 De-Shu Chen
De-Shu Chen Jing Yan1,2,3*
Jing Yan1,2,3*- 1Department of Cardiology, Laboratory of Heart Center, Zhujiang Hospital, Southern Medical University, Guangzhou, China
- 2Heart Center of Zhujiang Hospital, Guangdong Provincial Biomedical Engineering Technology Research Center for Cardiovascular Disease, Guangzhou, China
- 3Heart Center of Zhujiang Hospital, Sino-Japanese Cooperation Platform for Translational Research in Heart Failure, Guangzhou, China
Left ventricular (LV) mass loss is prevalent in doxorubicin (DOX)-induced cardiotoxicity and is responsible for the progressive decline of cardiac function. Comparing with the well-studied role of cell death, the part of cardiomyocyte atrophy (CMA) playing in the LV mass loss is underestimated and the knowledge of the underlying mechanism is still limited. In this review, we summarized the recent advances in the DOX-induced CMA. We found that the CMA caused by DOX is associated with the upregulation of FOXOs and “atrogenes,” the activation of transient receptor potential canonical 3-NADPH oxidase 2 (TRPC3-Nox2) axis, and the suppression of IGF-1-PI3K signaling pathway. The imbalance of anabolic and catabolic process may be the common final pathway of these mechanisms. At last, we provided some strategies that have been demonstrated to alleviate the DOX-induced CMA in animal models.
Introduction
Doxorubicin (DOX), the most prescribed anthracycline chemotherapy agent, remains one of the most commonly used anti-cancer drugs among the world while its clinical application is limited by its cumulative dose-dependent cardiotoxicity (1, 2). The congestive heart failure (CHF) is the end stage of DOX-induced cardiotoxicity (DIC) and predicts poor prognosis. The incidence of DOX-related CHF reaches to 26% in patients received DOX at a cumulative dose of 550 mg/m2 (3). The health of patients with cancer and cancer survivors is threatened by the DIC, unfortunately, the number of both is large. For cancer survivors only, it was reported that there are more than 16.9 million cancer survivors until January 1, 2019 in the United States; this number is estimated to reach more than 22.1 million in the next decade based on the growth and aging of the population alone (4). Nowadays, several strategies are recommended for patients planning to receive high-dose anthracyclines to prevent DIC, such as the use of dexrazoxane or liposomal formulation of doxorubicin, continuous doxorubicin infusion (evidence based; strength of recommendation: moderate) (5). However, there is still lack of evidence to confirm whether these strategies are safe and effective for all patients with cancer receiving chemotherapy (5–7). Although small clinical trials have revealed that conventional drug of heart failure therapy may be beneficial against DIC (8, 9). Cardinale et al. reported that with conventional heart failure therapy, only 11% patients showed complete recovery from DIC in a heterogeneous cohort study of 2,625 patients (10). Therefore, it is vital to uncover the key mechanism of DIC and find a new approach to prevent it.
Several studies have revealed that anthracycline-based chemotherapy (ANbC) accounts for the left ventricular (LV) mass loss in patients with cancer and cancer survivors. In patients receiving ANbC, the LV mass decrease was detected as early as 1 month after the initiation of the therapy (11) and a 5% reduction in LV mass in 6 month was found based on the cardiovascular magnetic resonance detection (12). In the studies focusing on the pediatric and adult cancer survivors, the LV mass reduction still exists more than 20 years after the ANbC therapy (13–17). The severity of LV mass loss is correlative with the cumulative dose of DOX (14, 18). Further, there may not be a safe dose of DOX to avoid cardiotoxicity for that the cardiac abnormalities, such as significantly reduced LV mass and dimension was found in patients who received as low as 45 mg/m2 cumulative dose (18).
Although there are multi-factors that can be resulted into the LV mass loss, for example, cancer-associated cachexia like food intake reduction and excess catabolism (19), heart load alteration (20), denervation (21), and bed rest (22). Jordan et al. found that the reduction of LV mass is not necessarily accompanied with the decline of body weight and the heart failure (HF) symptom is not associated with the body weight decrease in patients who received ANbC, indicating a process other than cancer-associated cachexia leads to LV mass loss (12). Consistent finding was reported in animal models, DOX itself caused the heart weight loss in healthy mice and the heart weight (HW)/body weight (BW) index decreases in a dose-dependent fashion of DOX treatment, implicating the possibility that the HW loss is out of portion of BW loss and is caused by the chemotherapy (11). Intriguingly, Pietzsch et al. reported that the cardiac dysfunction induced by cancer alone would nearly recover to the base line, while tumor-bearing mice with DOX treatment showed lower survival rate in the acute phase and long-lasting damage in the gene expression system (23).
The LV mass loss is correlative to the decline of life quality (12) and the increase of major adverse cardiovascular events, such as cardiovascular death, implantable cardioverter-defibrillator therapy, and decompensated heart failure (14). Generally, the LV afterload decreases, the LV mass reduces (20). However, a high afterload was paradoxically found in ANbC-treated patient (12). The same phenomenon was found in animal models. Matsumura et al. found that DOX caused the cardiac atrophy and induced higher blood pressure after angiotensin II treatment (24). In addition, DOX-treated juvenile mice failed to develop cardiac hypertrophic response to late-onset hypertension induced by angiotensin II, which resulted into higher blood pressure, cardiac output decline, and overt mortality (24). Maayah et al. also reported that DOX treatment led to the impairment of the adaptive hypertrophic response to hypertrophic stimuli (25). Insufficient ventricular mass plus high chronic afterload contributes to the progressive contractile deficit, decreased cardiac output, and the establishment of cardiomyopathy (18). These may explain why hypertension markedly increased the risk for coronary artery disease, HF, valvular disease, and arrhythmia in aging adult survivors of childhood cancer (26).
The LV mass loss derives from both cell death (27, 28) and cardiomyocyte atrophy (CMA) (11), leads to cardiac atrophy. It should be noted that cardiac atrophy is different from CMA. The term of cardiac atrophy generally defined as an acquired reduction in the size and mass of the heart (29), is usually evaluated by HW, HW/BW ratio, or HW/tibia length (TL) ratio in animal DIC model. A great number of studies have revealed that DOX caused cardiac atrophy, as indicated by the decrease of HW, HW/TL ratio or HW/BW ratio (30–35). However, several studies reported that DOX caused a reduction of HW and BW, did not affect or increase the HW/BW ratio (36–38). It was reported that the delivery of DOX through intraperitoneal route resulted into peritoneal damage, which interfered the food intake and absorption and caused BW loss (39). Therefore, the preserved or increased HW/BW ratio may originate from the greater BW reduction. Therefore, it may be more appropriate to evaluate the cardiac atrophy by HW/TL ratio or HW alone, which is more evident mostly.
Despite numerous studies focusing on the cell death, less attention was paid on the CMA in DIC studies. However, the weight of cardiomyocyte apoptosis in DIC might be overstated (40). Several studies demonstrated that the contribution of cardiomyocyte apoptosis is low in acute DIC model. Willis et al. reported that CMA rather than cell death determines the cardiac atrophy in acute DIC mice model. They sacrificed mice 7 days after injected with DOX (20 mg/kg) and found that there were barely no increase of serum Troponin-I level and TUNEL-stained cell number in DOX treated mice, however, a 44% reduction of cell cross-section area and an obvious cardiac atrophy were detected (11). Little doxorubicin-induced apoptotic effect in acute DIC model was reported by other groups (41–44). However, it was also reported that DOX caused a great amount of apoptotic cardiomyocyte in an acute DIC model (45–47). Maybe apoptosis plays less important role in cardiac atrophy of acute DIC than we thought. While in a chronic DIC model, cardiomyocyte may undergo a hypertrophy response in a compensated manner (48), CMA was also found in a chronic DIC model (49–51). The controversial results will require further research to clarify, and the role of CMA in the DIC model should be evaluated. In a study including 27 women with breast cancer, patients received the cardiac magnetic resonance image exam at 351–700 days after anthracycline therapy (240 mg/m2). Ferreira et al. found that the LV mass index in these patients is correlated with intracellular water lifetime (τ ic; a cardiomyocyte size maker) other than with extracellular volume (ECV), indicating that the cardiac atrophy originates from CMA (52). Cell size shrinkage alone accounted for an ~44% reduction in LV mass, while the increased ECV may attenuate the LV mass loss (52). Except for apoptosis, other forms of cell death had been found and demonstrated to participate in DIC (27), the relative contribution of cell death and CMA in DOX-induced cardiac atrophy needs further studies to illustrate.
Here, we aim to emphasize the importance of CMA in cardiac atrophy, summarize the current knowledge of the effect of DOX on CMA, and provide insight into the underlying molecular mechanism of it, finally discuss some approaches that have been identified to protect it.
Molecular Mechanism
Forkhead Box O1 (FOXO1)
Forkhead box O (FOXO) proteins are transcription factors regulating multi physiological and pathological processes included in cardiovascular system. The family contains four members in human, FOXO1, FOXO3, FOXO4, and FOXO6 (53). FOXOs are key regulators in maintaining the muscle mass (54). Depletion of FOXOs has been reported to prevent the muscle loss and weakness through suppressing autophagy–lysosome systems (ALS) and ubiquitin–proteasome systems (UPS) via inhibiting the AKT activity (55). Sengupta et al. reported that FOXOs activation may reduce the cardiomyocyte size by promoting autophagy (56). Additionally, Skurk et al. (57) reported that AKT-FOXO3a signaling regulates cardiomyocyte cell size against hypertrophy via mediating the expression of atrophy-related genes “atrogenes”. Actually, FOXOs regulates half of the atrogenes by binding their promoters, such as muscle RING finger 1 (MuRF1), muscle atrophy gene-1 (atrogin-1/MAFbx), and Bcl-2 19-kDa interacting protein 3 (Bnip3) (55). Atrogin-1 and MuRF-1 are two members of E3 ubiquitin ligases mastering the ubiquitin-mediated protein degradation (58). Bnip3, an autophagy-related gene, has been reported to regulate CMA in a model of mechanical unloading (59).
It has been reported that high dose (20 mg/kg) of DOX treatment activated FOXO1 phosphorylation at Ser-249 and upregulated nuclear FOXO1 levels, accompanied with the increased expression of its target gene, MuRF1 within 24 h. Pharmacological inhibition of FOXO1 with AS1842856 decreased MuRF1 and prevented DOX-induced CMA and LV mass loss (60). Consistently, Willis et al. reported that DOX treatment resulted into a significant upregulation of MuRF1 and Bnip3, while MuRF1 depletion reversed DOX-induced cardiac atrophy in mice (11). Yamamoto et al. reported that DOX-induced CMA was abrogated by MG-132, a proteasome inhibitor, indicating that the atrophy response is involved in the UPS (61). Wang et al. reported that 3-MA, an autophagy inhibitor, alleviated the DOX-induced CMA in vitro and Ghrelin, a multi-functional peptide hormone, attenuated DOX-induced CMA by inhibiting the excessive autophagy (62) (Figure 1).
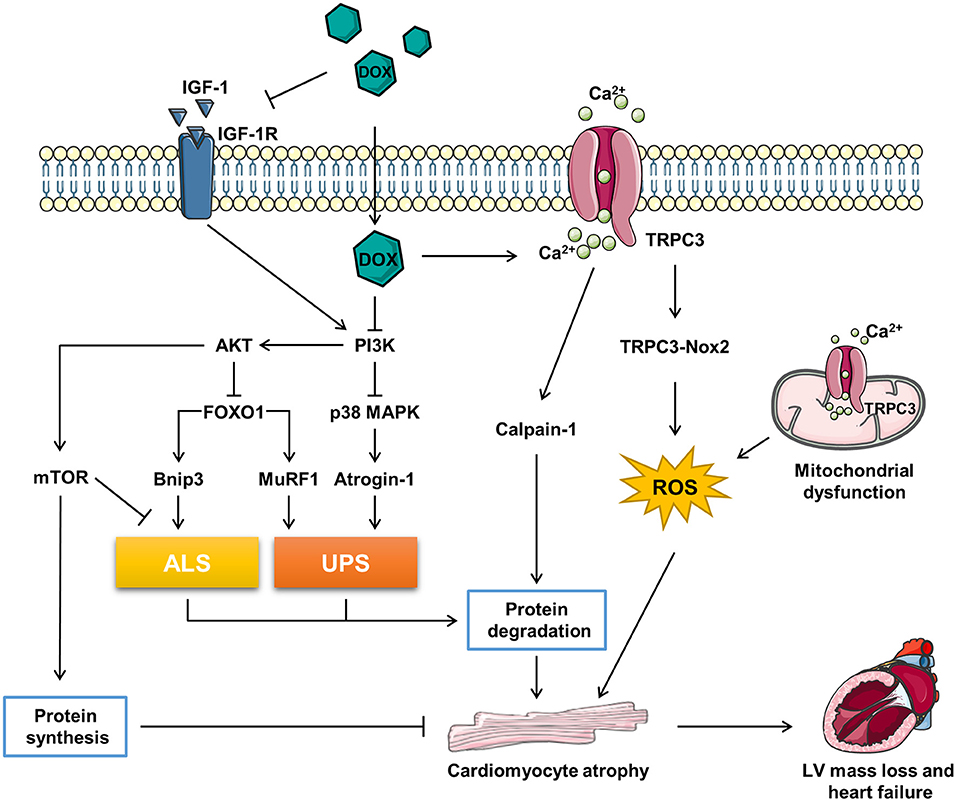
Figure 1. The related molecular mechanism of doxorubicin (DOX)-induced cardiomyocyte atrophy (CMA). ALS, autophagy–lysosome systems; atrogin-1/MAFbx, muscle atrophy gene-1; Bnip3, Bcl-2 19-kDa interacting protein 3; DOX, doxorubicin; FOXO1, forkhead box O1; IGF-1, insulin-like growth factor; IGF-1R, IGF-1 receptor; MuRF1, muscle RING finger 1; mTOR, mammalian target of rapamycin; Nox2, NADPH oxidase 2; PI3K, phosphoinositide 3-kinase; p38 MAPK, p38 mitogen-activated protein kinase; TRPC3, transient receptor potential canonical 3; UPS, ubiquitin–proteasome systems.
The expression of FOXO1 and its target genes might be induced by DOX in a time- and dose-dependent manner. Low dose (5 mg/kg) of DOX failed to induce MuRF1 expression at 24 h (60). In addition, the mRNA levels of FOXO1and Atrogin-1 were not upregulated in mice 7 days after injected with 20 mg/kg DOX (11).
In conclusion, DOX triggers catabolic process involving the induction of ALS and UPS via activating FOXO1 and its target genes, which contributes to the CMA. However, FOXOs are classified as tumor suppressor genes (63), inhibition of FOXOs may compromise the anti-tumor effect of DOX. Therefore, more precise and comprehensive studies need to be conducted to figure out if FOXOs inhibition is benefit in DIC therapy in patients with cancer (Figure 1).
Transient Receptor Potential Canonical 3 (TRPC3)-NADPH Oxidase 2 (Nox2) Axis
Transient receptor potential canonical (TRPC) proteins, regulating intracellular Ca2+, K+, and Na+, are involved in a variety of physiological and pathological processes in cardiovascular system (64). It has been reported that TRPC3 is a risk factor deteriorating the pathological cardiac remodeling (65, 66). TRPC3 was upregulated underlying the DOX-induced hypoxia stress, silence of TRPC3 ameliorated DOX-induced CMA (29). NADPH oxidase 2 (Nox2) is a key regulator accounting for the major reactive oxygen species (ROS) generation in response to cardiac injury. Nox2 knock-out mice exhibited ameliorated CMA and improved the cardiac function against accumulative DOX toxicity, which may be associated with the decrease of NADPH oxidase activity and oxidation (67).
Transient receptor potential canonical 3 (TRPC3) protects Nox2 from proteasome-dependent degradation via interacting with it at the specific C-terminal sites and promotes its activation by regulating Ca2+ entry (65). The functional interaction of TRPC3 and Nox2 is required for DOX-induced CMA, as the supplement of the TRPC3-C terminal fragment peptide, which disrupted the TRPC3-Nox2 complex without affecting the TRPC3 channel activity, attenuated DOX-induced CMA (29). Further, pharmacological inhibition of TRPC3-Nox2 complex by pyrazole-3 (Pyr3) abrogated DOX-induced CMA and ameliorated cardiotoxicity (29).
However, the downstream mechanism of TRPC3-Nox2 in DOX-induced CMA remains poorly known. It was reported that the mitochondrial dysfunction promoted muscle disuse atrophy by increasing oxidation stress, impairing Ca2+ handling, and activating associated cellular degradation processes (68, 69). TRPC3 was found to translocate to the mitochondria to mediate mitochondrial Ca2+ homeostasis and regulate the mitochondrial function (70). The number of evidence has revealed that the TRPC3-induced ROS emission and mitochondrial dysfunction participate in cardiac remodeling (65, 66, 71). Ca2+ overload is one of the major causes of DIC, Chen et al. reported that the upregulation of TRPC3 and TRPC6 contributed to the Ca2+ overload in DIC (72). Calmodulin is a ubiquitously expressed calcium binding protein which plays a key role in transducing intracellular calcium signal (73). Trifluoperazine, a strong calmodulin antagonist, was found to alleviate myofibril degeneration and cardiac atrophy induced by DOX (74). Calpains are Ca2+-activated neutral cysteine proteases and comprise two major molecules, calpain-1 and calpain-2 (75). Min et al. reported that DOX-induced skeleton and cardiac atrophy requiring the increased mitochondrial emission of ROS and calpain activation (76). Therefore, it can be speculated that DOX might induce CMA through TPRC3-Nox2 axis by disrupting the mitochondrial function, increasing Ca2+ entry, and activating the Ca2+-associated calpain protein degradation system (Figure 1).
Phosphoinositide 3-Kinase (PI3K)
Insulin-Like Growth Factor 1 (IGF-1) and PI3K
Insulin-like growth factor 1 (IGF-1), a key growth factor controlling both anabolic and catabolic pathways, plays a critical role in modulating the muscle size and function (76). IGF-1 binding to IGF-1 receptor (IGF-1R) leads to increased phosphorylation of insulin receptor substrate-1 (ISR-1), which recruits phosphoinositide 3-kinase (PI3K) and activates downstream the AKT signaling pathway (77). Besides, IGF binding protein (IGFBP) regulated IGF-1 activity by keeping it away from IGF-1R (78). DOX was reported to impair IGF-1R and upregulate IGFBP via p53 activation in H9C2 cells (79, 80). Restoration of IGF-1R-PI3K-AKT signaling pathway increased the cell survival ability against DIC (79, 80). Apart from that, exogenous IGF-1 (81) or insulin (82) were reported to alleviated DOX-induced cardiomyocyte apoptosis via stimulating PI3K-AKT. Interestingly, Mousa et al. discovered that the co-treatment of human umbilical cord blood mesenchymal stem cells (hUCB-MSCs) and carvedilol alleviated DOX-induced decrease of cardiac muscle fiber diameter, which is accompanied with the elevation of IGF-1, GATA-binding protein 4 (GATA-4), and vascular endothelial growth factor (VEGF) (83). Studies have uncovered that IGF-1 is a pro-hypertrophic inducer in cardiomyocyte (84, 85). Further, exogenous IGF-1 reversed cisplatin-induced skeleton muscle atrophy through inhibiting PI3K-AKT-FOXO mediated UPS (86). Overexpression of IGF-1 was also found to ameliorate cardiac atrophy in spinal muscular atrophy mice (87). However, whether IGF-1/IGF-1R have the potent to rescue CMA induced by DOX remains unknown (Figure 1).
PI3K and AKT
The PI3K-AKT signaling pathway plays a vital role in regulating the muscle hypertrophy and atrophy response (77). Studies have revealed that DOX inhibited PI3K-AKT activity both in vivo and in vitro (79, 88–90). PI3K, a lipid kinase family transducing receptor tyrosine kinase signaling, is aberrantly upregulated in human cancers frequently (91). Though targeting PI3K is effective in cancer therapy, inadvertently increases its side effect on the heart (92). PI3K is a key note in growth factor signaling as well as a modulator in heart muscle mass and contractility (93). Brent et al. found that specific inhibition of PI3Kα by BYL719 decreased the cross-sectional area of cardiomyocyte and induced cardiac atrophy (94). The use of PI3K inhibitor enhanced the anti-tumor effect of chemotherapy drugs, such as DOX (95, 96), while cotreatment of DOX and BYL719 aggravated CMA and cardiotoxicity compared with DOX alone (94). In addition, several studies shed the relationship of PI3K-AKT and cardiac atrophy as well. For example, Chen et al. found that total flavonoids stimulated PI3K-AKT and attenuated DOX-induced HW loss, while inhibition of PI3K-AKT abrogated the protection of total flavonoids against DIC (97). Meeran et al. revealed that nerolidol, a sesquiterpene from the essential oils of aromatic plants, alleviated DOX-induced cardiac atrophy possibly via the PI3K-AKT pathway (88). Intriguingly, both upregulation and downregulation of PI3K-AKT triggered by DOX has been reported (98, 99). The discrepancy may be explained by different DIC models and detected time. Interestingly, Cao et al. found that AKT activity was induced by DOX in the beginning, while this was suppressed in the long term (100). In addition, it was reported that PI3Kγ inhibition ameliorated DOX-induced CMA and cardiotoxicity as well as reduced tumor growth (101). Therefore, the role of PI3K-AKT in DIC requires deeper research to clarify and subunit specific inhibition of PI3K might be a promising idea.
Phosphoinositide 3-kinase-AKT activation promotes FOXOs to transport from nucleus to cytoplasm, where FOXOs are sequestered by 14-3-3 proteins and stay inactive (102). Several studies have revealed that the inhibition of PI3K-AKT signaling pathway promoted muscle atrophy via FOXOs-mediated activation of UPS (103–105). Moreover, Spurthi et al. reported that toll-like receptor 2 deficiency suppressed PI3K-AKT and activated FOXO1-atrogin-1/MuRF1, which resulted into cardiac atrophy in aging mice (106). Ni et al. found that angiotensin II induced cardiac hypertrophy via PI3K-AKT-FOXO pathway (107). Therefore, DOX-induced CMA may be associated with PI3K-AKT-FOXO pathway, which need further exploration. Worth to mention, Yamamoto et al. reported that DOX treatment induced a rapid increase of atrogin-1 mRNA expression via activation of p38 mitogen-activated protein kinase (MAPK) pathway without modulating the AKT-FOXO pathway (61).
Mammalian target of rapamycin (mTOR), acts as a serine/threonine kinase, plays an important role in regulating the protein synthesis and modulating autophagy by phosphorylating p70S6K and 4E-BP and Ulk-1, respectively (108, 109). The activity of mTOR regulates the cell growth and organ size (110). The AKT-mTOR axis has been reported to be involved in cardiac hypertrophy during volume overload (111). Further, the PI3K-AKT-mTOR signaling pathway has been found to participate in the DOX-induced skeleton muscle atrophy and cancer cachexia-related cardiac atrophy (112, 113). DOX was reported to impair AKT-mTOR axis by several research (82, 114–117). As reported, β2-agonist formoterol was reported to decrease protein degradation partially through inhibiting PI3K-AKT-mTOR mediated ALS, which prevented the muscle mass loss in fasted mice (118). Apart from that, the activation of PI3K-AKT signaling pathway prevented muscle atrophy via mTOR-mediated inhibition of ALS (119, 120). Wang et al. found that ghrelin ameliorated DOX-induced CMA by inhibiting excess autophagy via stimulating mTOR (62). Additionally, Hullin et al. revealed that enalapril protected against cardiotoxicity and CMA caused by DOX possibly through activating the PI3K-AKT-mTOR pathway (50). To sum up, DOX might cause CMA via inhibiting protein synthesis and activating ALS by suppressing the PI3K-AKT-mTOR pathway (Figure 1).
PI3K and p38 MAPK
The p38 MAPK family, which responses to the stress stimuli, plays an important role in cardiac development and function (121). The in vivo and in vitro evidence has shown that DOX activated the p38 MAPK pathway, which contributed to the DIC (89, 100, 122, 123). McLean et al. reported that suppression of PI3Kα with BYL719 or DOX activated p38 MAPK (94). The stimulation of p38 MAPK is correlative with the muscle wasting. Puigserver et al. found that p38 MAPK activation led to mitochondrial uncoupling and energy expenditure in muscle wasting (124). In addition, Fukawa et al. reported that cancer-secreted inflammatory factors resulted into the excessive fatty acid oxidation and the activation of p38 MAPK, which led to muscle atrophy (125). Several studies have revealed that the activation of p38 MAPK was responsible for DOX-induced CMA. Szeto-Schiller 31 (SS31), an antioxidant peptide, inhibited p38 MAPK phosphorylation and CMA induced by DOX (122). Diosgenin, a steroidal saponin of Dioscorea opposite, alleviated DOX-induced HW and HW/BW ratio reduction possibly via suppressing p38 MAPK (123). Further, therapeutic inhibition of p38 MAPK signaling mitigated DOX-induced CMA (94). However, the mechanism that downstream the p38 MAPK in DOX-induced CMA is beyond well established. It was reported that p38 MAPK activation resulted into the upregulation of atrogin-1 and the activation of catabolic process in cancer-induced muscle wasting (126). Pharmacological inhibition of p38 MAPK blunted DOX-induced atrogin-1 upregulation in cardiomyocytes and overexpression of atrogin-1 resulted into CMA (61). Besides, Odeh et al. reported that compromised p38 MAPK activity prevented the denervation-induced muscle atrophy through inhibiting UPS, decreasing oxidation stress, and increased clearance of damaged mitochondria (127). Ding et al. found that Activin A induced skeleton muscle atrophy via p38 mediated activation of UPS and autophagy, shown by the upregulation of atrogin-1 and LC3II (128). Therefore, DOX may induce CMA by activating catabolic process though PI3K-p38-atrogin-1 signaling pathway (Figure 1).
Therapy Strategies
Exercise
Appropriate exercise has been demonstrated to be beneficial for alleviating the muscle atrophy and improving the muscle strength (129). Wang et al. reported that moderate aerobic exercise decreased DOX exposure in cardiac tissue without altering the microvascular density (130). They found that moderate aerobic exercise during DOX treatment counteracted heart mass loss and cardiac function decline in juvenile tumor-bearing nude mice, while failed to preserve the cardiac function when exercise started after the closure of chemotherapy (130). Gomes-Santos et al. (131) found that aerobic exercise training prevented CMA, ameliorated cardiac atrophy, and attenuated exercise intolerance in mice developed with chronic DIC. While the LVEF reduction and fibrosis were not mitigated by it. Several studies have revealed the molecular mechanism underlying the effect of exercise in ameliorating DOX-induced CMA. Activation of TRPC3-Nox2 pathway contributes to the DOX-induced CMA, it was reported that voluntary exercise downregulated TRPC3 and Nox2 in a posttranslational manner (29). Further, it was reported that exercise upregulated IGF-1 mRNA expression (132) and activated PI3K-AKT impaired by DOX (133). Additionally, Kavazis et al. reported that the short-term endurance exercise training attenuated mRNA expression of some negative regulators of cardiac mass, such as FOXO1, MuRF1, myostatin but not atrogin-1, and Bnip3, which was probably associated with the activation of AMPK/PGC-1α pathway (134).
Non-Coding RNA (NcRNA)
Non-coding RNA (ncRNA), such as microRNA, small interference RNA (siRNA), long non-coding RNA (lncRNA), and circular RNA (cirRNA), plays an important role in regulating the cardiovascular system (135). Hu et al. reported that DOX treatment resulted into miR-200a downregulation both in vivo and in vitro, overexpression of miR-200a alleviated DOX-induced cardiac atrophy and cardiac dysfunction via nuclear factor (erythroid-derived 2)-like 2 (Nrf2) activation (136). Li et al. (137) also found that DOX caused elevation of miR-451 expression and miR-451 inhibition prevented the whole body wasting and cardiac atrophy and alleviated cardiotoxicity through AMPK signaling pathway in DIC mice. Moreover, Gupta et al. found that miR-212/132, a pro-hypertrophic cluster, ameliorated DOX-induced CMA and improved cardiac function by inhibiting downstream fat storage-inducing transmembrane protein 2 (Fitm2) (138). In addition, they found that Quaking, an RNA-binding protein, exerted cardiac protective effect against DOX-induced CMA and cardiotoxicity via mediating cardiac cirRNAs derived from Titin (Ttn), Formin homology 2 domain containing 3 (Fhod3), and Striatin calmodulin-binding protein 3 (Strn3) (139). It seems that interfering with ncRNAs may provide a new strategy in reversing the DOX-induced CMA, however, the related studies remain limited.
Hormones and Growth Factors
Growing evidence has demonstrated that part of endogenous hormones and growth factors have protective effect in cardiovascular diseases (140–143). Vascular endothelial growth factor-B (VEGF-B), one of the five known members of VEGF that regulate endothelial function (144), has been demonstrated to show potent in promoting coronary arteriogenesis and physiological cardiac hypertrophy (145). Räsänen et al. reported that overexpression of VEGF-B reversed CMA and cardiac mass loss through protecting endothelial in DOX-treated mice without compromising the anti-tumor effect of DOX (146). Li et al. (44) reported that exogenous supplementation of erythropoietin ameliorated DOX-induced CMA and cardiac dysfunction. The same team found that the atrophic response was attenuated by giving granulocyte colony-stimulating factor (G-CSF) in acute DIC mice in their following study (43). Interestingly, Esaki et al. reported that artificial upregulation of hepatocyte growth factor (HGF) at 2 weeks after the establishment of acute DIC model mitigated DOX-induced CMA and cardiac dysfunction (42). The related mechanism underlies the anti-atrophic effect of erythropoietin, G-CSF, and HGF might be similar, which was related to the activation of extracellular signal-regulated kinase (ERK) as well as the restoration of the expression of GATA-4 and its downstream 3 sarcomeric proteins, myosin heavy chain, troponin I, and desmin (42–44). GATA-4, a member of the GATA family of zinc finger transcription factors, is a major transcription factor regulating sarcomeric genes (147). DOX treatment caused a decrease in the level of GATA-4 DNA-binding activity as a result of downregulation of GATA-4 (148), which downregulated the sarcomeric proteins, and resulted into the degeneration of myofibrils in response to DOX.
Polyphenolic Compounds
The plant-derived polyphenolic compounds exert powerful antioxidant activity and have showed their beneficial effects in cardiovascular disease, such as DIC (149). The polyphenolic compounds can be classified as flavonoids, stilbenes, phenolic acids, and lignans based on the molecular structure (150). Rutin, a polyphenolic flavonoid, prevented DOX-induced cardiac atrophy and dysfunction via inhibiting excessive autophagy, reducing apoptosis, and restoring AKT activity (151). Isorhapontigenin, a new derivative of stilbene, alleviated CMA and cardiac atrophy caused by DOX, which is associated with the upregulation of yes-associated protein 1 expression (31). Resveratrol (3,5,4′-trihydroxy-trans-stilbene, RES), a natural polyphenol which can be found mainly in grapes, red wine, soy, and peanuts, has been well studied in DIC protection (152). Earlier, Zhang et al. found that RES prevented DOX-induced HW, BW, HW/BW ratio reduction, and cardiotoxicity via sirtuin 1(SIRT1)-p53 pathway (153). Furthermore, Arafa et al. revealed that RES was capable of alleviating cardiac atrophy caused by DOX (154). Several studies have implicated the possible molecular mechanism of the protection of RES on DOX-induced cardiac atrophy. It was reported that RES inhibited DOX-induced catabolic process as indicated by the downregulation of MuRF1 and ubiquitin-specific protease 7 (USP7) via increasing the deacetylase activity of SIRT1 in young mice (155). RES was reported to suppress DOX-induced p38 MAPK activation (24, 156) and restore VEGF-B and AKT impaired by DOX (157). Recently, Maayah et al. reported that RES ameliorated DOX-induced cardiac atrophy and cardiotoxicity through inhibiting nucleotide-binding domain-like receptor protein-3 (NLRP3) and systemic inflammation in juvenile mice (25). Interestingly, they found that RES restored DOX-induced deficiency of compensated hypertrophic response to the late-onset hypertension, as indicated by the alleviated CMA and increased heart wall thickness (25). Of note, some polyphenolic compounds have shown the effectiveness against cancer cells both in vivo and in vitro (158).
Clinical Drugs
The advantage of clinical drugs is the proved relative safety and the convenience for application. Here, we presented several studies about the protective effect of clinical drugs in DOX-induced CMA. Although the results of clinical study showed that only 11% patients showed complete recovery from DIC receiving conventional HF drugs (10), which may be associated with the underlying mechanism of DIC is cardiac atrophy rather than pathological hypertrophy. Losartan, a clinical used AT1 receptor antagonist, exerted cardioprotective effect against DOX-induced CMA possibly by inhibiting the Nox2 activity (67). Controversial studies about the effect of eplerenone on DIC were reported (50, 159). Enalapril, an angiotensin converting enzyme inhibitor (ACEI), attenuated DOX-induced CMA possibly via stimulating the PI3K-AKT-mTOR pathway and maintaining the normal levels of connective tissue growth factor (50). So, it reminds us that is it possible for some specific group population to benefit from the conventional HF drugs in DIC therapy? Oral supplementation of folic acid prevented myofibrils disruption, ameliorated DOX-induced CMA, and improved cardiac function (160). Of note, Durham et al. reported that upregulation of high-density lipoprotein (HDL) by overexpressing apolipoprotein A1 abrogated DOX-induce CMA in mice, which was required for the high-affinity HDL receptor, scavenger receptor class B type 1 (49). This study implicates that a lipid-lowering therapy may be beneficial for DOX-induced CMA.
The phosphodiesterase 5 (PDE5) inhibitors, such as tadalafil, sildenafil, and vardenafil, have been demonstrated to show protection in cardiovascular system (161). Koka et al. revealed that tadalafil, a long-acting selective inhibitor of cGMP-specific PDE5, improved cardiac function, reduced oxidation stress, attenuated apoptosis, and prevented cardiac atrophy in DIC mice (162). Prysyazhna et al. found that tadalafil protected against DOX-induced LV mass loss via attenuating protein kinase G Iαoxidation (163). Moreover, Jin et al. reported that tadalafil ameliorated the downregulation of 3 sarcomeric proteins, myosin heavy chain, troponin I, desmin, and alleviated CMA caused by DOX in mice (41). Another PDE5 inhibitor, sildenafil, has been verified to attenuate cardiac dysfunction, apoptosis, mitochondrial damage, and myofibrillar disarray induced by DOX (164). Multiple studies have reported that the administration of PDE5 inhibitors did not affect the anticancer effect but enhanced chemotherapeutic efficacy of DOX in animal tumor models (165–168). However, Poklepovic et al. found that sildenafil was safe, but did not show cardiac protection following DOX treatment in a small randomized clinical trial (169). The effect of sildenafil in DIC will require deeper research to verify. Worth to mention, several studies have shed light into the cardiac protective effect of other PDE inhibitors against DIC. Nishiyama et al. found that ibudilast, a PDE4 inhibitor already used in clinic, exerted cardioprotective effect against DOX-induced CMA by interfering the TRPC3-Nox2 complex without affecting the TRPC3 activity (170). Recently, Chen et al. reported that PDE10A deficiency ameliorated DOX-induced CMA and cardiotoxicity via cGMP and cAMP, and PDE10A inhibition antagonized tumor growth (171). Inspiringly, the safety of several PDE10A inhibitors have been demonstrated in phase I clinical trial (171). Zhang et al. revealed that PDE1C deficiency or suppression of ameliorated DOX-induced cardiac atrophy and improved cardiac function via adenosine A2 receptor stimulation (172). Cilostazol, a potent PDE3 inhibitor, also alleviated HW loss in DIC (173).
Discussion
In this review, we pointed out the importance of CMA in DIC and then, summarized recent advances in the molecular mechanism and the promising therapy strategies of DOX-induced CMA. Here, we paid more attention to the studies involving DOX-induced CMA, but not merely cardiac atrophy. Cardiac atrophy is a common finding and a major cause in the DIC. The weight of CMA in cardiac atrophy might be greater than we thought before. In addition, the reversibility of DIC also supports it (174). We are not going to say that we should downgrade the role of cell death yet. Although several studies have reported that little apoptotic effect was found in acute DIC models, the part of cardiomyocyte necrosis was not evaluated (11, 42–44). The apoptotic rate may be underestimated due to the secondary necrosis (175, 176). So, the relative contribution of CMA and cell death in DOX-induced cardiac atrophy is worth to elucidate in the future study. Inhibiting cellular degradation processes and promoting synthesis processes might be the key idea in preventing the DOX-induced CMA. The DOX-induced CMA is a degenerated process, which explains the protective effect of pro-growth therapy, such as exercise and supplementation of growth factors. Pathological hypertrophy is found in multi cardiovascular diseases; however, appropriate hypertrophy can be helpful for alleviating the DOX-induced CMA as proved by Gupta et al. (138). Considering that the cardiac regeneration technology is still far from application in clinic nowadays (177), reversing CMA serves an alternative and promising strategy in DIC therapy.
Author Contributions
D-SC collected the literature and wrote the manuscript. JY and P-ZY conceived the idea and supervised the manuscript. All authors agree to be accountable for the content of the work. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by the National Natural Science Foundation of China (Grant Nos. 82070247 and 82000249) and the Guangdong Basic and Applied Basic Research Foundation (Grant No. 2020A1515111028).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Chang HM, Moudgil R, Scarabelli T, Okwuosa TM, Yeh E. Cardiovascular complications of cancer therapy: best practices in diagnosis, prevention, and management: part 1. J Am Coll Cardiol. (2017) 70:2536–51. doi: 10.1016/j.jacc.2017.09.1096
2. Chang HM, Okwuosa TM, Scarabelli T, Moudgil R, Yeh E. Cardiovascular complications of cancer therapy: best practices in diagnosis, prevention, and management: part 2. J Am Coll Cardiol. (2017) 70:2552–65. doi: 10.1016/j.jacc.2017.09.1095
3. Swain SM, Whaley FS, Ewer MS. Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer-Am Cancer Soc. (2003) 97:2869–79. doi: 10.1002/cncr.11407
4. Miller KD, Nogueira L, Mariotto AB, Rowland JH, Yabroff KR, Alfano CM, et al. Cancer treatment and survivorship statistics, 2019. CA Cancer J Clin. (2019) 69:363–85. doi: 10.3322/caac.21565
5. Armenian SH, Lacchetti C, Barac A, Carver J, Constine LS, Denduluri N, et al. Prevention and monitoring of cardiac dysfunction in survivors of adult cancers: american society of clinical oncology clinical practice guideline. J Clin Oncol. (2017) 35:893–911. doi: 10.1200/JCO.2016.70.5400
6. Upshaw JN. Cardioprotective strategies to prevent cancer treatment-related cardiovascular toxicity: a review. Curr Oncol Rep. (2020) 22:72. doi: 10.1007/s11912-020-00923-w
7. Vejpongsa P, Yeh ET. Prevention of anthracycline-induced cardiotoxicity: challenges and opportunities. J Am Coll Cardiol. (2014) 64:938–45. doi: 10.1016/j.jacc.2014.06.1167
8. Lee M, Chung WB, Lee JE, Park CS, Park WC, Song BJ, et al. Candesartan and carvedilol for primary prevention of subclinical cardiotoxicity in breast cancer patients without a cardiovascular risk treated with doxorubicin. Cancer Med. (2021) 10:3964–73. doi: 10.1002/cam4.3956
9. Gupta V, Kumar SS, Agrawal V, Bali ST. Role of ace inhibitors in anthracycline-induced cardiotoxicity: a randomized, double-blind, placebo-controlled trial. Pediatr Blood Cancer. (2018) 65:e27308. doi: 10.1002/pbc.27308
10. Cardinale D, Colombo A, Bacchiani G, Tedeschi I, Meroni CA, Veglia F, et al. Early detection of anthracycline cardiotoxicity and improvement with heart failure therapy. Circulation. (2015) 131:1981–8. doi: 10.1161/CIRCULATIONAHA.114.013777
11. Willis MS, Parry TL, Brown DI, Mota RI, Huang W, Beak JY, et al. Doxorubicin exposure causes subacute cardiac atrophy dependent on the striated muscle-specific ubiquitin ligase murf1. Circ Heart Fail. (2019) 12:e5234. doi: 10.1161/CIRCHEARTFAILURE.118.005234
12. Jordan JH, Castellino SM, Meléndez GC, Klepin HD, Ellis LR, Lamar Z, et al. Left ventricular mass change after anthracycline chemotherapy. Circ Heart Fail. (2018) 11:e4560. doi: 10.1161/CIRCHEARTFAILURE.117.004560
13. Tham EB, Haykowsky MJ, Chow K, Spavor M, Kaneko S, Khoo NS, et al. Diffuse myocardial fibrosis by t1-mapping in children with subclinical anthracycline cardiotoxicity: relationship to exercise capacity, cumulative dose and remodeling. J Cardiovasc Magn Reson. (2013) 15:48. doi: 10.1186/1532-429X-15-48
14. Neilan TG, Coelho-Filho OR, Pena-Herrera D, Shah RV, Jerosch-Herold M, Francis SA, et al. Left ventricular mass in patients with a cardiomyopathy after treatment with anthracyclines. Am J Cardiol. (2012) 110:1679–86. doi: 10.1016/j.amjcard.2012.07.040
15. Armstrong GT, Plana JC, Zhang N, Srivastava D, Green DM, Ness KK, et al. Screening adult survivors of childhood cancer for cardiomyopathy: comparison of echocardiography and cardiac magnetic resonance imaging. J Clin Oncol. (2012) 30:2876–84. doi: 10.1200/JCO.2011.40.3584
16. Iarussi D, Galderisi M, Ratti G, Tedesco MA, Indolfi P, Casale F, et al. Left ventricular systolic and diastolic function after anthracycline chemotherapy in childhood. Clin Cardiol. (2001) 24:663–9. doi: 10.1002/clc.4960241006
17. De Wolf D, Suys B, Maurus R, Benoit Y, Verhaaren H, Matthijs D, et al. Dobutamine stress echocardiography in the evaluation of late anthracycline cardiotoxicity in childhood cancer survivors. Pediatr Res. (1996) 39:504–12. doi: 10.1203/00006450-199603000-00020
18. Lipshultz SE, Lipsitz SR, Sallan SE, Dalton VM, Mone SM, Gelber RD, et al. Chronic progressive cardiac dysfunction years after doxorubicin therapy for childhood acute lymphoblastic leukemia. J Clin Oncol. (2005) 23:2629–36. doi: 10.1200/JCO.2005.12.121
19. Baracos VE, Martin L, Korc M, Guttridge DC, Fearon K. Cancer-associated cachexia. Nat Rev Dis Primers. (2018) 4:17105. doi: 10.1038/nrdp.2017.105
20. Cooper GT, Kent RL, Mann DL. Load induction of cardiac hypertrophy. J Mol Cell Cardiol. (1989) 21:11–30. doi: 10.1016/0022-2828(89)90768-2
21. Takano H, Ozawa H, Kobayashi I, Hamaoka S, Nakajima A, Nakamura T, et al. Atrophic nerve fibers in regions of reduced mibg uptake in doxorubicin cardiomyopathy. J Nucl Med. (1995) 36:2060–1.
22. Dorfman TA, Levine BD, Tillery T, Peshock RM, Hastings JL, Schneider SM, et al. Cardiac atrophy in women following bed rest. J Appl Physiol. (1985) (2007) 103: 8-16. doi: 10.1152/japplphysiol.01162.2006
23. Pietzsch S, Wohlan K, Thackeray JT, Heimerl M, Schuchardt S, Scherr M, et al. Anthracycline-free tumor elimination in mice leads to functional and molecular cardiac recovery from cancer-induced alterations in contrast to long-lasting doxorubicin treatment effects. Basic Res Cardiol. (2021) 116:61. doi: 10.1007/s00395-021-00902-7
24. Matsumura N, Zordoky BN, Robertson IM, Hamza SM, Parajuli N, Soltys CM, et al. Co-administration of resveratrol with doxorubicin in young mice attenuates detrimental late-occurring cardiovascular changes. Cardiovasc Res. (2018) 114:1350–9. doi: 10.1093/cvr/cvy064
25. Maayah ZH, Alam AS, Takahara S, Soni S, Ferdaoussi M, Matsumura N, et al. Resveratrol reduces cardiac nlrp3-inflammasome activation and systemic inflammation to lessen doxorubicin-induced cardiotoxicity in juvenile mice. Febs Lett. (2021) 595:1681–95. doi: 10.1002/1873-3468.14091
26. Armstrong GT, Oeffinger KC, Chen Y, Kawashima T, Yasui Y, Leisenring W, et al. Modifiable risk factors and major cardiac events among adult survivors of childhood cancer. J Clin Oncol. (2013) 31:3673–80. doi: 10.1200/JCO.2013.49.3205
27. Christidi E, Brunham LR. Regulated cell death pathways in doxorubicin-induced cardiotoxicity. Cell Death Dis. (2021) 12:339. doi: 10.1038/s41419-021-03614-x
28. Zhang YW, Shi J, Li YJ, Wei L. Cardiomyocyte death in doxorubicin-induced cardiotoxicity. Arch Immunol Ther Exp (Warsz). (2009) 57:435–45. doi: 10.1007/s00005-009-0051-8
29. Shimauchi T, Numaga-Tomita T, Ito T, Nishimura A, Matsukane R, Oda S, et al. Trpc3-nox2 complex mediates doxorubicin-induced myocardial atrophy. JCI Insight. (2017) 2. doi: 10.1172/jci.insight.93358
30. Hou K, Shen J, Yan J, Zhai C, Zhang J, Pan JA, et al. Loss of trim21 alleviates cardiotoxicity by suppressing ferroptosis induced by the chemotherapeutic agent doxorubicin. Ebiomedicine. (2021) 69:103456. doi: 10.1016/j.ebiom.2021.103456
31. Wang P, Wang M, Hu Y, Chen J, Cao Y, Liu C, et al. Isorhapontigenin protects against doxorubicin-induced cardiotoxicity via increasing yap1 expression. Acta Pharm Sin B. (2021) 11:680–93. doi: 10.1016/j.apsb.2020.10.017
32. Tadokoro T, Ikeda M, Ide T, Deguchi H, Ikeda S, Okabe K, et al. Mitochondria-dependent ferroptosis plays a pivotal role in doxorubicin cardiotoxicity. JCI Insight. (2020) 5. doi: 10.1172/jci.insight.132747
33. Hu C, Zhang X, Wei W, Zhang N, Wu H, Ma Z, et al. Matrine attenuates oxidative stress and cardiomyocyte apoptosis in doxorubicin-induced cardiotoxicity via maintaining ampkα/ucp2 pathway. Acta Pharm Sin B. (2019) 9:690–701. doi: 10.1016/j.apsb.2019.03.003
34. Fang X, Wang H, Han D, Xie E, Yang X, Wei J, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. (2019) 116:2672–80. doi: 10.1073/pnas.1821022116
35. Lu J, Li J, Hu Y, Guo Z, Sun D, Wang P, et al. Chrysophanol protects against doxorubicin-induced cardiotoxicity by suppressing cellular parylation. Acta Pharm Sin B. (2019) 9:782–93. doi: 10.1016/j.apsb.2018.10.008
36. Singla DK, Johnson TA, Tavakoli DZ. Exosome treatment enhances anti-inflammatory m2 macrophages and reduces inflammation-induced pyroptosis in doxorubicin-induced cardiomyopathy. Cells-Basel. (2019) 8. doi: 10.3390/cells8101224
37. Sharifiaghdam Z, Dalouchi F, Sharifiaghdam M, Shaabani E, Ramezani F, Nikbakht F, et al. Curcumin-coated gold nanoparticles attenuate doxorubicin-induced cardiotoxicity via regulating apoptosis in a mouse model. Clin Exp Pharmacol Physiol. (2022) 49:70–83. doi: 10.1111/1440-1681.13579
38. Abu-Khudir R, Ibrahim WM, Shams ME, Salama AF. Trehalose alleviates doxorubicin-induced cardiotoxicity in female swiss albino mice by suppression of oxidative stress and autophagy. J Biochem Mol Toxicol. (2021) 35:e22859. doi: 10.1002/jbt.22859
39. Li DL, Wang ZV, Ding G, Tan W, Luo X, Criollo A, et al. Doxorubicin blocks cardiomyocyte autophagic flux by inhibiting lysosome acidification. Circulation. (2016) 133:1668–87. doi: 10.1161/CIRCULATIONAHA.115.017443
40. Kankeu C, Clarke K, Passante E, Huber HJ. Doxorubicin-induced chronic dilated cardiomyopathy-the apoptosis hypothesis revisited. J Mol Med (Berl). (2017) 95:239–48. doi: 10.1007/s00109-016-1494-0
41. Jin Z, Zhang J, Zhi H, Hong B, Zhang S, Guo H, et al. Beneficial effects of tadalafil on left ventricular dysfunction in doxorubicin-induced cardiomyopathy. J Cardiol. (2013) 62:110–6. doi: 10.1016/j.jjcc.2013.03.018
42. Esaki M, Takemura G, Kosai K, Takahashi T, Miyata S, Li L, et al. Treatment with an adenoviral vector encoding hepatocyte growth factor mitigates established cardiac dysfunction in doxorubicin-induced cardiomyopathy. Am J Physiol Heart Circ Physiol. (2008) 294:H1048–57. doi: 10.1152/ajpheart.01102.2007
43. Li L, Takemura G, Li Y, Miyata S, Esaki M, Okada H, et al. Granulocyte colony-stimulating factor improves left ventricular function of doxorubicin-induced cardiomyopathy. Lab Invest. (2007) 87:440–55. doi: 10.1038/labinvest.3700530
44. Li L, Takemura G, Li Y, Miyata S, Esaki M, Okada H, et al. Preventive effect of erythropoietin on cardiac dysfunction in doxorubicin-induced cardiomyopathy. Circulation. (2006) 113:535–43. doi: 10.1161/CIRCULATIONAHA.105.568402
45. Arunachalam S, Nagoor MM, Azimullah S, Sharma C, Goyal SN, Ojha S. Nerolidol attenuates oxidative stress, inflammation, and apoptosis by modulating nrf2/mapk signaling pathways in doxorubicin-induced acute cardiotoxicity in rats. Antioxidants (Basel). (2021) 10. doi: 10.3390/antiox10060984
46. Zhang WB, Zheng YF, Wu YG. Inhibition of mir-128-3p attenuated doxorubicin-triggered acute cardiac injury in mice by the regulation of ppar-γ. Ppar Res. (2021) 2021:7595374. doi: 10.1155/2021/7595374
47. Ma ZG, Kong CY, Wu HM, Song P, Zhang X, Yuan YP, et al. Toll-like receptor 5 deficiency diminishes doxorubicin-induced acute cardiotoxicity in mice. Theranostics. (2020) 10:11013–25. doi: 10.7150/thno.47516
48. Jafarinezhad Z, Rafati A, Ketabchi F, Noorafshan A, Karbalay-Doust S. Cardioprotective effects of curcumin and carvacrol in doxorubicin-treated rats: stereological study. Food Sci Nutr. (2019) 7:3581–8. doi: 10.1002/fsn3.1210
49. Durham KK, Chathely KM, Mak KC, Momen A, Thomas CT, Zhao YY, et al. Hdl protects against doxorubicin-induced cardiotoxicity in a scavenger receptor class b type 1-, pi3k-, and akt-dependent manner. Am J Physiol Heart Circ Physiol. (2018) 314:H31–44. doi: 10.1152/ajpheart.00521.2016
50. Hullin R, Métrich M, Sarre A, Basquin D, Maillard M, Regamey J, et al. Diverging effects of enalapril or eplerenone in primary prevention against doxorubicin-induced cardiotoxicity. Cardiovasc Res. (2018) 114:272–81. doi: 10.1093/cvr/cvx162
51. Zhu W, Shou W, Payne RM, Caldwell R, Field LJ. A mouse model for juvenile doxorubicin-induced cardiac dysfunction. Pediatr Res. (2008) 64:488–94. doi: 10.1203/PDR.0b013e318184d732
52. Ferreira DST, Quinaglia ACST, Osorio CF, Shah R, Neilan TG, Velloso L, et al. Anthracycline therapy is associated with cardiomyocyte atrophy and preclinical manifestations of heart disease. JACC Cardiovasc Imaging. (2018) 11:1045–55. doi: 10.1016/j.jcmg.2018.05.012
53. Calissi G, Lam EW, Link W. Therapeutic strategies targeting foxo transcription factors. Nat Rev Drug Discov. (2021) 20:21–38. doi: 10.1038/s41573-020-0088-2
54. Sanchez AM, Candau RB, Bernardi H. Foxo transcription factors: their roles in the maintenance of skeletal muscle homeostasis. Cell Mol Life Sci. (2014) 71:1657–71. doi: 10.1007/s00018-013-1513-z
55. Milan G, Romanello V, Pescatore F, Armani A, Paik JH, Frasson L, et al. Regulation of autophagy and the ubiquitin-proteasome system by the foxo transcriptional network during muscle atrophy. Nat Commun. (2015) 6:6670. doi: 10.1038/ncomms7670
56. Sengupta A, Molkentin JD, Yutzey KE. Foxo transcription factors promote autophagy in cardiomyocytes. J Biol Chem. (2009) 284:28319–31. doi: 10.1074/jbc.M109.024406
57. Skurk C, Izumiya Y, Maatz H, Razeghi P, Shiojima I, Sandri M, et al. The foxo3a transcription factor regulates cardiac myocyte size downstream of akt signaling. J Biol Chem. (2005) 280:20814–23. doi: 10.1074/jbc.M500528200
58. Gumucio JP, Mendias CL. Atrogin-1, murf-1, and sarcopenia. Endocrine. (2013) 43:12–21. doi: 10.1007/s12020-012-9751-7
59. Cao DJ, Jiang N, Blagg A, Johnstone JL, Gondalia R, Oh M, et al. Mechanical unloading activates foxo3 to trigger bnip3-dependent cardiomyocyte atrophy. J Am Heart Assoc. (2013) 2:e16. doi: 10.1161/JAHA.113.000016
60. Xia P, Chen J, Liu Y, Fletcher M, Jensen BC, Cheng Z. Doxorubicin induces cardiomyocyte apoptosis and atrophy through cyclin-dependent kinase 2-mediated activation of forkhead box o1. J Biol Chem. (2020) 295:4265–76. doi: 10.1074/jbc.RA119.011571
61. Yamamoto Y, Hoshino Y, Ito T, Nariai T, Mohri T, Obana M, et al. Atrogin-1 ubiquitin ligase is upregulated by doxorubicin via p38-map kinase in cardiac myocytes. Cardiovasc Res. (2008) 79:89–96. doi: 10.1093/cvr/cvn076
62. Wang X, Wang XL, Chen HL, Wu D, Chen JX, Wang XX, et al. Ghrelin inhibits doxorubicin cardiotoxicity by inhibiting excessive autophagy through ampk and p38-mapk. Biochem Pharmacol. (2014) 88:334–50. doi: 10.1016/j.bcp.2014.01.040
63. Farhan M, Wang H, Gaur U, Little PJ, Xu J, Zheng W. Foxo signaling pathways as therapeutic targets in cancer. Int J Biol Sci. (2017) 13:815–27. doi: 10.7150/ijbs.20052
64. Bon RS, Wright DJ, Beech DJ, Sukumar P. Pharmacology of trpc channels and its potential in cardiovascular and metabolic medicine. Annu Rev Pharmacol Toxicol. (2021) doi: 10.1146/annurev-pharmtox-030121-122314
65. Kitajima N, Numaga-Tomita T, Watanabe M, Kuroda T, Nishimura A, Miyano K, et al. Trpc3 positively regulates reactive oxygen species driving maladaptive cardiac remodeling. Sci Rep. (2016) 6:37001. doi: 10.1038/srep37001
66. Numaga-Tomita T, Kitajima N, Kuroda T, Nishimura A, Miyano K, Yasuda S, et al. Trpc3-gef-h1 axis mediates pressure overload-induced cardiac fibrosis. Sci Rep. (2016) 6:39383. doi: 10.1038/srep39383
67. Zhao Y, McLaughlin D, Robinson E, Harvey AP, Hookham MB, Shah AM, et al. Nox2 nadph oxidase promotes pathologic cardiac remodeling associated with doxorubicin chemotherapy. Cancer Res. (2010) 70:9287–97. doi: 10.1158/0008-5472.CAN-10-2664
68. Ji LL, Yeo D. Mitochondrial dysregulation and muscle disuse atrophy. F1000Res. (2019) 8. doi: 10.12688/f1000research.19139.1
69. Hyatt H, Deminice R, Yoshihara T, Powers SK. Mitochondrial dysfunction induces muscle atrophy during prolonged inactivity: a review of the causes and effects. Arch Biochem Biophys. (2019) 662:49–60. doi: 10.1016/j.abb.2018.11.005
70. Feng S, Li H, Tai Y, Huang J, Su Y, Abramowitz J, et al. Canonical transient receptor potential 3 channels regulate mitochondrial calcium uptake. Proc Natl Acad Sci U S A. (2013) 110:11011–6. doi: 10.1073/pnas.1309531110
71. Ma T, Lin S, Wang B, Wang Q, Xia W, Zhang H, et al. Trpc3 deficiency attenuates high salt-induced cardiac hypertrophy by alleviating cardiac mitochondrial dysfunction. Biochem Biophys Res Commun. (2019) 519:674–81. doi: 10.1016/j.bbrc.2019.09.018
72. Chen RC, Sun GB, Ye JX, Wang J, Zhang MD, Sun XB. Salvianolic acid b attenuates doxorubicin-induced er stress by inhibiting trpc3 and trpc6 mediated ca(2+) overload in rat cardiomyocytes. Toxicol Lett. (2017) 276:21–30. doi: 10.1016/j.toxlet.2017.04.010
73. Sorensen AB, Søndergaard MT, Overgaard MT. Calmodulin in a heartbeat. Febs J. (2013) 280:5511–32. doi: 10.1111/febs.12337
74. Goda AE, Elenany AM, Elsisi AE. Novel in vivo potential of trifluoperazine to ameliorate doxorubicin-induced cardiotoxicity involves suppression of nf-κb and apoptosis. Life Sci. (2021) 283:119849. doi: 10.1016/j.lfs.2021.119849
75. Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. (2003) 83:731–801. doi: 10.1152/physrev.00029.2002
76. Min K, Kwon OS, Smuder AJ, Wiggs MP, Sollanek KJ, Christou DD, et al. Increased mitochondrial emission of reactive oxygen species and calpain activation are required for doxorubicin-induced cardiac and skeletal muscle myopathy. J Physiol. (2015) 593:2017–36. doi: 10.1113/jphysiol.2014.286518
77. Yoshida T, Delafontaine P. Mechanisms of igf-1-mediated regulation of skeletal muscle hypertrophy and atrophy. Cells-Basel. (2020) 9. doi: 10.3390/cells9091970
78. Ren J, Samson WK, Sowers JR. Insulin-like growth factor i as a cardiac hormone: physiological and pathophysiological implications in heart disease. J Mol Cell Cardiol. (1999) 31:2049–61. doi: 10.1006/jmcc.1999.1036
79. Alzahrani AM, Rajendran P, Veeraraghavan VP, Hanieh H. Cardiac protective effect of kirenol against doxorubicin-induced cardiac hypertrophy in h9c2 cells through nrf2 signaling via pi3k/akt pathways. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms22063269
80. Fabbi P, Spallarossa P, Garibaldi S, Barisione C, Mura M, Altieri P, et al. Doxorubicin impairs the insulin-like growth factor-1 system and causes insulin-like growth factor-1 resistance in cardiomyocytes. Plos ONE. (2015) 10:e124643. doi: 10.1371/journal.pone.0124643
81. Chae HJ, Kim HR, Bae J, Chae SU, Ha KC, Chae SW. Signal transduction of the protective effect of insulin like growth factor-1 on adriamycin-induced apoptosis in cardiac muscle cells. Arch Pharm Res. (2004) 27:324–33. doi: 10.1007/BF02980068
82. Lee BS, Oh J, Kang SK, Park S, Lee SH, Choi D, et al. Insulin protects cardiac myocytes from doxorubicin toxicity by sp1-mediated transactivation of survivin. Plos One. (2015) 10:e135438. doi: 10.1371/journal.pone.0135438
83. Mousa H, Abdel AS, Abbas N. Umbilical cord blood-mesenchymal stem cells and carvedilol reduce doxorubicin- induced cardiotoxicity: possible role of insulin-like growth factor-1. Biomed Pharmacother. (2018) 105:1192–204. doi: 10.1016/j.biopha.2018.06.051
84. Fernández C, Torrealba N, Altamirano F, Garrido-Moreno V, Vásquez-Trincado C, Flores-Vergara R, et al. Polycystin-1 is required for insulin-like growth factor 1-induced cardiomyocyte hypertrophy. Plos One. (2021) 16:e255452. doi: 10.1371/journal.pone.0255452
85. Ito H, Hiroe M, Hirata Y, Tsujino M, Adachi S, Shichiri M, et al. Insulin-like growth factor-i induces hypertrophy with enhanced expression of muscle specific genes in cultured rat cardiomyocytes. Circulation. (1993) 87:1715–21. doi: 10.1161/01.CIR.87.5.1715
86. Sakai H, Asami M, Naito H, Kitora S, Suzuki Y, Miyauchi Y, et al. Exogenous insulin-like growth factor 1 attenuates cisplatin-induced muscle atrophy in mice. J Cachexia Sarcopenia Muscle. (2021) doi: 10.1002/jcsm.12760
87. Tsai LK, Chen CL, Ting CH, Lin-Chao S, Hwu WL, Dodge JC, et al. Systemic administration of a recombinant aav1 vector encoding igf-1 improves disease manifestations in sma mice. Mol Ther. (2014) 22:1450–9. doi: 10.1038/mt.2014.84
88. Meeran M, Azimullah S, Mamoudh HH, Sharma C, Kumar S, Goyal SN, et al. Nerolidol, a sesquiterpene from the essential oils of aromatic plants, attenuates doxorubicin-induced chronic cardiotoxicity in rats. J Agric Food Chem. (2021) 69:7334–43. doi: 10.1021/acs.jafc.0c05667
89. Venkatesan B, Prabhu SD, Venkatachalam K, Mummidi S, Valente AJ, Clark RA, et al. Wnt1-inducible signaling pathway protein-1 activates diverse cell survival pathways and blocks doxorubicin-induced cardiomyocyte death. Cell Signal. (2010) 22:809–20. doi: 10.1016/j.cellsig.2010.01.005
90. Sahu R, Dua TK, Das S, De Feo V, Dewanjee S. Wheat phenolics suppress doxorubicin-induced cardiotoxicity via inhibition of oxidative stress, map kinase activation, nf-κb pathway, pi3k/akt/mtor impairment, and cardiac apoptosis. Food Chem Toxicol. (2019) 125:503–19. doi: 10.1016/j.fct.2019.01.034
91. Yang Q, Jiang W, Hou P. Emerging role of pi3k/akt in tumor-related epigenetic regulation. Semin Cancer Biol. (2019) 59:112–24. doi: 10.1016/j.semcancer.2019.04.001
92. McMullen JR, Jay PY. Pi3k(p110alpha) inhibitors as anti-cancer agents: minding the heart. Cell Cycle. (2007) 6:910–3. doi: 10.4161/cc.6.8.4124
93. Crackower MA, Oudit GY, Kozieradzki I, Sarao R, Sun H, Sasaki T, et al. Regulation of myocardial contractility and cell size by distinct pi3k-pten signaling pathways. Cell. (2002) 110:737–49. doi: 10.1016/S0092-8674(02)00969-8
94. McLean BA, Patel VB, Zhabyeyev P, Chen X, Basu R, Wang F, et al. Pi3kα pathway inhibition with doxorubicin treatment results in distinct biventricular atrophy and remodeling with right ventricular dysfunction. J Am Heart Assoc. (2019) 8:e10961. doi: 10.1161/JAHA.118.010961
95. Mohlin S, Hansson K, Radke K, Martinez S, Blanco-Apiricio C, Garcia-Ruiz C, et al. Anti-tumor effects of pim/pi3k/mtor triple kinase inhibitor ibl-302 in neuroblastoma. Embo Mol Med. (2019) 11:e10058. doi: 10.15252/emmm.201810058
96. Kim SJ, Jung KH, Son MK, Park JH, Yan HH, Fang Z, et al. Tumor vessel normalization by the pi3k inhibitor hs-173 enhances drug delivery. Cancer Lett. (2017) 403:339–53. doi: 10.1016/j.canlet.2017.06.035
97. Chen RC, Xu XD, Zhi LX, Sun GB, Zhu YD, Dong X, et al. Total flavonoids from clinopodium chinense (benth.) O. Ktze protect against doxorubicin-induced cardiotoxicity in vitro and in vivo. Evid Based Complement Alternat Med. (2015) 2015:472565. doi: 10.1155/2015/472565
98. Li L, Li J, Wang Q, Zhao X, Yang D, Niu L, et al. Shenmai injection protects against doxorubicin-induced cardiotoxicity via maintaining mitochondrial homeostasis. Front Pharmacol. (2020) 11:815. doi: 10.3389/fphar.2020.00815
99. Kalantary-Charvadeh A, Sanajou D, Hemmati-Dinarvand M, Marandi Y, Khojastehfard M, Hajipour H, et al. Micheliolide protects against doxorubicin-induced cardiotoxicity in mice by regulating pi3k/akt/nf-kb signaling pathway. Cardiovasc Toxicol. (2019) 19:297–305. doi: 10.1007/s12012-019-09511-2
100. Cao Y, Ruan Y, Shen T, Huang X, Li M, Yu W, et al. Astragalus polysaccharide suppresses doxorubicin-induced cardiotoxicity by regulating the pi3k/akt and p38mapk pathways. Oxid Med Cell Longev. (2014) 2014:674219. doi: 10.1155/2014/674219
101. Li M, Sala V, De Santis MC, Cimino J, Cappello P, Pianca N, et al. Phosphoinositide 3-kinase gamma inhibition protects from anthracycline cardiotoxicity and reduces tumor growth. Circulation. (2018) 138:696–711. doi: 10.1161/CIRCULATIONAHA.117.030352
102. Ronnebaum SM, Patterson C. The foxo family in cardiac function and dysfunction. Annu Rev Physiol. (2010) 72:81–94. doi: 10.1146/annurev-physiol-021909-135931
103. Stitt TN, Drujan D, Clarke BA, Panaro F, Timofeyva Y, Kline WO, et al. The igf-1/pi3k/akt pathway prevents expression of muscle atrophy-induced ubiquitin ligases by inhibiting foxo transcription factors. Mol Cell. (2004) 14:395–403. doi: 10.1016/S1097-2765(04)00211-4
104. Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, et al. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell. (2004) 117:399–412. doi: 10.1016/S0092-8674(04)00400-3
105. Liu L, Hu R, You H, Li J, Liu Y, Li Q, et al. Formononetin ameliorates muscle atrophy by regulating myostatin-mediated pi3k/akt/foxo3a pathway and satellite cell function in chronic kidney disease. J Cell Mol Med. (2021) 25:1493–506. doi: 10.1111/jcmm.16238
106. Spurthi KM, Sarikhani M, Mishra S, Desingu PA, Yadav S, Rao S, et al. Toll-like receptor 2 deficiency hyperactivates the foxo1 transcription factor and induces aging-associated cardiac dysfunction in mice. J Biol Chem. (2018) 293:13073–89. doi: 10.1074/jbc.RA118.001880
107. Ni YG, Berenji K, Wang N, Oh M, Sachan N, Dey A, et al. Foxo transcription factors blunt cardiac hypertrophy by inhibiting calcineurin signaling. Circulation. (2006) 114:1159–68. doi: 10.1161/CIRCULATIONAHA.106.637124
108. Mizushima N. The role of the atg1/ulk1 complex in autophagy regulation. Curr Opin Cell Biol. (2010) 22:132–9. doi: 10.1016/j.ceb.2009.12.004
109. Wang X, Proud CG. The mtor pathway in the control of protein synthesis. Physiology (Bethesda). (2006) 21:362–9. doi: 10.1152/physiol.00024.2006
110. Csibi A, Blenis J. Hippo-yap and mtor pathways collaborate to regulate organ size. Nat Cell Biol. (2012) 14:1244–5. doi: 10.1038/ncb2634
111. Ikeda M, Ide T, Fujino T, Matsuo Y, Arai S, Saku K, et al. The akt-mtor axis is a pivotal regulator of eccentric hypertrophy during volume overload. Sci Rep. (2015) 5:15881. doi: 10.1038/srep15881
112. Hiensch AE, Bolam KA, Mijwel S, Jeneson J, Huitema A, Kranenburg O, et al. Doxorubicin-induced skeletal muscle atrophy: elucidating the underlying molecular pathways. Acta Physiol (Oxf). (2020) 229:e13400. doi: 10.1111/apha.13400
113. Murphy KT. The pathogenesis and treatment of cardiac atrophy in cancer cachexia. Am J Physiol Heart Circ Physiol. (2016) 310:H466–77. doi: 10.1152/ajpheart.00720.2015
114. Nie L, Liu M, Chen J, Wu Q, Li Y, Yi J, et al. Hydrogen sulfide ameliorates doxorubicin-induced myocardial fibrosis in rats via the pi3k/akt/mtor pathway. Mol Med Rep. (2021) 23. doi: 10.3892/mmr.2021.11938
115. Zhang J, Wang M, Ding W, Zhao M, Ye J, Xu Y, et al. Resolvin e1 protects against doxorubicin-induced cardiotoxicity by inhibiting oxidative stress, autophagy and apoptosis by targeting akt/mtor signaling. Biochem Pharmacol. (2020) 180:114188. doi: 10.1016/j.bcp.2020.114188
116. Zhang X, Hu C, Kong CY, Song P, Wu HM, Xu SC, et al. Fndc5 alleviates oxidative stress and cardiomyocyte apoptosis in doxorubicin-induced cardiotoxicity via activating akt. Cell Death Differ. (2020) 27:540–55. doi: 10.1038/s41418-019-0372-z
117. Bharathi PL, Baskaran R, Huang CY, Vijaya PV. Neferine modulates igf-1r/nrf2 signaling in doxorubicin treated h9c2 cardiomyoblasts. J Cell Biochem. (2018) 119:1441–52. doi: 10.1002/jcb.26305
118. Gonçalves DA, Silveira WA, Manfredi LH, Graça FA, Armani A, Bertaggia E, et al. Insulin/igf1 signalling mediates the effects of β(2) -adrenergic agonist on muscle proteostasis and growth. J Cachexia Sarcopenia Muscle. (2019) 10:455–75. doi: 10.1002/jcsm.12395
119. Tang G, Du Y, Guan H, Jia J, Zhu N, Shi Y, et al. Butyrate ameliorate skeletal muscle atrophy in diabetic nephropathy via enhancing gut barrier function and ffa2-mediated pi3k/akt/mtor signals. Br J Pharmacol. (2021) doi: 10.22541/au.160688559.90642598/v1
120. Yin D, Lin D, Xie Y, Gong A, Jiang P, Wu J. Neuregulin-1β alleviates sepsis-induced skeletal muscle atrophy by inhibiting autophagy via akt/mtor signaling pathway in rats. Shock. (2021) doi: 10.1097/SHK.0000000000001860
121. Romero-Becerra R, Santamans AM, Folgueira C, Sabio G. P38 mapk pathway in the heart: new insights in health and disease. Int J Mol Sci. (2020) 21. doi: 10.3390/ijms21197412
122. Zhang L, Feng M, Wang X, Zhang H, Ding J, Cheng Z, et al. Peptide szeto-schiller 31 ameliorates doxorubicin-induced cardiotoxicity by inhibiting the activation of the p38 mapk signaling pathway. Int J Mol Med. (2021) 47. doi: 10.3892/ijmm.2021.4896
123. Chen CT, Wang ZH, Hsu CC, Lin HH, Chen JH. In vivo protective effects of diosgenin against doxorubicin-induced cardiotoxicity. Nutrients. (2015) 7:4938–54. doi: 10.3390/nu7064938
124. Puigserver P, Rhee J, Lin J, Wu Z, Yoon JC, Zhang CY, et al. Cytokine stimulation of energy expenditure through p38 map kinase activation of ppargamma coactivator-1. Mol Cell. (2001) 8:971–82. doi: 10.1016/S1097-2765(01)00390-2
125. Fukawa T, Yan-Jiang BC, Min-Wen JC, Jun-Hao ET, Huang D, Qian CN, et al. Excessive fatty acid oxidation induces muscle atrophy in cancer cachexia. Nat Med. (2016) 22:666–71. doi: 10.1038/nm.4093
126. Zhang G, Jin B, Li YP. C/ebpβ mediates tumour-induced ubiquitin ligase atrogin1/mafbx upregulation and muscle wasting. Embo J. (2011) 30:4323–35. doi: 10.1038/emboj.2011.292
127. Odeh M, Tamir-Livne Y, Haas T, Bengal E. P38α mapk coordinates the activities of several metabolic pathways that together induce atrophy of denervated muscles. Febs J. (2020) 287:73–93. doi: 10.1111/febs.15070
128. Ding H, Zhang G, Sin KW, Liu Z, Lin RK, Li M, et al. Activin a induces skeletal muscle catabolism via p38β mitogen-activated protein kinase. J Cachexia Sarcopenia Muscle. (2017) 8:202–12. doi: 10.1002/jcsm.12145
129. He N, Ye H. Exercise and muscle atrophy. Adv Exp Med Biol. (2020) 1228:255–67. doi: 10.1007/978-981-15-1792-1_17
130. Wang F, Iskra B, Kleinerman E, Alvarez-Florez C, Andrews T, Shaw A, et al. Aerobic exercise during early murine doxorubicin exposure mitigates cardiac toxicity. J Pediatr Hematol Oncol. (2018) 40:208–15. doi: 10.1097/MPH.0000000000001112
131. Gomes-Santos IL, Jordão CP, Passos CS, Brum PC, Oliveira EM, Chammas R, et al. Exercise training preserves myocardial strain and improves exercise tolerance in doxorubicin-induced cardiotoxicity. Front Cardiovasc Med. (2021) 8:605993. doi: 10.3389/fcvm.2021.605993
132. Werner C, Hanhoun M, Widmann T, Kazakov A, Semenov A, Pöss J, et al. Effects of physical exercise on myocardial telomere-regulating proteins, survival pathways, and apoptosis. J Am Coll Cardiol. (2008) 52:470–82. doi: 10.1016/j.jacc.2008.04.034
133. Sequeira CM, Martins MA, Alves R, Nascimento A, Botti G, Rocha VN, et al. Aerobic exercise training attenuates doxorubicin-induced ultrastructural changes in rat ventricular myocytes. Life Sci. (2021) 264:118698. doi: 10.1016/j.lfs.2020.118698
134. Kavazis AN, Smuder AJ, Powers SK. Effects of short-term endurance exercise training on acute doxorubicin-induced foxo transcription in cardiac and skeletal muscle. J Appl Physiol. (1985) 117:223–30. doi: 10.1152/japplphysiol.00210.2014
135. Poller W, Dimmeler S, Heymans S, Zeller T, Haas J, Karakas M, et al. Non-coding rnas in cardiovascular diseases: diagnostic and therapeutic perspectives. Eur Heart J. (2018) 39:2704–16. doi: 10.1093/eurheartj/ehx165
136. Hu X, Liu H, Wang Z, Hu Z, Li L. Mir-200a attenuated doxorubicin-induced cardiotoxicity through upregulation of nrf2 in mice. Oxid Med Cell Longev. (2019) 2019:1512326. doi: 10.1155/2019/1512326
137. Li J, Wan W, Chen T, Tong S, Jiang X, Liu W. Mir-451 silencing inhibited doxorubicin exposure-induced cardiotoxicity in mice. Biomed Res Int. (2019) 2019:1528278. doi: 10.1155/2019/1528278
138. Gupta SK, Garg A, Avramopoulos P, Engelhardt S, Streckfuss-Bömeke K, Batkai S, et al. Mir-212/132 cluster modulation prevents doxorubicin-mediated atrophy and cardiotoxicity. Mol Ther. (2019) 27:17–28. doi: 10.1016/j.ymthe.2018.11.004
139. Gupta SK, Garg A, Bär C, Chatterjee S, Foinquinos A, Milting H, et al. Quaking inhibits doxorubicin-mediated cardiotoxicity through regulation of cardiac circular RNA expression. Circ Res. (2018) 122:246–54. doi: 10.1161/CIRCRESAHA.117.311335
140. Räsänen M, Sultan I, Paech J, Hemanthakumar KA, Yu W, He L, et al. Vegf-b promotes endocardium-derived coronary vessel development and cardiac regeneration. Circulation. (2021) 143:65–77. doi: 10.1161/CIRCULATIONAHA.120.050635
141. Gallo S, Sala V, Gatti S, Crepaldi T. Cellular and molecular mechanisms of hgf/met in the cardiovascular system. Clin Sci (Lond). (2015) 129:1173–93. doi: 10.1042/CS20150502
142. Lipsic E, Schoemaker RG, van der Meer P, Voors AA, van Veldhuisen DJ, van Gilst WH. Protective effects of erythropoietin in cardiac ischemia: from bench to bedside. J Am Coll Cardiol. (2006) 48:2161–7. doi: 10.1016/j.jacc.2006.08.031
143. Harada M, Qin Y, Takano H, Minamino T, Zou Y, Toko H, et al. G-csf prevents cardiac remodeling after myocardial infarction by activating the jak-stat pathway in cardiomyocytes. Nat Med. (2005) 11:305–11. doi: 10.1038/nm1199
144. Bry M, Kivelä R, Leppänen VM, Alitalo K. Vascular endothelial growth factor-b in physiology and disease. Physiol Rev. (2014) 94:779–94. doi: 10.1152/physrev.00028.2013
145. Bry M, Kivelä R, Holopainen T, Anisimov A, Tammela T, Soronen J, et al. Vascular endothelial growth factor-b acts as a coronary growth factor in transgenic rats without inducing angiogenesis, vascular leak, or inflammation. Circulation. (2010) 122:1725–33. doi: 10.1161/CIRCULATIONAHA.110.957332
146. Räsänen M, Degerman J, Nissinen TA, Miinalainen I, Kerkelä R, Siltanen A, et al. Vegf-b gene therapy inhibits doxorubicin-induced cardiotoxicity by endothelial protection. Proc Natl Acad Sci U S A. (2016) 113:13144–9. doi: 10.1073/pnas.1616168113
147. Suzuki YJ, Evans T. Regulation of cardiac myocyte apoptosis by the gata-4 transcription factor. Life Sci. (2004) 74:1829–38. doi: 10.1016/j.lfs.2003.10.002
148. Kim Y, Ma AG, Kitta K, Fitch SN, Ikeda T, Ihara Y, et al. Anthracycline-induced suppression of gata-4 transcription factor: implication in the regulation of cardiac myocyte apoptosis. Mol Pharmacol. (2003) 63:368–77. doi: 10.1124/mol.63.2.368
149. Alotaibi BS, Ijaz M, Buabeid M, Kharaba ZJ, Yaseen HS, Murtaza G. Therapeutic effects and safe uses of plant-derived polyphenolic compounds in cardiovascular diseases: a review. Drug Des Devel Ther. (2021) 15:4713–32. doi: 10.2147/DDDT.S327238
150. Michalska M, Gluba A, Mikhailidis DP, Nowak P, Bielecka-Dabrowa A, Rysz J, et al. The role of polyphenols in cardiovascular disease. Med Sci Monit. (2010) 16:A110–9.
151. Ma Y, Yang L, Ma J, Lu L, Wang X, Ren J, et al. Rutin attenuates doxorubicin-induced cardiotoxicity via regulating autophagy and apoptosis. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:1904–11. doi: 10.1016/j.bbadis.2016.12.021
152. Hu LF, Lan HR, Li XM, Jin KT. A systematic review of the potential chemoprotective effects of resveratrol on doxorubicin-induced cardiotoxicity: focus on the antioxidant, antiapoptotic, and anti-inflammatory activities. Oxid Med Cell Longev. (2021) 2021:2951697. doi: 10.1155/2021/2951697
153. Zhang C, Feng Y, Qu S, Wei X, Zhu H, Luo Q, et al. Resveratrol attenuates doxorubicin-induced cardiomyocyte apoptosis in mice through sirt1-mediated deacetylation of p53. Cardiovasc Res. (2011) 90:538–45. doi: 10.1093/cvr/cvr022
154. Arafa MH, Mohammad NS, Atteia HH, Abd-Elaziz HR. Protective effect of resveratrol against doxorubicin-induced cardiac toxicity and fibrosis in male experimental rats. J Physiol Biochem. (2014) 70:701–11. doi: 10.1007/s13105-014-0339-y
155. Sin TK, Tam BT, Yung BY, Yip SP, Chan LW, Wong CS, et al. Resveratrol protects against doxorubicin-induced cardiotoxicity in aged hearts through the sirt1-usp7 axis. J Physiol. (2015) 593:1887–99. doi: 10.1113/jphysiol.2014.270101
156. Ruan Y, Dong C, Patel J, Duan C, Wang X, Wu X, et al. Sirt1 suppresses doxorubicin-induced cardiotoxicity by regulating the oxidative stress and p38mapk pathways. Cell Physiol Biochem. (2015) 35:1116–24. doi: 10.1159/000373937
157. Tian W, Yang L, Liu Y, He J, Yang L, Zhang Q, et al. Resveratrol attenuates doxorubicin-induced cardiotoxicity in rats by up-regulation of vascular endothelial growth factor b. J Nutr Biochem. (2020) 79:108132. doi: 10.1016/j.jnutbio.2019.01.018
158. Kubczak M, Szustka A, Rogalińska M. Molecular targets of natural compounds with anti-cancer properties. Int J Mol Sci. (2021) 22. doi: 10.3390/ijms222413659
159. Lother A, Bergemann S, Kowalski J, Huck M, Gilsbach R, Bode C, et al. Inhibition of the cardiac myocyte mineralocorticoid receptor ameliorates doxorubicin-induced cardiotoxicity. Cardiovasc Res. (2018) 114:282–90. doi: 10.1093/cvr/cvx078
160. Octavia Y, Kararigas G, de Boer M, Chrifi I, Kietadisorn R, Swinnen M, et al. Folic acid reduces doxorubicin-induced cardiomyopathy by modulating endothelial nitric oxide synthase. J Cell Mol Med. (2017) 21:3277–87. doi: 10.1111/jcmm.13231
161. Das A, Durrant D, Salloum FN, Xi L, Kukreja RC. Pde5 inhibitors as therapeutics for heart disease, diabetes and cancer. Pharmacol Ther. (2015) 147:12–21. doi: 10.1016/j.pharmthera.2014.10.003
162. Koka S, Das A, Zhu SG, Durrant D, Xi L, Kukreja RC. Long-acting phosphodiesterase-5 inhibitor tadalafil attenuates doxorubicin-induced cardiomyopathy without interfering with chemotherapeutic effect. J Pharmacol Exp Ther. (2010) 334:1023–30. doi: 10.1124/jpet.110.170191
163. Prysyazhna O, Burgoyne JR, Scotcher J, Grover S, Kass D, Eaton P. Phosphodiesterase 5 inhibition limits doxorubicin-induced heart failure by attenuating protein kinase g iα oxidation. J Biol Chem. (2016) 291:17427–36. doi: 10.1074/jbc.M116.724070
164. Fisher PW, Salloum F, Das A, Hyder H, Kukreja RC. Phosphodiesterase-5 inhibition with sildenafil attenuates cardiomyocyte apoptosis and left ventricular dysfunction in a chronic model of doxorubicin cardiotoxicity. Circulation. (2005) 111:1601–10. doi: 10.1161/01.CIR.0000160359.49478.C2
165. Booth L, Roberts JL, Cruickshanks N, Conley A, Durrant DE, Das A, et al. Phosphodiesterase 5 inhibitors enhance chemotherapy killing in gastrointestinal/genitourinary cancer cells. Mol Pharmacol. (2014) 85:408–19. doi: 10.1124/mol.113.090043
166. Li Q, Shu Y. Pharmacological modulation of cytotoxicity and cellular uptake of anti-cancer drugs by pde5 inhibitors in lung cancer cells. Pharm Res. (2014) 31:86–96. doi: 10.1007/s11095-013-1134-0
167. Das A, Durrant D, Mitchell C, Mayton E, Hoke NN, Salloum FN, et al. Sildenafil increases chemotherapeutic efficacy of doxorubicin in prostate cancer and ameliorates cardiac dysfunction. Proc Natl Acad Sci U S A. (2010) 107:18202–7. doi: 10.1073/pnas.1006965107
168. Chang JF, Hsu JL, Sheng YH, Leu WJ, Yu CC, Chan SH, et al. Phosphodiesterase type 5 (pde5) inhibitors sensitize topoisomerase ii inhibitors in killing prostate cancer through pde5-independent impairment of HR and NHEJ DNA repair systems. Front Oncol. (2018) 8:681. doi: 10.3389/fonc.2018.00681
169. Poklepovic A, Qu Y, Dickinson M, Kontos MC, Kmieciak M, Schultz E, et al. Randomized study of doxorubicin-based chemotherapy regimens, with and without sildenafil, with analysis of intermediate cardiac markers. Cardiooncology. (2018) 4. doi: 10.1186/s40959-018-0033-2
170. Nishiyama K, Numaga-Tomita T, Fujimoto Y, Tanaka T, Toyama C, Nishimura A, et al. Ibudilast attenuates doxorubicin-induced cytotoxicity by suppressing formation of trpc3 channel and nadph oxidase 2 protein complexes. Br J Pharmacol. (2019) 176:3723–38. doi: 10.1111/bph.14777
171. Chen S, Chen J, Shi H, Mickelsen D, Yan C. Abstract mp265: the role of phosphodiesterase 10a in tumor growth and doxorubicin-induced cardiotoxicity. Circ Res. (2021) 129:P265. doi: 10.1161/res.129.suppl_1.MP265
172. Zhang Y, Knight W, Chen S, Mohan A, Yan C. Multiprotein complex with trpc (transient receptor potential-canonical) channel, pde1c (phosphodiesterase 1c), and a2r (adenosine a2 receptor) plays a critical role in regulating cardiomyocyte camp and survival. Circulation. (2018) 138:1988–2002. doi: 10.1161/CIRCULATIONAHA.118.034189
173. Koh JS, Yi CO, Heo RW, Ahn JW, Park JR, Lee JE, et al. Protective effect of cilostazol against doxorubicin-induced cardiomyopathy in mice. Free Radic Biol Med. (2015) 89:54–61. doi: 10.1016/j.freeradbiomed.2015.07.016
174. Narezkina A, Nasim K. Anthracycline cardiotoxicity. Circ Heart Fail. (2019) 12:e5910. doi: 10.1161/CIRCHEARTFAILURE.119.005910
175. Silva MT. Secondary necrosis: the natural outcome of the complete apoptotic program. Febs Lett. (2010) 584:4491–9. doi: 10.1016/j.febslet.2010.10.046
176. Leist M, Single B, Castoldi AF, Kühnle S, Nicotera P. Intracellular adenosine triphosphate (atp) concentration: a switch in the decision between apoptosis and necrosis. J Exp Med. (1997) 185:1481–6. doi: 10.1084/jem.185.8.1481
Keywords: doxorubicin, cardiotoxicity, cardiomyocyte atrophy, left ventricular mass loss, cell death
Citation: Chen D-S, Yan J and Yang P-Z (2022) Cardiomyocyte Atrophy, an Underestimated Contributor in Doxorubicin-Induced Cardiotoxicity. Front. Cardiovasc. Med. 9:812578. doi: 10.3389/fcvm.2022.812578
Received: 10 November 2021; Accepted: 11 January 2022;
Published: 25 February 2022.
Edited by:
Nazish Sayed, Stanford University, United StatesReviewed by:
Beshay Zordoky, University of Minnesota Twin Cities, United StatesAnindita Das, Virginia Commonwealth University, United States
Copyright © 2022 Chen, Yan and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ping-Zhen Yang, eV9waW5nemhlbiYjeDAwMDQwOzEyNi5jb20=; Jing Yan, eWFudmUxMDA4JiN4MDAwNDA7MTI2LmNvbQ==