
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med. , 08 April 2022
Sec. Cardiovascular Genetics and Systems Medicine
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.839862
This article is part of the Research Topic Genome-Wide Association Studies in Cardiovascular Disease View all 8 articles
 Mahdi Hesaraki1,2†
Mahdi Hesaraki1,2† Ugur Bora3,4†
Ugur Bora3,4† Sara Pahlavan2
Sara Pahlavan2 Najmeh Salehi5,6
Najmeh Salehi5,6 Seyed Ahmad Mousavi2Maryam Barekat7Seyed Javad Rasouli8Hossein Baharvand1,2
Seyed Ahmad Mousavi2Maryam Barekat7Seyed Javad Rasouli8Hossein Baharvand1,2 Gunes Ozhan3,4*
Gunes Ozhan3,4* Mehdi Totonchi2,5,6*
Mehdi Totonchi2,5,6*Cardiomyopathies are a group of common heart disorders that affect numerous people worldwide. Left ventricular non-compaction (LVNC) is a structural disorder of the ventricular wall, categorized as a type of cardiomyopathy that mostly caused by genetic disorders. Genetic variations are underlying causes of developmental deformation of the heart wall and the resultant contractile insufficiency. Here, we investigated a family with several affected members exhibiting LVNC phenotype. By whole-exome sequencing (WES) of three affected members, we identified a novel heterozygous missense variant (c.1963C>A:p.Leu655Met) in the gene encoding myosin heavy chain 7 (MYH7). This gene is evolutionary conserved among different organisms. We identified MYH7 as a highly enriched myosin, compared to other types of myosin heavy chains, in skeletal and cardiac muscles. Furthermore, MYH7 was among a few classes of MYH in mouse heart that highly expresses from early embryonic to adult stages. In silico predictions showed an altered actin-myosin binding, resulting in weaker binding energy that can cause LVNC. Moreover, CRISPR/Cas9 mediated MYH7 knockout in zebrafish caused impaired cardiovascular development. Altogether, these findings provide the first evidence for involvement of p.Leu655Met missense variant in the incidence of LVNC, most probably through actin-myosin binding defects during ventricular wall morphogenesis.
Left ventricular non-compaction (LVNC) is a structural abnormality of the cardiac muscle that presents in the ventricular wall, especially in the left ventricle, leading to extended trabeculation (1), and ultimately resulting in cardiomyopathies. LVNC follows an autosomal dominant inheritance pattern (2, 3), and could develop into both hypertrophic cardiomyopathy (HCM) and dilated cardiomyopathy (DCM) (4). Often, a genetic cause leads to LVNC that can be traced in a family which affects only the heart. The age of diagnosis varies between individuals, and ranges from birth to late adulthood. The precise cellular and molecular mechanisms of some LVNC subtypes are presently unknown. However, the time of onset and severity of the disease might be influenced by genetic conditions or developmental events such as tension heterogeneity-induced cardiac trabeculation (5, 6). Previously, mutations in various sarcomeric genes such as MYH7 (OMIM# 160760), MYBPC3 (OMIM# 600958), or TNNT2 (OMIM# 1910450) were associated to LVNC and introduced as major causes for this phenotype (4, 7). However, the myocardial wall morphogenesis should be also taken into account which has been shown to be tightly regulated by several signaling pathways, including TGFβ signaling (5, 8), NOTCH signaling (5, 9), Neuregulin/Erbb signaling (5, 8), and FGF signaling (8). Given the importance of sarcomeric structure and its function in cardiac contractility, any defects in sarcomeric components could result in cardiomyopathy leading to abnormalities in cardiovascular system (10). However, pathogenic variations in sarcomeric genes specifically actin and myosin, might also influence cardiac wall development (5) and cause cardiomyopathy.
The MYH7 gene known as the myosin heavy chain beta (MHC-β) which is classified as a type I fiber and also known as slow-twitch fiber (11). This protein is a critical component of sarcomeric structure and has several interactions with other key proteins such as actin TNN, MYBPC3 (12, 13).
Here, we report a novel heterozygous missense variant in the MYH7 gene in an Iranian family with a high prevalence of LVNC. In silico analysis was performed to check the availability and developmental presence of cardiac MYH7 transcripts. Moreover, the effect of this missense variation in MYH7-actin binding was modeled which suggests its potential impact on the development of LVNC in this family. Using CRISPR/Cas9 genome editing system, a zebrafish model of MYH7 was generated which showed developmental defects in early stages of heart formation. To our knowledge, this is the first MYH7 knockout model of zebrafish that also has an apparent cardiovascular malformation.
The Institutional Review Board of the Royan Institute Research Center and the Royan Ethics Committee (Tehran, Iran) approved this study. Written informed consent was obtained from all members of this family or their guardians. Zebrafish are maintained in accordance with the guidelines of the Izmir Biomedicine and Genome Center's Animal Care and Use Committee. All animal experiments were performed with the approval of the Animal Experiments Local Ethics Committee of Izmir Biomedicine and Genome Center (IBG-AELEC). All methods were performed in accordance with the declaration of Helsinki 1975.
This study was performed between 2018 and 2019 in Iran and comprised 20 genetically dependent members diagnosed with LVNC, who were between 12 and 53 years of age. The patients were diagnosed at the Royan Institute Cardiac Clinic (Tehran-Iran) by physical examination, electrocardiogram (ECG), and echocardiogram (Echo) assessments. All affected members had severe deep recesses and extended trabeculations in their left ventricular myocardia, and had normal heart valve function without additional symptoms related to other disorders (Table 1).
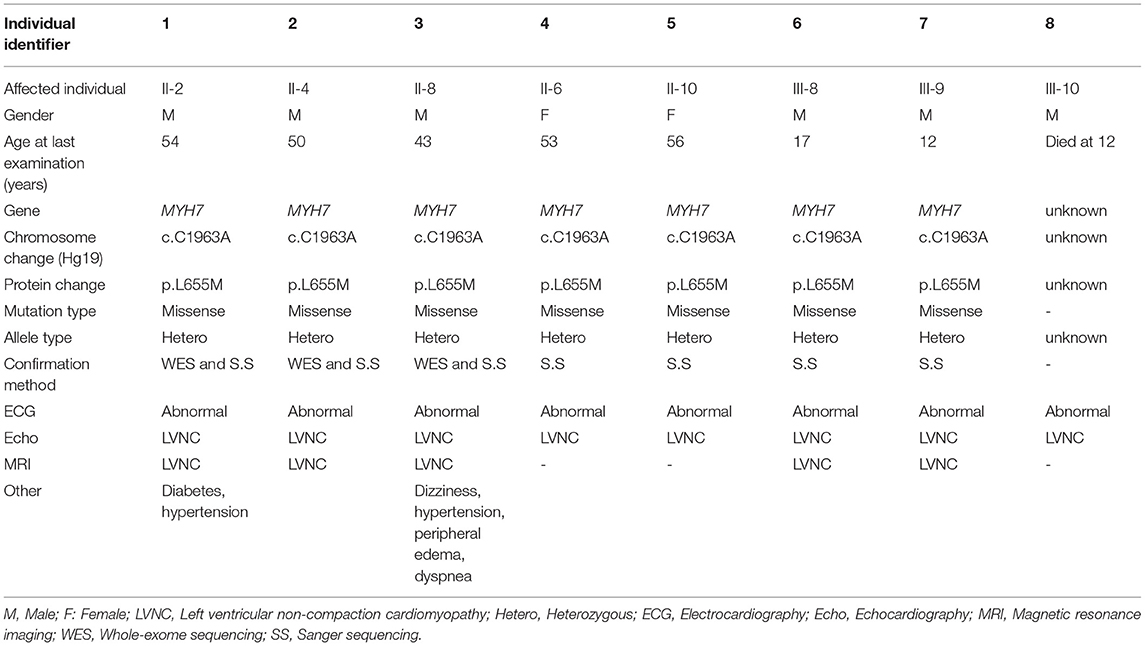
Table 1. Overview of clinical features of LVNC in the affected family members.
Peripheral blood samples were obtained for molecular analysis from all members of the family (n = 20). All genomic DNA was isolated by the salting out DNA extraction method and quantified with a NanoDrop 1000 Spectrophotometer (Thermo Fisher). The inheritance of LVNC in this pedigree follows an autosomal dominant pattern. Thus, we included 3 brothers of second generation in the WES study and skipped others. WES was performed using the Illumina NovaSeq platform and generated paired-end reads of 150 base pairs with an average coverage of 100X. Reads were aligned to genome reference consortium human 19 (GrCh37) with the Burrows-Wheeler Aligner (BWA, V.0.7.17) (14). The duplicated reads were removed by the Sequencing Picard tool. The Genome Analysis Toolkit (GATK, V4), followed by the best practice pipeline (15), was used for preprocessing the alignment results, detection and removal of duplicate reads, base recalibration, and variant calling. The VCF Filter toolkit was used for post-processing the variant calling results and variants that had a minimum depth of 12, mapping quality of 40, and quality of 30 were retained. The wANNOVAR website (http://wannovar.wglab.org/) was used for annotations (16). After the annotation process, all synonymous variations and those with ExAC frequency >0.01 were filtered by PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), the prediction algorithms. The sorting intolerant from tolerant (SIFT) (http://sift.bii.a-star.edu.sg/) and MutationTaster (http://www.mutationtaster.org) were used to evaluate candidate variations.
DNA samples from 20 members of the family were screened for the MYH7 candidate variant via the semi-nested PCR and Sanger sequencing methods. The primers used in this study were: F1: 5-ACTGTCGTGGGCTTGTATCA-3; F2: 5-CTGTCTCCTTGGTGCATTCG-3; and R1: 5-TGGTGGTAGGTAGGGAGATG-3. All PCRs were carried out in 25 μl volumes that consisted of a 12.5 μl PCR master mix, ~100 ng DNA, 1 μL of each primer (10 μM), and ~15 μL double distilled water (DDW). Cycling conditions involved denaturation (94 °C, 10 min) followed by 30 cycles of 94°C for 40 s, 54/57°C for 35 s, 72°C for 45/35 s, and a final extension at 72°C for 5 min. Finally, the sequencing results were aligned to the reference sequence in the CLC Main Workbench 5.5 software.
The expressions of the MYH protein classes in different tissues were compared using public databases. The BioGPS (http://biogps.org/) expression database was the best to evaluate all MYHs expression patterns in the human tissues. In the next step, the expression pattern of MYH7 during developmental stages of the mouse heart (E10.5-Old) was checked in a high-throughput expression data set (GEO: GSE93271, expression profiling by array,) and a p < 0.05 was considered as significantly different in the signal intensity (17). Then, the expression level of MYH7 (also known as ventricle myosin heavy chain like (vmhcl) as a zebrafish homolog of MYH7 in human) was checked in zebrafish heart at different developmental stages using a high throughput expression dataset (GEO: GSE120236, RNA-sequencing) (18) and a p < 0.05 on normalized counts was considered as significantly different. All expression analyses were performed using pheatmap package in RStudio.
The 3D structure of MYH7 was retrieved from the RCSB database (PDB ID:5TBY) for structural analysis. The p.Leu655Met mutation of the protein structure was induced by the psfgen plugin of VMD 1.9.3 (19). Both the wild type and mutant structures were minimized for 20 000 steps by the conjugate gradient method. Minimizations were done in NAMD 2.12 package (20) with CHARMM27 force field (21). The HADDOCK2.2 web server, which is an information-driven flexible docking approach (22, 23), was used to evaluate the effect of the p.Leu655Met mutation on the interaction between MYH7 and actin. The actin binding regions in MYH7 (655 to 677 and 757 to 771 residues) were set as the “active residues.” The surface neighbors of the active residues were selected as the “passive residues” that were automatically determined by HADDOCK2.2. The actin 3D structure and binding residues in interaction with MYH7 (residues of 3, 23, 24, 28, 30, 146–148, 333–345, 348, 350 from PDB ID of 4A7F) and the common binding site for actin-binding proteins on the actin surface (residues of 143, 144, 146, 148, 168, 341, 345, 346, 349, 351, 355) (24) were set as the “active residues” in interaction with MYH7. All other HADDOCK settings were kept at the default values. The docking results were ranked based on the HADDOCK score, which is a weighted sum of intermolecular electrostatic (Elec), van der Waals (vdW), desolvation (Dsolv), and buried surface area (BSA) (22, 23). All complex conformations were shown and analyzed by VMD 1.9.32.
Target sequence of MYH7 gene was selected by using the cloud-based informatics platform “Benchling” (https://www.benchling.com/crispr/). GRCz11 genome dataset was selected for determination of the target and guanine nucleotide was added to the 5′ of the target sequence to increase the efficiency of in vitro transcription. Template was generated by PCR using Phusion High-Fidelity DNA Polymerase (ThermoFisher Scientific) with the forward primer containing T7 promoter and targeted sgRNA sequence and the reverse primer encoding for the standard chimeric sgRNA scaffold (Supplementary Table 1). Scrambled (scr) gRNA was used as control. DNA template was purified with NucleoSpin Gel and PCR Clean up Kit (Macherey-Nagel GmbH & Co KG). sgRNAs were transcribed in vitro by using HiScribe™ T7 High Yield RNA Synthesis Kit (New England Biolabs). Transcription products were purified with RNA Clean & Concentrator-25 kit (Zymo Research, CA). RNA quantity was determined by NanoDrop™ OneC (Thermo Fischer Scientific).
sgRNA and Guide-it™ Recombinant Cas9 (Takara Bio Inc) were mixed at 1:1 (500/500 ng/μL) and incubated at room temperature for 5 min to generate the RNP complex. 0.05% phenol red was added to the mixture for visualization of injection. 1–1.5 nL of solution was injected directly into the cytoplasm of 1-cell stage zebrafish embryos.
Embryos injected with RNPs were used for genotyping at 5 dpf. Ten larvae were transferred individually to 1.5 mL tubes in 50 μL DNA extraction buffer (10 mM Tris HCl pH:8.0, 50 mM KCI, 0.3% Tween 20, 1 mM EDTA) and incubated at 98°C for 10 min. Next, 3 μl of proteinase K solution (5 mg/mL; Sigma Aldrich) was added and incubated at 55°C overnight. Samples were incubated at 98°C for 10 min to inactivate proteinase K and centrifuged at 14.000 rpm for 4 min. Supernatants were transferred into new tubes, mixed with 50 μL ddH2O, 10 μL 3M sodium acetate, 180 μl isopropanol, vortexed and kept on ice for 10 min. Samples were centrifuged at 14,000 rpm for 15 min at 4°C. Supernatants were discarded and pellets were washed with ice-cold 70% ethanol and air-dried for 15 min. Pellets were resuspended in 10 μL nuclease-free water. Genomic DNA quantity was determined by NanoDrop™ OneC. Targeted MYH7 loci that were amplified with PCR, used for Sanger sequencing. Genomic DNA was used as template at 100 ng/μL concentration and mixed with CloneAmp HiFi PCR Premix (Takara Bio Inc). Primers were designed to contain the mutation area (Supplementary Table 1). Final volume was adjusted to 25 μL with nuclease-free water. PCR was performed in SimpliAmp Thermal Cycler (Thermo Fischer Scientific) using the following conditions: 98°C for 1 min; 35 cycles of 98°C for 30 s, 62°C for 30 s and 72°C for 20 s; 72°C for 10 min. 1 μL of each sample was loaded into 1% agarose gel and electrophoresed. PCR products were cleaned up with NucleoSpin Gel and PCR Clean-up Kit. Products were quantified by using NanoDrop™ OneC.
Phenotypic evaluation of embryos and larvae was performed using an Olympus SZX2-ILLB stereomicroscope (Olympus Corporation, Japan). Images were captured with an Olympus SC50 microscope camera (Olympus Corporation, Japan).
Larvae were collected at 3 dpf and 5dpf and fixed in 4% PFA for 48 h. Larvae were washed twice with PBT, twice with methanol for 5 min each at RT and stored at −20°C for further use. Antisense RNA probes for MYH7, MYH7l, and mylk3 were synthesized. Briefly, a forward primer and two reverse primers were designed for each gene. One of the reverse primers were with T7 promoter sequence for in vitro transcription. First round of PCR was performed for amplification of the template. Second round of PCR was carried out to add T7 promoter. Probes were transcribed in vitro by using T7 RNA Polymerase (Thermo Fischer Scientific). Larvae were rehydrated by serial washes in 75% to 25% methanol/PBT for 5 min each followed by four PBT washes. Next, larvae were treated with Proteinase K (40 μg/μL) for 30 min, re-fixed in 4% PFA for 20 min and thoroughly washed afterwards. WMISH was performed as described previously (25, 26). Following prehybridization in hybridization buffer (Hyb) for 5 h at 67°C, samples were incubated with antisense probes in Hyb o/n at 67°C. The next morning, Hyb with probes were replaced with Hyb and incubated at 67°C for 20 min. Serial washes at 67°C were performed as follows: Three times in 2X 50% SSCT/50% formamide for 20 min, twice in 2X SSCT for 20 min and four times in 0.2X SSCT for 30 min. After a brief wash in PBT at RT, larvae were blocked with 5% sheep serum (Sigma-Aldrich) and 10 mg/ml BSA in PBT for 2 h and incubated in anti-Digoxigenin-AP Fab fragments o/n at 4°C (Merck&Co). After the removal of the antibody, larvae were washed in PBT over 3 h with 7–8 changes. Larvae were washed three times with NTMT buffer for 5 min, transferred into a 24 well-plate, stained with BCIP/NBT in NTMT buffer. Larvae were washed twice with PBT, incubated in stop solution for 1 h, washed twice with PBT and kept in 80% glycerol/20% stop solution until imaging (26, 27).
The Iranian family examined in this study, consisted of individuals with an autosomal dominant inheritance pattern. The proband was 55 years old with diabetes mellitus and hypertension since age 34 due to multivessel nonsignificant coronary artery disease and moderate left ventricular dysfunction (LVEF: 35–40%). He received anti-ischemic therapy without any LVNC diagnosis. The second brother was 44 years old and had repeated non-angina chest pain since age 36. We recruited a 46-year-old brother as the third member, who was asymptomatic until age 37 when his symptoms started to manifest with dizziness and hypertension. Currently, he suffers from occasional non-anginal chest pain, intermittent dyspnea on exertion and peripheral edema. He is in New York Heart Association (NYHA) class II. The last brother, who is 35 years old (III-11), was examined twice and found to have a normal left ventricular wall without any signs of LVNC. All affected siblings had late onset symptoms of LVNC that occurred around the third decade of life. One offspring (III-10) died at the age of 12 with LVNC phenotype (Figure 1A and Table 1).
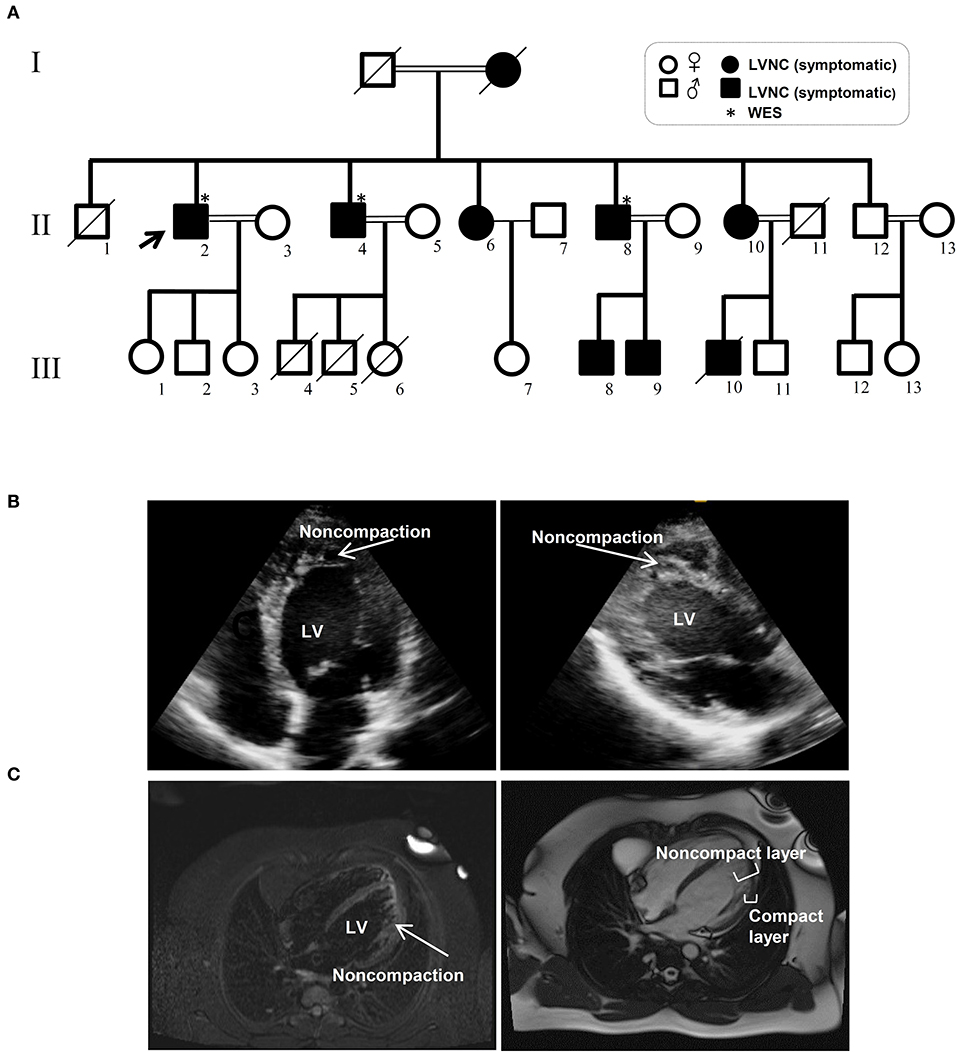
Figure 1. Pedigree and clinical manifestation of the left ventricular non-compaction (LVNC) in the affected members included in this study. (A) A pedigree of the family shows the number of affected members (affected members: shaded, WT: unshaded). A black arrow indicates the proband. Genotypes for the proband and all other family members are shown. (B) Echocardiograph (ECG) images of affected individuals with LVNC of the myocardium. (C) Magnetic resonance imaging (MRI) of thin epicardial and trabeculated endocardial layers of the ventricular wall.
WES was performed using the Illumina NovaSeq platform, which generated paired-end reads of 150 base pairs with an average coverage of 100X for the three affected, well-characterized brothers (II-2, II-4, and II-8) who had clinically proven disease (Figures 1B,C and Table 1).
A novel non-synonymous variant in MYH7 was identified (c.1963C>A:p.Leu655Met, GenBank: NM_000257) (Figure 2A). Myosin heavy chain beta (MHC-β) or MYH7 protein is encoded by the MYH7 gene on chromosome 14. The identified variant was a pathogenic missense variation. All three affected individuals were heterozygotes, which was further confirmed by Sanger sequencing (Figure 2B). Other variations in this gene are associated with HCM (13), DCM (28, 29) and LVNC (30–32). The resultant protein is composed of a myosin N-terminal SH3-like domain (residues 32–81), a myosin motor domain (residues 85–778) and an IQ domain (residues 781–810). MYH7 is an actin based motor protein that exhibits ATPase activity (22, 24). On the other hand, this protein has two actin-binding regions (655–677 and 757–771) and one nucleotide-binding region (178–185) (22, 24).
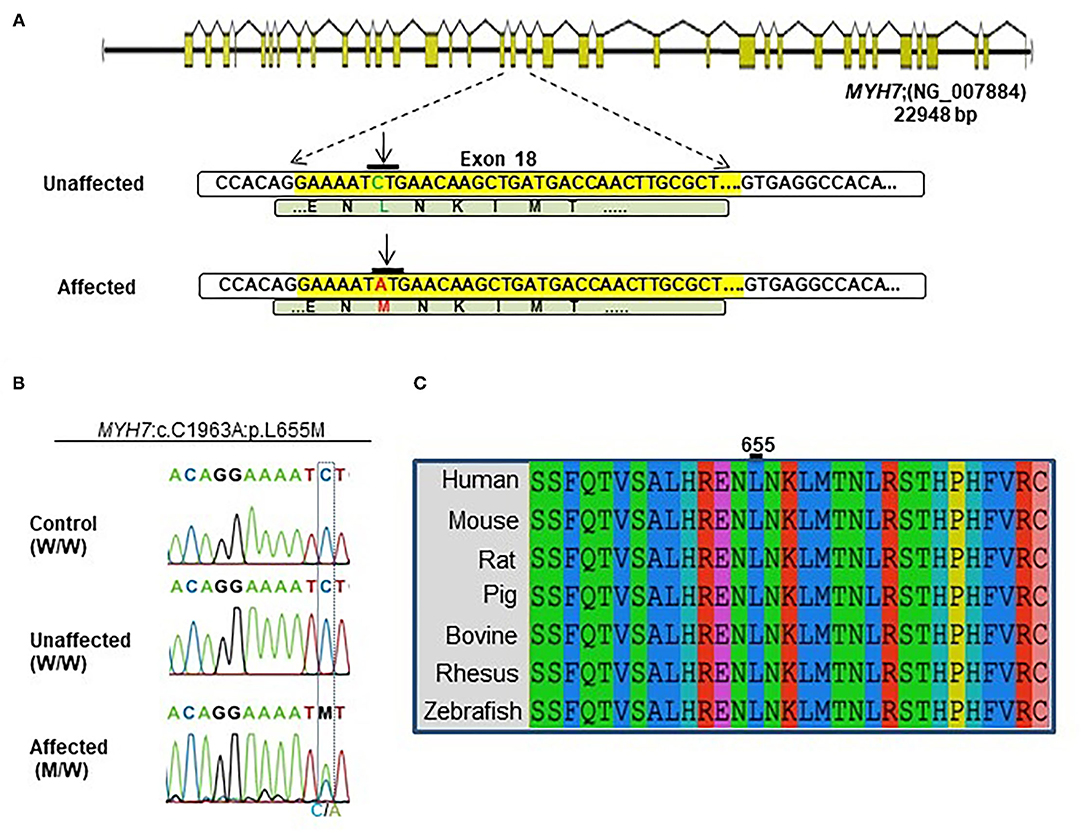
Figure 2. Identification of the MYH7 heterozygous variant in the selected Iranian family. (A) Schematic diagram shows the human MYH7 locus and the enlarged sequences of exon 18 of this locus, along with the sequences of the MYH7 variation. The yellow region is the exon 18 sequences from MYH7. Green letters are wild-type DNA and the amino acid motif in the unaffected family members. Red letters indicate the substituted (C–A) target nucleotide in the MYH7 and the p.Leu655Met alleles in the affected family members. (B) Sequence chromatograms of the MYH7 novel variant loci which were found in this study. The results of Sanger sequencing from affected, unaffected, and control DNA samples confirmed that the variation was inherited from one of the parents in a heterozygous pattern. (C) Evolutionary conservation of MYH7 in the actin-binding site domain shows that this region of protein is highly conserved throughout all species, especially in Leu655.
Segregation analysis of all family members (20 members) confirmed the presence of heterozygous alleles (MYH7W/M) in all seven affected members and wild type alleles (MYH7W/W) within the healthy members (Figure 2B). In silico prediction tools were used for protein-protein interaction analysis to predict the functional consequences of p.Leu655Met at the structural level. Phylogenetically, this variant is located in a conserved region among mammalians, and zebrafish and belongs to the motor domain area of the MYH7 protein (Figure 2C). Several prediction tools suggested this variant as a pathogenic one; MutationTaster2 predicted that it is a disease-causing mutation with a score of 0.489. SIFT showed a damaging score of 0.912 and Polymorphism Phenotyping v2 (PolyPhen-2) predicted a score of 0.971.
In preliminary studies, two boys in the third generation (III-8 and III-9) showed no specific clinical symptoms and appeared healthy at the initial consultation. However, after genotyping of all family members, it was found that III-8 and III-9 also carry this variation. To confirm the genotyping results, III-8 and III-9 were referred to a specialist, and they were diagnosed with mild LVNC symptoms.
The expression patterns of all myosin heavy chain classes were explored using a bio GPS database in order to investigate the tissue specificity of this protein. The results revealed high MYH7 protein expression levels in the heart, skeletal muscle, and fetal thyroid (Supplementary Figure 1). This result further confirmed the phenotypic presentations of the affected members, which was cardiac dysfunction and mild muscle weakness. These data suggested that MYH7 protein might be one of the major myosin heavy chain classes in the heart and skeletal muscle, and thus its mutations could result in cardiac and skeletal manifestations. Comparison of RNA sequencing data from various developmental stages of mouse heart revealed that, while all myosin classes were expressed, MYH7 had the highest expression level in all embryonic stages as well as in the adult and aged mouse hearts (GEO: GSE93271) (Figure 3A). Furthermore, the in silico analysis revealed that various myosin classes are expressed during different cardiac developmental stages of zebrafish [at 24, 48, and 72 h post fertilization (hpf)] (GEO: GSE120236). And MYH7 expression pattern showed the highest level among all myosin heavy chain classes in every stage (Supplementary Figure 2).
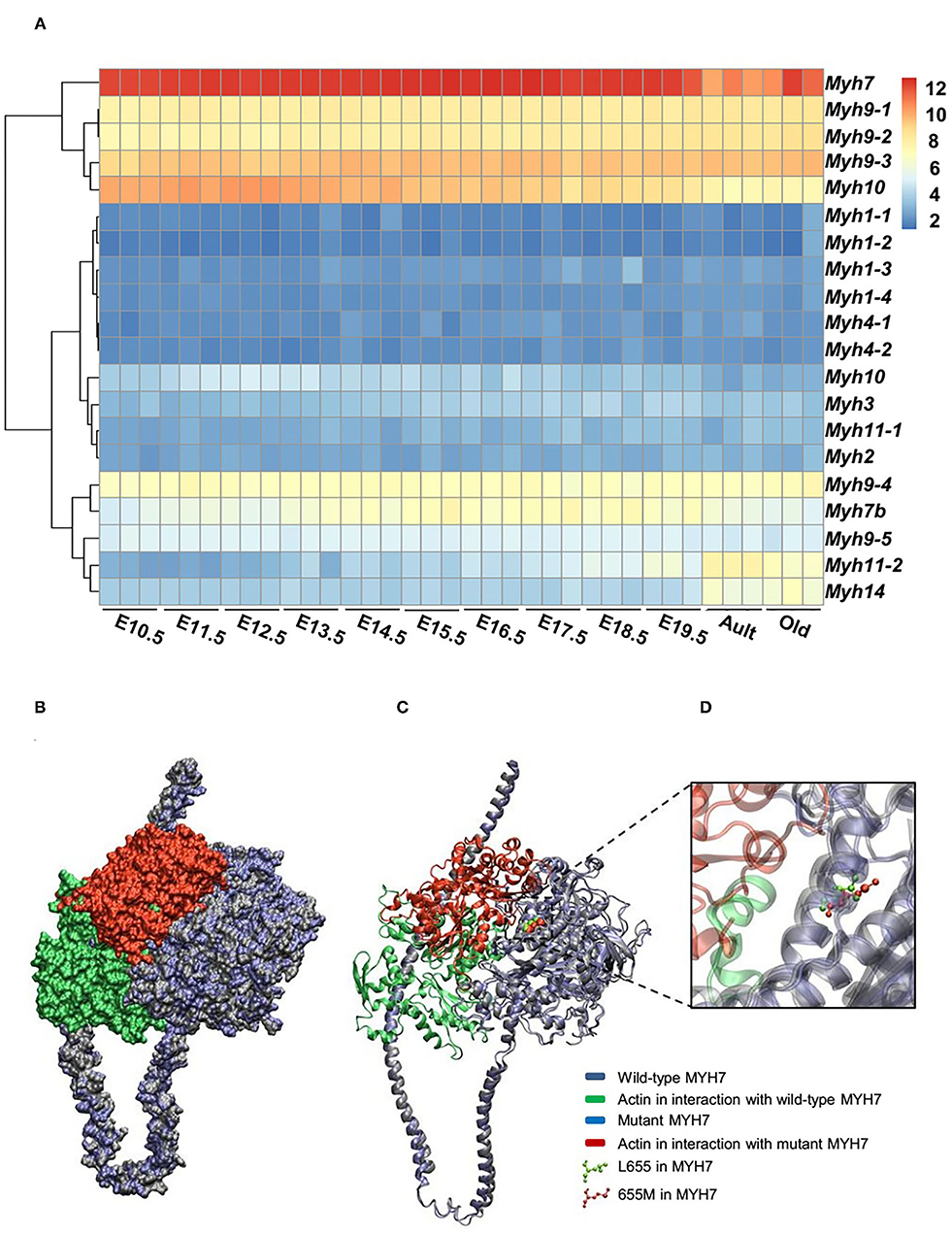
Figure 3. Expression patterns of all MYH superfamily members during cardiac development. (A) Differential expression pattern of MYH7 in the mouse heart during 12 stages of development indicates that the expression of this gene plays an important role in the mouse cardiogenesis, especially during embryonic life. (B) Modeling scheme of the protein-protein interaction between native MYH7/actin and MYH7 p.Leu655Met/actin at the surface. Actin in the wild-type position is shown in green and in the mutant position, is shown in red. (C) Secondary structure cartoon representation. Actin in the wild-type position is shown in green, and in the mutant position, is shown in red. (D) The Leu655Met mutation is highlighted. Wild MYH7 protein (Leu 655) is shown in green, and the protein that has the mutated residue (Met 655) is shown in red.
Contractile machinery of cardiac muscle is mainly dependent on actin and myosin proteins and their function; thus, it is likely that any structural associated dysfunction of these proteins would greatly affect cardiac performance.
For evaluating the effect of the variation (p.Leu655Met) on MYH7 function, the wild type and mutant minimized structures of MYH7 were docked with actin through the HADDOCK2.2 web server. The most reliable complex conformation of the wild type and mutant MYH7 interactions with actin were selected based on the HADDOCK2.2 results and presented here. Both complexes were aligned based on the MYH7 structure and shown in surface (Figure 3B) and secondary structure cartoon (Figure 3C) representations. The averaged HADDOCK2.2 score over the top four members of the best cluster was −125.1 ± 13.7 for the wild type MYH7-actin complex, which increased for the mutant MYH7-actin complex (HADDOCK2.2 score = −115.1 ± 13.4). This result revealed that the mutant complex was relatively more unstable than the wild type complex. In addition, a comparison of the actin protein position in interaction with the mutant MYH7 (red in Figures 3B–D) and wild type (green in Figures 3B–D) showed a mean root mean square deviation (RMSD value) of 48.05 Å, which indicated rotation and transition of this protein. In the p.Leu655Met mutation, a leucine amino acid is changed to methionine, which is a longer hydrophobic amino acid with an S-methyl thioether side chain. As it can be seen in Figure 3D, the amino acid at position 655 tends to the inner side of the protein. The methionine in this position would stabilize the structure with its longer hydrophobic side chain and S/π interactions between the side chain sulfur atom and aromatic amino acids. Thus, we postulate that this conformational change leads to a weaker binding energy of the actin protein to the 655M residue and subsequent functional insufficiency.
WES data revealed that p.Leu655Met point mutation in the MYH7 gene is related with LVNC. Next, to investigate the role of MYH7 in vertebrate heart development in vivo, we exploited clustered regularly interspaced short palindromic repeats/CRISPR associated protein 9 (CRISPR/Cas9) technology to generate a MYH7 knockout (KO) zebrafish model (33, 34). We specifically designed a sgRNA to target exon 18, similar loci where the missense mutation occurred in patients. Following the microinjection of ribonucleoprotein (RNP) complex, we monitored the embryos daily under a stereomicroscope and observed a pronounced phenotype in majority of the embryos starting from 3 days post-fertilization (dpf). Next, we extracted genomic DNA from individual larvae at 5 dpf and proceeded with Sanger sequencing in order to determine insertions and deletions (indels). Sequencing results confirmed indels in MYH7 gene with an efficiency of approximately 90% (data not shown). We further characterized MYH7 KO zebrafish by using fluorescence microscopy to examine heart development and whole-mount in situ hybridization (WMISH) to detect mRNA expression of cardiac marker genes.
Daily observation of MYH7 KO zebrafish larvae revealed cardiovascular defects such as pericardial edema, chamber formation at pericardial sac and misshapen heart that became visible starting 3 dpf compared to the control sgRNA injected larvae. We also observed hemorrhage at various parts of the body near the heart and the vasculature system (Figure 4A). Pericardial and yolk sac/abdominal edema continued to expand until 5 dpf. At day five post fertilization, MYH7 KO fish showed thickened heart with abnormal development of atrial and ventricular chambers as well as disturbed S-looping process (13), resulting in nearly unified heart (Figure 4A). Supplementary Video 1 shows a 5-dpf MYH7 KO zebrafish larva with complications in heart contraction and blood flow, which are likely to be responsible for the hemorrhage. In addition, mutant fish exhibited reduced swimming ability and mobility (data not shown). Cardiac malformations advanced over time, where most of the larvae were dead at 7 dpf due to failure of proper cardiovascular system development. These data suggest that MYH7 gene is essential for cardiovascular development.
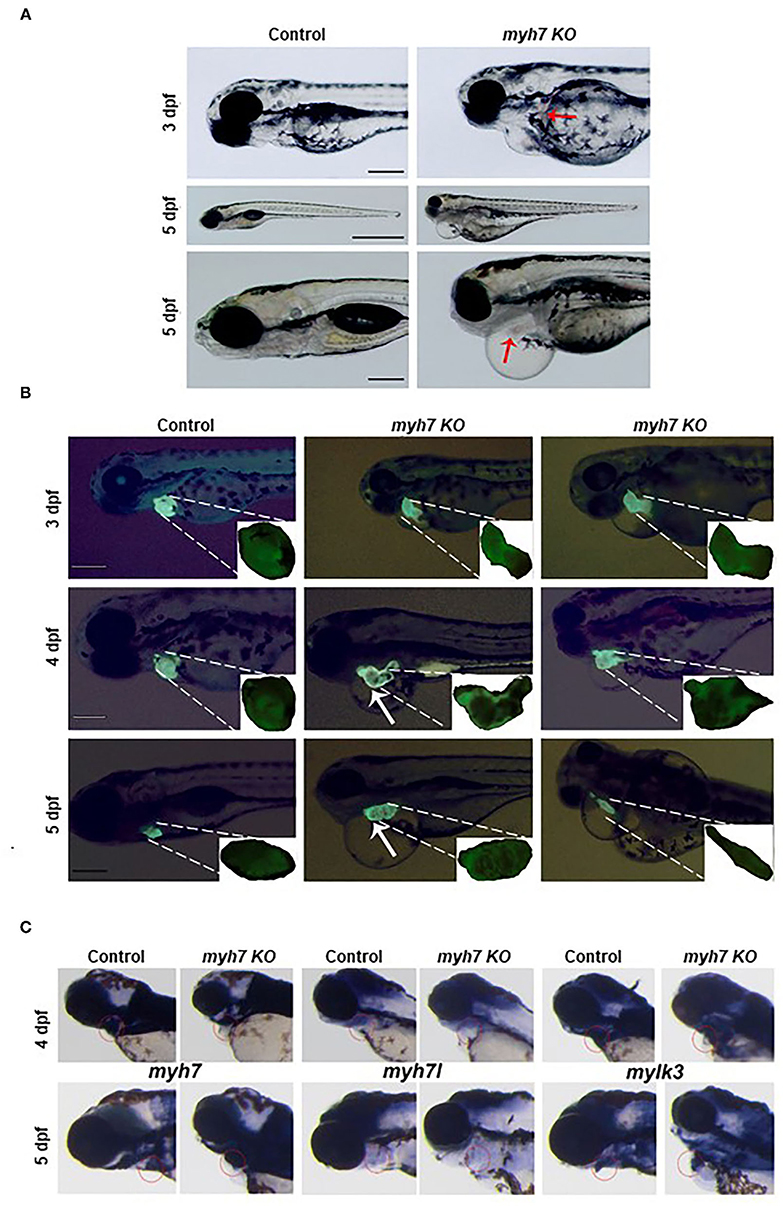
Figure 4. The MYH7 deficiency causes cardiovascular defects in zebrafish. (A) Loss of MYH7 disrupts heart development. Lateral view of 3 and 5 dpf scramble gRNA injected control group and MYH7 KO larvae. Cardiac edema is clearly noticeable. Arrow at 3 dpf shows hemorrhage near heart. Edema accumulated around heart and spread to the abdominal area at 5 dpf. Arrow at 5 dpf shows enlarged heart. Scale bars: 200 μm in 3 and 5 dpf, 1 mm in middle 5 dpf. (B) The MYH7 knockout zebrafish has distorted cardiac chamber formation and looping as shown by fluorescence imaging of the Tg(actb2:hyper3) transgenic zebrafish reporter injected with scramble or MYH7 sgRNA. Representative images of control and mutant zebrafish are shown at 3, 4, and 5 dpf. Two different MYH7 KO fish at 3 dpf showed abnormal looping. Two different MYH7 KO fish at 4 dpf displayed enlarged and deformed ventricles. At 5 dpf, myh7 KO fish showed enlarged and granulated ventricle. Furthermore, a MYH7 mutant zebrafish with tubular heart is shown. Scale bars: 200 μm. (C) MYH7 is essential for proper formation of the cardiac ventricle and the pericardial cavity as shown by WMISH. Expression of MYH7, MYH7l, and mylk3 mRNAs is strongly reduced in MYH7 KO zebrafish at 4 dpf. MYH7 expression expands to the pericardial cavity and ventricular chamber in MYH7 KO fish at 5 dpf. MYH7l expression is reduced in MYH7 KO fish at 5 dpf. Expression of mylk3 in the cardiac ventricle of MYH7 KO larvae is disrupted at 5 dpf.
The zebrafish heart has a tubular form at 1 dpf, later, it undergoes morphogenesis including looping and chamber ballooning at 2 dpf and forms the functional heart at 5 dpf (35). To understand how these processes are affected by MYH7 loss-of-function, we injected MYH7 sgRNA into the embryos of Tg(actb2:hyper3), a transgenic zebrafish reporter that contains the GFP driven by the cmlc2 promoter (cmlc2:egfp) transgenesis marker within HyPer3 (36). The MYH7 mutant zebrafish exhibited disorganized heart with enlarged atrium and ventricle along with abnormal looping at 3 dpf (Figure 4B). We observed several heart morphological defects in MYH7 KO zebrafish. Majority of the larvae displayed elongated and granulated ventricle at 4 dpf (Figure 4B). At 5 dpf, loss of MYH7 function was visible as bloated heart with thickened ventricle and altered S-looping formation (Figure 4B). Moreover, some larvae exhibited a tubular heart phenotype with visible separation between atrium and ventricle but no S-loop formation, resulting in a drastic reduction of the heart volume, ventricular contraction and blood flow rate (Figure 4B and Supplementary Video 2). These results indicate that MYH7 is necessary for proper establishment of the heart at the morphological level and its contractile function.
Next, to investigate how MYH7 mutation affected cardiac ventricular-related gene expression, we exploited WMISH by using the antisense RNA probes for MYH7 and two other cardiac genes myosin heavy chain 7-like (MYH7l) and myosin light chain kinase 3 (mylk3) on control and MYH7 KO zebrafish larvae at 3 and 5 dpf. In control larvae, MYH7, MYH7l, and mylk3 were properly expressed at the cardiac ventricle at 3 and 5 dpf (Figure 4C). In contrast, their expression levels were significantly reduced in MYH7 KO larvae at 3 dpf. At 5 dpf, MYH7 expression appeared to expand in parallel to the widening pericardial cavity and ventricular chamber (Figure 4C). The MYH7l was hardly detectable in the mutant larvae at 5 dpf. Expression of mylk3 was also severely distorted in the cardiac ventricle of MYH7 KO larvae, in accordance with its key role in enhancement of contraction force via regulation of actin-myosin interaction (Figure 4C) (37). These results suggest the MYH7 role in heart development, especially in proper formation of the cardiac ventricle and the pericardial cavity.
MYH7 or cardiac muscle beta isoform, the protein encoded by MYH7, is a member of the myosin superfamily of proteins expressed in muscle cells. MYH7 contributes to movement and contraction, especially in cardiac cells, and plays a vital role in the sarcomere structure and heartbeat. Any mutation in MHY7 can influence contractile properties and form-function relationship (32, 38) by affecting its binding to actin, MYBPC3, and TNNT2. The normal MYH7 can also regulate ATP consumption and protein-protein interaction, and as a result, the binding regions of this protein and accompanying proteins are crucial to the function of myosin (39).
In this study, we used WES to elucidate the cause of LVNC in a highly affected Iranian family. WES analysis revealed a novel variation in the MYH7 gene. Segregation analysis confirmed the variation in the affected members. In the next step, the expression pattern of myosin heavy chain classes was investigated in human tissues and in developmental stages of mice and zebrafish hearts. These analyses highlighted higher expression of MYH7 in the heart and skeletal muscle, the entire mouse and zebrafish cardiogenesis, in addition to its presence in adult mice hearts. According to this finding, MYH7 activity starts in the early embryonic stages of heart development. Due to its important role in the heart, its expression is distinct and excessive in this tissue. MYH7 is also expressed differently from other myosin classes in the heart. A previous report also indicated higher expression levels of MYH7 during the fetal stage of mice and its conditional depletion led to mild adulthood lethality (40, 41). The current RNAseq data analysis also suggested an important developmental role for myosins, especially for MYH7, as they are expressed ubiquistously during mouse cardiac organogenesis.
A novel missense variant was identified (c.1963C>A:p.Leu655Met) in the MYH7 gene of a rare cardiomyopathy condition called LVNC by using WES analysis, a mutation that affected the MYH7-actin interaction. SIFT data showed that the region in which the new variant is located, is sensitive to amino acid changes and would impair MYH7-actin binding and its function. Thus, WES analysis provides great opportunity for researchers to find the correlation between the phenotype and genotype in inherited diseases. The results of MYH7 protein alignment revealed that the original Leu655 is a conserved amino acid between several disciplines of vertebrates. These findings further highlight the importance of this protein's originality and the reason underlying its Leu655 conservation over the course of evolution. Although several researchers have investigated different variations in the vicinity of the actin-binding domain with the HCM phenotype (13, 42–44), This study suggested, for the first time, a novel variation of MYH7 which might affect the actin-binding site that causes LVNC (45–47).
In mammalian muscles, actin-myosin machinery requires a regular cycle of ATP molecules and the frequent binding of two proteins, actin and myosin. This process is regulated by the myosin light chain protein (48, 49). Any malfunction of this regulator will reduce the contractile strength and loss of actin filament mobility (50, 51). The 3D analysis of myosin heavy chain classes by crystallography (52) and cryo-electron micrographs of actin-myosin complexes (53) provide platforms for studying this complex under different conditions (39). In the current study, we investigated the interaction between MYH7 and actin at the new variation site. Three-dimensional modeling revealed that the MYH7 mutant, which causes an amino acid exchange in the actin-binding site, resulted in a change in protein-protein interaction by actin rotation and transition. Interestingly, in other reports, similar variations near the actin-interacting domain were associated with different stages of HCM (13, 42, 54). However, in our affected family members, the pathogenicity is an LVNC phenotype, which is another type of cardiomyopathy. This difference between our clinical report and other studies needs to be investigated in more detail to find the appropriate correlation between these two phenotype manifestations.
An investigation to the effect of the uncovered variant(s) in terms of protein structure and function, may provide more information on the specific cellular events during an organ function as well as development. In the early stages of heart development, delamination of compact layer cardiomyocytes is the result of apical-basal polarization. Enrichment of the actomyosin network on the apical side of the delaminating cardiomyocytes has been reported (5). We suggest that the current MHY7 variation may have altered the delamination process by affecting the frequency of apical-basal polarization and excessive response to tension heterogeneity. As a result, this variation induces more delamination signaling or more proliferation in delaminating cardiomyocytes, which eventually leads to irregular trabecular structures and deep recesses.
Here, we have generated the zebrafish knockout model of MYH7 by using CRISPR/Cas9 genome editing technology and examined the influence of MYH7 on cardiac morphogenesis and ventricular contraction. Cardiomyopathies have been modeled by using a variety of genetic or chemical manipulation techniques. The most abundant phenotypic indicators for zebrafish cardiomyopathy are (i) edema in the heart and the pericardial sac (ii) disrupted S-looping morphogenesis, and (iii) deformation of the heart anatomy. These phenotypic characteristics are consistent with those observed in other cardiomyopathy models (13, 55, 56).
Genetic screening of LVNC patients has identified several variations in genes encoding for sarcomeric proteins including MYH7, α-cardiac actin, cardiac troponin T, tafazzin, α-dystrobrevin, lamin A/C and dystrophin (30, 57, 58). Most of these genes have been associated with contractile function. Moreover, a mouse model of LVNC generated by a missense variation in the cardiac muscle gene dystrobrevin-α shows multiple trabeculations, expanded left ventricle and cardiac contractile dysfunction (59). Our MYH7 KO zebrafish displays abnormally developed heart with pericardial edema, severe hemorrhage, distorted chambers and contraction problems. The correlation of features between these models suggest the use of our KO zebrafish as a platform to investigate the molecular mechanisms of MYH7-related disorder such as non-compaction cardiomyopathies.
The MYH7 is major sarcomeric protein that is mainly expressed in the zebrafish heart. There are nine zebrafish homologs of the human MYH gene and seven of them share more than 80% protein homology with human MYH7 (60). Being a component of the myosin complex, zebrafish MYH7 is expressed in a variety of structures, including the heart primordium, the pericardial region and the musculature system during embryogenesis. The cardiac ventricle is a prominent structure where we detected high levels of MYH7 in zebrafish embryos. Thus, disrupted heart formation and ultimate death of MYH7 KO larvae suggests its key role in cardiovascular system development.
In summary, this study introduced a novel genetic variation in MYH7 (c.1963C>A:p.Leu655Met) and its corresponding clinical outcome was associated with LVNC. Data analysis and segregation study allowed us to predict new patients in the pedigree. Genotyping results combined with clinical phenotypes can confirm the function of any genetic variation and identify all affected, asymptomatic members in the pedigree for earlier and more effective treatment strategies. In silico and in vivo studies suggested actin-myosin binding changes as the underlying mechanism for MYH7 variation phenotype.
The accession number for the variant c.C1963A reported in this paper is [ClinVar]: [SCV000886483.1]. The data presented in the study is available per request from corresponding author.
The studies involving human participants were reviewed and approved by Royan Institute, Tehran, Iran. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. The animal study was reviewed and approved by the Animal Experiments Local Ethics Committee of Izmir Biomedicine and Genome Center (IBG-AELEC). Written informed consent was obtained from the individual(s), and minor(s)' legal guardian/next of kin, for the publication of any potentially identifiable images or data included in this article.
MH, UB, SP, MB, and HB contributed to the conception and design of the study. SAM, NS, and MH performed bioinformatics analysis and functional analysis. MH and SP wrote the first draft of the manuscript. SJR, MT, and GO revised the manuscript. All authors contributed to the revisions and approved the submitted manuscript.
This work was supported by a grant from Royan Institute, Tehran, Iran and the Scientific and Technological Research Council of Turkey (TUBITAK, grant no. 217Z123). GO Lab was funded by an EMBO Installation Grant (IG 3024).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors thank the proband and his family members for participating in this study. They also thank Izmir Biomedicine and Genome Center Vivarium-Zebrafish Core Facility and Optical Imaging Core Facility for providing zebrafish care and microscope facility support, respectively.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2022.839862/full#supplementary-material
Supplementary Figure 1. Expression patterns of MYH7 in different human tissues. Comparison of MYH7 expression patterns in different tissues shows that this protein has elevated expression in skeletal and heart muscles.
Supplementary Figure 2. Expression patterns of myh superfamily members during zebrafish cardiac development in 24, 48, and 72 hpf.
Supplementary Table 1. Primer sequences for MYH7 targeting sgRNA, scrambled control RNA, MYH7 sequencing and RNA probes.
Supplementary Video 1. A 5 dpf MYH7 KO zebrafish larva with complications in heart contraction and blood flow.
Supplementary Video 2. The MYH7 KO zebrafish heart contraction and blood flow rate.
1. Bennett CE, Freudenberger R. The current approach to diagnosis and management of left ventricular noncompaction cardiomyopathy: review of the literature. Cardiol Res Pract. (2016) 2016:5172308. doi: 10.1155/2016/5172308
2. Sasse-Klaassen S, Gerull B, Oechslin E, Jenni R, Thierfelder L. Isolated noncompaction of the left ventricular myocardium in the adult is an autosomal dominant disorder in the majority of patients. Am J Med Genet A. (2003) 119A:162–7. doi: 10.1002/ajmg.a.20075
3. Postma AV, Van Engelen K, Van De Meerakker J, Rahman T, Probst S, Baars MJ, et al. Mutations in the sarcomere gene MYH7 in Ebstein anomaly. Circ Cardiovasc Genet. (2011) 4:43–50. doi: 10.1161/CIRCGENETICS.110.957985
4. Gaertner A, Klauke B, Brodehl A, Milting H. RBM20 mutations in left ventricular non-compaction cardiomyopathy. Pediatr Investig. (2020) 4:61–3. doi: 10.1002/ped4.12184
5. Priya R, Allanki S, Gentile A, Mansingh S, Uribe V, Maischein HM, et al. Tension heterogeneity directs form and fate to pattern the myocardial wall. Nature. (2020) 588:130–4. doi: 10.1038/s41586-020-2946-9
6. Vasilescu C, Ojala TH, Brilhante V, Ojanen S, Hinterding HM, Palin E, et al. Genetic basis of severe childhood-onset cardiomyopathies. J Am Coll Cardiol. (2018) 72:2324–38. doi: 10.1016/j.jacc.2018.08.2171
7. Van Waning JI, Caliskan K, Hoedemaekers YM, Van Spaendonck-Zwarts KY, Baas AF, Boekholdt SM, et al. Clinical features, and long-term outcome of noncompaction cardiomyopathy. J Am Coll Cardiol. (2018) 71:711–22. doi: 10.1016/j.jacc.2017.12.019
8. Uribe V, Ramadass R, Dogra D, Rasouli SJ, Gunawan F, Nakajima H, et al. In vivo analysis of cardiomyocyte proliferation during trabeculation. Development. (2018) 145:dev164194. doi: 10.1242/dev.164194
9. Luxan G, Casanova JC, Martinez-Poveda B, Prados B, D'amato G, Macgrogan D, et al. Mutations in the NOTCH pathway regulator MIB1 cause left ventricular noncompaction cardiomyopathy. Nat Med. (2013) 19:193–201. doi: 10.1038/nm.3046
10. Van Der Velden J, Stienen GJ.M. Cardiac disorders and pathophysiology of sarcomeric proteins. Physiol Rev. (2019) 99:381–426. doi: 10.1152/physrev.00040.2017
11. Stuart CA, Stone WL, Howell ME, Brannon MF, Hall HK, Gibson AL, et al. Myosin content of individual human muscle fibers isolated by laser capture microdissection. Am J Physiol Cell Physiol. (2016) 310:C381–9. doi: 10.1152/ajpcell.00317.2015
12. Soetkamp D, Gallet R, Parker SJ, Holewinski R, Venkatraman V, Peck K, et al. Myofilament phosphorylation in stem cell treated diastolic heart failure. Circ Res. (2021) 129:1125–40. doi: 10.1161/CIRCRESAHA.119.316311
13. Cohn R, Thakar K, Lowe A, Ladha FA, Pettinato AM, Romano R, et al. A contraction stress model of hypertrophic cardiomyopathy due to sarcomere mutations. Stem Cell Reports. (2019) 12:71–83. doi: 10.1016/j.stemcr.2018.11.015
14. Li H, Durbin R. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics. (2009) 25:1754–60. doi: 10.1093/bioinformatics/btp324
15. Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. (2010) 38:e164. doi: 10.1093/nar/gkq603
16. Chang X, Wang K. wANNOVAR: annotating genetic variants for personal genomes via the web. J Med Genet. (2012) 49:433–6. doi: 10.1136/jmedgenet-2012-100918
17. Sabour D, Machado RSR, Pinto JP, Rohani S, Sahito RGA, Hescheler J, et al. Parallel genome-wide profiling of coding and non-coding RNAs to identify novel regulatory elements in embryonic and maturated heart. Mol Ther Nucleic Acids. (2018) 12:158–73. doi: 10.1016/j.omtn.2018.04.018
18. Pawlak M, Kedzierska KZ, Migdal M, Nahia KA, Ramilowski JA, Bugajski L, et al. Dynamics of cardiomyocyte transcriptome and chromatin landscape demarcates key events of heart development. Genome Res. (2019) 29:506–19. doi: 10.1101/gr.244491.118
19. Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graphics. (1996) 14:33–8. doi: 10.1016/0263-7855(96)00018-5
20. Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, et al. Scalable molecular dynamics with NAMD. J Comput Chem. (2005) 26:1781–802. doi: 10.1002/jcc.20289
21. Mackerell AD Jr, Bashford D, Bellott M, Dunbrack RL Jr, Evanseck JD, Field MJ, et al. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J Phys Chem B. (1998) 102:3586–616. doi: 10.1021/jp973084f
22. Dominguez C, Boelens R, Bonvin AM. HADDOCK: a protein– protein docking approach based on biochemical or biophysical information. J Am Chem Soc. (2003) 125:1731–7. doi: 10.1021/ja026939x
23. Van Zundert G, Rodrigues J, Trellet M, Schmitz C, Kastritis P, Karaca E, et al. The HADDOCK2. 2 web server: user-friendly integrative modeling of biomolecular complexes. J Mol Biol. (2016) 428:720–5. doi: 10.1016/j.jmb.2015.09.014
24. Dominguez R. A common binding site for actin-binding proteins on the actin surface. In: Actin-Monomer-Binding Proteins. Springer (2007). pp.107–15.
25. Jowett T, Lettice L. Whole-mount in situ hybridizations on zebrafish embryos using a mixture of digoxigenin- and fluorescein-labelled probes. Trends Genet. (1994) 10:73–4. doi: 10.1016/0168-9525(94)90220-8
26. Ozalp O, Cark O, Azbazdar Y, Haykir B, Cucun G, Kucukaylak I, et al. Nradd acts as a negative feedback regulator of wnt/beta-catenin signaling and promotes apoptosis. Biomolecules. (2021) 11:100. doi: 10.3390/biom11010100
27. Thisse C, Thisse B. High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat Protoc. (2008) 3:59–69. doi: 10.1038/nprot.2007.514
28. Kamisago M, Sharma SD, Depalma SR, Solomon S, Sharma P, Mcdonough B, et al. Mutations in sarcomere protein genes as a cause of dilated cardiomyopathy. N Engl J Med. (2000) 343:1688–96. doi: 10.1056/NEJM200012073432304
29. Gifford CA, Ranade SS, Samarakoon R, Salunga HT, De Soysa TY, Huang Y, et al. Oligogenic inheritance of a human heart disease involving a genetic modifier. Science. (2019) 364:865–70. doi: 10.1126/science.aat5056
30. Klaassen S, Probst S, Oechslin E, Gerull B, Krings G, Schuler P, et al. Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation. (2008) 117:2893–901. doi: 10.1161/CIRCULATIONAHA.107.746164
31. Probst S, Oechslin E, Schuler P, Greutmann M, Boye P, Knirsch W, et al. Sarcomere gene mutations in isolated left ventricular noncompaction cardiomyopathy do not predict clinical phenotype. Circ Cardiovasc Genet. (2011) 4:367–74. doi: 10.1161/CIRCGENETICS.110.959270
32. Kolokotronis K, Kuhnisch J, Klopocki E, Dartsch J, Rost S, Huculak C, et al. Biallelic mutation in MYH7 and MYBPC3 leads to severe cardiomyopathy with left ventricular noncompaction phenotype. Hum Mutat. (2019) 40:1101–111. doi: 10.1002/humu.23757
33. Tessadori F, Roessler HI, Savelberg SMC, Chocron S, Kamel SM, Duran KJ, et al. Effective CRISPR/Cas9-based nucleotide editing in zebrafish to model human genetic cardiovascular disorders. Dis Model Mech. (2018) 11:dmm035469. doi: 10.1242/dmm.035469
34. Varshney GK, Carrington B, Pei W, Bishop K, Chen Z, Fan C, et al. A high-throughput functional genomics workflow based on CRISPR/Cas9-mediated targeted mutagenesis in zebrafish. Nat Protoc. (2016) 11:2357–75. doi: 10.1038/nprot.2016.141
35. Derrick CJ, Noel ES. The ECM as a driver of heart development and repair. Development. (2021) 148:dev191320. doi: 10.1242/dev.191320
36. Bilan DS, Pase L, Joosen L, Gorokhovatsky AY, Ermakova YG, Gadella TW, et al. HyPer-3: a genetically encoded H(2)O(2) probe with improved performance for ratiometric and fluorescence lifetime imaging. ACS Chem Biol. (2013) 8:535–42. doi: 10.1021/cb300625g
37. Tobita T, Nomura S, Morita H, Ko T, Fujita T, Toko H, et al. Identification of MYLK3 mutations in familial dilated cardiomyopathy. Sci Rep. (2017) 7:17495. doi: 10.1038/s41598-017-17769-1
38. Mori AA, Castro LR, Bortolin RH, Bastos GM, Oliveira VF, Ferreira GM, et al. Association of variants in MYH7, MYBPC3 and TNNT2 with sudden cardiac death-related risk factors in Brazilian patients with hypertrophic cardiomyopathy. Forensic Sci Int Genet. (2021) 52:102478. doi: 10.1016/j.fsigen.2021.102478
39. Guhathakurta P, Prochniewicz E, Thomas DD. Actin-myosin interaction: structure, function and drug discovery. Int J Mol Sci. (2018) 19:2628. doi: 10.3390/ijms19092628
40. Yue P, Xia S, Wu G, Liu L, Zhou K, Liao H, et al. Attenuation of cardiomyocyte hypertrophy via depletion MYH7 using CASAAV. Cardiovasc Toxicol. (2021) 21:255–64. doi: 10.1007/s12012-020-09617-y
41. Guo Y, Pu WT. Cardiomyocyte maturation: new phase in development. Circ Res. (2020) 126:1086–106. doi: 10.1161/CIRCRESAHA.119.315862
42. Nag S, Sommese RF, Ujfalusi Z, Combs A, Langer S, Sutton S, et al. Contractility parameters of human beta-cardiac myosin with the hypertrophic cardiomyopathy mutation R403Q show loss of motor function. Sci Adv. (2015) 1:e1500511. doi: 10.1126/sciadv.1500511
43. Sommese RF, Sung J, Nag S, Sutton S, Deacon JC, Choe E, et al. Molecular consequences of the R453C hypertrophic cardiomyopathy mutation on human beta-cardiac myosin motor function. Proc Natl Acad Sci USA. (2013) 110:12607–12. doi: 10.1073/pnas.1309493110
44. Geisterfer-Lowrance AA, Kass S, Tanigawa G, Vosberg H-P, Mckenna W, Seidman CE, et al. A molecular basis for familial hypertrophic cardiomyopathy: a β cardiac myosin heavy chain gene missense mutation. Cell. (1990) 62:999–1006. doi: 10.1016/0092-8674(90)90274-I
45. Mohiddin SA, Begley DA, Mclam E, Cardoso JP, Winkler JB, Sellers JR, et al. Utility of genetic screening in hypertrophic cardiomyopathy: prevalence and significance of novel and double (homozygous and heterozygous) beta-myosin mutations. Genet Test. (2003) 7:21–7. doi: 10.1089/109065703321560895
46. Van Driest SL, Jaeger MA, Ommen SR, Will ML, Gersh BJ, Tajik AJ, et al. Comprehensive analysis of the beta-myosin heavy chain gene in 389 unrelated patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. (2004) 44:602–10. doi: 10.1016/j.jacc.2004.04.039
47. Richard P, Charron P, Carrier L, Ledeuil C, Cheav T, Pichereau C, et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation. (2003) 107:2227–32. doi: 10.1161/01.CIR.0000066323.15244.54
48. Guhathakurta P, Prochniewicz E, Roopnarine O, Rohde JA, Thomas DD. A cardiomyopathy mutation in the myosin essential light chain alters actomyosin structure. Biophys J. (2017) 113:91–100. doi: 10.1016/j.bpj.2017.05.027
49. Sweeney HL, Bowman BF, Stull JT. Myosin light chain phosphorylation in vertebrate striated muscle: regulation and function. Am J Physiol. (1993) 264:C1085–95. doi: 10.1152/ajpcell.1993.264.5.C1085
50. Lowey S, Waller GS, Trybus KM. Skeletal muscle myosin light chains are essential for physiological speeds of shortening. Nature. (1993) 365:454–6. doi: 10.1038/365454a0
51. Vanburen P, Waller GS, Harris DE, Trybus KM, Warshaw DM, Lowey S. The essential light chain is required for full force production by skeletal muscle myosin. Proc Natl Acad Sci USA. (1994) 91:12403–7. doi: 10.1073/pnas.91.26.12403
52. Sweeney HL, Houdusse A. Structural and functional insights into the myosin motor mechanism. Ann Rev Biophys. (2010) 39:539–57. doi: 10.1146/annurev.biophys.050708.133751
53. Wulf SF, Ropars V, Fujita-Becker S, Oster M, Hofhaus G, Trabuco LG, et al. Force-producing ADP state of myosin bound to actin. Proc Natl Acad Sci USA. (2016) 113:E1844–52. doi: 10.1073/pnas.1516598113
54. Chuan P, Sivaramakrishnan S, Ashley EA, Spudich JA. Cell-intrinsic functional effects of the alpha-cardiac myosin Arg-403-Gln mutation in familial hypertrophic cardiomyopathy. Biophys J. (2012) 102:2782–90. doi: 10.1016/j.bpj.2012.04.049
55. Lu S, Hu M, Wang Z, Liu H, Kou Y, Lyu Z, et al. Generation and application of the zebrafish heg1 mutant as a cardiovascular disease model. Biomolecules. (2020) 10:1542. doi: 10.3390/biom10111542
56. Kustermann M, Manta L, Paone C, Kustermann J, Lausser L, Wiesner C, et al. Loss of the novel Vcp (valosin containing protein) interactor Washc4 interferes with autophagy-mediated proteostasis in striated muscle and leads to myopathy in vivo. Autophagy. (2018) 14:1911–27. doi: 10.1080/15548627.2018.1491491
57. Xing Y, Ichida F, Matsuoka T, Isobe T, Ikemoto Y, Higaki T, et al. Genetic analysis in patients with left ventricular noncompaction and evidence for genetic heterogeneity. Mol Genet Metab. (2006) 88:71–7. doi: 10.1016/j.ymgme.2005.11.009
58. Zhang W, Chen H, Qu X, Chang CP, Shou W. Molecular mechanism of ventricular trabeculation/compaction and the pathogenesis of the left ventricular noncompaction cardiomyopathy (LVNC). Am J Med Genet C Semin Med Genet. (2013) 163C:144–56. doi: 10.1002/ajmg.c.31369
59. Cao Q, Shen Y, Liu X, Yu X, Yuan P, Wan R, et al. Phenotype and functional analyses in a transgenic mouse model of left ventricular noncompaction caused by a DTNA mutation. Int Heart J. (2017) 58:939–47. doi: 10.1536/ihj.16-019
Keywords: cardiomyopathy, LVNC, WES, myosin heavy chain 7, zebrafish
Citation: Hesaraki M, Bora U, Pahlavan S, Salehi N, Mousavi SA, Barekat M, Rasouli SJ, Baharvand H, Ozhan G and Totonchi M (2022) A Novel Missense Variant in Actin Binding Domain of MYH7 Is Associated With Left Ventricular Noncompaction. Front. Cardiovasc. Med. 9:839862. doi: 10.3389/fcvm.2022.839862
Received: 20 December 2021; Accepted: 22 February 2022;
Published: 08 April 2022.
Edited by:
Mads Kjolby, Aarhus University, DenmarkReviewed by:
Keiichi Hirono, University of Toyama, JapanCopyright © 2022 Hesaraki, Bora, Pahlavan, Salehi, Mousavi, Barekat, Rasouli, Baharvand, Ozhan and Totonchi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gunes Ozhan, Z3VuZXMub3poYW5AaWJnLmVkdS50cg==; Mehdi Totonchi, bS50b3RvbmNoaUByb3lhbmluc3RpdHV0ZS5vcmc=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.