 Martina B. Lorey
Martina B. Lorey Katariina Öörni
Katariina Öörni Petri T. Kovanen
Petri T. Kovanen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med., 03 March 2022
Sec. Lipids in Cardiovascular Disease
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.841545
This article is part of the Research TopicChronic Inflammation, Oxidative Stress and Lipoprotein Metabolism in Cardio-Pulmonary ContinuumView all 7 articles
Circulating apolipoprotein B-containing lipoproteins, notably the low-density lipoproteins, enter the inner layer of the arterial wall, the intima, where a fraction of them is retained and modified by proteases, lipases, and oxidizing agents and enzymes. The modified lipoproteins and various modification products, such as fatty acids, ceramides, lysophospholipids, and oxidized lipids induce inflammatory reactions in the macrophages and the covering endothelial cells, initiating an increased leukocyte diapedesis. Lipolysis of the lipoproteins also induces the formation of cholesterol crystals with strong proinflammatory properties. Modified and aggregated lipoproteins, cholesterol crystals, and lipoproteins isolated from human atherosclerotic lesions, all can activate macrophages and thereby induce the secretion of proinflammatory cytokines, chemokines, and enzymes. The extent of lipoprotein retention, modification, and aggregation have been shown to depend largely on differences in the composition of the circulating lipoprotein particles. These properties can be modified by pharmacological means, and thereby provide opportunities for clinical interventions regarding the prevention and treatment of atherosclerotic vascular diseases.
Atherosclerosis develops in the inner layer of the arterial wall, the intima. Atherosclerosis-prone locations, such as bifurcations of the coronary arteries, are characterized by a thick extracellular matrix containing collagen, elastin, and proteoglycans, and the presence of increased numbers of lipid-laden macrophages since childhood (1–3). Apolipoprotein B (apoB)-containing lipoproteins, i.e., LDL and remnant lipoproteins, such as small VLDL, IDL, and chylomicron remnants, cross the endothelial cell layer from the circulation into the intima (4), where they can become entrapped by the dense extracellular matrix (5–7).
A fraction of the lipoproteins can be modified in the intima by oxidizing agents and enzymes, proteases, and lipases secreted by local cells (5, 6). Such modifications of the lipoprotein structure have been shown to induce aggregation and fusion of lipoprotein particles (8, 9). Atherosclerotic lesions contain lipoproteins that show signs of extensive oxidation, proteolysis, and lipolysis of the various lipid molecules and that are often aggregated [recently reviewed in (10)]. The modified lipoproteins can be taken up by macrophages, which are converted into foam cells. The initial lesions of atherosclerosis, the fatty streaks, are characterized by the presence of numerous foam cells and can appear at any age, independent of risk factors for atherosclerotic cardiovascular disease (11). Moreover, in contrast to more advanced lesions, the fatty streaks are potentially fully reversible (12).
During atherogenesis, macrophages, dendritic cells, mast cells, and T lymphocytes are recruited into the intima in increasing numbers (13, 14). In the developing lesions, the macrophage foam cells die whereby the remains of the dead cells and the lipids they contained form a “necrotic lipid core.” As the necrotic core of the atherosclerotic plaque grows, the lumen of the coronary artery progressively narrows, and the myocardial tissue becomes progressively hypoxic (15). Simultaneously, the intimal layer between the endothelium and the necrotic lipid core, called the fibrous or collagenous cap, becomes progressively thinner (16) due to a decrease in the numbers of extracellular matrix producing smooth muscle cells (SMCs) and due to enhanced secretion of collagen-degrading enzymes by macrophages and macrophage foam cells. A thin-cap atherosclerotic lesion is vulnerable to rupture and it often causes an acute occluding thrombus in the coronary artery with subsequent ischemic myocardial damage, i.e., an acute myocardial infarction (17).
Modification of lipoproteins in the intima is one of the first steps in atherogenesis. The modified lipoproteins and the various proinflammatory molecules generated during lipoprotein modification influence the local cells and can initiate a vicious cycle of lipoprotein modification, lipid accumulation, and inflammation [reviewed in (18)]. In this review we discuss the distinct types of lipoprotein modifications and the consequences of the lipoprotein modifications on local cells as well as the development and progression of atherosclerosis.
To cross the endothelium, apoB-containing lipoproteins can bind to activin receptor-like kinase 1 and scavenger receptor B1 on the surface of the endothelial cells (ECs) lining the vessels after which they are transported by transcytosis into the intima [reviewed in (19)]. In the intima, the lipoproteins bind via ionic interactions to the negatively charged proteoglycans of the extracellular matrix (5–7) and can be attacked by various proteases, lipases, and oxidizing agents and enzymes, as evidenced by lipoproteins isolated from atherosclerotic lesions having oxidized epitopes and showing signs of proteolysis and lipolysis [reviewed in (8, 10)]. Of note, the oxidatively modified particles from human atherosclerotic lesions are more extensively modified than in the corresponding plasma, raising the possibility that the changes have been initiated already in the plasma (20). Also, each of the above-listed types of modification has been shown to induce lipoprotein aggregation in vitro (8). Aggregation and fusion of lipoprotein particles increase their affinities of their affinities to proteoglycans its and induce foam cell formation (8). Moreover, the aggregation-susceptibility of LDL particles has been linked with future cardiovascular events in patients having established atherosclerosis (21, 22). The relative importance of the various types of lipoprotein modification in atherogenesis remains to be determined, but regarding the inflammatory potential of the modified lipoproteins, the products generated during lipoprotein modification (Figure 1) may be particularly relevant, as will be discussed in this review.
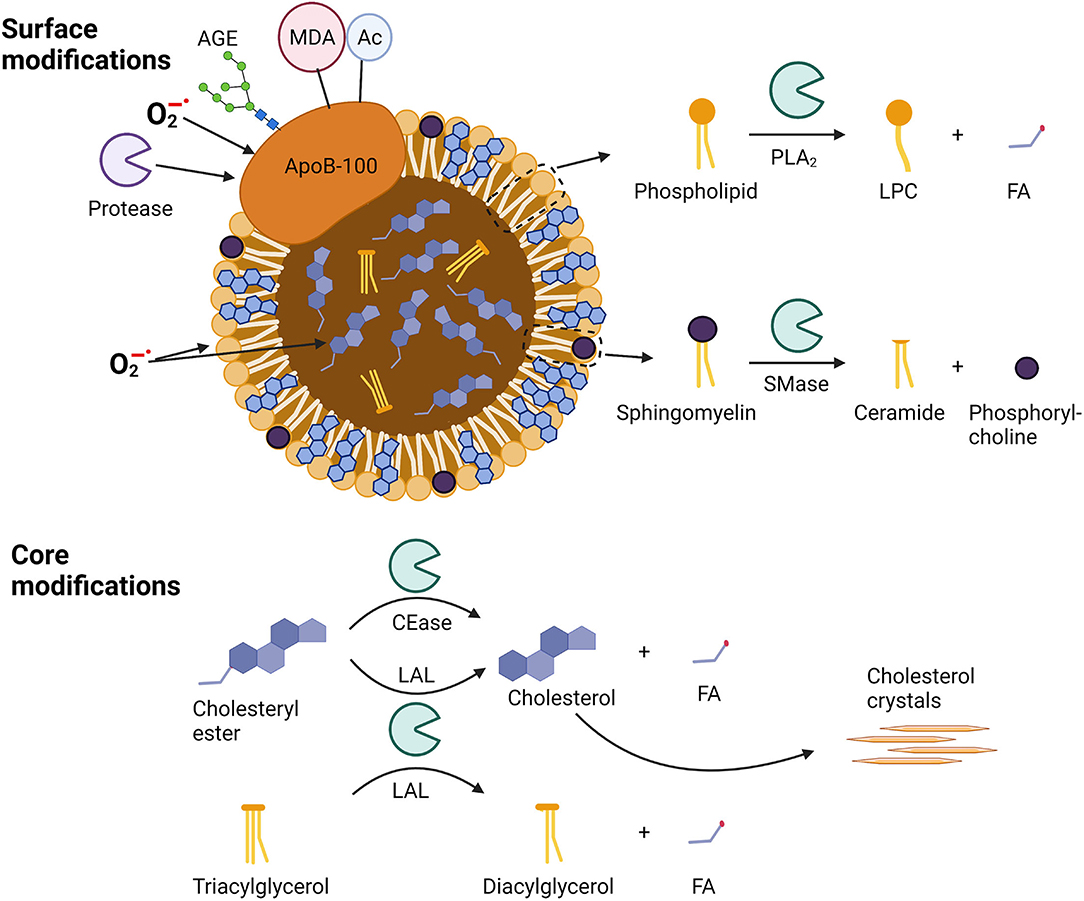
Figure 1. Sites of modification in apoB-100-containing lipoproteins. Surface modifications include modifications of apo-B100 by proteases, oxidation by superoxide anion radicals (), glycosylation by advanced glycosylation end-products (AGEs), binding of malondialdehyde (MDA) adducts, or acetylation (Ac). Phospholipids can be oxidized as well as hydrolyzed by phospholipase A2 (PLA2) to lysophosphocholine (LPC) and a fatty acid (FA), while sphingomyelins can be hydrolyzed by sphingomyelinase (SMase) to yield ceramides and phosphorylcholines. Core modifications include cholesteryl ester oxidation and well as hydrolysis by cholesterol esterase (CEase) or lysosomal acid lipase (LAL) to yield unesterified cholesterol and a fatty acid (FA). Triacylglycerol can be hydrolyzed by LAL to diacylglycerol and a fatty acid (FA).
Atherosclerotic lesions contain both neutral and acidic proteases and lipases that can modify lipoprotein particles. Among the proteases are serine proteases, cathepsins, and metalloproteinases that have been shown to degrade apoB-100 (23–26). Of note, these proteases may also degrade the structural proteins like collagens in the extracellular matrix and may thus lead to cap-thinning of the plaques [reviewed in (27)]. The extent of proteolytic degradation of the protein components of the lipoproteins depends on the protease: plasmin, for example, can induce fragmentation of apoB-100 of LDL and does not induce LDL aggregation and fusion, while cathepsins proteolyze apoB-100 of LDL into small fragments and induce the generation of large, fused particles (24, 28, 29). Proteolysis can also promote lipolytic modifications of the lipoproteins (30). An example of the combination of proteolysis and lipolysis is enzymatically modified LDL (E-LDL), which is generated by treating LDL with a protease, such as matrix metalloproteinase 9 or cathepsin H, and cholesteryl esterase (31–33).
Extensive oxidation can result in the fragmentation of apoB (34). Oxidation also leads to changes in the lipoprotein lipids, leads to the generation of malondialdehyde adducts in the lysine residues of the proteins, and induces the generation of oxidation-specific epitopes on the lipoproteins. Unlike proteolytic and lipolytic modifications, oxidation decreases the interaction of lipoproteins with proteoglycans. However, oxidized LDL (oxLDL) has been shown to bind to decorin-coated collagen via lipoprotein lipase thereby leading to extracellular accumulation of oxLDL (35). OxLDL also induces foam cell formation via interaction with various scavenger receptors on macrophages (36, 37).
Phospholipase A2 (PLA2) is an enzyme that hydrolyzes phospholipids generating non-esterified fatty acids (NEFAs) and lysophospholipids. PLA2 comprises several subtypes and can be found extracellularly as secreted PLA2s, intracellularly as cytosolic PLA2s, and in the bloodstream as lipoprotein-associated PLA2 (Lp-PLA2) (38). Lp-PLA2 is produced by hepatocytes, and also by inflammatory cells such as macrophages, foam cells, and mast cells, as well as T-lymphocytes in atherosclerotic plaques, and in the circulation it is mainly associated with LDL (39, 40). The various PLA2s have distinct specificities, group IIa, group V, and group X PLA2s being able to avidly hydrolyze phosphatidylcholine on LDL particles, while Lp-PLA2 preferably hydrolyzes oxidized phospholipids. Lp-PLA2 and several types of secretory PLA2s have been found in atherosclerotic plaques and in the normal arterial intima (38, 41–43). Modification of LDL, IDL, and VLDL with PLA2 increases their affinities to proteoglycans in vitro (44, 45), and thereby potentially enhances the retention of LDL particles in the vessel wall. PLA2-modified LDL also promotes macrophage foam cell formation (46, 47).
Treatment of LDL with an enzyme secreted by different cell types within the arterial intima, namely the sphingomyelinase (SMase), leads to conformational changes in apoB-100 and induces lipoprotein particle aggregation with ensuing increased proteoglycan binding (44, 48, 49). Secretory SMase implicated in atherogenesis has an acidic pH optimum, but can hydrolyze oxidized, PLA2-treated, or proteolyzed LDL at neutral pH (30, 49). In addition, VLDL particles appear to be very susceptible to hydrolysis and aggregation by SMase (50, 51). Interestingly, ceramide, the lipolytic product of SMase action, is found exclusively in very large aggregates (49), such as those formed in vitro by modification of LDL by SMase at pH 5.5-6 (48). Generally, the sizes of LDL aggregates after in vitro modification differ substantially depending on the type of modification and the conditions used (48, 52). Although the aggregate size can affect aggregate uptake by, e.g., leukocytes (53), no studies have been performed to systematically study the effect of aggregate size on their pro-inflammatory properties either in vitro or in vivo.
Hydrolysis of the core lipids (cholesteryl esters and triglycerides) of lipoproteins has the potential to induce the generation of large amounts of NEFAs. The lipolytic hydrolysis is enhanced if the surface of the particles is also modified, e.g., by proteolysis, which allows penetration of some of the core lipids into the surface membrane of the lipoproteins (54). The E-LDL is an example of such multiply modified lipoprotein and antibodies generated against E-LDL bind to extracellular lipids in atherosclerotic lesions (55). Hydrolysis of cholesteryl esters has the potential to generate large amounts of cholesterol and lead to the generation of cholesterol crystals (56), which are a typical feature of very advanced atherosclerotic lesions (14). More recently, small cholesterol crystals have been identified also in early atherosclerotic lesions (57).
The complement system is a tightly regulated proteolytic cascade, a key component of innate immunity comprising over 30 soluble and membrane-bound proteins that responds rapidly to clear invading pathogens and damaged host cells, as well as to limit tissue destruction and initiate tissue healing. Complement activation is triggered by exposure to endogenous danger molecules that are released from damaged or dying cells and called danger-associated molecular patterns (DAMPs) (58). Depending on the initiation, the complement cascade follows either the lectin, classical, or alternative pathway. While these pathways have different activation cascades, they all converge downstream with the formation of their respective Complement (C) 3 convertases and the generation of C3a and C3b. If the complement activation is of sufficient magnitude, the membrane attack complex C5b-9 is formed. This important innate immune effector has pore-forming, lytic properties designed to destroy pathogens and damaged cells (59–61).
Circulating C3 levels were found to be significantly elevated in patients having familial hypercholesterolemia with subclinical coronary atherosclerosis, however, there was no correlation between circulating C3 levels and increased plaque burden, indicating a local regulation of the C3 in atherosclerotic arteries (62). Indeed, active components of the C3 complement are found within the extracellular matrix of human arteries (63). Complement components, including C3 were also identified in extracellular lipoproteins isolated from human carotid atherosclerotic lesions (56), and E-LDL was shown to colocalize with C5b-9 in human atherosclerotic lesions (55). Potential triggers of complement activation in the arterial wall include modified lipoproteins, cholesterol crystals, antigen-antibody immune complexes, C-reactive protein (CRP), and apoptotic cells [reviewed in (59, 63–65)]. Importantly, aggregated and fused lipoproteins isolated from human atherosclerotic lesions possess complement-activating properties (66). These particles do not contain immunoreactive apoB-100 but contain both esterified and unesterified cholesterol, and have sizes ranging from 100 to 500 nm (33). E-LDL with or without bound CRP, oxLDL, malondialdehyde-LDL, and oxLDL-immune complexes have been shown to induce complement activation [reviewed in (67)]. E-LDL, but not oxLDL, can trigger C1 activation and bind complement components (68, 69). Factor H, a major complement inhibitor, colocalizes in human atherosclerotic lesions with C3d in the subendothelial proteoglycan-rich layer, while C5b-9 is found in deeper areas of atherosclerotic lesions suggesting that in different areas of the lesions the components may differ in their abilities to regulate complement activation (63).
Foam cells are the histological hallmark of atherosclerotic lesions from the very beginning of the lesion development, and their contribution to atherogenesis remains throughout the lesion progression (13, 15). They are derived mostly from macrophages that take up modified and aggregated lipoproteins and lipoprotein-immune complexes via several different mechanisms (36). In addition to the macrophages, also vascular SMCs take up modified lipoproteins and can become foam cells (70, 71). The various uptake mechanisms include scavenger receptors that are important particularly in the uptake of oxLDL, Fc-gamma receptors interacting with immune complexes, cell surface proteoglycan-aided uptake of PLA2-modified LDL, and phagocytosis of aggregated LDL and even cholesterol crystals (46, 72–75). Native VLDL remnant particles have been shown to induce foam cell formation through the VLDL receptor pathway in macrophages, and modification of VLDL particles by acetylation leads to macrophage foam cell formation via scavenger receptor-mediated uptake of the modified particles (76).
A specific uptake mechanism in macrophages has been reported for large LDL aggregates, such as SMase-modified LDL particles. Thus, aggregated LDL triggers the formation of extracellular surface-connected compartments into which the macrophages (77) and dendritic cells (78) secrete lysosomal enzymes, such as lysosomal acid lipase (LAL) (78–80). The process leads to a partial hydrolysis of the aggregated LDL outside the cells and to internalization of the partially hydrolyzed lipoproteins into lysosomes, where they are fully hydrolyzed. Actin polymerization and Toll-like receptor (TLR) 4 activation drive the intracellular catabolism of the aggregates (80, 81). In addition, treatment of LDL with macrophage-conditioned media containing catalytically active LAL induces LDL fusion and lipid droplet formation in VSMCs (54). LAL activity may be particularly prominent in the macrophage-rich acidic microenvironments observed in human atherosclerotic lesions (82).
Once lipoproteins have reached the lysosomes, their components are completely hydrolyzed (Figure 2). Importantly, cholesteryl ester hydrolysis by LAL results in the formation of unesterified cholesterol, which will then be re-esterified in the cytoplasm and packaged into intracellular lipid droplets characteristic of foam cells (83). Uptake of aggregated LDL has been shown to lead to accumulation of ceroid, a product of lipid oxidation consisting of protein-lipid complexes, in lysosomes (84). Lysosomal oxidation is likely mediated by iron and can be inhibited by cysteamine (85). On the other hand, uptake of oxLDL can result in lipid accumulation in the lysosomes (86, 87) and lead to the formation of cholesterol crystals in the lysosomes (88). However, the transformation of macrophages into foam cells may reduce their atheroinflammatory potential (89).
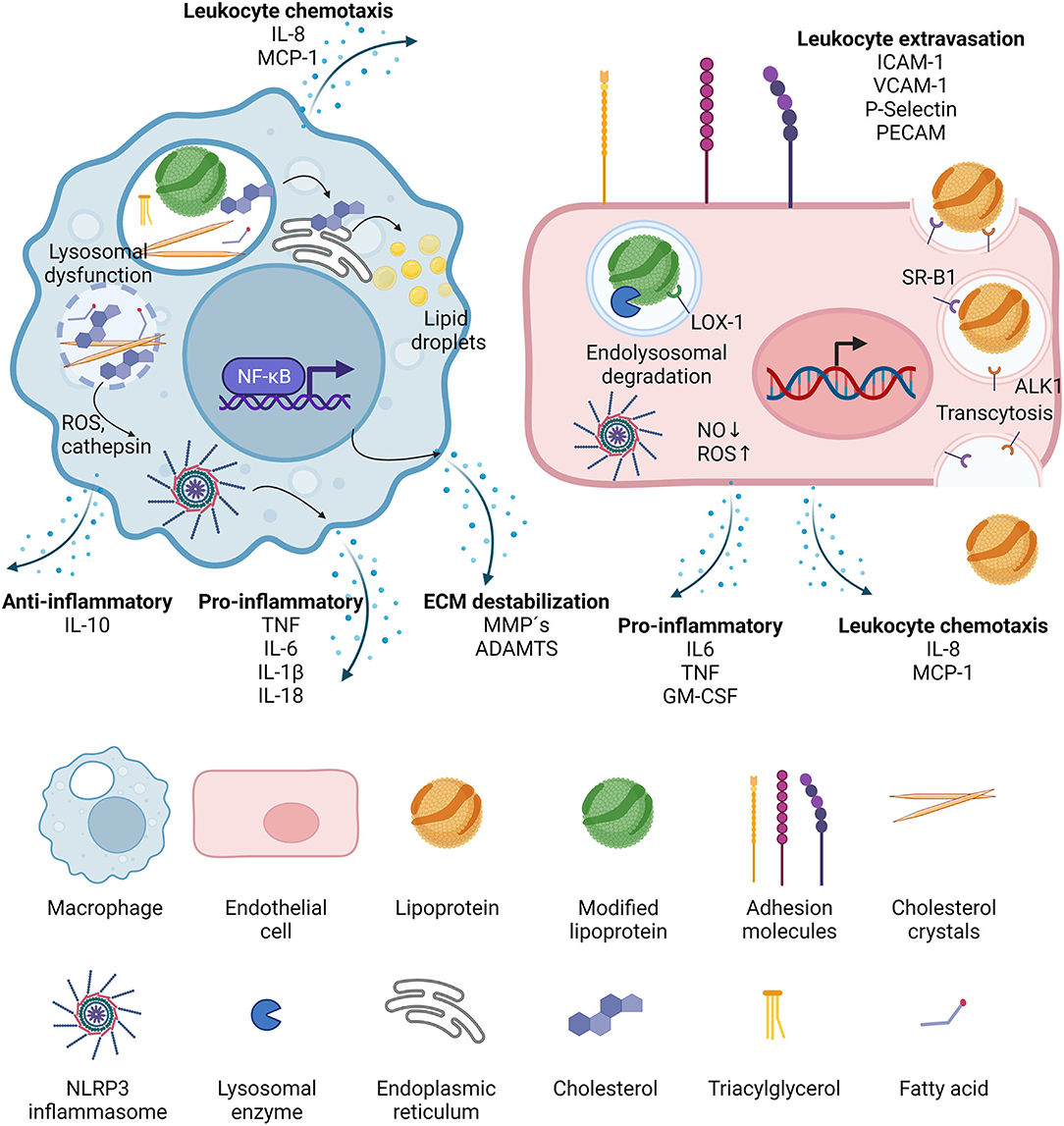
Figure 2. Effects of lipoproteins on macrophages and endothelial cells. Upon phagocytosis, modified lipoproteins are hydrolyzed in the lysosomes of macrophages. Cholesterol is then transported to the ER where it is packaged in lipid droplets for storage. It can also crystalize in the lysosomes, leading to lysosomal dysfunction and the release of reactive oxygen species (ROS) and cathepsins, which in turn activate cytosolic inflammasomes. NF-κB signaling is induced and the activated macrophages secrete leukocyte chemotactic molecules, pro- as well as anti-inflammatory cytokines, and proteases. In endothelial cells, lipoproteins can be transported through the cell via transcytosis, mediated by binding to scavenger receptor B1 (SR-B1) and activin-like kinase 1 (ALK1). If the lipoproteins have been modified, they can also bind to lectin-type oxidized LDL receptor 1 (LOX-1) and then targeted to the lysosomes after internalization. LOX-1 activation leads to decrease in nitric oxide (NO) and increase in reactive oxygen species (ROS), the latter being able to trigger NLRP3 inflammasome activation. Activated endothelial cells increase the expression of adhesion molecules which accelerates leukocyte extravasation, and they also secrete leukocyte chemotactic factors, as well as pro-inflammatory cytokines.
In vascular SMCs, foam cell formation due to uptake of native and modified VLDL was shown to depend on the activity of lipoprotein lipase (90), while the LDL receptor-related protein 1 plays a role in the uptake of aggregated LDL (91). Aggregated LDL particles increase the release of the soluble form of LDL receptor-related protein 1 from cultured vascular SMCs, and from human atherosclerotic plaques in an ex vivo model (92). Interestingly, the level of the soluble LDL receptor-related protein 1 in the circulating blood is increased in patients with coronary artery disease (92, 93). When compared with cultured human macrophages, cultured human SMCs express relatively low levels of LAL (94). Accordingly, in SMC foam cells, the ingested neutral lipids tend to accumulate within lysosomes, while in macrophages they appear as cytosolic droplets.
While complement activation is usually regarded to be pro-atherogenic, complement plays also a smoothing role in mediating the effects of foam cell formation. Thus, while the complement component C1q recognizes and opsonizes modified forms of LDL and promotes phagocytosis and efferocytosis in macrophage foam cells (95, 96), when bound to acLDL or oxLDL it reduces the release of proinflammatory cytokines from macrophage foam cells and atherosclerosis in mouse models (97). In addition, C1q downregulates both the levels and activities of apoptosis-related proteins in human and mouse macrophage foam cells, thereby leading to a measurable increase in the survival of these cells, which, again, could slow down the build-up of the necrotic core in the atherosclerotic plaques (95). Further, factor H, the main regulator of the alternative complement activation pathway, has been shown to bind to human monocytes and macrophage foam cells and has the potential to reduce complement activation and inflammation, and to increase the binding of apoE to these cells. It further increased cholesterol efflux, and cholesterol-loaded macrophages displayed reduced transcription of proinflammatory/proatherogenic factors and increased transcription of anti-inflammatory/anti-atherogenic factors, thereby indicating that apoE and factor H interact with monocytic cells in a concerted action and that this interaction tends to reduce complement activation and inflammation in atherosclerotic lesions (98).
Continued accumulation of immunogenic modified lipoproteins and the attendant pro-inflammatory reactions in the arterial intima result in the development of atherosclerotic lesions (99). Thus, atherosclerosis is driven by a chronic, low-grade inflammation of the vascular wall that continuously attracts immune cells into the developing atherosclerotic plaques (100).
As the first step in atherogenesis, native lipoproteins enter the intima by transcytosis, get trapped and modified by enzymes secreted by resident macrophages and mast cells, as described above. Minimally modified lipoproteins from the circulation or modified lipoproteins from the intima can be internalized by ECs via the lectin-like oxidized LDL receptor 1 (LOX-1) and be destined for lysosomal degradation, which activates the ECs and initiates endothelial dysfunction [Figure 2, reviewed in (101)]. ECs also get activated by secreted cytokines and chemoattractants derived from activated intimal macrophages (101). The activated endothelium then secretes chemoattractants and increases the expression of adhesion molecules so aiding the tethering, rolling, and adhesion steps of the leukocyte diapedesis process. Within the intima, monocytes differentiate into macrophages or DCs, and the naïve T cells mature. Macrophages can be of pro-inflammatory (M1) or anti-inflammatory/pro-resolving (M2) phenotype, and their phenotype can change in response to alterations in their microenvironmental conditions [reviewed in (102)]. The above widely used dichotomous classification of macrophages into pro- and anti-inflammatory phenotypes provides a useful framework, but only represents the opposite ends of a wide spectrum of phenotypes which includes an ever-increasing number of subtypes, as revealed by modern single-cell omics technologies. The polarization of macrophages is mediated by a plethora of extrinsic factors such as cytokines, and also by intrinsic regulatory mechanisms, such as increased glycolysis, all of which in various combinations may alter the gene expression profile and reactivity of the macrophages involved [reviewed in (102, 103)]. The metabolic changes in macrophages can be altered by ligand-activated transcription factors such as peroxisome proliferator-activated receptors (PPARs) and liver X receptors. Once activated, these receptors inhibit the pro-inflammatory transcription factors NF-κB and AP-1 and thus downregulate the expression of pro-inflammatory cytokines such as interleukin (IL)-1β, IL-6, and tumor necrosis factor (TNF) (104).
DCs and macrophages phagocytose modified LDL, which the DCs then present to T cells, while the activated macrophages start to secrete cytokines, chemokines, and enzymes, thus further activating the endothelium and leading to increasingly diverse and severe modifications of the trapped lipoproteins (Figure 2).
The modifications of the trapped lipoproteins lead to the formation of DAMPs, which are recognized by pattern recognition receptors (PRRs) located on the surface of antigen-presenting cells, and also on the surfaces of non-immune cells, such as the ECs. The membrane-bound PRRs include toll-like receptors (TLRs), scavenger receptors (72), and the receptor for advanced glycation end-products (RAGE) (105). When activated, the TLRs recruit adapter proteins to activate two main pathways, a MyD88-dependent and a TRIF-dependent pathway, that lead to the production of inflammatory cytokines or type I interferons, respectively (106). Several TLRs have been shown to play important roles in atherogenesis [reviewed in (106)].
Another set of PRRs, the NOD-like receptors (NLRs), are located in the cytoplasmic compartment of cells. Once a cell senses intracellular damage-associated events such as membrane damage, lysosomal rupture, or mitochondrial damage, it initiates the assembly of the NLR family pyrin domain containing 3 (NLRP3) inflammasome (107). Inflammasomes are large cytosolic multiprotein complexes, and once assembled, they induce the activation of inflammatory responses, including proteolytic activation and secretion of the proinflammatory cytokines IL-1β and IL-18 (108). Especially the NLRP3 inflammasome plays a role in atherosclerotic plaque formation [reviewed extensively in (108)]. Inflammasome-mediated activation of caspase-1 can also initiate a form of lytic and highly inflammatory cell death called pyroptosis, in which the protein gasdermin D is cleaved, after which its N-terminus oligomerizes and forms pores in the cell membrane (109). Crystalline cholesterol induces lysosomal damage, and it was the first NLRP3 activator associated with atherosclerosis (57, 75). Crystalline cholesterol can be formed extracellularly from lipoprotein aggregates in the intima, and intracellularly during the lysosomal degradation of modified lipoproteins.
Several studies have shown that modified lipoproteins can activate leukocytes and trigger a pro-inflammatory response (18, 110). This response is leading to the secretion of a plethora of cytokines, chemokines, growth factors, and lipids. The most important ones in the context of this review are shown in Table 1, and the individual responses of cell types to specific modified lipoproteins discussed below are summarized in Table 2.
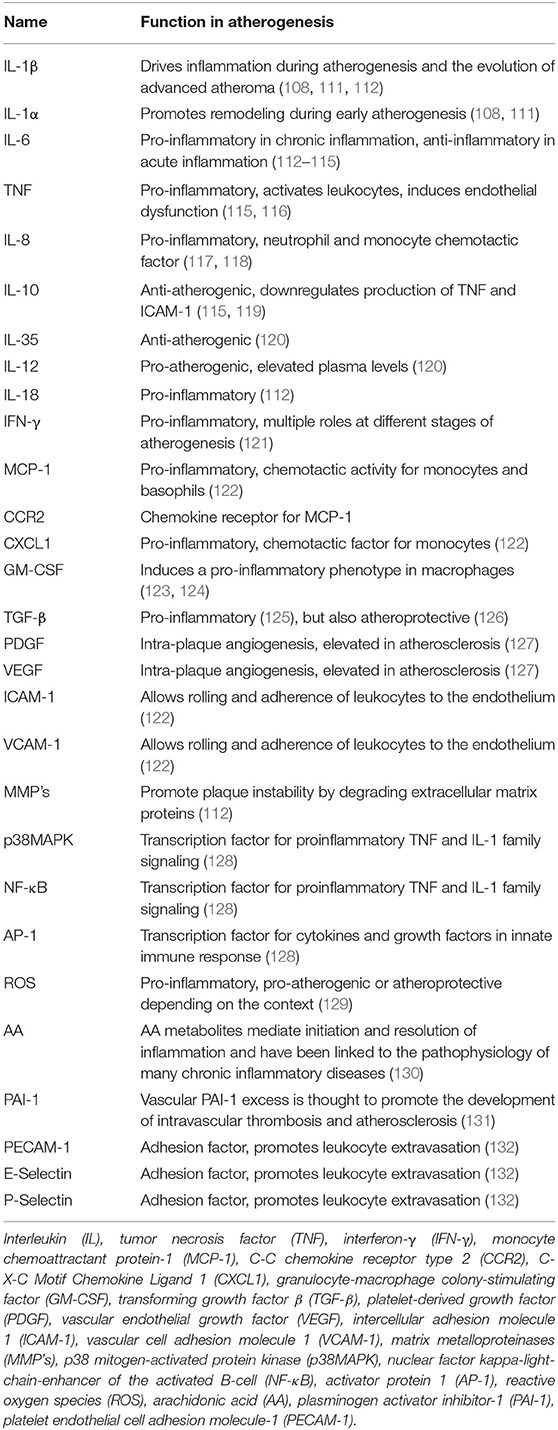
Table 1. Main cellular mediators of atheroinflammation.
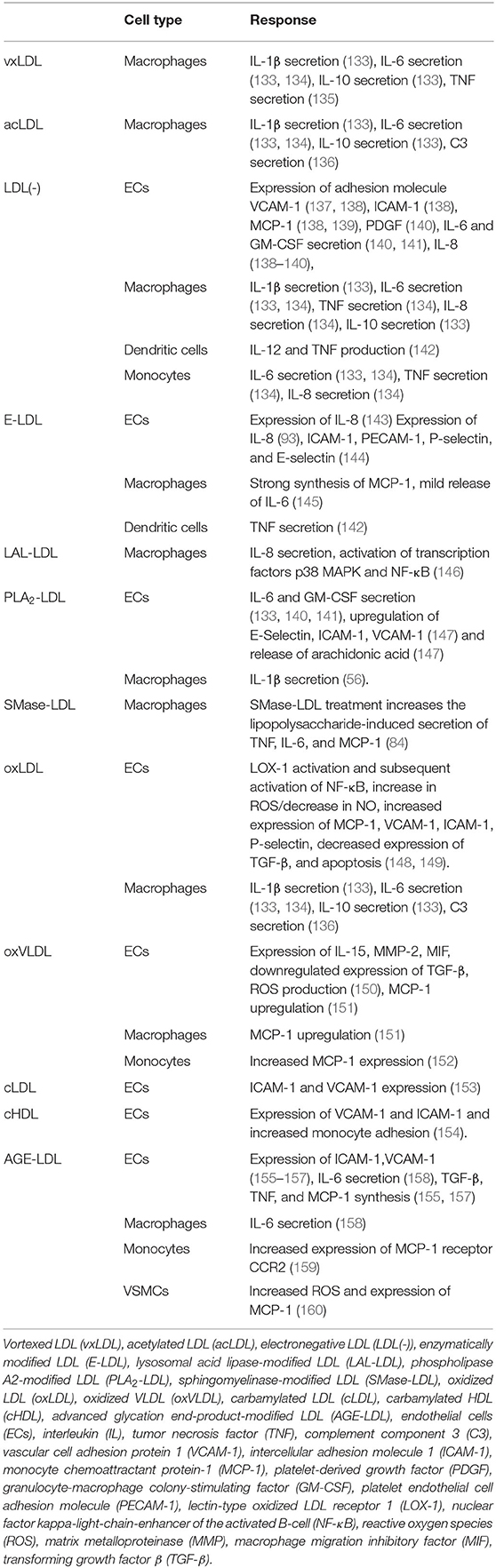
Table 2. Cellular responses to selected modified lipoproteins.
Acetylated LDL (acLDL) and vortexed LDL (vxLDL) are models of modified lipoproteins that have been used to study intracellular lipid accumulation in macrophages, SMCs, and ECs (161–165). Vortexing of LDL leads to aggregation of the particles, but these modifications do not change the lipid moiety of LDL particles and, therefore, experiments in which they have been used provide information merely on their effects on lipid loading on macrophages. Treatment of macrophages with modified LDLs that induce foam cell formation, including acLDL, results in up-regulation of genes involved in the inflammation and immune responses (110).
VxLDL induces mitochondrial dysfunction in macrophages, a known activator of the NLRP3 inflammasome (166). Persson et al. studied the basal and inducible cytokine expression in primary human macrophages loaded with cholesterol using vxLDL and they observed that the cholesterol contained in the vxLDL did not affect IL-1β secretion, while the secretion of TNF, IL-6, and IL-8 were significantly decreased (167). However, the amount or composition of neutral lipids in the vxLDL did not affect cellular activation by exogenous TNF, making it likely that lipid loading attenuates cytokine secretion during basal conditions, and that the effects can be overruled by TNF during an acute inflammation (167). Sabeva et al., however, reported that IL-1 and IL-6 signaling can be induced by vxLDL in THP-1-derived macrophages (135). Depending on the macrophage polarization, inflammatory responses to cholesterol loading can vary vastly, as recently shown by comparing the inflammatory responses in M1-like and M2-like macrophages upon acLDL loading (89).
Electronegative LDL (LDL(-)), carbamylated LDL (cLDL) or HDL, glycosylated LDL (AGE-LDL), and mildly oxidized LDL are found in the circulation, and they can initiate foam cell formation in cell culture [reviewed in (168–170)].
LDL(-) is a naturally occurring, minor form of modified LDL in plasma, and it can be isolated based on its differing size, density, and charge. Compared to native LDL, LDL(-) has similar oxidation levels, but it differs in its lipid and protein compositions and in its apoB-100 conformation. LDL(-) also has higher phospholipolytic activities, higher aggregation levels, and higher proteoglycan binding affinity than LDL (133).
In ECs, LDL(-) increases TNF-induced inflammatory responses, such as the expression of transcription factors nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and Activator Protein 1, the expression of Vascular Cell Adhesion Molecule 1 (VCAM-1) (137), and the secretion of the chemoattractants IL-8 and Monocyte Chemotactic Protein 1 (MCP-1) (139), C-X-C Motif Chemokine Ligand 1, the cytokine IL-6, and the growth factors platelet-derived growth factor, Granulocyte-Macrophage Colony-Stimulating Factor (GM-CSF) and Vascular Endothelial Growth Factor (138, 140, 141). In monocytes and macrophages, LDL(-) directly induces priming, inflammasome activation with subsequent IL-1β release (171, 172), as well as secretion of IL-6, IL-8, and TNF (134). Puig et al. compared the effects of oxLDL, vxLDL, and acLDL with those of LDL(-) on THP1-derived macrophages and found that while oxLDL induced a more pronounced IL-1β release, LDL(-) induced a stronger release of IL-6, IL-10, and GM-CSF than did other modified LDLs (133). Interestingly, unlike other modified LDL preparations, LDL(-) induced a very strong triglyceride (TG) accumulation in lipid droplets of macrophages (133), a finding which may have clinical significance in that macrophages isolated from human atherosclerotic aorta show significant accumulation of TGs (173).
Protein carbamylation is a post-translational modification of proteins involved in a variety of disease states including inflammation, impaired renal function in chronic kidney disease, and, moreover, elevated levels of carbamylated proteins are present in smokers. Carbamylation has been mechanistically linked to atherosclerosis [reviewed in (174)], especially in patients with chronic renal failure who have vastly higher levels of carbamylated LDL in serum and a higher ASCVD risk (175). It has also been shown to induce endothelial dysfunction (176), increase the expression of adhesion molecules (153), the oxidative stress, and DNA damage in endothelial progenitor cells (177). Also carbamylated HDL increases monocyte adhesion to ECs via inducing NF-κB activation and the expression of VCAM-1 and the intercellular adhesion molecule-1 (ICAM-1) (154). Carbamylation also renders LDL more prone to oxidation, and LDL with both modifications has more pro-atherogenic effects on ECs and macrophages than either modification alone (178).
Non-enzymatic modifications of proteins by glucose are called advanced glycation end-products (AGEs), and they are formed at accelerated rates in hyperglycemic diabetic patients. AGEs can also form on lipids, as evidenced by the observation of lipid-linked AGEs in LDL particles derived from individuals with or without diabetes (179). The AGE-products include circulating AGE-peptides which derive from the catabolism of AGE-modified tissue proteins, and, moreover, AGE-modified LDL is formed by direct reaction between native LDL and circulating reactive AGE-peptides (179). The AGEs, among them also the AGE-LDL, damage vascular cells, one pathway being their interaction with their purported receptors on vascular ECs, SMCs, and macrophages. Among the ensuing implicated diverse pro-atherogenic processes, those leading to endothelial dysfunction have received much attention (155). The adverse endothelial effects include apoptosis (180), reduced nitric oxide synthesis, increased production of reactive oxygen species, and increased expression of (VCAM)-1 (156). Moreover, AGE-LDL particles induce IL-6 secretion by ECs and macrophages (158).
The lipolytic products generated by the action of PLA2, fatty acids and lysophospholipids, induce proinflammatory responses in cultured human umbilical vein ECs (181). PLA2-treated LDL induces exocytosis of the Weibel-Palade Bodies, an early-phase endothelial inflammatory response from ECs in a lysophosphatidylcholine (LPC)-dependent manner (182). LPC acts as a find-me/eat-me signal, which belongs to a group of soluble mediators released by apoptotic cells that attract phagocytes into the tissue (183). PLA2-LDL has also been shown to upregulate the adhesion factors E-selectin, ICAM-1, and VCAM-1 in ECs (147). In primed human macrophages, PLA2-treated LDL and especially LDL and VLDL treated with both PLA2 and LAL have shown to elicit a strong induction of NLRP3-dependent IL-1β secretion (56).
Treatment of LDL particles with SMase, which is secreted by several cell types within the arterial wall, leads to the generation of ceramide and to conformational changes in apoB-100 that induce particle aggregation and increased proteoglycan binding (48–50). These effects could be alleviated by treatment of the LDLs with an apoA-I mimetic peptide prior to the hydrolysis (184). SMase-LDL has been shown to induce the secretion of TNF, IL-1β, IL-6, and the chemoattractant MCP-1 from human macrophages derived from cultured THP-1 monocytes (56, 84). LDL which has been lipolytically modified by secretory PLA2 or SMase potentiates the cellular release of proinflammatory lipid mediator arachidonic acid, and triggers activation of the cytoplasmic PLA2 in human THP-1 monocytes (185).
Regarding SMase, another point to consider is that it is expressed by macrophages. Endogenous sphingomyelins on the cell membrane can be hydrolyzed, and the generated ceramides can accumulate or be converted into numerous metabolites [reviewed in (186)]. These metabolites are involved in a variety of processes connected to cardiovascular health, they modulate the signaling and metabolic pathways driving insulin resistance, TG production, apoptosis, and fibrosis [reviewed in (187)]. Cellular uptake of SMase-modified LDL brings substantial amounts of ceramides into lysosomes, and, moreover, as LDL contains significant amounts of SM, uptake of modified LDL can also in other ways affect the sphingomyelin pool in macrophages and thus the concentrations of ceramides in them. Kinscherf et al. showed in their early work that loading of macrophages with oxLDL and acLDL delivers a substantial amounts of sphingomyelin into the cells and also stimulates ceramide formation in them (188).
Modifications of the lipoprotein core at neutral pH are more difficult to perform due to the protective surface monolayer of the lipoproteins. Therefore, the core-modifying treatments are usually preceded by a modification of the surface. To generate enzymatically modified LDL (E-LDL), LDL is first treated with a protease and subsequently with cholesterol esterase. Such multiply-modified LDL has been detected by immunohistochemical means in human atherosclerotic lesions at all stages of lesion development (55). Treatment of ECs with E-LDL leads to the expression of IL-8 (143), induces the adhesion of monocytes and T lymphocytes to EC monolayers, and stimulates the upregulation of the adhesion factors ICAM-1, platelet-endothelial cell adhesion molecule-1 (PECAM-1), P-selectin, and E-selectin (144). In aortic SMCs, E-LDL upregulates the expression of ICAM-1, which correlates with increased adhesion of T lymphocytes (144). It also strongly induces SMC foam cell formation and upregulates the expression of lectin-like oxLDL receptor 1 (LOX-1) with ensuing increase in the uptake of oxLDL (189).
In macrophages, E-LDL induces strong secretion of MCP-1 and mild release of IL-6 (145). It also promotes monocytic differentiation into dendritic cells, and, when dendritic cells are treated with CRP and/or E-LDL, the production of IL-12 and TNF is significantly increased, and a strong Th1 reaction and T cell proliferation are elicited (142).
LAL hydrolyzes TGs and cholesteryl esters in the core of the lipoprotein particles. LAL is an essential lysosomal enzyme and its deficiency leads to cholesteryl ester storage disease (190). Expression of LAL has been shown to become downregulated in several cell types when the cells are loaded with modified LDL (191, 192). LAL is also secreted from activated macrophages and can then act on lipoproteins also extracellularly (54). Treatment of macrophages with LAL-LDL activates the transcription factors p38 MAPK and NF-κB, induces the secretion of IL-8 (146), and may also promote vascular dysfunction and atherogenesis [reviewed in (193)]. LDL and VLDL, when treated first with enzymes that hydrolyze the particle surface before treatment with LAL are able to trigger a robust, NLRP3-mediated IL-1β secretion in human macrophages (56).
VLDL remnants treated with lipoprotein lipase to hydrolyze the TGs have been shown to induce increased expression of TNF, IL-1β, and IL-8 over native VLDL or lipoprotein lipase, with concurrent activation of NF-κB and activator protein 1 in peripheral blood mononuclear cells and THP-1 monocytes (194).
Oxidation is the most studied form of lipoprotein modification, and the pro-inflammatory effects of oxidized phospholipids in inflammation and various inflammatory diseases are well established (195).
OxLDL acts as a DAMP and triggers sterile inflammatory responses (195). In ECs it causes endothelial dysfunction and induces a pro-inflammatory and pro-thrombotic state (196). It also enhances monocyte chemotaxis and adhesion to EC in culture (197). The uptake of oxLDL by macrophages is mediated by scavenger receptors such as CD36, and it induces the assembly of a TLR4/TLR6 heterodimer, and results in NF-κB signaling and priming of these cells for inflammasome activation (198). OxLDL uptake by CD36 additionally results in intracellular cholesterol crystallization and can thereby directly activate the NLRP3 inflammasome in macrophages (88). Both minimally oxidized LDL and more highly oxidized LDL cause the secretion of pro-inflammatory cytokines by macrophages (70, 71) by activating TLR4 (199, 200). Li et al. showed in THP-1 monocytes that oxLDL and oxHDL increases the expression of NLRP3 and activates caspase-1 and induces IL-1β and IL-18 secretion in a dose-dependent manner (201). However, other studies have shown that oxLDL inhibits the production of inflammatory cytokines by macrophages in response to inflammatory stimuli, such as LPS (202). More recently, it was shown that oxLDL, as well as oxHDL3, reduces the secretion of mature IL-1β by inhibiting the activation of the NLRP3 inflammasome induced by known inflammasome activators including the acute-phase serum amyloid A protein, ATP, nigericin, and monosodium urate crystals (203). By binding to LOX-1 on SMCs, oxLDL can also induce SMC transition to an inflammatory phenotype. Thus, treating cultured SMCs with oxLDL stimulates the expression of the chemokines MCP-1 and C-X-C Motif Chemokine Ligand 1, the cytokine TNF, and the cell adhesion molecule VCAM-1 [reviewed in (204)].
OxLDL can also induce pyroptosis in macrophages (205) and ECs (206), and oxLDL-induced pyroptosis has also been implicated in endothelial dysfunction and vascular SMC foam cell formation [reviewed in (109)].
OxLDL is also able to induce a novel form of cell death coined ferroptosis, which is linked to atherogenesis (207, 208). Bai et al. showed that some of the key morphological features of ferroptosis, such as mitochondrial shrinkage, increased iron content, increased release of lactate dehydrogenase, and reduced expression of the solute carrier family 7 member 11 and glutathione peroxidase 4 were induced by oxLDL in mouse aortic ECs to a comparable amount achieved with the ferroptosis inducer erastin; the phenotype was rescued by co-stimulation with ferroptosis inhibitor ferrostatin-1 (209). Further, oxLDL- and erastin-treated cells had similarly increased levels of markers of intracellular lipid peroxidation, such as total ROS, lipid ROS, lipid peroxide, and malondialdehyde, while treatment with ferrostatin-1 downregulated the generation of these lipid peroxidation products (209). Similar induction of ferroptosis by oxLDL was observed in human coronary artery ECs (210).
While minimally oxidized LDL is found in circulation, the vast majority of circulating oxLDL is bound to antibodies and found as oxLDL immune complexes, which can prime dendritic cells and macrophages for inflammasome activation (211). While the circulating immune complexes may be too large to cross an intact endothelium, immune complexes can also be formed within the arterial intima (212). Interestingly, immune complexes composed of oxLDL and anti-LDL IgG antibodies can trigger activation of cultured human mast cells, an innate immune type of cell present in human atherosclerotic lesions, and, when activated, capable of proteolytically modifying LDL and HDL particles (213, 214).
In ECs, oxidized VLDL initiates the expression of the fibrinolysis inhibitor plasminogen activator inhibitor-1 and stress response protein heat shock factor 1 compared to the effects obtained with VLDL or LDL (215), and induces endothelial apoptosis (216). It also induces expression of IL-15, matrix metalloproteinase-2, macrophage migration inhibitory factor, as well as downregulated expression of transforming growth factor beta (TGF-β) (150). In macrophages, both oxidized VLDL and its remnants induce foam cell formation that can be attenuated by pre-incubation with TGF-β (217). In both monocytes and macrophages, oxidized VLDL induces the expression of MCP-1 (151, 152).
Oxidized HDL (oxHDL) plasma levels have been shown to correlate with ASCVD and coronary artery calcification [recently reviewed in (218)]. OxHDL induces endothelial dysfunction in ECs by a positive feedback mechanism in which an oxidative burst generates oxHDL from native HDL, activates LOX-1 which in turn increases the expression of NADPH oxidase 2, TNF, and LOX-1 receptor at the endothelial plasma membrane (219). It has also been shown to decrease the expression of CD63 in macrophages, thus attenuating the ability of the cell to ingest modified lipoproteins (220, 221). OxHDL also binds to CD36 on platelets and induces proinflammatory and procoagulant effects (222).
Another innate immune pathway contributing to vascular inflammation is neutrophil extracellular trap (NET) formation, also called NETosis (223). NETs are structures of chromatin filaments coated with histones, proteases, as well as granular and cytosolic proteins, which are ejected by activated neutrophils to immobilize and eliminate pathogens as a part of the first-line immune defense.
Recent research has revealed that NETosis is associated with the initiation and progression of various noninfectious diseases (224), and NETosis has also been shown to promote thrombosis (225). NETs have been found in vascular lesions such as atherosclerotic plaques (226), and infiltration of neutrophils into arteries during the early stages of atherosclerosis has been observed in hypercholesterolemic mice (227). One theory about the reason for NETs in plaques is that periodontal pathogens enter the bloodstream from severely infected periodontal tissues, then reach the inflamed atherosclerotic lesions where they can directly contribute to the development of the lesions [reviewed in (228)].
Non-infectious triggers such as LDL-derived cholesterol crystals might also induce NETosis in atherosclerosis. NETs prime macrophages, which in turn start to produce pro-IL-1β and IL-8 (117). Cholesterol crystals can bind to CD36 on the macrophages and thereby trigger inflammasome activation with the ensuing secretion of mature IL-1β. Thus, NETs may contribute to atherogenesis by initially activating macrophages (229, 230). Studies in apoE-/- mice with deletions of two neutrophil proteases showed attenuated NETosis as well as smaller atherosclerotic lesions, and lower systemic IL-1β levels, when compared to ApoE–/– mice expressing these proteases (230), indicating a potential role for NETs and/or these proteases in this process.
Kato et al. demonstrated the appearance of oxLDL in vascular tissues and circulation even prior to atherosclerotic lesion development in apoE-KO mice (231). OxLDL as well as its various oxidized phospholipid components, especially oxPCs and LPC, mediate NETs formation and subsequent endothelial inflammatory responses in both HL-60-derived neutrophils and human polymorphonuclear neutrophils (232, 233).
Adaptive immunity is heavily involved in atherogenesis [reviewed in (234)] and comprises a humoral component of specific antibodies produced by B cell-derived plasma cells, and a cellular component with T cells that either activate B cells or differentiate into effector T cells, which are matured by being presented antigens by antigen-presenting cells. Of the T cells, those expressing CD8 mature into cytotoxic CD8+ T cells and those expressing CD4 into helper CD4+ T cells or regulatory T cells. Both CD4+ and CD8+ T cells are recruited into atherosclerotic lesions and display signs of activation (235). On the other hand, the regulatory T cells comprise the only T cell subset that has only negative regulatory effects on the autoimmune response, and therefore also a dampening influence on atheroinflammation. The recruitment of regulatory T cells into the arterial intima is promoted by the activation of the nuclear receptor peroxisome proliferator-activated receptor γ (PPAR-γ), which can further exert anti-atherosclerotic effects by inhibiting the expression of various pro-inflammatory factors, improving the function of endothelial cells, inhibiting MCP-1 expression, and restraining the differentiation of monocytes into macrophages [reviewed in (236)].
T cells with a specificity for apoB-derived epitopes have been identified, so linking adaptive immune responses to the vascular retention of LDL (237). Vaccination of humanized atherosclerotic mice with two epitopes from human native apoB-100 that trigger T-cell activation protected the mice against atherosclerosis (238). T cells constitute about 10% of all cells in human plaques, and 70% of them have been described to be CD4+, and the remaining largely CD8+ T cells (239).
The final stage of B cell maturation into antibody-producing plasma cells occurs after their activation. There are two main subtypes, B1 cells which produce the majority of natural IgM and to a lesser extent IgG, and B2 cells which can differentiate directly into memory B cells (240). T cells and macrophages are found in all stages of atherosclerosis, whereas B cells are only occasionally found in the atherosclerotic plaques themselves. Rather, a larger number of B cells may be found in the adventitial layer of the arterial wall, where tertiary lymphoid organs can be formed (241). Resident B2-type lymphocytes isolated from human carotid atherosclerotic walls have been found to be mainly activated plasmablasts lacking terminal differentiation to plasma cells, and preferentially expressing IgG and IgA (242).
Functionally, the effects of B and T cells in atherosclerosis, the primary site of lipoprotein modification, are mixed. In a recent consensus paper (6) the involvement of lymphocytes is described as follows: Dendritic cells take up modified LDL and present specific epitopes such as apoB peptides to naive T cells, inducing their differentiation into CD4+ helper T cells (243). These CD4+ T cells, together with the specific cytokines that they secrete, provide help to B cells, and regulate the activity of other T cell subtypes. The pro-atherogenic role of interferon gamma (IFN-γ)-secreting Th1 cells and the anti-atherogenic effect of IL-10/TGF-β-secreting T regulatory cells are well established (244). Cytotoxic CD8+ T cells have been shown to promote atherogenesis (245). Hermansson et al. studied the mechanism of T cell recognition of LDL particles by generating T cell hybridomas from human apoB-100 transgenic mice that were immunized with human oxLDL, and found that none of the hybridomas responded to oxLDL, but, instead, to native LDL and isolated apoB-100. However, sera from these immunized mice contained antibodies against oxLDL, suggesting that T cell responses to apoB-100 aid B cells to generate antibodies against the epitopes present in both native LDL and oxLDL. ApoB-100-responding CD4+ T cell hybridomas expressed a single T cell receptor variable β chain, and immunization of humanized atherosclerotic mice with a peptide derived from that T cell receptor-induced antibodies that blocked T cell recognition of apoB-100, significantly reduced atherosclerosis with a concomitant reduction of macrophage infiltration in the lesions (246). Ruuth et al. observed similar inhibition of T cell activation by oxLDL, but they found that SMase-LDL activated T cells and that the degree of LDL aggregation induced by SMase associated positively with the degree of T cell activation, as measured by the secretion of interleukin-2 (22). Up to 10% of CD4+ T cells isolated from human atherosclerotic plaques displayed specificity for oxLDL (247).
If incubated with whole blood, native and vxLDL elicit vastly different T cell activation, native LDL activating CD4+ cells only to a small extent, and vxLDL potently activating CD8+ cells (248). The response might be indicative of in vivo formed intimal aggregation of modified LDL.
Distinct roles for different B cell subsets have been reported, anti-oxLDL immunoglobulin IgM antibodies produced by B1 cells being atheroprotective, and anti-oxLDL IgG antibodies produced by B2-cell subsets likely being pro-atherogenic (237, 249, 250). Although only small numbers of B cells are found in atherosclerotic lesions, both IgG and IgM antibodies derived from such cells accumulate in the lesions. Recently, Upadhye et al. identified bone marrow B-1a cells that contribute abundantly to IgM production with a unique IgM pool, which includes anti- malondialdehyde-modified LDL (MDA-LDL) antibodies. They also showed that expression of the chemokine receptor CXCR4 is a critical factor for the B-1a localization and production of IgM against oxidation-specific epitopes. Expression of CXCR4 on human B-1 cells was greater in humans with low coronary artery plaque burden, suggesting an antiatherogenic function for the cells (251).
Natural antibodies are antibodies that spontaneously arise without prior infection or defined immune exposure. Most serum antibodies at birth are IgM natural antibodies and, as such, they represent a primitive innate-like “layer” of the adaptive immune system (252, 253). Antigens that are specifically recognized in atherosclerosis include microbial antigens, endogenous heat shock proteins, β-2 glycoprotein I, and modified LDL, especially oxLDL (253). The degree of oxidation of LDL in humans is variable, and two major modifications prepared in vitro—copper-oxidized LDL and MDA-LDL—are recognized by human autoantibodies. Both forms share MDA-lysine as their main oxidation-specific epitope but MDA-LDL contains roughly a 10-fold higher amount of the epitope (254).
IgG and IgM antibodies against oxidation-specific epitopes of oxLDL such as MDA-modified lysine residues or the phosphorylcholine headgroups of oxidized phospholipids have also been found in the plasma, and oxLDL-immune complexes are present in plasma (255) as well as in atherosclerotic lesions of humans and animals (212). Several clinical studies investigated the association of oxLDL autoantibodies with atherosclerosis progression, and in most, atherosclerosis or its progression was directly associated with oxLDL IgG levels, and inversely associated with IgM (256, 257) [reviewed in (240)]. While oxidation-specific epitopes are also commonly found on surfaces of apoptotic cells, microvesicles, and bacteria (195), antibodies against the epitopes were shown to have the capacity to block oxLDL uptake (258), the case for their role in atherogenesis being strong.
Antibodies against carbamylated LDL have also been found in individuals with uremia or tobacco smoking, and these antibodies also cross-react with MDA- and malondialdehyde acetaldehyde-LDL (259, 260). Further, also IgG against AGE-LDL has been found in human serum and to be associated with diabetes mellitus (261).
Inflammation is usually followed by its resolution, an active process that reduces leukocyte recruitment, stimulates efferocytosis, repairs tissue damage, and ultimately restores tissue homeostasis. It is partly mediated by proteins such as the anti-inflammatory cytokine IL-10 (119), endogenous gases such as nitric oxide (262), and also by regulatory T cells (263). In recent years, specialized pro-resolving lipid mediators, among others, were discovered to play major roles in mediating the resolution of atherosclerotic lesions [reviewed in (264)]. In the progressing atherosclerotic lesions, efferocytosis of lipid-laden apoptotic cells is impaired, a malfunction that leads to chronic inflammation and a constant influx of circulating monocytes, thus promoting the growth of a necrotic lipid core (263). In macrophage foam cells of atherosclerotic plaques, the lipid mediators, such as 5-lipoxygenase, show nuclear localization, thereby shifting their metabolism to promote the generation of pro-inflammatory leukotrienes and to decrease the production of specialized pro-resolving mediators (265).
OxLDL has been shown to impair efferocytosis in mouse macrophages and to induce apoptosis (266), potentially due to oxidized phospholipids [reviewed in (267)] and cholesterol (268) which are potent mediators of inflammation and influence the ability of the macrophages to perform this function of cell removal. Copper-oxidized LDL was also found to contain lipofuscin-like fluorophores (269), known inducers of the NLRP3-inflammasome (270) and of necroptosis (271). Oleic and linoleic acids associated with E-LDL induced IL-8 expression in ECs (143). Ceramides arising from SMase activity have been shown to inhibit efferocytosis by alveolar macrophages (272, 273). PLA2-LDL induces the release of arachidonic acid from ECs (147). Arachidonic acid can be converted into prostaglandins, thromboxanes, and leukotrienes, all of which are potent modulators of inflammation. PLA2 treatment also releases LPC which is a well-known chemotactic eat-me signal during apoptosis, and which also increases the expression of ICAM-1 and VCAM-1 in ECs and enhances monocyte binding to the endothelium (274). Thus, modified lipoproteins and their cargo directly contribute to impaired resolution in atherogenesis.
HDLs are considered antiatherogenic [reviewed in (275)]. Firstly, low levels of HDL-C are present in many conditions that are associated with an increased ASCVD risk. The most prominent antiatherogenic property associated with HDL is their ability to induce macrophage cholesterol efflux. HDL also enhances the endothelial synthesis of the vasodilator nitric oxide and thus can also ameliorate endothelial dysfunction. Furthermore, HDL reduces coronary atherosclerosis by decreasing the expression of adhesion molecules on ECs and thereby reducing the infiltration of pro-inflammatory cells and LDL into the subendothelial space. HDL also inhibits lipid oxidation and exerts anti-inflammatory and antiapoptotic actions [reviewed in (275)].
However, oxidative stress can modify HDL. Within the subendothelial space, apoA-I can be oxidized on multiple residues by the proinflammatory enzyme myeloperoxidase (275, 276), and the resulting dysfunctional HDL is associated with an increased incidence of cardiovascular events [reviewed in (218)]. During inflammatory states, inflammatory cytokines induce hepatic expression of the acute-phase serum amyloid A (277) and group IIa sPLA2 (278), which then leads to the formation of HDL particles that are relatively enriched in SAA, while depleted of apoA-I and phospholipids. Also, increased oxidative stress during inflammation generates dysfunctional HDL that contains oxidatively modified or carbamylated apoA-I.
HDL remodeling during inflammation also leads to the loss of HDL-associated enzymes such as serum paraoxonase and arylesterases 1 and 3 that normally impede peroxidation of LDL particles; so, the loss of these enzymes decreases the antioxidative and anti-inflammatory capacity of HDL (279).
ApoE-/- mice with dysfunctional HDL displayed an increased cholesterol efflux capacity and lipoprotein peroxidation when treated with an apoA-I mimetic peptide (280). This peptide has also been able to attenuate LDL aggregation after hydrolysis by SMase provided the peptide was added prior to hydrolysis (184).
Impaired cholesterol efflux capacity of HDL correlates with the severity of atherosclerosis, including the early stages of disease development (281, 282). Thus, cholesterol efflux and resulting foam cell formation can be attributed to the dysfunctional HDL rather than the alterations of circulating levels of normal HDL particles, making dysfunctional HDL a better biomarker of atherosclerosis than the level of HDL-cholesterol (168). Mast cells are present in atherosclerotic lesions where they become activated to secrete heparin-bound neutral proteases capable of degrading apoA-I (283). The mast cell proteases avidly degrade lipid-poor HDL particles and thereby prevent their ability to induce cholesterol efflux from macrophage foam cells, i.e., the first step of reverse cholesterol transport (283). We consider that the HDL-modifying enzymes and agents present in inflamed atherosclerotic lesions are the most critical components of the malfunctioning reverse cholesterol transport system. Accordingly, emerging knowledge and understanding of the various steps of reverse cholesterol transport, particularly of the initiation step when macrophage foam cells and HDL particles interact in an atherosclerotic lesion, is crucially important when attempting to develop novel HDL-targeted therapies (284).
As discussed in this Review, the apoB-containing lipoprotein particles and the lipids contained in them are critical players in atherogenesis. A large and overwhelming body of evidence strengthens the causal role for the cholesterol contained in the LDL particles throughout all stages of atherogenesis (285). Indeed, without cholesterol there is no atherosclerosis. Of particular clinical significance are the clinical observations on lifelong high or low concentrations of LDL-cholesterol (LDL-C). Thus, in patients with familial hypercholesterolemia, the genetic absence of LDL-receptors, which normally remove LDL particles from the circulation, leads to high LDL-C concentrations since birth and inevitably results in accelerated development of coronary atherosclerosis and its clinical sequelae, such as acute myocardial infarction (286). In contrast, individuals with a life-long history of low LDL-C levels due to genetic defects in the function of the LDL-receptor-suppressing PCSK9 molecule have a dramatic reduction in coronary heart disease even in the presence of other risk factors (287). Finally, the ability of therapeutic lowering of LDL-C has proven the essential role of cholesterol in atherosclerosis and its clinical sequelae (288).
Considering the multitude of inflammatory pathways involved in atherogenesis as discussed here, we wish to add that there is no atherosclerosis without superimposed inflammation in the arterial wall. The contribution of inflammation to the development of atherosclerosis has received support from the findings that statin drugs exert anti-inflammatory effects on atherosclerotic lesions and reduce clinical events due to coronary atherosclerosis, and that part of these beneficial effects may be independent of LDL-C lowering (289). The clinical trials have also demonstrated that many patients remain at increased risk because of persistent elevation in high-sensitivity CRP despite significant reductions in LDL-C level, and that this “residual inflammatory risk” is a viable pharmacologic target of ASCVD (290).
Recent trials have successfully demonstrated the ability of anti-inflammatory interventions to reduce recurrent cardiovascular events even in patients with optimal control of LDL-C level (291). In the Canakinumab Anti-inflammatory Thrombosis Outcomes Study (CANTOS) (292), the pro-inflammatory cytokine IL-1β was inhibited by administering systemically the monoclonal antibody canakinumab to patients after myocardial infarction, and a reduction of recurrent major adverse cardiovascular events was observed (292). Since then, clinical trials testing anti-inflammatory compounds have shown beneficial effects on cardiovascular outcomes. Reduction of cardiovascular events has been observed in several trials involving, in addition to the state-of-the-art lipid-lowering drug regimen, the broad anti-inflammatory drug colchicine. Thus, colchicine was used in the Colchicine Cardiovascular Outcomes Trial (COLCOT) in individuals with a recent myocardial infarction (293), and in the low-dose colchicine trials LoDoCo1 (294) and LoDoCo2 involving individuals with clinically chronic stable coronary disease (295). The pro-inflammatory cytokine IL-6 was targeted by the specific antibody tocilizumab in the ASSessing the effect of Anti-IL-6 treatment in MI (ASSAIL-MI) trial (296, 297), and the treatment attenuated the inflammatory response and the degree of myocardial damage in patients with non-ST-elevation myocardial infarction. Moreover, after acute myocardial infarction hydroxychlorine reduced the levels of IL-6 at least for a period of 32 months suggesting that this anti-inflammatory drug may reduce cardiovascular events after such an event (298). However, other anti-inflammatory drugs, like the immunosuppressant methotrexate (299) or the p38 mitogen-activated protein kinase (MAPK) inhibitor Losmapimod (300) did not show any effects revealing the specificity of the clinically relevant anti-inflammatory pathways. Of concern are the observations that systemic anti-inflammatory treatments with IL-1β antibodies increased the individual's susceptibility to fatal infections (301), and that the COLCOT colchicine trial saw a small increase in pneumonia (291).
Interestingly, the adverse effects of air pollutants may occur to a large part via generation of ROS and reactive nitrogen species from organic chemicals present in the inhaled particulate matter (302). Pulmonary oxidative stress and inflammation also results from the activation of pulmonary epithelial cells, macrophages, neutrophils, and ECs by the particulate matter deposited in the lungs. Moreover, the ultrafine particulate matter can reach the systemic circulation where the particles have the potential to directly oxidize HDL or other circulating lipoproteins. Taken together, the pollutants promote the generation of oxLDL, oxHDL, and induce a prothrombotic dysfunction of the coronary endothelium. By causing systemic oxidative stress and inflammation, the air pollutants form one arm in the “common soil” of the cardio-pulmonary continuum (303).
Regarding the development of diabetic vascular injury, the cellular responses to AGEs are thought to largely depend on the interaction between the AGE and AGE-specific cell surface receptors, the RAGEs, which via activation of the transcription factor NF-κB leads to a range of proinflammatory reactions and induction of oxidant stress in the cells involved (304). Since AGE formation may lead to oxidative stress and oxidative stress may accelerate the formation of specific AGEs, both AGE-LDL and ox-LDL could be involved in the accelerated development of atherosclerosis in patients with type 2 diabetes (305, 306). Based on the above findings, we may postulate that the observations of specific AGEs being associated with the severity of subclinical atherosclerosis and macrovascular complications in patients with long-standing type 2 diabetes, and the requirement of long-standing glucose-lowering (for 10 years) to lower cardiovascular risk, both could at least partly depend on the generation of AGE-LDL in these patients (307, 308). Indeed, among the molecular mechanisms of how hyperglycemia promotes atherosclerosis, the direct protein-modifying effects of glucose appear to be prominent, the glycated LDL particles being both pro-inflammatory and foam cell-forming species of modified lipoproteins (309).
The atherogenic potential of cholesterol in TG-rich lipoprotein particles (TGRLs) and their remnants is being increasingly acknowledged (310–313). Indeed, elevated remnant cholesterol associates with increased risk for acute myocardial infarction, ischemic stroke and, even stronger for peripheral arterial disease, which is also considered to have a strong atherosclerotic component in its pathogenesis (314). The clinical significance of the TGRL remnants has been mostly assigned to their ability to enter the arterial intima and to contribute to the formation of foam cells. However, the mechanisms by which postprandial lipemia induces atherosclerosis also involve induction of endothelial dysfunction, oxidative stress, and inflammation (315). Importantly, the lipoprotein lipase -mediated hydrolytic modification of the TGRLs takes place not only in capillaries but also in atherosclerosis-susceptible arterial segments where it results in high local concentrations of lipolytic products, such as oxidized free fatty acids which jointly with the TGRLs cause a multitude of inflammatory reactions in the ECs and the cells within the intima (316, 317).
Modified lipoproteins are the main drivers of atherogenic inflammation of the arterial wall (Figure 3). To decrease the vascular wall inflammation due to modified lipoprotein several concepts are being discussed. Without the entry of cholesterol-containing apoB-lipoproteins into the arterial wall, there is no atherosclerosis. Moreover, it is the current consensus that local inflammation in the evolving atherosclerotic lesions is an inherent part of atherogenesis. Regarding the therapeutic options of atherosclerotic cardiovascular diseases, the lipid-lowering and anti-inflammatory therapies do not compete, but they are complementary and compatible. Currently, a myriad of new treatment options of both dyslipidemia and inflammation in ASCVD is emerging, to mention the availability of RNA-based inhibitors of the hepatic synthesis of apoB-containing lipoproteins which could be administered even once a year (318). Moreover, ASCVD-related events could be substantially reduced by prescription-grade eicosapentaenoic acid in patients with hypertriglyceridemia (319). In that study, the beneficial effect likely at least to some extent resulted from lowering cholesterol accumulation and reduction of inflammation in the arterial wall via changes in composition of lipoproteins and cells.
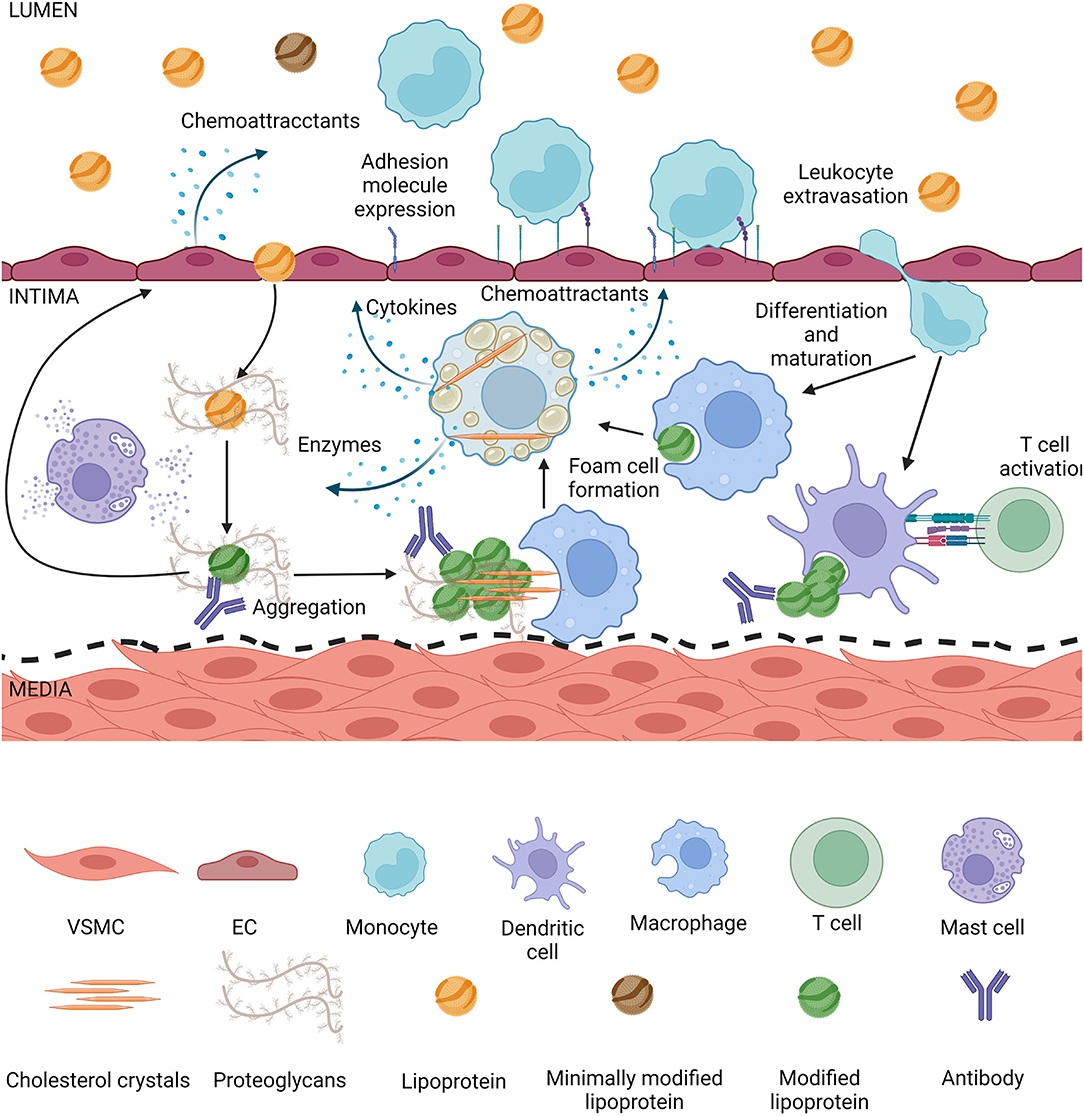
Figure 3. Lipoproteins modified in the intima induce inflammation. Lipoproteins smaller than 80 nm can enter the intima by transcytosis, they get trapped by proteoglycans and during retention they are exposed to enzymes secreted by intimal cells. Modified lipoproteins tend to aggregate, depending on the modification cholesterol crystals can form. Modified lipoproteins also are recognized by antibodies and form immunocomplexes in the intima. The aggregates and crystals can be phagocytized by macrophages or dendritic cells, which can form foam cells or activate T cells, respectively. Mast cells are degranulated during atherogenesis, leading to matrix degradation and lipoprotein degradation. Activated macrophage foam cells secrete more enzymes thus increasing the rate of modification, they secrete cytokines inducing an inflammatory state in the intima, as well as chemoattractants. Endothelial cells either directly by contact with modified lipoproteins or due to pro-inflammatory cytokines from foam cells themselves secrete chemoattractants and increase expression of adhesion molecules, increasing the rate of leukocyte extravasation into the intima. Endothelial activation and dysfunction also lead to a higher rate of lipoproteins entering the intima.
Finally, the perception that inflammation would replace traditional risk factors of atherosclerosis poses a false dichotomy (320). Currently, lowering the concentration of the apoB-containing lipoproteins and their remnants either by dietary or pharmacological means is the mainstream approach to combat atherosclerosis both in the primary and secondary prevention settings, while the anti-inflammatory therapies serve as adjunct therapies in the setting of secondary prevention strategies. This chronology adopts our traditional understanding of cholesterol accumulation being the primary event and an inflammatory reaction being a secondary event taking place in the arterial intima, the concept originating in the early work dealing with animal models of atherosclerosis. This basic concept is also reflected in the present-day clinical approach in which the inflammation is considered to present a residual cardiovascular risk after very efficient lowering of LDL-C in patients with clinically manifest ASCVD (321). However, as discussed in this review in-depth, we may consider the cholesterol- and TG-containing lipoproteins entering the arterial intima as innocent bystanders, who are attacked by the “healthy” resident macrophages which have been present in the intima lifelong, and which are performing their physiological patrol tasks in this tissue like they do in any tissue (322). Then, it is, after all the close encounter of the two which ignites and sustains the atherogenic process in which cholesterol accumulation and inflammation coexist. This novel understanding is necessary when aiming at developing new anti-atherosclerotic therapies aiming at primary prevention of ASCVD, as it dictates that the inflammatory component is not only a residual risk but a risk existing throughout the development of atherosclerosis. Recent results obtained in mice with early atherosclerosis revealed that inhibition of IL-1β and NLRP3 inflammasome reduces leukocyte accumulation in atherosclerotic aortas (323). Thus, we may predict that cardiovascular interventions utilizing lowering the levels of circulating apoB-containing lipoproteins, prevention of their modifications, and reduction of the numbers of lesional macrophages already at an initial or early stage of the long subclinical asymptomatic phase of atherosclerosis may ultimately provide us the tools required for full eradication of the clinically manifest ASCVD.
ML, KÖ, and PK designed, wrote, and edited the manuscript. All authors contributed to the article and approved the submitted version.
Wihuri Research Institute was maintained by the Jenny and Antti Wihuri Foundation. This study was also supported by grants from the Academy of Finland (#332564), Novo Nordisk Fonden (#NNF19OC0057411), the Finnish Foundation for Cardiovascular Research, and the Aarne Koskelo Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Stary HC. Lipid and macrophage accumulations in arteries of children and the development of atherosclerosis. Am J Clin Nutr. (2000) 72(5 Suppl):1297s–306. doi: 10.1093/ajcn/72.5.1297s
2. Stary HC. Macrophages, macrophage foam cells, and eccentric intimal thickening in the coronary arteries of young children. Atherosclerosis. (1987) 64:91–108. doi: 10.1016/0021-9150(87)90234-6
3. Stary HC, Blankenhorn DH, Chandler AB, Glagov S, Insull W Jr., Richardson M, et al. A definition of the intima of human arteries and of its atherosclerosis-prone regions. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Arterioscler Thromb. (1992) 12:120–34. doi: 10.1161/01.ATV.12.1.120
4. Borén J, Williams KJ. The central role of arterial retention of cholesterol-rich apolipoprotein-B-containing lipoproteins in the pathogenesis of atherosclerosis: a triumph of simplicity. Curr Opin Lipidol. (2016) 27:473–83. doi: 10.1097/MOL.0000000000000330
5. Williams KJ, Tabas I. The response-to-retention hypothesis of early atherogenesis. Arterioscler Thromb Vasc Biol. (1995) 15:551–61. doi: 10.1161/01.ATV.15.5.551
6. Borén J, Chapman MJ, Krauss RM, Packard CJ, Bentzon JF, Binder CJ, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. (2020) 41:2313–30. doi: 10.1093/eurheartj/ehz962
7. Skålén K, Gustafsson M, Rydberg EK, Hultén LM, Wiklund O, Innerarity TL, et al. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature. (2002) 417:750–4. doi: 10.1038/nature00804
8. Öörni K, Pentikäinen MO, Ala-Korpela M, Kovanen PT. Aggregation, fusion, and vesicle formation of modified low density lipoprotein particles: molecular mechanisms and effects on matrix interactions. J Lipid Res. (2000) 41:1703–14. doi: 10.1016/S0022-2275(20)31964-7
9. Lu M, Gursky O. Aggregation and fusion of low-density lipoproteins in vivo and in vitro. Biomol Concepts. (2013) 4:501–18. doi: 10.1515/bmc-2013-0016
10. Öörni K, Kovanen PT. Aggregation susceptibility of low-density lipoproteins-A novel modifiable biomarker of cardiovascular risk. J Clin Med. (2021) 10:1769. doi: 10.3390/jcm10081769
11. Stary HC. Macrophage foam cells in the coronary artery intima of human infants. Ann N Y Acad Sci. (1985) 454:5–8. doi: 10.1111/j.1749-6632.1985.tb11839.x
12. Stary HC. Evolution and progression of atherosclerotic lesions in coronary arteries of children and young adults. Arteriosclerosis. (1989) 9(1 Suppl):I19–32.
13. Stary HC, Chandler AB, Glagov S, Guyton JR, Insull W Jr., Rosenfeld ME, et al. A definition of initial, fatty streak, and intermediate lesions of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. (1994) 89:2462–78. doi: 10.1161/01.CIR.89.5.2462
14. Stary HC, Chandler AB, Dinsmore RE, Fuster V, Glagov S, Insull W Jr., et al. A definition of advanced types of atherosclerotic lesions and a histological classification of atherosclerosis. A report from the Committee on Vascular Lesions of the Council on Arteriosclerosis, American Heart Association. Circulation. (1995) 92:1355–74. doi: 10.1161/01.CIR.92.5.1355
15. Fuster V, Moreno PR, Fayad ZA, Corti R, Badimon JJ. Atherothrombosis and high-risk plaque: part I: evolving concepts. J Am Coll Cardiol. (2005) 46:937–54. doi: 10.1016/j.jacc.2005.03.074
16. Virmani R, Burke AP, Kolodgie FD, Farb A. Pathology of the thin-cap fibroatheroma: a type of vulnerable plaque. J Interv Cardiol. (2003) 16:267–72. doi: 10.1034/j.1600-0854.2003.8042.x
17. Bentzon JF, Otsuka F, Virmani R, Falk E. Mechanisms of plaque formation and rupture. Circ Res. (2014) 114:1852–66. doi: 10.1161/CIRCRESAHA.114.302721
18. Pentikäinen MO, Öörni K, Ala-Korpela M, Kovanen PT. Modified LDL – trigger of atherosclerosis and inflammation in the arterial intima. J Intern Med. (2000) 247:359–70. doi: 10.1046/j.1365-2796.2000.00655.x
19. Jang E, Robert J, Rohrer L, von Eckardstein A, Lee WL. Transendothelial transport of lipoproteins. Atherosclerosis. (2020) 315:111–25. doi: 10.1016/j.atherosclerosis.2020.09.020
20. Tailleux A, Torpier G, Caron B, Fruchart JC, Fievet C. Immunological properties of apoB-containing lipoprotein particles in human atherosclerotic arteries. J Lipid Res. (1993) 34:719–28. doi: 10.1016/S0022-2275(20)39693-0
21. Heffron SP, Ruuth MK, Xia Y, Hernandez G, Äikäs L, Rodriguez C, et al. Low-density lipoprotein aggregation predicts adverse cardiovascular events in peripheral artery disease. Atherosclerosis. (2021) 316:53–7. doi: 10.1016/j.atherosclerosis.2020.11.016
22. Ruuth M, Nguyen SD, Vihervaara T, Hilvo M, Laajala TD, Kondadi PK, et al. Susceptibility of low-density lipoprotein particles to aggregate depends on particle lipidome, is modifiable, and associates with future cardiovascular deaths. Eur Heart J. (2018) 39:2562–73. doi: 10.1093/eurheartj/ehy319
23. Maaninka K, Nguyen SD, Mäyränpää MI, Plihtari R, Rajamäki K, Lindsberg PJ, et al. Human mast cell neutral proteases generate modified LDL particles with increased proteoglycan binding. Atherosclerosis. (2018) 275:390–9. doi: 10.1016/j.atherosclerosis.2018.04.016
24. Oorni K, Sneck M, Bromme D, Pentikainen MO, Lindstedt KA, Mayranpaa M, et al. Cysteine protease cathepsin F is expressed in human atherosclerotic lesions, is secreted by cultured macrophages, and modifies low density lipoprotein particles in vitro. J Biol Chem. (2004) 279:34776–84. doi: 10.1074/jbc.M310814200
25. Galis ZS, Sukhova GK, Lark MW, Libby P. Increased expression of matrix metalloproteinases and matrix degrading activity in vulnerable regions of human atherosclerotic plaques. J Clin Invest. (1994) 94:2493–503. doi: 10.1172/JCI117619
26. Sukhova GK, Zhang Y, Pan JH, Wada Y, Yamamoto T, Naito M, et al. Deficiency of cathepsin S reduces atherosclerosis in LDL receptor-deficient mice. J Clin Invest. (2003) 111:897–906. doi: 10.1172/JCI200314915
27. Newby AC. Metalloproteinase expression in monocytes and macrophages and its relationship to atherosclerotic plaque instability. Arterioscler Thromb Vasc Biol. (2008) 28:2108–14. doi: 10.1161/ATVBAHA.108.173898
28. Piha M, Lindstedt L, Kovanen PT. Fusion of proteolyzed low-density lipoprotein in the fluid phase: a novel mechanism generating atherogenic lipoprotein particles. Biochemistry. (1995) 34:10120–9. doi: 10.1021/bi00032a004
29. Kruth HS. The fate of lipoprotein cholesterol entering the arterial wall. Curr Opin Lipidol. (1997) 8:246–52. doi: 10.1097/00041433-199710000-00002
30. Plihtari R, Hurt-Camejo E, Öörni K, Kovanen PT. Proteolysis sensitizes LDL particles to phospholipolysis by secretory phospholipase A2 group V and secretory sphingomyelinase. J Lipid Res. (2010) 51:1801–9. doi: 10.1194/jlr.M003103
31. Bhakdi S, Dorweiler B, Kirchmann R, Torzewski J, Weise E, Tranum-Jensen J, et al. On the pathogenesis of atherosclerosis: enzymatic transformation of human low density lipoprotein to an atherogenic moiety. J Exp Med. (1995) 182:1959–71. doi: 10.1084/jem.182.6.1959
32. Torzewski M, Suriyaphol P, Paprotka K, Spath L, Ochsenhirt V, Schmitt A, et al. Enzymatic modification of low-density lipoprotein in the arterial wall. Arterioscler Thromb Vasc Biol. (2004) 24:2130–6. doi: 10.1161/01.ATV.0000144016.85221.66
33. Seifert PS, Hugo F, Tranum-Jensen J, Zâhringer U, Muhly M, Bhakdi S. Isolation and characterization of a complement-activating lipid extracted from human atherosclerotic lesions. J Exp Med. (1990) 172:547–57. doi: 10.1084/jem.172.2.547
34. Esterbauer H, Gebicki J, Puhl H, Jürgens G. The role of lipid peroxidation and antioxidants in oxidative modification of LDL. Free Radic Biol Med. (1992) 13:341–90. doi: 10.1016/0891-5849(92)90181-F
35. Pentikäinen MO, Oörni K, Kovanen PT. Lipoprotein lipase (LPL) strongly links native and oxidized low density lipoprotein particles to decorin-coated collagen. Roles for both dimeric and monomeric forms of LPL. J Biol Chem. (2000) 275:5694–701. doi: 10.1074/jbc.275.8.5694
37. Kaplan M, Aviram M. Retention of oxidized LDL by extracellular matrix proteoglycans leads to its uptake by macrophages: an alternative approach to study lipoproteins cellular uptake. Arterioscler Thromb Vasc Biol. (2001) 21:386–93. doi: 10.1161/01.ATV.21.3.386
38. Santoso A, Heriansyah T, Rohman MS. Phospholipase A2 is an inflammatory predictor in cardiovascular diseases: is there any spacious room to prove the causation? Curr Cardiol Rev. (2020) 16:3–10. doi: 10.2174/1573403X15666190531111932
39. Epps KC, Wilensky RL. Lp-PLA2- a novel risk factor for high-risk coronary and carotid artery disease. J Intern Med. (2011) 269:94–106. doi: 10.1111/j.1365-2796.2010.02297.x
40. Tellis CC, Tselepis AD. The role of lipoprotein-associated phospholipase A2 in atherosclerosis may depend on its lipoprotein carrier in plasma. Biochimica et Biophysica Acta (BBA) Mol Cell Biol Lipids. (2009) 1791:327–38. doi: 10.1016/j.bbalip.2009.02.015
41. Kolodgie FD, Burke AP, Skorija KS, Ladich E, Kutys R, Makuria AT, et al. Lipoprotein-associated phospholipase A2 protein expression in the natural progression of human coronary atherosclerosis. Arterioscler Thromb Vasc Biol. (2006) 26:2523–9. doi: 10.1161/01.ATV.0000244681.72738.bc
42. Rosengren B, Peilot H, Umaerus M, Jönsson-Rylander AC, Mattsson-Hultén L, Hallberg C, et al. Secretory phospholipase A2 group V: lesion distribution, activation by arterial proteoglycans, and induction in aorta by a Western diet. Arterioscler Thromb Vasc Biol. (2006) 26:1579–85. doi: 10.1161/01.ATV.0000221231.56617.67
43. Yamamoto K, Isogai Y, Sato H, Taketomi Y, Murakami M. Secreted phospholipase A2, lipoprotein hydrolysis, and atherosclerosis: integration with lipidomics. Anal Bioanal Chem. (2011) 400:1829–42. doi: 10.1007/s00216-011-4864-z
44. Öörni K, Hakala JK, Annila A, Ala-Korpela M, Kovanen PT. Sphingomyelinase induces aggregation and fusion, but phospholipase A2 only aggregation, of low density lipoprotein (LDL) particles: two distinct mechanisms leading to increased binding strength of LDL to human aortic proteoglycans. J Biol Chem. (1998) 273:29127–34. doi: 10.1074/jbc.273.44.29127
45. Lähdesmäki K, Plihtari R, Soininen P, Hurt-Camejo E, Ala-Korpela M, Öörni K, et al. Phospholipase A2-modified LDL particles retain the generated hydrolytic products and are more atherogenic at acidic pH. Atherosclerosis. (2009) 207:352–9. doi: 10.1016/j.atherosclerosis.2009.04.031
46. Boyanovsky BB, van der Westhuyzen DR, Webb NR. Group V secretory phospholipase A2-modified low density lipoprotein promotes foam cell formation by a SR-A- and CD36-independent process that involves cellular proteoglycans. J Biol Chem. (2005) 280:32746–52. doi: 10.1074/jbc.M502067200
47. Menschikowski M, Lattke P, Bergmann S, Jaross W. Exposure of macrophages to PLA2-modified lipoproteins leads to cellular lipid accumulations. Anal Cell Pathol. (1995) 9:113–21.
48. Sneck M, Nguyen SD, Pihlajamaa T, Yohannes G, Riekkola M-L, Milne R, et al. Conformational changes of apoB-100 in SMase-modified LDL mediate formation of large aggregates at acidic pH. J Lipid Res. (2012) 53:1832–9. doi: 10.1194/jlr.M023218
49. Schissel SL, Tweedie-Hardman J, Rapp JH, Graham G, Williams KJ, Tabas I. Rabbit aorta and human atherosclerotic lesions hydrolyze the sphingomyelin of retained low-density lipoprotein. Proposed role for arterial-wall sphingomyelinase in subendothelial retention and aggregation of atherogenic lipoproteins. J Clin Invest. (1996) 98:1455–64. doi: 10.1172/JCI118934
50. Oorni K. Sphingomyelinase induces aggregation and fusion of small very low-density lipoprotein and intermediate-density lipoprotein particles and increases their retention to human arterial proteoglycans. Arterioscler Thromb Vasc Biol. (2005) 25:1678–83. doi: 10.1161/01.ATV.0000168912.42941.60
51. Marathe S, Choi Y, Leventhal AR, Tabas I. Sphingomyelinase converts lipoproteins from apolipoprotein E knockout mice into potent inducers of macrophage foam cell formation. Arterioscler Thromb Vasc Biol. (2000) 20:2607–13. doi: 10.1161/01.ATV.20.12.2607
52. Han CY, Pak YK. Oxidation-dependent effects of oxidized LDL: proliferation or cell death. Exp Mol Med. (1999) 31:165–73. doi: 10.1038/emm.1999.27
53. Walters MJ, Wrenn SP. Size-selective uptake of colloidal low density lipoprotein aggregates by cultured white blood cells. J Colloid Interface Sci. (2010) 350:494–501. doi: 10.1016/j.jcis.2010.06.059
54. Hakala JK, Oksjoki R, Laine P, Du H, Grabowski GA, Kovanen PT, et al. Lysosomal enzymes are released from cultured human macrophages, hydrolyze LDL in vitro, and are present extracellularly in human atherosclerotic lesions. Arterioscler Thromb Vasc Biol. (2003) 23:1430–6. doi: 10.1161/01.ATV.0000077207.49221.06
55. Torzewski M, Klouche M, Hock J, Meßner M, Dorweiler B, Torzewski J, et al. Immunohistochemical demonstration of enzymatically modified human LDL and its colocalization with the terminal complement complex in the early atherosclerotic lesion. Arterioscler Thromb Vasc Biol. (1998) 18:369–78. doi: 10.1161/01.ATV.18.3.369
56. Lehti S, Nguyen SD, Belevich I, Vihinen H, Heikkilä HM, Soliymani R, et al. Extracellular lipids accumulate in human carotid arteries as distinct three-dimensional structures and have proinflammatory properties. Am J Pathol. (2018) 188:525–38. doi: 10.1016/j.ajpath.2017.09.019
57. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflamasomes are required for atherogenesis and activated by cholesterol crystals that form early in disease. Nature. (2010) 464:1357–61. doi: 10.1038/nature08938
58. Roh JS, Sohn DH. Damage-associated molecular patterns in inflammatory diseases. Immune Netw. (2018) 18:e27. doi: 10.4110/in.2018.18.e27
59. Kim H, Conway EM. Platelets and complement cross-talk in early atherogenesis. Front Cardiovasc Med. (2019) 6:131. doi: 10.3389/fcvm.2019.00131
60. Baines AC, Brodsky RA. Complementopathies. Blood Rev. (2017) 31:213–23. doi: 10.1016/j.blre.2017.02.003
61. Elieh Ali Komi D, Shafaghat F, Kovanen PT, Meri S. Mast cells and complement system: ancient interactions between components of innate immunity. Allergy. (2020) 75:2818–28. doi: 10.1111/all.14413
62. Garcia-Arguinzonis M, Diaz-Riera E, Peña E, Escate R, Juan-Babot O, Mata P, et al. Alternative C3 complement system: lipids and atherosclerosis. Int J Mol Sci. (2021) 22:5122. doi: 10.3390/ijms22105122
63. Oksjoki R, Kovanen PT, Pentikäinen MO. Role of complement activation in atherosclerosis. Curr Opin Lipidol. (2003) 14:477–82. doi: 10.1097/00041433-200310000-00008
64. Martin-Ventura JL, Martinez-Lopez D, Roldan-Montero R, Gomez-Guerrero C, Blanco-Colio LM. Role of complement system in pathological remodeling of the vascular wall. Mol Immunol. (2019) 114:207–15. doi: 10.1016/j.molimm.2019.06.016
65. Oksjoki R, Kovanen PT, Meri S, Pentikainen MO. Function and regulation of the complement system in cardiovascular diseases. Front Biosci. (2007) 12:4696–708. doi: 10.2741/2419
66. Wieland E, Dorweiler B, Bonitz U, Lieser S, Walev I, Bhakdi S. Complement activation by oxidatively modified low-density lipoproteins. Eur J Clin Invest. (1999) 29:835–41. doi: 10.1046/j.1365-2362.1999.00548.x
67. Torzewski M. Enzymatically modified LDL, atherosclerosis and beyond: paving the way to acceptance. Front Biosci. (2018) 23:1257–71. doi: 10.2741/4642
68. Arlaud GJ, Biro A, Ling WL. Enzymatically modified low-density lipoprotein is recognized by c1q and activates the classical complement pathway. J Lipids. (2011) 2011:376092. doi: 10.1155/2011/376092
69. Torzewski M. The initial human atherosclerotic lesion and lipoprotein modification-a deep connection. Int J Mol Sci. (2021) 22:11488 doi: 10.3390/ijms222111488
70. Allahverdian S, Chehroudi AC, McManus BM, Abraham T, Francis GA. Contribution of intimal smooth muscle cells to cholesterol accumulation and macrophage-like cells in human atherosclerosis. Circulation. (2014) 129:1551–9. doi: 10.1161/CIRCULATIONAHA.113.005015
71. Pryma CS, Ortega C, Dubland JA, Francis GA. Pathways of smooth muscle foam cell formation in atherosclerosis. Curr Opin Lipidol. (2019) 30:117–24. doi: 10.1097/MOL.0000000000000574
72. Mineo C. Lipoprotein receptor signalling in atherosclerosis. Cardiovasc Res. (2020) 116:1254–74. doi: 10.1093/cvr/cvz338
73. Lopes-Virella MF, Binzafar N, Rackley S, Takei A, La Via M, Virella G. The uptake of LDL-IC by human macrophages: predominant involvement of the Fc gamma RI receptor. Atherosclerosis. (1997) 135:161–70. doi: 10.1016/S0021-9150(97)00157-3
74. Kruth HS. Sequestration of aggregated low-density lipoproteins by macrophages. Curr Opin Lipidol. (2002) 13:483–8. doi: 10.1097/00041433-200210000-00003
75. Rajamäki K, Lappalainen J, Öörni K, Välimäki E, Matikainen S, Kovanen PT, et al. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS ONE. (2010) 5:e11765. doi: 10.1371/journal.pone.0011765
76. Öörni K, Lehti S, Sjövall P, Kovanen PT. Triglyceride-rich lipoproteins as a source of proinflammatory lipids in the arterial wall. Curr Med Chem. (2018) 25:1–10. doi: 10.2174/0929867325666180530094819
77. Haka AS, Grosheva I, Chiang E, Buxbaum AR, Baird BA, Pierini LM, et al. Macrophages create an acidic extracellular hydrolytic compartment to digest aggregated lipoproteins. (2009) 20:4932–40. doi: 10.1091/mbc.e09-07-0559
78. Haka AS, Singh RK, Grosheva I, Hoffner H, Capetillo-Zarate E, Chin HF, et al. Monocyte-derived dendritic cells upregulate extracellular catabolism of aggregated low-density lipoprotein on maturation, leading to foam cell formationsignificance. (2015) 35:2092–103. doi: 10.1161/ATVBAHA.115.305843
79. Singh RK, Barbosa-Lorenzi VC, Lund FW, Grosheva I, Maxfield FR, Haka AS. Degradation of aggregated LDL occurs in complex extracellular sub-compartments of the lysosomal synapse. (2016) 129:1072–82. doi: 10.1242/jcs.181743
80. Grosheva I, Haka AS, Qin C, Pierini LM, Maxfield FR. Aggregated LDL in contact with macrophages induces local increases in free cholesterol levels that regulate local actin polymerization. Arterioscler Thromb Vasc Biol. (2009) 29:1615–21. doi: 10.1161/ATVBAHA.109.191882
81. Singh RK, Haka AS, Asmal A, Barbosa-Lorenzi VC, Grosheva I, Chin HF, et al. TLR4 (toll-like receptor 4)-dependent signaling drives extracellular catabolism of ldl (low-density lipoprotein) aggregates. Arterioscler Thromb Vasc Biol. (2020) 40:86–102. doi: 10.1161/ATVBAHA.119.313200
82. Öörni K, Rajamäki K, Nguyen SD, Lähdesmäki K, Plihtari R, Lee-Rueckert M, et al. Acidification of the intimal fluid: the perfect storm for atherogenesis. J Lipid Res. (2015) 56:203–14. doi: 10.1194/jlr.R050252
83. Brown MS, Goldstein JL. Lipoprotein metabolism in the macrophage: implications for cholesterol deposition in atherosclerosis. Annu Rev Biochem. (1983) 52:223–61. doi: 10.1146/annurev.bi.52.070183.001255
84. Ahmad F, Leake DS. Lysosomal oxidation of LDL alters lysosomal pH, induces senescence, and increases secretion of pro-inflammatory cytokines in human macrophages. J Lipid Res. (2019) 60:98–110. doi: 10.1194/jlr.M088245
85. Ahmad F, Mitchell RD, Houben T, Palo A, Yadati T, Parnell AJ, et al. Cysteamine decreases low-density lipoprotein oxidation, causes regression of atherosclerosis, and improves liver and muscle function in low-density lipoprotein receptor-deficient mice. J Am Heart Assoc. (2021) 10:e017524. doi: 10.1161/JAHA.120.017524
86. Yancey PG, Jerome WG. Lysosomal sequestration of free and esterified cholesterol from oxidized low density lipoprotein in macrophages of different species. J Lipid Res. (1998) 39:1349–61. doi: 10.1016/S0022-2275(20)32515-3
87. Brown AJ, Mander EL, Gelissen IC, Kritharides L, Dean RT, Jessup W. Cholesterol and oxysterol metabolism and subcellular distribution in macrophage foam cells. Accumulation of oxidized esters in lysosomes. J Lipid Res. (2000) 41:226–37. doi: 10.1016/S0022-2275(20)32056-3
88. Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, et al. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol. (2013) 14:812–20. doi: 10.1038/ni.2639
89. Lappalainen J, Yeung N, Nguyen SD, Jauhiainen M, Kovanen PT, Lee-Rueckert M. Cholesterol loading suppresses the atheroinflammatory gene polarization of human macrophages induced by colony stimulating factors. Sci Rep. (2021) 11:4923. doi: 10.1038/s41598-021-84249-y
90. Argmann CA, Sawyez CG, Li S, Nong Z, Hegele RA, Pickering JG, et al. Human smooth muscle cell subpopulations differentially accumulate cholesteryl ester when exposed to native and oxidized lipoproteins. Arterioscler Thromb Vasc Biol. (2004) 24:1290–6. doi: 10.1161/01.ATV.0000131260.80316.37
91. Llorente-Cortés V, Martínez-González J, Badimon L. LDL Receptor–related protein mediates uptake of aggregated LDL in human vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. (2000) 20:1572–9. doi: 10.1161/01.ATV.20.6.1572
92. De Gonzalo-Calvo D, Cenarro A, Martínez-Bujidos M, Badimon L, Bayes-Genis A, Ordonez-Llanos J, et al. Circulating soluble low-density lipoprotein receptor-related protein 1 (sLRP1) concentration is associated with hypercholesterolemia: a new potential biomarker for atherosclerosis. Int J Cardiol. (2015) 201:20–9. doi: 10.1016/j.ijcard.2015.07.085
93. De Gonzalo-Calvo D, Elosua R, Vea A, Subirana I, Sayols-Baixeras S, Marrugat J, et al. Soluble low-density lipoprotein receptor-related protein 1 as a biomarker of coronary risk: predictive capacity and association with clinical events. Atherosclerosis. (2019) 287:93–9. doi: 10.1016/j.atherosclerosis.2019.06.904
94. Dubland JA, Allahverdian S, Besler KJ, Ortega C, Wang Y, Pryma CS, et al. Low LAL (lysosomal acid lipase) expression by smooth muscle cells relative to macrophages as a mechanism for arterial foam cell formation. Arterioscler Thromb Vasc Biol. (2021) 41:e354–68. doi: 10.1161/ATVBAHA.120.316063
95. Pulanco MC, Cosman J, Ho M-M, Huynh J, Fing K, Turcu J, et al. Complement protein C1q enhances macrophage foam cell survival and efferocytosis. J Immunol. (2017) 198:472–80. doi: 10.4049/jimmunol.1601445
96. Biró A, Thielens NM, Cervenák L, Prohászka Z, Füst G, Arlaud GJ. Modified low density lipoproteins differentially bind and activate the C1 complex of complement. Mol Immunol. (2007) 44:1169–77. doi: 10.1016/j.molimm.2006.06.013
97. Bhatia VK, Yun S, Leung V, Grimsditch DC, Benson GM, Botto MB, et al. Complement C1q reduces early atherosclerosis in low-density lipoprotein receptor-deficient mice. Am J Pathol. (2007) 170:416–26. doi: 10.2353/ajpath.2007.060406
98. Nissilä E, Hakala P, Leskinen K, Roig A, Syed S, Van Kessel KPM, et al. Complement factor H and apolipoprotein E participate in regulation of inflammation in THP-1 macrophages. Front Immunol. (2018) 9:2701. doi: 10.3389/fimmu.2018.02701
99. Miller YI, Choi SH, Fang L, Tsimikas S. Lipoprotein modification and macrophage uptake: role of pathologic cholesterol transport in atherogenesis. Subcell Biochem. (2010) 51:229–51. doi: 10.1007/978-90-481-8622-8_8
100. Rocha VZ, Libby P. Obesity, inflammation, and atherosclerosis. Nat Rev Cardiol. (2009) 6:399–409. doi: 10.1038/nrcardio.2009.55
101. Xu S, Ogura S, Chen J, Little PJ, Moss J, Liu P. LOX-1 in atherosclerosis: biological functions and pharmacological modifiers. Cell Mol Life Sci. (2013) 70:2859–72. doi: 10.1007/s00018-012-1194-z
102. Murray PJ. Macrophage polarization. Annu Rev Physiol. (2017) 79:541–66. doi: 10.1146/annurev-physiol-022516-034339
103. Tabas I, Bornfeldt KE. Intracellular and intercellular aspects of macrophage immunometabolism in atherosclerosis. Circ Res. (2020) 126:1209–27. doi: 10.1161/CIRCRESAHA.119.315939
104. Rigamonti E, Chinetti-Gbaguidi G, Staels B. Regulation of macrophage functions by PPAR-alpha, PPAR-gamma, and LXRs in mice and men. Arterioscler Thromb Vasc Biol. (2008) 28:1050–9. doi: 10.1161/ATVBAHA.107.158998
105. Hudson BI, Lippman ME. Targeting RAGE signaling in inflammatory disease. Annu Rev Med. (2018) 69:349–64. doi: 10.1146/annurev-med-041316-085215
106. Goulopoulou S, McCarthy CG, Webb RC. Toll-like receptors in the vascular system: sensing the dangers within. Pharmacol Rev. (2016) 68:142–67. doi: 10.1124/pr.114.010090
107. Guo H, Callaway JB, Ting JPY. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. (2015) 21:677–87. doi: 10.1038/nm.3893
108. Grebe A, Hoss F, Latz E. NLRP3 inflammasome and the IL-1 pathway in atherosclerosis. Circ Res. (2018) 122:1722–40. doi: 10.1161/CIRCRESAHA.118.311362
109. He X, Fan X, Bai B, Lu N, Zhang S, Zhang L. Pyroptosis is a critical immune-inflammatory response involved in atherosclerosis. Pharmacol Res. (2021) 165:105447. doi: 10.1016/j.phrs.2021.105447
110. Orekhov AN, Oishi Y, Nikiforov NG, Zhelankin AV, Dubrovsky L, Sobenin IA, et al. Modified LDL particles activate inflammatory pathways in monocyte-derived macrophages: transcriptome analysis. Curr Pharmaceut Design. (2018) 24:3143–51. doi: 10.2174/1381612824666180911120039
111. Vromman A, Ruvkun V, Shvartz E, Wojtkiewicz G, Santos Masson G, Tesmenitsky Y, et al. Stage-dependent differential effects of interleukin-1 isoforms on experimental atherosclerosis. Eur Heart J. (2019) 40:2482–91. doi: 10.1093/eurheartj/ehz008
112. Ridker PM. Anticytokine agents: targeting interleukin signaling pathways for the treatment of atherothrombosis. Circ Res. (2019) 124:437–50. doi: 10.1161/CIRCRESAHA.118.313129
113. Tyrrell DJ, Goldstein DR. Ageing and atherosclerosis: vascular intrinsic and extrinsic factors and potential role of IL-6. Nat Rev Cardiol. (2021) 18:58–68. doi: 10.1038/s41569-020-0431-7
114. Gabay C. Interleukin-6 and chronic inflammation. Arthritis Res Ther. (2006) 8 (Suppl 2):S3. doi: 10.1186/ar1917
115. Tousoulis D, Oikonomou E, Economou EK, Crea F, Kaski JC. Inflammatory cytokines in atherosclerosis: current therapeutic approaches. Eur Heart J. (2016) 37:1723–32. doi: 10.1093/eurheartj/ehv759
116. McKellar GE, McCarey DW, Sattar N, McInnes IB. Role for TNF in atherosclerosis? Lessons from autoimmune disease. Nat Rev Cardiol. (2009) 6:410–7. doi: 10.1038/nrcardio.2009.57
117. An Z, Li J, Yu J, Wang X, Gao H, Zhang W, et al. Neutrophil extracellular traps induced by IL-8 aggravate atherosclerosis via activation NF-κB signaling in macrophages. Cell Cycle. (2019) 18:2928–38. doi: 10.1080/15384101.2019.1662678
118. Apostolakis S, Vogiatzi K, Amanatidou V, Spandidos DA. Interleukin 8 and cardiovascular disease. Cardiovasc Res. (2009) 84:353–60. doi: 10.1093/cvr/cvp241
119. Han X, Boisvert WA. Interleukin-10 protects against atherosclerosis by modulating multiple atherogenic macrophage function. Thromb Haemost. (2015) 113:505–12. doi: 10.1160/TH14-06-0509
120. Ye J, Wang Y, Wang Z, Liu L, Yang Z, Wang M, et al. Roles and mechanisms of interleukin-12 family members in cardiovascular diseases: opportunities and challenges. Front Pharmacol. (2020) 11:129. doi: 10.3389/fphar.2020.00129
121. Elyasi A, Voloshyna I, Ahmed S, Kasselman LJ, Behbodikhah J, De Leon J, et al. The role of interferon-γ in cardiovascular disease: an update. Inflamm Res. (2020) 69:975–88. doi: 10.1007/s00011-020-01382-6
122. van der Vorst EPC, Döring Y, Weber C. Chemokines and their receptors in atherosclerosis. J Mol Med. (2015) 93:963–71. doi: 10.1007/s00109-015-1317-8
123. Hamilton JA, Cook AD, Tak PP. Anti-colony-stimulating factor therapies for inflammatory and autoimmune diseases. Nat Rev Drug Discov. (2017) 16:53–70. doi: 10.1038/nrd.2016.231
124. Tabas I, Bornfeldt KE. Macrophage phenotype and function in different stages of atherosclerosis. Circ Res. (2016) 118:653–67. doi: 10.1161/CIRCRESAHA.115.306256
125. Chen P-Y, Qin L, Li G, Wang Z, Dahlman JE, Malagon-Lopez J, et al. Endothelial TGF-β signalling drives vascular inflammation and atherosclerosis. Nat Metab. (2019) 1:912–26. doi: 10.1038/s42255-019-0102-3
126. Toma I, McCaffrey TA. Transforming growth factor-β and atherosclerosis: interwoven atherogenic and atheroprotective aspects. Cell Tissue Res. (2012) 347:155–75. doi: 10.1007/s00441-011-1189-3
127. Shoeibi S, Mozdziak P, Mohammadi S. Important signals regulating coronary artery angiogenesis. Microvasc Res. (2018) 117:1–9. doi: 10.1016/j.mvr.2017.12.002
128. Youn SW, Park KK. Small-nucleic-acid-based therapeutic strategy targeting the transcription factors regulating the vascular inflammation, remodeling and fibrosis in atherosclerosis. Int J Mol Sci. (2015) 16:11804–33. doi: 10.3390/ijms160511804
129. Burtenshaw D, Kitching M, Redmond EM, Megson IL, Cahill PA. Reactive oxygen species (ROS), intimal thickening, and subclinical atherosclerotic disease. Front Cardiovasc Med. (2019) 6:89. doi: 10.3389/fcvm.2019.00089
130. Sonnweber T, Pizzini A, Nairz M, Weiss G, Tancevski I. Arachidonic acid metabolites in cardiovascular and metabolic diseases. Int J Mol Sci. (2018) 19:3285. doi: 10.3390/ijms19113285
131. Vaughan DE. PAI-1 and atherothrombosis. J Thromb Haemost. (2005) 3:1879–83. doi: 10.1111/j.1538-7836.2005.01420.x
132. Tavares JC, Muscará MN. Chapter 14 - adhesion molecules and endothelium. In: Da Luz PL, Libby P, Chagas ACP, Laurindo FRM, editors. Endothelium and Cardiovascular Diseases. Academic Press (2018). p. 189–201.
133. Puig N, Montolio L, Camps-Renom P, Navarra L, Jiménez-Altayó F, Jiménez-Xarrié E, et al. Electronegative LDL promotes inflammation and triglyceride accumulation in macrophages. Cells. (2020) 9:583. doi: 10.3390/cells9030583
134. Chang C-K, Chen P-K, Lan J-L, Chang S-H, Hsieh T-Y, Liao P-J, et al. Association of electronegative LDL with macrophage foam cell formation and CD11c expression in rheumatoid arthritis patients. Int J Mol Sci. (2020) 21:5883. doi: 10.3390/ijms21165883
135. Sabeva NS, McPhaul CM, Li X, Cory TJ, Feola DJ, Graf GA. Phytosterols differentially influence ABC transporter expression, cholesterol efflux and inflammatory cytokine secretion in macrophage foam cells. J Nutr Biochem. (2011) 22:777–83. doi: 10.1016/j.jnutbio.2010.07.002
136. Mogilenko DA, Kudriavtsev IV, Trulioff AS, Shavva VS, Dizhe EB, Missyul BV, et al. Modified low density lipoprotein stimulates complement C3 expression and secretion via liver X receptor and Toll-like receptor 4 activation in human macrophages. J Biol Chem. (2012) 287:5954–68. doi: 10.1074/jbc.M111.289322
137. Ziouzenkova O, Asatryan L, Sahady D, Orasanu G, Perrey S, Cutak B, et al. Dual roles for lipolysis and oxidation in peroxisome proliferation-activator receptor responses to electronegative low density lipoprotein. J Biol Chem. (2003) 278:39874–81. doi: 10.1074/jbc.M306786200
138. Abe Y, Fornage M, Yang CY, Bui-Thanh NA, Wise V, Chen HH, et al. L5, the most electronegative subfraction of plasma LDL, induces endothelial vascular cell adhesion molecule 1 and CXC chemokines, which mediate mononuclear leukocyte adhesion. Atherosclerosis. (2007) 192:56–66. doi: 10.1016/j.atherosclerosis.2006.06.012
139. Sánchez-Quesada JL, Benítez S, Pérez A, Wagner AM, Rigla M, Carreras G, et al. The inflammatory properties of electronegative low-density lipoprotein from type 1 diabetic patients are related to increased platelet-activating factor acetylhydrolase activity. Diabetologia. (2005) 48:2162–9. doi: 10.1007/s00125-005-1899-8
140. de Castellarnau C, Bancells C, Benítez S, Reina M, Ordóñez-Llanos J, Sánchez-Quesada JL. Atherogenic and inflammatory profile of human arterial endothelial cells (HUAEC) in response to LDL subfractions. Clin Chim Acta. (2007) 376:233–6. doi: 10.1016/j.cca.2006.07.024
141. Benítez S, Camacho M, Bancells C, Vila L, Sánchez-Quesada JL, Ordóñez-Llanos J. Wide proinflammatory effect of electronegative low-density lipoprotein on human endothelial cells assayed by a protein array. Biochim Biophys Acta. (2006) 1761:1014–21. doi: 10.1016/j.bbalip.2006.03.020
142. He W, Ren Y, Wang X, Chen Q, Ding S. C reactive protein and enzymatically modified LDL cooperatively promote dendritic cell-mediated T cell activation. Cardiovasc Pathol. (2017) 29:1–6. doi: 10.1016/j.carpath.2017.03.009
143. Suriyaphol P, Fenske D, Zähringer U, Han S-R, Bhakdi S, Husmann M. Enzymatically modified nonoxidized low-density lipoprotein induces interleukin-8 in human endothelial cells. Circulation. (2002) 106:2581–7. doi: 10.1161/01.CIR.0000038366.11851.D0
144. Klouche M, May AE, Hemmes M, Messner M, Kanse SM, Preissner KT, et al. Enzymatically modified, nonoxidized LDL induces selective adhesion and transmigration of monocytes and T-lymphocytes through human endothelial cell monolayers. Arterioscler Thromb Vasc Biol. (1999) 19:784–93. doi: 10.1161/01.ATV.19.3.784
145. Klouche M, Gottschling S, Gerl V, Hell W, Husmann M, Dorweiler B, et al. Atherogenic properties of enzymatically degraded LDL: selective induction of MCP-1 and cytotoxic effects on human macrophages. Arterioscler Thromb Vasc Biol. (1998) 18:1376–85. doi: 10.1161/01.ATV.18.9.1376
146. Hakala JK, Lindstedt KA, Kovanen PT, Pentikäinen MO. Low-density lipoprotein modified by macrophage-derived lysosomal hydrolases induces expression and secretion of IL-8 via p38 MAPK and NF-kappaB by human monocyte-derived macrophages. Arterioscler Thromb Vasc Biol. (2006) 26:2504–9. doi: 10.1161/01.ATV.0000245796.97133.ad
147. Karabina S-A, Brochériou I, Le Naour G, Agrapart M, Durand H, Gelb M, et al. Atherogenic properties of LDL particles modified by human group X secreted phospholipase A2 on human endothelial cell function. FASEB J. (2006) 20:2547–9. doi: 10.1096/fj.06-6018fje
148. Chen M, Masaki T, Sawamura T. LOX-1, the receptor for oxidized low-density lipoprotein identified from endothelial cells: implications in endothelial dysfunction and atherosclerosis. Pharmacol Ther. (2002) 95:89–100. doi: 10.1016/S0163-7258(02)00236-X
149. Chen H, Li D, Saldeen T, Mehta JL. Transforming growth factor-beta(1) modulates oxidatively modified LDL-induced expression of adhesion molecules: role of LOX-1. Circ Res. (2001) 89:1155–60. doi: 10.1161/hh2401.100598
150. Norata GD, Pirillo A, Callegari E, Hamsten A, Catapano AL, Eriksson P. Gene expression and intracellular pathways involved in endothelial dysfunction induced by VLDL and oxidised VLDL. Cardiovasc Res. (2003) 59:169–80. doi: 10.1016/S0008-6363(03)00335-3
151. Wang GP, Deng ZD, Ni J, Qu ZL. Oxidized low density lipoprotein and very low density lipoprotein enhance expression of monocyte chemoattractant protein-1 in rabbit peritoneal exudate macrophages. Atherosclerosis. (1997) 133:31–6. doi: 10.1016/S0021-9150(97)00109-3
152. Wang G, Deng Z, Ni J. Expression of monocyte chemoattractant protein-1 in monocytes and effects of native and oxidized very low density lipoproteins. J Tongji Med Univ. (1997) 17:203–5. doi: 10.1007/BF02895619
153. Apostolov EO, Shah SV, Ok E, Basnakian AG. Carbamylated low-density lipoprotein induces monocyte adhesion to endothelial cells through intercellular adhesion molecule-1 and vascular cell adhesion molecule-1. Arterioscler Thromb Vasc Biol. (2007) 27:826–32. doi: 10.1161/01.ATV.0000258795.75121.8a
154. Chen Z, Ding S, Wang YP, Chen L, Mao JY, Yang Y, et al. Association of carbamylated high-density lipoprotein with coronary artery disease in type 2 diabetes mellitus: carbamylated high-density lipoprotein of patients promotes monocyte adhesion. J Transl Med. (2020) 18:460. doi: 10.1186/s12967-020-02623-2
155. Toma L, Stancu CS, Sima AV. Endothelial dysfunction in diabetes is aggravated by glycated lipoproteins; novel molecular therapies. Biomedicines. (2020) 9:18. doi: 10.3390/biomedicines9010018
156. Khan BV, Parthasarathy SS, Alexander RW, Medford RM. Modified low density lipoprotein and its constituents augment cytokine-activated vascular cell adhesion molecule-1 gene expression in human vascular endothelial cells. J Clin Invest. (1995) 95:1262–70. doi: 10.1172/JCI117776
157. Zhao R, Ren S, Moghadasain MH, Rempel JD, Shen GX. Involvement of fibrinolytic regulators in adhesion of monocytes to vascular endothelial cells induced by glycated LDL and to aorta from diabetic mice. J Leukocyte Biol. (2014) 95:941–9. doi: 10.1189/jlb.0513262
158. Hodgkinson CP, Laxton RC, Patel K, Ye S. Advanced glycation end-product of low density lipoprotein activates the toll-like 4 receptor pathway implications for diabetic atherosclerosis. Arterioscler Thromb Vasc Biol. (2008) 28:2275–81. doi: 10.1161/ATVBAHA.108.175992
159. Isoda K, Folco E, Marwali MR, Ohsuzu F, Libby P. Glycated LDL increases monocyte CC chemokine receptor 2 expression and monocyte chemoattractant protein-1-mediated chemotaxis. Atherosclerosis. (2008) 198:307–12. doi: 10.1016/j.atherosclerosis.2007.10.035
160. Toma L, Stancu CS, Botez GM, Sima AV, Simionescu M. Irreversibly glycated LDL induce oxidative and inflammatory state in human endothelial cells; added effect of high glucose. Biochem Biophys Res Commun. (2009) 390:877–82. doi: 10.1016/j.bbrc.2009.10.066
161. Ryu J-H, Ge M, Merscher S, Rosenberg AZ, Desante M, Roshanravan H, et al. APOL1 renal risk variants promote cholesterol accumulation in tissues and cultured macrophages from APOL1 transgenic mice. PLoS ONE. (2019) 14:e0211559. doi: 10.1371/journal.pone.0211559
162. Khoo JC, Miller E, McLoughlin P, Steinberg D. Enhanced macrophage uptake of low density lipoprotein after self-aggregation. Arteriosclerosis. (1988) 8:348–58. doi: 10.1161/01.ATV.8.4.348
163. Brown MS, Goldstein JL, Krieger M, Ho YK, Anderson RG. Reversible accumulation of cholesteryl esters in macrophages incubated with acetylated lipoproteins. J Cell Biol. (1979) 82:597–613. doi: 10.1083/jcb.82.3.597
164. Schmitz G, Grandl M. Endolysosomal phospholipidosis and cytosolic lipid droplet storage and release in macrophages. Biochimica et Biophysica Acta (BBA) Mol Cell Biol Lipids. (2009) 1791:524–39. doi: 10.1016/j.bbalip.2008.12.007
165. Zhao B, Huang W, Zhang W-Y, Ishii I, Kruth HS. Retention of aggregated LDL by cultured human coronary artery endothelial cells. Biochem Biophys Res Commun. (2004) 321:728–35. doi: 10.1016/j.bbrc.2004.07.017
166. Sanda GM, Stancu CS, Deleanu M, Toma L, Niculescu LS, Sima AV. Aggregated LDL turn human macrophages into foam cells and induce mitochondrial dysfunction without triggering oxidative or endoplasmic reticulum stress. PLoS ONE. (2021) 16:e0245797. doi: 10.1371/journal.pone.0245797
167. Persson J, Nilsson J, Lindholm MW. Cytokine response to lipoprotein lipid loading in human monocyte-derived macrophages. Lipids Health Dis. (2006) 5:17. doi: 10.1186/1476-511X-5-17
168. Orekhov AN, Sobenin IA. Modified lipoproteins as biomarkers of atherosclerosis. Front Biosci Landmark. (2018) 23:1422–44. doi: 10.2741/4653
169. Sánchez–Quesada JL, Estruch M, Benìtez S, Ordóñez-Llanos J. Electronegative LDL: a useful biomarker of cardiovascular risk? Clin Lipidol. (2012) 7:345–59. doi: 10.2217/clp.12.26
170. Shiu Sammy WM, Xiao S-M, Wong Y, Chow W-S, Lam Karen SL, Tan KCB. Carbamylation of LDL and its relationship with myeloperoxidase in Type 2 diabetes mellitus. Clin Sci. (2013) 126:175–81. doi: 10.1042/CS20130369
171. Estruch M, Rajamäki K, Sanchez-Quesada JL, Kovanen PT, Öörni K, Benitez S, et al. Electronegative LDL induces priming and inflammasome activation leading to IL-1β release in human monocytes and macrophages. Biochimica et Biophysica Acta (BBA) Mol Cell Biol Lipids. (2015) 1851:1442–9. doi: 10.1016/j.bbalip.2015.08.009
172. Yang TC, Chang PY, Lu SC. L5-LDL from ST-elevation myocardial infarction patients induces IL-1β production via LOX-1 and NLRP3 inflammasome activation in macrophages. Am J Physiol Heart Circ Physiol. (2017) 312:H265–74. doi: 10.1152/ajpheart.00509.2016
173. Mattsson L, Johansson H, Ottosson M, Bondjers G, Wiklund O. Expression of lipoprotein lipase mRNA and secretion in macrophages isolated from human atherosclerotic aorta. J Clin Invest. (1993) 92:1759–65. doi: 10.1172/JCI116764
174. Verbrugge FH, Tang WHW, Hazen SL. Protein carbamylation and cardiovascular disease. Kidney Int. (2015) 88:474–8. doi: 10.1038/ki.2015.166
175. Reis SE, Olson MB, Fried L, Reeser V, Mankad S, Pepine CJ, et al. Mild renal insufficiency is associated with angiographic coronary artery disease in women. Circulation. (2002) 105:2826–9. doi: 10.1161/01.CIR.0000021597.63026.65
176. Speer T, Owala FO, Holy EW, Zewinger S, Frenzel FL, Stähli BE, et al. Carbamylated low-density lipoprotein induces endothelial dysfunction. Eur Heart J. (2014) 35:3021–32. doi: 10.1093/eurheartj/ehu111
177. Carracedo J, Merino A, Briceño C, Soriano S, Buendía P, Calleros L, et al. Carbamylated low-density lipoprotein induces oxidative stress and accelerated senescence in human endothelial progenitor cells. FASEB J. (2011) 25:1314–22. doi: 10.1096/fj.10-173377
178. Apostolov EO, Ok E, Burns S, Nawaz S, Savenka A, Shah S, et al. Carbamylated-oxidized LDL: proatherosclerotic effects on endothelial cells and macrophages. J Atheroscler Thromb. (2013) 20:878–92. doi: 10.5551/jat.14035
179. Bucala R, Makita Z, Vega G, Grundy S, Koschinsky T, Cerami A, et al. Modification of low density lipoprotein by advanced glycation end products contributes to the dyslipidemia of diabetes and renal insufficiency. Proc Natl Acad Sci USA. (1994) 91:9441–5. doi: 10.1073/pnas.91.20.9441
180. Artwohl M, Graier WF, Roden M, Bischof M, Freudenthaler A, Waldhäusl W, et al. Diabetic LDL triggers apoptosis in vascular endothelial cells. Diabetes. (2003) 52:1240–7. doi: 10.2337/diabetes.52.5.1240
181. Gora S, Maouche S, Atout R, Wanherdrick K, Lambeau G, Cambien F, et al. Phospholipolyzed LDL induces an inflammatory response in endothelial cells through endoplasmic reticulum stress signaling. FASEB J. (2010) 24:3284–97. doi: 10.1096/fj.09-146852
182. Nguyen SD, Korhonen EA, Lorey MB, Hakanpää L, Mäyränpää MI, Kovanen PT, et al. Lysophosphatidylcholine in phospholipase A(2)-modified LDL triggers secretion of angiopoietin 2. Atherosclerosis. (2021) 327:87–99. doi: 10.1016/j.atherosclerosis.2021.04.007
183. Lauber K, Bohn E, Kröber SM, Xiao YJ, Blumenthal SG, Lindemann RK, et al. Apoptotic cells induce migration of phagocytes via caspase-3-mediated release of a lipid attraction signal. Cell. (2003) 113:717–30. doi: 10.1016/S0092-8674(03)00422-7
184. Nguyen SD, Javanainen M, Rissanen S, Zhao H, Huusko J, Kivelä AM, et al. Apolipoprotein A-I mimetic peptide 4F blocks sphingomyelinase-induced LDL aggregation. J Lipid Res. (2015) 56:1206–21. doi: 10.1194/jlr.M059485
185. Oestvang J, Bonnefont-Rousselot D, Ninio E, Hakala JK, Johansen B, Anthonsen MW. Modification of LDL with human secretory phospholipase A(2) or sphingomyelinase promotes its arachidonic acid-releasing propensity. J Lipid Res. (2004) 45:831–8. doi: 10.1194/jlr.M300310-JLR200
186. Kolesnick R, Fuks Z. Radiation and ceramide-induced apoptosis. Oncogene. (2003) 22:5897–906. doi: 10.1038/sj.onc.1206702
187. Summers SA, Chaurasia B, Holland WL. Metabolic messengers: ceramides. Nat Metab. (2019) 1:1051–8. doi: 10.1038/s42255-019-0134-8
188. Kinscherf R, Claus R, Deigner HP, Nauen O, Gehrke C, Hermetter A, et al. Modified low density lipoprotein delivers substrate for ceramide formation and stimulates the sphingomyelin-ceramide pathway in human macrophages. FEBS Lett. (1997) 405:55–9. doi: 10.1016/S0014-5793(97)00157-9
189. Chellan B, Reardon CA, Getz GS, Hofmann Bowman MA. Enzymatically modified low-density lipoprotein promotes foam cell formation in smooth muscle cells via macropinocytosis and enhances receptor-mediated uptake of oxidized low-density lipoprotein. Arterioscler Thromb Vasc Biol. (2016) 36:1101–13. doi: 10.1161/ATVBAHA.116.307306
190. Li F, Zhang H. Lysosomal acid lipase in lipid metabolism and beyond. Arterioscler Thromb Vasc Biol. (2019) 39:850–6. doi: 10.1161/ATVBAHA.119.312136
191. Heltianu C, Robciuc A, Botez G, Musina C, Stancu C, Sima AV, et al. Modified low density lipoproteins decrease the activity and expression of lysosomal acid lipase in human endothelial and smooth muscle cells. Cell Biochem Biophys. (2011) 61:209–16. doi: 10.1007/s12013-011-9190-8
192. Ismael FO, Barrett TJ, Sheipouri D, Brown BE, Davies MJ, Hawkins CL. Role of myeloperoxidase oxidants in the modulation of cellular lysosomal enzyme function: a contributing factor to macrophage dysfunction in atherosclerosis? PLoS ONE. (2016) 11:e0168844. doi: 10.1371/journal.pone.0168844
193. Zhang H. Lysosomal acid lipase and lipid metabolism: new mechanisms, new questions, and new therapies. Curr Opin Lipidol. (2018) 29:218–23. doi: 10.1097/MOL.0000000000000507
194. den Hartigh LJ, Altman R, Norman JE, Rutledge JC. Postprandial VLDL lipolysis products increase monocyte adhesion and lipid droplet formation via activation of ERK2 and NFκB. Am J Physiol Heart Circ Physiol. (2014) 306:H109–20. doi: 10.1152/ajpheart.00137.2013
195. Binder CJ, Papac-Milicevic N, Witztum JL. Innate sensing of oxidation-specific epitopes in health and disease. Nat Rev Immunol. (2016) 16:485–97. doi: 10.1038/nri.2016.63
196. Luchetti F, Crinelli R, Nasoni MG, Benedetti S, Palma F, Fraternale A, et al. LDL receptors, caveolae and cholesterol in endothelial dysfunction: oxLDLs accomplices or victims? Br J Pharmacol. (2021) 178:3104–14. doi: 10.1111/bph.15272
197. Berliner JA, Territo MC, Sevanian A, Ramin S, Kim JA, Bamshad B, et al. Minimally modified low density lipoprotein stimulates monocyte endothelial interactions. J Clin Invest. (1990) 85:1260–6. doi: 10.1172/JCI114562
198. Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. (2010) 11:155–61. doi: 10.1038/ni.1836
199. Miller YI, Viriyakosol S, Worrall DS, Boullier A, Butler S, Witztum JL. Toll-like receptor 4-dependent and -independent cytokine secretion induced by minimally oxidized low-density lipoprotein in macrophages. Arterioscler Thromb Vasc Biol. (2005) 25:1213–9. doi: 10.1161/01.ATV.0000159891.73193.31
200. Bekkering S, Quintin J, Joosten LA, van der Meer JW, Netea MG, Riksen NP. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler Thromb Vasc Biol. (2014) 34:1731–8. doi: 10.1161/ATVBAHA.114.303887
201. Li WL, Hua LG, Qu P, Yan WH, Ming C, Jun YD, et al. NLRP3 inflammasome: a novel link between lipoproteins and atherosclerosis. Arch Med Sci. (2016) 12:950–8. doi: 10.5114/aoms.2016.61356
202. Thai SF, Lewis JG, Williams RB, Johnson SP, Adams DO. Effects of oxidized LDL on mononuclear phagocytes: inhibition of induction of four inflammatory cytokine gene RNAs, release of NO, and cytolysis of tumor cells. J Leukoc Biol. (1995) 57:427–33. doi: 10.1002/jlb.57.3.427
203. Nurmi K, Niemi K, Kareinen I, Silventoinen K, Lorey MB, Chen Y, et al. Native and oxidised lipoproteins negatively regulate the serum amyloid A-induced NLRP3 inflammasome activation in human macrophages. Clin Transl Immunol. (2021) 10:e1323. doi: 10.1002/cti2.1323
204. Orr AW, Hastings NE, Blackman BR, Wamhoff BR. Complex regulation and function of the inflammatory smooth muscle cell phenotype in atherosclerosis. J Vasc Res. (2010) 47:168–80. doi: 10.1159/000250095
205. Lin J, Shou X, Mao X, Dong J, Mohabeer N, Kushwaha KK, et al. Oxidized low density lipoprotein induced caspase-1 mediated pyroptotic cell death in macrophages: implication in lesion instability? PLoS ONE. (2013) 8:e62148. doi: 10.1371/journal.pone.0062148
206. Zhaolin Z, Jiaojiao C, Peng W, Yami L, Tingting Z, Jun T, et al. OxLDL induces vascular endothelial cell pyroptosis through miR-125a-5p/TET2 pathway. J Cell Physiol. (2019) 234:7475–91. doi: 10.1002/jcp.27509
207. Ajoolabady A, Aslkhodapasandhokmabad H, Libby P, Tuomilehto J, Lip GYH, Penninger JM, et al. Ferritinophagy and ferroptosis in the management of metabolic diseases. Trends Endocrinol Metab. (2021) 32:444–62. doi: 10.1016/j.tem.2021.04.010
208. Conrad M, Pratt DA. The chemical basis of ferroptosis. Nat Chem Biol. (2019) 15:1137–47. doi: 10.1038/s41589-019-0408-1
209. Bai T, Li M, Liu Y, Qiao Z, Wang Z. Inhibition of ferroptosis alleviates atherosclerosis through attenuating lipid peroxidation and endothelial dysfunction in mouse aortic endothelial cell. Free Radical Biol Med. (2020) 160:92–102. doi: 10.1016/j.freeradbiomed.2020.07.026
210. Yang K, Song H, Yin D. PDSS2 inhibits the ferroptosis of vascular endothelial cells in atherosclerosis by activating Nrf2. J Cardiovasc Pharmacol. (2021) 77:767–76. doi: 10.1097/FJC.0000000000001030
211. Rhoads JP, Lukens JR, Wilhelm AJ, Moore JL, Mendez-Fernandez Y, Kanneganti T-D, et al. Oxidized low-density lipoprotein immune complex priming of the Nlrp3 inflammasome involves TLR and FcγR cooperation and is dependent on CARD9. J Immunol. (2017) 198:2105–14. doi: 10.4049/jimmunol.1601563
212. Ylä-Herttuala S, Palinski W, Butler SW, Picard S, Steinberg D, Witztum JL. Rabbit and human atherosclerotic lesions contain IgG that recognizes epitopes of oxidized LDL. Arterioscler Thromb. (1994) 14:32–40. doi: 10.1161/01.ATV.14.1.32
213. Lappalainen J, Lindstedt KA, Oksjoki R, Kovanen PT. OxLDL-IgG immune complexes induce expression and secretion of proatherogenic cytokines by cultured human mast cells. Atherosclerosis. (2011) 214:357–63. doi: 10.1016/j.atherosclerosis.2010.11.024
214. Kovanen PT. Mast cells as potential accelerators of human atherosclerosis-from early to late lesions. Int J Mol Sci. (2019) 20:4479. doi: 10.3390/ijms20184479
215. Zhao R, Ma X, Shen GX. Transcriptional regulation of plasminogen activator inhibitor-1 in vascular endothelial cells induced by oxidized very low density lipoproteins. Mol Cell Biochem. (2008) 317:197–204. doi: 10.1007/s11010-008-9851-0
216. Au-Yeung KK, O K, Choy PC, Zhu DY, Siow YL. Magnesium tanshinoate B protects endothelial cells against oxidized lipoprotein-induced apoptosis. Can J Physiol Pharmacol. (2007) 85:1053–62. doi: 10.1139/Y07-096
217. Argmann CA, Van Den Diepstraten CH, Sawyez CG, Edwards JY, Hegele RA, Wolfe BM, et al. Transforming growth factor-beta1 inhibits macrophage cholesteryl ester accumulation induced by native and oxidized VLDL remnants. Arterioscler Thromb Vasc Biol. (2001) 21:2011–8. doi: 10.1161/hq1201.099426
218. Itabe H, Sawada N, Makiyama T, Obama T. Structure and dynamics of oxidized lipoproteins in vivo: roles of high-density lipoprotein. Biomedicines. (2021) 9:655. doi: 10.3390/biomedicines9060655
219. Pérez L, Vallejos A, Echeverria C, Varela D, Cabello-Verrugio C, Simon F. OxHDL controls LOX-1 expression and plasma membrane localization through a mechanism dependent on NOX/ROS/NF-κB pathway on endothelial cells. Lab Invest. (2019) 99:421–37. doi: 10.1038/s41374-018-0151-3
220. Ren J, Jin W, Chen H. oxHDL decreases the expression of CD36 on human macrophages through PPARgamma and p38 MAP kinase dependent mechanisms. Mol Cell Biochem. (2010) 342:171–81. doi: 10.1007/s11010-010-0481-y
221. Kunjathoor VV, Febbraio M, Podrez EA, Moore KJ, Andersson L, Koehn S, et al. Scavenger receptors class A-I/II and CD36 are the principal receptors responsible for the uptake of modified low density lipoprotein leading to lipid loading in macrophages. J Biol Chem. (2002) 277:49982–8. doi: 10.1074/jbc.M209649200
222. Assinger A, Koller F, Schmid W, Zellner M, Babeluk R, Koller E, et al. Specific binding of hypochlorite-oxidized HDL to platelet CD36 triggers proinflammatory and procoagulant effects. Atherosclerosis. (2010) 212:153–60. doi: 10.1016/j.atherosclerosis.2010.05.010
223. Delgado-Rizo V, Martínez-Guzmán MA, Iñiguez-Gutierrez L, García-Orozco A, Alvarado-Navarro A, Fafutis-Morris M. Neutrophil extracellular traps and its implications in inflammation: an overview. Front Immunol. (2017) 8:81. doi: 10.3389/fimmu.2017.00081
224. Castanheira FVS, Kubes P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood. (2019) 133:2178–85. doi: 10.1182/blood-2018-11-844530
225. Kapoor S, Opneja A, Nayak L. The role of neutrophils in thrombosis. Thromb Res. (2018) 170:87–96. doi: 10.1016/j.thromres.2018.08.005
226. Döring Y, Libby P, Soehnlein O. Neutrophil extracellular traps participate in cardiovascular diseases: recent experimental and clinical insights. Circ Res. (2020) 126:1228–41. doi: 10.1161/CIRCRESAHA.120.315931
227. Drechsler M, Megens RT, van Zandvoort M, Weber C, Soehnlein O. Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation. (2010) 122:1837–45. doi: 10.1161/CIRCULATIONAHA.110.961714
228. Zardawi F, Gul S, Abdulkareem A, Sha A, Yates J. Association between periodontal disease and atherosclerotic cardiovascular diseases: revisited. Front Cardiovasc Med. (2021) 7:625579. doi: 10.3389/fcvm.2020.625579
229. Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med. (2017) 23:279–87. doi: 10.1038/nm.4294
230. Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. (2015) 349:316–20. doi: 10.1126/science.aaa8064
231. Kato R, Mori C, Kitazato K, Arata S, Obama T, Mori M, et al. Transient increase in plasma oxidized LDL during the progression of atherosclerosis in apolipoprotein E knockout mice. Arterioscler Thromb Vasc Biol. (2009) 29:33–9. doi: 10.1161/ATVBAHA.108.164723
232. Obama T, Ohinata H, Takaki T, Iwamoto S, Sawada N, Aiuchi T, et al. Cooperative action of oxidized low-density lipoproteins and neutrophils on endothelial inflammatory responses through neutrophil extracellular trap formation. Front Immunol. (2019) 10:1899. doi: 10.3389/fimmu.2019.01899
233. Awasthi D, Nagarkoti S, Kumar A, Dubey M, Singh AK, Pathak P, et al. Oxidized LDL induced extracellular trap formation in human neutrophils via TLR-PKC-IRAK-MAPK and NADPH-oxidase activation. Free Radic Biol Med. (2016) 93:190–203. doi: 10.1016/j.freeradbiomed.2016.01.004
234. Ketelhuth DF, Hansson GK. Adaptive response of T and B cells in atherosclerosis. Circ Res. (2016) 118:668–78. doi: 10.1161/CIRCRESAHA.115.306427
235. Tabas I, Lichtman AH. Monocyte-macrophages and T cells in atherosclerosis. Immunity. (2017) 47:621–34. doi: 10.1016/j.immuni.2017.09.008
236. Gao Z, Xu X, Li Y, Sun K, Yang M, Zhang Q, et al. Mechanistic insight into PPARγ and Tregs in atherosclerotic immune inflammation. Front Pharmacol. (2021) 12:750078. doi: 10.3389/fphar.2021.750078
237. Wolf D, Ley K. Immunity and inflammation in atherosclerosis. Circ Res. (2019) 124:315–27. doi: 10.1161/CIRCRESAHA.118.313591
238. Gisterå A, Hermansson A, Strodthoff D, Klement ML, Hedin U, Fredrikson GN, et al. Vaccination against T-cell epitopes of native ApoB100 reduces vascular inflammation and disease in a humanized mouse model of atherosclerosis. J Intern Med. (2017) 281:383–97. doi: 10.1111/joim.12589
239. Jonasson L, Holm J, Skalli O, Bondjers G, Hansson GK. Regional accumulations of T cells, macrophages, and smooth muscle cells in the human atherosclerotic plaque. Arteriosclerosis. (1986) 6:131–8. doi: 10.1161/01.ATV.6.2.131
240. Suciu CF, Prete M, Ruscitti P, Favoino E, Giacomelli R, Perosa F. Oxidized low density lipoproteins: the bridge between atherosclerosis and autoimmunity. Possible implications in accelerated atherosclerosis and for immune intervention in autoimmune rheumatic disorders. Autoimmun Rev. (2018) 17:366–75. doi: 10.1016/j.autrev.2017.11.028
241. Mohanta SK, Yin C, Peng L, Srikakulapu P, Bontha V, Hu D, et al. Artery tertiary lymphoid organs contribute to innate and adaptive immune responses in advanced mouse atherosclerosis. Circ Res. (2014) 114:1772–87. doi: 10.1161/CIRCRESAHA.114.301137
242. Hamze M, Desmetz C, Berthe ML, Roger P, Boulle N, Brancherau P, et al. Characterization of resident B cells of vascular walls in human atherosclerotic patients. J Immunol. (2013) 191:3006–16. doi: 10.4049/jimmunol.1202870
243. Kimura T, Kobiyama K, Winkels H, Tse K, Miller J, Vassallo M, et al. Regulatory CD4(+) T cells recognize major histocompatibility complex class II molecule-restricted peptide epitopes of apolipoprotein B. Circulation. (2018) 138:1130–43. doi: 10.1161/CIRCULATIONAHA.117.031420
244. Ait-Oufella H, Sage AP, Mallat Z, Tedgui A. Adaptive (T and B cells) immunity and control by dendritic cells in atherosclerosis. Circ Res. (2014) 114:1640–60. doi: 10.1161/CIRCRESAHA.114.302761
245. Kyaw T, Winship A, Tay C, Kanellakis P, Hosseini H, Cao A, et al. Cytotoxic and proinflammatory CD8+ T lymphocytes promote development of vulnerable atherosclerotic plaques in apoE-deficient mice. Circulation. (2013) 127:1028–39. doi: 10.1161/CIRCULATIONAHA.112.001347
246. Hermansson A, Ketelhuth DF, Strodthoff D, Wurm M, Hansson EM, Nicoletti A, et al. Inhibition of T cell response to native low-density lipoprotein reduces atherosclerosis. J Exp Med. (2010) 207:1081–93. doi: 10.1084/jem.20092243
247. Stemme S, Faber B, Holm J, Wiklund O, Witztum JL, Hansson GK. T lymphocytes from human atherosclerotic plaques recognize oxidized low density lipoprotein. Proc Natl Acad Sci. (1995) 92:3893–7. doi: 10.1073/pnas.92.9.3893
248. Arneth B. Activation of T-lymphocytes by LDL-cholesterol. Lipids. (2009) 44:311–6. doi: 10.1007/s11745-008-3273-3
249. Tsiantoulas D, Diehl CJ, Witztum JL, Binder CJ. B cells and humoral immunity in atherosclerosis. Circ Res. (2014) 114:1743–56. doi: 10.1161/CIRCRESAHA.113.301145
250. Sage AP, Tsiantoulas D, Binder CJ, Mallat Z. The role of B cells in atherosclerosis. Nat Rev Cardiol. (2019) 16:180–96. doi: 10.1038/s41569-018-0106-9
251. Upadhye A, Srikakulapu P, Gonen A, Hendrikx S, Perry HM, Nguyen A, et al. Diversification and CXCR4-dependent establishment of the bone marrow B-1a cell pool governs atheroprotective IgM production linked to human coronary atherosclerosis. Circ Res. (2019) 125:e55–70. doi: 10.1161/CIRCRESAHA.119.315786
252. Baumgarth N, Herman OC, Jager GC, Brown L, Herzenberg LA, Herzenberg LA. Innate and acquired humoral immunities to influenza virus are mediated by distinct arms of the immune system. Proc Natl Acad Sci. (1999) 96:2250–5. doi: 10.1073/pnas.96.5.2250
253. Binder CJ, Silverman GJ. Natural antibodies and the autoimmunity of atherosclerosis. Springer Semin Immunopathol. (2005) 26:385–404. doi: 10.1007/s00281-004-0185-z
254. Virella G, Wilson K, Elkes J, Hammad SM, Rajab HA, Li Y, et al. Immune complexes containing malondialdehyde (MDA) LDL induce apoptosis in human macrophages. Clin Immunol. (2018) 187:1–9. doi: 10.1016/j.clim.2017.06.010
255. Binder CJ, Hartvigsen K, Chang M-K, Miller M, Broide D, Palinski W, et al. IL-5 links adaptive and natural immunity specific for epitopes of oxidized LDL and protects from atherosclerosis. J Clin Invest. (2004) 114:427–37. doi: 10.1172/JCI200420479
256. Tsimikas S, Brilakis ES, Lennon RJ, Miller ER, Witztum JL, McConnell JP, et al. Relationship of IgG and IgM autoantibodies to oxidized low density lipoprotein with coronary artery disease and cardiovascular events. J Lipid Res. (2007) 48:425–33. doi: 10.1194/jlr.M600361-JLR200
257. Ravandi A, Boekholdt SM, Mallat Z, Talmud PJ, Kastelein JJ, Wareham NJ, et al. Relationship of IgG and IgM autoantibodies and immune complexes to oxidized LDL with markers of oxidation and inflammation and cardiovascular events: results from the EPIC-Norfolk Study. J Lipid Res. (2011) 52:1829–36. doi: 10.1194/jlr.M015776
258. Boullier A, Gillotte KL, Hörkkö S, Green SR, Friedman P, Dennis EA, et al. The binding of oxidized low density lipoprotein to mouse CD36 is mediated in part by oxidized phospholipids that are associated with both the lipid and protein moieties of the lipoprotein. J Biol Chem. (2000) 275:9163–9. doi: 10.1074/jbc.275.13.9163
259. Kummu O, Turunen SP, Wang C, Lehtimäki J, Veneskoski M, Kastarinen H, et al. Carbamyl adducts on low-density lipoprotein induce IgG response in LDLR-/- mice and bind plasma autoantibodies in humans under enhanced carbamylation. Antioxid Redox Signal. (2013) 19:1047–62. doi: 10.1089/ars.2012.4535
260. Kummu O, Turunen SP, Prus P, Lehtimäki J, Veneskoski M, Wang C, et al. Human monoclonal Fab and human plasma antibodies to carbamyl-epitopes cross-react with malondialdehyde-adducts. Immunology. (2014) 141:416–30. doi: 10.1111/imm.12204
261. Virella G, Thorpe SR, Alderson NL, Stephan EM, Atchley D, Wagner F, et al. Autoimmune response to advanced glycosylation end-products of human LDL. J Lipid Res. (2003) 44:487–93. doi: 10.1194/jlr.M200370-JLR200
262. Shaw CA, Taylor EL, Megson IL, Rossi AG. Nitric oxide and the resolution of inflammation: implications for atherosclerosis. Mem Inst Oswaldo Cruz. (2005) 100(Suppl 1):67–71. doi: 10.1590/S0074-02762005000900012
263. Proto JD, Doran AC, Gusarova G, Yurdagul A Jr., Sozen E, Subramanian M, et al. Regulatory T cells promote macrophage efferocytosis during inflammation resolution. Immunity. (2018) 49:666–77.e6. doi: 10.1016/j.immuni.2018.07.015
264. Bäck M, Yurdagul A, Tabas I, Öörni K, Kovanen PT. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol. (2019) 16:389–406. doi: 10.1038/s41569-019-0169-2
265. Fredman G, Hellmann J, Proto JD, Kuriakose G, Colas RA, Dorweiler B, et al. An imbalance between specialized pro-resolving lipid mediators and pro-inflammatory leukotrienes promotes instability of atherosclerotic plaques. Nat Commun. (2016) 7:12859. doi: 10.1038/ncomms12859
266. Liu B, Zhang B, Guo R, Li S, Xu Y. Enhancement in efferocytosis of oxidized low-density lipoprotein-induced apoptotic RAW264.7 cells through Sirt1-mediated autophagy. Int J Mol Med. (2014) 33:523–33. doi: 10.3892/ijmm.2013.1609
267. Freigang S. The regulation of inflammation by oxidized phospholipids. Eur J Immunol. (2016) 46:1818–25. doi: 10.1002/eji.201545676
268. Lu SM, Fairn GD. 7-Ketocholesterol impairs phagocytosis and efferocytosis via dysregulation of phosphatidylinositol 4,5-bisphosphate. Traffic. (2018) 19:591–604. doi: 10.1111/tra.12576
269. Itakura K, Oya-Ito T, Osawa T, Yamada S, Toyokuni S, Shibata N, et al. Detection of lipofuscin-like fluorophore in oxidized human low-density lipoprotein. FEBS Lett. (2000) 473:249–53. doi: 10.1016/S0014-5793(00)01539-8
270. Anderson OA, Finkelstein A, Shima DT. A2E induces IL-1ß production in retinal pigment epithelial cells via the NLRP3 inflammasome. PLoS ONE. (2013) 8:e67263. doi: 10.1371/journal.pone.0067263
271. Pan C, Banerjee K, Lehmann GL, Almeida D, Hajjar KA, Benedicto I, et al. Lipofuscin causes atypical necroptosis through lysosomal membrane permeabilization. Proc Natl Acad Sci. (2021) 118:e2100122118. doi: 10.1073/pnas.2100122118
272. Petrusca DN, Gu Y, Adamowicz JJ, Rush NI, Hubbard WC, Smith PA, et al. Sphingolipid-mediated inhibition of apoptotic cell clearance by alveolar macrophages. J Biol Chem. (2010) 285:40322–32. doi: 10.1074/jbc.M110.137604
273. Justice MJ, Petrusca DN, Rogozea AL, Williams JA, Schweitzer KS, Petrache I, et al. Effects of lipid interactions on model vesicle engulfment by alveolar macrophages. Biophys J. (2014) 106:598–609. doi: 10.1016/j.bpj.2013.12.036
274. Kume N, Cybulsky MI, Gimbrone MA Jr. Lysophosphatidylcholine, a component of atherogenic lipoproteins, induces mononuclear leukocyte adhesion molecules in cultured human and rabbit arterial endothelial cells. J Clin Invest. (1992) 90:1138–44. doi: 10.1172/JCI115932
275. Rosenson RS, Brewer HB Jr., Ansell BJ, Barter P, Chapman MJ, Heinecke JW, et al. Dysfunctional HDL and atherosclerotic cardiovascular disease. Nat Rev Cardiol. (2016) 13:48–60. doi: 10.1038/nrcardio.2015.124
276. Baldus S, Eiserich JP, Mani A, Castro L, Figueroa M, Chumley P, et al. Endothelial transcytosis of myeloperoxidase confers specificity to vascular ECM proteins as targets of tyrosine nitration. J Clin Invest. (2001) 108:1759–70. doi: 10.1172/JCI200112617
277. Coetzee GA, Strachan AF, van der Westhuyzen DR, Hoppe HC, Jeenah MS, de Beer FC. Serum amyloid A-containing human high density lipoprotein 3. Density, size, and apolipoprotein composition. J Biol Chem. (1986) 261:9644–51. doi: 10.1016/S0021-9258(18)67562-3
278. Tietge UJ, Maugeais C, Cain W, Grass D, Glick JM, de Beer FC, et al. Overexpression of secretory phospholipase A(2) causes rapid catabolism and altered tissue uptake of high density lipoprotein cholesteryl ester and apolipoprotein A-I. J Biol Chem. (2000) 275:10077–84. doi: 10.1074/jbc.275.14.10077
279. Van Lenten BJ, Wagner AC, Nayak DP, Hama S, Navab M, Fogelman AM. High-density lipoprotein loses its anti-inflammatory properties during acute influenza a infection. Circulation. (2001) 103:2283–8. doi: 10.1161/01.CIR.103.18.2283
280. Navab M, Anantharamaiah GM, Reddy ST, Hama S, Hough G, Grijalva VR, et al. Oral D-4F causes formation of pre-beta high-density lipoprotein and improves high-density lipoprotein-mediated cholesterol efflux and reverse cholesterol transport from macrophages in apolipoprotein E-null mice. Circulation. (2004) 109:3215–20. doi: 10.1161/01.CIR.0000134275.90823.87
281. Ogura M, Hori M, Harada-Shiba M. Association between cholesterol efflux capacity and atherosclerotic cardiovascular disease in patients with familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. (2016) 36:181–8. doi: 10.1161/ATVBAHA.115.306665
282. Monette JS, Hutchins PM, Ronsein GE, Wimberger J, Irwin AD, Tang C, et al. Patients with coronary endothelial dysfunction have impaired cholesterol efflux capacity and reduced HDL particle concentration. Circ Res. (2016) 119:83–90. doi: 10.1161/CIRCRESAHA.116.308357
283. Lee-Rueckert M, Kovanen PT. The mast cell as a pluripotent HDL-modifying effector in atherogenesis: from in vitro to in vivo significance. Curr Opin Lipidol. (2015) 26:362–8. doi: 10.1097/MOL.0000000000000224
284. Lee-Rueckert M, Escola-Gil JC, Kovanen PT. HDL functionality in reverse cholesterol transport–Challenges in translating data emerging from mouse models to human disease. Biochim Biophys Acta. (2016) 1861:566–83. doi: 10.1016/j.bbalip.2016.03.004
285. Ference BA, Ginsberg HN, Graham I, Ray KK, Packard CJ, Bruckert E, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. (2017) 38:2459–72. doi: 10.1093/eurheartj/ehx144
286. Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, et al. Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. (2013) 34:3478–90a. doi: 10.1093/eurheartj/eht273
287. Cohen JC, Boerwinkle E, Mosley TH, Hobbs HH. Sequence variations in PCSK9, Low LDL, and protection against coronary heart disease. N Engl J Med. (2006) 354:1264–72. doi: 10.1056/NEJMoa054013
288. Goldstein JL, Brown MS. A century of cholesterol and coronaries: from plaques to genes to statins. Cell. (2015) 161:161–72. doi: 10.1016/j.cell.2015.01.036
289. Ridker PM, Cannon CP, Morrow D, Rifai N, Rose LM, McCabe CH, et al. C-reactive protein levels and outcomes after statin therapy. N Engl J Med. (2005) 352:20–8. doi: 10.1056/NEJMoa042378
290. Aday AW, Ridker PM. Targeting residual inflammatory risk: a shifting paradigm for atherosclerotic disease. Front Cardiovasc Med. (2019) 6:16. doi: 10.3389/fcvm.2019.00016
291. Soehnlein O, Libby P. Targeting inflammation in atherosclerosis - from experimental insights to the clinic. Nat Rev Drug Discov. (2021) 20:589–610. doi: 10.1038/s41573-021-00198-1
292. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
293. Tardif JC, Kouz S, Waters DD, Bertrand OF, Diaz R, Maggioni AP, et al. Efficacy and safety of low-dose colchicine after myocardial infarction. N Engl J Med. (2019) 381:2497–505. doi: 10.1056/NEJMoa1912388
294. Nidorf SM, Eikelboom JW, Budgeon CA, Thompson PL. Low-dose colchicine for secondary prevention of cardiovascular disease. J Am Coll Cardiol. (2013) 61:404–10. doi: 10.1016/j.jacc.2012.10.027
295. Nidorf SM, Fiolet ATL, Mosterd A, Eikelboom JW, Schut A, Opstal TSJ, et al. Colchicine in patients with chronic coronary disease. N Engl J Med. (2020) 383:1838–47. doi: 10.1056/NEJMoa2021372
296. Anstensrud AK, Woxholt S, Sharma K, Broch K, Bendz B, Aakhus S, et al. Rationale for the ASSAIL-MI-trial: a randomised controlled trial designed to assess the effect of tocilizumab on myocardial salvage in patients with acute ST-elevation myocardial infarction (STEMI). Open Heart. (2019) 6:e001108. doi: 10.1136/openhrt-2019-001108
297. Kleveland O, Kunszt G, Bratlie M, Ueland T, Broch K, Holte E, et al. Effect of a single dose of the interleukin-6 receptor antagonist tocilizumab on inflammation and troponin T release in patients with non-ST-elevation myocardial infarction: a double-blind, randomized, placebo-controlled phase 2 trial. Eur Heart J. (2016) 37:2406–13. doi: 10.1093/eurheartj/ehw171
298. Ulander L, Tolppanen H, Hartman O, Rissanen TT, Paakkanen R, Kuusisto J, et al. Hydroxychloroquine reduces interleukin-6 levels after myocardial infarction: the randomized, double-blind, placebo-controlled OXI pilot trial. Int J Cardiol. (2021) 337:21–7. doi: 10.1016/j.ijcard.2021.04.062
299. Ridker PM, Everett BM, Pradhan A, MacFadyen JG, Solomon DH, Zaharris E, et al. Low-dose methotrexate for the prevention of atherosclerotic events. N Engl J Med. (2019) 380:752–62. doi: 10.1056/NEJMoa1809798
300. O'Donoghue ML, Glaser R, Cavender MA, Aylward PE, Bonaca MP, Budaj A, et al. Effect of losmapimod on cardiovascular outcomes in patients hospitalized with acute myocardial infarction: a randomized clinical trial. JAMA. (2016) 315:1591–9. doi: 10.1001/jama.2016.3609
301. Baylis RA, Gomez D, Mallat Z, Pasterkamp G, Owens GK. The CANTOS trial: one important step for clinical cardiology but a giant leap for vascular biology. Arterioscler Thromb Vasc Biol. (2017) 37:e174–7. doi: 10.1161/ATVBAHA.117.310097
302. Delfino RJ, Staimer N, Vaziri ND. Air pollution and circulating biomarkers of oxidative stress. Air Qual Atmos Health. (2011) 4:37–52. doi: 10.1007/s11869-010-0095-2
303. Ukena C, Mahfoud F, Kindermann M, Kindermann I, Bals R, Voors AA, et al. The cardiopulmonary continuum systemic inflammation as “common soil” of heart and lung disease. Int J Cardiol. (2010) 145:172–6. doi: 10.1016/j.ijcard.2010.04.082
304. Schmidt AM, Hori O, Cao R, Yan SD, Brett J, Wautier JL, et al. RAGE: a novel cellular receptor for advanced glycation end products. Diabetes. (1996) 45(Suppl 3):S77–80. doi: 10.2337/diab.45.3.S77
305. Vlassara H. Advanced glycation end-products and atherosclerosis. Ann Med. (1996) 28:419–26. doi: 10.3109/07853899608999102
306. Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation. (2006) 114:597–605. doi: 10.1161/CIRCULATIONAHA.106.621854
307. Koska J, Saremi A, Howell S, Bahn G, De Courten B, Ginsberg H, et al. Advanced glycation end products, oxidation products, and incident cardiovascular events in patients with type 2 diabetes. Diabetes Care. (2018) 41:570–6. doi: 10.2337/dc17-1740
308. Saremi A, Howell S, Schwenke DC, Bahn G, Beisswenger PJ, Reaven PD. Advanced glycation end products, oxidation products, and the extent of atherosclerosis during the VA diabetes trial and follow-up study. Diabetes Care. (2017) 40:591–8. doi: 10.2337/dc16-1875
309. Aronson D, Rayfield EJ. How hyperglycemia promotes atherosclerosis: molecular mechanisms. Cardiovasc Diabetol. (2002) 1:1. doi: 10.1186/1475-2840-1-1
310. Botham KM, Wheeler-Jones CP. Postprandial lipoproteins and the molecular regulation of vascular homeostasis. Prog Lipid Res. (2013) 52:446–64. doi: 10.1016/j.plipres.2013.06.001
311. Ginsberg HN, Packard CJ, Chapman MJ, Borén J, Aguilar-Salinas CA, Averna M, et al. Triglyceride-rich lipoproteins and their remnants: metabolic insights, role in atherosclerotic cardiovascular disease, and emerging therapeutic strategies-a consensus statement from the European Atherosclerosis Society. Eur Heart J. (2021) 42:4791–806. doi: 10.1093/eurheartj/ehab551
312. Packard CJ, Boren J, Taskinen MR. Causes and consequences of hypertriglyceridemia. Front Endocrinol. (2020) 11:252. doi: 10.3389/fendo.2020.00252
313. Sandesara PB, Virani SS, Fazio S, Shapiro MD. The forgotten lipids: triglycerides, remnant cholesterol, and atherosclerotic cardiovascular disease risk. Endocr Rev. (2019) 40:537–57. doi: 10.1210/er.2018-00184
314. Wadström BN, Wulff AB, Pedersen KM, Jensen GB, Nordestgaard BG. Elevated remnant cholesterol increases the risk of peripheral artery disease, myocardial infarction, and ischaemic stroke: a cohort-based study. Eur Heart J. (2021). doi: 10.1093/eurheartj/ehab705. [Epub ahead of print].
315. Zhao Y, Liu L, Yang S, Liu G, Pan L, Gu C, et al. Mechanisms of atherosclerosis induced by postprandial lipemia. Front Cardiovasc Med. (2021) 8:636947. doi: 10.3389/fcvm.2021.636947
316. Pentikainen MO. Lipoprotein lipase in the arterial wall: linking LDL to the arterial extracellular matrix and much more. (2002) 22:211–7. doi: 10.1161/hq0102.101551
317. Toth PP. Triglyceride-rich lipoproteins as a causal factor for cardiovascular disease. Vasc Health Risk Manag. (2016) 12:171–83. doi: 10.2147/VHRM.S104369
318. Miname MH, Rocha VZ, Santos RD. The role of RNA-targeted therapeutics to reduce ASCVD risk: what have we learned recently? Curr Atheroscler Rep. (2021) 23:40. doi: 10.1007/s11883-021-00936-1
319. Bhatt DL, Steg PG, Miller M, Brinton EA, Jacobson TA, Ketchum SB, et al. Cardiovascular risk reduction with icosapent ethyl for hypertriglyceridemia. N Engl J Med. (2018) 380:11–22. doi: 10.1056/NEJMoa1812792
320. Strandberg TE, Libby P, Kovanen PT. A tale of two therapies lipid-lowering vs. anti-inflammatory therapy: a false dichotomy? Eur Heart J Cardiovasc Pharmacother. (2021) 7:238–41. doi: 10.1093/ehjcvp/pvaa131
321. Pradhan AD, Aday AW, Rose LM, Ridker PM. Residual inflammatory risk on treatment with PCSK9 inhibition and statin therapy. Circulation. (2018) 138:141–9. doi: 10.1161/CIRCULATIONAHA.118.034645
322. Randolph GJ. Tissue macrophages break dogma. Nat Rev Immunol. (2021) 21:625. doi: 10.1038/s41577-021-00620-0
Keywords: modified lipoproteins, inflammation, atherogenesis, endothelial dysfunction, foam cell
Citation: Lorey MB, Öörni K and Kovanen PT (2022) Modified Lipoproteins Induce Arterial Wall Inflammation During Atherogenesis. Front. Cardiovasc. Med. 9:841545. doi: 10.3389/fcvm.2022.841545
Received: 22 December 2021; Accepted: 26 January 2022;
Published: 03 March 2022.
Edited by:
Günther Silbernagel, Medical University of Graz, AustriaReviewed by:
Mark Borja, California State University, United StatesCopyright © 2022 Lorey, Öörni and Kovanen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katariina Öörni, a2F0YXJpaW5hLm9vcm5pQGhlbHNpbmtpLmZp
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.