 Lucie Pothen1
Lucie Pothen1 Roxane Verdoy1
Roxane Verdoy1 Delphine De Mulder1Hrag Esfahani1Charlotte Farah1Lauriane Y. M. Michel1
Delphine De Mulder1Hrag Esfahani1Charlotte Farah1Lauriane Y. M. Michel1 Flavia Dei Zotti1
Flavia Dei Zotti1 Bertrand Bearzatto2
Bertrand Bearzatto2 Jerome Ambroise2
Jerome Ambroise2 Caroline Bouzin3
Caroline Bouzin3 Chantal Dessy1
Chantal Dessy1 Jean-Luc Balligand1*
Jean-Luc Balligand1*- 1Institute of Experimental and Clinical Research (IREC), Pole of Pharmacology and Therapeutics (FATH), Cliniques Universitaires St-Luc and Université Catholique de Louvain (UCLouvain), Brussels, Belgium
- 2Institute of Experimental and Clinical Research (IREC), Centre des Technologies Moléculaires Appliquées (CTMA), Cliniques Universitaires St-Luc and Université Catholique de Louvain (UCLouvain), Brussels, Belgium
- 3Institute of Experimental and Clinical Research (IREC), Imaging Platform (2IP), Cliniques Universitaires St-Luc and Université Catholique de Louvain (UCLouvain), Brussels, Belgium
Background: Activation of the renin-angiotensin-aldosterone system (RAAS) plays a critical role in the development of hypertension. Published evidence on a putative “memory effect” of AngII on the vascular components is however scarce.
Aim: To evaluate the long-term effects of transient exposure to AngII on the mouse heart and the arterial tissue.
Methods: Blood pressure, cardiovascular tissue damage and remodeling, and systemic oxidative stress were evaluated in C57/B6/J mice at the end of a 2-week AngII infusion (AngII); 2 and 3 weeks after the interruption of a 2-week AngII treatment (AngII+2W and AngII +3W; so-called “memory” conditions) and control littermate (CTRL). RNAseq profiling of aortic tissues was used to identify potential key regulated genes accounting for legacy effects on the vascular phenotype. RNAseq results were validated by RT-qPCR and immunohistochemistry in a reproduction cohort of mice. Key findings were reproduced in a homotypic cell culture model.
Results: The 2 weeks AngII infusion induced cardiac hypertrophy and aortic damage that persisted beyond AngII interruption and despite blood pressure normalization, with a sustained vascular expression of ICAM1, infiltration by CD45+ cells, and cell proliferation associated with systemic oxidative stress. RNAseq profiling in aortic tissue identified robust Acta2 downregulation at transcript and protein levels (α-smooth muscle actin) that was maintained beyond interruption of AngII treatment. Among regulators of Acta2 expression, the transcription factor Myocardin (Myocd), exhibited a similar expression pattern. The sustained downregulation of Acta2 and Myocd was associated with an increase in H3K27me3 in nuclei of aortic sections from mice in the “memory” conditions. A sustained downregulation of ACTA2 and MYOCD was reproduced in the cultured human aortic vascular smooth muscle cells upon transient exposure to Ang II.
Conclusion: A transient exposure to Ang II produces prolonged vascular remodeling with robust ACTA2 downregulation, associated with epigenetic imprinting supporting a “memory” effect despite stimulus withdrawal.
Introduction
Hypertension is a well recognized cardiovascular risk factor, causing up to 7.6 million deaths per year worldwide (13.5% of total deaths) (1). Usually, it develops as a slow and gradual increase in blood pressure, with occasional acute hypertensive peaks. This silent disease can remain unrecognized until potentially fatal complications occur, such as hypertrophic cardiomyopathy, strokes, or ischemic heart disease.
Hypertension is conventionally associated with a neurohormonal activation from the sympathetic nervous and the renin-angiotensin-aldosterone systems (RAAS) (2, 3). The RAAS is involved in numerous physiological functions, including vasoconstriction, fluid volume regulation, cardiac remodeling, cell growth, and vascular wall integrity. Angiotensin II (AngII), as the main product and effector of the RAAS, is a potent regulator of blood pressure (4, 5). As such, it is also a key player in hypertension development, mainly through activation of the type 1 AngII receptors (AT1R), that triggers structural remodeling and inflammation in the heart and vascular tissue (6). In particular, cellular processes underlying vascular injury include, among others, impaired endothelial function and a phenotypic switch of vascular smooth muscle cells (VSMCs), characterized by a reduced expression of myofibrillar proteins and contractility, evolving toward a more proliferative and synthetic state, with increased production, e.g., of proteoglycans (7–9).
Usually, activation of the RAAS accompanies the development of other cardiovascular risk factors than hypertension, such as diabetes or metabolic syndrome, including hypercholesterolemia. The resulting cardiovascular pathologies evolve with sustained deleterious effects despite the removal of the pathogenic stimulus (e.g., hyperglycemia), a phenomenon that has been coined “metabolic memory” (10). In the previous models of diabetes, temporary exposure to hyperglycemia leads to an epigenetic imprinting in endothelial cells, with sustained mitochondrial oxidative stress despite glycemic normalization (11, 12). Similar epigenetic mechanisms might explain the enduring increase in cardiovascular risk, despite glycemic control in the clinical studies, named as legacy effect (13, 14). The specific participation of the RAAS, particularly of AngII, in these enduring effects is less clear. Intriguingly, other clinical trials suggest that treatement with a RAAS inhibitor, e.g., sartans that inhibit AngII binding to the AT1R, could prevent organ damage and reduce cardiovascular events through protective effects beyond blood pressure lowering in hypertensive patients (15). Published evidence on a putative “memory effect” of AngII is scarce. One previous study observed a sustained vascular injury with persistent activation of multiple signaling pathways (JNK1/2, STAT3, and NF-κB) and increase in the reactive oxygen species (ROS) production up to 1 week after withdrawal of an initial AngII infusion in mice; the data suggested a link with persistent NADPH oxidase activation. However, the upstream mechanism for this sustained oxidative stress was not established (16, 17).
Surprisingly, while transcriptomic profiles of AngII-treated organs/tissues are available in the kidneys (18), heart (19), or abdominal aortic aneurysm in ApoE-/- mice (20), the AngII treatment protocol varies widely between studies, and transcriptomic data on the arterial tissue of WT mice were rarely reported (21–23), with no attempt to analyze any legacy effect.
In this study, we developed an AngII “memory” model in which we examined long-term effects of temporary exposure to AngII on heart and arterial tissue, including cardiovascular remodeling and VSMC phenotypic switch. Longitudinal transcriptomic profiling of aortic tissues after AngII withdrawal identified uniquely regulated genes potentially involved in legacy effects on the vascular phenotype.
Materials and Methods
Animal Experimental Protocol
11-week male C57B6/J mice were implanted with osmotic minipumps delivering of 2.8 mg/kg/d of AngII (Sigma, A9525) for 2 weeks. To confirm the effect of the pharmacological treatment, blood pressure signals from aortic arch before (CTRL); under AngII up to 2 weeks (AngII); and 2 and 3 weeks after the interruption of AngII (AngII+2W; AngII+3w, respectively) were recorded in selected, conscious, and unrestrained animals, with surgically implanted miniaturized telemetry devices (DSI, USA) as described previously (24). Mice were sacrificed after anesthesia at the same time points and age-matched littermate served as control (Figure 1A). All the investigations conformed to the Guide for the Care and Use of Laboratory Animals (NIH Publications no. 8023, revised 2011) and were approved by the Institutional Animal Care and Research Advisory Committee of the Université Catholique de Louvain.
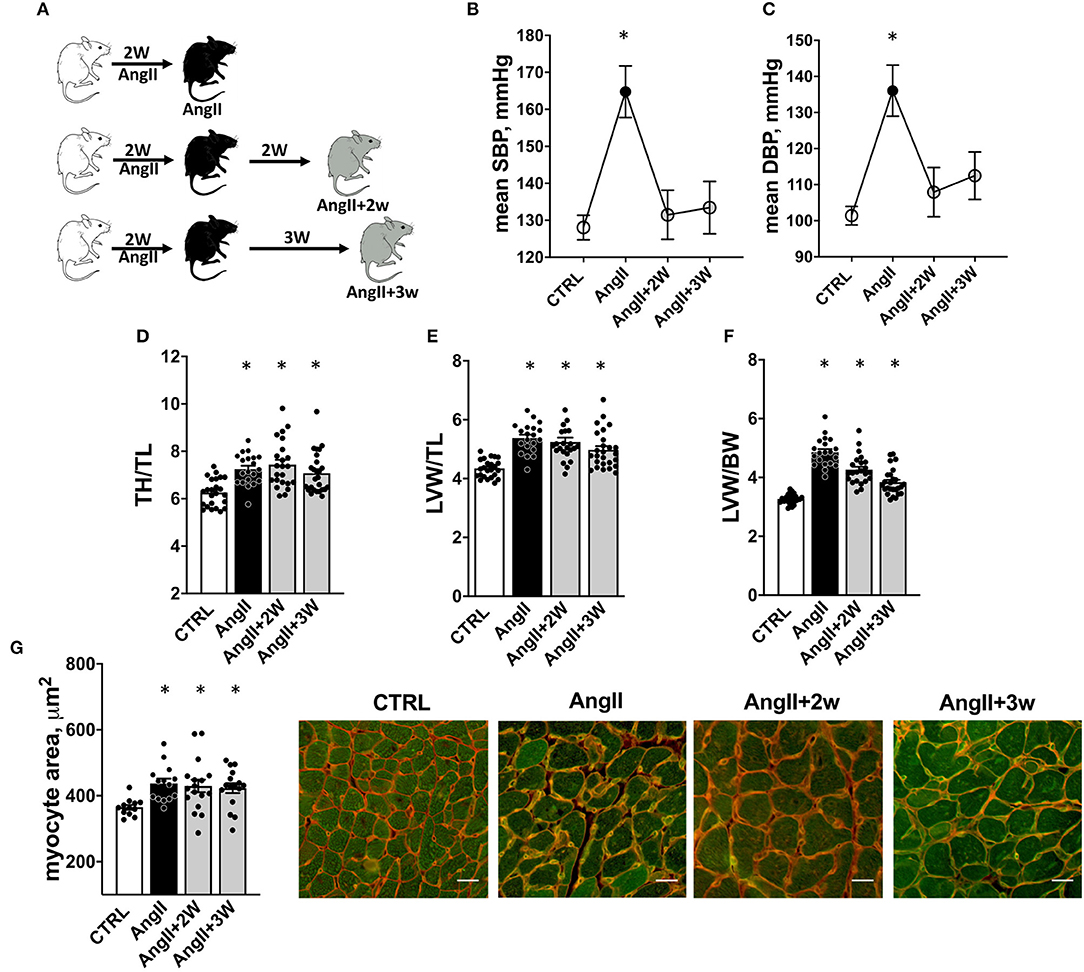
Figure 1. Experimental procedure and cardiac hypertrophy. Experimental procedure: (A) C57Bl6 mice were implanted with osmotic minipump delivering 2.8mg/kg/d of AngII during 2 weeks. One group was sacrificed at the end of the treatment (AngII), 2 others were sacrificed, respectively, 2 weeks (AngII+2W) and 3 weeks (AngII+3W) after the end of the minipump, and were considered as “memory groups”. Control mice (CTRL) were littermate age-matched mice sacrificed at each time point (2 weeks of AngII, 2 weeks of AngII + 2 weeks, 2 weeks of AngII +3 weeks). (B) Systolic and (C) Diastolic blood pressure measured with implanted telemetry devices. n = 19; *p < 0.05, one-way ANOVA for repeated measures, mixed-effect analysis followed by Tukey's multiple comparison test. (D–G) Cardiac Hypertrophy: (D) Total heart/tibial length ratio (TH/TL); (E) Left ventricular weight/tibial length ratio (LVW/TL); and (F) Left ventricular weight/body weight ratio (LVW/BW). n = 93 (initial + replication cohorts); * p < 0.05, one-way ANOVA followed by Holm-Sidak's multiple comparisons test. (G) Quantification of myocyte size area by isolectin/WGA staining and representative pictures (white bar scale = 20 μm). n = 59; *p < 0.05, one-way ANOVA followed by Dunett's multiple comparisons test.
Histomorphometric Analysis of Hearts
Morphometric and histologic measurements were obtained from hearts arrested in diastole in KCl solution, washed, subsequently fixed with 4% formaldehyde and paraffin embedded. To assess cardiac myocyte transverse area, tissue was costained with wheat germ agglutinin (WGA; for plasma membrane staining, rhodamine-conjugated) and isolectin B4 (GS-IB4; for endothelial staining, biotin-conjugated and revealed with fluorescein-conjugated streptavidin). Cell area from 150 to 200 cells per slide was determined using AxioVision software 4.8.2.0.
Immunostaining on Carotid Arteries and Aortas
Vessels (carotid arteries and aortas in totality) were carefully dissected, washed in cold PBS, and divided in several pieces for further experiments. One piece was subsequently fixed with 4% formaldehyde and paraffin embedded. To assess endothelial activation, carotid sections were stained with ICAM-1 primary antibody (R&D #AF796, 1/1000). On the aortic tissue, to evaluate inflammatory cells infiltration and proliferation, sections were stained with anti-CD45 primary antibody (BD Biosciences #55053P, 1/50) or rabbit anti-Ki67 primary antibody (CST #12202, 1/200), respectively. Smooth muscle actin and H3K27me3 stainings were performed using anti-αSMA primary antibody (CST #19245, 1/400) and anti-tri-methyl-histone H3 antibody (CST #9733, 1/200). Primary antibodies were revealed with EnVision-HRP systems (Agilent) and DAB (Agilent). Finally, nuclei were counterstained with H&E (Agilent). Slides were digitized with a slide scanner (SCN400 Leica) and blindly analyzed with QuPath software (University of Edinburgh) (25).
RNA-seq on Aortic Tissue
One-third of aorta was immediately frozen in liquid nitrogen in dry Eppendorf. Because of low yield of RNA extracted in preliminary experiments, and to ensure sufficient amount and quality of RNA for sequencing, tissues from two animals were pooled in 1 ml of tri-reagent (TR118, MRC) for subsequent homogenization with the use of a Precellys Evolution homogenizer (Bertin Instrument, France). Total RNA was extracted with the PureLink™ RNA Micro Scale Kit (Invitrogen) according to the manufacturer's instructions, including a DNase step. In total, RNA from 8 samples, corresponding to 16 mice, were quantified by Qubit RNA BR assay kit (Thermo Fisher Scientific, Q10211) on a Qubit 4 Fluorometer (Thermo Fisher Scientific). RNA integrity was evaluated on the Agilent 2100 Bioanalyzer using the RNA 6000 nanokit (Agilent, 5067-1511). All the samples had RNA integrity number values between 6.8 and 7.7.
Libraries were prepared starting from 150 ng of total RNA using the KAPA RNA HyperPrep Kit with RiboErase (HMR) (KAPA Biosystems, KK8560) following the manufacturer's recommendations (KR1351—version 1.16). Libraries were equimolarly pooled and sequenced on a single lane on an Illumina NovaSeq 6000 platform. All the libraries were paired end (2 × 100 bp reads) sequenced and a minimum of 35 million paired end reads were generated per sample.
All the sequencing data were analyzed using the Automated Reproducible MOdular workflow for preprocessing and differential analysis of RNA-seq data (ARMOR) pipeline v.1.2.0 (26). In this pipeline, reads underwent a quality check using FastQC v0.11.7 (27). Quantification and quality control results were summarized in a MultiQC report (28) before being mapped using Salmon (29) to the transcriptome index which was built using all Ensembl cDNA sequences obtained in the Mus_musculus.GRCm38.cdna.all.fa (release 101) file (30). Then, the estimated transcript abundances from Salmon were imported into R using the tximeta (1.7.14) package (31, 32) and analyzed for differential gene expression with edgeR (3.31.4) package, in which p-value was adjusted using Benjamini-Hochberg method (33). Accordingly, each experimental group (AngII, AngII+2W, and AngII+3W) was compared against the control group (CTRL), thereby producing 3 lists of differentially expressed genes. Differential gene expression results from edgeR were used to conduct Over-Representation Analysis (ORA) and Gene Set Enrichment Analysis (GSEA) with the WebGestaltR (v.0.4.3) package (34). These analyses were conducted on the Kyoto Encyclopedia of Genes and Genomes (KEGG) and Reactome database. RNA-seq full data are available in the NCBI Gene Expression Omnibus (GEO) database under the study accession code GSE175588.
RT-qPCR on Aortas in a Replication Cohort
Total RNA was extracted from one-third of the total aorta in a replication cohort of mice, with the same protocol and extraction procedure, i.e., freeze drying, further homogenization in Trizol with Precellys Evolution homogenizer, and extraction with PureLink™ RNA Micro Scale Kit. Extracted RNA was reverse-transcribed and analyzed by quantitative polymerase chain reaction (qPCR) with GAPDH as housekeeping gene. The primer sequences used for qPCR are presented in Table I in Supplemental Material.
Human Aortic Vascular Smooth Muscle Cells (HVSMCs) Culture and in-vitro AngII Memory Model
Human aortic vascular smooth muscle cells (HAVSMC) were purchased at ScienCell Research Laboratories. Cells were grown in full Smooth Muscle Cell medium (ScienCell #1101), in T75 flask coated with polylysine (2 μg/cm2); medium was renewed every 24 h as cells reached 80% confluence. After serum starvation (0.1% FBS for 18 h) cells were treated with AngII 1μM for 72 h (AngII), or 48 h and then 24 h in control, serum-deprived medium (MemAngII); and compared with the cells maintained 72 h in control, serum-deprived medium (CTRL). Cells between passages 4 and 8 were used for experiments. Total RNA was extracted with Maxwell Kit (Promega, #AS1340). Extracted RNA was reverse-transcribed and analyzed by quantitative polymerase chain reaction (qPCR) with GAPDH as housekeeping gene. The primer sequences used for qPCR are presented in Table I in Supplemental Material. For protein extraction, cells were scrapped in RIPA buffer containing proteinases and phosphates inhibitors. Denatured proteins (in Laemmli buffer) were separated by SDS-PAGE and transferred on PVDF membrane. Membranes were then blocked 1 h in 5% non-fat dry milk in TBS-Tween and incubated overnight at 4°C in 1% milk Tween-TBS with primary antibodies. Antibodies were αSMA (CST #19245, 1/10000) and HSP90 (BD Biosciences #610419, 1/2500). Membranes were visualized by enhanced chemiluminescence on CL-Xposure film (Thermo Fisher Scientific).
Statistics
Statistical tests were performed using GraphPadPrism (GraphPad Software Incorporation, San Diego, California, USA). Results are reported as mean and standard error of the mean. Statistical analysis was performed using parametric or non-parametric tests where appropriate after verifying normality of values distribution. P < 0.05 is considered as significant with * meaning P < 0.05.
Results
The experimental design is illustrated in Figure 1A. Blood pressure was recorded longitudinally in each mouse by telemetry at four time points: at baseline (CTRL); at the end of the 2-week treatment with Ang II (AngII); and 2 and 3 weeks after the end of the Ang II infusion (AngII+2W and AngII+3W, respectively); the last two composing the “memory” groups. As expected, we observed an increase in systolic and diastolic blood pressure during AngII infusion (Figures 1B,C). Importantly, blood pressure values reverted to normal levels after the end of the 2-week treatment, confirming treatment interruption and transient exposure to Ang II, as planned. We next evaluated specific parameters of cardiovascular remodeling classically affected by AngII and their eventual persistence at longer time points.
Cardiac Hypertrophy
As expected (4, 35), we observed a significant increase in total heart/tibial length (TH/TL), left ventricular weight/tibial length (LVW/TL), and left ventricular weight/body weight (LVW/BW) ratios in the AngII group (Figures 1D–F). This hypertrophic phenotype was sustained in time with ratios significantly increased in the AngII+2W and AngII+3W groups (Figures 1D–F). This was reflected by concordant increases in cardiac myocyte transverse area in the AngII group (Figure 1G), which remained significantly elevated in the AngII+2W and AngII+3W groups.
Vascular Remodeling
Similar analyses were performed on the vascular phenotype. Histological analysis of carotid sections in the AngII group revealed an increased endothelial expression of ICAM-1, reflective of endothelial activation. ICAM-1 expression remained elevated in the AngII+2W and AngII+3W group (Figure 2A). Consistently, CD45 labeling was increased in the aortic tissue of the AngII group, reflective of inflammation that was also sustained in time in the AngII+2W and AngII+3W groups (Figure 2B). In line with the known proliferative effect of AngII in vascular tissue (36, 37), Ki67 labeling of aortic tissue was increased in the AngII group and, again, maintained in the AngII+3W group (Figure 2C).
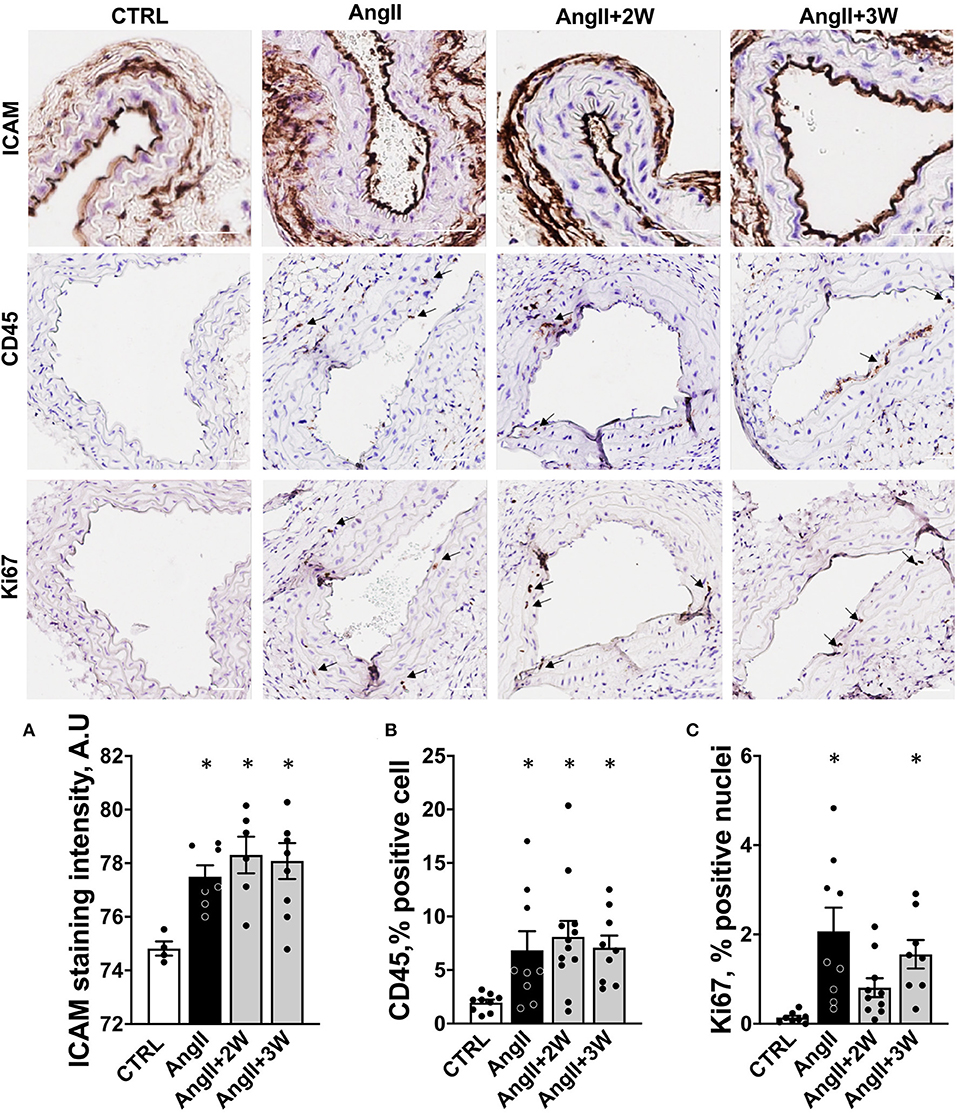
Figure 2. Memory effect on vascular endothelial activation, cell proliferation, and inflammation. (A) Quantification of endothelial staining for ICAM-1 in paraffin embedded sections of carotid arteries, expressed in arbitrary units of staining intensity, n = 26. (B) Quantification of staining for CD45 in paraffin embedded sections of aortas, expressed as % of stained cell. (C) Quantification of staining for Ki67 in paraffin-embedded sections of aortas, expressed as % of stained cells. *p < 0.05; n = 39 (B,C); One-way ANOVA followed by Dunett's multiple comparisons test. Arrows indicate specific staining for CD45 and Ki67, scale bar = 100 μm.
Oxidative Stress
AngII stimulates tissue nicotinamide adenine dinucleotide phosphate (NADPH) oxidases in cardiac and vascular cells to produce superoxide anions and, upon dismutation with extracellular SOD, the secondary oxidizing product H2O2 (38). Accordingly, plasma hydroperoxides were elevated in the AngII group, and also in the AngII+2W group (Supplementary Figure 1), reflecting persistent systemic oxidant stress at least up to 2 weeks after Ang II removal in our model.
Comparative Transcriptomic Profiling of AngII vs. AngII “Memory” Conditions
To gain further mechanistic insight into the sustained effect of AngII on this vascular phenotype, we used an unbiased approach through whole aortic tissue RNA-sequencing. We first assessed the differential expression of genes and underlying signaling pathways in the AngII group compared with control, untreated mice. Next, we performed a similar comparison between the AngII+2W and AngII+3W groups (i.e., “memory” conditions) vs. control, untreated mice. We also compared the resulting list of genes and pathways to identify transcripts similarly modulated in AngII and “memory” groups, assuming that these sustained up and/or downregulated transcripts may be related to the observed “memory” phenotype.
Volcano plots in Figures 3A–C represent differential expression of gene data sets comparing each condition to CTRL, considering FDR < 0.05 as a cutoff. As shown in Figures 3A,C, we found 808 genes significantly differentially expressed in AngII and 13 genes in AngII+3W memory condition. Unlike the AngII condition (Figure 3A) in which 55% of genes were upregulated, we found a vast majority of significantly underexpressed genes in AngII+3W memory condition (Figure 3C). As no significant differentially expressed gene was found with this FDR cutoff in the AngII+2W group (Figure 3B), we focused on the AngII+3W memory group compared with AngII for further analysis.
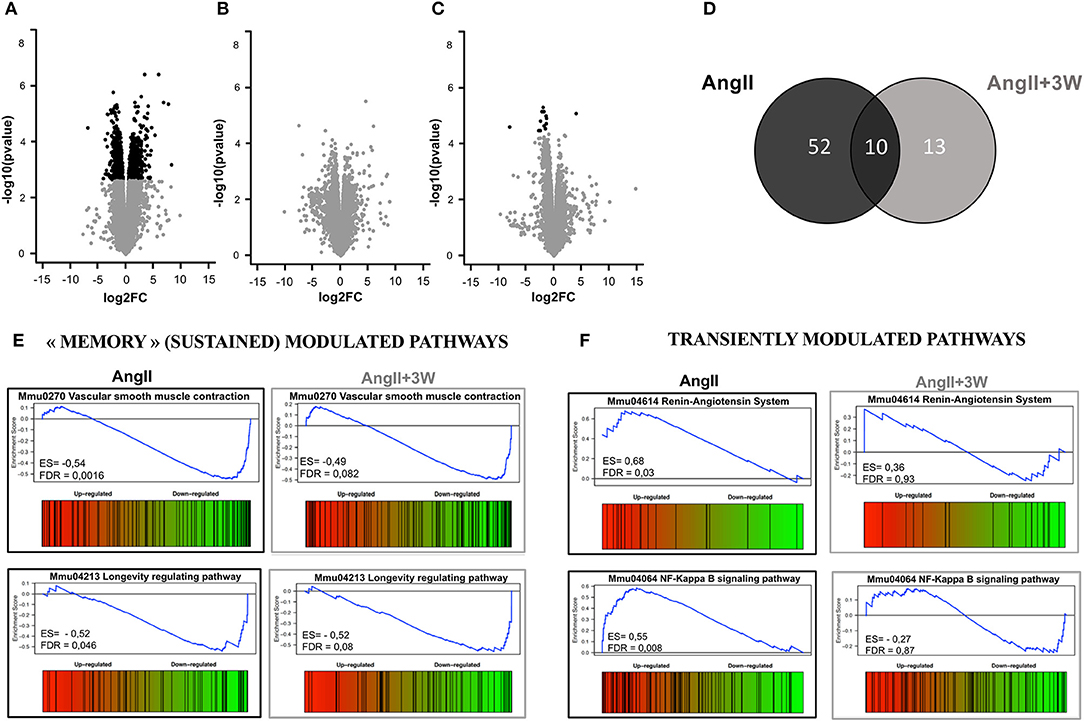
Figure 3. Transcriptomic profiling of aortic tissue from AngII memory model. Volcano plot representing the analysis of differential expression of genes in (A) AngII vs. CTRL; (B) AngII+2W vs. CTRL and (C) AngII+3W vs. CTRL data sets. Black points mark the genes with significantly (FDR < 0.05) increased or decreased expression compared to CTRL. The x-axis shows log2fold-changes in expression and the y-axis the –log10(p-value). (D) Gene set enrichment analysis (GSEA) from compiled KEGG pathways. Venn diagram showing the number of gene sets differentially modulated in each condition, and the overlap for 10 of them (FDR value < 0.1) (E) Enrichment score diagram illustrating the expression pattern of 2 of the 10 commonly regulated pathways (e.g., present in AngII and in AngII+3w, and modulated in the same direction) “vascular smooth muscle contraction” and “longevity regulating pathway”. (F) Enrichment score diagram illustrating the expression pattern of 2 transiently modulated pathways by Angiotensin II infusion: “Renin angiotensin System” and “NF KappaB signaling pathway”.
Gene set enrichment analysis of the 2 data sets is summarized in Figures 3D–F. A total of 62 KEGG pathways were significantly differentially modulated in AngII group, and 23 in AngII+3W group, using FDR < 0.1 as a cut-off. The Venn diagram in Figure 3D shows that 10 of them were commonly modulated pathways between the 2 groups. Among these 10 pathways, we next searched for those modulated in the same direction, e.g., up- or downregulated. Four of the 10 commonly modulated pathways were similarly downregulated, and identified as “dilated cardiomyopathy” (mmu05414); “hypertrophic cardiomyopathy” (mmu05410); “longevity regulating pathway” (mmu04213) and, notably, “vascular smooth muscle contraction” (mmu04270) (Figure 3E; Supplementary Figure 2). Conversely, other pathways known to be regulated by AngII, such as, “Renin angiotensin system” (mmu044614), “NF-KappaB signaling” (mmmu04064) and “cGMP signaling” (mmu04022) were, as expected, up (for the first two) or downregulated (for the latter) in the AngII group, but returned to normal level of enrichment in the AngII+3W “memory” group (Figure 3F; Supplementary Figure 2).
We next compared the gene lists to identify transcripts commonly regulated in both AngII and AngII+3W conditions. A total of 13 genes were commonly differentially expressed in both conditions, i.e., 13 genes similarly and significantly modulated both in the “memory” group vs. control, untreated mice and in the AngII group vs. control, untreated mice. Figure 4A represents a heat map with these 13 common transcripts, illustrating changes in the expression level in each condition (AngII and AngII+3W) compared with control, untreated condition (see also Supplementary Figure 3).
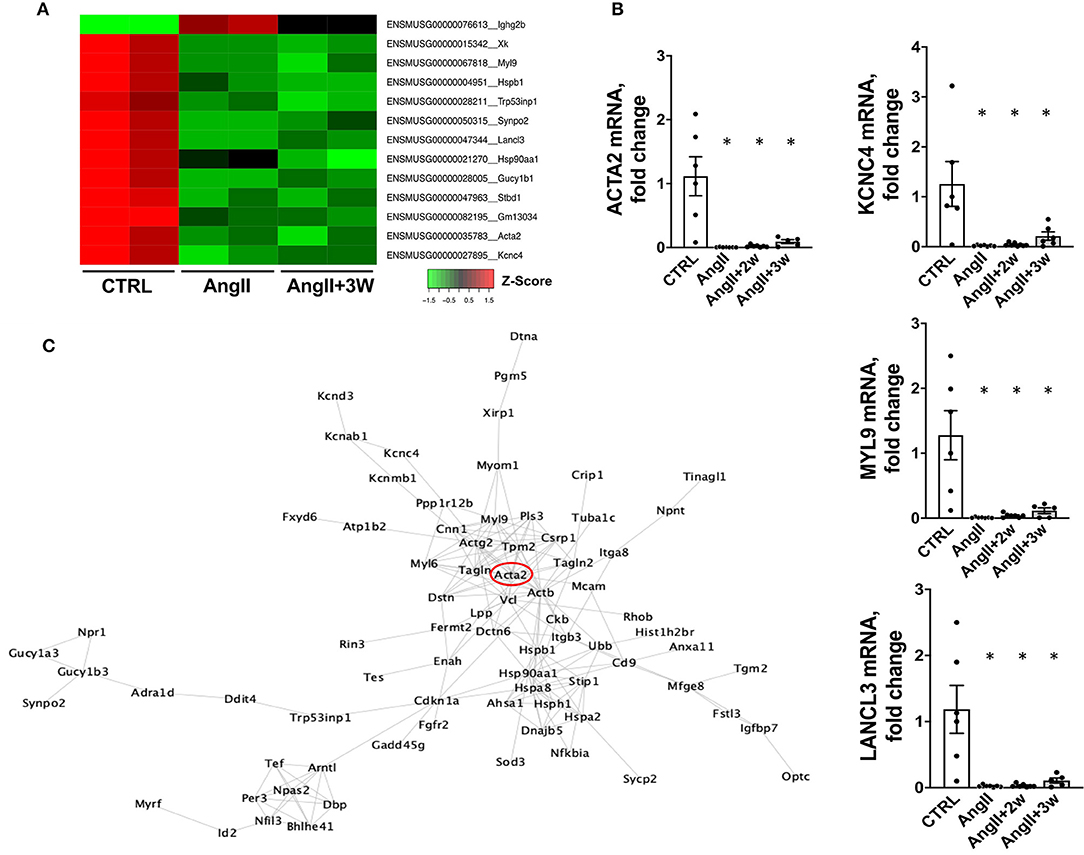
Figure 4. Common downregulation of specific genes in aortic tissue from AngII and “memory model” and corresponding interactome mapping. (A) HeatMap representing expression level (Z-score) of the 13 identified commonly modulated transcripts in AngII+3W and AngII vs. CTRL conditions. (B) RT qPCR on a replication group of mice confirming RNA seq expression data on selected genes: Lancl3, Kcnc4, Myl9, and Acta2, n = 25; *p < 0,05, one way ANOVA followed by Holm's-Sidak comparison test. (C) Protein–protein interaction network corresponding to differentially expressed genes in aorta in the “memory” condition (AngII+3w) vs. CTRL. To build a comprehensive picture, genes with FDR up to < 0,1 were included. Based on Cytoscape String Network database.
Among these, our attention was drawn to Acta2. Acta2 is a gene coding for alpha-smooth muscle actin (αSMA), a protein of the cytoskeleton mainly expressed in smooth muscle cells that is involved in vascular contractility and blood pressure homeostasis. Mutations in this gene cause a variety of vascular diseases, such as thoracic dilated aortic disease, coronary artery disease, stroke, and Moyamoya disease (39), and also multisytemic smooth muscle dysfunction syndrome (40). Downregulation of αSMA expression induced by AngII has also been described in vascular SMC in vitro (41, 42).
To better delineate the putative role of Acta2 in the context of our pathway enrichment analysis, we drew a virtual protein–protein interaction network using the Cytoscape software (fed from the String database). We only included the list of genes that were differentially expressed in AngII+3W memory condition (compared with CTRL), with enlarged FDR cut-off of < 0.1 for a more comprehensive picture (Figure 4C). Note that this interactome did not consider the direction of modulation (e.g., if a transcript was either up or downregulated, to the extent that protein abundance is modulated similarly). Interestingly, we observed that Acta2 was located at a central hub of this protein–protein interaction network.
Based on the earlier observations, we focused on Acta2 as a potential driver of our phenotype.
Sustained Downregulation of Acta2 in a Replication Cohort: Putative Role of Myocardin and Histone Methylation
We confirmed our RNAseq data in a replication cohort of identically treated mice, in which we found a downregulation of Acta2 mRNA expression by RT-qPCR in AngII, and both AngII+ 2W and AngII+3W groups, compared with CTRL (Figure 4B); among the 13 genes identified in the RNAseq data, downregulation of Myl9, Kcnc4, and Lancl3 mRNA expression were also confirmed (Figure 4B). αSMA protein levels were also evaluated through quantitative immunostaining on aortic tissue. We confirmed a significant decrease in αSMA protein expression in the AngII group, which was significantly maintained in both “memory” conditions, AngII+2W and AngII+3W group (Figure 5A). Remarkably, the cytoskeleton in smooth muscle cells of the arterial media in these two groups was profoundly altered, with important structural defects and disorganization (see representative pictures in Figure 5).
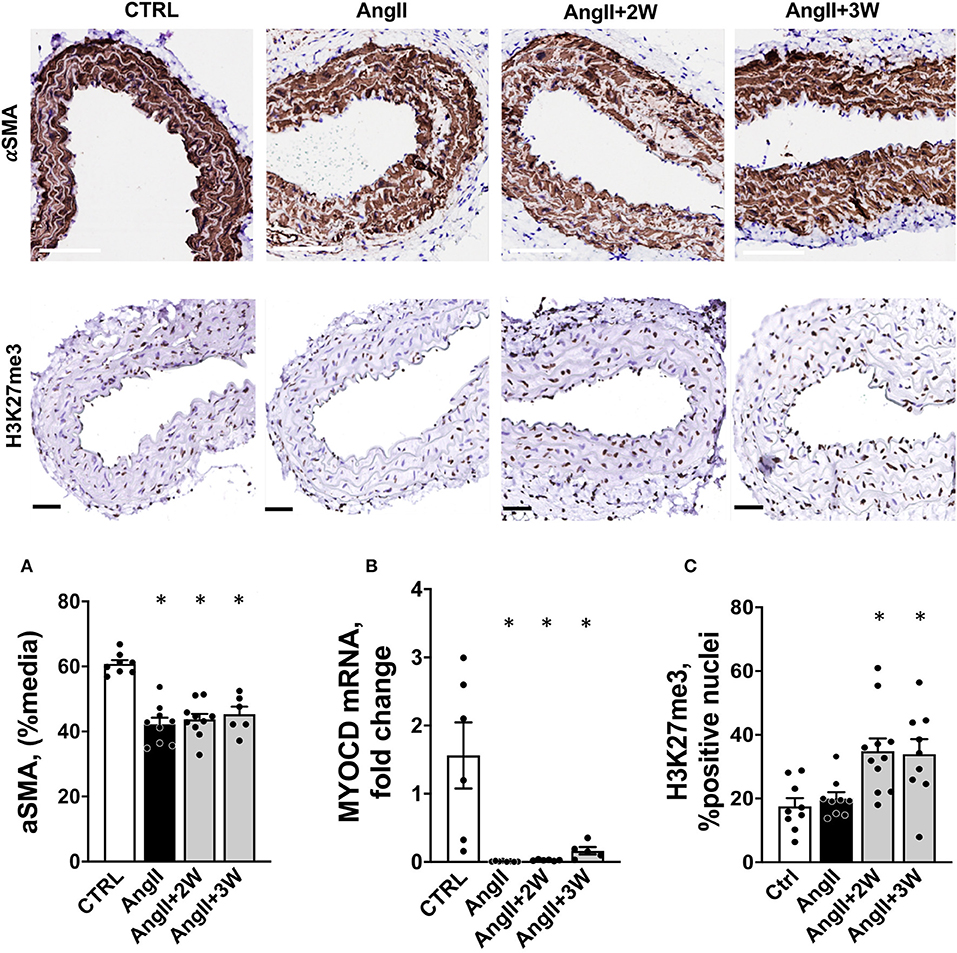
Figure 5. Coordinated regulation of Alpha-Smooth Muscle Actin, its transcription factor Myocd and histone trimethylation in aortic tissue from the Ang II “memory” model. (A) Sustained down regulation of αSMA protein in aortic tissue from a replication cohort by immunostaining (white bar = 100 μm). n = 33; * = p < 0.05, one way ANOVA followed by Dunett's multiple comparison test. (B) Myocd mRNA expression by RTqPCR in mouse aortas from a replication cohort, n = 26; *p < 0.05, one way ANOVA followed by Holm's-Sidak comparison test. (C) H3K27me3 staining and quantification in paraffin-embedded sections of aortas, expressed as % of positive nuclei (black bar = 50 μm), n = 39; *p < 0.05, one way ANOVA followed by Dunett's multiple comparison test.
Among factors controling αSMA expression are transcription factors Myocardin (Myocd) and Serum Response Factor (SRF). Altogether they form a complex which binds CArG (CCA/T(rich)GG) sequence motif upstream Acta2 (and other contractile genes), SRF serving as doking platform for Myocd activity, leading to active contractile transcription machinery. In our RNAseq data, we observed that Myocd and SRF transcripts were significantly downregulated under AngII (−2,07 log2FC, FDR = 0,02), with the same trend in the memory condition for Myocd (−1,42 log2FC, FDR= 0,13). We decided to evaluate Myocd expression by RT qPCR in our replication group of mice. We observed that this transcription factor was robustly repressed under AngII infusion and that this repression was sustained in time despite the end of the pharmacological stimulation (Figure 5B).
To gain further understanding of the sustained downregulation of Acta2 and Myocd, we examined putative epigenetic regulatory mechanisms. Indeed, our pathway analysis using ORA or GSEA, identified significant enrichement of several epigenetic pathways under AngII such as “HDAC's deacetylase histone” (R-MMU-3214815), “HATs acetylate histone” (R-MMU-3214847), and also “PRC2 methylates histone and DNA”(R-MMU-21230) (see Table I in Supplemental Material, Reactome T1 ORA, Reactome T1 GSEA). Interstingly, PRC2 is a protein complex that keeps transcriptionally silent genes in a repressed state by trimethylating histone H3 on lysine 27. We then evaluated the status of this epigenetic mark in our model, using immunostaining on aortic sections. Notably, this revealed a significant increase in H3K27me3, only in the 2 memory conditions, AngII+2W and AngII+3W (Figure 5C).
In vitro AngII Memory Model in Human Aortic Vascular Smooth Muscle Cells
To verify these observations in a homotypic cell system of human origin, we developed an in vitro model of AngII memory on cultured human aortic VSMCs. Cells were exposed to continuous AngII at 1μM for 72 h (AngII); or to the same serum-deprived control medium for 72 h (CTRL), or 48 h of AngII, followed by 24 h of control medium (MemAngII). First, we observed the same downregulation of ACTA2 under AngII stimulation, as depicted by mRNA expression and protein levels (Figures 6A,C). Notably, this downregulation was also maintained in the “memory” condition. This was paralleled with a downregulation of mRNA expression of the transcription factor MYOCD, upon continuous AngII, as well as in the “memory” condition (Figure 6B).
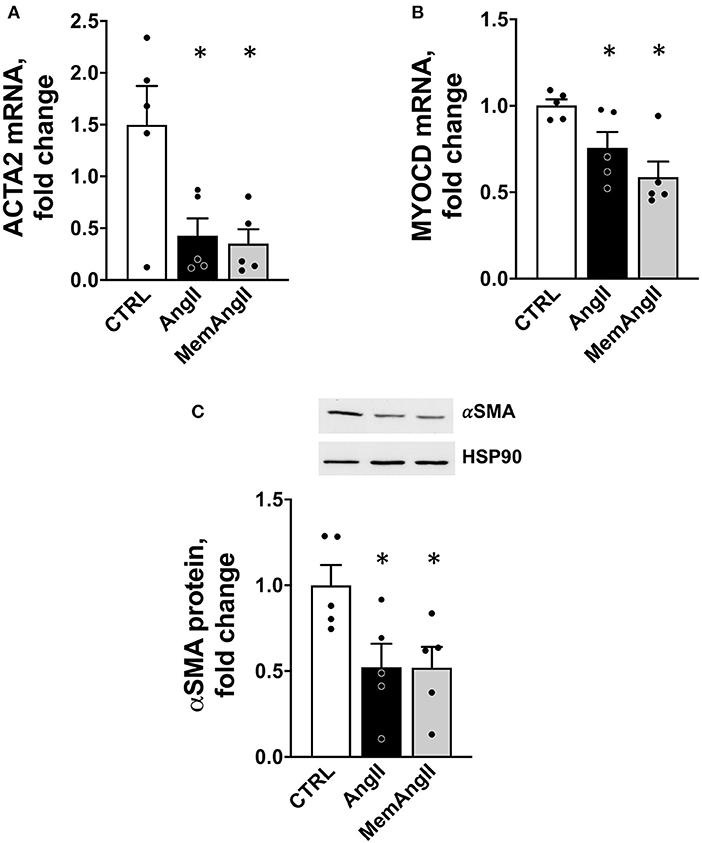
Figure 6. Downregulation of ACTA2 and MYOCD in a homotypic cell culture model of human vascular smooth muscle cells reproducing the AngII “memory” effect in vitro. (A) ACTA2 mRNA expression (by RT qPCR) in HAVSMC exposed continuously (AngII) or transiently (MemAngII) to AngII compared with the control cells maintained in serum-deprived control media. (B) MYOCD mRNA expression in the same in vitro model (C) aSMA protein level in the same model, N = 5 independent experiments, *p < 0.05 one way ANOVA followed by Sidak's multiple comparisons test.
Discussion
The main findings of this study can be summarized as follows: (i) an initial 2-week exposure to AngII induces profound changes in cardiac and vascular remodeling, including endothelial activation, vascular inflammation and oxidant stress, all of which are maintained up to 3 weeks after AngII withdrawal; notably, this phenotype is sustained despite early normalization of blood pressure after AngII withdrawal, a proxy to a “legacy” or “memory” effect in this mouse model; (ii) comparison of the transcriptomic profiles at the end of the 2-week Ang II treatment (AngII group) or 3 weeks after Ang II withdrawal (AngII+3 weeks group) identified 13 commonly regulated transcripts (1 up and 12 down), and a set of 4 commonly modulated pathways by GSEA, some of which point to altered structural or contractile properties of the arterial wall. Conversely, many other transcripts classically associated with AngII effects and (mostly) upregulated in the AngII group, are not persistently regulated in the AngII+3 weeks group; this highlights the above 13 genes, corresponding to the sustained downregulated transcripts, as potential targets for a “memory” effect; (iii) among these, Acta2 is a likely candidate, as first confirmed in a replication cohort, including at the protein level, but also from our observation of striking downregulation of Myocardin (Myocd), its transcriptional coregulator and from changes in histone methylation, as corresponding epigenetic repressive marks, in aortas of both the memory groups, AngII+2W and AngII+3W groups.
Ours is one of the few studies examining the effect of AngII on the full transcriptomic profile of mouse aortic or arterial tissues in “wild-type” (C57Bl6/J) mice. In a microarray study on ApoE-/- mice-treated with AngII, Rush et al. identified genes overexpressed in mice that did not develop aneurysms under pharmacological Ang II stimulation, i.e., protective against abdominal aortic aneurysms (20). Consistent with their results, a majority of those transcripts were significantly downregulated in our AngII group of mice, such as Sost, Dstn, or Hspaa1a (see Supplementary Data). Our profiles are also in line with results by Spin et al. identifying genes decreased in prematurely ruptured anevrysm in AngII-treated ApoE-/- mice (e.g., Bmp6, Ltbp1, Rock1) (43). Another transcriptomic analysis of 3 differential arterial beds of C57Bl6 mice treated with AngII identified Sphk1 as commonly modulated transcript in the 3 types of tissue. This transcript is also upregulated under AngII in our RNAseq data, albeit not significant with an FDR cutoff of 0.05. Other transcripts such as Thy1 or Htatip2, described to be overexpressed in thoracic aorta, are similarly regulated in our AngII group. Notably, as we did, the same study identified a downregulation of Gucy1b3 transcripts (corresponding to the beta subunit of soluble guanylyl cyclase) in thoracic and abdominal aortic tissue, expected to be associated with a reduced vascular relaxation (21). Finally, in a recent study in C57Bl6 mice treated for a longer time with lower dose of AngII, Lv et al. identified 773 genes differentially expressed in aortas of hypertensive mice (22). As in this study, under AngII, mostly were upregulated genes (22). Among identified transcripts, we confirmed the overexpression of IGF1 in our RNAseq data in AngII-infused mice.
Even fewer studies have attempted to develop an AngII “memory” model (16, 17, 44) and in none of them was a transcriptomic profiling performed. In concordance with the only 2 studies in mice found in the literature, we observed sustained macroscopic and microscopic cardiac hypertrophy (16); ICAM-1 endothelial expression, inflammation in the vascular wall (e.g., CD45-positive cell infiltration), and oxidative stress (17) that persisted after AngII withdrawal. A major difference in the aforementionned study compared with ours is that blood pressure remained elevated 1 week after the end of AngII infusion, the only “remote” time-point examined (17). This could simply be explained by the different AngII dosage and timing for the “memory” condition (1 week vs. 2 weeks after the end of AngII infusion). Although the distinction between differential delays for phenotype reversal (e.g., slower for tissue remodeling, more rapid for blood pressure) vs. “memory” effect could be argued, the present model shows the persistence of remote effects (up to 3 weeks) despite clear evidence of termination of the initial stimulus (normalization of blood pressure). At the very least, and contrary to the previous study, it excludes that the long-lasting effects on remodeling result from sustained high blood pressure.
Another strengh of this study is the use of an unbiased approach to understand our phenotype through transcriptomic profiling. While differential transcript regulation did not reach the more stringent significance level in the AngII+2W group probably because of insufficient statistical power, the genes differentially expressed in the AngII+3W group at FDR < 0.05 were similary regulated in AngII+2W (Supplementary Data), suggesting a continuum in their regulation.
AngII, among other factors, promotes a switch in vascular smooth muscle cells from a contractile to “secretrory” phenotype (45). Through AT1R, AngII stimulation produces changes in the contractile machinery, constisting in decreased expression of contractiles markers such as αSMA (ACTA2), SM22a (TAGLN), and SMMHC (MYH11) (9, 46). As others (41, 47), in parallel with vascular remodeling, we observed significant changes in contractile markers expression in aortas under AngII infusion, with downregulation of “vascular smooth muscle contraction” pathway, and ACTA2 expression, but also TAGLN and MYH11 (see raw data). In addition, this phenotypic switch was maintained in time despite the end of AngII stimulation, for the “vascular smooth muscle contraction pathway”, and also for αSMA expression, both at mRNA and protein levels (Figures 4B, 5A). Among factors controling VSMC phenotypic plasticity are transcription factors Myocardin (Myocd) and Serum Response Factor (SRF). Altogether they form a complex which binds CArG (CCA/T(rich)GG) sequence motif upstream contractile genes, with SRF serving as doking platform for Myocd activity, leading to active contractile transcription machinery. Conversely, Krupple-like Factor 4 (KLF4) and ETS domain containign protein-1 (ELK1) binding to G/C repressor element, inhibit Myocd/SRF complex, leading to decressed expression of SMC differentiation markers (46). In our RNAseq data, we observed that Myocd and SRF transcripts were significantly downregulated under AngII (- 2,07 log2FC, FDR = 0,02), with the same trend in the AngII+3W memory condition for (-1,42 log2FC, FDR = 0,13). This was not the case for KLF4 and ELK1 (see raw data). We confirmed this downregulation of Myocd transcripts by RT-qPCR, including in the 2 memory conditions (AngII+2W and AngII+3W group) (Figure 5B). Interestingly, we could reproduce same results in HAVSMC in culture, in which we observed a sustained down expression of ACTA2 and protein αSMA induced by AngII, associated with reduced expression of transcription factor Myocd; and despite removal of the pharmacologic stimulus. This makes MYOCD a likely candidate for upstream, sustained downregulation of ACTA2 expression in AngII “memory” context.
Epigenetic regulation may also account for the sustained alteration of the vascular phenotype in the “memory” groups in vivo. Indeed, Histones H3 and H4 associated with CArG-containing regulatory elements of SM-MHCs (MYH11), SM22a (TAGLN), and αSMA (ACTA2) have been shown to be acetylated (a characteristic of chromatin accessibility) in contractile SMCs, facilitating SRF binding to the CArG box (48). Increased histone acetyltransferase (HAT) activity stimulates SM22a expression whereas increased histone deacetylases (HDACs) prevent SM22a expression (49). Consistently, our pathway analysis using ORA or GSEA, identified significant enrichement of several epigenetic pathways under AngII (such as “HDACs deacetylase histone” (R-MMU-3214815), “HATs acetylate histone” (R-MMU-3214847).
Other modalities of epigenetic regulation may also be at play. H3 histone dimethylation in lysine 4 (H3K4me2) is a marker of differentiated SMCs and is maintained even if SMCs undergo phenotypic modulation (50). DNA demethylation by TET2 increases DNA accessibility to transcription factors resulting in increased SMC differentiation marker expression (51). Of interest, the ACTA2 promoter was shown to be hypermethylated (a usual mark of repressed expression) in genome-wide methylation studies in human atherosclerotic aortas (52, 53). As we observed sustained downregulation of Acta2 transcripts in our “memory” condition, an AngII-induced methylation leading to repressive imprinting and phenotypic switch might well be involved. Again, our RNAseq pathway analysis suggests the involvement of epigenetic regulators involved in methylation, such as “PRC2 methylates histone and DNA”(R-MMU-21230) (see Supplementary Data, Reactome T1 ORA, Reactome T1 GSEA). In particular, PCR2 complex catalyzes trimethylation of histone H3 on lysine 27 (H3K27me3), a histone mark necessary for maintaining transcriptional repression during multicellular development. Cell type-specific patterns of H3K27me3 are crucial for preserving cell identity (54). Consistent with this analysis, we observed a significant increase in H3K27me3 epigenetic mark in the aortic tissue and, intriguingly, only in both memory conditions (Figure 5C). However, the signaling elements involved in this response to AngII remain to be studied in more details.
Conclusion
Altogether, our observations support a “memory” effect sustained beyond AngII-induced hypertension and leading to downregulation of specific gene expression, such as Acta2, and vascular injury. Future characterization of the underlying AngII-dependent signaling might unveil new targets for its therapeutic modulation and reversal of this adverse legacy effect.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE175588.
Ethics Statement
The animal study was reviewed and approved by Institutional Animal Care and Research Advisory Committee of the Université Catholique de Louvain.
Author Contributions
LP, CD, and J-LB designed project and experiments. LP wrote manuscript and designed figures under J-LB supervision. J-LB reviewed and corrected, and other authors reviewed and commented. CB performed immunohistochemistry experiments and analysis. LP, BB, and JA performed RNAseq anaylsis (LP: RNA extraction and interactome, BB: pre-analytic and libraries, JA: bioinformatic analysis). LP, RV, DD, HE, CF, and LM performed in in vivo and in vitro experiments. HE especially for telemetries. All authors contributed to the article and approved the submitted version.
Funding
This study was funded by Belgian Fond National de la Recherche Scientifique (FNRS, CDR J.309.21) and Fondation Saint Luc (Grant Pierre de Merre).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank M De Beukelaer for its precious assistance throughout immunohistochemistry experiments.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2022.854361/full#supplementary-material
References
1. Lawes CM, Vander Hoorn S, Rodgers A. International society of, global burden of blood-pressure-related disease, 2001. Lancet. (2008) 371:1513–8. doi: 10.1016/S0140-6736(08)60655-8
2. Guyenet PG. The sympathetic control of blood pressure. Nat Rev Neurosci. (2006) 7:335–46. doi: 10.1038/nrn1902
3. Takimoto-Ohnishi E, Murakami K. Renin-angiotensin system research: from molecules to the whole body. J Physiol Sci. (2019) 69:581–7. doi: 10.1007/s12576-019-00679-4
4. Forrester SJ, Booz GW, Sigmund CD, Coffman TM, Kawai T, Rizzo V, et al. Angiotensin II signal transduction: an update on mechanisms of physiology and pathophysiology. Physiol Rev. (2018) 98:1627–738. doi: 10.1152/physrev.00038.2017
5. Karnik SS, Unal H, Kemp JR, Tirupula KC, Eguchi S, Vanderheyden PM, et al. International union of basic and clinical pharmacology. XCIX. angiotensin receptors: interpreters of pathophysiological angiotensinergic stimuli [corrected]. Pharmacol Rev. (2015) 67:754–819. doi: 10.1124/pr.114.010454
6. Carey RM, Siragy HM. Newly recognized components of the renin-angiotensin system: potential roles in cardiovascular and renal regulation. Endocr Rev. (2003) 24:261–71. doi: 10.1210/er.2003-0001
7. Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. (2004) 84:767–801. doi: 10.1152/physrev.00041.2003
8. Mulvihill ER, Jaeger J, Sengupta R, Ruzzo WL, Reimer C, Lukito S, et al. Atherosclerotic plaque smooth muscle cells have a distinct phenotype. Arterioscler Thromb Vasc Biol. (2004) 24:1283–9. doi: 10.1161/01.ATV.0000132401.12275.0c
9. Montezano AC A., Nguyen Dinh Cat, Rios FJ, Touyz RM. Angiotensin II and vascular injury. Curr Hypertens Rep. (2014) 16:431. doi: 10.1007/s11906-014-0431-2
10. Ihnat MA, Thorpe JE, Ceriello A. Hypothesis: the 'metabolic memory', the new challenge of diabetes. Diabet Med. (2007) 24:582–6. doi: 10.1111/j.1464-5491.2007.02138.x
11. Paneni F, Volpe M, Luscher TF, Cosentino F. SIRT1, p66(Shc), and Set7/9 in vascular hyperglycemic memory: bringing all the strands together. Diabetes. (2013) 62:1800–7. doi: 10.2337/db12-1648
12. Villeneuve LM, Reddy MA, Natarajan R. Epigenetics: deciphering its role in diabetes and its chronic complications. Clin Exp Pharmacol Physiol. (2011) 38:451–9. doi: 10.1111/j.1440-1681.2011.05497.x
13. Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med (2008) 359:1577–89. doi: 10.1056/NEJMoa0806470
14. Diabetes C I. Complications trial /epidemiology of diabetes, and G. complications study research, intensive diabetes treatment and cardiovascular outcomes in type 1 diabetes: the dcct/edic study 30-year follow-up. Diabetes Care. (2016) 39:686–93. doi: 10.2337/dc15-1990
15. Dahlof B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, de Faire U, et al. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet. (2002) 359:995–1003. doi: 10.1016/S0140-6736(02)08089-3
16. Wang HX, Yang H, Han QY, Li N, Jiang X, Tian C, et al. NADPH oxidases mediate a cellular “memory” of angiotensin II stress in hypertensive cardiac hypertrophy. Free Radic Biol Med. (2013) 65:897–907. doi: 10.1016/j.freeradbiomed.2013.08.179
17. Li WJ, Liu Y, Wang JJ, Zhang YL, Lai S, Xia YL, et al. “Angiotensin II memory” contributes to the development of hypertension and vascular injury via activation of NADPH oxidase. Life Sci. (2016) 149:18–24. doi: 10.1016/j.lfs.2016.02.037
18. Makhanova NA, Crowley SD, Griffiths RC, Coffman TM. Gene expression profiles linked to AT1 angiotensin receptors in the kidney. Physiol Genomics 42A. (2010) 211–8. doi: 10.1152/physiolgenomics.00063.2010
19. Tsushima K, Osawa T, Yanai H, Nakajima A, Takaoka A, Manabe I, et al. IRF3 regulates cardiac fibrosis but not hypertrophy in mice during angiotensin II-induced hypertension. FASEB J. (2011) 25:1531–43. doi: 10.1096/fj.10-174615
20. Rush C, Nyara M, Moxon JV, Trollope A, Cullen B, Golledge J. Whole genome expression analysis within the angiotensin II-apolipoprotein E deficient mouse model of abdominal aortic aneurysm. BMC Genomics. (2009) 10:298. doi: 10.1186/1471-2164-10-298
21. Siedlinski M, Nosalski R, Szczepaniak P, Ludwig-Galezowska AH, Mikolajczyk T, Filip M, et al. Vascular transcriptome profiling identifies Sphingosine kinase 1 as a modulator of angiotensin II-induced vascular dysfunction. Sci Rep. (2017) 7:44131. doi: 10.1038/srep44131
22. Lv SJ, Ding YN, Pei XY, Zhao X, Hao L, Zhang ZQ, et al. Vascular transcriptome profiling reveals aging-related genes in angiotensin -induced hypertensive mouse aortas. Chin Med Sci J. (2020) 35:43–53.
23. Tan N, Zhang Y, Zhang Y, Li L, Zong Y, Han W, et al. Berberine ameliorates vascular dysfunction by a global modulation of lncRNA and mRNA expression profiles in hypertensive mouse aortae. PLoS ONE. (2021) 16:e0247621. doi: 10.1371/journal.pone.0247621
24. Desjardins F, Lobysheva I, Pelat M, Gallez B, Feron O, Dessy C, et al. Control of blood pressure variability in caveolin-1-deficient mice: role of nitric oxide identified in vivo through spectral analysis. Cardiovasc Res. (2008) 79:527–36. doi: 10.1093/cvr/cvn080
25. Bankhead P, Loughrey MB, Fernandez JA, Dombrowski Y, McArt DG, Dunne PD, et al. QuPath: Open source software for digital pathology image analysis. Sci Rep. (2017) 7:16878. doi: 10.1038/s41598-017-17204-5
26. Orjuela S, Huang R, Hembach KM, Robinson MD, Soneson C. ARMOR: An automated reproducible modular workflow for preprocessing and differential analysis of RNA-seq Data. G3 (Bethesda). (2019) 9:2089–2096. doi: 10.1534/g3.119.400185
28. Ewels P, Magnusson M, Lundin S, Kaller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. (2016) 32:3047–8. doi: 10.1093/bioinformatics/btw354
29. Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat Methods. (2017) 14:417–19. doi: 10.1038/nmeth.4197
30. Cunningham F, Achuthan P, Akanni W, Allen J, Amode MR, Armean IM, et al. Ensembl 2019. Nucleic Acids Res. (2019) 47:D745–51. doi: 10.1093/nar/gky1113
31. Soneson C, Love MI, Robinson MD. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. (2015) 4:1521. doi: 10.12688/f1000research.7563.1
32. Love M, Patro R, Hickey P, Soneson C. TranscriptQuantification import with automatic metadata. R package version1.1.16. tximeta. (2019) doi: 10.1371/journal.pcbi.1007664
33. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. (2010) 26:139–40. doi: 10.1093/bioinformatics/btp616
34. Liao Y, Wang J, Jaehnig EJ, Shi Z, Zhang B. WebGestalt 2019: gene set analysis toolkit with revamped UIs and APIs. Nucleic Acids Res. (2019) 47:W199–W205. doi: 10.1093/nar/gkz401
35. Ferrario CM, Cardiac remodelling and RAS inhibition. Ther Adv Cardiovasc Dis (2016) 10:162–71. doi: 10.1177/1753944716642677
36. Ozasa Y, Akazawa H, Qin Y, Tateno K, Ito K, Kudo-Sakamoto Y, et al. Notch activation mediates angiotensin II-induced vascular remodeling by promoting the proliferation and migration of vascular smooth muscle cells. Hypertens Res. (2013) 36:859–65. doi: 10.1038/hr.2013.52
37. Moraes JA, Frony AC, Dias AM, Renovato-Martins M, Rodrigues G, Marcinkiewicz C, et al. Alpha1beta1 and integrin-linked kinase interact and modulate angiotensin II effects in vascular smooth muscle cells. Atherosclerosis. (2015) 243:477–85. doi: 10.1016/j.atherosclerosis.2015.09.026
38. Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. (2007) 292:C82–97. doi: 10.1152/ajpcell.00287.2006
39. Guo DC, Papke CL, Tran-Fadulu V, Regalado ES, Avidan N, Johnson RJ, et al. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, Moyamoya disease. along with thoracic aortic disease. Am J Hum Genet. (2009) 84:617–27. doi: 10.1016/j.ajhg.2009.04.007
40. Milewicz DM, Ostergaard JR, Ala-Kokko LM, Khan N, Grange DK, Mendoza-Londono R, et al. De novo ACTA2 mutation causes a novel syndrome of multisystemic smooth muscle dysfunction. Am J Med Genet A. (2010) 152:2437–43. doi: 10.1002/ajmg.a.33657
41. Chen S, Chen H, Zhong Y, Ge Y, Li C, Qiao Z, et al. Insulin-like growth factor-binding protein 3 inhibits angiotensin II-induced aortic smooth muscle cell phenotypic switch and matrix metalloproteinase expression. Exp Physiol. (2020) doi: 10.1113/EP088927
42. He X, Deng J, Yu XJ, Yang S, Yang Y, Zang WJ. Activation of m3achr (type 3 muscarinic acetylcholine receptor) and nrf2 (nuclear factor erythroid 2-related factor 2) signaling by choline alleviates vascular smooth muscle cell phenotypic switching and vascular remodeling. Arterioscler Thromb Vasc Biol. (2020) 40:2649–64. doi: 10.1161/ATVBAHA.120.315146
43. Spin JM, Hsu M, Azuma J, Tedesco MM, Deng A, Dyer JS, et al. Transcriptional profiling and network analysis of the murine angiotensin II-induced abdominal aortic aneurysm. Physiol Genomics. (2011) 43:993–1003. doi: 10.1152/physiolgenomics.00044.2011
44. Togashi N, Maeda T, Yoshida H, Koyama M, Tanaka M, Furuhashi M, et al. Angiotensin II receptor activation in youth triggers persistent insulin resistance and hypertension–a legacy effect? Hypertens Res. (2012) 35:334–40. doi: 10.1038/hr.2011.206
45. Liu M, Gomez D. Smooth Muscle Cell Phenotypic Diversity. Arterioscler Thromb Vasc Biol. (2019) 39:1715–23. doi: 10.1161/ATVBAHA.119.312131
46. Allahverdian S, Chaabane C, Boukais K, Francis GA, Bochaton-Piallat ML. Smooth muscle cell fate and plasticity in atherosclerosis. Cardiovasc Res. (2018) 114:540–50. doi: 10.1093/cvr/cvy022
47. Lu W, Zhou Y, Zeng S, Zhong L, Zhou S, Song H, et al. Loss of FoxO3a prevents aortic aneurysm formation through maintenance of VSMC homeostasis. Cell Death Dis. (2021) 12:378. doi: 10.1038/s41419-021-03659-y
48. McDonald OG, Wamhoff BR, Hoofnagle MH, Owens GK. Control of SRF binding to CArG box chromatin regulates smooth muscle gene expression in vivo. J Clin Invest. (2006) 116:36–48. doi: 10.1172/JCI26505
49. Gomez D, Swiatlowska P, Owens GK. Epigenetic control of smooth muscle cell identity and lineage memory. Arterioscler Thromb Vasc Biol. (2015) 35:2508–16. doi: 10.1161/ATVBAHA.115.305044
50. Alexander MR, Owens GK. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu Rev Physiol. (2012) 74:13–40. doi: 10.1146/annurev-physiol-012110-142315
51. Liu R, Jin Y, Tang WH, Qin L, Zhang X, Tellides G, et al. Ten-eleven translocation-2 (TET2) is a master regulator of smooth muscle cell plasticity. Circulation. (2013) 128:2047–57. doi: 10.1161/CIRCULATIONAHA.113.002887
52. Lacey M, Baribault C, Ehrlich KC, Ehrlich M. Atherosclerosis-associated differentially methylated regions can reflect the disease phenotype and are often at enhancers. Atherosclerosis. (2019) 280:183–91. doi: 10.1016/j.atherosclerosis.2018.11.031
53. Lacey M, Baribault C, Ehrlich KC, Ehrlich M. Data showing atherosclerosis-associated differentially methylated regions are often at enhancers. Data Brief. (2019) 23:103812. doi: 10.1016/j.dib.2019.103812
Keywords: angiotensin II, aortic tissue, VSMC, memory, ACTA2, smooth muscle actin (SMA)
Citation: Pothen L, Verdoy R, De Mulder D, Esfahani H, Farah C, Michel LYM, Dei Zotti F, Bearzatto B, Ambroise J, Bouzin C, Dessy C and Balligand J-L (2022) Sustained Downregulation of Vascular Smooth Muscle Acta2 After Transient Angiotensin II Infusion: A New Model of “Vascular Memory”. Front. Cardiovasc. Med. 9:854361. doi: 10.3389/fcvm.2022.854361
Received: 13 January 2022; Accepted: 14 February 2022;
Published: 14 March 2022.
Edited by:
Marco Mongillo, University of Padua, ItalyReviewed by:
Michele Ciccarelli, University of Salerno, ItalyTlili Barhoumi, King Abdullah International Medical Research Center (KAIMRC), Saudi Arabia
Copyright © 2022 Pothen, Verdoy, De Mulder, Esfahani, Farah, Michel, Dei Zotti, Bearzatto, Ambroise, Bouzin, Dessy and Balligand. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jean-Luc Balligand, amVhbi1sdWMuYmFsbGlnYW5kQHVjbG91dmFpbi5iZQ==