
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med., 04 April 2022
Sec. Cardiovascular Genetics and Systems Medicine
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.798033
This article is part of the Research TopicGenomic Architecture of Cardiovascular DiseasesView all 15 articles
 Rui Peng1,2†
Rui Peng1,2† Binbin Li3,4†
Binbin Li3,4† Shuxia Chen1,2
Shuxia Chen1,2 Zhiwen Shi1,2Liwei Yu1,5
Zhiwen Shi1,2Liwei Yu1,5 Yunqian Gao1,6
Yunqian Gao1,6 Xueyan Yang1,2
Xueyan Yang1,2 Lei Lu1,2*
Lei Lu1,2* Hongyan Wang1,2*
Hongyan Wang1,2*The Glioma-associated oncogene (Gli) family members of zinc finger DNA-binding proteins are core effectors of Sonic hedgehog (SHH) signaling pathway. Studies in model organisms have identified that the Gli genes play critical roles during organ development, including the heart, brain, kidneys, etc. Deleterious mutations in GLI genes have previously been revealed in several human developmental disorders, but few in congenital heart disease (CHD). In this study, the mutations in GLI1-3 genes were captured by next generation sequencing in human cohorts composed of 412 individuals with CHD and 213 ethnically matched normal controls. A total of 20 patient-specific nonsynonymous rare mutations in coding regions of human GLI1-3 genes were identified. Functional analyses showed that GLI1 c.820G> T (p.G274C) is a gain-of-function mutation, while GLI1 c.878G>A (p.R293H) and c.1442T>A (p.L481X) are loss-of-function mutations. Our findings suggested that deleterious rare mutations in GLI1 gene broke the balance of the SHH signaling pathway regulation and may constitute a great contribution to human CHD, which shed new light on understanding genetic mechanism of embryo cardiogenesis regulated by SHH signaling.
Congenital heart disease (CHD) is the most common developmental anomaly and the leading non-infectious cause of mortality in newborns (1). In China, the incidence of CHD has been increased from 0.201‰ in 1980–1984 to 4.905‰ in 2015–2019 (2). Ventricular septal defect (VSD) and atrial septal defect (ASD) are the most common congenital heart defects subtypes among the offspring. The etiology of CHD is complicated, and genetic factors play important roles in CHD occurrence (3–5). However, identification of these factors has been historically slow due to technical limitations and short understanding of signaling pathways regulating embryonic cardiovascular development (5, 6).
In mammals, the Hedgehog (HH) family genes of Sonic hedgehog (SHH), Indian hedgehog (IHH) and Desert hedgehog (DHH) encode evolutionarily conserved ligand proteins initiating pathways crucial for embryogenesis (7). The SHH signaling pathway is transduced by the seven-transmembrane G-protein-coupled receptor (GPCR)-like protein Smoothened (Smo), leading to the activation of Glioma-associated oncogene (Gli) family of transcription factors and downstream target genes transcription (8–10). In vertebrates SHH signaling pathway, Gli gene family contains three members of Gli1, Gli2, and Gli3 (11). Gli1 protein acts as a transcriptional activator and provides a positive feedback loop of signaling, whereas Gli2 and Gli3 serve as both transcriptional activators and repressors, depending on specific post-translational modifications and proteolysis processes (12, 13).
SHH signaling pathway has recently been implicated in the specification of early embryonic cardiac progenitor fate. SHH signaling specifies atrial septum from non-septum atrial progenitors (14). Dysregulated SHH signaling pathway involves in numerous human diseases, including birth defects and cancers (15). Various cardiac malformations are observed in Shh−/− mouse embryos (16). The critical roles of GLI1-3 in embryonic development have been well established (17, 18). GLI1 participates in differentiation and development of several organs in humans through SHH signaling pathway (19, 20). According to a recent study, GLI1 nonsynonymous variants were identified among 25 heterotaxy syndrome (HS) patients with CHD (21). Moreover, Gli2−/− and Gli3+/− double mutant mice show a full complement of VACTERL syndrome including cardiac defects (22). These results raise the possibilities that functional rare mutations in GLI1-3 may also affect human embryo cardiogenesis.
To understand the association between GL1-3 variants and the risk of CHD in humans, we screened 5′-untranslated region (UTR), 3′-UTR and coding regions of GLI1-3 genes in a Chinese cohort with 412 cases and 213 matched controls by next generation sequencing. As a result, a total of 20 patient-specific nonsynonymous rare mutations in coding regions of human GLI1-3 genes were identified. Our in vitro and in vivo functional analyses showed that GLI1 c.820G>T (p.G274C) is a gain-of-function mutation, GLI1 c.878G>A (p.R293H) and c.1442T>A (p.L481X) are loss-of-function mutations. Thus, our data implicate the association between dysregulated SHH signaling pathway and CHD occurrence, and broaden the current knowledge of human embryonic cardiogenesis.
Sample collection was performed as described previously (23–25). Blood samples from 412 CHD patients (mean age 2.9 ± 2.7 years, 55.6% males) were collected from the Cardiovascular Disease Institute of Jinan Military Command (Jinan, China). The patients were diagnosed by echocardiography, and some were further confirmed surgically. Patients with clinical features of developmental anomalies, positive family history of CHD in a first-degree relative, maternal diabetes mellitus, maternal exposure to known teratogens or any therapeutic drugs during gestation were excluded. The 213 controls (mean age 7.1 ± 3.7 years, 49.8% males) were ethnically and gender-matched, unrelated healthy volunteers recruited from the same geographical area. Sample collection and protocols used in this study were reviewed and approved by the Ethics Committee of the School of Life Sciences, Fudan University and local ethics committees before the start of the present study. All procedures were in accordance with the Declaration of Helsinki. Informed consents were signed by the parents or guardians of the children. Detailed information of the samples was shown in Table 1.
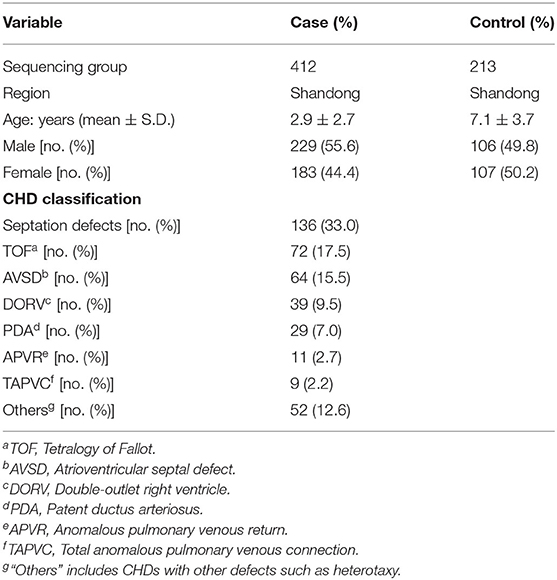
Table 1. Information of CHD cases and controls.
Peripheral blood samples were collected. Genomic DNA was extracted and target-sequenced was conducted as described previously (23–25). The genomic structures of human GLI1-3 genes were determined using NCBI Genebank (mRNA references are NM_001166045, NM_005270, NM_000168, respectively). The 5′ -UTR, 3′ -UTR and coding regions in GLI1-3 were screened. Identified variants were filtered using the dbSNP database (http://www.ncbi.nlm.nih.gov/projects/SNP), the 1000 genomes projects (http://www.1000genomes.org/), the Genome Aggregation Database (gnomAD, http://gnomad-sg.org/), and the HuaBiao Database (https://www.biosino.org/wepd) (26). All the patient-specific nonsynonymous mutations were confirmed by Sanger sequencing, the PCR primers were listed in Supplementary Table S1.
Human GLI1-3 ORF without stop codon were amplified by PCR (Supplementary Table S2) using cDNA reverse transcripted from human total RNA. GLI1 and GLI3 were subcloned into SgfI/MluI restriction enzyme sites. GLI2 was subcloned into SgfI/NotI restriction enzyme sites of pCMV6-AC-HA (Origene, #PS100004). All plasmid were verified by Sanger sequencing. We then performed the site-directed mutagenesis using QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent, #210518) according to manufacturer's instruction. For GLI1 c.1442T>A (p.L481X) stop-gain mutation, sequence after premature stop codon (including stop codon) was removed for C-terminal tag fusion. The pGMGLI-Lu firefly luciferase SHH signaling pathway reporter plasmid was obtained from Genomeditech (#GM-021024). The constitutive Renilla luciferase reporter pRL-TK was from Promega (#E6921).
HEK 293T cells (ATCC, #CRL-3216) and HeLa cells (ATCC, #CCL-2) were cultured in high-glucose Dulbecco's Modified Eagle Medium (DMEM, Thermo Fisher Scientific, #11995065) supplemented with 10% FBS (Thermo Fisher Scientific, #A3840001) at 37°C with 5% CO2. The cells were seeded and maintained overnight to reach ~80% confluency at the time of transfection by Lipofectamine 2000 Transfection Reagent (Thermo Fisher Scientific, #11668019) and Lipofectamine 3000 Transfection Reagent (Thermo Fisher Scientific, #L3000015) separately according to manufacturer's protocols.
Dual-luciferase reporter assay was performed as described previously (24, 27). Briefly, in a 24-well plate well, 300 ng of empty vector or GLI1 wild-type/mutant constructs, 200 ng of pGMGLI-Lu firefly luciferase reporter plasmid, 10 ng of Renilla luciferase plasmid serving as an internal control were transfected. Reporter assay was performed with the Dual-Luciferase Reporter Assay System (Promega, #E1910) on GloMax Navigator Microplate Luminometer (Promega, #GM2010) 24 h post transfection. At least three independent biological repeats were performed, and data were presented as the mean ± S.D. P-values were calculated by Student's t-test and considered significant when <0.05.
C-terminal HA-tagged wild-type or mutant GLI1 expressing plasmids were transfected into HEK293T cells. Forty-eight hours later, cells were lysed with Cell lysis buffer for Western and IP (Beyotime, #P0013) containing a cocktail of protease inhibitors (ThermoFisher Scientific, #1862209) and heated for 10 min at 100°C. Cell lysates were loaded and separated on a 10% SDS-polyacrylamide gel electrophoresis and transferred onto a PVDF membrane (Merck Millipore, #ISEQ00010). After blocking for 1 h with 5% non-fat milk, the membrane was incubated with mouse HA-tag antibody (Proteintech, #66006-2-Ig) and mouse anti-GAPDH antibody (Proteintech, #66004-1-Ig) at 4°C overnight. Horseradish peroxidase-conjugated anti-mouse IgG was served as secondary antibody (Cell signaling Technology, #7076) for 2 h at room temperature and visualized through the ECL Detection System (Tanon, Shanghai, China) (25). Three independent experiments were performed and representative results were shown.
The probes (F: 5′-AGCTACCTGGGTGGTCTCT-3′, R: 5′-TCGAAGAGACCACCCAGGT-3′) were designed according to the consensus GLI-binding site (5′-TGGGTGGTC-3′) from PTCH1 promoter region (Supplementary Table S3) (28). The DNA-binding affinity of the different GLI proteins was determined by EMSA according to previous description (29). Briefly, nuclear extracts were prepared from HEK 293T cells transfected with empty vector or GLI1 wild-type/mutation constructs using the NE-PER Nuclear and Cytoplasmic Extraction kit (ThermoFisher Scientific, #78835) and stored at −80°C before use. Protein concentrations were determined by Pierce BCA Protein Assay Kit (ThermoFisher Scientific, #23227). Nuclear proteins were mixed and incubated with indicated probes and subsequently processed using LightShift Chemiluminescent EMSA Kit (ThermoFisher Scientific, #20148) following manufacturer's instruction.
HeLa cells were plated into 35 mm glass bottom dishes and cultured overnight to reach ~80% confluency. Then cells were transiently transfected with HA-tagged GLI1 wild-type, p.G274C, p.R293H, or p.L481X constructs. 24 h later, cells were fixed in 4% PFA for 15 min at room temperature, then immunofluorescence was performed using HA antibody (MilliporeSigma, #H6908) as described previously (25), and images were captured on a Zeiss LSM700 confocal microscope under 40x objective lens. Experiment was repeated at least in triplicates and representative result was presented.
Wild-type AB and Cmlc2-mCherry strain zebrafish (Danio rerio) were maintained under standard conditions (25, 30). All plasmids of the empty vector, GLI1 wild-type, p.G274C, p.R293H, and p.L481X were extracted by Endo-free Mini Plasmid Kit II (Tiangen Biotech, #DP118), and diluted in nuclease-free water. In this study, each plasmid was injected into >200 zebrafish embryos at 1–2 cell stage. From each plasmid 2–3 nl was injected at a concentration of 40 ng/μl. 72 h post injection, photographs were taken using Leica MZ95 stereo microscope (30) or Olmpus IX83 fluorescence inverted microscope and the percentages of pericardial abnormal embryos were calculated (25). Phenotype distribution differences compared with wild-type group were calculated using χ2 analysis (25).
A Chinese CHD cohort with ethnically and gender-matched healthy controls was used in this study. The 5′-UTR, 3′-UTR and coding regions of GLI1-3 genes were sequenced in all patients and controls. CHD is one of severe disorders that impacts mortality and reproductive fitness. A very large negative selection eliminates highly deleterious common mutations from the human population. Thus the existing common variants like single nucleotide polymorphism (SNP) may make less contributions to this disease. Therefore, it is most likely that novel nonsynonymous rare mutations make significant contributions to population prevalence of this defect, especially for sporadic cases. Based on this hypothesis, we sorted out the variants with minor allele frequencies (MAF) < 1% as rare variants.
A total of 94 variants in the GLI1-3 genes were identified in CHD patients (these data were not shown). 20 nonsynonymous patient-specific rare mutations were identified (Table 2, Figure 1) and further confirmed by Sanger sequencing (Supplementary Figure S1). The effect of rare missense variants on the protein function was predicted in silico using SIFT, PolyPhen-2 and CADD (Table 2) (31–34). We also evaluated the evolutionary conservativeness of these mutations with Genomic Evolutionary Rate Profiling (GERP, the higher of the GERP score means that the site is more conserved) (Table 2) (35), as well as alignment assay across several species (Supplementary Figure S2). For GLI1 gene, we identified one nonsense stop-gain rare mutation and five missense rare mutations. Of which, the stop-gain mutation of c.1442T>A (p.L481X) leads to loss of several crucial domains including the transcriptional activation domain. Mutations of c.820G>T (p.G274C), c.878G>A (p.R293H), c.1776A>T (p.R592S) and c.1924C>T (p.R642S) are predicted as harmful mutations by both SIFT and PolyPhen-2. In addition, mutations of p.G274C and p.R293H locate in the zinc finger domain, which probably interfere with the protein-DNA affinity.
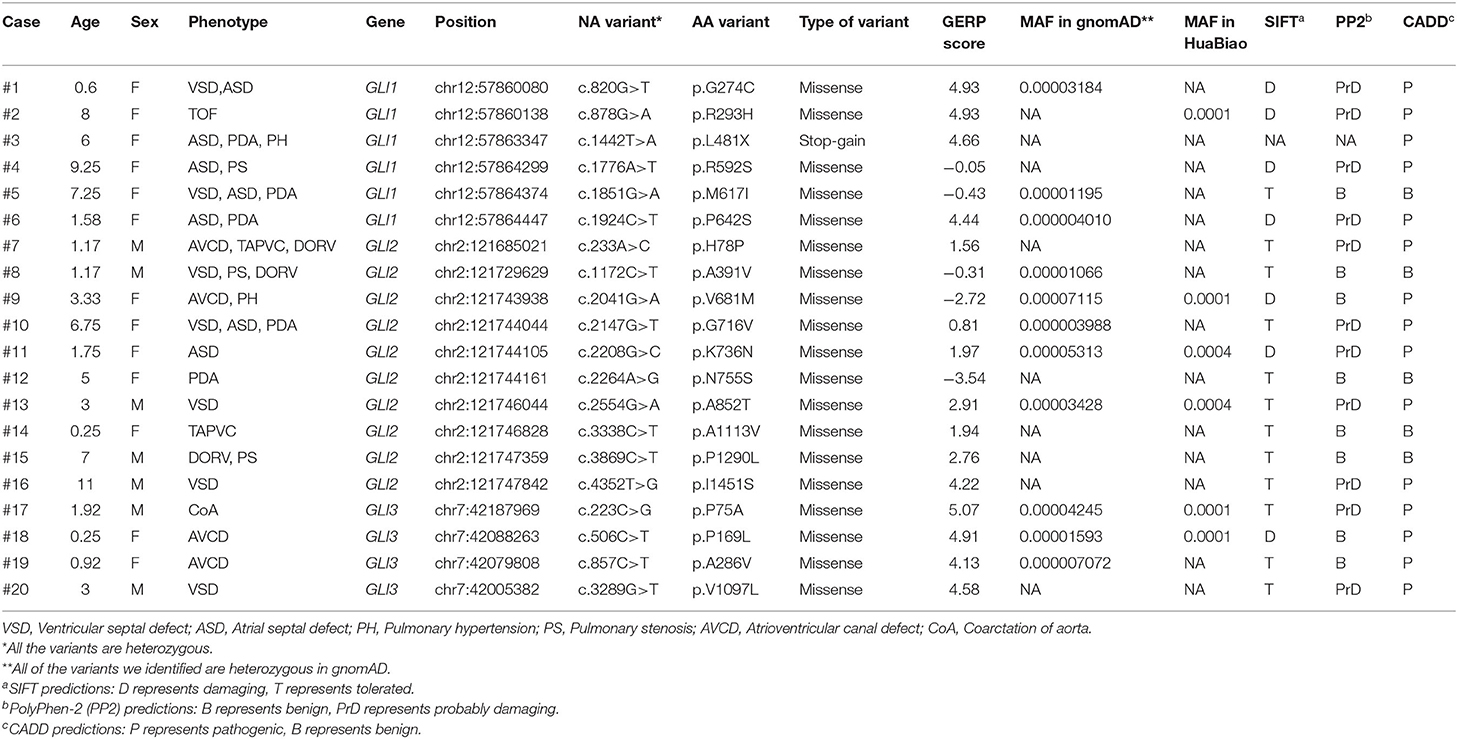
Table 2. Bioinformatics analysis of patient-specific nonsynonymous rare mutations identified in GLI1-3.
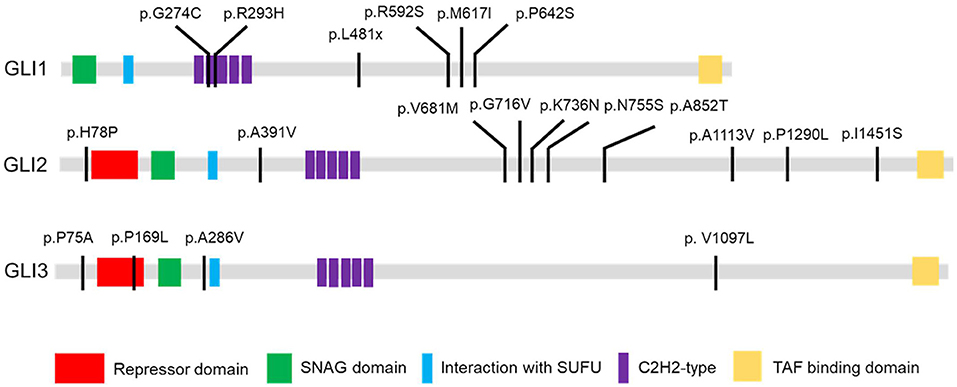
Figure 1. GLI1-3 mutations were identified in CHD patients. Schematic diagram of case-specific mutations in GLI1, GLI2 and GLI3 protein.
As GLI1 protein only serves as transcriptional activator whereas GLI2 and GLI3 can be processed into both transcriptional activator and repressor under specific condition, we first put our attentions on studying GLI1 mutations. To explore the effect of these nonsynonymous rare variants of GLI1 on regulating SHH signaling pathway, we conducted dual-luciferase reporter assay using pGMGLI Lu reporter containing GLI binding sites upstream of firefly luciferase, and Renilla luciferase plasmid as internal control. As expected, wild-type GLI1 dramatically activates the signaling compared with the empty vector group, whereas the p.G274C mutation shows increased activation, in contrast, p.R293H and p.L481X result in significantly decreased activation of the signaling in HEK293T cells. The result indicates that p.G274C is a gain-of-function mutation, whereas p.R293H and p.L481X are loss-of-function mutations. As predicted, the nonsense mutation p.L481X generating a truncated protein (Supplementary Figure S3) almost completely loses its ability in SHH signaling pathway activation (Figure 2).
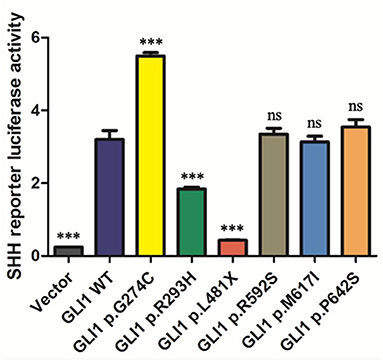
Figure 2. The statistical analyses of luciferase relative activity for HEK293T cells co-transfected with expressing plasmids as indicated and Sonic hedgehog (SHH) signaling pathway reporter plasmids. Activation of SHH signaling pathway pGMGLI-Lu firefly luciferase reporter by human GLI1 wild-type or mutants 24 h post-transfection in HEK 293T cells. Constitutively expressed Renilla Reniformis Luciferase served as an internal control (n ≥ 3, *P < 0.05, **P < 0.01, ***P < 0.001, ns, not significant, compared with wild-type group).
As there is no doubt that the nonsense mutation of p.L481X disrupts protein function due to the loss of large portion of several critical domains (Figure 1), we focused on the other two harmful missense mutations of p.G274C and p.R293H as indicated above. Immunoblot analysis revealed no significant protein stability change made by two mutations (Supplementary Figure S3). Both mutations are mapped into the zinc finger domain of GLI1 protein (Figure 1), raising the possibility that they probably affect the protein binding ability with DNA. To this end, we performed electrophoretic mobility shift assay (EMSA) using nuclear extract from GLI1 wild-type/mutation constructs transfected cells and synthesized probes containing GLI protein bind motif from PTCH1 promoter. The result demonstrates that, the mutation of p.G274C results in significantly higher binding ability with GLI-Probe than wild-type (Figure 3), which probably accounts for its enhanced SHH signaling activation. Slightly higher binding ablility with GLI-Probe was also found in p.R293H (Figure 3).
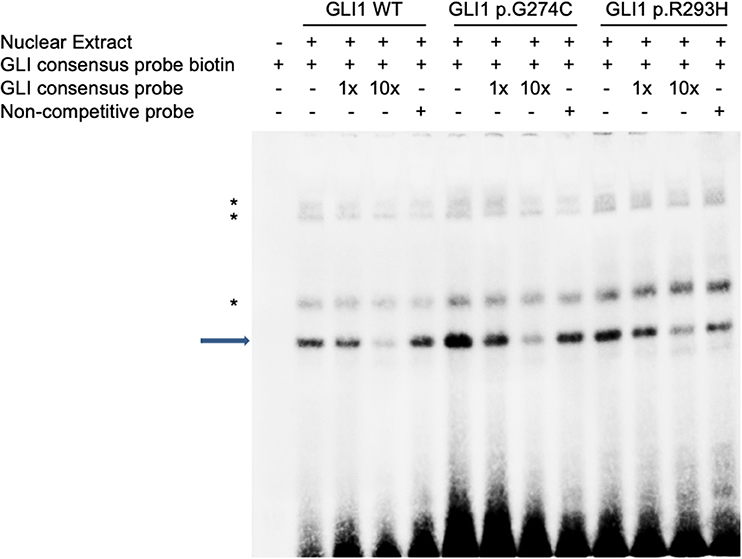
Figure 3. GLI1 mutations affect protein binding affinity with DNA in Electrophoretic Mobility Shift Assay (EMSA). HEK293T cells were transfected with wild-type or mutant GLI1 expression constructs. Nuclear extract was prepared and incubated with specific DNA oligos as indicated. Arrow highlights specific GLI1-DNA binding signal. Asterisks represent non-specific signals.
GLI1 protein has two putative nuclear localization signals (NLS), including a monopartite signal NLS1 (aa 79-84) and a bipartite signal NLS2 (aa 383-401) (36). In addition, GLI1 possesses a leucine-rich nuclear export signal (NES) (aa 496-504), which is fully conserved in other vertebrate species (36, 37). In order to evaluate the functional consequences of the mutations, HeLa cells were transfected using Lipofectamine 3000 with various GLI1 plasmid constructs. No significant differences in level of expression were detected between these GLI1 variants. Both proteins were readily detectable in the nucleus of transfected cells (Figure 4). However, compared to wild-type GLI1 protein, p.L481X leads to more accumulation in the nuclei, whereas p.G274C and p.R293H don't affect protein subcellular localization obviously.
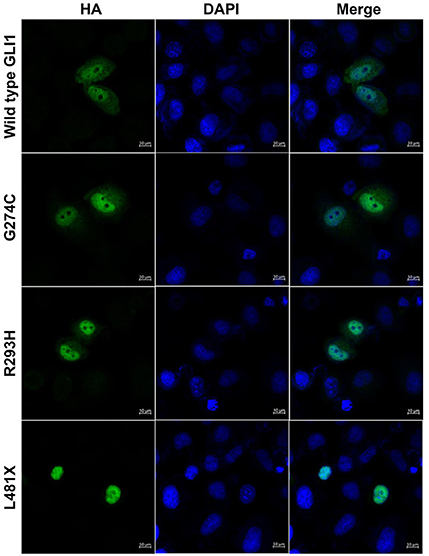
Figure 4. GLI1 p.L481X affect protein subcellular localization. HeLa cells were transfected with HA-tagged wild-type or mutant GLI1 expression constructs. Immunofluorescence was performed using anti-HA antibody (Green). Nuclei were counterstained with Hoechst (Blue). Scale bars are 10 μm.
The Sonic hedgehog (SHH) signaling pathway is evolutionarily conserved and plays critical roles in organogenesis, particularly of the embryonic heart. Both elevated and repressed core factors in signal transduction will cause failure of heart development as precise spacial and temporal signaling regulation is crucial (38, 39). Hence, we performed plasmid microinjection into one to two cell stage fertilized zebrafish embryos to evaluate the dominant negative teratogenic effect of human GLI1 overexpression on zebrafish cardiogenesis. Indeed, we observed multiple cardiovascular abnormalities as a result of wild-type human GLI1 gene overexpression in zebrasish embryos, including malformations on atrium, ventricle, atrioventricular septal and heart blood flow and beat rate (Figure 5C, Supplementary Videos S1–S5). We then calculated the enlarged pericardium (Figure 5A) which is a common phenotype of zebrafish heart malformations (25, 40–42) to evaluate GLI1 function alteration by mutations. As shown in Figure 5B, compared to the uninjected and empty vector groups, p.G274C resulted in the most heart malformation occurence, indicating its enhanced protein function over the wild-type form. Mutations p.R293H and p.L481X generated less abnormal embryos than wild-type GLI1 protein, showed the protein function loss by those two mutations. This result further confirmed that the function of GLI1 gene was seriously affected by those three mutations.
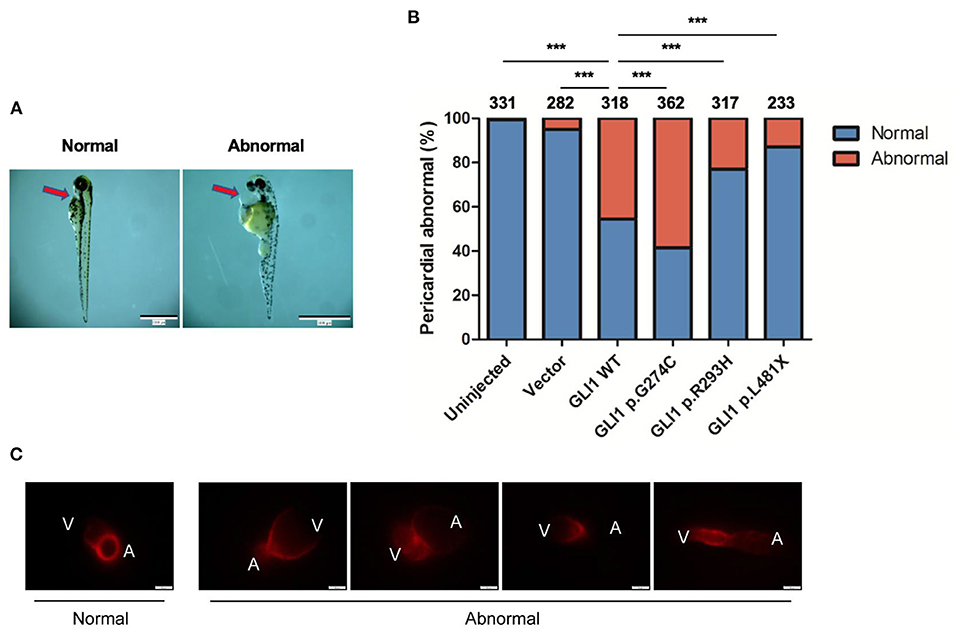
Figure 5. Effect of overexpressed human GLI1 wild-type or mutant on zebrafish heart development. Empty vector or GLI1 wild-type/mutant expression construct were injected into 1-2 cell stage fertilized zebrafish embryos, and the teratogenic effect in zebrafish cardiogenesis was calculated 72 h post injection. (A) Red arrows indicate the pericardial cavity of zebrafish embryos. (B) The frequencies of pericardial abnormalities. (n ≥ 200, *P < 0.05, **P < 0.01, ***P < 0.001, compared with wild-type group). (C) Phenotype of Cmlc2-mCherry zebrafish after overexpressing wild-type human GLI1. GLI1 wild-type expression construct were injected into 1-2 cell stage fertilized Cmlc2-mCherry zebrafish embryos. Images were captured with Olmpus IX83 72 hours post injection. V, ventricle; A, atrium.
SHH signaling exists in a variety of animals and plays a fundamental role in regulating accurate organization of the body plan, and its abnormal activation occurs in a variety of tumor cells (17). The GLI proteins are the main effectors of SHH signal and characterized as DNA-binding transcription factors (11, 43). The critical roles of GLI1-3 genes in embryonic development has been well established and is manifested by the clinical features of patients with mutations in these genes (17, 18). However, the contribution of these genes on human cardiovascular system formation remains less studied. Herein, we conducted genetic screening in CHD patients and identified several case specific nonsynonymous rare mutations.
Previous studies have shown that perturbations of Hedgehog pathway can lead to developmental errors presenting partially overlapping clinical manifestations and atrioventricular canal defects (AVCD) as a common denominator. Moreover, Shh pathway also involved in cardiac outflow tract and neural crest development and, therefore, Shh−/− embryos display conotruncal and pharyngeal arch artery defects (44, 45). Some variants of GLI1-3 genes have been found in several diseases including polydactyly, holoprosencephaly, Pallister-Hall syndrome and others (18, 46–51). However, our CHD patients are sporadic and non-syndromic cases in which ones showing additional symptoms such as polydactyly or holoprosencephaly should be excluded already. All the variants of GLI1-3 genes we identified are heterozygous, and showed much stronger connection with AVSD and PDA than other subtypes of CHD. While this phenotype-genotype correlation deserves further validation by expanding the sample size in rare mutation study.
Dual-luciferase reporter assay revealed p.G274C mutation enhanced transcription activation, p.R293H decreased transcription activation, whereas p.L481X almost completely loses transcription activation of SHH signaling pathway. EMSA showed p.G274C increased DNA-binding affinity and so did the mutant p.R293H. However, the luciferase assay showed that mutant p.G274C gains stronger function and mutant p.R293H decreases activation of the signaling compared to the function of wild GLI1 protein. Then we defined mutant p.G274C as gain-of-function and mutant p.R293H as loss-of-function. The fact that p.R293H shows loss-of-function does not necessarily need to be associated with decreased DNA-binding ability. The mutation specific characteristics may determine that its impact on the reduction of function is much greater than its impact on the ability to bind DNA. Therefore, the comprehensive performance of mutant p.G293H is that the function is reduced and the DNA-binding ability is reversely increased. Alternatively, the loss-of-function gained by mutant p.G293H might be too prominent to be balanced by the reverse effect of increased DNA-binding ability.
Usually, stop-gain mutations with aberrant translation termination leads to the degradation of the mRNA via activation of nonsense-mediated mRNA decay (NMD) and fail to produce a stable truncated protein (52). In response to NMD of mutated allele, wild-type allele always increasingly expressed to avoid haploinsufficiency. However, our data have demonstrated that the L481X truncated protein can exist stably and the truncated protein is largely produced in vivo (Supplementary Figure S3). The truncated protein not only failed to activate SHH signaling, but also placed additional inhibitory effects on SHH target genes (Figures 2, 4). Therefore, mutation p.L481X resulting haploinsufficiency was supported by the evidence that the heterozygous mutation p.L481X caused reverse phenotypes when the wild-type allele expression was strictly limited in half by the existed truncated allele. Because the result that heterozygous mutation p.L481X caused reverse phenotypes was partially attributed to the truncated allele, it's also hard to conclude that what we observed was merely caused by the half-expressed wild-type allele.
The alterations of critical genes in the early stage of cardiac development, such as de novo mutations, copy number variants, common variants, noncoding mutations and epigenetic modification, have contributed to the occurrence of CHD (5, 53–55). The in vitro function assays indicated that compared to the normal function of wild-type GLI1 protein, mutation p.R293H shows loss of function effect and mutation p.L481X also causes severely loss-of-function effect due to the stop-gain, while, mutation p.R274C shows gain of function. When we used overexpression system to test the cardiac morphology in zebrafish, as expected, the overexpressed wild-type GLI1 gene would lead to observed cardiac anomalies because of the dosage imbalance in addition to the existed endogenous GLI1 working normally. Similarly, compared to the wild-type GLI1 protein, the overexpressed p.R293H mutated GLI1 or p.L481X mutated GLI1 protein would result in less reverse effect in vivo because they are partially or maximum lose the normal function as wild-type GLI1 protein. Supposed mutation p.R274C gains of the original function, the most severe reverse effect on zebrafish phenotypes would be imaginable. In other words, the in vitro and in vivo function assays kept consistent although the outcomes looked a sign of contradiction.
Our functional study demonstrated some of those mutations significantly interfere with the protein function, thus lead to dysregulated SHH signaling pathway. Our study suggests the strong association of GLI1 gene mutations with the CHD occurrence. Interestingly, both gain and loss-of-function nonsynonymous rare mutations in GLI1 gene were identified in the CHD patients. As GLI1 protein simply serves as a transcriptional activator in SHH signaling pathway, our results suggest that either elevated or repressed GLI1 protein activity caused imbalanced signaling transduction could be a causative factor of CHD. This fits the fact that temporally and spatially tight regulation of key factors in fundamental developmental pathways underlies the normal embryogenesis.
However, since our samples are all sporadic and lack pedigrees, the mutations we found might not be the only causative factor leading to CHD. It is possible that the patients with GLI1-3 mutations may also carry harmful mutation(s) in other gene(s). We presume that single deleterous mutation contributes partially to the CHD and multiple such mutations help individual approaches the threshold of CHD occurrence, as our group demonstrated in another congenital malformation recently (56).
In conclusion, in present study, we systematically identified a set of novel rare mutations in GLI1-3 genes in human CHD that have the potential to be used diagnostically. Our data support our hypothesis that in GLI1, both gain and loss-of-function mutations are likely to be associated with CHD. Further understanding of the regulation and balance of SHH signaling pathway is crucial for understanding CHD etiology.
The data in the present study are available from the corresponding authors upon reasonable request. The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by Ethics Committee of the School of Life Sciences, Fudan University. Written informed consent to participate in this study was provided by the participants' legal guardian/next of kin. The animal study was reviewed and approved by Ethics Committee of the School of Life Sciences, Fudan University.
RP, BL, LL, and HW designed the project and prepared and edited the manuscript. RP, BL, and LL performed the experiments and analyzed the data. SC, YG, and XY contributed to the zebrafish embryo injection experiment. ZS and LY contributed to the clinical specimens collection and sequencing. All authors contributed to the article and approved the submitted version.
This work was supported by grants from National Key R&D Program of China (2021YFC2701101, HW) and National Natural Science Foundation of China (81930036 and 82150008, HW) and the Commission for Science and Technology of Shanghai Municipality (20ZR1404800, LL).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We sincerely thank all the individuals who participated in this study and the clinicians who helped in recruiting the volunteers.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2022.798033/full#supplementary-material
1. Triedman JK, Newburger JW. Trends in congenital heart disease: the next decade. Circulation. (2016) 133:2716–33. doi: 10.1161/CIRCULATIONAHA.116.023544
2. Zhao LJ, Chen LZ, Yang TB, Wang TT, Zhang SM, Chen LT, et al. Birth prevalence of congenital heart disease in China, 1980–2019: a systematic review and meta-analysis of 617 studies. Eur J Epidemiol. (2020) 35:631–42. doi: 10.1007/s10654-020-00653-0
3. Botto LD, Mulinare J, Erickson JD. Occurrence of congenital heart defects in relation to maternal multivitamin use. Am J Epidemiol. (2000) 151:878–84. doi: 10.1093/oxfordjournals.aje.a010291
4. Botto LD, Mulinare J, Erickson JD. Do multivitamin or folic acid supplements reduce the risk for congenital heart defects? Evidence and gaps. Am J Med Genet A. (2003) 121A:95–101. doi: 10.1002/ajmg.a.20132
5. Gelb BD, Chung WK. Complex genetics and the etiology of human congenital heart disease. Cold Spring Harb Perspect Med. (2014) 4:a013953. doi: 10.1101/cshperspect.a013953
6. Blue GM, Krik EP, Giannoulatou E, Sholler GF, Dunwoodie SL, Harvey RP, et al. Advances in the genetics of congenital heart disease: a clinician's guide. J Am Coll Cardiol. (2017) 69:859–70. doi: 10.1016/j.jacc.2016.11.060
7. Ingham PW, Nakano Y, Seger C. Mechanisms and functions of Hedgehog signalling across the metazoa. Nat Rev Genet. (2011) 12:393–406. doi: 10.1038/nrg2984
8. Carpenter D, Stone DM, Brush J, Ryan A, Armanini M, Frantz G, et al. Characterization of two patched receptors for the vertebrate hedgehog protein family. Proc Natl Acad Sci USA. (1998) 95:13630–4. doi: 10.1073/pnas.95.23.13630
9. Zaphiropoulos PG, Undén AB, Rahnama F, Hollingsworth RE, Toftgård RE. PTCH2, a novel human patched gene, undergoing alternative splicing and up-regulated in basal cell carcinomas. Cancer Res. (1999) 59:787–92.
10. Altaba ARI, Sánchez P, Dahmane N. Gli and hedgehog in cancer: tumours, embryos and stem cells. Nat Rev Cancer. (2002) 2:361–72. doi: 10.1038/nrc796
11. Altaba ARI. Gli proteins encode context-dependent positive and negative functions: implications for development and disease. Development. (1999) 126:3205–16. doi: 10.1007/s004290050265
12. Hynes M, Stone DM, Dowd M, Pitts-Meek S, Goddard A, Gurney A, et al. Control of cell pattern in the neural tube by the zinc finger transcription factor and oncogene Gli-1. Neuron. (1997) 19:15–26. doi: 10.1016/S0896-6273(00)80344-X
13. Persson M, Despina S, Pascal TW, Elisabet A, Jens BS, Ulrich R, et al. Dorsal-ventral patterning of the spinal cord requires Gli3 transcriptional repressor activity. Genes Dev. (2002) 16:2865. doi: 10.1101/gad.243402
14. Hoffmann AD, Peterson MA, Friedland-Little JM, Anderson SA, Moskowitz IP. Sonic hedgehog is required in pulmonary endoderm for atrial septation. Development. (2009) 136:1761–70. doi: 10.1242/dev.034157
15. Jiang J, Hui CC. Hedgehog signaling in development and cancer. Dev Cell. (2008) 15:801–12. doi: 10.1016/j.devcel.2008.11.010
16. Bijlsma MF, Peppelenbosch MP, Spek CA. Hedgehog morphogen in cardiovascular disease. Circulation. (2006) 114:1985–91. doi: 10.1161/CIRCULATIONAHA.106.619213
17. Riddle RD, Johnson RL, Laufer E, Tabin C. Sonic hedgehog mediates the polarizing activity of the ZPA. Cell. (1993) 75:1401–16. doi: 10.1016/0092-8674(93)90626-2
18. Palencia-Campos A, Ullah A, Nevado J, Yildirim R, Unal E, Ciorraga M, et al. GLI1 Inactivation is associated with Developmental Phenotypes Overlapping with Ellis-Van Creveld Syndrome. Hum Mol Genet. (2017) 26:4556–71. doi: 10.1093/hmg/ddx335
19. Goel HL, Underwood JM, Nickerson JA, Hsieh CC, Languino LR. β1 integrins mediate cell proliferation in three-dimensional cultures by regulating expression of the sonic hedgehog effector protein, GLI1. J Cell Physiol. (2010) 224:210–7. doi: 10.1002/jcp.22116
20. Palle K, Mani C, Tripathi K, Athar M. Aberrant GLI1 activation in DNA damage response, carcinogenesis and chemoresistance. Cancers (Basel). (2015) 7:2330–51. doi: 10.3390/cancers7040894
21. Liang SZ, Shi X, Yu CX, Shao XL, Zhou HT Li XY, et al. Identification of novel candidate genes in heterotaxy syndrome patients with congenital heart diseases by whole exome sequencing. Biochim Biophys Acta Mol Basis Dis. (2020) 1866:165906. doi: 10.1016/j.bbadis.2020.165906
22. Kim PCW, Mo R, Hui CC. Murine models of VACTERL syndrome: role of sonic hedgehog signaling pathway. J Pediatr Surg. (2001) 36:381–4. doi: 10.1053/jpsu.2001.20722
23. Qiao XJ, Liu YH Li PQ, Chen ZZ Li HL, Yang XY, et al. Genetic analysis of rare coding mutations of CELSR1-3 in congenital heart and neural tube defects in Chinese people. Clin Sci (Lond). (2016) 130:2329–40. doi: 10.1042/CS20160686
24. Li BB, Yu LW, Liu D, Yang XY, Zheng YF, Gui YH, et al. MIB1 mutations reduce Notch signaling activation and contribute to congenital heart disease. Clin Sci (Lond). (2018) 132:2483–91. doi: 10.1042/CS20180732
25. Zhu MJ, Ma XY, Ding PC, Tang HF, Peng R, Lu L, et al. Novel mutations of AXIN2 identified in a Chinese congenital heart disease cohort. J Hum Genet. (2019) 64:427–35. doi: 10.1038/s10038-019-0572-x
26. Hao M, Pu WL, Li Y, Wen SQ, Sun C, Ma YY, et al. The huabiao project: whole-exome sequencing of 5,000 han chinese individuals. J Genet Genomics. (2021) doi: 10.1016/j.jgg.2021.07.013
27. Li BB, Niswander LA. TMEM132A, a novel wnt signaling pathway regulator through wntless (WLS) interaction. Front Cell Dev Biol. (2020) 8:599890. doi: 10.3389/fcell.2020.599890
28. Agren M, Kogerman P, Kleman MI, Wessling M, Toftgård R. Expression of the PTCH1 tumor suppressor gene is regulated by alternative promoters and a single functional Gli-binding site. Gene. (2004) 330:101–14. doi: 10.1016/j.gene.2004.01.010
29. Yu LW, Wang F, Yang XY, Sun SN, Zheng YF Li BB, et al. Mild decrease in TBX20 promoter activity is a potentially protective factor against congenital heart defects in the Han Chinese population. Sci Rep. (2016) 6:23662. doi: 10.1038/srep23662
30. Ye JH, Tong YL, Lv JS, Peng R, Chen SX, Kuang LL, et al. Rare mutations in the autophagy-regulating gene AMBRA1 contribute to human neural tube defects. Hum Mutat. (2020) 41:1383–93. doi: 10.1002/humu.24028
31. Ng PC, Henikoff S SIFT. Predicting amino acid changes that affect protein function. Nucleic Acids Res. (2003) 31:3812–4. doi: 10.1086/175612
32. Adzhubei I, Jordan DM, Sunyaev SR. Predicting functional effect of human missense mutations using PolyPhen-2. Curr Protoc Hum Genet. (2013). doi: 10.1002/0471142905.hg0720s76
33. Rentzsch P, Witten D, Cooper GM, Shendure J. Kircher. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. (2019) 47:D886–94. doi: 10.1093/nar/gky1016
34. van der Velde KJ, de Boer EN, van Diemen CC, Sikkema-Raddatz B, Abbott KM, Knopperts A, et al. GAVIN: Gene-Aware Variant INterpretation for medical sequencing. Genome Biol. (2017) 18:6. doi: 10.1186/s13059-016-1141-7
35. Cooper GM, Stone EA, Asimenos G, Green ED, Batzoglou S, Sidow A. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. (2005) 15:901–13. doi: 10.1101/gr.3577405
36. Barnfield PC, Zhang XY, Thanabalasingham V, Yoshida M, Hui CC. Negative regulation of Gli1 and Gli2 activator function by Suppressor of fused through multiple mechanisms. Differentiation. (2005) 73:397–405. doi: 10.1111/j.1432-0436.2005.00042.x
37. Kogerman P, Grimm T, Kogerman L, Krause D, Undén AB, Sandstedt B, et al. Mammalian suppressor-of-fused modulates nuclear-cytoplasmic shuttling of Gli-1. Nat Cell Biol. (1999) 1:312–9. doi: 10.1038/13031
38. Olson EN. Gene regulatory networks in the evolution and development of the heart. Science. (2006) 313:1922–7. doi: 10.1126/science.1132292
39. Verma MK, Lenka N. Temporal and contextual orchestration of cardiac fate by WNT-BMP synergy and threshold. J Cell Mol Med. (2010) 14:2094–108. doi: 10.1111/j.1582-4934.2009.00774.x
40. Nasevicius A, Larson J, Ekker SC. Distinct requirements for zebrafish angiogenesis revealed by a VEGF-A morphant. Yeast. (2000) 17:294–301. doi: 10.1002/1097-0061(200012)17:4<294::AID-YEA54>3.0.CO
41. Wang WD, Huang CJ, Lu YF, Hsin JP, Prabhakar VR, Cheng CF, et al. Heart-targeted overexpression of Nip3a in zebrafish embryos causes abnormal heart development and cardiac dysfunction. Biochem Biophys Res Commun. (2006) 347:979–87. doi: 10.1016/j.bbrc.2006.06.174
42. Gou DZ, Zhou J, Song QX, Wang ZJ, Bai XM, Zhang YD, et al. Mog1 knockout causes cardiac hypertrophy and heart failure by downregulating tbx5-cryab-hspb2 signalling in zebrafish. Acta Physiol (Oxf). (2021) 231:e13567. doi: 10.1111/apha.13567
43. Hui CC, Angers S. Gli proteins in development and disease. Annu Rev Cell Dev Biol. (2011) 27:513–37. doi: 10.1146/annurev-cellbio-092910-154048
44. Digilio MC, Pugnaloni F, Luca AD, Calcagni G, Baban A, Dentici ML, et al. Atrioventricular canal defect and genetic syndromes: The unifying role of sonic hedgehog. Clin Genet. (2019) 95:268–76. doi: 10.1111/cge.13375
45. Washington Smoak IW, Byrd NA, Abu-Issa R, Goddeeris MM, Anderson R, Morris J, et al. Sonic hedgehog is required for cardiac outflow tract and neural crest cell development. Dev Biol. (2005) 283:357–72. doi: 10.1016/j.ydbio.2005.04.029
46. Ullah A, Umair M, Majeed AI., Abdullah Jan A, Ahmad W. A novel homozygous sequence variant in GLI1 underlies first case of autosomal recessive pre-axial polydactyly. Clin Genet. (2019) 95:540–1. doi: 10.1111/cge.13495
47. Palencia-Campos A, Martínez-Fernández M, Altunoglu U, Soto-Bielicka P, Torres A, Marín P, et al. Heterozygous pathogenic variants in GLI1 are a common finding in isolated postaxial polydactyly A/B. Hum Mutat. (2020) 41:265–76. doi: 10.1002/humu.23921
48. Yousaf M, Ullah A, Azeem Z, Majeed AI, Memon MI, Ghous T, et al. Novel heterozygous sequence variant in the GLI1 underlies postaxial polydactyly. Congenit Anom (Kyoto). (2020) 60:115–9. doi: 10.1111/cga.12361
49. Umair M, Ahmad F, Ahmad S, Alam Q, Rehan M, Alqosaibi AI, et al. A novel homozygous missense mutation in the zinc finger DNA binding domain of GLI1 causes recessive post-axial polydactyly. Front Genet. (2021) 12:746949. doi: 10.3389/fgene.2021.746949
50. Arnhold IJP, França MM, Carvalho LR, Mendonca BB, Jorge AAL. Role of GLI2 in hypopituitarism phenotype. J Mol Endocrinol. (2015) 54:R141–50. doi: 10.1530/JME-15-0009
51. Andreu-Cervera A, Catala M, Schneider-Maunoury S. Cilia, ciliopathies and hedgehog-related forebrain developmental disorders. Neurobiol Dis. (2021) 150:105236. doi: 10.1016/j.nbd.2020.105236
52. Karousis ED, Mühlemann O. Nonsense-Mediated mRNA Decay Begins Where Translation Ends. Cold Spring Harb Perspect Biol. (2019) 11:a032862. doi: 10.1101/cshperspect.a032862
53. Pierpont ME, Basson CT, Benson DW, Gelb BD, Giglia TM, Goldmuntz E, et al. Genetic basis for congenital heart defects: current knowledge: a scientific statement from the American Heart Association Congenital Cardiac Defects Committee, Council on Cardiovascular Disease in the Young: endorsed by the American Academy of Pediatrics. Circulation. (2007) 115:3015–38. doi: 10.1161/CIRCULATIONAHA.106.183056
54. Bentham J. Bhattacharya. Genetic mechanisms controlling cardiovascular development. Ann N Y Acad Sci. (2008) 1123:10–9. doi: 10.1196/annals.1420.003
55. Ordovás JM, Smith CE. Epigenetics and cardiovascular disease. Nat Rev Cardiol. (2010) 7:510–9. doi: 10.1038/nrcardio.2010.104
Keywords: congenital heart disease (CHD), GLI1, genetic variant, rare mutation, Sonic hedgehog (SHH) signaling pathway
Citation: Peng R, Li B, Chen S, Shi Z, Yu L, Gao Y, Yang X, Lu L and Wang H (2022) Deleterious Rare Mutations of GLI1 Dysregulate Sonic Hedgehog Signaling in Human Congenital Heart Disease. Front. Cardiovasc. Med. 9:798033. doi: 10.3389/fcvm.2022.798033
Received: 19 October 2021; Accepted: 03 March 2022;
Published: 04 April 2022.
Edited by:
Seitaro Nomura, The University of Tokyo, JapanReviewed by:
Bruno Marino, Sapienza University of Rome, ItalyCopyright © 2022 Peng, Li, Chen, Shi, Yu, Gao, Yang, Lu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lei Lu, bHVsZWlAZnVkYW4uZWR1LmNu; Hongyan Wang, d2FuZ2h5bGFiQGZ1ZGFuLmVkdS5jbg==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.