 Deqiang Yuan
Deqiang Yuan Jiapeng Chu
Jiapeng Chu Hao Lin
Hao Lin Guoqi Zhu
Guoqi Zhu Fei Chen
Fei Chen
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 16 January 2023
Sec. Atherosclerosis and Vascular Medicine
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.1109445
Homocysteine (Hcy) is an intermediate amino acid formed during the conversion from methionine to cysteine. When the fasting plasma Hcy level is higher than 15 μmol/L, it is considered as hyperhomocysteinemia (HHcy). The vascular endothelium is an important barrier to vascular homeostasis, and its impairment is the initiation of atherosclerosis (AS). HHcy is an important risk factor for AS, which can promote the development of AS and the occurrence of cardiovascular events, and Hcy damage to the endothelium is considered to play a very important role. However, the mechanism by which Hcy damages the endothelium is still not fully understood. This review summarizes the mechanism of Hcy-induced endothelial injury and the treatment methods to alleviate the Hcy induced endothelial dysfunction, in order to provide new thoughts for the diagnosis and treatment of Hcy-induced endothelial injury and subsequent AS-related diseases.
Hyperhomocysteinemia (HHcy) is an important risk factor for atherosclerosis (AS) and is defined as fasting plasma homocysteine (Hcy) higher than 15 μmol/L (1). Hcy is formed during the conversion of an essential amino acid methionine to cysteine, the factors that affect the level of Hcy in plasma include genetics, nutrition, age, sex, drugs, disease state. Hcy as an intermediate amino acid affects many cellular biological processes, such as cellular methylation status, cell metabolism, and cell injury.
Endothelial cells (ECs) are located in the innermost layer of blood vessels and form the vascular intima, which is important for maintaining vascular homeostasis and normal blood circulation. Pro-atherosclerotic risk factors in the blood can damage the endothelium, which acts as the initiation of AS and causes its progression. In recent years, numerous studies have shown that Hcy can damage the endothelium through various mechanisms, which may be a key way for it to promote AS-related diseases (2). Although the specific mechanism of Hcy injury to the endothelium is still not fully clear, it may be related to the induction of inflammation and cell death, interference with nitric oxide (NO) production, reactive oxygen species (ROS) accumulation and oxidative stress, cellular hypomethylation, protein homocysteinylation, and abnormal lipid metabolism (2). Hcy can cause intimal damage through these mechanisms and aggravate the progression of AS, and more in-depth mechanisms need to be studied. In terms of treatment, besides dietary therapy and supplementation of vitamin B12/B6 and folate to reduce plasma Hcy level, many drugs that can alleviate Hcy-induced endothelial damage have been studied, such as melatonin (3), estrogen (4), some lipid-lowering (5), and hypoglycemic (6) drugs. It is of great scientific and clinical significance for the prevention and treatment of endothelial injury and subsequent AS caused by HHcy to further explore the mechanism and mitigation methods of Hcy damage to the endothelium. This review mainly summarizes the mechanism of Hcy damage to ECs and related treatment, aiming to provide some new perspectives for research and clinical practice in this direction.
Homocysteine is an intermediate amino acid formed during the conversion from an essential sulfur-containing amino acid methionine to cysteine (7). The source of methionine is dietary protein, and the liver is considered to play an important role in the methionine and Hcy metabolism because of its full complement of related enzymes to regulate plasma Hcy level (8). Hcy is produced via transmethylation and removed through either the remethylation or the trans-sulfuration pathways (9). First, methionine is converted into the high-energy sulfonium compound S-adenosylmethionine (SAM, AdoMet), which is an important methyl donor for nearly all cell transmethylation process (10), the reaction is catalyzed by methionine adenosyltransferase (MAT) and provided the adenosyl moiety by adenosine triphosphate (ATP) (11). Next, SAM is demethylated to form S-adenosylhomocysteine (SAH, AdoHcy), which is then hydrolyzed to adenosine and Hcy (9). Hcy then experiences the transsulfuration pathway to form cysteine and glutathione or the remethylation cycle to back to methionine (12). The transsulfuration pathway tends to occur through upregulating the vitamin B6-dependent enzyme, cystathionine β-synthase (CBS) and cystathionine-gamma-lyase (CSE), and downregulating the remethylation pathway in the situations of excess methionine (13). Therefore, the lack of CBS will lead to the abnormal accumulation of Hcy and the occurrence of related diseases such as homocystinuria, homocysteinemia, and hypermethioninemia (14). The cysteine that is formed from Hcy by transsulfuration is finally oxidized to the sulfate and is excreted in the urine (15). On the other hand, Hcy is remethylated to methionine by using 5-methyltetrahydrofolic acid (5-methylTHF) or betaine as a methyl donor (16). These two different reactions are more likely to occur in methionine deficiency. In the first reaction, Hcy is catalyzed by methylcobalamin-containing methionine synthase (MS) to form methionine and tetrahydrofolate (THF) via receiving methyl from 5-methylTHF (16). Thus, Hcy metabolism generates cross-link with intracellular folate metabolism (17), which is also an important reason for folate level affect Hcy metabolism. In the second reaction, betaine-dependent remethylation backs Hcy to methionine by betaine-homocysteine methyltransferase (BHMT) (18). Several B vitamins exert important role in Hcy metabolism, vitamin B12 is the cofactor for MS (19), vitamin B6 is the coenzyme for CBS (20), so these B vitamins deficiencies can lead to Hcy accumulation. The mechanism of Hcy metabolism described above is shown in Figure 1.
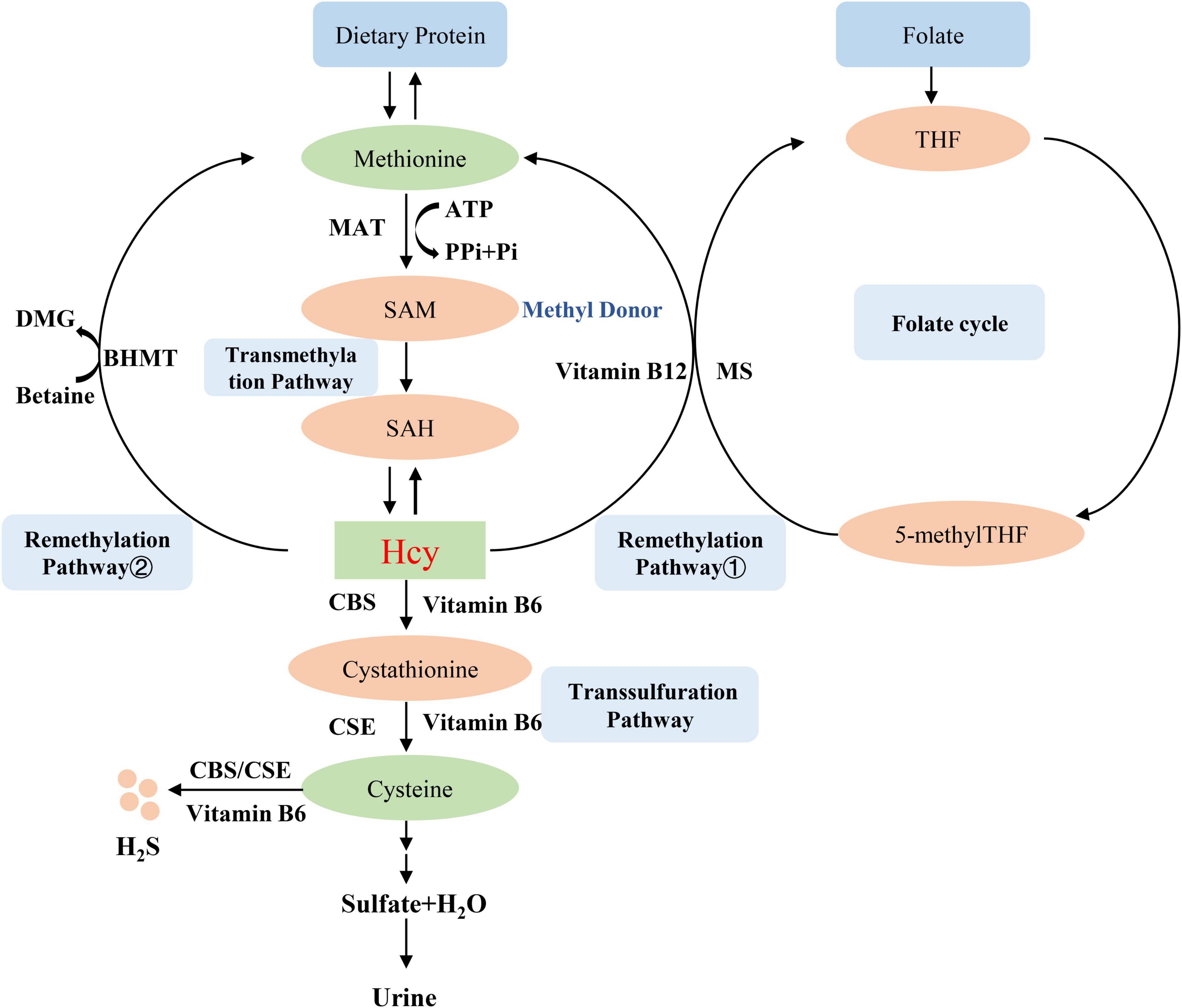
Figure 1. Homocysteine metabolism. MAT, methionine adenosyltransferase; SAM, S-adenosylmethionine, AdoMet; SAH, S-adenosylhomocysteine, AdoHcy; CBS, cystathionine β-synthase; BHMT, betaine-homocysteine methyltransferase; MS, methionine synthase; THF, tetrahydrofolate; 5-methylTHF, 5-methyltetrahydrofolic acid; CSE, cystathionine-gamma-lyase.
Intracellular concentration of Hcy is often under tight control by the above-mentioned reactions and by controlling its export from the cell (2). Plasma Hcy can easily act on ECs located in the innermost layer of blood vessels, and has significant effects on ECs function (21). There are three different forms of plasma Hcy, which including free Hcy, protein-bound Hcy and oxidized forms of Hcy (22). The Hcy concentration currently measured in the clinic is representative of the total plasma Hcy concentration. The normal range for plasma Hcy is 5–15 μmol/L measured in the fasting state, so HHcy is defined as plasma Hcy higher than 15 μmol/L (23). And HHcy is categorized into three classes as mild, moderate and severe HHcy with plasma HCy levels ranging from 15 to 30, 31 to 100, and >100 μmol/L, respectively.
Plasma Hcy level is affected by a variety of factors, as shown in Figure 2. Such as genetics, nutrition, age, sex, drugs, disease states, which are described in detail below. The exploration of the causes of elevated plasma Hcy level is of great significance for the treatment of HHcy and various injuries caused by it.
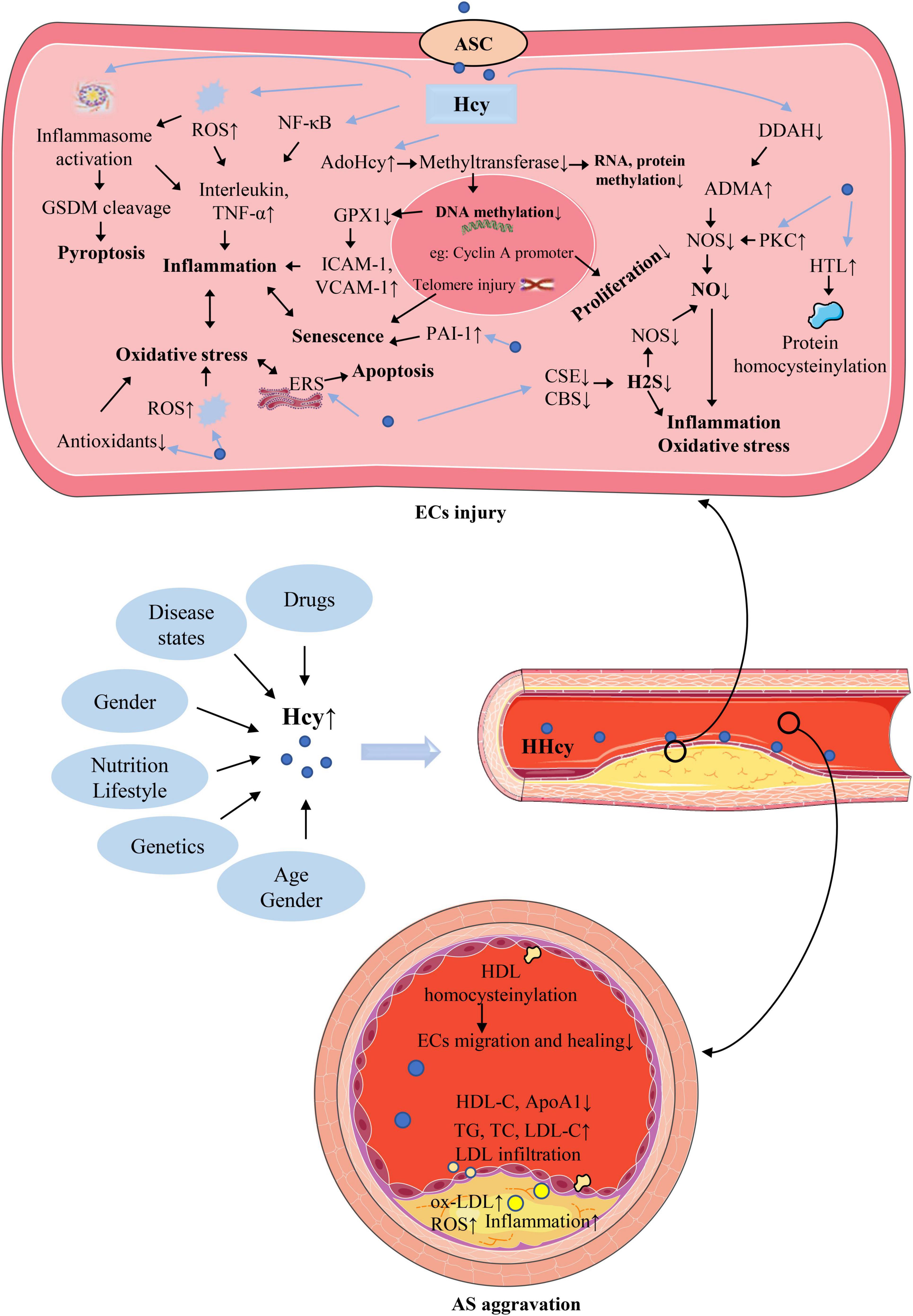
Figure 2. The causes of hyperhomocysteinemia (HHcy) and the mechanism of homocysteine (Hcy)–mediated endothelial cells (ECs) injury and its consequences for atherosclerosis (AS). The abbreviations in this figure are shown in the “Glossary” section.
Severe elevations of Hcy concentration in plasma is rare and often caused by homozygous mutations in enzymes involved in its metabolism, such as MS, methylene tetrahydrofolate reductase, and CBS. Numerous studies have investigated the changes in plasma concentration of Hcy and related disease progression in experimental animals after CBS knockout. The researchers used CRISPR/Cas9 to knock out the CBS gene in rabbits and found that the plasma Hcy level in knockout rabbits (50.73 μmol/L) was almost twice as high as that in controls (27.93 μmol/L) (24). And severe HHcy was observed in CBS–/– (289 ± 58 μM) but not in CBS± or control mice (<10 μM) after knocking out CBS in mice (25). The clinical manifestations of homozygous HHcy often include psychiatric symptoms and abnormalities of the hair, skin, joints, bones, and cardiovascular system. However, HHcy caused by heterozygous mutations in related enzymes is often asymptomatic, with only moderately elevated or normal plasma Hcy (26).
As mentioned above, B vitamins and folate exert important role in the metabolism of Hcy. Therefore, plasma Hcy concentration can be significantly increased in the deficiency of essential cofactor Vitamin B12/B6 or cosubstrate folate (27). Asian populations are prone to insufficient folate intake due to dietary and cooking habits, which may partly account for incidence of folate deficiency and HHcy in Asian populations is much higher than that in Western populations (28, 29). The incidence of HHcy in American society is just 5–7%, in Chinese 27.5% and in Indians 52–84% (30). Inappropriate lifestyle, such as high intake of coffee (31), alcohol, obesity and smoking can also lead to HHcy (32), which may be related to vitamin malabsorption.
Plasma Hcy is generally believed to increase with age (33). The specific mechanism of this phenomenon is still unclear. The possible reasons include the reduction of Hcy metabolizing enzyme activity, the impairment of renal function, hormonal changes, and the reduction of vitamin B12/B6 level as cofactor (34).
Plasma Hcy concentration in premenopausal women is typically 20% lower than in men of similar age. This may be related to the higher creatinine concentration and more muscle mass in men (34). However, female menopause is comparable to male plasma Hcy level. This suggests that Hcy is related to the change of estrogen level, and the decrease of Hcy level by estrogen may be related to the increase of CBS activity (35).
Some lipid-lowering drugs such as fibrates (36) and niacin can increase plasma Hcy. Mechanistically, fenofibrate may significantly increase plasma Hcy levels by reducing renal function (37). Therefore, the benefits of treating elevated cholesterol concentrations with these drugs should be weighed against the possible long-term risks of elevated Hcy. Methotrexate inhibits the conversion of dihydrofolate to tetrahydrofolate with physiological activity by inhibiting dihydrofolate reductase (DHFR), thereby increasing plasma Hcy (38, 39).
Studies have shown that plasma Hcy level is elevated in some disease states, such as various cancers (40), psoriasis (41), hypothyroidism (42), diabetes (43), and renal dysfunction (16). Among them, the disease state most closely related to HHcy is chronic renal failure. Plasma Hcy level is positively correlated with creatinine level (44), and the specific mechanism remains to be further studied. The possible mechanism is that chronic renal failure inhibits the activity or inactivates the key enzymes of Hcy metabolism, such as CBS (45) and BHMT (46), resulting in the inhibition of Hcy metabolism and accumulation of Hcy. HHcy occurs in the early stage of chronic renal failure and further increases with the progression of renal failure. Elevated Hcy can further aggravate renal injury by inducing oxidative stress (47), insufficient autophagy and renal aging (48), forming a negative feedback loop. Elevated plasma Hcy level in patients with malignant tumors may be related to changes in methionine metabolism in tumor cells (49). The relationship between diabetes and HHcy is relatively complex. It is generally believed that secondary pathological changes caused by diabetes, such as renal dysfunction, often lead to elevated plasma Hcy level, and the impact of diabetes itself on Hcy needs further study (50). The possible mechanism is that glucose metabolism disorder and Hcy metabolism worsen each other, resulting in the increase of Hcy level (32, 51).
Vascular endothelium is a homeostatic barrier to circulating blood and vessel walls like a gatekeeper of cardiovascular health (52). The vascular intima plays an important role in the homeostasis regulation of the vascular wall, including vascular tone, coagulation, inflammation, and permeability (53). As the innermost layer of the blood vessel wall, ECs are directly exposed to circulating blood, so they are easily injured by various risk factors and lead to their dysfunction and endothelial barrier damage (54). Impaired endothelial function and barrier can lead to a series of cascade reactions, such as inflammatory response (55), monocyte recruitment (56), plaque formation (57), structural remodeling (58), thrombosis (59). The results of cultured ECs and animal models of HHcy indicate that Hcy can injury ECs and cause endothelial dysfunction (60, 61). Several studies have confirmed that plasma Hcy concentration is positively correlated with AS. Clinical studies have shown that plasma Hcy level in patients with coronary artery disease is significantly higher than that in angiographic normal controls (62), and high plasma Hcy level with only 12% of the upper limit of normal are associated with a 3.4-fold increased risk of myocardial infarction (63). The above description is the newly proposed homocysteine theory of AS. H-type hypertension is a special type of hypertension combined with elevated plasma Hcy on the basis of hypertension. The risk of stroke caused by this kind of hypertension is obviously higher than that of simple hypertension (64), which may be related to Hcy damage to the vascular endothelium. This suggests that Hcy-damaged endothelium plays a central role in its promotion of vascular diseases. Therefore, exploring the mechanism of Hcy injury to vascular endothelium is of great significance for the diagnosis and treatment of AS-related diseases.
Homocysteine can cause ECs damage through various intracellular mechanisms. Such as induction of inflammation and cell death, interference with NO production, ROS accumulation and oxidative stress, cellular hypomethylation. There is a complex interaction between these mechanisms, which leads to a series of reactions in the local and circulation of AS lesions. In addition, abnormal lipoprotein metabolism major as an extracellular mechanism also causes ECs damage and promotes AS progression. Protein homocysteinylation can cause endothelial damage through both intracellular and extracellular mechanisms. A brief illustration of these mechanisms is shown in Figure 2 and described in detail below.
Endothelial inflammation is a crucial driver of AS. When atherosclerotic risk factors act on ECs, damaged ECs secrete a large number of cytokines, adhesion molecules, and chemokines (65). These secreted factors recruit circulating monocytes to the site of endothelial injury and induce the transformation of monocytes into pro-inflammatory macrophages, which in turn develop into foam cells, promoting the formation of atherosclerotic plaques at the site of injury (66). Experimental studies on cultured ECs have shown that Hcy can induce inflammation by inducing a variety of inflammatory cytokines, such as interleukin (IL)-1β (67), IL-6 (68), IL-8 (69, 70), IL-18 (71), and tumor necrosis factor (TNF)-α (72), which may be due to the ROS accumulation, inflammasome activation, nuclear factor kappa-B (NF-κB) activation. Hcy can promote ECs senescence by upregulating plasminogen activator inhibitor-1 (PAI-1) (73), and telomere shortening and dysfunction may also be the cause of HCy-induced ECs senescence (74). ECs senescence further accelerates inflammation and endothelial injury by senescence-associated secretory phenotype (SASP). When the damage of Hcy to ECs is further aggravated, the cells may go toward death, resulting in severe endometrial damage. Numerous studies have shown that Hcy can cause various forms of ECs death, such as apoptosis (75), pyroptosis (76), and ferroptosis (77). Pyroptosis is a newly discovered form of programed cell death, which depends on gasdermin family proteins such as gasdermin D (GSDMD), GSDMB, GSDME. Both in vivo and in vitro studies have shown that Hcy can induce ECs pyroptosis and release of inflammatory factors such as IL-1β and IL-18 via caspase-1-dependent inflammasome activation through the accumulation of intracellular ROS (76). ECs death causes cell death on the one hand, and some inflammatory cell death forms, such as pyroptosis, can cause strong local and circulatory inflammatory responses and accelerate the progression of AS.
Nitric oxide is a key signaling molecule in endothelium to act as vasodilator factor, generated by nitric oxide synthase (NOS) (78). Endothelial production of NO can inhibit multiple events during AS, such as inhibiting ECs activation, macrophage infiltration (79), foam cell formation/migration (80), platelet aggregation (81), inflammation (82), thrombosis (83), vascular wall remodeling (84), and mediating vasodilation (85). The mechanism of Hcy disturbance of NO synthesis is relatively complex, asymmetric dimethylarginine (ADMA) plays an important role in it, which is an endogenous inhibitor of NOS. Specifically, Hcy post-translationally inhibits dimethylarginine dimethylaminohydrolase (DDAH) activity, the enzyme that degrades ADMA (86). Therefore, Hcy can cause ADMA to accumulate and inhibit NO synthesis. Hcy can also inhibit NOS and reduce NO synthesis in ECs by activating protein kinase C (PKC). Reduced NO synthesis causes endothelial injury by aggravating oxidative stress and inflammation (87).
Major producing systems of ROS in ECs include reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, xanthine oxidase, the mitochondrial electron transport chain, and uncoupled endothelial NOS (79). Moderate concentrations of ROS have important signaling functions, while excessive accumulation of ROS can defeat the antioxidant system, leading to oxidative stress. A large number of studies illustrate that oxidative stress plays an important role in Hcy-induced ECs dysfunction and AS. Specifically, Hcy can induce oxidative stress in ECs by mediating ROS production or impairing the antioxidant system (2). Hcy also can induce ECs NADPH oxidase upregulation to accumulate ROS (2). An important mechanism of Hcy-induced ECs injury is endoplasmic reticulum stress (ERS), which is a response to dangerous stimulation that leads to the accumulation of unfolded or abnormally folded proteins in the endoplasmic reticulum (ER) (88). Cell and animal model results found that Hcy upregulates ER oxidoreductin-1α (Ero-1α) expression by promoting binding of hypoxia-inducible factor-1α (HIF-1α) to the Ero-1α promoter and downregulates the antioxidant pathway mediated by ER glutathione peroxidase (GPX) 7 to induce ERS and disrupt ER homeostasis (89). Oxidative stress and ERS aggravate each other. When ECs suffer from oxidative stress, the redox equilibrium of ER is broken, thereby disrupting ER function and triggering ERS. Similarly, when ERS occurs, a large amount of ROS will be produced to aggravate oxidative stress. Moderate ERS is a protective response, but excessive ERS can lead to cellular dysfunction, inflammatory responses and apoptosis (90). Therefore, oxidative stress caused by Hcy is closely related to ECs inflammation and death. On the other hand, Hcy also can cause oxidative stress by inhibiting ECs non-enzymatic antioxidants and damages enzymatic antioxidants (2).
We already know that Hcy is metabolized to cysteine through the transsulfurization pathway, which is then decomposed into sulfate and excreted in urine. This pathway is regulated by two key enzymes, CBS and CSE, furthermore, these two enzymes can also use cysteine as a substrate and are dependent on pyridoxal-5’-phosphate to produce dihydrogen sulfide as shown in the Figure 1. Dihydrogen sulfide is also known as hydrogen sulfide (H2S) or sulfane (91, 92). H2S is considered to be an endogenously produced gasotransmitter that acts on various targeted signaling pathways to play an important role in vascular homeostasis (93, 94). Downregulation of the CSE/H2S pathway has an important pathological role in the development of AS (95). H2S is reported to regulate multiple endothelial functions such as angiogenesis, proliferation and migration (96). The study found that H2S pretreatment of ECs can improve mitochondrial function and cell viability after hypoxia treatment by activating extracellular regulated protein kinases (ERK) 1/2, as well as enhance ECs migration and angiogenesis, thereby protecting ECs from ischemia/reperfusion injury and gradually reducing cardiac damage (97). It has also been found that exogenous H2S treatment or CSE overexpression can rescue high glucose-induced ECs migration impairment by upregulating microRNA (miR)-126-3p (98). Although the role of H2S on inflammation is complex, it is very noteworthy that recent studies have suggested that H2S can inhibit ECs inflammation (99), such as inhibiting NF-κB pathway and scavenging ROS (2). Interestingly, H2S also has a heterotypic interaction with another important signaling molecule, NO, for example, H2S can promote endothelial NO production via activating NOS phosphorylation (100). The complex interaction between H2S and NO might serve as an important regulator for endothelial homeostasis.
Hyperhomocysteinemia can cause downregulation of CSE and CBS, thereby causing production of H2S is injury (2). Lack of H2S protection in ECs can cause endothelial dysfunction and damage, and gradually lead to HHcy-related vascular disease (96). The study has shown that exogenous H2S treatment can alleviate Hcy-induced endothelial dysfunction by inhibiting mitochondrial toxicity (101). Therefore, Hcy can impair endothelial homeostasis by impairing the H2S pathway, and maintaining the normal function of this pathway is important for Hcy-induced endothelial dysfunction.
S-adenosylmethionine, formed when methionine is converted into Hcy, is the methyl donor for almost all transmethylation reactions of DNA, RNA, protein and other components in cells (102, 103). Therefore, the methylation reactions of various cellular components can be significantly affected by Hcy metabolism. It is worth mentioning that, hydrolysis of S-AdoHcy to Hcy is invertible, and the synthesis of S-AdoHcy from SAM/AdoMet is thermodynamically more favored (104). The reaction toward hydrolysis direction because of rapid clearance of Hcy by cellular export and metabolic conversion under normal conditions. However, When Hcy levels are abnormally elevated, which can cause elevated AdoHcy. Importantly, AdoHcy is a critically endogenous inhibitor of cellular methyltransferases, HHcy is easy to form hypomethylating environment, which further impairs methylation reactions related to intimal homeostasis (105). ECs hypomethylation can reduce aquaporin-1 levels, leading to impaired water permeability and endothelial dysfunction (106). ECs hypomethylation can also upregulate the expression of the adhesion molecules intercellular adhesion molecule-1 (ICAM-1) and vascular adhesion molecule-1(VCAM-1) by suppressing GPX1 protein expression, causing ECs inflammation and injury (107). The epigenetic study has found that Hcy induces DNA hypomethylation of the cyclin A promoter by downregulating the expression of DNA methyltransferase 1 (DNMT1) to inhibit endothelial progenitor cells (EPCs) proliferation (108). These results suggest that Hcy-induced hypomethylation of DNA, RNA, protein in ECs leads to endothelial injury by impairing water permeability, inducing inflammation, and inhibiting proliferation.
Homocysteinylation mainly targets proteins and is classified as S-homocysteinylation and N-homocysteinylation, N-homocysteinylation is mainly discussed here. When plasma Hcy level is elevated, N-homocysteinylation of numerous proteins in cells occurs due to the interaction between the highly reactive homocysteine thiolactone (HTL) and lysine residues of a target protein. Also, when HHcy is present in atherosclerotic patients, protein homocysteinylation is enhanced (22). HTL is a cyclic thioester generated during protein biosynthesis because of error-editing reaction of Hcy with methionyl-tRNA synthetase (MetRS) (7). The N-homocysteinylation of proteins can lead to abnormal protein structure and biochemical function, which is closely involved in the injury of vascular intima caused by HHcy (109). Cellular protein quality control (PQC) is an important event in maintaining cellular homeostasis to maintain proteome integrity and cell viability, which include unfolded protein response (UPR), autophagy, ubiquitin-proteasome system (UPS), and chaperones (110–113). Numerous studies suggest that HHcy can damage PQC by activating UPR, impairing autophagy, and reducing chaperone level (114).
High-density lipoproteins is considered to have endothelial repair function, and then it was found that when HDL is homocysteinylated by N-homocysteine, it reduces ECs migration and attenuates HDL-mediated endothelial healing compared with control (109). One study found that angiotensin-converting enzyme (ACE) could be directly homocysteinylated by Hcy, which enhances ACE reactivity toward angiotensin II (ANG II)-NADPH oxidase-superoxide-dependent endothelial injury (115). Therefore, finding more proteins with specific and sensitive homocysteine changes in endothelial injury is of great value for the diagnosis and treatment of AS.
Some metabolic changes outside ECs caused by HHcy can also damage the endothelium and promote AS. Abnormal lipid and lipoprotein metabolism is an important risk factor for AS (116). Lipoproteins such as Low-density lipoproteins (LDL) and high-density lipoproteins (HDL) are mainly responsible for the transport of lipids such as cholesterol and triglycerides (117). Several clinical and experimental studies have shown that HHcy affects lipid metabolism. A clinical study involving 7,898 subjects showed that plasma Hcy was negatively associated with high-density lipoprotein cholesterol (HDL-C) and apolipoprotein A1 (ApoA1) and positively associated with triglyceride (TG) (118). Moreover, a study (24) has found that the blood lipid levels of HHcy rabbits are significantly higher than that of the controls, the TG level showed almost 52-fold increase (3746.7/71.31 mg/dL). The total cholesterol (TC) (540.8 mg/dL) and low-density lipoprotein cholesterol (LDL-C) (72.02 mg/dL) in HHcy rabbits were almost 4.4-and 2.5-fold higher that in WT controls, respectively. These provides some evidences for HHcy disordered lipid metabolism. HDL mainly transports cholesterol from peripheral tissues such as blood vessels to the liver for metabolic clearance, so plasma HDL has an anti-atherosclerotic effect. However, the reduction of HDL in HHcy patients indicates that the reverse transport of cholesterol is impaired, thereby disturbing lipid metabolism and promoting AS. When the endothelium is damaged, LDL can infiltrate into the intima and be oxidized to oxidized low density lipoprotein (ox-LDL), which can be phagocytosed by macrophages and vascular smooth muscle cells (VSMCs) to form foam cells and gradually form atherosclerotic plaques (119, 120). We already know that Hcy can induce ROS accumulation and generate oxidative stress, which can enhance the oxidation of LDL to promote AS (23, 121). Therefore, Hcy can damage the vascular intima and promote the development of AS by affecting the metabolic disorder of lipoproteins.
Due to the vascular damage effect of HHcy and the great harm to the cardiovascular and cerebrovascular, the timely monitoring, reduction and assessment of the blood Hcy level in HHcy patients is a topic worthy of discussion and research. When the plasma Hcy level is found to be elevated, dietary therapy and lifestyle modification can be tried first. Most HHcy is caused by chronic low level of folate and vitamin B12 (122). Correcting the deficiencies of folate and vitamin B12 can reduce Hcy level (123). Fruits, vegetables, and low-fat dairy products are rich in folate and B12 (124), and low-saturated fatty acid and low-fat meals can also reduce Hcy level. In addition, methionine intake should be limited. Another to be introduced is drug therapy, which includes folate, betaine, and vitamin supplements such as vitamin B12 and B6. It is generally accepted that all patients should also be treated with B-complex vitamins to reduce peripheral neuropathy. In terms of mechanism, folate and vitamin B6/B12 can alleviate Hcy-induced ECs apoptosis, oxidative stress, and mitochondrial dysfunction (125), folate also can modulate DNA methylation to delay AS (126). However, the therapeutic challenge is that even though B vitamins reduce Hcy level, which do not seem to reduce the risk of cardiovascular disease (127). The exact mechanism remains to be investigated. One possible explanation is that the beneficial effects of lowering Hcy are offset by the direct adverse effects of B vitamins supplementation (particularly with high-dose folate), or are associated with the pro-inflammatory and pro-proliferative effects of B vitamins on advanced AS lesions (128). It may also be related to the fact that although the plasma Hcy level is reduced, the intracellular Hcy concentration do not change significantly (87). Even so, B vitamins and folate can alleviate the progression of AS by reducing plasma Hcy level and improving epigenetic factors (129). HHcy can also cause extensive damage to a variety of organs, so reducing Hcy is still widely recommended.
In recent years, there have been many studies on alleviating Hcy-induced endothelial damage, which has important implications for the treatment of cardiovascular diseases caused by HHcy. Studies have found that melatonin can protect ECs by alleviating oxidative stress caused by Hcy (3), specifically, melatonin can decrease ROS and lipid peroxidation(LPO) levels induced by Hcy to exert anti-oxidants protection (130). Melatonin also can increase cell migration and downregulate pro-apoptotic proteins caspase-3, caspase-9, cytochrome c (Cyt C), and B-cell lymphoma-2 assaciated X protein (Bax), but upregulate anti-apoptotic protein B-cell lymphoma-2 (Bcl-2) to against Hcy-induced ECs apoptosis (130). Propofol can alleviate inflammation and apoptosis in Hcy-induced human umbilic vein endothelial cells (HUVECs) by inhibiting ERS, manifested as propofol increases cell viability, suppresses NF-κB signaling pathway activation and decreases the expression of inflammatory factors (131). It has been reported that the glucagon-like peptide 1 (GLP-1) analog exendin-4 lowers ERS and enhances protein folding to ameliorate Hcy-induced endothelial dysfunction in vivo and in vitro (6). Epigallocatechin gallate (EGCG), a well-known anti-oxidant in green tea, can reduce Hcy-induced oxidative damage and apoptosis by enhancing the silent information regulator 1 (SIRT1)/AMP-activated protein kinase (AMPK) survival signaling pathway (132). Estrogen supplementation ameliorates pyroptosis and inflammation in Hcy-treated HUVECs (71), moreover, estradiol-17β can inhibit Hcy mediated damage by promoting H2S production via upregulating CBS and CSE expression in HUVECs (4). Nicorandil for coronary heart disease alleviates Hcy-induced human coronary artery endothelial cells (HCAECs) dysfunction via regulating PI3K/Akt/NOS pathway in vivo and in vitro (133). L-cystathionine, an amino acid mainly produced during the conversion of methionine to cysteine, which can protect against Hcy-induced mitochondria-dependent apoptosis in HUVECs (75). The lipid-lowering drug atorvastatin can also alleviate ECs damage caused by Hcy (5). To sum up, it is of great significance to further explore the treatment and mechanism of alleviating Hcy-induced endothelial injury for the prevention and treatment of the pro-AS effect of HHcy.
As an important pro-AS risk factor, HHcy promotes the occurrence and development of cardiovascular disease. Causes of elevated plasma Hcy level include genetics, nutritional deficiencies, smoking, drugs, demographics, disease states. HHcy damage to the vascular endothelium is the core link of its promotion of AS, and the study of its mechanism is of great significance for the prevention and treatment of HHcy-induced endothelial damage and subsequent AS-related diseases. At present, the mechanisms of Hcy-induced endothelial injury include induction of inflammation and cell death, disturbance of NO production, ROS accumulation and oxidative stress, dysfunction of H2S signaling pathway, cellular hypomethylation, protein homocysteinylation, and lipid metabolism disorder. A novel concept is that Hcy-methionine cycle is a metabolic sensor system for methylation-regulated pathological signaling (134). The traditional view is that risk factors such as damage-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) produce pathological signals by acting on pattern recognition receptors (PRRs) on the cell surface/intracellular/nuclear. However, for metabolism-associated danger signal, probably through receptor-independent recognition by the metabolic sensor system of Hcy-methionine cycle. Further regulating SAM/SAH-dependent methylation in disease conditions and that hypomethylation on frequently modified histone residues may be a key mechanism for cardiovascular disease. This is an important research point of Hcy damage endothelium and cause AS related diseases. In addition, the mechanism of Hcy uptake by ECs is one of the research difficulties. One study found that alanine-serine-cysteine (ASC) transporter systems and lysosome function play an important role in Hcy transport in ECs (135). The specific mechanism needs to be further studied. Exploring more in-depth mechanisms of Hcy damage to the endothelium will help to further discover effective therapeutic targets and methods. The methods of treating HHcy include dietary therapy and supplementation of vitamin B12/B6 and folate. However, a controversial issue is that it has not been proven that lowering Hcy by B vitamins supplementation can reduce the risk of cardiovascular disease. Therefore, the causal relationship between HHcy and B vitamins deficiency and AS remains unclear and requires further investigation. Although lowering Hcy using B vitamins has no beneficial effect on secondary prevention of cardiovascular disease, the role of Hcy in primary disease prevention has not been fully studied. Therefore, more interventions and experimental studies are needed to address the existing knowledge gaps (74). In addition, some studies have found some drugs that can alleviate Hcy-induced endothelial damage by alleviating oxidative stress, ERS, inflammation, such as melatonin, propofol, estrogen, nicorandil, L-cystathionine. In the future, more effective therapeutic drugs need to be further studied. When patients have the following conditions, special attention should be paid Hcy level. Patients with coronary heart disease, cerebrovascular disease or peripheral AS, or with risk factors for cardiovascular and cerebrovascular diseases, such as hypertension, diabetes, obesity, smoking, or a family history of coronary heart disease or AS. In conclusion, the mechanism of Hcy injury to the endothelium and the promotion of AS is a direction worthy of further research, which is of great significance for reducing the occurrence and development of AS-related diseases.
DY, FC, and XL contributed to the conception, writing, and editing of this manuscript. JC, HL, GZ, JQ, YY, TY, and FP put forward some amendments to the article. All authors contributed to the article and approved the submitted version.
This study was supported by the National Natural Science Foundation of China (Grant Nos. 82170346, 81670403, and 81370390) and Grant of Shanghai Science and Technology Committee (No. 22Y11909800).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Paganelli F, Mottola G, Fromonot J, Marlinge M, Deharo P, Guieu R, et al. Hyperhomocysteinemia and cardiovascular disease: is the adenosinergic system the missing link? Int J Mol Sci. (2021) 22:1690. doi: 10.3390/ijms22041690
2. Esse R, Barroso M, Tavares DA I, Castro R. The contribution of homocysteine metabolism disruption to endothelial dysfunction: state-of-the-art. Int J Mol Sci. (2019) 20:867. doi: 10.3390/ijms20040867
3. Karolczak K, Watala C. Melatonin as a reducer of neuro- and vasculotoxic oxidative stress induced by homocysteine. Antioxidants. (2021) 10:1178. doi: 10.3390/antiox10081178
4. Zhang D, Hong X, Wang J, Jiang Y, Zhang Y, Chen J, et al. Estradiol-17beta inhibits homocysteine mediated damage by promoting H2 S production via upregulating CBS and CSE expression in human umbilical vein endothelial cells. J Cell Biochem. (2019). [Epub ahead of print]. doi: 10.1002/jcb.29527
5. Jia F, Wu C, Chen Z, Lu G. Atorvastatin inhibits homocysteine-induced endoplasmic reticulum stress through activation of AMP-activated protein kinase. Cardiovasc Ther. (2012) 30:317–25. doi: 10.1111/j.1755-5922.2011.00287.x
6. Cheng CK, Luo JY, Lau CW, Cho WC, Ng CF, Ma RCW, et al. A GLP-1 analog lowers ER stress and enhances protein folding to ameliorate homocysteine-induced endothelial dysfunction. Acta Pharmacol Sin. (2021) 42:1598–609. doi: 10.1038/s41401-020-00589-x
7. Chen N, Liu J, Qiao Z, Liu Y, Yang Y, Jiang C, et al. Chemical proteomic profiling of protein N-homocysteinylation with a thioester probe. Chem Sci. (2018) 9:2826–30. doi: 10.1039/C8SC00221E
8. Baloula V, Fructuoso M, Kassis N, Gueddouri D, Paul JL, Janel N. Homocysteine-lowering gene therapy rescues signaling pathways in brain of mice with intermediate hyperhomocysteinemia. Redox Biol. (2018) 19:200–9. doi: 10.1016/j.redox.2018.08.015
9. Xu Q, Li Y, Gao X, Kang K, Williams JG, Tong L, et al. HNF4alpha regulates sulfur amino acid metabolism and confers sensitivity to methionine restriction in liver cancer. Nat Commun. (2020) 11:3978. doi: 10.1038/s41467-020-17818-w
10. Mendel M, Chen KM, Homolka D, Gos P, Pandey RR, Mccarthy AA, et al. Methylation of Structured RNA by the m(6)A writer METTL16 is essential for mouse embryonic development. Mol Cell. (2018) 71:986–1000.e11. doi: 10.1016/j.molcel.2018.08.004
11. Greco CM, Cervantes M, Fustin JM, Ito K, Ceglia N, Samad M, et al. S-adenosyl-l-homocysteine hydrolase links methionine metabolism to the circadian clock and chromatin remodeling. Sci Adv. (2020) 6:eabc5629. doi: 10.1126/sciadv.abc5629
12. Ye C, Sutter BM, Wang Y, Kuang Z, Tu BP. A metabolic function for phospholipid and histone methylation. Mol Cell. (2017) 66:180–193.e8. doi: 10.1016/j.molcel.2017.02.026
13. Pusceddu I, Herrmann W, Kleber ME, Scharnagl H, Hoffmann MM, Winklhofer-Roob BM, et al. Subclinical inflammation, telomere shortening, homocysteine, vitamin B6, and mortality: the ludwigshafen risk and cardiovascular health study. Eur J Nutr. (2020) 59:1399–411. doi: 10.1007/s00394-019-01993-8
14. Ismail HM, Krishnamoorthy N, Al-Dewik N, Zayed H, Mohamed NA, Giacomo VD, et al. In silico and in vivo models for Qatari-specific classical homocystinuria as basis for development of novel therapies. Hum Mutat. (2019) 40:230–40. doi: 10.1002/humu.23682
15. Kriebitzsch C, Verlinden L, van Schoor NM, Van S, Swart K, Lips P, et al. 1,25-dihydroxyvitamin D3 influences cellular homocysteine levels in murine preosteoblastic MC3T3-E1 cells by direct regulation of cystathionine beta-synthase. J Bone Miner Res. (2011) 26:2991–3000. doi: 10.1002/jbmr.493
16. Gylling B, Myte R, Ulvik A, Ueland PM, Midttun O, Schneede J, et al. One-carbon metabolite ratios as functional B-vitamin markers and in relation to colorectal cancer risk. Int J Cancer. (2019) 144:947–56. doi: 10.1002/ijc.31606
17. Xiang Y, Liang B, Zhang X, Qiu X, Deng Q, Yu L, et al. Atheroprotective mechanism by which folic acid regulates monocyte subsets and function through DNA methylation. Clin Epigenetics. (2022) 14:32. doi: 10.1186/s13148-022-01248-0
18. Li J, Li F, Yu N, Liu Z. The betaine-dependent remethylation pathway is a homocysteine metabolism pathway associated with the carnivorous feeding habits of spiders. Insect Sci. (2021) 29:1047–58. doi: 10.1111/1744-7917.12976
19. Lurz E, Horne RG, Maattanen P, Wu RY, Botts SR, Li B, et al. Vitamin B12 deficiency alters the gut microbiota in a murine model of colitis. Front Nutr. (2020) 7:83. doi: 10.3389/fnut.2020.00083
20. Zhang X, Chen M, Ni X, Wang Y, Zheng X, Zhang H, et al. Metabolic reprogramming of sulfur in hepatocellular carcinoma and sulfane sulfur-triggered anti-cancer strategy. Front Pharmacol. (2020) 11:571143. doi: 10.3389/fphar.2020.571143
21. Jan M, Cueto R, Jiang X, Lu L, Sardy J, Xiong X, et al. Molecular processes mediating hyperhomocysteinemia-induced metabolic reprogramming, redox regulation and growth inhibition in endothelial cells. Redox Biol. (2021) 45:102018. doi: 10.1016/j.redox.2021.102018
22. Fu Y, Wang X, Kong W. Hyperhomocysteinaemia and vascular injury: advances in mechanisms and drug targets. Br J Pharmacol. (2018) 175:1173–89. doi: 10.1111/bph.13988
23. Li H, Liu Z, Liu L, Li W, Cao Z, Song Z, et al. Vascular protection of TPE-CA on hyperhomocysteinemia-induced vascular endothelial dysfunction through AA metabolism modulated CYPs pathway. Int J Biol Sci. (2019) 15:2037–50. doi: 10.7150/ijbs.35245
24. Zhang T, Lu R, Chen Y, Yuan Y, Song S, Yan K, et al. Hyperhomocysteinemia and dyslipidemia in point mutation G307S of cystathionine beta-synthase-deficient rabbit generated using CRISPR/Cas9. Lipids Health Dis. (2020) 19:224. doi: 10.1186/s12944-020-01394-5
25. Nakladal D, Lambooy SPH, Misuth S, Cepcova D, Joschko CP, van Buiten A, et al. Homozygous whole body Cbs knockout in adult mice features minimal pathology during ageing despite severe homocysteinemia. FASEB J. (2022) 36:e22260. doi: 10.1096/fj.202101550R
26. Cui X, Navneet S, Wang J, Roon P, Chen W, Xian M, et al. Analysis of MTHFR, CBS, glutathione, taurine, and hydrogen sulfide levels in retinas of hyperhomocysteinemic mice. Invest Ophthalmol Vis Sci. (2017) 58:1954–63. doi: 10.1167/iovs.16-21247
27. Zheng Z, Liu L, Zhou K, Ding L, Zeng J, Zhang W. Anti-oxidant and anti-endothelial dysfunctional properties of nano-selenium in vitro and in vivo of hyperhomocysteinemic rats. Int J Nanomedicine. (2020) 15:4501–21. doi: 10.2147/IJN.S255392
28. Paul L, Jacques PF, Aviv A, Vasan RS, D’Agostino RB, Levy D, et al. High plasma folate is negatively associated with leukocyte telomere length in framingham offspring cohort. Eur J Nutr. (2015) 54:235–41. doi: 10.1007/s00394-014-0704-1
29. Morellato AE, Umansky C, Pontel LB. The toxic side of one-carbon metabolism and epigenetics. Redox Biol. (2021) 40:101850. doi: 10.1016/j.redox.2020.101850
30. Qureshi SS, Gupta JK, Goyal A, Narayan YH. A novel approach in the management of hyperhomocysteinemia. Med Hypotheses. (2019) 129:109245. doi: 10.1016/j.mehy.2019.109245
31. Calcaterra V, Larizza D, De Giuseppe R, De Liso F, Klersy C, Albertini R, et al. Diet and lifestyle role in homocysteine metabolism in turner syndrome. Med Princ Pract. (2019) 28:48–55. doi: 10.1159/000494138
32. Cheng Z, Shen X, Jiang X, Shan H, Cimini M, Fang P, et al. Hyperhomocysteinemia potentiates diabetes-impaired EDHF-induced vascular relaxation: role of insufficient hydrogen sulfide. Redox Biol. (2018) 16:215–25. doi: 10.1016/j.redox.2018.02.006
33. Zhao G, Deng J, Shen Y, Zhang P, Dong H, Xie Z, et al. Hyperhomocysteinemia is key for increased susceptibility to PND in aged mice. Ann Clin Transl Neurol. (2019) 6:1435–44. doi: 10.1002/acn3.50838
34. Mayer EL, Jacobsen DW, Robinson K. Homocysteine and coronary atherosclerosis. J Am Coll Cardiol. (1996) 27:517–27. doi: 10.1016/0735-1097(95)00508-0
35. Bai J, Lechuga TJ, Makhoul J, Yan H, Major C, Hameed A, et al. ERalpha/ERbeta-directed CBS transcription mediates E2beta-stimulated hUAEC H2S production. J Mol Endocrinol. (2022). [Epub ahead of print]. doi: 10.1530/JME-22-0175
36. Raza-Iqbal S, Tanaka T, Anai M, Inagaki T, Matsumura Y, Ikeda K, et al. Transcriptome analysis of K-877 (a novel selective PPARalpha modulator (SPPARMalpha))-regulated genes in primary human hepatocytes and the mouse liver. J Atheroscler Thromb. (2015) 22:754–72. doi: 10.5551/jat.28720
37. Hamed AM, El-Kharashi OA, Boctor SS, Abd-Elaziz LF. Potential involvement of PPAR alpha activation in diminishing the hepatoprotective effect of fenofibrate in NAFLD: accuracy of non- invasive panel in determining the stage of liver fibrosis in rats. Biomed Pharmacother. (2017) 85:68–78. doi: 10.1016/j.biopha.2016.11.114
38. Cole PD, Vijayanathan V, Ali NF, Wagshul ME, Tanenbaum EJ, Price J, et al. Memantine protects rats treated with intrathecal methotrexate from developing spatial memory deficits. Clin Cancer Res. (2013) 19:4446–54. doi: 10.1158/1078-0432.CCR-13-1179
39. Sirichoat A, Anosri T, Kaewngam S, Aranarochana A, Pannangrong W, Wigmore P, et al. Neuroprotective properties of chrysin on decreases of cell proliferation, immature neurons and neuronal cell survival in the hippocampal dentate gyrus associated with cognition induced by methotrexate. Neurotoxicology. (2022) 92:15–24. doi: 10.1016/j.neuro.2022.06.010
40. Padovani D, Hessani A, Castillo FT, Liot G, Andriamihaja M, Lan A, et al. Sulfheme formation during homocysteine S-oxygenation by catalase in cancers and neurodegenerative diseases. Nat Commun. (2016) 7:13386. doi: 10.1038/ncomms13386
41. Lin X, Meng X, Song Z. Homocysteine and psoriasis. Biosci Rep. (2019) 39:BSR20190867. doi: 10.1042/BSR20190867
42. Rong D, Liu J, Jia X, Al-Nafisee D, Jia S, Sun G, et al. Hyperhomocysteinaemia is an independent risk factor for peripheral arterial disease in a Chinese Han population. Atherosclerosis. (2017) 263:205–10. doi: 10.1016/j.atherosclerosis.2017.05.006
43. Mohammad G, Radhakrishnan R, Kowluru RA. Hydrogen sulfide: a potential therapeutic target in the development of diabetic retinopathy. Invest Ophthalmol Vis Sci. (2020) 61:35. doi: 10.1167/iovs.61.14.35
44. Garcia-Alfaro P, Rodriguez I, Pascual MA. Evaluation of the relationship between homocysteine levels and bone mineral density in postmenopausal women. Climacteric. (2022) 25:179–85. doi: 10.1080/13697137.2021.1921729
45. Aminzadeh MA, Vaziri ND. Downregulation of the renal and hepatic hydrogen sulfide (H2S)-producing enzymes and capacity in chronic kidney disease. Nephrol Dial Transplant. (2012) 27:498–504. doi: 10.1093/ndt/gfr560
46. McGregor DO, Dellow WJ, Lever M, George PM, Robson RA, Chambers ST. Dimethylglycine accumulates in uremia and predicts elevated plasma homocysteine concentrations. Kidney Int. (2001) 59:2267–72. doi: 10.1046/j.1523-1755.2001.00743.x
47. Gao N, Zhang Y, Li L, Lei L, Cao P, Zhao X, et al. Hyperhomocysteinemia-induced oxidative stress aggravates renal damage in hypertensive rats. Am J Hypertens. (2020) 33:1127–35. doi: 10.1093/ajh/hpaa086
48. Zhang S, Zhang Y, Zhang X, Luo C, Cao Y, Ji D, et al. Nitrative stress-related autophagic insufficiency participates in hyperhomocysteinemia-induced renal aging. Oxid Med Cell Longev. (2020) 2020:4252047. doi: 10.1155/2020/4252047
49. Sedillo JC, Cryns VL. Targeting the methionine addiction of cancer. Am J Cancer Res. (2022) 12:2249–76.
50. Ndrepepa G, Kastrati A, Braun S, Koch W, Kolling K, Mehilli J, et al. Circulating homocysteine levels in patients with type 2 diabetes mellitus. Nutr Metab Cardiovasc Dis. (2008) 18:66–73. doi: 10.1016/j.numecd.2006.03.007
51. Cheng Z, Jiang X, Pansuria M, Fang P, Mai J, Mallilankaraman K, et al. Hyperhomocysteinemia and hyperglycemia induce and potentiate endothelial dysfunction via mu-calpain activation. Diabetes. (2015) 64:947–59. doi: 10.2337/db14-0784
52. Luk C, Haywood NJ, Bridge KI, Kearney MT. Paracrine role of the endothelium in metabolic homeostasis in health and nutrient excess. Front Cardiovasc Med. (2022) 9:882923. doi: 10.3389/fcvm.2022.882923
53. Qiu T, Zhou H, Li S, Tian N, Li Z, Wang R, et al. Effects of saccharides from Arctium lappa L. Root on FeCl3-induced arterial thrombosis via the ERK/NF-kappaB signaling pathway. Oxid Med Cell Longev. (2020) 2020:7691352. doi: 10.1155/2020/7691352
54. Jiang H, Zhou Y, Nabavi SM, Sahebkar A, Little PJ, Xu S, et al. Mechanisms of oxidized LDL-mediated endothelial dysfunction and its consequences for the development of atherosclerosis. Front Cardiovasc Med. (2022) 9:925923. doi: 10.3389/fcvm.2022.925923
55. Dagvadorj J, Shimada K, Chen S, Jones HD, Tumurkhuu G, Zhang W, et al. Lipopolysaccharide Induces alveolar macrophage necrosis via CD14 and the P2X7 receptor leading to interleukin-1alpha release. Immunity. (2015) 42:640–53. doi: 10.1016/j.immuni.2015.03.007
56. Wei X, Ying M, Dehaini D, Su Y, Kroll AV, Zhou J, et al. Nanoparticle functionalization with platelet membrane enables multifactored biological targeting and detection of atherosclerosis. ACS Nano. (2018) 12:109–16. doi: 10.1021/acsnano.7b07720
57. Tan W, Wang Y, Wang K, Wang S, Liu J, Qin X, et al. Improvement of endothelial dysfunction of berberine in atherosclerotic mice and mechanism exploring through TMT-based proteomics. Oxid Med Cell Longev. (2020) 2020:8683404. doi: 10.1155/2020/8683404
58. Zhang Y, Li C, Huang Y, Zhao S, Xu Y, Chen Y, et al. EOFAZ inhibits endothelialtomesenchymal transition through downregulation of KLF4. Int J Mol Med. (2020) 46:300–10. doi: 10.3892/ijmm.2020.4572
59. Kamanna VS, Ganji SH, Shelkovnikov S, Norris K, Vaziri ND. Iron sucrose promotes endothelial injury and dysfunction and monocyte adhesion/infiltration. Am J Nephrol. (2012) 35:114–9. doi: 10.1159/000334939
60. Chen LT, Xu TT, Qiu YQ, Liu NY, Ke XY, Fang L, et al. Homocysteine induced a calcium-mediated disruption of mitochondrial function and dynamics in endothelial cells. J Biochem Mol Toxicol. (2021) 35:e22737. doi: 10.1002/jbt.22737
61. Rodionov RN, Dayoub H, Lynch CM, Wilson KM, Stevens JW, Murry DJ, et al. Overexpression of dimethylarginine dimethylaminohydrolase protects against cerebral vascular effects of hyperhomocysteinemia. Circ Res. (2010) 106:551–8. doi: 10.1161/CIRCRESAHA.109.200360
62. Kang SS, Wong PW, Malinow MR. Hyperhomocyst(e)inemia as a risk factor for occlusive vascular disease. Annu Rev Nutr. (1992) 12:279–98. doi: 10.1146/annurev.nu.12.070192.001431
63. Stampfer MJ, Malinow MR, Willett WC, Newcomer LM, Upson B, Ullmann D, et al. A prospective study of plasma homocyst(e)ine and risk of myocardial infarction in US physicians. JAMA. (1992) 268:877–81. doi: 10.1001/jama.268.7.877
64. Li T, Zhu J, Fang Q, Duan X, Zhang M, Diao S, et al. Association of H-type hypertension with stroke severity and prognosis. Biomed Res Int. (2018) 2018:8725908. doi: 10.1155/2018/8725908
65. Jiang T, Zhang W, Wang Z. Laquinimod protects against TNF-alpha-induced attachment of monocytes to human aortic endothelial cells (HAECs) by increasing the expression of KLF2. Drug Des Devel Ther. (2020) 14:1683–91. doi: 10.2147/DDDT.S243666
66. Chhour P, Naha PC, O’Neill SM, Litt HI, Reilly MP, Ferrari VA, et al. Labeling monocytes with gold nanoparticles to track their recruitment in atherosclerosis with computed tomography. Biomaterials. (2016) 87:93–103.
67. Leng Y, Chen R, Chen R, He S, Shi X, Zhou X, et al. HMGB1 mediates homocysteine-induced endothelial cells pyroptosis via cathepsin V-dependent pathway. Biochem Biophys Res Commun. (2020) 532:640–6. doi: 10.1016/j.bbrc.2020.08.091
68. Han S, Wu H, Li W, Gao P. Protective effects of genistein in homocysteine-induced endothelial cell inflammatory injury. Mol Cell Biochem. (2015) 403:43–9. doi: 10.1007/s11010-015-2335-0
69. Hammons AL, Summers CM, Jochems J, Arora JS, Zhang S, Blair IA, et al. Pemetrexed alters folate phenotype and inflammatory profile in EA.hy 926 cells grown under low-folate conditions. Eur J Pharmacol. (2012) 696:12–7. doi: 10.1016/j.ejphar.2012.08.008
70. Poddar R, Sivasubramanian N, Dibello PM, Robinson K, Jacobsen DW. Homocysteine induces expression and secretion of monocyte chemoattractant protein-1 and interleukin-8 in human aortic endothelial cells: implications for vascular disease. Circulation. (2001) 103:2717–23. doi: 10.1161/01.CIR.103.22.2717
71. Meng Q, Li Y, Ji T, Chao Y, Li J, Fu Y, et al. Estrogen prevent atherosclerosis by attenuating endothelial cell pyroptosis via activation of estrogen receptor alpha-mediated autophagy. J Adv Res. (2021) 28:149–64. doi: 10.1016/j.jare.2020.08.010
72. Li W, Yuan W, Zhang D, Cai S, Luo J, Zeng K. LCZ696 possesses a protective effect against homocysteine (Hcy)-induced impairment of blood-brain barrier (BBB) integrity by increasing occludin, mediated by the inhibition of Egr-1. Neurotox Res. (2021) 39:1981–90. doi: 10.1007/s12640-021-00414-1
73. Sun T, Ghosh AK, Eren M, Miyata T, Vaughan DE. PAI-1 contributes to homocysteine-induced cellular senescence. Cell Signal. (2019) 64:109394. doi: 10.1016/j.cellsig.2019.109394
74. Herrmann W, Herrmann M. The controversial role of HCY and vitamin B deficiency in cardiovascular diseases. Nutrients. (2022) 14:1412. doi: 10.3390/nu14071412
75. Wang X, Wang Y, Zhang L, Zhang D, Bai L, Kong W, et al. L-cystathionine protects against homocysteine-induced mitochondria-dependent apoptosis of vascular endothelial cells. Oxid Med Cell Longev. (2019) 2019:1253289. doi: 10.1155/2019/1253289
76. Xi H, Zhang Y, Xu Y, Yang WY, Jiang X, Sha X, et al. Caspase-1 inflammasome activation mediates homocysteine-induced pyrop-apoptosis in endothelial cells. Circ Res. (2016) 118:1525–39. doi: 10.1161/CIRCRESAHA.116.308501
77. Wang Y, Kuang X, Yin Y, Han N, Chang L, Wang H, et al. Tongxinluo prevents chronic obstructive pulmonary disease complicated with atherosclerosis by inhibiting ferroptosis and protecting against pulmonary microvascular barrier dysfunction. Biomed Pharmacother. (2022) 145:112367. doi: 10.1016/j.biopha.2021.112367
78. Kopacz A, Kloska D, Proniewski B, Cysewski D, Personnic N, Piechota-Polanczyk A, et al. Keap1 controls protein S-nitrosation and apoptosis-senescence switch in endothelial cells. Redox Biol. (2020) 28:101304. doi: 10.1016/j.redox.2019.101304
79. Forstermann U, Xia N, Li H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ Res. (2017) 120:713–35. doi: 10.1161/CIRCRESAHA.116.309326
80. Huang H, Koelle P, Fendler M, Schrottle A, Czihal M, Hoffmann U, et al. Induction of inducible nitric oxide synthase (iNOS) expression by oxLDL inhibits macrophage derived foam cell migration. Atherosclerosis. (2014) 235:213–22. doi: 10.1016/j.atherosclerosis.2014.04.020
81. Degjoni A, Campolo F, Stefanini L, Venneri MA. The NO/cGMP/PKG pathway in platelets: the therapeutic potential of PDE5 inhibitors in platelet disorders. J Thromb Haemost. (2022) 20:2465–74. doi: 10.1111/jth.15844
82. Ning K, Wang MJ, Lin G, Zhang YL, Li MY, Yang BF, et al. eNOS-Nitric oxide system contributes to a novel antiatherogenic effect of leonurine via inflammation inhibition and plaque stabilization. J Pharmacol Exp Ther. (2020) 373:463–75. doi: 10.1124/jpet.119.264887
83. Zhang B, Qin Y, Wang Y. A nitric oxide-eluting and REDV peptide-conjugated coating promotes vascular healing. Biomaterials. (2022) 284:121478. doi: 10.1016/j.biomaterials.2022.121478
84. Cai D, Chen SY. Nanoparticle endothelial delivery of PGC-1alpha attenuates hypoxia-induced pulmonary hypertension by attenuating EndoMT-caused vascular wall remodeling. Redox Biol. (2022) 58:102524. doi: 10.1016/j.redox.2022.102524
85. Lucas-Herald AK, Touyz RM. Androgens and androgen receptors as determinants of vascular sex differences across the lifespan. Can J Cardiol. (2022) 38:1854–64. doi: 10.1016/j.cjca.2022.09.018
86. Gopu CL, Hari PR, George R, Harikrishnan S, Sreenivasan K. Simultaneous determination of homocysteine and asymmetric dimethylarginine in human urine by liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. (2013) 939:32–7. doi: 10.1016/j.jchromb.2013.09.010
87. Smith DE, Hornstra JM, Kok RM, Blom HJ, Smulders YM. Folic acid supplementation does not reduce intracellular homocysteine, and may disturb intracellular one-carbon metabolism. Clin Chem Lab Med. (2013) 51:1643–50. doi: 10.1515/cclm-2012-0694
88. Zhu H, Bhatt B, Sivaprakasam S, Cai Y, Liu S, Kodeboyina SK, et al. Ufbp1 promotes plasma cell development and ER expansion by modulating distinct branches of UPR. Nat Commun. (2019) 10:1084. doi: 10.1038/s41467-019-08908-5
89. Wu X, Zhang L, Miao Y, Yang J, Wang X, Wang CC, et al. Homocysteine causes vascular endothelial dysfunction by disrupting endoplasmic reticulum redox homeostasis. Redox Biol. (2019) 20:46–59. doi: 10.1016/j.redox.2018.09.021
90. Sprenkle NT, Sims SG, Sanchez CL, Meares GP. Endoplasmic reticulum stress and inflammation in the central nervous system. Mol Neurodegener. (2017) 12:42. doi: 10.1186/s13024-017-0183-y
91. Rahman MA, Glasgow JN, Nadeem S, Reddy VP, Sevalkar RR, Lancaster JR JR., et al. The role of host-generated H2S in microbial pathogenesis: new perspectives on tuberculosis. Front Cell Infect Microbiol. (2020) 10:586923. doi: 10.3389/fcimb.2020.586923
92. Zhao L, Xiao Y, Weng RX, Liu X, Zhang PA, Hu CY, et al. Neonatal colonic inflammation increases spinal transmission and cystathionine beta-synthetase expression in spinal dorsal horn of rats with visceral hypersensitivity. Front Pharmacol. (2017) 8:696. doi: 10.3389/fphar.2017.00696
93. Li H, Teng X, Yang R, Guo Q, Xue H, Xiao L, et al. Hydrogen sulfide facilitates the impaired sensitivity of carotid sinus baroreflex in rats with vascular calcification. Front Pharmacol. (2017) 8:629. doi: 10.3389/fphar.2017.00629
94. Foster JC, Carrazzone RJ, Spear NJ, Radzinski SC, Arrington KJ, Matson JB. Tuning H2S release by controlling mobility in a micelle core. Macromolecules. (2019) 52:1104–11. doi: 10.1021/acs.macromol.8b02315
95. Sun HJ, Wu ZY, Nie XW, Bian JS. Role of endothelial dysfunction in cardiovascular diseases: the link between inflammation and hydrogen sulfide. Front Pharmacol. (2019) 10:1568. doi: 10.3389/fphar.2019.01568
96. Citi V, Martelli A, Gorica E, Brogi S, Testai L, Calderone V. Role of hydrogen sulfide in endothelial dysfunction: pathophysiology and therapeutic approaches. J Adv Res. (2021) 27:99–113. doi: 10.1016/j.jare.2020.05.015
97. Zicola E, Arrigo E, Mancardi D. H2S pretreatment is promigratory and decreases ischemia/reperfusion injury in human microvascular endothelial cells. Oxid Med Cell Longev. (2021) 2021:8886666. doi: 10.1155/2021/8886666
98. Xue WL, Chen RQ, Zhang QQ, Li XH, Cao L, Li MY, et al. Hydrogen sulfide rescues high glucose-induced migration dysfunction in HUVECs by upregulating miR-126-3p. Am J Physiol Cell Physiol. (2020) 318:C857–69. doi: 10.1152/ajpcell.00406.2019
99. Wen YD, Wang H, Zhu YZ. The drug developments of hydrogen sulfide on cardiovascular disease. Oxid Med Cell Longev. (2018) 2018:4010395. doi: 10.1155/2018/4010395
100. Xiao L, Dong JH, Teng X, Jin S, Xue HM, Liu SY, et al. Hydrogen sulfide improves endothelial dysfunction in hypertension by activating peroxisome proliferator-activated receptor delta/endothelial nitric oxide synthase signaling. J Hypertens. (2018) 36:651–65. doi: 10.1097/HJH.0000000000001605
101. Kamat PK, Kalani A, Tyagi SC, Tyagi N. Hydrogen sulfide epigenetically attenuates homocysteine-induced mitochondrial toxicity mediated through NMDA receptor in mouse brain endothelial (bEnd3) Cells. J Cell Physiol. (2015) 230:378–94. doi: 10.1002/jcp.24722
102. Bian Y, Li W, Kremer DM, Sajjakulnukit P, Li S, Crespo J, et al. Cancer SLC43A2 alters T cell methionine metabolism and histone methylation. Nature. (2020) 585:277–82. doi: 10.1038/s41586-020-2682-1
103. Liu Q, Lin B, Tao Y. Improved methylation in E. coli via an efficient methyl supply system driven by betaine. Metab Eng. (2022) 72:46–55. doi: 10.1016/j.ymben.2022.02.004
104. Esse R, Imbard A, Florindo C, Gupta S, Quinlivan EP, Davids M, et al. Protein arginine hypomethylation in a mouse model of cystathionine beta-synthase deficiency. FASEB J. (2014) 28:2686–95. doi: 10.1096/fj.13-246579
105. Lupu DS, Orozco LD, Wang Y, Cullen JM, Pellegrini M, Zeisel SH. Altered methylation of specific DNA loci in the liver of Bhmt-null mice results in repression of Iqgap2 and F2rl2 and is associated with development of preneoplastic foci. FASEB J. (2017) 31:2090–103. doi: 10.1096/fj.201601169R
106. da Silva IV, Barroso M, Moura T, Castro R, Soveral G. Endothelial aquaporins and hypomethylation: potential implications for atherosclerosis and cardiovascular disease. Int J Mol Sci. (2018) 19:130. doi: 10.3390/ijms19010130
107. Barroso M, Florindo C, Kalwa H, Silva Z, Turanov AA, Carlson BA, et al. Inhibition of cellular methyltransferases promotes endothelial cell activation by suppressing glutathione peroxidase 1 protein expression. J Biol Chem. (2014) 289:15350–62. doi: 10.1074/jbc.M114.549782
108. Zhang HP, Wang YH, Ma SC, Zhang H, Yang AN, Yang XL, et al. Homocysteine inhibits endothelial progenitor cells proliferation via DNMT1-mediated hypomethylation of Cyclin A. Exp Cell Res. (2018) 362:217–26. doi: 10.1016/j.yexcr.2017.11.021
109. Kameda T, Horiuchi Y, Shimano S, Yano K, Lai SJ, Ichimura N, et al. Effect of myeloperoxidase oxidation and N-homocysteinylation of high-density lipoprotein on endothelial repair function. Biol Chem. (2022) 403:265–77. doi: 10.1515/hsz-2021-0247
110. Samant RS, Livingston CM, Sontag EM, Frydman J. Distinct proteostasis circuits cooperate in nuclear and cytoplasmic protein quality control. Nature. (2018) 563:407–11. doi: 10.1038/s41586-018-0678-x
111. Xu N, Gulick J, Osinska H, Yu Y, Mclendon PM, Shay-Winkler K, et al. Ube2v1 positively regulates protein aggregation by modulating ubiquitin proteasome system performance partially through K63 ubiquitination. Circ Res. (2020) 126:907–22.
112. Varanda AS, Santos M, Soares AR, Vitorino R, Oliveira P, Oliveira C, et al. Human cells adapt to translational errors by modulating protein synthesis rate and protein turnover. RNA Biol. (2020) 17:135–49. doi: 10.1080/15476286.2019.1670039
113. Khandros E, Thom CS, D’Souza J, Weiss MJ. Integrated protein quality-control pathways regulate free alpha-globin in murine beta-thalassemia. Blood. (2012) 119:5265–75. doi: 10.1182/blood-2011-12-397729
114. Reddy VS, Trinath J, Reddy GB. Implication of homocysteine in protein quality control processes. Biochimie. (2019) 165:19–31. doi: 10.1016/j.biochi.2019.06.017
115. Huang A, Pinto JT, Froogh G, Kandhi S, Qin J, Wolin MS, et al. Role of homocysteinylation of ACE in endothelial dysfunction of arteries. Am J Physiol Heart Circ Physiol. (2015) 308:H92–100. doi: 10.1152/ajpheart.00577.2014
116. Wu Z, Jin F, Wang L, Zhao Y, Jiang Y, Li J, et al. Antioxidant effects of baoyuan decoction on dysfunctional erythrocytes in high-fat diet-induced hyperlipidemic ApoE(-/-) Mice. Oxid Med Cell Longev. (2019) 2019:5172480. doi: 10.1155/2019/5172480
117. Yang Q, Civelek M. Transcription factor KLF14 and metabolic syndrome. Front Cardiovasc Med. (2020) 7:91. doi: 10.3389/fcvm.2020.00091
118. Zhou L, Liu J, An Y, Wang Y, Wang G. Plasma homocysteine level is independently associated with conventional atherogenic lipid profile and remnant cholesterol in adults. Front Cardiovasc Med. (2022) 9:898305. doi: 10.3389/fcvm.2022.898305
119. Zhang X, Bishawi M, Zhang G, Prasad V, Salmon E, Breithaupt JJ, et al. Modeling early stage atherosclerosis in a primary human vascular microphysiological system. Nat Commun. (2020) 11:5426. doi: 10.1038/s41467-020-19197-8
120. Ma C, Xia R, Yang S, Liu L, Zhang J, Feng K, et al. Formononetin attenuates atherosclerosis via regulating interaction between KLF4 and SRA in apoE(-/-) mice. Theranostics. (2020) 10:1090–106. doi: 10.7150/thno.38115
121. Benowitz NL, Fraiman JB. Cardiovascular effects of electronic cigarettes. Nat Rev Cardiol. (2017) 14:447–56. doi: 10.1038/nrcardio.2017.36
122. Zaric BL, Obradovic M, Bajic V, Haidara MA, Jovanovic M, Isenovic ER. Homocysteine and Hyperhomocysteinaemia. Curr Med Chem. (2019) 26:2948–61. doi: 10.2174/0929867325666180313105949
123. Chernyavskiy I, Veeranki S, Sen U, Tyagi SC. Atherogenesis: hyperhomocysteinemia interactions with LDL, macrophage function, paraoxonase 1, and exercise. Ann N Y Acad Sci. (2016) 1363:138–54. doi: 10.1111/nyas.13009
124. Hou L, Wang H, Sartori S, Gawron A, Lissowska J, Bollati V, et al. Blood leukocyte DNA hypomethylation and gastric cancer risk in a high-risk Polish population. Int J Cancer. (2010) 127:1866–74. doi: 10.1002/ijc.25190
125. Cui S, Li W, Wang P, Lv X, Gao Y, Huang G. Folic acid inhibits homocysteine-induced cell apoptosis in human umbilical vein endothelial cells. Mol Cell Biochem. (2018) 444:77–86. doi: 10.1007/s11010-017-3232-5
126. Cui S, Li W, Lv X, Wang P, Gao Y, Huang G. Folic acid supplementation delays atherosclerotic lesion development by modulating MCP1 and VEGF DNA methylation levels in vivo and in vitro. Int J Mol Sci. (2017) 18:990. doi: 10.3390/ijms18050990
127. Marti-Carvajal AJ, Sola I, Lathyris D. Homocysteine-lowering interventions for preventing cardiovascular events. Cochrane Database Syst Rev. (2015) 1:CD006612. doi: 10.1002/14651858.CD006612.pub4
128. Smulders YM, Blom HJ. The homocysteine controversy. J Inherit Metab Dis. (2011) 34:93–9. doi: 10.1007/s10545-010-9151-1
129. Hou H, Zhao H. Epigenetic factors in atherosclerosis: DNA methylation, folic acid metabolism, and intestinal microbiota. Clin Chim Acta. (2021) 512:7–11. doi: 10.1016/j.cca.2020.11.013
130. Aykutoglu G, Tartik M, Darendelioglu E, Ayna A, Baydas G. Melatonin and vitamin E alleviate homocysteine-induced oxidative injury and apoptosis in endothelial cells. Mol Biol Rep. (2020) 47:5285–93. doi: 10.1007/s11033-020-05607-z
131. Ji C, Yi H, Huang J, Zhang W, Zheng M. Propofol alleviates inflammation and apoptosis in HCYinduced HUVECs by inhibiting endoplasmic reticulum stress. Mol Med Rep. (2021) 23:333. doi: 10.3892/mmr.2021.11972
132. Pai PY, Chou WC, Chan SH, Wu SY, Chen HI, Li CW, et al. Epigallocatechin gallate reduces homocysteine-caused oxidative damages through modulation SIRT1/AMPK pathway in endothelial cells. Am J Chin Med. (2021) 49:113–29. doi: 10.1142/S0192415X21500063
133. Zhan B, Xu Z, Zhang Y, Wan K, Deng H, Wang D, et al. Nicorandil reversed homocysteine-induced coronary microvascular dysfunction via regulating PI3K/Akt/eNOS pathway. Biomed Pharmacother. (2020) 127:110121. doi: 10.1016/j.biopha.2020.110121
134. Shen W, Gao C, Cueto R, Liu L, Fu H, Shao Y, et al. Homocysteine-methionine cycle is a metabolic sensor system controlling methylation-regulated pathological signaling. Redox Biol. (2020) 28:101322. doi: 10.1016/j.redox.2019.101322
135. Jiang X, Yang F, Brailoiu E, Jakubowski H, Dun NJ, Schafer AI, et al. Differential regulation of homocysteine transport in vascular endothelial and smooth muscle cells. Arterioscler Thromb Vasc Biol. (2007) 27:1976–83. doi: 10.1161/ATVBAHA.107.148544
HHCY, hyperhomocysteinemia; AS, atherosclerosis; HCY, homocysteine; ECs, endothelial cells; NO, nitric oxide; ROS, reactive oxygen species; SAM, AdoMet, S-adenosylmethionine; MAT, methionine adenosyltransferase; ATP, adenosine triphosphate; SAH, AdoHcy, S-adenosylhomocysteine; CBS, cystathionine β-synthase; CSE, cystathionine-gamma-lyase; 5-methylTHF, 5-methyltetrahydrofolic acid; MS, methionine synthase; THF, tetrahydrofolate; BHMT, betaine-homocysteine methyltransferase; DHFR, dihydrofolate reductase; IL, interleukin; TNF, tumor necrosis factor; NF-κB, nuclear factor kappa-B; PAI-1, plasminogen activator inhibitor-1; SASP, senescence-associated secretory phenotype; NOS, nitric oxide synthase; ADMA, asymmetric dimethylarginine; DDAH, dimethylarginine dimethylaminohydrolase; PKC, protein kinase C; NADPH, nicotinamide adenine dinucleotide phosphate; ERS, endoplasmic reticulum stress; ER, endoplasmic reticulum; Ero-1α, endoplasmic reticulum oxidoreductin-1α; HIF-1α, hypoxia-inducible factor-1α; GPX, glutathione peroxidase; H2S, hydrogen sulfide; ERK, extracellular regulated protein kinases; miR, microRNA; LDL: low-density lipoproteins; HDL, high-density lipoproteins; HDL-C, high-density lipoprotein cholesterol; ApoA1, apolipoprotein A1; TG, triglyceride; TC, total cholesterol; LDL-C, low-density lipoprotein cholesterol; ox-LDL, oxidized low density lipoprotein; VSMCs, vascular smooth muscle cells; ICAM-1, intercellular adhesion molecule-1; VCAM-1, vascular adhesion molecule-1; DNMT1, DNA methyltransferase 1; EPCs, endothelial progenitor cells; HTL, homocysteine thiolactone; MetRS, methionyl-tRNA synthetase; PQC, cellular protein quality control; UPR, unfolded protein response; UPS, ubiquitin-proteasome system; ACE, angiotensin-converting enzyme; ANG II, angiotensin II; LPO, lipid peroxidation; Cyt C, cytochrome c; Bax, B-cell lymphoma-2 associated X protein; Bcl-2, B-cell lymphoma-2; HUVECs, human umbilic vein endothelial cells; GLP-1, glucagon-like peptide 1; EGCG, epigallocatechin gallate; SIRT1, silent information regulator 1; AMPK, AMP-activated protein kinase; Nano-Se, nanoscale selenium; HCAECs, human coronary artery endothelial cells; DAMPs, damage-associated molecular patterns; PAMPs, pathogen-associated molecular patterns; PRRs, pattern recognition receptors; ASC, alanine-serine-cysteine.
Keywords: homocysteine, endothelial cells, hyperhomocysteinemia, atherosclerosis, inflammation
Citation: Yuan D, Chu J, Lin H, Zhu G, Qian J, Yu Y, Yao T, Ping F, Chen F and Liu X (2023) Mechanism of homocysteine-mediated endothelial injury and its consequences for atherosclerosis. Front. Cardiovasc. Med. 9:1109445. doi: 10.3389/fcvm.2022.1109445
Received: 27 November 2022; Accepted: 28 December 2022;
Published: 16 January 2023.
Edited by:
Liming Yu, The University of Texas Health Science Center at San Antonio, United StatesReviewed by:
Yuanli Chen, Hefei University of Technology, ChinaCopyright © 2023 Yuan, Chu, Lin, Zhu, Qian, Yu, Yao, Ping, Chen and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xuebo Liu,  bGl1eHVlYm83MEAxMjYuY29t; Fei Chen, cml2ZXJhcHRAMTI2LmNvbQ==
bGl1eHVlYm83MEAxMjYuY29t; Fei Chen, cml2ZXJhcHRAMTI2LmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.