T, Nishizaki F, Hanada K, Yokota T, et al. Prognostic impact of body mass index and culprit lesion calcification in patients with acute myocardial infarction. Heart Vessels. (2019) 34:1909–16. doi: 10.1007/s00380-019-01439-5
30. Lindman BR, Clavel MA, Mathieu P, Iung B, Lancellotti P, Otto CM, et al. Calcific aortic stenosis. Nat Rev Dis Primers. (2016) 2:16006.
31. Snow T, Semple T, Duncan A, Barker S, Rubens M, DiMario C, et al. ‘Porcelain aorta’: a proposed definition and classification of ascending aortic calcification. Open Heart. (2018) 5:e000703. doi: 10.1136/openhrt-2017-000703
32. Abramowitz Y, Jilaihawi H, Chakravarty T, Mack MJ, Makkar RR. Porcelain aorta: a comprehensive review. Circulation. (2015) 131:827–36. doi: 10.1161/CIRCULATIONAHA.114.011867
33. Demer LL, Tintut Y. Inflammatory, metabolic, and genetic mechanisms of vascular calcification. Arterioscler Thromb Vasc Biol. (2014) 34:715–23. doi: 10.1161/ATVBAHA.113.302070
34. Sigrist MK, Taal MW, Bungay P, McIntyre CW. Progressive vascular calcification over 2 years is associated with arterial stiffening and increased mortality in patients with stages 4 and 5 chronic kidney disease. Clin J Am Soc Nephrol. (2007) 2:1241–8. doi: 10.2215/CJN.02190507
35. Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, et al. Cellular senescence: defining a path forward. Cell. (2019) 179:813–27. doi: 10.1016/j.cell.2019.10.005
36. Owens WA, Walaszczyk A, Spyridopoulos I, Dookun E, Richardson GD. Senescence and senolytics in cardiovascular disease: promise and potential pitfalls. Mech Ageing Dev. (2021) 198:111540. doi: 10.1016/j.mad.2021.111540
37. Dai L, Schurgers LJ, Shiels PG, Stenvinkel P. Early vascular ageing in chronic kidney disease: impact of inflammation, vitamin K, senescence and genomic damage. Nephrol Dial Transplant. (2020) 35(‘. 2):ii31–7. doi: 10.1093/ndt/gfaa006
38. Carracedo J, Ramírez-Carracedo R, Alique M, Ramírez-Chamond, R. Endothelial cell senescence in the pathogenesis of endothelial dysfunction. In: Lenasi H editor. Endothelial Dysfunction - Old Concepts and New Challenges [Internet]. London: IntechOpen (2018). doi: 10.5772/intechopen.73024
39. Carracedo J, Alique M, Ramirez-Carracedo R, Bodega G, Ramirez R. Endothelial extracellular vesicles produced by senescent cells: pathophysiological role in the cardiovascular disease associated with all types of diabetes mellitus. Curr Vasc Pharmacol. (2018). 17:447–454. doi: 10.2174/1570161116666180820115726
40. Virani SS, Alonso A, Aparicio HJ, Benjamin EJ, Bittencourt MS, Callaway CW, et al. Heart disease and stroke statistics-2021 update: a report from the American heart association. Circulation. (2021) 143:e254–743. doi: 10.1161/CIR.0000000000000950
41. Sangiorgi G, Rumberger JA, Severson A, Edwards WD, Gregoire J, Fitzpatrick LA, et al. Arterial calcification and not lumen stenosis is highly correlated with atherosclerotic plaque burden in humans: a histologic study of 723 coronary artery segments using nondecalcifying methodology. J Am Coll Cardiol. (1998) 31:126–33. doi: 10.1016/s0735-1097(97)00443-9
42. Rosito GA, Massaro JM, Hoffmann U, Ruberg FL, Mahabadi AA, Vasan RS, et al. Pericardial fat, visceral abdominal fat, cardiovascular disease risk factors, and vascular calcification in a community–based sample: the Framingham heart study. Circulation. (2008) 117:605–13. doi: 10.1161/CIRCULATIONAHA.107.743062
43. Tamashiro M, Iseki K, Sunagawa O, Inoue T, Higa S, Afuso H, et al. Significant association between the progression of coronary artery calcification and dyslipidemia in patients on chronic hemodialysis. Am J Kidney Dis. (2001) 38:64–9. doi: 10.1053/ajkd.2001.25195
44. Anand DV, Lim E, Darko D, Bassett P, Hopkins D, Lipkin D, et al. Determinants of progression of coronary artery calcification in type 2 diabetes role of glycemic control and inflammatory/vascular calcification markers. J Am Coll Cardiol. (2007) 50:2218–25. doi: 10.1016/j.jacc.2007.08.032
45. Nitta K, Akiba T, Uchida K, Otsubo S, Otsubo Y, Takei T, et al. Left ventricular hypertrophy is associated with arterial stiffness and vascular calcification in hemodialysis patients. Hypertens Res. (2004) 27:47–52. doi: 10.1291/hypres.27.47
46. Tesauro M, Mauriello A, Rovella V, Annicchiarico-Petruzzelli M, Cardillo C, Melino G, et al. Arterial ageing: from endothelial dysfunction to vascular calcification. J Intern Med. (2017) 281:471–82. doi: 10.1111/joim.12605
47. Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, Franceschi C, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med. (2019) 25:1822–32. doi: 10.1038/s41591-019-0675-0
48. Kraus MA, Kalra PA, Hunter J, Menoyo J, Stankus N. The prevalence of vascular calcification in patients with end-stage renal disease on hemodialysis: a cross-sectional observational study. Ther Adv Chronic Dis. (2015) 6:84–96. doi: 10.1177/2040622315578654
49. López-Mejías R, González-Gay MA. IL-6: linking chronic inflammation and vascular calcification. Nat Rev Rheumatol. (2019) 15:457–9. doi: 10.1038/s41584-019-0259-x
50. Shantsila E, Lip GY. Systemic inflammation as a driver of vascular calcification: a proof of concept. J Intern Med. (2009) 266:453–6. doi: 10.1111/j.1365-2796.2009.02138.x
51. Benz K, Hilgers KF, Daniel C, Amann K. Vascular calcification in chronic kidney disease: the role of inflammation. Int J Nephrol. (2018) 2018:4310379.
52. Bourebaba L, Marycz K. Pathophysiological implication of fetuin-A glycoprotein in the development of metabolic disorders: a concise review. J Clin Med. (2019) 8:2033. doi: 10.3390/jcm8122033
53. Yin L, Li X, Ghosh S, Xie C, Chen J, Huang H. Role of gut microbiota-derived metabolites on vascular calcification in CKD. J Cell Mol Med. (2021) 25:1332–41. doi: 10.1111/jcmm.16230
54. Filipska I, Winiarska A, Knysak M, Stompór T. Contribution of gut microbiota-derived uremic toxins to the cardiovascular system mineralization. Toxins (Basel). (2021) 13:274. doi: 10.3390/toxins13040274
55. Abbasian N. Vascular calcification mechanisms: updates and renewed insight into signaling pathways involved in high phosphate-mediated vascular smooth muscle cell calcification. Biomedicines. (2021) 9:804. doi: 10.3390/biomedicines9070804
56. Byon CH, Javed A, Dai Q, Kappes JC, Clemens TL, Darley-Usmar VM, et al. Oxidative stress induces vascular calcification through modulation of the osteogenic transcription factor Runx2 by AKT signaling. J Biol Chem. (2008) 283:15319–27. doi: 10.1074/jbc.M800021200
57. Wei R, Enaka M, Muragaki Y. Activation of KEAP1/NRF2/P62 signaling alleviates high phosphate-induced calcification of vascular smooth muscle cells by suppressing reactive oxygen species production. Sci Rep. (2019) 9:10366. doi: 10.1038/s41598-019-46824-2
58. Reynolds JL, Joannides AJ, Skepper JN, McNair R, Schurgers LJ, Proudfoot D, et al. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J Am Soc Nephrol. (2004) 15:2857–67. doi: 10.1097/01.ASN.0000141960.01035.28
59. Müller KH, Hayward R, Rajan R, Whitehead M, Cobb AM, Ahmad S, et al. Poly(ADP-Ribose) links the DNA damage response and biomineralization. Cell Rep. (2019) 27:3124–38.e13. doi: 10.1016/j.celrep.2019.05.038
60. Kapustin AN, Chatrou ML, Drozdov I, Zheng Y, Davidson SM, Soong D, et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ Res. (2015) 116:1312–23. doi: 10.1161/CIRCRESAHA.116.305012
61. Chen NX, O’Neill KD, Dominguez JM, Moe SM. Regulation of reactive oxygen species in the pathogenesis of matrix vesicles induced calcification of recipient vascular smooth muscle cells. Vasc Med. (2021) 26:585–94. doi: 10.1177/1358863X211024721
62. van Deursen JM. The role of senescent cells in ageing. Nature. (2014) 509:439–46. doi: 10.1038/nature13193
63. Schroth J, Thiemermann C, Henson SM. Senescence and the aging immune system as major drivers of chronic kidney disease. Front Cell Dev Biol. (2020) 8:564461. doi: 10.3389/fcell.2020.564461
64. Zhang YX, Tang RN, Wang LT, Liu BC. Role of crosstalk between endothelial cells and smooth muscle cells in vascular calcification in chronic kidney disease. Cell Prolif. (2021) 54:e12980. doi: 10.1111/cpr.12980
65. Khaidakov M, Wang X, Mehta JL. Potential involvement of LOX-1 in functional consequences of endothelial senescence. PLoS One. (2011) 6:e20964. doi: 10.1371/journal.pone.0020964
66. Ketteler M, Wanner C, Metzger T, Bongartz P, Westenfeld R, Gladziwa U, et al. Deficiencies of calcium-regulatory proteins in dialysis patients: a novel concept of cardiovascular calcification in uremia. Kidney Int Suppl. (2003) 63:S84–7. doi: 10.1046/j.1523-1755.63.s84.21.x
67. Schafer C, Heiss A, Schwarz A, Westenfeld R, Ketteler M, Floege J, et al. The serum protein alpha 2-heremans-schmid glycoprotein/fetuin-A is a systemically acting inhibitor of ectopic calcification. J Clin Invest. (2003) 112:357–66. doi: 10.1172/JCI17202
68. Leopold JA. Vascular calcification: an age-old problem of old age. Circulation. (2013) 127:2380–2. doi: 10.1161/CIRCULATIONAHA.113.003341
69. Ragnauth CD, Warren DT, Liu Y, McNair R, Tajsic T, Figg N, et al. Prelamin A acts to accelerate smooth muscle cell senescence and is a novel biomarker of human vascular aging. Circulation. (2010) 121:2200–10. doi: 10.1161/CIRCULATIONAHA.109.902056
70. Olive M, Harten I, Mitchell R, Beers JK, Djabali K, Cao K, et al. Cardiovascular pathology in Hutchinson-Gilford progeria: correlation with the vascular pathology of aging. Arterioscler Thromb Vasc Biol. (2010) 30:2301–9. doi: 10.1161/ATVBAHA.110.209460
71. Liu Y, Drozdov I, Shroff R, Beltran LE, Shanahan CM. Prelamin A accelerates vascular calcification via activation of the DNA damage response and senescence-associated secretory phenotype in vascular smooth muscle cells. Circ Res. (2013) 112:e99–109. doi: 10.1161/CIRCRESAHA.111.300543
72. Childs BG, Zhang C, Shuja F, Sturmlechner I, Trewartha S, Velasco RF, et al. Senescent cells suppress innate smooth muscle cell repair functions in atherosclerosis. Nat Aging. (2021) 1:698–714. doi: 10.1038/s43587-021-00089-5
73. Uryga AK, Bennett MR. Ageing induced vascular smooth muscle cell senescence in atherosclerosis. J Physiol. (2016) 594:2115–24. doi: 10.1113/JP270923
74. Song Y, Shen H, Schenten D, Shan P, Lee PJ, Goldstein DR. Aging enhances the basal production of IL-6 and CCL2 in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. (2012) 32:103–9. doi: 10.1161/ATVBAHA.111.236349
75. Yáñez-Mó M, Siljander PR, Andreu Z, Zavec AB, Borràs FE, Buzas EI, et al. Biological properties of extracellular vesicles and their physiological functions. J Extracell Vesicles. (2015) 4:27066.
76. Santos P, Almeida F. Exosome-based vaccines: history, current state, and clinical trials. Front Immunol. (2021) 12:711565. doi: 10.3389/fimmu.2021.711565
77. Rovira J, Diekmann F, Campistol JM, Ramírez-Bajo MJ. Therapeutic application of extracellular vesicles in acute and chronic renal injury. Nefrologia. (2017) 37:126–37. doi: 10.1016/j.nefro.2016.04.006
78. Lovren F, Verma S. Evolving role of microparticles in the pathophysiology of endothelial dysfunction. Clin Chem. (2013) 59:1166–74. doi: 10.1373/clinchem.2012.199711
79. Borras C, Mas-Bargues C, Sanz-Ros J, Román-Domínguez A, Gimeno-Mallench L, Inglés M, et al. Extracellular vesicles and redox modulation in aging. Free Radic Biol Med. (2020) 149:44–50. doi: 10.1016/j.freeradbiomed.2019.11.032
80. Picca A, Guerra F, Calvani R, Bucci C, Lo Monaco MR, Bentivoglio AR, et al. Mitochondrial dysfunction and aging: insights from the analysis of extracellular vesicles. Int J Mol Sci. (2019) 20:805. doi: 10.3390/ijms20040805
81. Benedikter BJ, Weseler AR, Wouters EFM, Savelkoul PHM, Rohde GGU, Stassen FRM. Redox-dependent thiol modifications: implications for the release of extracellular vesicles. Cell Mol Life Sci. (2018) 75:2321–37. doi: 10.1007/s00018-018-2806-z
82. Misawa T, Tanaka Y, Okada R, Takahashi A. Biology of extracellular vesicles secreted from senescent cells as senescence–associated secretory phenotype factors. Geriatr Gerontol Int. (2020) 20:539–46. doi: 10.1111/ggi.13928
83. Wallis R, Mizen H, Bishop CL. The bright and dark side of extracellular vesicles in the senescence-associated secretory phenotype. Mech Ageing Dev. (2020) 189:111263. doi: 10.1016/j.mad.2020.111263
84. Chiaradia E, Tancini B, Emiliani C, Delo F, Pellegrino RM, Tognoloni A, et al. Extracellular vesicles under oxidative stress conditions: biological properties and physiological roles. Cells. (2021) 10:1763. doi: 10.3390/cells10071763
85. Yarana C, St Clair DK. Chemotherapy-induced tissue injury: an insight into the role of extracellular vesicles-mediated oxidative stress responses. Antioxidants (Basel). (2017) 6:75. doi: 10.3390/antiox6040075
86. Tomaniak M, Ga̧secka A, Filipiak KJ. Cell-derived microvesicles in cardiovascular diseases and antiplatelet therapy monitoring – a lesson for future trials? Current evidence, recent progresses and perspectives of clinical application. Int J Cardiol. (2017) 226:93–102. doi: 10.1016/j.ijcard.2016.10.007
87. Amabile N, Cheng S, Renard JM, Larson MG, Ghorbani A, McCabe E, et al. Association of circulating endothelial microparticles with cardiometabolic risk factors in the Framingham heart study. Eur Heart J. (2014) 35:2972–9. doi: 10.1093/eurheartj/ehu153
88. Yin M, Loyer X, Boulanger CM. Extracellular vesicles as new pharmacological targets to treat atherosclerosis. Eur J Pharmacol. (2015) 763(Pt A):90–103. doi: 10.1016/j.ejphar.2015.06.047
89. Takahashi A, Okada R, Nagao K, Kawamata Y, Hanyu A, Yoshimoto S, et al. Exosomes maintain cellular homeostasis by excreting harmful DNA from cells. Nat Commun. (2017) 8:15287.
90. Belov L, Matic KJ, Hallal S, Best OG, Mulligan SP, Christopherson RI. Extensive surface protein profiles of extracellular vesicles from cancer cells may provide diagnostic signatures from blood samples. J Extracell Vesicles. (2016) 5:25355. doi: 10.3402/jev.v5.25355
91. Ekström K, Crescitelli R, Pétursson HI, Johansson J, Lässer C, Olofsson Bagge R. Characterization of surface markers on extracellular vesicles isolated from lymphatic exudate from patients with breast cancer. BMC Cancer. (2022) 22:50. doi: 10.1186/s12885-021-08870-w
92. Dickhout A, Koenen RR. Extracellular vesicles as biomarkers in cardiovascular disease; chances and risks. Front Cardiovasc Med. (2018) 5:113. doi: 10.3389/fcvm.2018.00113
93. Yuan C, Ni L, Zhang C, Hu X, Wu X. Vascular calcification: new insights into endothelial cells. Microvasc Res. (2021) 134:104105. doi: 10.1016/j.mvr.2020.104105
94. Alique M, Bodega G, Giannarelli C, Carracedo J, Ramírez R. MicroRNA-126 regulates hypoxia-inducible factor-1α which inhibited migration, proliferation, and angiogenesis in replicative endothelial senescence. Sci Rep. (2019) 9:7381. doi: 10.1038/s41598-019-43689-3
95. Freise C, Querfeld U, Ludwig A, Hamm B, Schnorr J, Taupitz M. Uraemic extracellular vesicles augment osteogenic transdifferentiation of vascular smooth muscle cells via enhanced AKT signalling and PiT-1 expression. J Cell Mol Med. (2021) 25:5602–14. doi: 10.1111/jcmm.16572
96. Rojas-Franco P, Franco-Colín M, Blas-Valdivia V, Melendez-Camargo ME, Cano-Europa E. Arthrospira maxima (Spirulina) prevents endoplasmic reticulum stress in the kidney through its C-phycocyanin. J Zhejiang Univ Sci B. (2021) 22:603–8. doi: 10.1631/jzus.B2000725
97. Yaker L, Kamel S, Ausseil J, Boullier A. Effects of chronic kidney disease and uremic toxins on extracellular vesicle biology. Toxins (Basel). (2020) 12:811. doi: 10.3390/toxins12120811
98. Tanaka Y, Takahashi A. Senescence-associated extracellular vesicle release plays a role in senescence-associated secretory phenotype (SASP) in age-associated diseases. J Biochem. (2021) 169:147–53. doi: 10.1093/jb/mvaa109
99. Khan SY, Awad EM, Oszwald A, Mayr M, Yin X, Waltenberger B, et al. Premature senescence of endothelial cells upon chronic exposure to TNFα can be prevented by N-acetyl cysteine and plumericin. Sci Rep. (2017) 7:39501. doi: 10.1038/srep39501
100. Aikawa E, Blaser MC. 2020 Jeffrey M. Hoeg award lecture: calcifying extracellular vesicles as building blocks of microcalcifications in cardiovascular disorders. Arterioscler Thromb Vasc Biol. (2021) 41:117–27. doi: 10.1161/ATVBAHA.120.314704
101. Furmanik M, van Gorp R, Whitehead M, Ahmad S, Bordoloi J, Kapustin A, et al. Endoplasmic reticulum stress mediates vascular smooth muscle cell calcification via increased release of Grp78 (glucose-regulated protein, 78 kDa)-loaded extracellular vesicles. Arterioscler Thromb Vasc Biol. (2021) 41:898–914. doi: 10.1161/atvbaha.120.315506
102. Liu Y, Bao S, Guo W, Liu W. Bone mesenchymal stem cell derived exosomes alleviate high phosphorus-induced calcification of vascular smooth muscle cells through the NONHSAT 084969.2/NF-κB axis. Aging (Albany NY). (2021) 13:16749–62. doi: 10.18632/aging.203195
103. Xu F, Zhong JY, Lin X, Shan SK, Guo B, Zheng MH, et al. Melatonin alleviates vascular calcification and ageing through exosomal miR-204/miR-211 cluster in a paracrine manner. J Pineal Res. (2020) 68:e12631. doi: 10.1111/jpi.12631
104. Lin X, Li S, Wang YJ, Wang Y, Zhong JY, He JY, et al. Exosomal Notch3 from high glucose-stimulated endothelial cells regulates vascular smooth muscle cells calcification/aging. Life Sci. (2019) 232:116582. doi: 10.1016/j.lfs.2019.116582
105. Li T, Yu H, Zhang D, Feng T, Miao M, Li J, et al. Matrix vesicles as a therapeutic target for vascular calcification. Front Cell Dev Biol. (2022) 10:825622. doi: 10.3389/fcell.2022.825622
106. Novais AA, Chuffa LGA, Zuccari DAPC, Reiter RJ. Exosomes and melatonin: where their destinies intersect. Front Immunol. (2021) 12:692022. doi: 10.3389/fimmu.2021.692022
107. Duan M, Zhao WL, Zhou L, Novák P, Zhu X, Yin K. Omics research in vascular calcification. Clin Chim Acta. (2020) 511:198–207. doi: 10.1016/j.cca.2020.10.021
108. Rogers MA, Aikawa E. Cardiovascular calcification: artificial intelligence and big data accelerate mechanistic discovery. Nat Rev Cardiol. (2019) 16:261–74. doi: 10.1038/s41569-018-0123-8
109. Kurabayashi M. [Molecular mechanism of vascular calcification.]. Clin Calcium. (2019) 29:157–63.
110. Viegas CSB, Simes DC. A dual role for GRP in cardiovascular disease. Aging (Albany NY). (2019) 11:1323–4. doi: 10.18632/aging.101851
111. Zhang C, Zhang K, Huang F, Feng W, Chen J, Zhang H, et al. Exosomes, the message transporters in vascular calcification. J Cell Mol Med. (2018) 22:4024–33. doi: 10.1111/jcmm.13692
112. Yamada S, Giachelli CM. Vascular calcification in CKD-MBD: roles for phosphate, FGF23, and Klotho. Bone. (2017) 100:87–93. doi: 10.1016/j.bone.2016.11.012
113. Smith ER. Vascular calcification in uremia: new-age concepts about an old-age problem. Methods Mol Biol. (2016) 1397:175–208. doi: 10.1007/978-1-4939-3353-2_13
Keywords: aging, aging-related diseases, extracellular vesicles, inflammation, medial arterial calcification, senescence, smooth vessel cells, vascular calcification
Citation: Mas-Bargues C, Borrás C and Alique M (2022) The Contribution of Extracellular Vesicles From Senescent Endothelial and Vascular Smooth Muscle Cells to Vascular Calcification. Front. Cardiovasc. Med. 9:854726. doi: 10.3389/fcvm.2022.854726
Received: 14 January 2022; Accepted: 11 March 2022;
Published: 15 April 2022.
Edited by:
Jona Benjamin Krohn, Heidelberg University Hospital, GermanyReviewed by:
Patrick Malcolm Siegel, University Heart Center Freiburg, GermanyCopyright © 2022 Mas-Bargues, Borrás and Alique. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Consuelo Borrás, Y29uc3VlbG8uYm9ycmFzQHV2LmVz; Matilde Alique, bWF0aWxkZS5hbGlxdWVAdWFoLmVz
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.