 Lisa Hasselbach
Lisa Hasselbach Johannes Weidner2
Johannes Weidner2 Gregor Theilmeier
Gregor Theilmeier- 1Division of Cardiology and Division of Perioperative Inflammation and Infection, Department Human Medicine, University of Oldenburg, Oldenburg, Germany
- 2Division of Perioperative Inflammation and Infection, Department Human Medicine, University of Oldenburg, Oldenburg, Germany
- 3Division of Cardiology, Department Human Medicine, University of Oldenburg, Oldenburg, Germany
Cardiovascular diseases continue to be the most imminent health care problems in the western world, accounting for numerous deaths per year. Heart failure (HF), namely the reduction of left ventricular function, is one of the major cardiovascular disease entities. It is chronically progressing with relapsing acute decompensations and an overall grave prognosis that is little different if not worse than most malignant diseases. Interestingly acute metabolically and/or immunologically challenging events like infections or major surgical procedures will cause relapses in the course of preexisting chronic heart failure, decrease the patients wellbeing and worsen myocardial function. HF itself and or its progression has been demonstrated to be driven at least in part by inflammatory pathways that are similarly turned on by infectious or non-infectious stress responses. These thus add to HF progression or relapse. TNF-α plasma levels are associated with disease severity and progression in HF. In addition, several cytokines (e.g., IL-1β, IL-6) are involved in deteriorating left ventricular function. Those observations are based on clinical studies using inhibitors of cytokines or their receptors or they stem from animal studies examining the effect of cytokine mediated inflammation on myocardial remodeling in models of heart failure. This short review summarizes the known underlying immunological processes that are shared by and drive all: chronic heart failure, select infectious diseases, and inflammatory stress responses. In conclusion the text provides a brief summary of the current development in immunomodulatory therapies for HF and their overlap with treatments of other disease entities.
Scope of the Review
Chronic heart failure (HF) is in part driven by inflammatory processes. Ventricular function in chronic HF patients worsens if the patient experiences acute illnesses or stresses. Some of these share inflammatory motifs with those active in progression of HF. We will focus on the overlap of key pathways in chronic inflammation in HF, sepsis, viral infection (i.e., COVID-19) and non-infectious stressors to shed light on their potentially synergistic adverse effects in HF progression and deterioration (Figure 1).
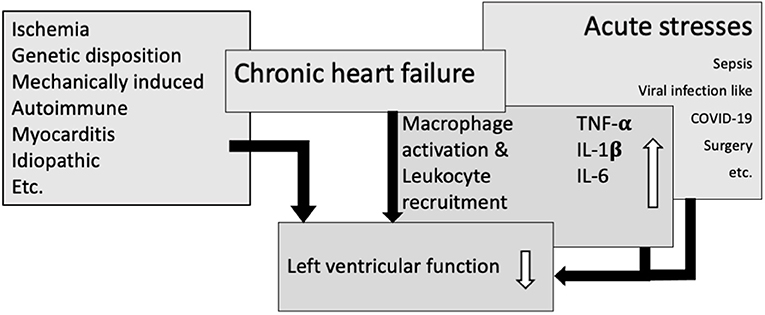
Figure 1. Overlapping immunological pathways in heart failure and acute stresses result in macrophage activation and leukocyte recruitment, leading to an increase in cytokines and thereby amplify impairment of left ventricular function.
Relevance of Heart Failure
HF can present as a chronic disease or in an acute setting with rapidly decreasing myocardial function either as the primary presentation or as acute decompensation of preexisting disease. Acute events (like operations or infections) can trigger a reduction of myocardial function that frequently will not return to prior levels (1).
HF has a prevalence of 11.8 % in people aged 60 or older (2). An observational study in 2,700 HF-patients showed an all-cause 5-year mortality of 53%. HF-patients over 65 years are even more likely to die (from cardiovascular—as well as non-cardiovascular causes). Hospitalization occurs about once per patient/year, mainly for non-cardiovascular causes (3).
The mainstay of HF is the loss of cardiomyocyte (functional) mass due to noxious stimuli ranging from ischemia via pressure overload-induced remodeling processes to viral infection and genetic causes (see Figure 1). Cardiomyocyte death sets off a remodeling cascade that aims to preserve left ventricular function as sufficient as possible. This remodeling cascade involves restructuring of the muscular tissue and reshuffling of its cellular and extracellular components to accommodate an optimized function in the face of the resulting new and disadvantageous ventricular geometry. The mechanisms active in this process are those of inflammatory responses, tissue breakdown, reorganization and replacement (4, 5).
Inflammation in Heart Failure
Recently, the recognition of the role of the immune system in chronic HF is evolving (6). The most common hypothesis explaining the development of HF, is an insufficient resolution of acute inflammation resulting in chronic inflammation. Recent data suggest, that a failure in resolution of inflammation could be due to a dysregulated system of specialized pro-resolving mediators (7), leading to a peak of interest in research influencing these receptors in cardiovascular diseases (8). An interesting phenomenon in this particular context is para-inflammation, that describes a low grade inflammatory response mainly carried by tissue macrophages and perpetuated by metabolic syndrome disease entities (9, 10). Para-inflammation may chronify if it is not resolved and could thereby precipitate chronic myocardial inflammation and HF (11).
After an ischemic insult macrophages infiltrate the site of injury and alter their polarization around 1 week after the acute injury from classical proinflammatory to alternative anti-inflammatory phenotypic characteristics, the latter expressing IL-10. Inflammatory macrophages express tumor necrosis factor a (TNF-α), interleukin-6 (IL-6) and interleukin-1β (IL-1β) (12) (see Figure 1). The immune receptor responsible for production and release of these cytokines is the toll-like receptor 4 (TLR4) a receptor steering the innate immune response to tissue damage (13). Pathogen– and damage-associated molecular patterns (PAMPS and DAMPS), the ligands for TL-receptors are released from necrotic cardiomyocytes. They evoke the secretion of cytokines from resident macrophages as well as the activation and recruitment of neutrophils. TLR4 is thus actively fueling inflammation after acute myocardial injury. Its expression is regulated by cytokines, among others by IL-1β (14). TLR4 is present and upregulated in chronic HF (15).
One of the cytokines that has been shown to play an important role in HF is TNF-α since Levine et al. showed that HF patients have increased levels of circulating TNF-α (16). TNF-α- and IL-6-levels increase in episodes of acute decompensation (17). The TNF-α level correlates with worse prognosis in HF (18). In vivo animal experiments have shown that overexpression of TNF-α leads to chronic inflammation in the heart, to dilatative cardiomyopathy and in the end to HF (19). Blocking TNF-α showed promising results in rats reducing cardiac inflammation and adverse myocardial remodeling (20, 21).
IL-1β is upregulated in animal models of HF of different etiologies (22, 23). The knockdown of the IL-1β receptor in in vivo experiments prompted a reduced inflammatory response and less fibrosis after myocardial infarction in mice (24). Studies performed in animals have suggested, that IL-1β might be a good candidate molecule to influence HF. Anakinra, a receptor antagonist for IL-1β, reduced myocardial remodeling after myocardial infarction in mice (25). Targeting IL-1β with an antibody has resulted in reduced post-infarction remodeling and an improved LVEF in rats (26).
In addition are elevated levels of IL-6 associated with a increased risk of developing HF over time (27). Since IL-6 is also important in the pathophysiology of rheumatoid arthritis (RA), tocilizumab, an antibody against the IL-6 receptor has been shown to reduce pro-BNP and improve LV-function in RA patients without previously known HF (28, 29). This might imply a role for blocking IL-6 in a preventive way because patients with RA often develop HF during their course (30). Studies on the effect of blocking IL-6 in patients with known HF have not yet been conducted.
Inflammatory Response to Non-cardiac Surgery Exemplifies the Effects of Non-infectious Stresses on Chronic Heart Failure
Surgical stress induces proinflammatory responses targeted to remove destroyed tissue, as such inducing wound healing and ultimately recovery of organ function (31–38). The prevalence of HF in the VA Surgical Quality Affairs population was 7.9% in patients undergoing non-cardiac surgery and preexisting HF resulted in increased mortality with an odd's ratio of 1.69 (CI 1.57–1.76) (39). Patients with preexisting HF undergoing non-cardiac surgery in other very large retrospective cohort studies (AHRQ-NIS) with less selected patient populations likewise carried dramatically increased mortality and rehospitalization risks (40). Preexisting HF causes rehospitalization due to CV and non-CV causes in more than 10% of the patients compared to 2.5% (41). Pre-existing chronic diseases, such as HF weaken the ability of an organism to balance pro- and anti-inflammatory responses, in short its resilience (42). In this setting, the pro-inflammatory response evoked by surgical stress may promote deterioration of left ventricular function and MI (43). Surgery induces a cytokine based inflammatory response, predominantly reflected by increases in IL-6 (38). We have shown, using a mouse model with preexisting atherosclerosis exposed to perioperative stress, that IL-6 neutralizing antibodies but not inhibition of its trans-signaling protects plaque stability, proving that IL-6 here is causal for vascular effects of surgical stress (44). In addition, surgery induces an adrenergic stress response originating from pain and tissue destruction and translating into hemodynamic changes such as increased blood pressure and heart rate. Augmented adrenergic responses will trigger more inflammation and increased blood pressure and heart rate increase the risk for deterioration of LV function (40, 42, 45).
Neutrophilia and monocytosis are hallmarks of the stress response to surgery during the first postoperative days (35–37). Neutrophils and monocytes propagate inflammatory responses targeted to remove destroyed tissue, which induces wound healing and ultimately leads to preservation of organ function (34). However, enhanced neutrophil and monocyte infiltration in inflamed myocardium may worsen HF (43, 46, 47). Hence, HF relapses after surgery may be due to elevated IL-6, monocytosis, and neutrophilia, or alternatively or in addition, hypertension and tachycardia (48).
Heart Failure and Infection
One of the main reasons for hospitalization in patients with HF are infections. One out of four HF patients dies because of sepsis (a dysregulated host reaction of the immune system to severe infections) (49), mostly due to respiratory tract infections (50). In that context the risk of hospitalization during the influenza season is significantly increased in HF patients (51). Contracting infections on the other hand also carries a high risk to aggravate existing HF (52, 53).
There are parallels between the cytokine profile in HF patients and patients suffering from bacterial sepsis. TNF-α is elevated in septic patients (54). Survivors compared to non-survivors have significantly more TNF-α and IL-1β in their plasma (55). IL-6 levels are also elevated in sepsis and directly correlate with mortality (56). TLR4, the immune receptor responsible for the cytokine release in HF is upregulated in animal models of sepsis induced myocardial dysfunction (57), hinting at its central role in mediating what is called the “cytokine storm.”
The increased susceptibility of HF patients to infection is conveyed through several, partly independent, partly overlapping mechanisms in addition to circulating cytokines. Infections increase the metabolic demand, leading to a mismatch of oxygen demand to supply. In HF the metabolic alterations are increased oxidative stress (58), energy deficiency (59) and loss of metabolic flexibility (60) resulting in what is called sepsis-induced cardiomyopathy after infections (61). Another mainstay of sepsis is impaired renal function with volume overload and congestion that occurs particularly in HF patients. Acute kidney injury is more common in sepsis patients with preexisting HF (62).
One of the key therapeutic measures for patients in septic shock is however maintenance of intravascular volume status by infusion of fluids. Patients with preexisting HF are at high risk of acute decompensation with development of pulmonary edema due to fluid substitution. HF patients in septic shock have been shown to receive less fluids than non-HF patients. Among the HF patients those who received less fluids have a higher mortality (63). On the other hand homeostasis of intra- and extravascular fluids and electrolyte balance is important for the activation status and phenotypic characteristics of tissue macrophages. Macrophage polarity has been demonstrated to be affected by the tonicity of extravascular fluids and in turn contribute to blood pressure regulation (64, 65). If this axis is active and potentially detrimental in HF remains to be investigated.
Taken together these data suggest that it is important for HF patients to prevent infection altogether. Said that, all measures to avoid infections are effective. A very effective way to do so is vaccination. Vaccination against influenza reduces all-cause mortality and rate of hospitalization due to cardiovascular events as well as to all-causes (66–68) which has thus been recommended by international cardiology societies accordingly (69, 70).
Heart Failure and COVID-19
Since 2020 the world has been confronted with the SARS-CoV2 pandemic. Preexisting HF increases mortality of COVID-19 (71). Exacerbation of HF can even be the only sign of a SARS-CoV2-Infection (72). Lymphocytopenia, one of the classical changes in white blood counts during COVID-19 correlates with prognosis (73), and is associated with severe HF during the early disease (74). Troponin-T showing myocardial injury, correlates with disease severity and mortality of COVID-19 even without preexisting HF (75, 76).
Like in HF and sepsis macrophage-derived cytokines are increased in COVID-19 (77, 78). Analysis of monocytes from COVID-19 patients suggests that the inflammatory response just like in HF and sepsis might be mediated through TLR4 (79). TNF-α has proven to be an independent predictor of COVID-19 severity and mortality (80).
There is contradicting data about the levels of IL-1β in COVID-19 patients. A small study showed an increase in IL-1β in COVID-19 patients correlating with the severity of the disease (81) but a larger study of roughly 1500 patients in New York has not found significant contributions of IL-1β (80).
IL-6 is significantly elevated in severe COVID-19 according to a meta-analysis and levels of IL-6 like in sepsis correlated with adverse outcomes (82). Especially the IL-6 to α1-Antitrypsin ratio is predictive for the course of the disease (81).
Post-acute sequelae of COVID-19, also called Long-COVID, is as HF potentially caused by incomplete resolution of inflammation and characterized by a long lasting elevation of IL-1β, IL-6 and TNF-α (83).
Cardiomyocytes can be directly infected with SARS-CoV2 via the angiotensin-converting enzyme type 2 (ACE2) which is then cleaved by transmembrane protease serine 2 (TMPRSS2) (84). Due to the abundant expression of ACE2 and TMPRSS2 cardiomyocytes are, apart from alveolar cells and macrophages, the main target cells for an infection with SARS-CoV2 (85). In the later course of the infection the inflammatory cytokines upregulate ACE2 resulting in a self-reinforcing process that worsens the disease (86). Because of the upregulation of ACE2 through ACE-inhibitors, an adverse effect of ACE-Inhibitors in COVID-19 has been controversially discussed (87).
ACE2 counterbalances the activated RAAS in HF by transforming Angiotensin II to angiotensin 1-7. Increased ACE2 levels thus even prevent or improve preexisting HF (88). The effect of reduced ACE2 bioavailability due to cleavage after binding SARS-CoV2 could augment RAAS-system activity and thereby blood pressure, vasoconstriction and sodium-retention, eventually leading to worsening of LV function. A small study has shown that ACE-inhibitors decrease IL-6 levels and peak viral load in COVID-19 patients (89) which has led to a change in antihypertensive management in COVID-19 patients. The optimal medical treatment for HF should be continued in COVID-Patients if the hemodynamic situation allows it (90).
Autopsies have shown that both the innate and the adaptive immune systems are activated in the heart of patients with SARS-CoV2-Infection (91). An autopsy study on 40 COVID-19 patients showed acute cardiomyocyte necrosis and apoptosis along with cardiac fibrosis in all samples. The virus itself could only be confirmed in hearts of few patients (92). Damage to the heart thus seems to be possible directly by viral infection of cardiomyocytes as well as indirectly in the course of systemic inflammatory response to local infection.
Using Cytokine Directed Immunomodulatory Therapies to Treat Heart Sfailure
One of the cytokines indicted in inflammation in HF as well as in sepsis is TNF-α. Contrary to the promising animal experiments the use of infliximab, a TNF-α antibody resulted in a reduced cardiac output in patients with rheumatoid arthritis without known HF (93). The ATTACH trial used infliximab in patients with at least moderate HF, as well as the RENEWAL study (a combined analysis of the RENAISSANCE and RECOVER trial that used etanercept) both demonstrated that blocking TNF-α in HF patients does not yield a clinical benefit, but exerts adverse effects at higher doses (94, 95).
Numerous studies have examined the effect of monoclonal and polyclonal antibodies against TNF-α in septic patients. Both reduce over-all mortality in patients with severe sepsis. Monoclonal anti-TNF-α-antibodies improve survival of patients in septic shock (96). In COVID-19 the use of adalimumab, a TNF-α-antibody did not reduce mortality or beneficially alter the course of the disease (97).
These results suggest that even though TNF-α is upregulated in HF as well as in other infections, blocking it by using antibodies has negative effects on HF despite positive effects on some select infectious diseases.
In the CANTOS-trial canakinumab, an IL-1β antibody, in patients with myocardial infarction and active inflammation resulted in less all-cause mortality and a lower rate for HF hospitalization (98, 99). Also canakinumab did not result in a reduction of COVID-19 mortality (100). Anakinra, the IL-1β receptor antagonist which had proven effective in reducing adverse myocardial remodeling in mice also successfully improved left ventricular function after myocardial infarction in humans (101). It also reduced mortality in septic patients (102). In the REMAP-CAP trial anakinra did not have a positive effect on COVID-19 (103).
A positive effect of the anti-IL-6 antibody tocilizumab has been seen for the cardiac function of patients without HF (28, 29). Up to now no studies have been performed on the use of tocilizumab or other IL-6 Inhibitors in HF patients but a case report of a patient with HF after myocardial infarction who received tocilizumab because of a large vessel arteriitis has shown an improvement of myocardial function after the treatment (104). In the IMICA trial tocilizumab has reduced inflammation as well as myocardial damage in post-cardiac arrest syndrome (105). After a series of cases and small studies of a successful treatment of COVID-19 related HF and cytokine storm with tocilizumab had been reported (106, 107), the REMAP-CAP trial has evaluated the effect of the IL-6 receptor antibodies tocilizumab and sarilumab (103). There is now a recommendation to use tocilizumab or sarilumab in patients with severe COVID-19 without invasive ventilation (108). Whether the effect of anti-IL-6 receptor antibodies is due to the blocking of myocardial IL-6 receptors or other effector organs has still to be examined. In sepsis no large scale studies have been performed on blocking IL-6 but tocilizumab has proven to attenuate infection and improve survival in an animal model of sepsis (109). Of the examined cytokines IL-6 seems to offer the most potential for immunomodulatory therapy to affect both infections and HF.
In a broader attempt to address the immunological aspects in HF the ACCLAIM study showed better over-all survival in the mildly impaired (NYHA II) group after using a non-specific immunomodulatory therapy (using the Celacade system™) (110). Surprisingly, follow-up studies revealed that the circulating antibody amount is the same with or without receiving non-specific immunemodulation (111).
CytoResc, a recent, not yet published, trial uses an extracorporeal polystyrene-based hemoadsorber in COVID-19 patients (112). Prior to this study, a single-center study was performed in Freiburg using extracorporeal membrane oxygenation in combination with hemoadsorption, leading to no reduction in IL-6 and even decreased survival (113). Interestingly, even though resulting in clinically favorable outcome in septic patients (114) said hemoadsorber did not change the levels of IL-1β, IL-6 or TNF-α (115).
The use of corticosteroids is a treatment option for patients in septic shock if treatment with vasopressors and fluids is not successful in improving the hemodynamic situation (116). For a long time, using an unspecific immunosuppression with corticosteroids was the only effective treatment for COVID-19 patients (117). In HF use of corticosteroids is debated controversially: While it may decrease chronic inflammation it also affects the neurohormonal RAA system by inducing ACE (118). Modulation of the RAAS by using ACE-Inhibitors or angiotensin receptor neprilysin inhibitors (ANRI) is one of the main therapeutic components in HF. It reduces mortality and morbidity with the ANRI valsartan in combination with sacubitril showing superiority over Enalapril in the PARADIGM-HF study (119, 120). However, the CORT-AHF study did not show a difference in adverse events for patients with or without HF after corticosteroid treatment, this result was independent from use of ACE-Inhibitors or ANRI (121).
Conclusion
The aim of this short review was to delineate parallels in the immunological processes in HF and inflammatory responses to infections or non-infectious stresses, which can also be viewed as an opportunity for using immune based strategies to fight deteriorating HF in cases with such comorbidities.
Influencing the release and circulation of cytokines could be an interesting starting point for HF therapy but is not yet ready to be administered in a broad setting. Inhibition of IL-6-signaling seems to be one of the more promising strategies for treatment of HF as well as infections but it has to be considered, that there is a lot of contradictory data so that a clear recommendation for immunomodulatory therapy cannot be given yet.
TLR4 is involved in the development of various diseases connected to inflammation is. To date TLR4 signaling cannot be therapeutically targeted, while this may be a promising avenue to improve cardiac function in HF patients as well as to reduce myocardial dysfunction in infectious diseases (122).
One thing one should keep in mind while discussing immunomodulatory therapy is that these medications have a risk of increasing susceptibility for new infections necessitating a strict indication and close monitoring during and after application.
Author Contributions
The manuscript was written by LH and GT and revised by JW and AE. All authors contributed to the article and approved the submitted version.
Funding
Funding for LH was provided through the University of Oldenburg (Forschungspool-Projekt 2018-030).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Gheorghiade M, De Luca L, Fonarow GC, Filippatos G, Metra M, Francis GS. Pathophysiologic targets in the early phase of acute heart failure syndromes. Am J Cardiol. (2005) 96:11G−17G. doi: 10.1016/j.amjcard.2005.07.016
2. van Riet EE, Hoes AW, Wagenaar KP, Limburg A, Landman MA, Rutten FH. Epidemiology of heart failure: the prevalence of heart failure and ventricular dysfunction in older adults over time. A systematic review. Eur J Heart Fail. (2016) 18:242–52. doi: 10.1002/ejhf.483
3. Gerber Y, Weston SA, Redfield MM, Chamberlain AM, Manemann SM, Jiang R, et al. A contemporary appraisal of the heart failure epidemic in Olmsted County, Minnesota, 2000 to 2010. JAMA Intern Med. (2015) 175:996–1004. doi: 10.1001/jamainternmed.2015.0924
4. Frangogiannis NG. The mechanistic basis of infarct healing. Antioxid Redox Signal. (2006) 8:1907–39. doi: 10.1089/ars.2006.8.1907
5. Frantz S, Bauersachs J, Ertl G. Post-infarct remodelling: contribution of wound healing and inflammation. Cardiovasc Res. (2009) 81:474–81. doi: 10.1093/cvr/cvn292
6. Adamo L, Rocha-Resende C, Prabhu SD, Mann DL. Reappraising the role of inflammation in heart failure. Nat Rev Cardiol. (2020) 17:269–85. doi: 10.1038/s41569-019-0315-x
7. Serhan CN. Treating inflammation and infection in the 21st century: new hints from decoding resolution mediators and mechanisms. FASEB J. (2017) 31:1273–88. doi: 10.1096/fj.201601222R
8. Fu T, Mohan M, Brennan EP, Woodman OL, Godson C, Kantharidis P, et al. Therapeutic Potential of Lipoxin A4 in Chronic Inflammation: Focus on Cardiometabolic Disease. ACS Pharmacol Transl Sci. (2020) 3:43–55. doi: 10.1021/acsptsci.9b00097
9. Hotamisligil GS. Inflammation and metabolic disorders. Nature. (2006) 444:860–7. doi: 10.1038/nature05485
10. Van Linthout S, Tschope C. Inflammation - Cause or Consequence of Heart Failure or Both? Curr Heart Fail Rep. (2017) 14:251–65. doi: 10.1007/s11897-017-0337-9
11. Reina-Couto M, Pereira-Terra P, Quelhas-Santos J, Silva-Pereira C, Albino-Teixeira A, Sousa T. Inflammation in Human Heart Failure: Major Mediators and Therapeutic Targets. Front Physiol. (2021) 12:746494. doi: 10.3389/fphys.2021.746494
12. Troidl C, Mollmann H, Nef H, Masseli F, Voss S, Szardien S, et al. Classically and alternatively activated macrophages contribute to tissue remodelling after myocardial infarction. J Cell Mol Med. (2009) 13:3485–96. doi: 10.1111/j.1582-4934.2009.00707.x
13. Liang Q, Wu Q, Jiang J, Duan J, Wang C, Smith MD, et al. Characterization of sparstolonin B, a Chinese herb-derived compound, as a selective Toll-like receptor antagonist with potent anti-inflammatory properties. J Biol Chem. (2011) 286:26470–9. doi: 10.1074/jbc.M111.227934
14. Frantz S, Kobzik L, Kim YD, Fukazawa R, Medzhitov R, Lee RT, et al. Toll4 (TLR4) expression in cardiac myocytes in normal and failing myocardium. J Clin Invest. (1999) 104:271–80. doi: 10.1172/JCI6709
15. Liu L, Wang Y, Cao ZY, Wang MM, Liu XM, Gao T, et al. Up-regulated TLR4 in cardiomyocytes exacerbates heart failure after long-term myocardial infarction. J Cell Mol Med. (2015) 19:2728–40. doi: 10.1111/jcmm.12659
16. Levine B, Kalman J, Mayer L, Fillit HM, Packer M. Elevated circulating levels of tumor necrosis factor in severe chronic heart failure. N Engl J Med. (1990) 323:236–41. doi: 10.1056/NEJM199007263230405
17. Abernethy A, Raza S, Sun JL, Anstrom KJ, Tracy R, Steiner J, et al. Pro-Inflammatory Biomarkers in Stable Versus Acutely Decompensated Heart Failure With Preserved Ejection Fraction. J Am Heart Assoc. (2018) 7:e007385. doi: 10.1161/JAHA.117.007385
18. Dunlay SM, Weston SA, Redfield MM, Killian JM, Roger VL. Tumor necrosis factor-alpha and mortality in heart failure: a community study. Circulation. (2008) 118:625–31. doi: 10.1161/CIRCULATIONAHA.107.759191
19. Kubota T, McTiernan CF, Frye CS, Slawson SE, Lemster BH, Koretsky AP, et al. Dilated cardiomyopathy in transgenic mice with cardiac-specific overexpression of tumor necrosis factor-alpha. Circ Res. (1997) 81:627–35. doi: 10.1161/01.RES.81.4.627
20. Berry MF, Woo YJ, Pirolli TJ, Bish LT, Moise MA, Burdick JW, et al. Administration of a tumor necrosis factor inhibitor at the time of myocardial infarction attenuates subsequent ventricular remodeling. J Heart Lung Transplant. (2004) 23:1061–8. doi: 10.1016/j.healun.2004.06.021
21. Jobe LJ, Melendez GC, Levick SP, Du Y, Brower GL, Janicki JS. TNF-alpha inhibition attenuates adverse myocardial remodeling in a rat model of volume overload. Am J Physiol Heart Circ Physiol. (2009) 297:H1462–1468. doi: 10.1152/ajpheart.00442.2009
22. Dewald O, Ren G, Duerr GD, Zoerlein M, Klemm C, Gersch C, et al. Of mice and dogs: species-specific differences in the inflammatory response following myocardial infarction. Am J Pathol. (2004) 164:665–77. doi: 10.1016/S0002-9440(10)63154-9
23. Shioi T, Matsumori A, Kihara Y, Inoko M, Ono K, Iwanaga Y, et al. Increased expression of interleukin-1 beta and monocyte chemotactic and activating factor/monocyte chemoattractant protein-1 in the hypertrophied and failing heart with pressure overload. Circ Res. (1997) 81:664–71. doi: 10.1161/01.RES.81.5.664
24. Bujak M, Dobaczewski M, Chatila K, Mendoza LH, Li N, Reddy A, et al. Interleukin-1 receptor type I signaling critically regulates infarct healing and cardiac remodeling. Am J Pathol. (2008) 173:57–67. doi: 10.2353/ajpath.2008.070974
25. Abbate A, Salloum FN, Vecile E, Das A, Hoke NN, Straino S, et al. Anakinra, a recombinant human interleukin-1 receptor antagonist, inhibits apoptosis in experimental acute myocardial infarction. Circulation. (2008) 117:2670–83. doi: 10.1161/CIRCULATIONAHA.107.740233
26. Harouki N, Nicol L, Remy-Jouet I, Henry JP, Dumesnil A, Lejeune A, et al. The IL-1beta Antibody Gevokizumab Limits Cardiac Remodeling and Coronary Dysfunction in Rats With Heart Failure. JACC Basic Transl Sci. (2017) 2:418–30. doi: 10.1016/j.jacbts.2017.06.005
27. Chia YC, Kieneker LM, van Hassel G, Binnenmars SH, Nolte IM, van Zanden JJ, et al. Interleukin 6 and Development of Heart Failure With Preserved Ejection Fraction in the General Population. J Am Heart Assoc. (2021) 10:e018549. doi: 10.1161/JAHA.120.018549
28. Kobayashi H, Kobayashi Y, Giles JT, Yoneyama K, Nakajima Y, Takei M. Tocilizumab treatment increases left ventricular ejection fraction and decreases left ventricular mass index in patients with rheumatoid arthritis without cardiac symptoms: assessed using 3. 0 tesla cardiac magnetic resonance imaging. J Rheumatol. (2014) 41:1916–21. doi: 10.3899/jrheum.131540
29. Yokoe I, Kobayashi H, Kobayashi Y, Giles JT, Yoneyama K, Kitamura N, et al. Impact of tocilizumab on N-terminal pro-brain natriuretic peptide levels in patients with active rheumatoid arthritis without cardiac symptoms. Scand J Rheumatol. (2018) 47:364–70. doi: 10.1080/03009742.2017.1418424
30. Maradit-Kremers H, Nicola PJ, Crowson CS, Ballman KV, Gabriel SE. Cardiovascular death in rheumatoid arthritis: a population-based study. Arthritis Rheum. (2005) 52:722–32. doi: 10.1002/art.20878
31. Krohn CD, Reikeras O, Aasen AO. The cytokines IL-1beta and IL-1 receptor antagonist, IL-2 and IL-2 soluble receptor-alpha, IL-6 and IL-6 soluble receptor, TNF-alpha and TNF soluble receptor I, and IL10 in drained and systemic blood after major orthopaedic surgery. Eur J Surg. (1999) 165:101–9. doi: 10.1080/110241599750007261
32. Lahiri R, Derwa Y, Bashir Z, Giles E, Torrance HD, Owen HC, et al. Systemic Inflammatory Response Syndrome After Major Abdominal Surgery Predicted by Early Upregulation of TLR4 and TLR5. Ann Surg. (2016) 263:1028–37. doi: 10.1097/SLA.0000000000001248
33. Lin E, Calvano SE, Lowry SF. Inflammatory cytokines and cell response in surgery. Surgery. (2000) 127:117–26. doi: 10.1067/msy.2000.101584
34. Menger MD, Vollmar B. Surgical trauma: hyperinflammation versus immunosuppression? Langenbecks Arch Surg. (2004) 389:475–84. doi: 10.1007/s00423-004-0472-0
35. Toft P, Nielsen C, Tønnesen E, Hansen TG, Hokland M. Changes in adhesion molecule expression and oxidative burst activity of granulocytes and monocytes during open-heart surgery with cardiopulmonary bypass compared with abdominal surgery. Eur J Anaesthesiol. (1998) 15:345–53. doi: 10.1097/00003643-199805000-00018
36. Toft P, Svendsen P, Tonnesen E, Rasmussen JW, Christensen NJ. Redistribution of lymphocytes after major surgical stress. Acta Anaesthesiol Scand. (1993) 37:245–9. doi: 10.1111/j.1399-6576.1993.tb03708.x
37. Toft P, Tonnesen E, Helbo-Hansen HS, Lillevang ST, Rasmussen JW, Christensen NJ. Redistribution of granulocytes in patients after major surgical stress. APMIS. (1994) 102:43–8. doi: 10.1111/j.1699-0463.1994.tb04843.x
38. Decker D, Tolba R, Springer W, Lauschke H, Hirner A, von Ruecker A. Abdominal surgical interventions: local and systemic consequences for the immune system–a prospective study on elective gastrointestinal surgery. J Surg Res. (2005) 126:12–8. doi: 10.1016/j.jss.2005.01.006
39. Lerman BJ, Popat RA, Assimes TL, Heidenreich PA, Wren SM. Association of Left Ventricular Ejection Fraction and Symptoms With Mortality After Elective Noncardiac Surgery Among Patients With Heart Failure. JAMA. (2019) 321:572–9. doi: 10.1001/jama.2019.0156
40. Smilowitz NR, Banco D, Katz SD, Beckman JA, Berger JS. Association between heart failure and perioperative outcomes in patients undergoing non-cardiac surgery. Eur Heart J Qual Care Clin Outcomes. (2021) 7:68–75. doi: 10.1093/ehjqcco/qcz066
41. van Diepen S, Bakal JA, McAlister FA, Ezekowitz JA. Mortality and readmission of patients with heart failure, atrial fibrillation, or coronary artery disease undergoing noncardiac surgery: an analysis of 38 047 patients. Circulation. (2011) 124:289–96. doi: 10.1161/CIRCULATIONAHA.110.011130
42. Priebe HJ. Preoperative cardiac management of the patient for non-cardiac surgery: an individualized and evidence-based approach. Br J Anaesth. (2011) 107:83–96. doi: 10.1093/bja/aer121
43. Gualandro DM, Campos CA, Calderaro D, Yu PC, Marques AC, Pastana AF, et al. Coronary plaque rupture in patients with myocardial infarction after noncardiac surgery: frequent and dangerous. Atherosclerosis. (2012) 222:191–5. doi: 10.1016/j.atherosclerosis.2012.02.021
44. Janssen H, Wagner CS, Demmer P, Callies S, Sölter G., Loghmani-khouzani H, et al. Acute perioperative-stress-induced increase of atherosclerotic plaque volume and vulnerability to rupture in apolipoprotein-E-deficient mice is amenable to statin treatment and IL-6 inhibition. Dis Model Mech. (2015) 8:1071–80. doi: 10.1242/dmm.018713
45. Larmann J, Theilmeier G. Inflammatory response to cardiac surgery: cardiopulmonary bypass versus non-cardiopulmonary bypass surgery. Best Pract Res Clin Anaesthesiol. (2004) 18:425–38. doi: 10.1016/j.bpa.2003.12.004
46. Silvestre-Roig C, Braster Q, Wichapong K, Lee EY, Teulon JM, Berrebeh N, et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature. (2019) 569:236–40. doi: 10.1038/s41586-019-1167-6
47. Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, et al. Ly-6C hi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. (2007) 117:195–205. doi: 10.1172/JCI29950
48. Levy-Lambert D, Ramly EP, Kantar RS, Alfonso AR, Levine JP. Congestive Heart Failure Predicts Major Complications and Increased Length of Stay in Lower Extremity Pedicled Flap Reconstruction. Plast Reconstr Surg. (2020) 146:790e−795e. doi: 10.1097/PRS.0000000000007363
49. Singer M, Deutschman CS, Seymour CW, Shankar-Hari M, Annane D, Bauer M, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. (2016) 315:801–10. doi: 10.1001/jama.2016.0287
50. Walker AMN, Drozd M, Hall M, Patel PA, Paton M, Lowry J, et al. Prevalence and Predictors of Sepsis Death in Patients With Chronic Heart Failure and Reduced Left Ventricular Ejection Fraction. J Am Heart Assoc. (2018) 7:e009684. doi: 10.1161/JAHA.118.009684
51. Sandoval C, Walter SD, Krueger P, Smieja M, Smith A, Yusuf S, et al. Risk of hospitalization during influenza season among a cohort of patients with congestive heart failure. Epidemiol Infect. (2007) 135:574–82. doi: 10.1017/S095026880600714X
52. Kytomaa S, Hegde S, Claggett B, Udell JA, Rosamond W, Temte J, et al. Association of Influenza-like Illness Activity With Hospitalizations for Heart Failure: The Atherosclerosis Risk in Communities Study. JAMA Cardiol. (2019) 4:363–9. doi: 10.1001/jamacardio.2019.0549
53. Corrales-Medina VF, Alvarez KN, Weissfeld LA, Angus DC, Chirinos JA, Chang CC, et al. Association between hospitalization for pneumonia and subsequent risk of cardiovascular disease. JAMA. (2015) 313:264–74. doi: 10.1001/jama.2014.18229
54. Kumar S, Rizvi M. Serum tumor necrosis factor alpha and C-reactive protein in pediatric patients with sepsis and its correlation with microbiologic findings. Indian J Pathol Microbiol. (2010) 53:494–7. doi: 10.4103/0377-4929.68290
55. Mera S, Tatulescu D, Cismaru C, Bondor C, Slavcovici A, Zanc V, et al. Multiplex cytokine profiling in patients with sepsis. APMIS. (2011) 119:155–63. doi: 10.1111/j.1600-0463.2010.02705.x
56. Kellum JA, Kong L, Fink MP, Weissfeld LA, Yealy DM, Pinsky MR, et al. Understanding the inflammatory cytokine response in pneumonia and sepsis: results of the Genetic and Inflammatory Markers of Sepsis (GenIMS) Study. Arch Intern Med. (2007) 167:1655–63. doi: 10.1001/archinte.167.15.1655
57. Chang C, Hu L, Sun S, Song Y, Liu S, Wang J, et al. Regulatory role of the TLR4/JNK signaling pathway in sepsisinduced myocardial dysfunction. Mol Med Rep. (2021) 23:334. doi: 10.3892/mmr.2021.11973
58. Munzel T, Camici GG, Maack C, Bonetti NR, Fuster V, Kovacic JC. Impact of Oxidative Stress on the Heart and Vasculature: Part 2 of a 3-Part Series. J Am Coll Cardiol. (2017) 70:212–29. doi: 10.1016/j.jacc.2017.05.035
59. Neubauer S. The failing heart–an engine out of fuel. N Engl J Med. (2007) 356:1140–51. doi: 10.1056/NEJMra063052
60. Karwi QG, Uddin GM, Ho KL, Lopaschuk GD. Loss of Metabolic Flexibility in the Failing Heart. Front Cardiovasc Med. (2018) 5:68. doi: 10.3389/fcvm.2018.00068
61. L'Heureux M, Sternberg M, Brath L, Turlington J, Kashiouris MG. Sepsis-Induced Cardiomyopathy: a Comprehensive Review. Curr Cardiol Rep. (2020) 22:35. doi: 10.1007/s11886-020-01277-2
62. Bagshaw SM, Laupland KB, Doig CJ, Mortis G, Fick GH, Mucenski M, et al. Prognosis for long-term survival and renal recovery in critically ill patients with severe acute renal failure: a population-based study. Crit Care. (2005) 9:R700–709. doi: 10.1186/cc3879
63. Wardi G, Wali A, Sell R, Malhotra A, Beitler J. 1482: IMPACT OF FLUID RESUSCITATION ON SEPTIC PATIENTS WITH SYSTOLIC HEART FAILURE. Crit Care Med. (2016) 44:446. doi: 10.1097/01.ccm.0000510156.55790.66
64. Machnik A, Neuhofer W, Jantsch J, Dahlmann A, Tammela T, Machura K, et al. Macrophages regulate salt-dependent volume and blood pressure by a vascular endothelial growth factor-C-dependent buffering mechanism. Nat Med. (2009) 15:545–52. doi: 10.1038/nm.1960
65. Wenstedt EFE, Olde Engberink RH, Rorije NMG, van den Born BH, Claessen N, Aten J, et al. Salt-sensitive blood pressure rise in type 1 diabetes patients is accompanied by disturbed skin macrophage influx and lymphatic dilation-a proof-of-concept study. Transl Res. (2020) 217:23–32. doi: 10.1016/j.trsl.2019.12.001
66. Liu IF, Huang CC, Chan WL, Huang PH, Chung CM, Lin SJ, et al. Effects of annual influenza vaccination on mortality and hospitalization in elderly patients with ischemic heart disease: a nationwide population-based study. Prev Med. (2012) 54:431–3. doi: 10.1016/j.ypmed.2012.03.020
67. Mohseni H, Kiran A, Khorshidi R, Rahimi K. Influenza vaccination and risk of hospitalization in patients with heart failure: a self-controlled case series study. Eur Heart J. (2017) 38:326–33. doi: 10.1093/eurheartj/ehw411
68. Vardeny O, Claggett B, Udell JA, Packer M, Zile M, Rouleau J, et al. Influenza Vaccination in Patients With Chronic Heart Failure: The PARADIGM-HF Trial. JACC Heart Fail. (2016) 4:152–8. doi: 10.1016/j.jchf.2015.10.012
69. Davis MM, Taubert K, Benin AL, Brown DW, Mensah GA, Baddour LM, et al. Influenza vaccination as secondary prevention for cardiovascular disease: a science advisory from the American Heart Association/American College of Cardiology. J Am Coll Cardiol. (2006) 48:1498–502. doi: 10.1016/j.jacc.2006.09.004
70. McDonagh TA, Metra M, Adamo M, Gardner RS, Baumbach A, Bohm M, et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. (2021) 42:3599–726. doi: 10.1093/eurheartj/ehab368
71. Epidemiology Working Group for Ncip Epidemic Response CCfDC, Prevention. [The epidemiological characteristics of an outbreak of 2019 novel coronavirus diseases (COVID-19) in China]. Zhonghua Liu Xing Bing Xue Za Zhi. (2020) 41:145–51. doi: 10.3760/cma.j.issn.0254-6450.2020.02.003
72. Villanueva DH, Lusby HP, Islam SP, Gupte AA, Beatty NL. Heart failure exacerbation as only presenting sign of COVID-19. IDCases. (2020) 21:e00870. doi: 10.1016/j.idcr.2020.e00870
73. Tan L, Wang Q, Zhang D, Ding J, Huang Q, Tang YQ, et al. Lymphopenia predicts disease severity of COVID-19: a descriptive and predictive study. Signal Transduct Target Ther. (2020) 5:33. doi: 10.1038/s41392-020-0148-4
74. Yucel H, Refiker Ege M, Zorlu A, Kaya H, Beton O, Gungor H, et al. Lymphocytopenia is associated with poor NYHA functional class in chronic heart failure patients with reduced ejection fraction. Turk Kardiyol Dern Ars. (2015) 43:427–33. doi: 10.5543/tkda.2015.89439
75. Guo T, Fan Y, Chen M, Wu X, Zhang L, He T, et al. Cardiovascular Implications of Fatal Outcomes of Patients With Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. (2020) 5:811–8. doi: 10.1001/jamacardio.2020.1017
76. Shi S, Qin M, Shen B, Cai Y, Liu T, Yang F, et al. Association of Cardiac Injury With Mortality in Hospitalized Patients With COVID-19 in Wuhan, China. JAMA Cardiol. (2020) 5:802–10. doi: 10.1001/jamacardio.2020.0950
77. Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19). Front Immunol. (2020) 11:827. doi: 10.3389/fimmu.2020.00827
78. Liao M, Liu Y, Yuan J, Wen Y, Xu G, Zhao J, et al. Single-cell landscape of bronchoalveolar immune cells in patients with COVID-19. Nat Med. (2020) 26:842–4. doi: 10.1038/s41591-020-0901-9
79. Sohn KM, Lee SG, Kim HJ, Cheon S, Jeong H, Lee J, et al. COVID-19 Patients Upregulate Toll-like Receptor 4-mediated Inflammatory Signaling That Mimics Bacterial Sepsis. J Korean Med Sci. (2020) 35:e343. doi: 10.3346/jkms.2020.35.e343
80. Del Valle DM, Kim-Schulze S, Huang HH, Beckmann ND, Nirenberg S, Wang B, et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat Med. (2020) 26:1636–43. doi: 10.1038/s41591-020-1051-9
81. McElvaney OJ, McEvoy NL, McElvaney OF, Carroll TP, Murphy MP, Dunlea DM, et al. Characterization of the Inflammatory Response to Severe COVID-19 Illness. Am J Respir Crit Care Med. (2020) 202:812–21. doi: 10.1164/rccm.202005-1583OC
82. Coomes EA, Haghbayan H. Interleukin-6 in Covid-19: a systematic review and meta-analysis. Rev Med Virol. (2020) 30:1–9. doi: 10.1002/rmv.2141
83. Schultheiß C, Willscher E, Paschold L, Gottschick C, Klee B, Glasauer S, et al. From online data collection to identification of disease mechanisms: the IL-1ß, IL-6 and TNF-α cytokine triad is associated with post-acute sequelae of COVID-19 in a digital research cohort. SSRN Electron J. (2021). doi: 10.2139/ssrn.3963839
84. Hoffmann M, Kleine-Weber H, Schroeder S, Kruger N, Herrler T, Erichsen S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. (2020) 181:271–280 e278. doi: 10.1016/j.cell.2020.02.052
85. Zhou L, Niu Z, Jiang X, Zhang Z, Zheng Y, Wang Z, et al. Systemic analysis of tissue cells potentially vulnerable to SARS-CoV-2 infection by the protein-proofed single-cell RNA profiling of ACE2, TMPRSS2 and Furin proteases. bioRxiv [Preprint]. (2020) bioRxiv: 2020.04.06.028522. doi: 10.1101/2020.04.06.028522
86. Ziegler CGK, Allon SJ, Nyquist SK, Mbano IM, Miao VN, Tzouanas CN, et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell. (2020) 181:1016–1035 e1019. doi: 10.1016/j.cell.2020.04.035
87. Ferrario CM, Jessup J, Chappell MC, Averill DB, Brosnihan KB, Tallant EA, et al. Effect of angiotensin-converting enzyme inhibition and angiotensin II receptor blockers on cardiac angiotensin-converting enzyme 2. Circulation. (2005) 111:2605–10. doi: 10.1161/CIRCULATIONAHA.104.510461
88. Patel VB, Zhong JC, Grant MB, Oudit GY. Role of the ACE2/Angiotensin 1-7 Axis of the Renin-Angiotensin System in Heart Failure. Circ Res. (2016) 118:1313–26. doi: 10.1161/CIRCRESAHA.116.307708
89. Meng J, Xiao G, Zhang J, He X, Ou M, Bi J, et al. Renin-angiotensin system inhibitors improve the clinical outcomes of COVID-19 patients with hypertension. Emerg Microbes Infect. (2020) 9:757–60. doi: 10.1080/22221751.2020.1746200
90. Bozkurt B, Kovacs R, Harrington B. Joint HFSA/ACC/AHA Statement Addresses Concerns Re: Using RAAS Antagonists in COVID-19. J Card Fail. (2020) 26:370. doi: 10.1016/j.cardfail.2020.04.013
91. Schurink B, Roos E, Radonic T, Barbe E, Bouman CSC, de Boer HH, et al. Viral presence and immunopathology in patients with lethal COVID-19: a prospective autopsy cohort study. Lancet Microbe. (2020) 1:e290–9. doi: 10.1016/S2666-5247(20)30144-0
92. Dal Ferro M, Bussani R, Paldino A, Nuzzi V, Collesi C, Zentilin L, et al. SARS-CoV-2, myocardial injury and inflammation: insights from a large clinical and autopsy study. Clin Res Cardiol. (2021) 110:1694. doi: 10.1007/s00392-021-01919-7
93. Santos RC, Figueiredo VN, Martins LC, Moraes Cde H, Quinaglia T, Boer-Martins L, et al. Infliximab reduces cardiac output in rheumatoid arthritis patients without heart failure. Rev Assoc Med Bras. (2012) 58:698–702. doi: 10.1590/S0104-42302012000600015
94. Chung ES, Packer M, Lo KH, Fasanmade AA, Willerson JT, Anti TNFTACHFI. Randomized, double-blind, placebo-controlled, pilot trial of infliximab, a chimeric monoclonal antibody to tumor necrosis factor-alpha, in patients with moderate-to-severe heart failure: results of the anti-TNF Therapy Against Congestive Heart Failure (ATTACH) trial. Circulation. (2003) 107:3133–40. doi: 10.1161/01.CIR.0000077913.60364.D2
95. Mann DL, McMurray JJ, Packer M, Swedberg K, Borer JS, Colucci WS, et al. Targeted anticytokine therapy in patients with chronic heart failure: results of the Randomized Etanercept Worldwide Evaluation (RENEWAL). Circulation. (2004) 109:1594–602. doi: 10.1161/01.CIR.0000124490.27666.B2
96. Lv S, Han M, Yi R, Kwon S, Dai C, Wang R. Anti-TNF-alpha therapy for patients with sepsis: a systematic meta-analysis. Int J Clin Pract. (2014) 68:520–8. doi: 10.1111/ijcp.12382
97. Fakharian A, Barati S, Mirenayat M, Rezaei M, Haseli S, Torkaman P, et al. Evaluation of adalimumab effects in managing severe cases of COVID-19: A randomized controlled trial. Int Immunopharmacol. (2021) 99:107961. doi: 10.1016/j.intimp.2021.107961
98. Everett BM, Cornel JH, Lainscak M, Anker SD, Abbate A, Thuren T, et al. Anti-Inflammatory Therapy With Canakinumab for the Prevention of Hospitalization for Heart Failure. Circulation. (2019) 139:1289–99. doi: 10.1161/CIRCULATIONAHA.118.038010
99. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N Engl J Med. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
100. Caricchio R, Abbate A, Gordeev I, Meng J, Hsue PY, Neogi T, et al. Effect of Canakinumab vs Placebo on Survival Without Invasive Mechanical Ventilation in Patients Hospitalized With Severe COVID-19: A Randomized Clinical Trial. JAMA. (2021) 326:230–9. doi: 10.1001/jama.2021.9508
101. Buckley LF, Carbone S, Trankle CR, Canada JM, Erdle CO, Regan JA, et al. Effect of Interleukin-1 Blockade on Left Ventricular Systolic Performance and Work: A Post Hoc Pooled Analysis of 2 Clinical Trials. J Cardiovasc Pharmacol. (2018) 72:68–70. doi: 10.1097/FJC.0000000000000591
102. Shakoory B, Carcillo JA, Chatham WW, Amdur RL, Zhao H, Dinarello CA, et al. Interleukin-1 Receptor Blockade Is Associated With Reduced Mortality in Sepsis Patients With Features of Macrophage Activation Syndrome: Reanalysis of a Prior Phase III Trial. Crit Care Med. (2016) 44:275–81. doi: 10.1097/CCM.0000000000001402
103. Investigators TR-C, Derde LPG. Effectiveness of tocilizumab, sarilumab, and anakinra for critically ill patients with covid-19 the remap-cap covid-19 immune modulation therapy domain randomized clinical trial. medRxiv [Preprint]. (2021). medRxiv: 2021.06.18.21259133. doi: 10.1101/2021.06.18.21259133
104. Yano T, Osanami A, Shimizu M, Katano S, Nagano N, Kouzu H, et al. Utility and safety of tocilizumab in Takayasu arteritis with severe heart failure and muscle wasting. ESC Heart Fail. (2019) 6:894–7. doi: 10.1002/ehf2.12487
105. Meyer MAS, Wiberg S, Grand J, Meyer ASP, Obling LER, Frydland M, et al. Treatment Effects of Interleukin-6 Receptor Antibodies for Modulating the Systemic Inflammatory Response After Out-of-Hospital Cardiac Arrest (The IMICA Trial): A Double-Blinded, Placebo-Controlled, Single-Center, Randomized, Clinical Trial. Circulation. (2021) 143:1841–51. doi: 10.1161/CIRCULATIONAHA.120.053318
106. Chitturi KR, Thacker S, Al-Saadi MA, Kassi M. Successful treatment of acute heart failure in COVID-19-induced cytokine storm with tocilizumab: a case report. Eur Heart J Case Rep. (2020) 4:1–6. doi: 10.1093/ehjcr/ytaa188
107. Toniati P, Piva S, Cattalini M, Garrafa E, Regola F, Castelli F, et al. Tocilizumab for the treatment of severe COVID-19 pneumonia with hyperinflammatory syndrome and acute respiratory failure: A single center study of 100 patients in Brescia, Italy. Autoimmun Rev. (2020) 19:102568. doi: 10.1016/j.autrev.2020.102568
108. Rochwerg B, Agarwal A, Siemieniuk RA, Agoritsas T, Lamontagne F, Askie L, et al. A living WHO guideline on drugs for covid-19. BMJ. (2020) 370:m3379. doi: 10.1136/bmj.m3379
109. Ibrahim YF, Moussa RA, Bayoumi AMA, Ahmed AF. Tocilizumab attenuates acute lung and kidney injuries and improves survival in a rat model of sepsis via down-regulation of NF-kappaB/JNK: a possible role of P-glycoprotein. Inflammopharmacology. (2020) 28:215–30. doi: 10.1007/s10787-019-00628-y
110. Torre-Amione G, Anker SD, Bourge RC, Colucci WS, Greenberg BH, Hildebrandt P, et al. Results of a non-specific immunomodulation therapy in chronic heart failure (ACCLAIM trial): a placebo-controlled randomised trial. Lancet. (2008) 371:228–36. doi: 10.1016/S0140-6736(08)60134-8
111. Torre-Amione G, Sestier F, Radovancevic B, Young J. Effects of a novel immune modulation therapy in patients with advanced chronic heart failure: results of a randomized, controlled, phase II trial. J Am Coll Cardiol. (2004) 44:1181–6. doi: 10.1016/j.jacc.2004.06.047
112. Stockmann H, Keller T, Buttner S, Jorres A, Kindgen-Milles D, Kunz JV, et al. CytoResc - “CytoSorb” Rescue for critically ill patients undergoing the COVID-19 Cytokine Storm: a structured summary of a study protocol for a randomized controlled trial. Trials. (2020) 21:577. doi: 10.1186/s13063-020-04501-0
113. Supady A, Weber E, Rieder M, Lother A, Niklaus T, Zahn T, et al. Cytokine adsorption in patients with severe COVID-19 pneumonia requiring extracorporeal membrane oxygenation (CYCOV): a single centre, open-label, randomised, controlled trial. Lancet Respir Med. (2021) 9:755–62. doi: 10.1016/S2213-2600(21)00177-6
114. Rimmele T, Kellum JA. Clinical review: blood purification for sepsis. Crit Care. (2011) 15:205. doi: 10.1186/cc9411
115. Zuccari S, Damiani E, Domizi R, Scorcella C, D'Arezzo M, Carsetti A, et al. Changes in Cytokines, Haemodynamics and Microcirculation in Patients with Sepsis/Septic Shock Undergoing Continuous Renal Replacement Therapy and Blood Purification with CytoSorb. Blood Purif. (2020) 49:107–13. doi: 10.1159/000502540
116. Heming N, Sivanandamoorthy S, Meng P, Bounab R, Annane D. Immune Effects of Corticosteroids in Sepsis. Front Immunol. (2018) 9:1736. doi: 10.3389/fimmu.2018.01736
117. Group RC, Horby P, Lim WS, Emberson JR, Mafham M, Bell JL, et al. Dexamethasone in Hospitalized Patients with Covid-19. N Engl J Med. (2021) 384:693–704. doi: 10.1056/NEJMoa2021436
118. Fishel RS, Eisenberg S, Shai SY, Redden RA, Bernstein KE, Berk BC. Glucocorticoids induce angiotensin-converting enzyme expression in vascular smooth muscle. Hypertension. (1995) 25:343–9. doi: 10.1161/01.HYP.25.3.343
119. Garg R, Yusuf S. Overview of randomized trials of angiotensin-converting enzyme inhibitors on mortality and morbidity in patients with heart failure. Collaborative Group on ACE Inhibitor Trials. JAMA. (1995) 273:1450–6. doi: 10.1001/jama.1995.03520420066040
120. McMurray JJ, Packer M, Desai AS, Gong J, Lefkowitz MP, Rizkala AR, et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N Engl J Med. (2014) 371:993–1004. doi: 10.1056/NEJMoa1409077
121. Miro O, Takagi K, Gayat E, Llorens P, Martin-Sanchez FJ, Jacob J, et al. CORT-AHF Study: Effect on Outcomes of Systemic Corticosteroid Therapy During Early Management Acute Heart Failure. JACC Heart Fail. (2019) 7:834–45. doi: 10.1016/j.jchf.2019.04.022
Keywords: heart failure, inflammation, acute relapses, immunology, macrophages, infection, surgical stress response
Citation: Hasselbach L, Weidner J, Elsässer A and Theilmeier G (2022) Heart Failure Relapses in Response to Acute Stresses – Role of Immunological and Inflammatory Pathways. Front. Cardiovasc. Med. 9:809935. doi: 10.3389/fcvm.2022.809935
Received: 05 November 2021; Accepted: 23 March 2022;
Published: 25 April 2022.
Edited by:
Henrike Janssen, Heidelberg University Hospital, GermanyReviewed by:
Matthias Derwall, University Hospital RWTH Aachen, GermanyMartin Schick, Medical Center—University of Freiburg, Germany
Copyright © 2022 Hasselbach, Weidner, Elsässer and Theilmeier. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lisa Hasselbach, bGlzYS5oYXNzZWxiYWNoQHVuaS1vbGRlbmJ1cmcuZGU=