 Farah Cadour
Farah Cadour Franck Thuny3
Franck Thuny3 Joevin Sourdon
Joevin Sourdon- 1Aix-Marseille Université, CNRS, CRMBM, Marseille, France
- 2APHM, Hôpital Universitaire Timone, CEMEREM, Marseille, France
- 3Aix-Marseille University, University Mediterranean Center of Cardio-Oncology, Unit of Heart Failure and Valvular Heart Diseases, Department of Cardiology, North Hospital, Assistance Publique - Hôpitaux de Marseille, Centre for CardioVascular and Nutrition Research (C2VN), Inserm 1263, Inrae 1260, Marseille, France
Cardio-oncology requires a good knowledge of the cardiotoxicity of anticancer drugs, their mechanisms, and their diagnosis for better management. Anthracyclines, anti-vascular endothelial growth factor (VEGF), alkylating agents, antimetabolites, anti-human epidermal growth factor receptor (HER), and receptor tyrosine kinase inhibitors (RTKi) are therapeutics whose cardiotoxicity involves several mechanisms at the cellular and subcellular levels. Current guidelines for anticancer drugs cardiotoxicity are essentially based on monitoring left ventricle ejection fraction (LVEF). However, knowledge of microvascular and metabolic dysfunction allows for better imaging assessment before overt LVEF impairment. Early detection of anticancer drug-related cardiotoxicity would therefore advance the prevention and patient care. In this review, we provide a comprehensive overview of the cardiotoxic effects of anticancer drugs and describe myocardial perfusion, metabolic, and mitochondrial function imaging approaches to detect them before over LVEF impairment.
Introduction
Cancer therapy significantly improves patient survival but is sometimes accompanied by cardiotoxic effects. Cardiotoxic complications can range from myocardial abnormalities, valvular abnormalities, pericardial diseases, coronary artery disease (CAD), and alteration in left ventricle ejection fraction (LVEF).
Anthracyclines, one of the most used and oldest chemotherapies, are the archetypal cardiotoxic anticancer drug, ultimately leading to the heart failure (1). In addition, the emerging field of cardio-oncology has seen the development of new anticancer drugs such as antiangiogenics also leading to cardiotoxicity with endothelial dysfunction, forcing a reconsideration of the stages, timing, and levels of cardiotoxicity.
Initial evaluation of LVEF and subsequent evaluation under anticancer therapy is paramount as the most guidelines for cardiotoxicity are based on LVEF impairment (2). To date, echocardiography remains the most frequently used method to detect LVEF alteration, but also by assessment of left ventricle (LV) longitudinal strain evaluation that might identify early LVEF dysfunction (3). Although not considered the first-line method, cardiac magnetic resonance imaging (CMR) can assess cardiac anatomy, structure, and tissue properties in addition to LVEF.
These modalities have been able to detect impaired cardiac function in the later stages of cardiac side effects (4). Myocardial perfusion imaging and metabolic imaging are powerful approaches providing novel biomarkers that can improve early detection of cardiotoxicity before irreversible cardiac damage occurs. This review summarizes the alterations in cardiac perfusion and metabolism that occur in anticancer drug-related cardiotoxicity and the advantage of assessing perfusion and metabolism non-invasively in the beating heart with cardiac imaging.
Myocardial Vascular and Metabolic Effects of Anticancer Drugs
Overview of the Link Between Myocardial Circulation and Metabolism
There is a close relationship between myocardial blood circulation, which delivers oxygen and nutrients, tissue metabolism, and oxidative stress. The heart has a very high energy demand to sustain contractile function and synthesizes adenosine triphosphate (ATP) through oxidative metabolism of free fatty acids (FFA), glucose, ketones, and lactate (5).
The adult heart normally obtains 50–70% of its ATP from fatty acid β-oxidation in the presence of oxygen. However, it must adapt, switching from one substrate to another, to sustain demand depending upon the metabolic state and physical conditions at the time (5). Under well-perfused aerobic conditions, glucose and FFA are catabolized into pyruvate or acyl-CoA, respectively, both of which are catabolized to acetyl-CoA to enter the tricarboxylic acid (TCA, Krebs) cycle. Most of the energy supply is then derived from the mitochondrial oxidative phosphorylation system. The main cardiac energy reserve is phosphocreatine (PCr), which is maintained by the following creatine kinase (CK) reaction:
This system facilitates intracellular delivery of energy from mitochondria to cytoplasmic sites of ATP utilization and maintains a high level of ATP during changes in energy demand (6).
Direct damage to the mitochondria, blood supply, and myocardial metabolism will be responsible for abnormal production of reactive oxygen species (ROSs). ROS are reactive intermediates of the molecular oxygen that are essentially generated during mitochondrial oxidative phosphorylation (7). Cellular sources of ROSs are cardiomyocytes, endothelial cells, stromal cells, and inflammatory cells in the heart (8). One of the major ROSs is the proximal mitochondrial ROSs (superoxide anion), which can be generated by a loss of ATP production or when there is a high NADH/NAD+ ratio in the mitochondrial matrix (9). An imbalance between ROS production and antioxidant cell response leads to endothelial dysfunction, the release of proinflammatory cytokines, and vasoconstriction of epicardial and microvascular coronary arteries (10). The heart is particularly sensitive to oxidative stress because of its low-antioxidant resources (11–13). One of the main mechanisms of ROS leading to endothelial dysfunction is the uncoupling of endothelial nitric oxide (NO) synthase, which usually facilitates NO production (14), ultimately leading to reduce NO bioavailability. Indeed, the endothelium synthetizes the NO (15), which acts as a vasodilator, an antithrombotic, and an anti-atherosclerotic molecule (14). Endothelial nitric oxide synthase (eNOS) is the type III of NO synthases (NOS) that will lead to NO radicals synthesis from L-arginine and is expressed in endothelial cells. But in the inflammatory situation, the other NO synthases are neuronal NOS (type I) and inducible NOS (iNOS, type II). The latter will be expressed in blood vessels under pathological conditions such as inflammation or oxidative stress (16). Major cell structure and function damages will result reaction of NO with superoxide anion leading to peroxynitrite (17).
Interestingly, initial vascular injury also results in the production of ROSs species derived from NAD(P)H (18). Oxidative inflammation will ultimately cause adventitial fibrosis and smooth muscle hypertrophy (18). The latter phenomenon can also be observed in the media and intima through paracrine effects of adventitial inflammation. As a result, medial layers of vessels do not respond to NO to adapt blood flow and assure normal myocardial perfusion (19), resulting in impaired endothelium-dependent relaxation.
It is important to bear in mind that impaired myocardial perfusion and/or subsequent alteration of metabolic pathways, substrate preferences, and bioenergetics (i.e., reduced PCr/ATP ratio) might contribute to the development of several common cardiovascular diseases (20). For these reasons, perfusion and metabolic imaging are preferred methods to study early vascular and metabolic cardiotoxic effects.
Anticancer Drugs
The vascular and metabolic cardiotoxic effects of the various anticancer drugs are given in Table 1.
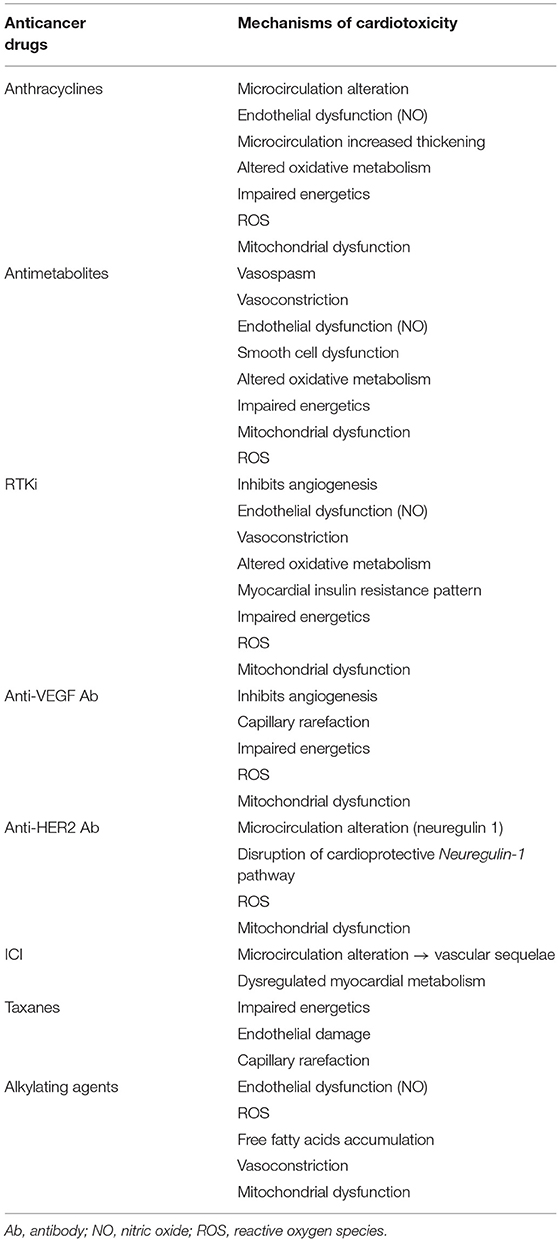
Table 1. Myocardial vascular and metabolic effects of common anticancer drugs.
Anthracyclines
Anthracyclines are a group of chemotherapy broadly used in cancer treatment, with doxorubicin (DOX) being one of the most widely used. Its cardiotoxicity is well-known with cumulative toxicity ultimately leading to permanent cardiac alteration (21). The initial alteration of this end state is thought to be at a microvascular level through ROS production (22–24), with mitochondrial superoxide production increasing with DOX dose (25).
Excessive production of ROS by DOX leads to apoptosis, cardiac function impairment, inflammation, and vascular injury (25, 26). Both the cardiomyocytes and arterial endothelial cells can experience mitochondrial dysfunction under anthracyclines (27, 28). These properties suggest that, in addition to its known direct effect on deoxyribonucleic acid through topoisomerase II beta inhibition (29), endothelial cells injury could be one cause of anthracycline cardiotoxicity. Although anthracyclines cardiotoxicity is usually detected at a stage of altered ejection (21), studies suggest that anthracyclines cardiotoxicity occurs in a continuum, challenging the hypothesis of irreversible cardiac injury (30, 31).
Current guidelines suggest monitoring of patients with cancer undergoing chemotherapy by echocardiography since most definitions of cardiotoxicity are based on LVEF decline (2), but the literature reports microcirculation changes long before any LVEF or contraction alterations occur (31, 32). This myocardial perfusion alteration could be the result of increased arterial walls thickening, which can occur early and even after a single DOX injection (31, 33), but is more overt with repeated injections (33). The increase in intima-media thickness under anthracyclines (34) is in part secondary to oxidative inflammation. Thus, anthracyclines cardiotoxicity appears at the histological level and these microcirculation alterations appear to be an early form of the well-known anthracyclines cardiotoxicity, suggesting modalities to assess the initial endothelial cell damage and better prevent its progression. Moreover, the combination of radiotherapy with anthracyclines potentiates heart damage. Radiotherapy has been reported as responsible for cardiac perfusion defect development, however, myocardial perfusion imaging of the combination of radiotherapy with anthracyclines remains poorly described (32).
Antimetabolites
5-Fluorouracil (5-FU) is a part of antimetabolite agents and is commonly used in the treatment of malignancies. One of the major cardiotoxicities of 5-FU is coronary vasospasm that can lead to ischemia. Its mechanism remains uncertain, with some suggesting an endothelial-dependent mechanism through endothelial dysfunction, but others an endothelium-independent with vasoconstriction of dysfunctional smooth muscle cells (35). Studies in animal models demonstrated that altered erythrocyte metabolism decreases erythrocyte ability to bring oxygen to the myocardium (36, 37). 5-FU reduces oxidative metabolism (38), impairs energetics (38), and induces mitochondrial uncoupling reducing aerobic efficiency (39). At a subcellular level, the toxicity of 5-FU and another antimetabolite drug, the capecitabine, have been shown to be mediated through oxidative stress with ROS generation leading to altered mitochondrial membrane potential in isolated rat cardiomyocytes (40).
Alkylating Agents
One of the main alkylating agents, mostly used in hematologic cancers, is cyclophosphamide, for which dose-mediated cardiotoxicity is one of the notable toxic effects. The metabolites of cyclophosphamide reported to be involved in cardiotoxicity are acrolein and 4-hydroxy-cyclophosphamide. These metabolites are involved in ROS generation (41, 42) that damage mitochondrial membrane by decreasing its detoxifying capacity, but also by disrupting normal vasotone response pathway through NO reduction or an increase in the vasoconstrictor endothelin-1 (23). In addition, cyclophosphamide is responsible for FFA accumulation and reduction of ATP production resulting in the release of proinflammatory cytokines (41). Cardiac microscopic findings of alkylating agents consist of interstitial damages, myocardial necrosis, vacuolar changes, and intramural changes in small coronary vessels (43). Similar disturbances have also been reported with cisplatin-based chemotherapy, another alkylating agent (44).
Taxanes
Taxanes are antimicrotubules whose main cardiotoxicity is disruption of cardiac rhythm and conduction. Heart failure (possibly in combination with DOX), ischemia, and microvascular rarefaction because of the endothelial damage might also occur (45).
Receptor Tyrosine Kinase Inhibitors
Receptor tyrosine kinase inhibitors (RTKi) include sorafenib, pazopanib, and sunitinib. As a part of antiangiogenic therapy, RTKi inhibits the tyrosine kinase activity of the vascular endothelial growth factor (VEGF) receptor, thereby blocking the VEGF pathway, but also platelet-derived growth factor receptors and c-kit (46). Oxidative stress and dysregulation of NO signaling have been proposed to mediate RTKi-induced hypertension, as they are known to be involved in the VEGF pathway (47, 48). However, sunitinib-induced hypertension has been associated with upregulation of the endothelin peptide (49–51), a potent vasconstrictor known to induce cardiac endothelial dysfunction (52). Experimental studies investigating the effects of VEGFR blockade on cardiac microvasculature did not reveal any changes in the number of capillaries (50, 53). Nevertheless, sunitinib induces a loss of coronary microvascular pericytes in mice (53), which might explain the impaired coronary flow reserve (CFR) of sunitnib-induced cardiotoxicity (49, 53).
Carbohydrate metabolism is altered in the myocardium of sunitinib-treated mice, which exhibits higher glucose uptake, higher gene expression of pyruvate dehydrogenase kinase, and of the pyruvate kinase isoform 2 (54), a signature of fetal myocardium in which the metabolism is mostly anaerobic. The sensor of cardiac energetic metabolism, AMP-activated protein kinase, is inhibited by sunitinib (55). Energy impairment because of the loss of mitochondrial membrane potential resulting in reduced ATP has been reported in the early stages of sunitinib-treated cardiomyocytes (56).
In a comparative study, only sorafenib among others RTKi directly impaired mitochondrial function and oxidative metabolism at clinically concentrations (57), but ROS generation was documented in several RTKi-treated myocardium (58, 59).
Anti-vascular Endothelial Growth Factor (VEGF) Monoclonal Antibody
Another antiangiogenic approach consists of blocking VEGF with a humanized monoclonal antibody, which traps endogenous VEGF and inhibits its binding with the receptor. Bevacizumab was the first anti-VEGF antibody with a rate of sytemic hypertension as high as 70%, probably because of the vascular resistance, endothelial dysfunction, and capillary rarefection (39). Bevacizumab induces mitochondrial dysfunction plus ROS formation in isolated rat heart (60, 61) and in isolated cardiomyocytes (62).
Anti-human Epidermal Growth Factor Receptor (HER 2)
Human epidermal growth factor receptor 2 is a receptor that promotes cell growth, proliferation, and repair in the body. Tumors can hijack these functions to proliferate. Therefore, one treatment option is to specifically target this receptor, with anti-HER2 therapy, led by Trastuzumab, which has revolutionized the treatment and prognostic of patients with HER2 positive breast cancer (63). Trastuzumab will result in ROS production, mitochondrial dysfunction, and proapoptotic signals release in cardiomyocytes (64). Unlike anthracyclines, cardiotoxicity of anti-HER2 is dose-independent and often reversible. However, it results in greater cardiotoxicity in the presence of or after anthracyclines (65).
Anti-HER2 might cause cardiomyocyte damage by disrupting the neuregulin-1 axis that normally activates protective pathways in response to stress (66), which could lead to LVEF decrease. Neuregulin-1 is a cardioactive growth factor that normally participates in the dimerization of HER receptors on cardiomyocytes to provide cell protection. However, the fact that neuregulin-1 is released from the endothelial cells in the heart leads to the question of whether the impaired LVEF is due to a direct impact of anti-HER2 on cardiomyocytes or an indirect impact via endothelial cells of the altered coronary microvasculature (67). Interestingly, a decrease in neuregulin-1 levels has been associated with CAD (68). The same neuregulin-1/HER pathway may also explain the increased susceptibility to anthracyclines cardiotoxicity when the two treatments are combined.
Immune Checkpoint Inhibitors (ICIs)
Immune checkpoint inhibitors are monoclonal antibodies that restore antitumor immunity by targeting inhibitory receptors on the lymphocytes surface, such as cytotoxic T-lymphocyte-associated protein 4, programmed cell death receptor 1 (PD1), and its ligand. By reactivating the immune response against the tumor, ICIs can lead to immune-related cardiovascular adverse events that, although rare, present a case-fatality rate as high as 50% (69). The most-reported cardiac complications of ICIs are ICI-induced myocarditis but also pericardial diseases, cardiomyopathy, myocardial fibrosis, and acute heart failure (70). Microvascular damage leading to vascular sequelae has also been reported with ICI (10). Furthermore, studies are needed to explore all the different pathways involved in the cardiotoxicity of ICIs with possible yet unknown microcirculation damage. A recent in vivo study in mice showed that anti-PD1 drugs cause myocardial dysfunction and altered myocardial metabolism, suggesting damage at a subcellular level (71).
Imaging
Imaging modalities in cardio-oncology and their assessment of anticancer-drug-related cardiotoxicity are given in Figure 1.
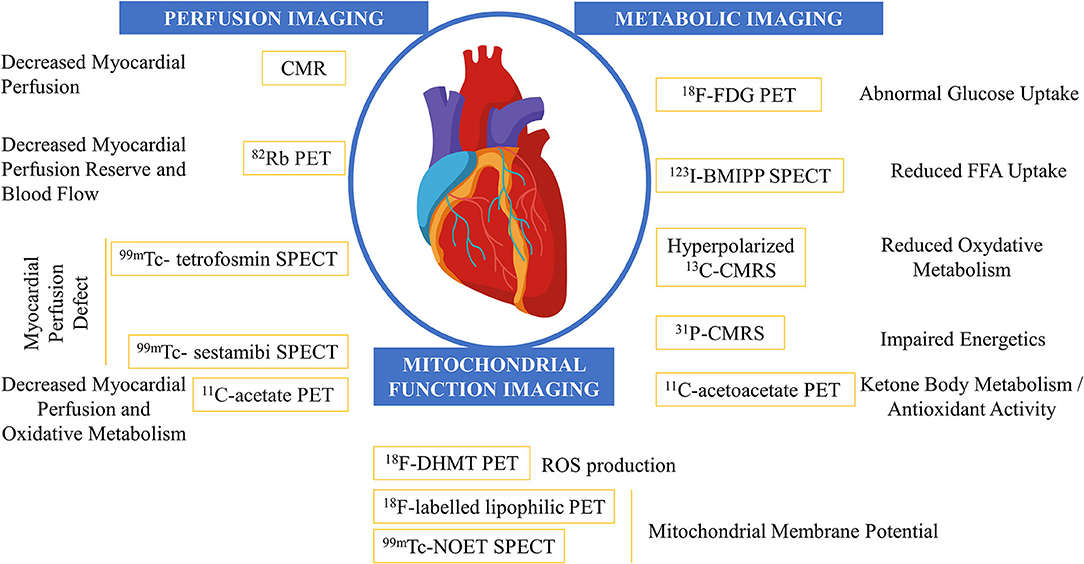
Figure 1. Imaging modalities in the field of cardio-oncology and their assessment of anticancer-drug related cardiotoxicity. CMR, cardiac MRI; CMRS, cardiac magnetic resonance sprectroscopy; FDG, fluoro-D-glucose; FFA, free fatty acid; SPECT, single-photon emission CT.
Perfusion Imaging
Perfusion imaging involves assessing the delivery of oxygen and nutrients to tissues through blood flow. It aims to describe microvasculature that can be altered under the effect of anticancer drugs. Since 1997, Hasdai et al. reported that coronary endothelial dysfunction may be associated with myocardial perfusion defects (72). Both radiotherapy and chemotherapy have shown to be associated with microvascular dysfunction (2), although the effect of non-radiation therapies on the latter is less well-described (31). Knowing the effects of anticancer drugs on myocardial microcirculation, myocardial perfusion imaging appears to be an attractive modality to detect anticancer drug-related myocardial toxicity. Moreover, by the time cardiotoxicity-associated LV dysfunction is detectable by echocardiography, it is often too late, emphasizing the need to assess the initial microvasculature dysfunction to better prevent it.
Symptomatic oxygen supply-demand mismatch can be evaluated invasively by invasive coronary angiography (ICA), but myocardial microcirculation disturbance can occur before any visible epicardial coronary on ICA (73), requiring blood flow measurements to assess myocardial function. Myocardial malperfusion can be unmasked through fractional flow reserve (FFR), which is an invasive measurement under hyperemia to determine the significance of an epicardial coronary artery stenosis, with an FFR ≤ 0.80 considered to be ischemia prone (74), and defined as the ratio of maximal blood flow distal to proximal to the stenosis. The invasive measurement of CFR is intended to study the vascular bed and describe the myocardial reserve capacity for vasodilatation, and is defined as the ratio of maximal hyperemic to the resting coronary blood flow (75).
Another interesting measure to evaluate coronary microvascular dysfunction is the index of microcirculatory resistance (IMR) (76) which is an index of coronary microvasculature and considered as abnormal if ≥25 independently of epicardial stenosis (77). However, these different parameters remain invasive, which could explain their low use in clinical practice for monitoring patients undergoing anticancer therapy, and should be discussed with respect to non-invasive techniques for the assessment of myocardial perfusion, which we review here.
Nuclear Imaging
Nuclear imaging techniques include single-photon emission computerized tomography (SPECT) and PET. These techniques are based on the detection of radioactive gamma rays and photons (after positrons annihilation) from an injected radioactive compound, respectively.
Single-Photon Emission CT (SPECT)
Impairment of epicardial arteries vasodilatation, by evaluation of change in coronary diameters under pharmacological stress, has been reported after DOX infusions on CT angiography suggesting dysfunction of smooth cells and the microvascular bed (78). However, the resolution of cardiac CT is insufficient to visually assess microvessels, underlining the need for cardiac perfusion CT to assess myocardial microcirculation by detecting hypoperfused territories. Coupling of metabolic information by traditional radiotracers 201Tl-chloride, 99mTc- sestamibi, and 99mTc- tetrofosmin, is obtained by myocardial perfusion SPECT. SPECT is performed at rest and under stress, which can be achieved by exercise or pharmacologically with vasodilators (79). The added value of SPECT is that the radiotracers will be delivered to the myocardium in proportion to flow and therefore be able to unmask a myocardial perfusion defect secondary to CAD. Territories with myocardial perfusion abnormalities may not only be secondary to CAD but reflect the myocardial cardiotoxicity at a microvascular level.
Studies have reported increased perfusion defects in DOX-treated patients with a history of radiotherapy (32, 80). Galluci et al., have suggested myocardial perfusion abnormalities, assessed by SPECT, without LVEF dysfunction in patients who had undergone chemotherapy and radiotherapy (32). However, this observational study could not strictly conclude that the findings were only due to the chemotherapy because of the lack of a control group before cancer treatment, and because of the inclusion of patients with a history of radiotherapy.
Some studies described LVEF dysfunction after the introduction of DOX in patients with cancer (81), but there are very little data on the incidence of SPECT perfusion defect in patients under DOX alone. One study on 36 patients with breast carcinoma evaluated before and after anthracyclines found no significant perfusion defect after anthracyclines (34), leaving the question of myocardial perfusion monitoring with 99mTc-sestamibi SPECT subject open to debate.
Positron Emission Tomography
Compared with SPECT, PET allows assessment of myocardial blood flow with better spatial resolution and sensitivity. CFR can be quantified as the ratio of myocardial blood flow between stress and rest on PET (82). The most commonly used and validated radionuclide for cardiac perfusion evaluation is rubidium-82 (82Rb) (82). Although 82Rb PET is often used for semiquantitative myocardial perfusion, it may assess coronary microvascular function by absolute quantification of myocardial perfusion and myocardial perfusion (or flow) reserve (MPR) (83). MPR is the ratio of stress flow to resting flow and describes the capacity of the coronary bed to maximize flow (84).
Myocardial perfusion reserve has been reported to be decreased after DOX exposure, representing a possible early marker of DOX myocardial cardiotoxicity (85). Detection of changes in mitochondrial function, estimation of myocardial blood flow and myocardial oxygen consumption, and thus, the ability of coronary arteries to respond to stress, can also be assessed by 11C-acetate rest stress PET. Using the latter, a decrease in myocardial perfusion and oxygen consumption reserve in DOX-treated rats compared with the control animals has been reported (86). 11C-acetate PET is not only used to investigate DOX cardiotoxicity but has also been evaluated in sunitinib-induced cardiotoxicity. Similarly, an in vivo study in rats described a decrease in myocardial perfusion, evaluated by 11C-acetate PET, as early as 5 days after treatment initiation (87).
Cardiac MR
Common practices remain the assessment of cardiotoxicity by echocardiography because of its ability and availability to detect LVEF alteration, which is the current standard for oncologic treatment cardiotoxicity (88). However, the gold standard in LVEF evaluation remains CMR imaging (89). But in addition to LVEF assessment, it is currently admitted that CMR with vasodilator stress perfusion should be performed to non-invasively investigate microvascular dysfunction (90). Yet, we know that anthracyclines may be responsible for myocardial damage at a histologic level long before any overt LVEF decrease (91). Although most studies of anthracyclines have focused on their effect on myocyte damages (92), more recent studies suggest that DOX cardiotoxicity may present as direct vascular injury and arterial damage with coronary arteriolar wall abnormalities (31, 33, 93, 94). Some mechanisms of microcirculation damage arise from increased thickening of microcirculatory arterioles and loss of smooth muscle cells, which may contribute to myocardial perfusion defects.
Thus, the literature reports that DOX cardiotoxicity results in microvascular dysfunction, and we know that microvascular can technically be assessed by myocardial perfusion on CMR. We had to wait until 2021 to finally find a study that proved in vivo that there was a reduction in myocardial perfusion well before any overt LVEF alteration. Indeed, to the best of our knowledge, Galán-Arriola et al. (31) were the first to describe in large animals the impact of DOX on coronary microcirculation, assessed by CMR but also by invasive measurement and histology, under different DOX protocols. In this study, the alteration of myocardial perfusion by CMR followed a similar pattern to that observed in the assessment of microcirculatory function by CFR. Indeed, they showed that in the early stages of DOX treatment, there was a decline in CMR perfusion. This decline in perfusion was present although LVEF, cardiac motion, cardiac contractility were not impaired; and was persistent as long-term changes with cumulative doses of DOX.
Myocardial perfusion assessment by CMR is a validated non-invasive assessment of microvascular CAD (95) and has been shown to outperform SPECT in detecting obstructive CAD (96–99). Newer CMR techniques that could quantitatively detect epicardial and microvascular CAD have correlated well with IMR and FFR measurements (77), and coronary sinus flow evaluation could be a good surrogate for CFR measurements (100). Although to the best of our knowledge, no study has yet reported myocardial perfusion CMR findings of anthracyclines-treated patients, it is legitimate to speculate that vasoconstriction and increased wall thickness of the heart microvasculature may reveal a myocardial perfusion defect and decreased myocardial blood flow reserve. Myocardial perfusion is acquired during the first pass of gadolinium-based contrast agents, based on an ECG-triggered fast T1-sensitive pulse sequences that can be acquired both at rest and with stress. The additional benefit of stress in CMR perfusion compared with resting perfusion alone is still debated but is theoretically used to unmask myocardial perfusion defect that could be compensated at rest (101). Indeed, stress could reveal insufficient coronary reserve resulting in decreased perfusion and ischemia in territories with thickened vessels walls and impaired ability to respond to stress-induced vasodilation. Although the mechanisms leading to 5-FU-related cardiotoxicity are numerous and detailed elsewhere (102), ischemia, especially secondary to vasospasm, can be imaged by perfusion defect in the coronary territory of the vasospasm (103, 104).
Regarding the evaluation of anti-VEGF myocardial cardiotoxicity with perfusion CMR, there are very sparse data in the literature. A small study on 9 patients evaluated both resting and stress perfusion with CMR before treatment and at 4 and 6 weeks of treatment (105). They were able to show a decrease in myocardial blood flow on resting perfusion after treatment introduction but no difference under stress, and an increase in vascular permeability. These preliminary findings suggest that anti-VEGF cardiotoxicity leads to microvascular constriction, which may, fortunately, be reversible, and that microvascular endothelial dysfunction may be responsible in part for impaired LVEF.
Metabolic Imaging
Metabolic imaging focuses and targets changes in metabolic pathways and energetics. It includes CMRS and nuclear imaging techniques such as SPECT and PET.
Cardiac Magnetic Resonance Spectroscopy
Cardiac magnetic resonance spectroscopy has several advantages for metabolic imaging since it is able of measuring several metabolic biomarkers without using ionizing radiation (106). Metabolites containing proton (1H) such as creatine or lipids; containing carbon (13C) such as glucose, and containing phosphorus (31P) such as PCr or ATP can be assessed by CMRS. In addition, the development of 31P saturation magnetic resonance spectroscopy allows the measurement of the metabolic rate of ATP production via the enzyme creatine kinase (= CK flux) (106, 107).
Early studies performed on isolated animal hearts have demonstrated several alterations in the cardiac metabolic. The injection of [1-13C]glucose into isolated perfused hearts treated for 10 weeks with anthracyclines highlighted altered glycolytic metabolism (108). Similarly, abnormal cardiac bioenergetics, as revealed by a reduced PCr/ATP ratio, was measured with 31P-CMRS in an isolated animal hearts of acute (109) and chronic (110–112) anthracycline-related cardiotoxicity. In addition, Bittner et al. showed that hearts chronically exposed to DOX failed to adapt metabolically, as evidenced by the delayed recovery of PCr after hemodynamic stress (113). Recently, Henderson et al. showed that acute and clinically relevant exposure to DOX in isolated, perfused rat hearts induced a reduction in energy reserve, as measured by a decrease in PCr, in response to the cardiac-stimulant isoproterenol (114). These studies demonstrated abnormal cardiac energetics production and utilization, even in the setting of acute anthracycline exposure. Interestingly, the myocardial PCr/ATP ratio was reduced after 6 weeks of anthracycline treatment without evidence of cardiac damage in an in vivo study (110). In addition, the authors showed a strong correlation between cardiac energetics and LV systolic and diastolic dysfunction after 8 and 10 weeks of treatment. The same group then demonstrated that the absolute concentration of PCr was decreased in DOX-treated mice and that 31P-CMRS also detected a reduced rate of ATP synthesis through CK reaction (115). Importantly, overexpression of cardiac-specific myofibrillar isoform of CK restored impaired PCr and CK flux, which was associated with improved LVEF and survival in DOX-treated mice (115), opening up a new possibility for preventive therapy.
Recent research has focused on improving the signal-to-noise ratio of conventional CMRS, with the development of hyperpolarization CMRS: the injection of hyperpolarized [1-13C]pyruvate and [2-13C]pyruvate enables measurement of the flux through the pyruvate dehydrogenase (PDH) complex and TCA flux, respectively (116). A decrease in PDH flux, representative of reduced oxidative mitochondrial carbohydrate metabolism, was observed in the myocardium of DOX-treated rats for 3 weeks without impairment of cardiac function (117). After 6 weeks of treatment, the authors showed, in addition to reduced PDH activity, a decrease of TCA cycle flux and impaired cardiac function. This altered carbohydrate metabolism reflected the loss of mitochondrial integrity, which was not because of the oxidative stress in this study, and preceded cardiac function impairment.
The exploration of cardiac energetics in the clinic has been recently proposed. The authors found no difference in cardiac PCr/ATP ratio of anthracycline-treated women despite a 5% reduction in LVEF between the start and end treatment (118). This could be explained, at least in part, by the small number of patients in whom CMRS was possible (11 patients).
Nuclear Imaging
Several radiopharmaceuticals can be used as biomarkers of myocardial metabolism using nuclear imaging, the two best known being iodine-123 betamethyl-iodophenyl-pentadecanoic acid (BMIPP) for the assessment of myocardial FFA uptake and 2'-deoxy-2'-[18F]fluoro-D-glucose (FDG) for the assessment of cardiac glucose uptake. Because myocardial metabolism is tightly regulated, the heart switches from FFA metabolism to glycolysis in high-insulin/glucose levels and low oxygen by increasing its glucose transporter protein translocation to the plasma membrane (119). Hence, PET with FDG under fasting condition is preferred for oncology study (minimize myocardial uptake) but is performed under fasted condition or with glucose load after an overnight fasting for cardiac study (maximize myocardial uptake).
Early studies conducted two decades ago showed a significantly lower myocardial BMIPP uptake in patients treated with DOX (120) and taxanes (121), but other studies showed that only one in four (122), and one in six (123) patients displayed hypomyocardial BMIPP accumulation. Importantly, modeling of kinetics, which was measured by the acquisition of dynamic time sequences in the latter study, revealed a significant decrease in BMIPP flux in DOX-treated patients (123). This analysis more accurately reflects the features of fatty acid metabolism disorders by measuring the metabolic flux of the tracer rather than its accumulation in the myocardium. The lower cardiac uptake of BMIPP, which is a biomarker of impaired fatty acid beta-oxidation, was predictive of LV dysfunction (120).
An exciting exploration in cardio-oncology is ketone body imaging. This has been proposed with cardiac 11C-acetoacetate PET. As a ketone body, acetoacetate can be used as a substrate by the heart and be involved in cardioprotection through its antioxidant activity plus mitochondrial membrane repair (124, 125). Greater uptake and retention of 11C-acetoacetate in the myocardium was found in non-fasted rats treated for 6 weeks with DOX, which may be associated with mitochondrial membrane alteration (126). Although it has been studied only once in this field, ketone body imaging may hold promise as a theranostic approach.
In 2012, Borde et al. first described enhanced 18F-FDG uptake in the myocardium of DOX-treated patients, highlighting the ability of PET to early detect cardiotoxicity (127). Similar observations have been reproduced by others attempting to better understand the increased myocardial 18F-FDG uptake in animals and patients treated with chemotherapy. First, DOX dose-dependently increased myocardial metabolic flux of 18F-FDG measured by dynamic PET in the fasted mice (128). The same group demonstrated that a low pretreatment 18F-FDG standardized uptake value (SUV) in Hodgkin's disease patients may predict the development of chemotherapy-induced cardiotoxicity, which was subsequently detected by a higher myocardial 18F-FDG SUV (128). Another study showed that 12% of 121 patients with breast cancer treated with anthracycline or trastuzumab had increased 18F-FDG uptake in the right ventricle, which was significantly associated with cardiotoxicity (129). Second, increased LV 18F-FDG uptake correlated with LVEF decline after two cycles and at the end of DOX therapy in a retrospective study including a cohort of 43 patients (130). Another interesting study explored 18F-FDG myocardial uptake and myocardial perfusion (through 99mTc-tetrofosmin SPECT) in a retrospective cohort of 332 patients followed for malignant disorders (131). As part of an oncologic PET protocol, patients were fasted to avoid myocardial 18F-FDG uptake: 36% of patients had no 18F-FDG uptake, 22.5% had diffuse 18F-FDG uptake, 8% had focal 18F-FDG uptake, and 30.5% had a focal uptake overlying the diffuse pattern 18F-FDG uptake. Among all the patients, multivariate logistic regression identified focal myocardial 18F-FDG uptake as a predictor of impaired LVEF and myocardial perfusion (131). It is important to bear in mind two interesting points. First, no direct mechanisms that could explain the increased cardiac 18F-FDG uptake have been explored in these reports. This could be because of the recruitment of inflammatory cells, switch to anaerobic glycolysis, or being associated with other pathological mechanisms. Second, the correlation between 18F-FDG uptake and LV function was made at the same time, which cannot directly prove the ability of early detection of cardiotoxicity before the decline of LV function. In terms of mechanisms and correlations, the increase in cardiac uptake of 18F-FDG seven days after DOX treatment in mice was directly correlated with oxidative stress and antioxidant mechanisms assessed by biochemical measurements (132). This is particularly interesting knowing the close relationship between metabolic imbalance (i.e., mismatch of oxidative metabolism plus reduced ATP production) and ROS generation in mitochondria (133, 134).
Chemotherapy-induced cardiotoxicity is not limited to an increase in 18F-FDG uptake. The SUV of 18F-FDG was significantly reduced in the fasted rats treated for 6 weeks (135) and in non-fasted rats treated for 4 weeks (136) with DOX. 18F-FDG PET could have detected a loss of cell viability and necrosis in these experimental models, which was associated with decreased LVEF (136). This supports the fact that dietary status is important in the cardiac 18F-FDG PET investigation.
With respect to antiangiogenic therapies, few reports have described the role of 18F-FDG PET. In 2011, a case report described decreased myocardial 18F-FDG uptake in patients treated with imatinib plus sorafenib who later developed a cardiac event (137). Later, O'Farrell et al. also showed an increase in 18F-FDG uptake 2–3 days after the introduction of sunitinib in mice and 5 days in rats (87). In another study, sunitnib induced higher 18F-FDG uptake after 1 week of treatment in fasted mice but not in non-fasted mice (138), highlighting once again a role of the dietary status on myocardial 18F-FDG uptake for further investigations. In both studies, this early side effect was associated with a switch from oxidative metabolism to glycolytic metabolism (138) and correlated with late myocardial hypertrophy measured after 6 weeks of treatment (139). Moreover, the metabolic flux of 18F-FDG from the blood to the cytoplasmatic glycolysis, measured by dynamic time sequence acquisition and kinetic modeling, was reduced after 3 weeks of treatment (87, 138) with sunitinib and was associated with an insulin resistance pattern (138).
Mitochondrial Function Imaging
In-vivo assessment of cardiotoxicity-induced ROS production is tempting as there is a close relationship between altered circulation, metabolism, and oxidative stress. 18F-labeled analog of dihydroethidium (18F-DHMT) is a radioactive compound that can assess free radicals because it is trapped in the cell when oxidized by ROS (140, 141). In an initial in-vivo study in mice, the authors reported a 2-fold increase in cardiac retention of 18F-DHMT after a single injection of DOX, which revealed ROS production compared with controls (141). This observation was later confirmed with an increased cardiac uptake of 18F-DHMT in DOX-treated rats following 4 and 6 weeks of treatment (142). Interestingly, no impairment of cardiac function was found after 4 weeks of treatment, but 6 weeks of DOX treatment induced a decrease in LVEF (142). In another study, dynamic time sequence 18F-DHMT PET and kinetic modeling confirmed higher absolute quantification of myocardial ROS production in beagle dogs following 2 weeks of DOX treatment (143).
Similarly, new radiopharmaceuticals have been developed to assess early DOX myocardial cardiotoxicity detection, such as 18F-labeled lipophilic cation PET tracers (144). Its principle is to image mitochondrial damage by 18F-labeled lipophilic tracers, which diffuse across mitochondrial membranes depending upon the mitochondrial membrane potential (144). The tracers will therefore accumulate in cardiac tissue in case of mitochondrial damage, which is one of the possible mechanisms of myocardial cardiotoxicity of DOX, allowing early detection of its cardiotoxicity.
In SPECT imaging, in the same perspective, the usual 99mTc- sestamibi, which is used to assess myocardial perfusion, is also a lipophilic cation and so its myocardial distribution depends on the mitochondrial membrane potential additionally to regional myocardial perfusion. Safee et al. recently demonstrated in a rat model a correction tool to free the 99mTc-sestamibi from its perfusion imaging, to assess only the mitochondrial potential, and thus, its possible perturbation by anthracyclines (145). They proposed to correct the 99mTc-sestamibi with a lipophilic uncharged radiotracer that would thus be a perfusion tracer independent of the mitochondrial membrane potential [the bis (N-ethoxy-N-ethyldithiocarbamato)nitrido 99mTc(V)]. The latter 99mTc-NOET would, therefore, be able to detect DOX cardiotoxicity through its mitochondrial damage.
Perspectives
We are convinced that the assessment of the mechanisms of anticancer drug cardiotoxicity by imaging is a cornerstone in the new era of cardio-oncology. Table 2 supports our assertion by summarizing studies that demonstrate DOX-induced cardiotoxicity early before overt LVEF impairment (Table 2).
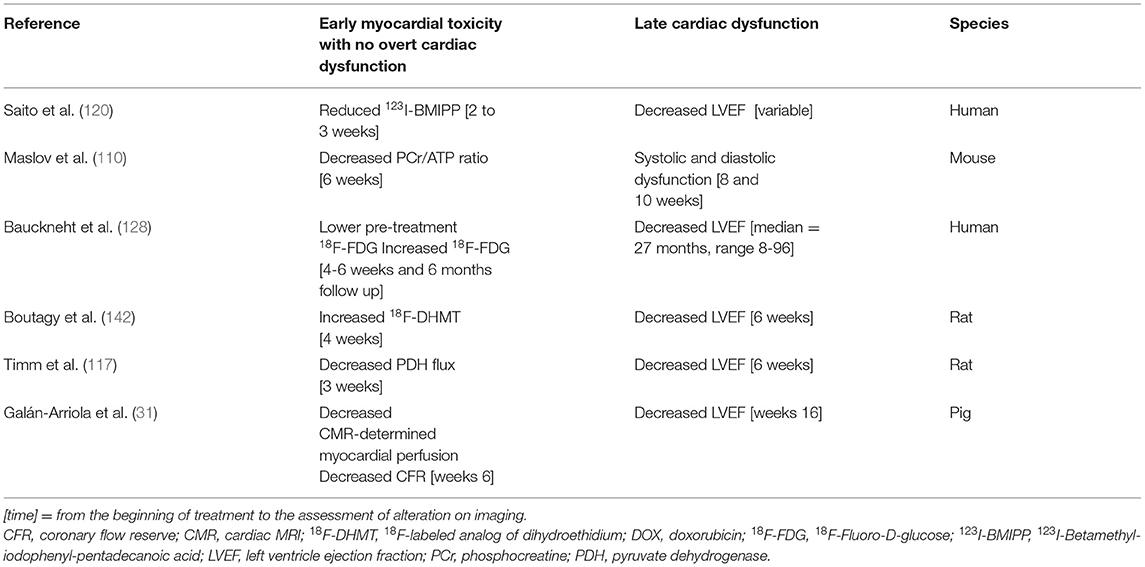
Table 2. This table summarizes early perfusion, metabolic and mitochondrial function imaging findings suggestive of DOX myocardial toxicity that subsequently revealed impaired left ventricle ejection fraction.
Imaging Opportunities
We have seen throughout this review that most studies have been conducted in animal models. We are confident that this research has been and will be of great importance for the development of a standardized protocol to predict drug-related cardiotoxicity and to test preventive interventions.
Early detection of metabolism and vascular alteration is paramount to prevent DOX-induced permanent cardiac dysfunction (Table 2) and could be extended to other anticancer drugs since several vascular and metabolic cardiotoxic effects have been described in this review (Table 1). The assessment of myocardial cardiotoxicity by CMR seems to be of interest, to seek other complications of oncologic therapies such as ICI-induced cardiotoxicity. The major cardiotoxicity reported in this therapeutic class is myocarditis, with CMR being of great importance when suspected (146). Although not a commonly used modality for myocardial inflammation (147), increased 18F-FDG uptake on PET could be found in myocarditis, including in ICI myocarditis (148). Interestingly, 18F-FDG uptake has also been reported as a marker of anthracyclines cardiotoxicity, either via inflammatory response or altered myocardial metabolism (149). Fusion between 18F-FDG and CMR have also been reported (148) for simultaneous vascular, metabolic, and functional imaging and may benefit from creatine measurement with proton CMRS (150) since creatine is decreased in both ischemic (151) and non-ischemic (152) cardiovascular disease.
Clinical Feasibilities
Because most studies of perfusion and metabolic imaging have been performed in animal models, their clinical relevance in routine practice is questionable. Anyhow, further clinical studies are required to ensure the utility of early detection of anticancer drugs.
Cardiac magnetic resonance imaging appears to be a non-invasive, radiation-free tool for monitoring patients with cancer, capable of imaging microcirculation, metabolism, and myocardial inflammation, which could be offered routinely before and after the introduction of an anticancer drug. We believe that CMR could be a justifiable perfusion approach as a part of standard patient care. Indeed, we have seen that altered myocardial perfusion in large animal models has been reported by resting myocardial perfusion on CMR (31). Multiple other CMR parameters have been reported to be related to cardiotoxicity of anticancer drugs (153–156), so the addition of a rapid perfusion sequence to the CMR protocol would be sufficient to obtain an argument for cardiotoxic effect. As the gold standard, CMR would also provide an accurate evaluation of LVEF. Unfortunately, LVEF assessment is so far performed in daily practice by echocardiography because of the lack of access to CMR. This would be the only limitation we see for its routine integration into the health care of patients with cancer.
We believe that the use of nuclear perfusion imaging in daily practice is difficult to justify. One of the main possible obstacles is the use of radiation and the cost of the technique that would allow assessment of myocardial perfusion without assessing oncologic follow-up. Nevertheless, it may be interesting to consider the integration of 18F-FDG PET in the follow-up of patients with cancer in order to assess tumor progression and, at the same time, to look for possible cardiotoxic effects. Indeed, the most PET scans for oncology monitoring use 18F-FDG, which is also, as mentioned earlier, sensitive to myocardial metabolic imbalance and also to myocardial inflammation. This capability of PET for whole-body imaging would be attractive in patients with cancer to concomitantly allow imaging of tumor progression in addition to an assessment of myocardial toxicity, thus providing a unique modality. We believe that further studies regarding the place of PET imaging in the future of cardio-oncology are required.
Conclusion
Early detection of cardiotoxicity is crucial and offers the opportunity for early therapeutic intervention. In this review, we have shown that perfusion imaging, metabolic imaging, and mitochondrial function imaging are capable of assessing myocardial cardiotoxic effects of cancer therapeutics before irreversible cardiac damage occurs (Figure 1, Table 2). Knowledge of these possible early imaging findings in anticancer drug-related myocardial toxicity could change the paradigm of “late-onset cardiotoxicity.” Earlier detection would allow for better prevention, with specific therapeutics attempting in part to reduce oxidative stress. Current guidelines on cardiotoxicity do not include myocardial and metabolic perfusion imaging, but in light of this review, it may be worthwhile to add these parameters to better detect and prevent dramatic progression.
Author Contributions
FC and JS contributed equally to this study and wrote the manuscript. FT did proofreading and provided useful advice. All authors contributed to the article and approved the submitted version.
Funding
This study was performed by a laboratory member of the France Life Imaging Network (grant ANR-11-INBS-0006).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Schwartz RG, McKenzie WB, Alexander J, Sager P, D'Souza A, Manatunga A, et al. Congestive heart failure and left ventricular dysfunction complicating doxorubicin therapy. Seven-year experience using serial radionuclide angiocardiography. Am J Med. (1987) 82:1109–18. doi: 10.1016/0002-9343(87)90212-9
2. Burrage MK, Ferreira VM. The use of cardiovascular magnetic resonance as an early non-invasive biomarker for cardiotoxicity in cardio-oncology. Cardiovasc Diagn Ther. (2020) 10:610–24. doi: 10.21037/cdt-20-165
3. Jordan JH, Todd RM, Vasu S, Hundley WG. Cardiovascular magnetic resonance in the oncology patient. JACC Cardiovasc Imaging. (2018) 11:1150–72. doi: 10.1016/j.jcmg.2018.06.004
4. Pizzino F, Vizzari G, Qamar R, Bomzer C, Carerj S, Zito C, et al. Multimodality imaging in cardiooncology. J Oncol. (2015) 2015:263950. doi: 10.1155/2015/263950
5. Taegtmeyer H, Golfman L, Sharma S, Razeghi P, van Arsdall M. Linking gene expression to function: metabolic flexibility in the normal and diseased heart. Ann N Y Acad Sci. (2004) 1015:202–13. doi: 10.1196/annals.1302.017
6. Saks V, Dzeja P, Schlattner U, Vendelin M, Terzic A, Wallimann T. Cardiac system bioenergetics: metabolic basis of the Frank-Starling law. J Physiol. (2006) 571:253–73. doi: 10.1113/jphysiol.2005.101444
7. Ray PD, Huang B-W, Tsuji Y. Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal. (2012) 24:981–90. doi: 10.1016/j.cellsig.2012.01.008
8. Varricchi G, Ameri P, Cadeddu C, Ghigo A, Madonna R, Marone G, et al. Antineoplastic drug-induced cardiotoxicity: a redox perspective. Front Physiol. (2018) 9:167. doi: 10.3389/fphys.2018.00167
9. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. (2009) 417:1–13. doi: 10.1042/BJ20081386
10. Soufer A, Baldassarre LA. The role of cardiac magnetic resonance imaging to detect cardiac toxicity from cancer therapeutics. Curr Treat Options Cardiovasc Med. (2019) 21:28. doi: 10.1007/s11936-019-0732-5
11. Minotti G, Menna P, Salvatorelli E, Cairo G, Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev. (2004) 56:185–229. doi: 10.1124/pr.56.2.6
12. Minotti G, Salvatorelli E, Menna P. Pharmacological foundations of cardio-oncology. J Pharmacol Exp Ther. (2010) 334:2–8. doi: 10.1124/jpet.110.165860
13. Sourdon J, Keceli G, Lindsey ML, Paolocci N. Death of an antioxidant brings heart failure with preserved ejection fraction to life: 5-oxoproline and post-ischaemic cardio-renal dysfunction. Cardiovasc Res. (2018) 114:1819–21. doi: 10.1093/cvr/cvy239
14. Tang X, Luo Y-X, Chen H-Z, Liu D-P. Mitochondria, endothelial cell function, and vascular diseases. Front Physiol. (2014) 5:175. doi: 10.3389/fphys.2014.00175
15. Chow AY, Chin C, Dahl G, Rosenthal DN. Anthracyclines cause endothelial injury in pediatric cancer patients: a pilot study. J Clin Oncol. (2006) 24:925–8. doi: 10.1200/JCO.2005.03.5956
16. Pautz A, Art J, Hahn S, Nowag S, Voss C, Kleinert H. Regulation of the expression of inducible nitric oxide synthase. Nitric Oxide. (2010) 23:75–93. doi: 10.1016/j.niox.2010.04.007
17. Heeringa P, van Goor H, Moshage H, Klok PA, Huitema MG, de Jager A, et al. Expression of iNOS, eNOS, and peroxynitrite-modified proteins in experimental anti-myeloperoxidase associated crescentic glomerulonephritis. Kidney Int. (1998) 53:382–93. doi: 10.1046/j.1523-1755.1998.00780.x
18. Csányi G, Taylor WR, Pagano PJ. NOX and inflammation in the vascular adventitia. Free Radic Biol Med. (2009) 47:1254–66. doi: 10.1016/j.freeradbiomed.2009.07.022
19. Ueno H, Kanellakis P, Agrotis A, Bobik A. Blood flow regulates the development of vascular hypertrophy, smooth muscle cell proliferation, and endothelial cell nitric oxide synthase in hypertension. Hypertension. (2000) 36:89–96. doi: 10.1161/01.HYP.36.1.89
20. Neubauer S. The failing heart–an engine out of fuel. N Engl J Med. (2007) 356:1140–51. doi: 10.1056/NEJMra063052
21. Hader SN, Zinkevich N, Norwood Toro LE, Kriegel AJ, Kong A, Freed JK, et al. Detrimental effects of chemotherapy on human coronary microvascular function. Am J Physiol Heart Circ Physiol. (2019) 317:H705–10. doi: 10.1152/ajpheart.00370.2019
22. Wolf MB, Baynes JW. The anti-cancer drug, doxorubicin, causes oxidant stress-induced endothelial dysfunction. Biochim Biophys Acta. (2006) 1760:267–71. doi: 10.1016/j.bbagen.2005.10.012
23. Soultati A, Mountzios G, Avgerinou C, Papaxoinis G, Pectasides D, Dimopoulos M-A, et al. Endothelial vascular toxicity from chemotherapeutic agents: preclinical evidence and clinical implications. Cancer Treat Rev. (2012) 38:473–83. doi: 10.1016/j.ctrv.2011.09.002
24. Wojcik T, Buczek E, Majzner K, Kolodziejczyk A, Miszczyk J, Kaczara P, et al. Comparative endothelial profiling of doxorubicin and daunorubicin in cultured endothelial cells. Toxicol In Vitro. (2015) 29:512–21. doi: 10.1016/j.tiv.2014.12.009
25. Mukhopadhyay P, Rajesh M, Bátkai S, Kashiwaya Y, Haskó G, Liaudet L, et al. Role of superoxide, nitric oxide, and peroxynitrite in doxorubicin-induced cell death in vivo and in vitro. Am J Physiol Heart Circ Physiol. (2009) 296:H1466–83. doi: 10.1152/ajpheart.00795.2008
26. Wang S, Kotamraju S, Konorev E, Kalivendi S, Joseph J, Kalyanaraman B. Activation of nuclear factor-kappaB during doxorubicin-induced apoptosis in endothelial cells and myocytes is pro-apoptotic: the role of hydrogen peroxide. Biochem J. (2002) 367:729–40. doi: 10.1042/bj20020752
27. Chaosuwannakit N, D'Agostino R, Hamilton CA, Lane KS, Ntim WO, Lawrence J, et al. Aortic stiffness increases upon receipt of anthracycline chemotherapy. J Clin Oncol. (2010) 28:166–72. doi: 10.1200/JCO.2009.23.8527
28. Pennell DJ, Sechtem UP, Higgins CB, Manning WJ, Pohost GM, Rademakers FE, et al. Clinical indications for cardiovascular magnetic resonance (CMR): consensus Panel report. J Cardiovasc Magn Reson. (2004) 6:727–65. doi: 10.1081/JCMR-200038581
29. Zhang S, Liu X, Bawa-Khalfe T, Lu L-S, Lyu YL, Liu LF, et al. Identification of the molecular basis of doxorubicin-induced cardiotoxicity. Nat Med. (2012) 18:1639–42. doi: 10.1038/nm.2919
30. Medeiros-Lima DJM, Carvalho JJ, Tibirica E, Borges JP, Matsuura C. Time course of cardiomyopathy induced by doxorubicin in rats. Pharmacol Rep. (2019) 71:583–90. doi: 10.1016/j.pharep.2019.02.013
31. Galán-Arriola C, Vílchez-Tschischke JP, Lobo M, López GJ, de Molina-Iracheta A, Pérez-Martínez C, et al. Coronary microcirculation damage in anthracycline cardiotoxicity. Cardiovasc Res. (2021) doi: 10.1093/cvr/cvab053. [Epub ahead of print].
32. Gallucci G, Coccaro M, Storto G, Lapadula L, Tartarone A, Nappi A, et al. The clinical impact of a cardiologic follow-up in breast cancer survivors: an observational study. Int J Immunopathol Pharmacol. (2010) 23:1221–7. doi: 10.1177/039463201002300426
33. Eckman DM, Stacey RB, Rowe R, D′Agostino R, Kock ND, Sane DC, et al. Weekly doxorubicin increases coronary arteriolar wall and adventitial thickness. PLoS ONE. (2013) 8:e57554. doi: 10.1371/journal.pone.0057554
34. Kalábová H, Melichar B, Ungermann L, DoleŽal J, Krčmová L, Kašparová M, et al. Intima-media thickness, myocardial perfusion and laboratory risk factors of atherosclerosis in patients with breast cancer treated with anthracycline-based chemotherapy. Med Oncol. (2011) 28:1281–7. doi: 10.1007/s12032-010-9593-1
35. Kinhult S, Albertsson M, Eskilsson J, Cwikiel M. Antithrombotic treatment in protection against thrombogenic effects of 5-fluorouracil on vascular endothelium: a scanning microscopy evaluation. Scanning. (2001) 23:1–8. doi: 10.1002/sca.4950230101
36. Tsibiribi P, Bui-Xuan C, Bui-Xuan B, Lombard-Bohas C, Duperret S, Belkhiria M, et al. Cardiac lesions induced by 5-fluorouracil in the rabbit. Hum Exp Toxicol. (2006) 25:305–9. doi: 10.1191/0960327106ht628oa
37. Kinhult S, Albertsson M, Eskilsson J, Cwikiel M. Effects of probucol on endothelial damage by 5-fluorouracil. Acta Oncol. (2003) 42:304–8. doi: 10.1080/02841860310004409
38. Matsubara I, Kamiya J, Imai S. Cardiotoxic effects of 5-fluorouracil in the guinea pig. Jpn J Pharmacol. (1980) 30:871–9. doi: 10.1016/S0021-5198(19)52945-6
39. Tamatsu H, Nakazawa M, Imai S, Watari H. 31P-topical nuclear magnetic resonance (31P-TMR) studies of cardiotoxic effects of 5-fluorouracil (5-FU) and 5'-deoxy-5-fluorouridine (5'-DFUR). Jpn J Pharmacol. (1984) 34:375–9. doi: 10.1016/S0021-5198(19)52291-0
40. Eskandari MR, Moghaddam F, Shahraki J, Pourahmad J. A comparison of cardiomyocyte cytotoxic mechanisms for 5-fluorouracil and its pro-drug capecitabine. Xenobiotica. (2015) 45:79–87. doi: 10.3109/00498254.2014.942809
41. Kurauchi K, Nishikawa T, Miyahara E, Okamoto Y, Kawano Y. Role of metabolites of cyclophosphamide in cardiotoxicity. BMC Res Notes. (2017) 10:406. doi: 10.1186/s13104-017-2726-2
42. Clarke L, Waxman DJ. Oxidative metabolism of cyclophosphamide: identification of the hepatic monooxygenase catalysts of drug activation. Cancer Res. (1989) 49:2344–50.
43. Colvin M. Alkylating Agents. Holland-Frei Cancer Medicine. 6th ed. (2003). Available online at: https://www.ncbi.nlm.nih.gov/books/NBK12772/ (accessed October 31, 2021).
44. Herradón E, González C, Uranga JA, Abalo R, Martín MI, López-Miranda V. Characterization of cardiovascular alterations induced by different chronic cisplatin treatments. Front Pharmacol. (2017) 8:196. doi: 10.3389/fphar.2017.00196
45. Hsu P-Y, Mammadova A, Benkirane-Jessel N, Désaubry L, Nebigil CG. Updates on anticancer therapy-mediated vascular toxicity and new horizons in therapeutic strategies. Front Cardiovasc Med. (2021) 8:694711. doi: 10.3389/fcvm.2021.694711
46. Hartmann JT, Kanz L. Sunitinib and periodic hair depigmentation due to temporary c-KIT inhibition. Arch Dermatol. (2008) 144:1525–6. doi: 10.1001/archderm.144.11.1525
47. Lévy BI. Blood pressure as a potential biomarker of the efficacy angiogenesis inhibitor. Ann Oncol. (2009) 20:200–3. doi: 10.1093/annonc/mdp018
48. Facemire CS, Nixon AB, Griffiths R, Hurwitz H, Coffman TM. Vascular endothelial growth factor receptor 2 controls blood pressure by regulating nitric oxide synthase expression. Hypertension. (2009) 54:652–8. doi: 10.1161/HYPERTENSIONAHA.109.129973
49. Kappers MHW, van Esch JHM, Sluiter W, Sleijfer S, Danser AHJ, van den Meiracker AH. Hypertension induced by the tyrosine kinase inhibitor sunitinib is associated with increased circulating endothelin-1 levels. Hypertension. (2010) 56:675–81. doi: 10.1161/HYPERTENSIONAHA.109.149690
50. Lankhorst S, Kappers MHW, van Esch JHM, Smedts FMM, Sleijfer S, Mathijssen RHJ, et al. Treatment of hypertension and renal injury induced by the angiogenesis inhibitor sunitinib: preclinical study. Hypertension. (2014) 64:1282–9. doi: 10.1161/HYPERTENSIONAHA.114.04187
51. Dhaun N, Webb DJ. Receptor tyrosine kinase inhibition, hypertension, and proteinuria: is endothelin the smoking gun? Hypertension. (2010) 56:575–7. doi: 10.1161/HYPERTENSIONAHA.110.155762
52. Lerman A, Holmes DR, Bell MR, Garratt KN, Nishimura RA, Burnett JC. Endothelin in coronary endothelial dysfunction and early atherosclerosis in humans. Circulation. (1995) 92:2426–31. doi: 10.1161/01.CIR.92.9.2426
53. Chintalgattu V, Rees ML, Culver JC, Goel A, Jiffar T, Zhang J, et al. Coronary microvascular pericytes are the cellular target of sunitinib malate induced cardiotoxicity. Sci Transl Med. (2013) 5:187ra69. doi: 10.1126/scitranslmed.3005066
54. Rees ML, Subramaniam J, Li Y, Hamilton DJ, Frazier OH, Taegtmeyer H. A PKM2 signature in the failing heart. Biochem Biophys Res Commun. (2015) 459:430–6. doi: 10.1016/j.bbrc.2015.02.122
55. Hasinoff BB, Patel D, O'Hara KA. Mechanisms of myocyte cytotoxicity induced by the multiple receptor tyrosine kinase inhibitor sunitinib. Mol Pharmacol. (2008) 74:1722–8. doi: 10.1124/mol.108.050104
56. Kerkela R, Woulfe KC, Durand J-B, Vagnozzi R, Kramer D, Chu TF, et al. Sunitinib-induced cardiotoxicity is mediated by off-target inhibition of AMP-activated protein kinase. Clin Transl Sci. (2009) 2:15–25. doi: 10.1111/j.1752-8062.2008.00090.x
57. Will Y, Dykens JA, Nadanaciva S, Hirakawa B, Jamieson J, Marroquin LD, et al. Effect of the multitargeted tyrosine kinase inhibitors imatinib, dasatinib, sunitinib, and sorafenib on mitochondrial function in isolated rat heart mitochondria and H9c2 cells. Toxicol Sci. (2008) 106:153–61. doi: 10.1093/toxsci/kfn157
58. Rainer PP, Doleschal B, Kirk JA, Sivakumaran V, Saad Z, Groschner K, et al. Sunitinib causes dose-dependent negative functional effects on myocardium and cardiomyocytes. BJU Int. (2012) 110:1455–62. doi: 10.1111/j.1464-410X.2012.11134.x
59. Ma W, Liu M, Liang F, Zhao L, Gao C, Jiang X, et al. Cardiotoxicity of sorafenib is mediated through elevation of ROS level and CaMKII activity and dysregulation of calcium homoeostasis. Basic Clin Pharmacol Toxicol. (2020) 126:166–80. doi: 10.1111/bcpt.13318
60. Sabet NS, Atashbar S, Khanlou EM, Kahrizi F, Salimi A. Curcumin attenuates bevacizumab-induced toxicity via suppressing oxidative stress and preventing mitochondrial dysfunction in heart mitochondria. Naunyn Schmiedebergs Arch Pharmacol. (2020) 393:1447–57. doi: 10.1007/s00210-020-01853-x
61. Mohammad Khanlou E, Atashbar S, Kahrizi F, Shokouhi Sabet N, Salimi A. Bevacizumab as a monoclonal antibody inhibits mitochondrial complex II in isolated rat heart mitochondria: ameliorative effect of ellagic acid. Drug Chem Toxicol. (2020) 1–8. doi: 10.1080/01480545.2020.1715423. [Epub ahead of print].
62. Li Y, Tian W, Yue D, Chen C, Li C, Zhang Z, et al. Bevacizumab-induced mitochondrial dysfunction, endoplasmic reticulum stress, and erk inactivation contribute to cardiotoxicity. Oxid Med Cell Longev. (2021) 2021:5548130. doi: 10.1155/2021/5548130
63. Bartsch R, Wenzel C, Steger GG. Trastuzumab in the management of early and advanced stage breast cancer. Biologics. (2007) 1:19–31.
64. ElZarrad MK, Mukhopadhyay P, Mohan N, Hao E, Dokmanovic M, Hirsch DS, et al. Trastuzumab alters the expression of genes essential for cardiac function and induces ultrastructural changes of cardiomyocytes in mice. PLoS ONE. (2013) 8:e79543. doi: 10.1371/journal.pone.0079543
65. Farolfi A, Melegari E, Aquilina M, Scarpi E, Ibrahim T, Maltoni R, et al. Trastuzumab-induced cardiotoxicity in early breast cancer patients: a retrospective study of possible risk and protective factors. Heart. (2013) 99:634–9. doi: 10.1136/heartjnl-2012-303151
66. Timolati F, Ott D, Pentassuglia L, Giraud M-N, Perriard J-C, Suter TM, et al. Neuregulin-1 beta attenuates doxorubicin-induced alterations of excitation-contraction coupling and reduces oxidative stress in adult rat cardiomyocytes. J Mol Cell Cardiol. (2006) 41:845–54. doi: 10.1016/j.yjmcc.2006.08.002
67. Giordano FJ, Gerber H-P, Williams S-P, VanBruggen N, Bunting S, Ruiz-Lozano P, et al. A cardiac myocyte vascular endothelial growth factor paracrine pathway is required to maintain cardiac function. Proc Natl Acad Sci USA. (2001) 98:5780–5. doi: 10.1073/pnas.091415198
68. Geisberg CA, Wang G, Safa RN, Smith HM, Anderson B, Peng X-Y, et al. Circulating neuregulin-1β levels vary according to the angiographic severity of coronary artery disease and ischemia. Coron Artery Dis. (2011) 22:577–82. doi: 10.1097/MCA.0b013e32834d3346
69. Salem J-E, Manouchehri A, Moey M, Lebrun-Vignes B, Bastarache L, Pariente A, et al. Cardiovascular toxicities associated with immune checkpoint inhibitors: an observational, retrospective, pharmacovigilance study. Lancet Oncol. (2018) 19:1579–89. doi: 10.1016/S1470-2045(18)30608-9
70. Heinzerling L, Ott PA, Hodi FS, Husain AN, Tajmir-Riahi A, Tawbi H, et al. Cardiotoxicity associated with CTLA4 and PD1 blocking immunotherapy. J Immunother Cancer. (2016) 4:50. doi: 10.1186/s40425-016-0152-y
71. Michel L, Helfrich I, Hendgen-Cotta UB, Mincu R-I, Korste S, Mrotzek SM, et al. Targeting early stages of cardiotoxicity from anti-PD1 immune checkpoint inhibitor therapy. Eur Heart J. (2021) ehab430. doi: 10.1093/eurheartj/ehab430. [Epub ahead of print].
72. Hasdai D, Gibbons RJ, Holmes DR, Higano ST, Lerman A. Coronary endothelial dysfunction in humans is associated with myocardial perfusion defects. Circulation. (1997) 96:3390–5. doi: 10.1161/01.CIR.96.10.3390
73. Behrenbeck TR, McCollough CH, Miller WL, Williamson EE, Leng S, Kline TL, et al. Early changes in myocardial microcirculation in asymptomatic hypercholesterolemic subjects: as detected by perfusion CT. Ann Biomed Eng. (2014) 42:515–25. doi: 10.1007/s10439-013-0934-z
74. Garcia D, Harbaoui B, van de Hoef TP, Meuwissen M, Nijjer SS, Echavarria-Pinto M, et al. Relationship between FFR, CFR and coronary microvascular resistance - Practical implications for FFR-guided percutaneous coronary intervention. PLoS ONE. (2019) 14:e0208612. doi: 10.1371/journal.pone.0208612
75. Gould KL, Kirkeeide RL, Buchi M. Coronary flow reserve as a physiologic measure of stenosis severity. J Am Coll Cardiol. (1990) 15:459–74. doi: 10.1016/S0735-1097(10)80078-6
76. Lee B-K, Lim H-S, Fearon WF, Yong AS, Yamada R, Tanaka S, et al. Invasive evaluation of patients with angina in the absence of obstructive coronary artery disease. Circulation. (2015) 131:1054–60. doi: 10.1161/CIRCULATIONAHA.114.012636
77. Kotecha T, Martinez-Naharro A, Boldrini M, Knight D, Hawkins P, Kalra S, et al. automated pixel-wise quantitative myocardial perfusion mapping by CMR to detect obstructive coronary artery disease and coronary microvascular dysfunction: validation against invasive coronary physiology. JACC Cardiovasc Imaging. (2019) 12:1958–69. doi: 10.1016/j.jcmg.2018.12.022
78. Feher A, Boutagy NE, Stendahl JC, Hawley C, Guerrera N, Booth CJ, et al. Computed tomographic angiography assessment of epicardial coronary vasoreactivity for early detection of doxorubicin-induced cardiotoxicity. JACC CardioOncol. (2020) 2:207–19. doi: 10.1016/j.jaccao.2020.05.007
79. Fathala A. Myocardial perfusion scintigraphy: techniques, interpretation, indications and reporting. Ann Saudi Med. (2011) 31:625–34. doi: 10.4103/0256-4947.87101
80. Hardenbergh PH, Munley MT, Bentel GC, Kedem R, Borges-Neto S, Hollis D, et al. Cardiac perfusion changes in patients treated for breast cancer with radiation therapy and doxorubicin: preliminary results. Int J Radiat Oncol Biol Phys. (2001) 49:1023–8. doi: 10.1016/S0360-3016(00)01531-5
81. Chernov VI, Kravchuk TL, Zelchan RV, Goldberg VE. [Radionuclide methods in the assessment of anthracycline-induced cardiotoxicity]. Kardiologiia. (2015) 55:57–62. doi: 10.18565/cardio.2015.7.57-62
82. Ghotbi AA, Kjaer A, Hasbak P. Review: comparison of PET rubidium-82 with conventional SPECT myocardial perfusion imaging. Clin Physiol Funct Imaging. (2014) 34:163–70. doi: 10.1111/cpf.12083
83. Hagemann CE, Ghotbi AA, Kjær A, Hasbak P. Quantitative myocardial blood flow with Rubidium-82 PET: a clinical perspective. Am J Nucl Med Mol Imaging. (2015) 5:457–68.
84. Ziadi MC, de Kemp R, Beanlands RSB, Small GR. Looking for trouble: reduced myocardial flow reserve following anthracyclines. J Nucl Cardiol. (2020) 27:1708–13. doi: 10.1007/s12350-018-01564-0
85. Laursen AH, Elming MB, Ripa RS, Hasbak P, Kjær A, Køber L, et al. Rubidium-82 positron emission tomography for detection of acute doxorubicin-induced cardiac effects in lymphoma patients. J Nucl Cardiol. (2020) 27:1698–07. doi: 10.1007/s12350-018-1458-6
86. Croteau E, Gascon S, Bentourkia M, Langlois R, Rousseau JA, Lecomte R, Bénard F. [11C]Acetate rest-stress protocol to assess myocardial perfusion and oxygen consumption reserve in a model of congestive heart failure in rats. Nucl Med Biol. (2012) 39:287–94. doi: 10.1016/j.nucmedbio.2011.07.010
87. O'Farrell AC, Evans R, Silvola JMU, Miller IS, Conroy E, Hector S, et al. A novel Positron Emission Tomography (PET) approach to monitor cardiac metabolic pathway remodeling in response to sunitinib malate. PLoS ONE. (2017) 12:e0169964. doi: 10.1371/journal.pone.0169964
88. Plana JC, Galderisi M, Barac A, Ewer MS, Ky B, Scherrer-Crosbie M, et al. Expert consensus for multimodality imaging evaluation of adult patients during and after cancer therapy: a report from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J Am Soc Echocardiogr. (2014) 27:911–39. doi: 10.1016/j.echo.2014.07.012
89. Bellenger NG, Burgess MI, Ray SG, Lahiri A, Coats AJ, Cleland JG, et al. Comparison of left ventricular ejection fraction and volumes in heart failure by echocardiography, radionuclide ventriculography and cardiovascular magnetic resonance; are they interchangeable? Eur Heart J. (2000) 21:1387–96. doi: 10.1053/euhj.2000.2011
90. Hubert A, Seitz A, Pereyra VM, Bekeredjian R, Sechtem U, Ong P. Coronary artery spasm: the interplay between endothelial dysfunction and vascular smooth muscle cell hyperreactivity. Eur Cardiol. (2020) 15:e12. doi: 10.15420/ecr.2019.20
91. Ewer MS, Ali MK, Mackay B, Wallace S, Valdivieso M, Legha SS, et al. A comparison of cardiac biopsy grades and ejection fraction estimations in patients receiving Adriamycin. J Clin Oncol. (1984) 2:112–7. doi: 10.1200/JCO.1984.2.2.112
92. Gianni L, Herman EH, Lipshultz SE, Minotti G, Sarvazyan N, Sawyer DB. Anthracycline cardiotoxicity: from bench to bedside. J Clin Oncol. (2008) 26:3777–84. doi: 10.1200/JCO.2007.14.9401
93. Bar-Joseph H, Ben-Aharon I, Tzabari M, Tsarfaty G, Stemmer SM, Shalgi R. In vivo bioimaging as a novel strategy to detect doxorubicin-induced damage to gonadal blood vessels. PLoS ONE. (2011) 6:e23492. doi: 10.1371/journal.pone.0023492
94. Murata T, Yamawaki H, Yoshimoto R, Hori M, Sato K, Ozaki H, et al. Chronic effect of doxorubicin on vascular endothelium assessed by organ culture study. Life Sci. (2001) 69:2685–95. doi: 10.1016/S0024-3205(01)01352-2
95. Hamirani YS, Kramer CM. Cardiac MRI assessment of myocardial perfusion. Future Cardiol. (2014) 10:349–58. doi: 10.2217/fca.14.18
96. Kwong RY, Ge Y, Steel K, Bingham S, Abdullah S, Fujikura K, et al. Cardiac magnetic resonance stress perfusion imaging for evaluation of patients with chest pain. J Am Coll Cardiol. (2019) 74:1741–55. doi: 10.1016/j.jacc.2019.07.074
97. Nagel E, Greenwood JP, McCann GP, Bettencourt N, Shah AM, Hussain ST, et al. Magnetic resonance perfusion or fractional flow reserve in coronary disease. N Engl J Med. (2019) 380:2418–28. doi: 10.1056/NEJMoa1716734
98. Schwitter J, Wacker CM, Wilke N, Al-Saadi N, Sauer E, Huettle K, et al. MR-IMPACT II: magnetic resonance imaging for myocardial perfusion assessment in coronary artery disease trial: perfusion-cardiac magnetic resonance vs. single-photon emission computed tomography for the detection of coronary artery disease: a comparative multicentre, multivendor trial. Eur Heart J. (2013) 34:775–81. doi: 10.1093/eurheartj/ehs022
99. Greenwood JP, Maredia N, Younger JF, Brown JM, Nixon J, Everett CC, et al. Cardiovascular magnetic resonance and single-photon emission computed tomography for diagnosis of coronary heart disease (CE-MARC): a prospective trial. Lancet. (2012) 379:453–60. doi: 10.1016/S0140-6736(11)61335-4
100. Indorkar R, Kwong RY, Romano S, White BE, Chia RC, Trybula M, et al. Global coronary flow reserve measured during stress cardiac magnetic resonance imaging is an independent predictor of adverse cardiovascular events. JACC Cardiovasc Imaging. (2019) 12:1686–95. doi: 10.1016/j.jcmg.2018.08.018
101. Kirkham AA, Virani SA, Campbell KL. The utility of cardiac stress testing for detection of cardiovascular disease in breast cancer survivors: a systematic review. Int J Womens Health. (2015) 7:127–40. doi: 10.2147/IJWH.S68745
102. Sara JD, Kaur J, Khodadadi R, Rehman M, Lobo R, Chakrabarti S, et al. 5-fluorouracil and cardiotoxicity: a review. Ther Adv Med Oncol. (2018) 10:1758835918780140. doi: 10.1177/1758835918780140
103. Pirozzolo G, Martínez Pereyra V, Hubert A, Guenther F, Sechtem U, Bekeredjian R, et al. Coronary artery spasm and impaired myocardial perfusion in patients with ANOCA: predictors from a multimodality study using stress CMR and acetylcholine testing. Int J Cardiol. (2021) 343:5–11. doi: 10.1016/j.ijcard.2021.09.003
104. Yilmaz A, Mahrholdt H, Athanasiadis A, Sechtem U. Non-invasive evaluation of coronary vasospasm using a combined hyperventilation and cold-pressure-test perfusion CMR protocol. J Cardiovasc Magn Reson. (2007) 9:759–64. doi: 10.1080/10976640701544662
105. Dobbin SJH, Mangion K, Berry C, Roditi G, Basak S, Sourbron S, et al. Cardiotoxicity and myocardial hypoperfusion associated with anti-vascular endothelial growth factor therapies: prospective cardiac magnetic resonance imaging in patients with cancer. Eur J Heart Fail. (2020) 22:1276–7. doi: 10.1002/ejhf.1847
106. Sourdon J, Lewsey SC, Schär M, Weiss RG. Measuring myocardial energetics with cardiovascular magnetic resonance spectroscopy. Heart Fail Clin. (2021) 17:149–56. doi: 10.1016/j.hfc.2020.08.011
107. Weiss RG, Gerstenblith G, Bottomley PA. ATP flux through creatine kinase in the normal, stressed, and failing human heart. Proc Natl Acad Sci USA. (2005) 102:808–13. doi: 10.1073/pnas.0408962102
108. Chatham JC, Hutchins GM, Glickson JD. Altered glucose metabolism in adriamycin-induced heart failure. Biochim Biophys Acta. (1992) 1138:1–5. doi: 10.1016/0925-4439(92)90143-B
109. Chatham JC, Cousins JP, Glickson JD. The relationship between cardiac function and metabolism in acute adriamycin-treated perfused rat hearts studied by 31P and 13C NMR spectroscopy. J Mol Cell Cardiol. (1990) 22:1187–97. doi: 10.1016/0022-2828(90)90082-D
110. Maslov MY, Chacko VP, Hirsch GA, Akki A, Leppo MK, Steenbergen C, et al. Reduced in vivo high-energy phosphates precede adriamycin-induced cardiac dysfunction. Am J Physiol Heart Circ Physio. (2010) 299:H332–7. doi: 10.1152/ajpheart.00727.2009
111. Dekker T, van Echteld CJ, Kirkels JH, Ruigrok TJ, van Hoesel QG, de Jong WH, et al. Chronic cardiotoxicity of adriamycin studied in a rat model by 31P NMR. NMR Biomed. (1991) 4:16–24. doi: 10.1002/nbm.1940040104
112. Nicolay K, Aue WP, Seelig J, van Echteld CJ, Ruigrok TJ, de Kruijff B. Effects of the anti-cancer drug adriamycin on the energy metabolism of rat heart as measured by in vivo 31P-NMR and implications for adriamycin-induced cardiotoxicity. Biochim Biophys Acta. (1987) 929:5–13. doi: 10.1016/0167-4889(87)90234-5
113. Bittner V, Reeves RC, Digerness SB, Caulfield JB, Pohost GM. 31P NMR spectroscopy in chronic adriamycin cardiotoxicity. Magn Reson Med. (1991) 17:69–81. doi: 10.1002/mrm.1910170112
114. Henderson KA, Borders RB, Ross JB, Abdulalil A, Gibbs S, Skowronek AJ, et al. Integration of cardiac energetics, function and histology from isolated rat hearts perfused with doxorubicin and doxorubicin-ol; a model for use in drug safety evaluations. J Pharmacol Toxicol Methods. (2018) 94:54–63. doi: 10.1016/j.vascn.2018.08.004
115. Gupta A, Rohlfsen C, Leppo MK, Chacko VP, Wang Y, Steenbergen C, et al. Creatine kinase-overexpression improves myocardial energetics, contractile dysfunction and survival in murine doxorubicin cardiotoxicity. PLoS ONE. (2013) 8:e74675. doi: 10.1371/journal.pone.0074675
116. Schroeder MA, Clarke K, Neubauer S, Tyler DJ. Hyperpolarized magnetic resonance: a novel technique for the in vivo assessment of cardiovascular disease. Circulation. (2011) 124:1580–94. doi: 10.1161/CIRCULATIONAHA.111.024919
117. Timm KN, Perera C, Ball V, Henry JA, Miller JJ, Kerr M, et al. Early detection of doxorubicin-induced cardiotoxicity in rats by its cardiac metabolic signature assessed with hyperpolarized MRI. Commun Biol. (2020) 3:692. doi: 10.1038/s42003-020-01440-z
118. Macnaught G, Oikonomidou O, Rodgers CT, Clarke W, Cooper A, McVicars H, et al. Cardiac energetics before, during, and after anthracycline-based chemotherapy in breast cancer patients using 31P magnetic resonance spectroscopy: a pilot study. Front Cardiovasc Med. (2021) 8:653648. doi: 10.3389/fcvm.2021.653648
119. Hue L, Beauloye C, Marsin A-S, Bertrand L, Horman S, Rider MH. Insulin and ischemia stimulate glycolysis by acting on the same targets through different and opposing signaling pathways. J Mol Cell Cardiol. (2002) 34:1091–7. doi: 10.1006/jmcc.2002.2063
120. Saito K, Takeda K, Okamoto S, Okamoto R, Makino K, Tameda Y, et al. Detection of doxorubicin cardiotoxicity by using iodine-123 BMIPP early dynamic SPECT: quantitative evaluation of early abnormality of fatty acid metabolism with the Rutland method. J Nucl Cardiol. (2000) 7:553–61. doi: 10.1067/mnc.2000.108351
121. Saito K, Takeda K, Imanaka-Yoshida K, Imai H, Sekine T, et al. Assessment of fatty acid metabolism in taxan-induced myocardial damage with iodine-123 BMIPP SPECT: comparative study with myocardial perfusion, left ventricular function, and histopathological findings. Ann Nucl Med. (2003) 17:481–8. doi: 10.1007/BF03006439
122. Nousiainen T, Vanninen E, Jantunen E, Remes J, Kuikka J, Hartikainen J. Anthracycline-induced cardiomyopathy: long-term effects on myocardial cell integrity, cardiac adrenergic innervation and fatty acid uptake. Clin Physiol. (2001) 21:123–8. doi: 10.1046/j.1365-2281.2001.00292.x
123. Kitagawa K, Takeda K, Saito K, Okamoto S, Makino K, Maeda H, et al. Differences in fatty acid metabolic disorder between ischemic myocardium and doxorubicin-induced myocardial damage: assessment using BMIPP dynamic SPECT with analysis by the Rutland method. J Nucl Med. (2002) 43:1286–94.
124. Maalouf M, Sullivan PG, Davis L, Kim DY, Rho JM. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience. (2007) 145:256–64. doi: 10.1016/j.neuroscience.2006.11.065
125. Fukao T, Lopaschuk GD, Mitchell GA. Pathways and control of ketone body metabolism: on the fringe of lipid biochemistry. Prostaglandins Leukot Essent Fatty Acids. (2004) 70:243–51. doi: 10.1016/j.plefa.2003.11.001
126. Croteau E, Tremblay S, Gascon S, Dumulon-Perreault V, Labbé SM, Rousseau JA, et al. [(11)C]-Acetoacetate PET imaging: a potential early marker for cardiac heart failure. Nuclear Med Biol. (2014) 41:863–70. doi: 10.1016/j.nucmedbio.2014.08.006
127. Borde C, Kand P, Basu S. Enhanced myocardial fluorodeoxyglucose uptake following Adriamycin-based therapy: evidence of early chemotherapeutic cardiotoxicity? World J Radiol. (2012) 4:220–3. doi: 10.4329/wjr.v4.i5.220
128. Bauckneht M, Ferrarazzo G, Fiz F, Morbelli S, Sarocchi M, Pastorino F, et al. Doxorubicin effect on myocardial metabolism as a prerequisite for subsequent development of cardiac toxicity: a translational 18F-FDG PET/CT observation. J Nucl Med. (2017) 58:1638–45. doi: 10.2967/jnumed.117.191122
129. Kim J, Cho S-G, Kang S-R, Yoo SW, Kwon SY, Min J-J, et al. Association between FDG uptake in the right ventricular myocardium and cancer therapy-induced cardiotoxicity. J Nucl Cardiol. (2020) 27:2154–63. doi: 10.1007/s12350-019-01617-y
130. Sarocchi M, Bauckneht M, Arboscello E, Capitanio S, Marini C, Morbelli S, et al. An increase in myocardial 18-fluorodeoxyglucose uptake is associated with left ventricular ejection fraction decline in Hodgkin lymphoma patients treated with anthracycline. J Transl Med. (2018) 16:295. doi: 10.1186/s12967-018-1670-9
131. Haider A, Bengs S, Schade K, Wijnen WJ, Portmann A, Etter D, Fröhlich S, et al. Myocardial 18F-FDG uptake pattern for cardiovascular risk stratification in patients undergoing oncologic PET/CT. J Clin Med. (2020) 9:E2279. doi: 10.3390/jcm9072279
132. Bauckneht M, Pastorino F, Castellani P, Cossu V, Orengo AM, Piccioli P, et al. Increased myocardial 18F-FDG uptake as a marker of Doxorubicin-induced oxidative stress. J Nucl Cardiol. (2020) 27:2183–94. doi: 10.1007/s12350-019-01618-x
133. Peoples JN, Saraf A, Ghazal N, Pham TT, Kwong JQ. Mitochondrial dysfunction and oxidative stress in heart disease. Exp Mol Med. (2019) 51:1–13. doi: 10.1038/s12276-019-0355-7
134. Zhou B, Tian R. Mitochondrial dysfunction in pathophysiology of heart failure. J Clin Invest. (2018) 128:3716–26. doi: 10.1172/JCI120849
135. Shen L-J, Lu S, Zhou Y-H, Li L, Xing Q-M, Xu Y-L. Developing a rat model of dilated cardiomyopathy with improved survival. J Zhejiang Univ Sci B. (2016) 17:975–83. doi: 10.1631/jzus.B1600257
136. Oudot A, Courteau A, Guillemin M, Vrigneaud J-M, Walker PM, Brunotte F, et al. [123I]MIBG is a better early marker of anthracycline cardiotoxicity than [18F]FDG: a preclinical SPECT/CT and simultaneous PET/MR study. EJNMMI Res. (2021) 11:92. doi: 10.1186/s13550-021-00835-1
137. Toubert M-E, Vercellino L, Faugeron I, Lussato D, Hindie E, Bousquet G. Fatal heart failure after a 26-month combination of tyrosine kinase inhibitors in a papillary thyroid cancer. Thyroid. (2011) 21:451–4. doi: 10.1089/thy.2010.0270
138. Sourdon J, Lager F, Viel T, Balvay D, Moorhouse R, Bennana E, et al. Cardiac metabolic deregulation induced by the tyrosine kinase receptor inhibitor sunitinib is rescued by endothelin receptor antagonism. Theranostics. (2017) 7:2757–74. doi: 10.7150/thno.19551
139. Sourdon J, Facchin C, Certain A, Viel T, Robin B, Lager F, et al. Sunitinib-induced cardiac hypertrophy and the endothelin axis. Theranostics. (2021) 11:3830–8. doi: 10.7150/thno.49837
140. Zhang W, Cai Z, Li L, Ropchan J, Lim K, Boutagy NE, et al. Optimized and automated radiosynthesis of [18F]DHMT for translational imaging of reactive oxygen species with positron emission tomography. Molecules. (2016) 21:E1696. doi: 10.3390/molecules21121696
141. Chu W, Chepetan A, Zhou D, Shoghi KI, Xu J, Dugan LL, et al. Development of a PET radiotracer for non-invasive imaging of the reactive oxygen species, superoxide, in vivo. Org Biomol Chem. (2014) 12:4421–31. doi: 10.1039/C3OB42379D
142. Boutagy NE, Wu J, Cai Z, Zhang W, Booth CJ, Kyriakides TC, et al. In vivo reactive oxygen species detection with a novel positron emission tomography tracer, 18F-DHMT, allows for early detection of anthracycline-induced cardiotoxicity in rodents. JACC Basic Transl Sci. (2018) 3:378–90. doi: 10.1016/j.jacbts.2018.02.003
143. Wu J, Boutagy NE, Cai Z, Lin S-F, Zheng M-Q, Feher A, et al. Feasibility study of PET dynamic imaging of [18F]DHMT for quantification of reactive oxygen species in the myocardium of large animals. J Nucl Cardiol. (2020). doi: 10.1007/s12350-020-02184-3. [Epub ahead of print].
144. McCluskey SP, Haslop A, Coello C, Gunn RN, Tate EW, Southworth R, et al. Imaging of chemotherapy-induced acute cardiotoxicity with 18F-labeled lipophilic cations. J Nucl Med. (2019) 60:1750–6. doi: 10.2967/jnumed.119.226787
145. Safee ZM, Baark F, Waters ECT, Veronese M, Pell VR, Clark JE, et al. Detection of anthracycline-induced cardiotoxicity using perfusion-corrected 99mTc sestamibi SPECT. Sci Rep. (2019) 9:216. doi: 10.1038/s41598-018-36721-5
146. Michel L, Rassaf T, Totzeck M. Cardiotoxicity from immune checkpoint inhibitors. Int J Cardiol Heart Vasc. (2019) 25:100420. doi: 10.1016/j.ijcha.2019.100420
147. Nensa F, Kloth J, Tezgah E, Poeppel TD, Heusch P, Goebel J, et al. Feasibility of FDG-PET in myocarditis: comparison to CMR using integrated PET/MRI. J Nucl Cardiol. (2018) 25:785–94. doi: 10.1007/s12350-016-0616-y
148. Chen Y, Jia Y, Liu Q, Shen Y, Zhu H, Dong X, et al. Myocarditis related to immune checkpoint inhibitors treatment: two case reports and literature review. Ann Palliat Med. (2021) 10:8512–7. doi: 10.21037/apm-20-2620
149. Bauckneht M, Morbelli S, Fiz F, Ferrarazzo G, Piva R, Nieri A, et al. A score-based approach to 18F-FDG PET images as a tool to describe metabolic predictors of myocardial doxorubicin susceptibility. Diagnostics. (2017) 7:E57. doi: 10.3390/diagnostics7040057
150. Sourdon J, Roussel T, Costes C, Viout P, Guye M, Ranjeva J-P, et al. Comparison of single-voxel 1H-cardiovascular magnetic resonance spectroscopy techniques for in vivo measurement of myocardial creatine and triglycerides at 3T. J Cardiovasc Magn Reson. (2021) 23:53. doi: 10.1186/s12968-021-00748-x
151. Bottomley PA, Weiss RG. Non-invasive magnetic-resonance detection of creatine depletion in non-viable infarcted myocardium. Lancet. (1998) 351:714–8. doi: 10.1016/S0140-6736(97)06402-7
152. Nakae I, Mitsunami K, Matsuo S, Inubushi T, Morikawa S, Tsutamoto T, et al. Myocardial creatine concentration in various nonischemic heart diseases assessed by 1H magnetic resonance spectroscopy. Circ J. (2005) 69:711–6. doi: 10.1253/circj.69.711
153. Drafts BC, Twomley KM, D'Agostino R, Lawrence J, Avis N, Ellis LR, et al. Low to moderate dose anthracycline-based chemotherapy is associated with early noninvasive imaging evidence of subclinical cardiovascular disease. JACC Cardiovasc Imaging. (2013) 6:877–85. doi: 10.1016/j.jcmg.2012.11.017
154. Neilan TG, Coelho-Filho OR, Shah RV, Feng JH, Pena-Herrera D, Mandry D, et al. Myocardial extracellular volume by cardiac magnetic resonance imaging in patients treated with anthracycline-based chemotherapy. Am J Cardiol. (2013) 111:717–22. doi: 10.1016/j.amjcard.2012.11.022
155. Fallah-Rad N, Lytwyn M, Fang T, Kirkpatrick I, Jassal DS. Delayed contrast enhancement cardiac magnetic resonance imaging in trastuzumab induced cardiomyopathy. J Cardiovasc Magn Reson. (2008) 10:5. doi: 10.1186/1532-429X-10-5
156. Jordan JH, Vasu S, Morgan TM, D'Agostino RB, Meléndez GC, Hamilton CA, et al. Anthracycline-associated T1 mapping characteristics are elevated independent of the presence of cardiovascular comorbidities in cancer survivors. Circ Cardiovasc Imaging. (2016) 9:e004325. doi: 10.1161/CIRCIMAGING.115.004325
Keywords: cardio-oncology, cardiotoxicity, perfusion, metabolism, mitochondria, magnetic resonance spectroscopy or MRS, magnetic resonance imaging, nuclear imaging
Citation: Cadour F, Thuny F and Sourdon J (2022) New Insights in Early Detection of Anticancer Drug-Related Cardiotoxicity Using Perfusion and Metabolic Imaging. Front. Cardiovasc. Med. 9:813883. doi: 10.3389/fcvm.2022.813883
Received: 12 November 2021; Accepted: 06 January 2022;
Published: 07 February 2022.
Edited by:
Feng Cao, People's Liberation Army General Hospital, ChinaReviewed by:
Michele Russo, University of Turin, ItalyRohit Moudgil, Cleveland Clinic, United States
Copyright © 2022 Cadour, Thuny and Sourdon. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joevin Sourdon, am9ldmluLnNvdXJkb25AdW5pdi1hbXUuZnI=