
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Cardiovasc. Med., 14 December 2022
Sec. Cardiac Rhythmology
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.1080608
This article is part of the Research TopicSudden Cardiac Death From Channelopathies and CardiomyopathiesView all 6 articles
 Francesca Girolami1*†
Francesca Girolami1*† Valentina Spinelli2†
Valentina Spinelli2† Niccolò Maurizi3
Niccolò Maurizi3 Martina Focardi3,4Gabriella Nesi3,5Vincenza Maio3,5Rossella Grifoni3,4Giuseppe Albora3
Martina Focardi3,4Gabriella Nesi3,5Vincenza Maio3,5Rossella Grifoni3,4Giuseppe Albora3 Bruno Bertaccini6Mattia Targetti3
Bruno Bertaccini6Mattia Targetti3 Raffaele Coppini2
Raffaele Coppini2 Silvia Favilli1Iacopo Olivotto1,7
Silvia Favilli1Iacopo Olivotto1,7 Elisabetta Cerbai2
Elisabetta Cerbai2Background: Sudden cardiac arrest (SCA) in young people represents a dramatic event, often leading to severe neurologic outcomes or sudden cardiac death (SCD), and is frequently caused by genetic heart diseases. In this study, we report the results of the Tuscany registry of sudden cardiac death (ToRSADE) registry, aimed at monitoring the incidence and investigating the genetic basis of SCA and SCD occurring in subjects < 50 years of age in Tuscany, Italy.
Methods and results: Creation of the ToRSADE registry allowed implementation of a repository for clinical, molecular and genetic data. For 22 patients, in whom a genetic substrate was documented or suspected, blood samples could be analyzed; 14 were collected at autopsy and 8 from resuscitated patients after SCA. Next generation sequencing (NGS) analysis revealed likely pathogenetic (LP) variants associated with cardiomyopathy (CM) or channelopathy in four patients (19%), while 17 (81%) carried variants of uncertain significance in relevant genes (VUS). In only one patient NGS confirmed the diagnosis obtained during autopsy: the p.(Asn480Lysfs*20) PKP2 mutation in a patient with arrhythmogenic cardiomyopathy (AC).
Conclusion: Systematic genetic screening allowed identification of LP variants in 19% of consecutive patients with SCA/SCD, including subjects carrying variants associated with hypertrophic cardiomyopathy (HCM) or AC who had SCA/SCD in the absence of structural cardiomyopathy phenotype. Genetic analysis combined with clinical information in survived patients and post-mortem evaluation represent an essential multi-disciplinary approach to manage juvenile SCD and SCA, key to providing appropriate medical and genetic assistance to families, and advancing knowledge on the basis of arrhythmogenic mechanisms in inherited cardiomyopathies and channelopathies.
Among all cases of out-of-hospital sudden cardiac arrest (SCA) or sudden cardiac death (SCD), those occurring at juvenile age are matters of social and clinical urgency. SCD is defined as a natural and unexpected cardiovascular collapse within 1 h of initial acute symptoms (1), affecting apparently healthy young people (≤ 40 years). SCD requires carefully designed, regional, or multicenter studies to generate accurate statistics regarding incidence and risk factors (2). In Italy, the incidence of SCD in the general population is 1/1,000 person/year (3) although in younger individuals the value is much lower (around 1/100,000 per year). These numbers are necessary to formulate useful public health policy for the early detection and prevention. In subjects below 35 years of age, no cause is identified in up to 30% of cases after clinical autopsy (4). Because major etiologies of SCD in the young are associated with inherited conditions, a genetic characterization and management of family members must often be conducted. Case series of SCD are valuable for defining etiology and mechanisms but they cannot quantify the denominator population (2). The creation of well-designed clinical registries is an emerging worldwide necessity; recently, many registries have been developed to monitor the occurrence of SCD/SCA in the young. SCD/SCA registries already created (49 of SCA and 15 of SCD) have been shown to support epidemiological analysis and ameliorate the attention between ambulance services, hospital procedures and clinical/molecular autopsy (4–10). In a pilot anonymized research study promoted by the Tuscany region, we outlined an algorithm suitable to monitor, collect, and investigate SCA or SCD in people aged from 18 to 50 years, referred to hospitals of the Florentine area.
The project planned to create and exploit the ToRSADE (Tuscany Registry of Sudden Cardiac Death) registry to (i) identify and record all patients deceased from SCD or resuscitated after SCA in Tuscany, (ii) establish a diagnostic-therapeutic plan and preventive-research strategy against SCD or SCA events in family members. We here describe the clinical records collected at Careggi University Hospital during the period 2016–2019.
The study was approved by the Local Ethical Committee (No. BIO.16.011) and conformed to the ethical guidelines of the declaration of Helsinki. The registry was based on the following inclusion and exclusion criteria.
– Age < 50 years;
– Patients who died in hospital due to brain death following cardiac arrest in whom autopsy was performed;
– Patient who died due to out-of-hospital cardiac arrest and in whom forensic autopsy was performed;
– Patients who survived cardiac arrest.
– Patients surviving cardiac arrest who denied informed consent;
– In-hospital or out-of-hospital death when cardiac arrest occurred as a consequence of traumatic injuries, toxicological causes, neoplastic or infectious disease and structural cardiac abnormalities.
Resuscitated patients were transferred to the emergency department of Careggi University Hospital for post-cardiac arrest care and clinical data collected where possible; blood samples were used for genetic testing. SCD subjects underwent full autopsy as described below.
According to the 2017 guidelines for autopsy investigation of SCD developed by the association for European cardiovascular pathology (AECVP) (11), a complete external and internal post-mortem examination was performed in all cases. The hearth was taken and fixed in formalin for subsequent examination. Evaluation of the conduction system, using a simplified method, was performed when no pathological findings were observed at gross or histological examination (11, 12). Myocardial samples of the ventricles and the major epicardial coronary arteries were then included in paraffin and cut with a microtome in micrometric slides. Hematoxylin and eosin staining was performed and observed under a microscope. If required, we also performed additional histological stains. Toxicological investigations were undertaken on peripheral venous blood. Finally, we sampled and stored blood (at −20°) and fresh tissues (heart, liver, and spleen at −80°) for long-term preservation.
A panel of 174 cardiac genes implicated in cardiomyopathies or channelopathies was analyzed by Next Generation Sequencing (NGS). Specifically, genomic DNA was automatically extracted from whole-blood samples, using QIAsymphony DSP DNA Kits in combination with the QIAsymphony SP (Qiagen, Hilden, Germany), following the manufacturer’s protocol. Gene libraries were prepared from DNA using an Illumina Nextera TruSight™ Cardio Sequencing Kit (Illumina Inc., San Diego, CA, USA). The sequencing step was performed on an Illumina MiSeq system, targeting for 151 bp pair-end reads, and a mean sequencing coverage averaging above 200×. Variant analysis was performed by Local Run Manager and Base Space Variant Interpreter software (Illumina Inc., San Diego, CA, USA). Sanger sequencing was used to confirm clinically relevant detected variants (forward and/or reverse strand). All variants were classified according to the American college of medical genetics (ACMG) classification (13) and the association for clinical genomic science (ACGS) (14, 15). Reanimated subjects received genetic counseling and gave informed consent before performing genetic tests (see Supplementary material).
The Figure 1 shows the genetic characterization of ToRSADE patients.
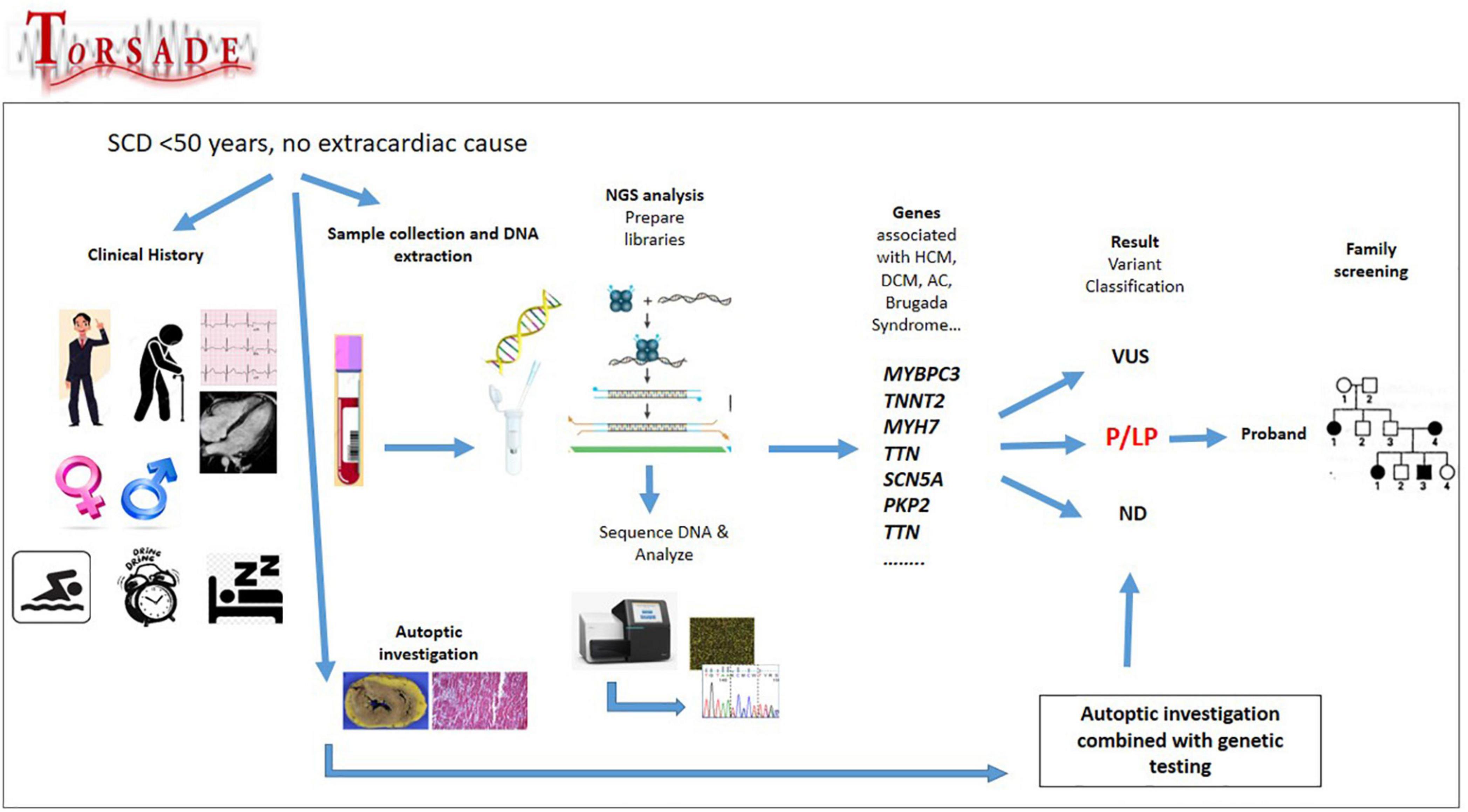
Figure 1. Genetic characterization of ToRSADE patients: an overview of ToRSADE workflow for the genetic characterization of diseases involved in sudden cardiac death.
Between July 2016 and February 2019, of 52 SCA/SCD victims seen at Careggi University Hospital (AOUC), 22 subjects met the inclusion criteria (15–18). Eight resuscitated patients underwent genetic analysis; the remaining 14, who died out of hospital or were brain-dead after SCA underwent clinical autopsy and genetic testing. For comparison, 34 subjects < 50 years were filed as out-of-hospital SCD by the local rescue service over the same period of time and referred to the district hospitals; however, none of them was genotyped and only three were annotated as receiving autopsy, but no results were available. The main features of the 22 study subjects are shown in Table 1. Data regarding past medical history and family history were available only for survived patients (Table 2). In 1 out of 14 cases, autopsy led to a diagnostic hypothesis of arrhythmogenic cardiomyopathy (AC) but, in the majority of cases, autoptic results were uncertain or inconclusive. Genetic investigation identified four likely pathogenic (LP) variants in deceased patients (submitted in ClinVar database1). Overall, 3/4 LP variants (75%) were found in genes associated with hypertrophic cardiomyopathy (HCM) (MYBPC3 c. 2441_2443del ClinVar ID#177700, TNNT2 c.517_519del ClinVar ID#43648, and TCAP c.360_361del ClinVar ID#1708038), one was detected in PKP2 c. 1440_1444del (ClinVar ID# 419977) associated with AC. Variants of uncertain significance (VUS) were found in 17 cases (eight resuscitated and nine dead) in genes associated with HCM or dilated cardiomyopathy (DCM), such as TTN, TNNT2, MYH6, DSP, ACTN2, CALR3, and MYH7. Of note, one SCD victim carried the SCN5A variant c.1398G > T (ClinVar ID#201453) associated with Brugada syndrome and classified as a VUS based on the ACGS guidelines (13, 14).
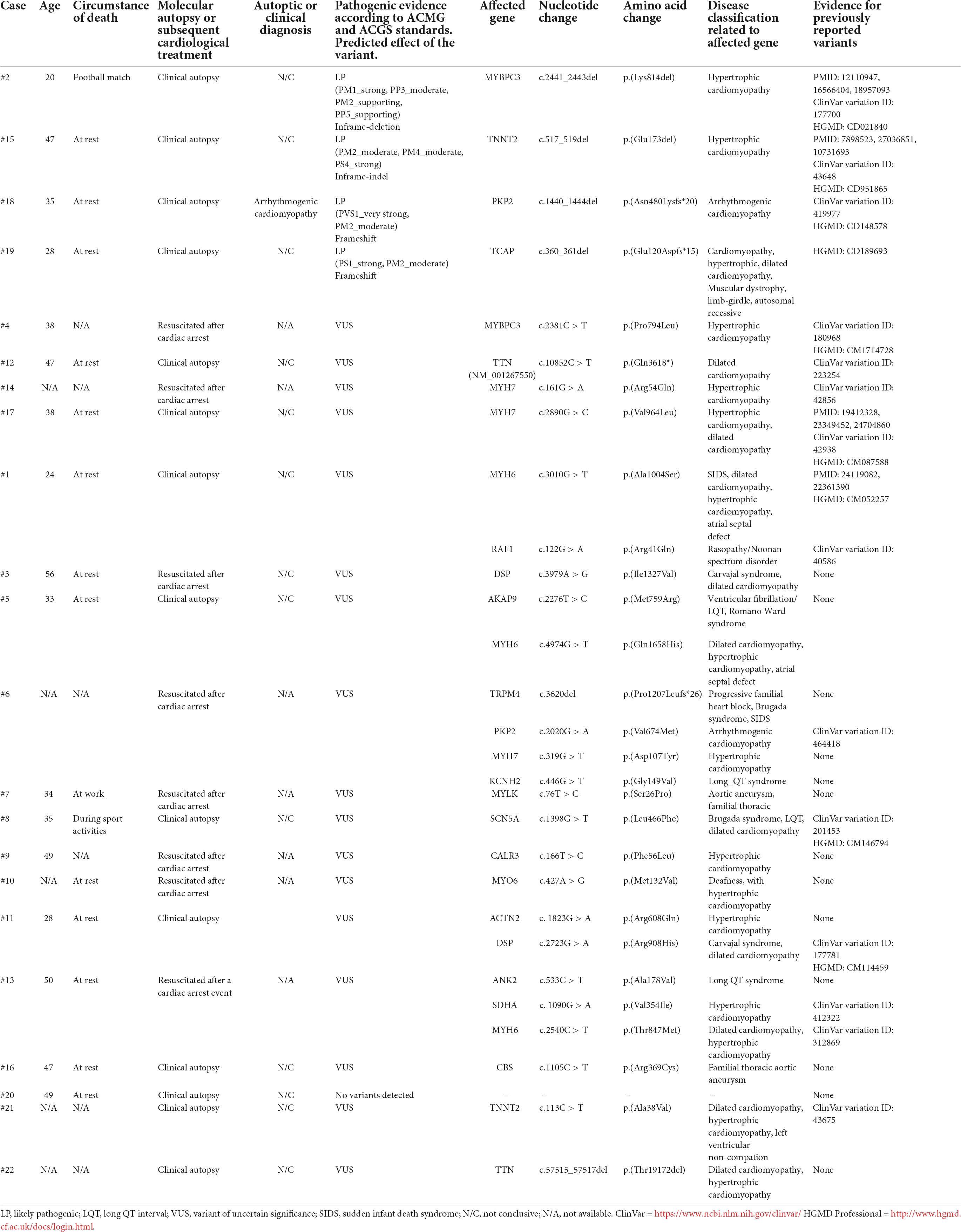
Table 1. Genetic findings in the Tuscany registry of sudden cardiac death (ToRSADE) registry.
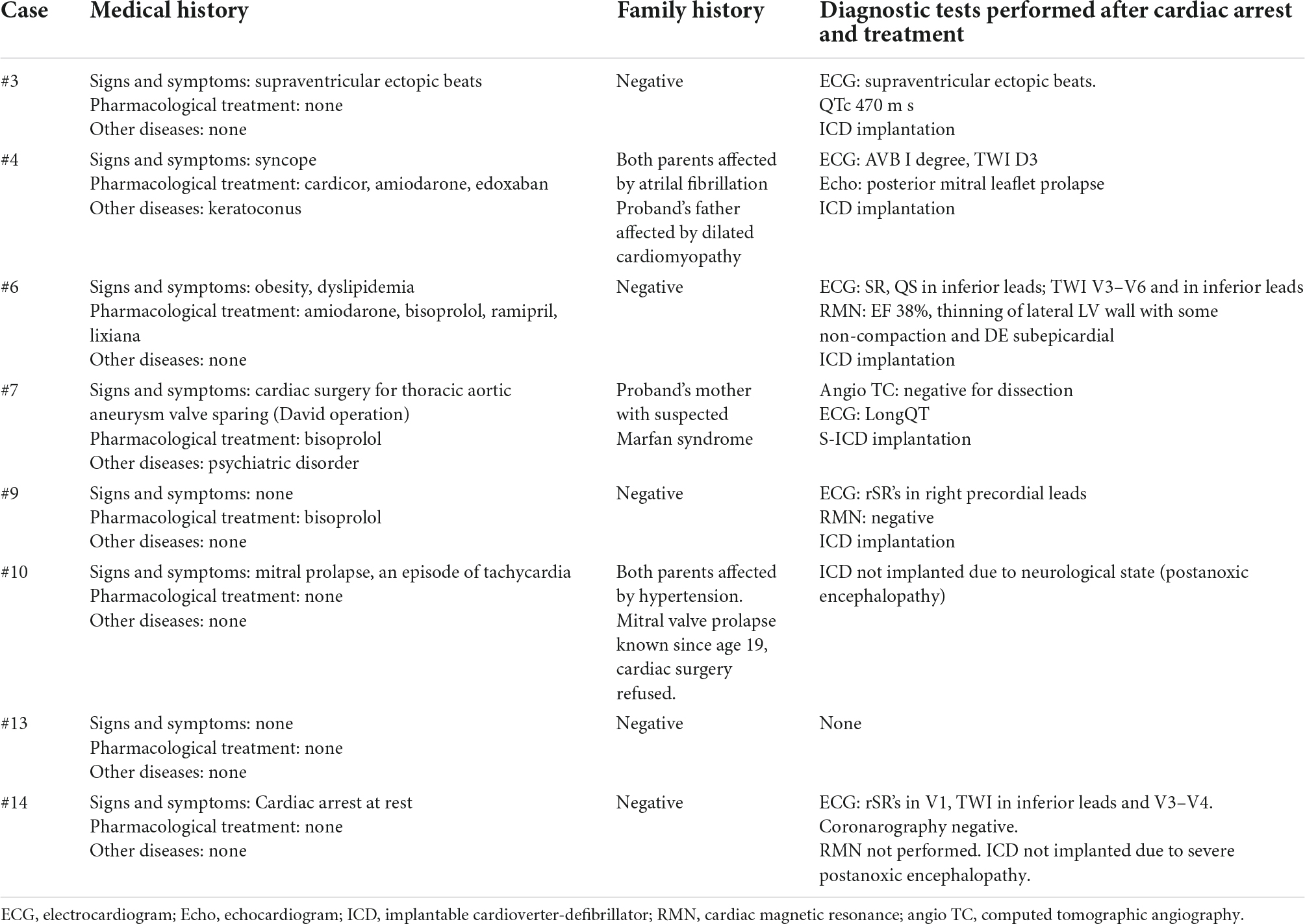
Table 2. Data regarding past medical history and family history of Tuscany registry of sudden cardiac death (ToRSADE) survived patients.
In 12 out of 14 cases, the autopsy results were inconclusive; however, combining autopsy and genetic testing allowed a definitive diagnosis in a 35-year-old woman (patient #18) who collapsed and died while at rest. At autopsy, cardiovascular examination showed a 350-g heart with multifocal areas of wall thinning and apparent fatty infiltration, ventricular thickness being within normal values. Histologically, myocytes loss and disorganization with replacement of fibro-fatty tissue was observed (Figure 2A). Myocardial inflammatory infiltrates were present, but granulomas or giant cells were absent. These findings were consistent with AC. Genetic testing detected a heterozygous deletion in the PKP2 gene (Genbank accession no. NM_004572.3), c.1440_1444del p.(Asn480Lysfs*20) (Figure 3A). This truncated variant has been previously described in patients with AC (19), annotated in ClinVar and in human gene mutation professional databases. The genome aggregation database (gnomAD)2 has reported this variant with an allele frequency of 0.00001105 in the general population. According to ACMG and ACGS guidelines (13, 14), this variant is classified as LP. Patient #15 was a 47-year-old man found unconscious outside his house. On arrival, the medical team proceeded to cardiopulmonary resuscitation but without success. At autopsy, external examination showed a lean male of 186 cm in height and 71 kg in weight. There was no evidence of external injuries. Internal examination revealed a 480-g heart with a body mass index (BMI) of 20.5. The thickness of the right and left ventricular walls was 5 and 15 mm, respectively. There was no sign of acute myocardial infarction. Histology showed myocyte disarray and myocytes exhibiting nuclear enlargement and hyperchromasia were occasionally seen. Masson trichrome stain displayed interstitial and replacement-type fibrosis (Figure 2B). Genetic analysis revealed a deletion in TNNT2 (Genbank accession no. NM_000364.2), c.517_519del p.(Glu173del) in heterozygous state (Figure 3B). This variant has been previously described in patients with HCM (20–22), annotated in ClinVar database and human gene mutation professional database, but not reported in the gnomAD (see text footnote 2). Since multiple tools of computational evidence support a deleterious effect on the gene or protein, the variant was classified as LP according to current ACMG and ACGS guidelines (13, 14). Finally, we report a case where a possible diagnosis was made solely on the basis of the genetic test. Patient #8 was a 35-year-old male professional athlete who died suddenly while swimming. At autopsy, both lungs appeared swollen and multi-organ congestion was evident. The heart (weight: 450 g; LV thickness: 1.2 cm; IVS thickness: 2.0 cm; RV thickness: 0.5 cm) showed no relevant gross findings. Similarly, analysis of conduction system and ventricular tissue did not reveal microscopic anomalies or inflammatory changes (Figure 2C). Genetic analysis showed a missense variant in the SCN5A gene (Genbank accession no. NM_198056), c.1398G > T p.(Leu466Phe) (Figure 3C). This variant is annotated in ClinVar and in human gene mutation professional databases in association with Brugada syndrome (23). The gnomAD (see text footnote 2) has reported this variant with an allele frequency of 0.00002413 on a total population and multiple tools of computational evidence supporting a deleterious effect on the gene or protein. The variant was classified as a VUS (13, 14).
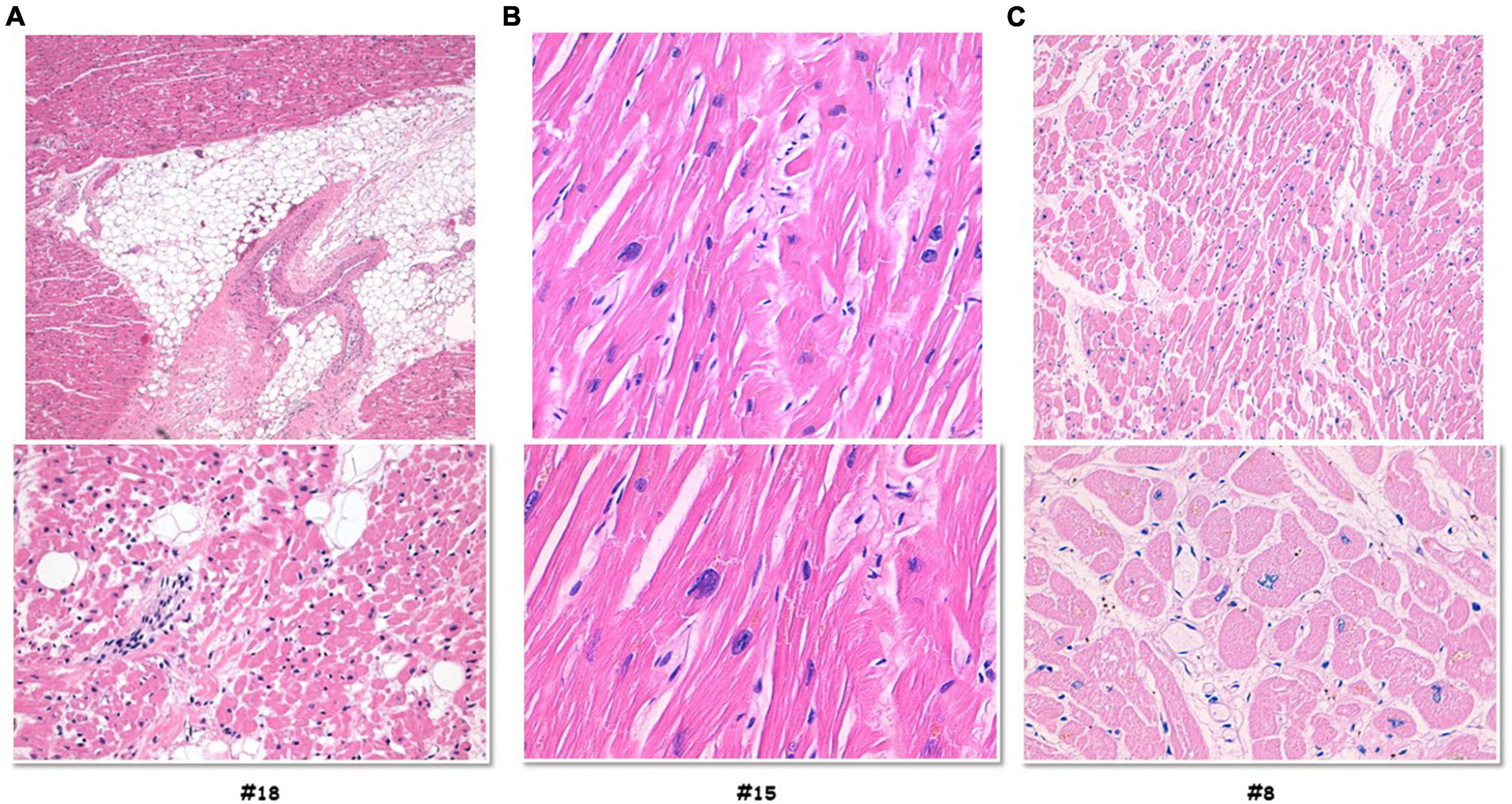
Figure 2. Histology investigation of three selected sudden cardiac death cases. Hematoxylin and eosin staining performed on myocardial samples of the ventricles. Case #18, (A) histology showed myocytes loss and disorganization with replacement of fibro-fatty tissue; myocardial inflammatory infiltrates were present, but granulomas or giant cells were absent. Case #15, (B) the images showed myocytes disarray with storiform pattern, and occasionally myocytes exhibiting nuclear enlargement and hyperchromasia. Masson trichrome stain displayed interstitial and replacement-type fibrosis. Case #8, (C) analysis of conduction system and ventricular tissue did not reveal microscopic anomalies or inflammatory changes.
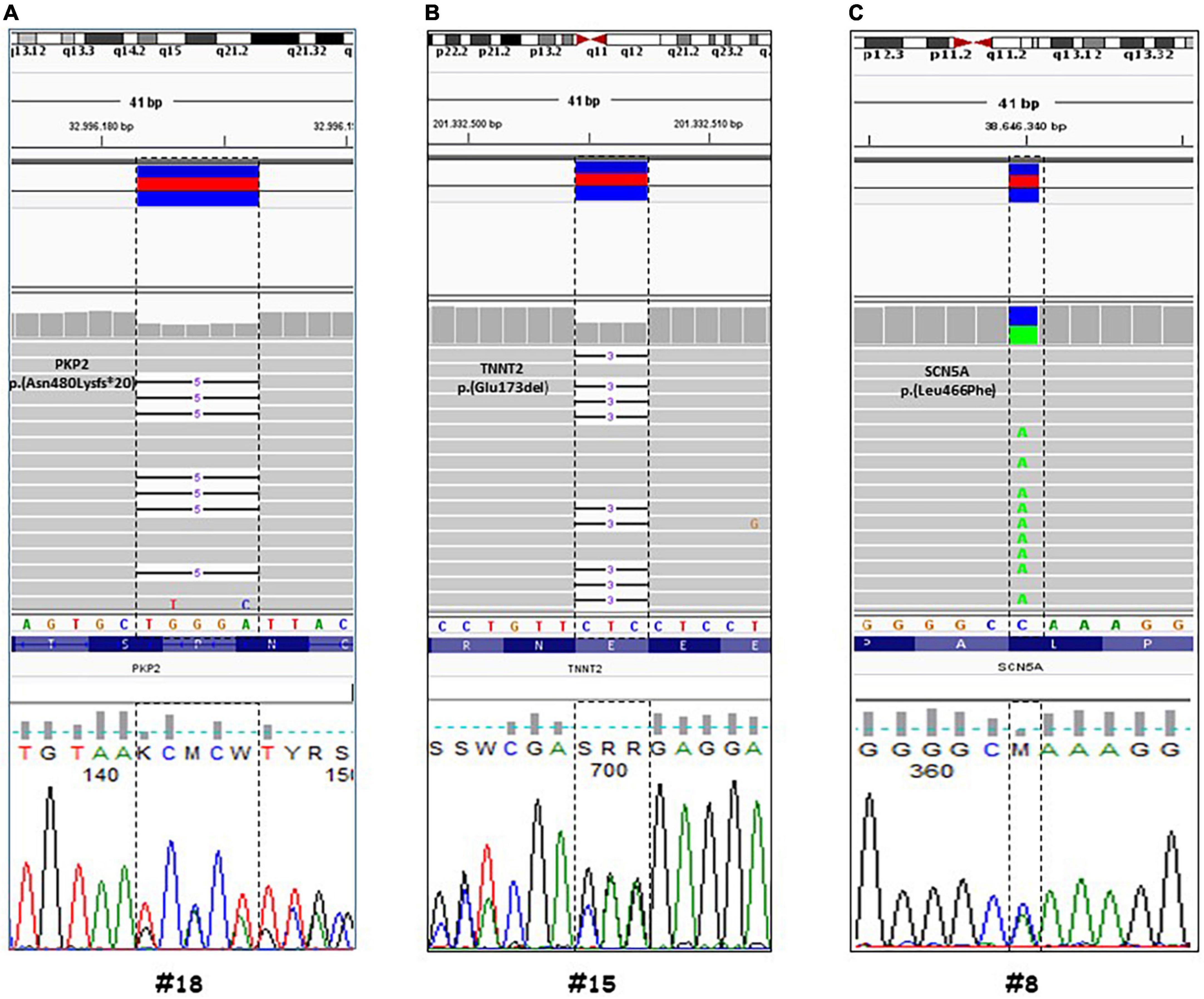
Figure 3. Genetic analysis by next generation sequencing (NGS) of three selected cases. Visualization by integrative genomic viewer (IGV) software of NGS data (through alignment of enriched sequences to Hg19). Case #18, (A) a heterozygous deletion in the PKP2 gene (Genbank accession no. NM_004572.3), c.1440_1444del p.(Asn480Lysfs*20). Case #15, (B) a deletion in TNNT2 (Genbank accession no. NM_000364.2), c.517_519del p.(Glu173del) in heterozygous state. Case #8, (C) a missense variant in the SCN5A gene (Genbank accession no. NM_198056), c.1398G > T p.(Leu466Phe). The detected variants were confirmed by Sanger sequencing.
Our study represents a systematic approach aimed at exploring the feasibility and relevance of a regional registry of SCD in Tuscany, starting from a defined geographical area (Florence) and during a 30-month pilot regional project. In our experience, the estimated incidence of juvenile sudden death was approximately 1/100,000 per year, in line with the incidence reported by Del Vecchio and Padeletti (3); there was a prevalence of male subjects (14 vs. 8) (24), and mean age was 39 years. Among the 22 victims, autopsy (in 14 victims) and clinical data in survived patients failed to identify the underlying disease in most. Likewise, subsequent genetic tests was often inconclusive. A negative genetic test is not surprising given the lack of definite genetic diagnosis in over 50% of patients with clinically symptomatic, likely inherited cardiomyopathies (25). In a recent study from our institution based on over 1,200 HCM patients, the contemporary yield of genetic screening is 47% (26). Notably, however, in the current study NGS identified four LP variants, mostly involved genes causing structural cardiomyopathies: all had a negative autopsy except one patient with evidence of AC. Overall, 19% of patients exhibited a clinically actionable variant, comparing favorably with the diagnostic yield of molecular autopsy in the literature, ranging from 20 to 35% (27). VUS were found in 17 cases. These inconclusive genetic testing results may be due to the bias to perform variants interpretation without a clear diagnosis and a clear pertinence of the phenotype to the affected gene and with a negative or inconclusive autopsy. In the process of genetic test interpretation the clinical diagnosis has a key role because represents one of the most important criteria (PP4 according to ACMG guidelines for variants classification) (13). Indeed, in the case of victims of SD the genetic result may be often inconclusive. In our study we did not report VUS likely benign or benign instead we have annotated VUS potentially pathogenic. To investigate the pathogenic role of the VUS identified we could perform in vivo or in vitro functional studies supportive of a damaging effect on the gene. In addition these VUS variants should be periodically revaluated on the basis of new literature/public databases data (i.e., ClinVar, PubMed, HGMD, Clin Gen) (13). In one case, with negative autopsy, genetic testing revealed the p.(Leu466Phe) variant in SCN5A, associated with Brugada syndrome (28). Notably, in four subjects carrying LP variants in genes associated with HCM (MYBPC3, MYH7 and TCAP), SCD occurred in the absence of clear structural disease manifestation at autopsy. Recent studies highlighted that cardiomyocyte functional abnormality, occurring as a direct consequence of sarcomere mutations, may occur before the onset of gross hypertrophy or dilatation, and may theoretically lead to increased arrhythmic risk in mutation carriers in the absence of hypertrophy (29, 30). Nevertheless, a clear link between these mutations and the SCA/SCD event is elusive, while arrhythmic risk seems particularly low in sarcomere mutation carriers without an HCM phenotype. Therefore, the clinical value of this anecdotal observation requires further investigation in larger series (31). All in all, our study supports the idea that genetic test represents a significant source of information to infer the cause of death, or resuscitated cardiac arrest, in otherwise healthy young people. Building of these promising observations, the use of more extended genetic analyses in genotype-negative SCA/SCD victims (exome, whole genome), paralleled by targeted in vitro studies, may offer further insight into the substrates of juvenile arrhythmias. From a public health perspective, the creation of a local but expandable registry, such as ToRSADE for Tuscany, allows clinicians to plan the clinical and genetic screening strategy for potentially affected relatives, both in the presence of a clear autoptic diagnosis or of non-conclusive autoptic results (3). Since the ToRSADE project was based on an anonymous registry, cascade family screening was not feasible at this stage. However, these results have led to implement a prospective clinical enrolment protocol, now active at our hospital, which now allows such policy. Partnering with a similar initiative in Pisa (The JUST project “JUvenile Sudden cardiac deaTh: JUST know and treat”), the plan is to extend this algorithm to the whole Tuscany region. At variance with previous studies performed in larger groups (32), we performed genetic testing only in subjects where clinical examination or autopsy excluded coronary artery disease. This is of particular relevance for two reasons. First, the number of LP variants retrieved in our samples was due to the pre-selection of cases of unexplained cardiac death excluding toxicological, infectious, oncological, trauma conditions or structural cardiac abnormalities. Second, the presence of cases of interest older than 40 sheds light on the occurrence of arrhythmic cardiac arrest due to non-ischemic causes in subjects usually excluded from epidemiology of juvenile SCD. ToRSADE is the first Tuscan registry to combine forensic and non-forensic autopsy, histology, clinical data evaluation (available only for survived patients) and genetic testing of SCA patients and SCD victims. Despite the time and geographic limitations, the rigorous multidisciplinary approach allowed identifying variants deserving further investigation and provided a further step toward the integration of local registries into a national one.
The most important limitations of ToRSADE project was based on an anonymous registry that did not allowed cascade family screening and the analysis of variants cosegregation in the family. For the same reason data collection regarding anamnestic symptoms in all potentially sudden death associated diseases such as HCM or Brugada syndrome that is essential (33–38), it was unfortunately not investigated except for survived patients (Table 2). However, these results have led to implement a prospective clinical enrolment protocol, now active at our hospital, in which patients selection is no more anonymous with important clinical consequences for sudden death relatives victims.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/clinvar/, SUB12103611.
The studies involving human participants were reviewed and approved by the Local Ethical Committee of Careggi University Hospital, Florence (No. BIO.16.011). The patients/participants provided their written informed consent to participate in this study.
FG and VS drafted the manuscript, made substantial contributions to the analysis and interpretation of genetic and clinical data, and took responsibility for all aspects of the reliability and freedom from bias of the data presented and their discussed interpretation. MF, VM, RG, and GN made substantial contributions to the analysis and interpretation of data from autopsy and pathological anatomy. MT, NM, and GA made substantial contributions to the analysis and interpretation of clinical data. NM, MT, RC, and BB contributed substantially to the conceptual design of the ToRSADE algorithm. GN and EC made contributions to the analysis and interpretation of data, and critically revised the manuscript for important intellectual content analyzed. EC, RF, and IO reviewed and revised the manuscript. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
This work was funded by a grant from Regione Toscana (FAS-Salute 2017 and ToRSADE project to EC). ToRSADE© all right reserved.
We are grateful to the district hospitals of Tuscany that collaborated to the creation of ToRSADE registry.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2022.1080608/full#supplementary-material
SCA, sudden cardiac arrest; SCD, sudden cardiac death; ToRSADE, Tuscany registry of sudden cardiac death; CMs, cardiomyopathies; HCM, hypertrophic cardiomyopathy; DCM, dilated cardiomyopathy; AC, arrhythmogenic cardiomyopathy; NGS, next generation sequencing.
1. Sollazzo F, Palmieri V, Gervasi SF, Cuccaro F, Modica G, Narducci ML, et al. Sudden cardiac death in athletes in italy during 2019: internet-based epidemiological research. Medicina (Kaunas). (2021) 1:57–61. doi: 10.3390/medicina57010061
2. Ackerman M, Atkins DL, Triedman JK. Sudden cardiac death in the young. Circulation. (2016) 133:1006–26. doi: 10.1161/circulationaha.115.020254
3. Del Vecchio M, Padeletti L. Cardiac sudden death in Italy. Dimensions, perceptions, policies and economic/financial impact. G Ital Cardiol. (2008) 9(Suppl. 1):5S–23S.
4. Paratz ED, Rowsell L, Zentner D, Parsons S, Morgan N, Thompson T, et al. Cardiac arrest and sudden cardiac death registries: a systematic review of global coverage. Open Heart. (2020) 7:e001195. doi: 10.1136/openhrt-2019-001195
5. de Vreede-Swagemakers JJM, Gorgels APM, Dubois-Arbouw WI, van Ree JW, Daemen MJAP, Houben LGE, et al. Out-of-hospital cardiac arrest in the 1990s: a population-based study in the Maastricht area on incidence, characteristics and survival. J Am Coll Cardiol. (1997) 30:1500–5. doi: 10.1016/S0735-1097(97)00355-0
6. Winkel BG, Holst AG, Theilade J, Kristensen IB, Thomsen JL, Ottesen GL, et al. Nationwide study of sudden cardiac death in persons aged 1–35 years. Eur Heart J. (2011) 32:983–90. doi: 10.1093/eurheartj/ehq428
7. Lynge TH, Risgaard B, Banner J, Nielsen JL, Jespersen T, Stampe NK, et al. Nationwide burden of sudden cardiac death: a study of 54,028 deaths in Denmark. Heart Rhythm. (2021) 8:1657–65. doi: 10.1016/j.hrthm.2021.05.005
8. Blom MT, van Hoeijen DA, Bardai A, Berdowski J, Souverein PC, De Bruin ML, et al. Genetic, clinical and pharmacological determinants of out-of-hospital cardiac arrest: rationale and outline of the AmsteRdam Resuscitation Studies (ARREST) registry. Open Heart. (2014) 1:e000112. doi: 10.1136/openhrt-2014-000112
9. Risgaard B, Winkel BG, Jabbari R, Behr ER, Ingemann-Hansen O, Thomsen JL, et al. Burden of sudden cardiac death in persons aged 1 to 49 years: nationwide study in Denmark. Circ Arrhythm Electrophysiol. (2014) 7:205–11. doi: 10.1161/CIRCEP.113.001421
10. Svane J, Lynge TH, Hansen CJ, Risgaard BG, Tfelt-Hansen J. Witnessed and unwitnessed sudden cardiac death: a nationwide study of persons aged 1-35 years. Europace. (2021) 23:898–906. doi: 10.1093/europace/euab017
11. Basso C, Aguilera B, Banner J, Cohle S, d’Amati G, Henriques de Gouveia R, et al. Guidelines for autopsy investigation of sudden cardiac death: 2017 update from the association for European cardiovascular pathology. Virchows Arch. (2017) 471:691–705. doi: 10.1007/s00428-017-2221-0
12. Michaud K, Romain N, Taroni F, Horisberger B, Mangin P. Evaluation of a simplified method of the conduction system analysis in 110 forensic cases. Forensic Sci Int. (2002) 130:13–24. doi: 10.1016/S0379-0738(02)00269-4
13. Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. ACMG laboratory quality assurance committee standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. (2015) 17:405–24. doi: 10.1038/gim.2015.30
14. Ellard S, Baple EL, Callaway A. ACGS Best Practice Guidelines for Variant Classification in Rare Disease 2020. Cambridge: Association for Clinical Genomic Science (2020).
15. Fellmann F, van El CG, Charron P, Michaud K, Howard HC, Boers SN, et al. European recommendations integrating genetic testing into multidisciplinary management of sudden cardiac death. Eur J Hum Genet. (2019) 27:1763–73. doi: 10.1038/s41431-019-0445-y
16. Priori SG, Marino M. Sudden cardiac death in the young: are we still missing the opportunity to prevent recurrences in the family? Heart Rhythm. (2021) 18:1645–6. doi: 10.1016/j.hrthm.2021.06.1179
17. Stiles MK, Wilde AAM, Abrams DJ, Ackerman MJ, Albert CM, Behr ER, et al. 2020 APHRS/HRS expert consensus statement on the investigation of decedents with sudden unexplained death and patients with sudden cardiac arrest, and of their families. J Arrhythmia. (2021) 37:481–534. doi: 10.1002/joa3.12449
18. Ommen SR, Mital S, Burke MA, Day SM, Deswal A, Elliott P, et al. 2020 AHA/ACC guideline for the diagnosis and treatment of patients with hypertrophic cardiomyopathy: a report of the American college of cardiology/American heart association joint committee on clinical practice guidelines. J Thorac Cardiovasc Surg. (2021) 162:e23–106.
19. Brun F, Barnes CV, Sinagra G, Slavov D, Barbati G, Zhu X, et al. Titin and desmosomal genes in the natural history of arrhythmogenic right ventricular cardiomyopathy. J Med Genet. (2014) 51:669–76. doi: 10.1136/jmedgenet-2014-102591
20. Watkins H, McKenna WJ, Thierfelder L, Suk HJ, Anan R, O’Donoghue A, et al. Mutations in the genes for cardiac troponin T and alpha-tropomyosin in hypertrophic cardiomyopathy. N Engl J Med. (1995) 332:1058–64. doi: 10.1056/NEJM199504203321603
21. Harada K, Takahashi-Yanaga F, Minakami R, Morimoto S, Ohtsuki I. Functional consequences of the deletion mutation deltaGlu160 in human cardiac troponin T. J Biochem. (2000) 127:263–8. doi: 10.1093/oxfordjournals.jbchem.a022603
22. Messer AE, Bayliss CR, El-Mezgueldi M, Redwood CS, Ward DG, Leung MC, et al. Mutations in troponin T associated with hypertrophic cardiomyopathy increase Ca2+-sensitivity and suppress the modulation of Ca(2+)-sensitivity by troponin I phosphorylation. Arch Biochem Biophys. (2016) 601:113–20. doi: 10.1016/j.abb.2016.03.027
23. Savastano S, Rordorf R, Vicentini A, Petracci B, Taravelli E, Castelletti S, et al. A comprehensive electrocardiographic, molecular, and echocardiographic study of Brugada syndrome: validation of the 2013 diagnostic criteria. Heart Rhythm. (2014) 11:1176–83. doi: 10.1016/j.hrthm.2014.04.010
24. Kim SK, Bennett R, Ingles J, Kumar S, Zaman S. Arrhythmia in cardiomyopathy: sex and gender differences. Curr Heart Fail Rep. (2021) 18:274–83. doi: 10.1007/s11897-021-00531-0
25. Girolami F, Vergaro G, Pieroni M, Passantino S, Giannotti G, Grippo G, et al. Clinical pathway for cardiomyopathies: a genetic testing strategy proposed by ANMCO in Tuscany. G Ital Cardiol. (2020) 21:926–34. doi: 10.1714/3472.34547
26. Mazzarotto F, Girolami F, Boschi B, Barlocco F, Tomberli A, Baldini K, et al. Defining the diagnostic effectiveness of genes for inclusion in panels: the experience of two decades of genetic testing for hypertrophic cardiomyopathy at a single center. Genet Med. (2019) 21:284–92. doi: 10.1038/s41436-018-0046-0
27. Castiglione V, Modena M, Aimo A, Chiti E, Botto N, Vittorini S, et al. Molecular autopsy of sudden cardiac death in the genomics era. Diagnostics (Basel). (2021) 11:1378. doi: 10.3390/diagnostics11081378
28. Wilde AAM, Semsarian C, Márquez MF, Sepehri Shamloo A, Ackerman MJ, Ashley EA, et al. European heart rhythm association (EHRA)/heart rhythm society (HRS)/Asia Pacific heart rhythm society (APHRS)/Latin American heart rhythm society (LAHRS) expert consensus statement on the state of genetic testing for cardiac diseases. Europace. (2022) 24:1307–67. doi: 10.1093/europace/euac030
29. Coppini R, Santini L, Olivotto I, Ackerman MJ, Cerbai E. Abnormalities in sodium current and calcium homoeostasis as drivers of arrhythmogenesis in hypertrophic cardiomyopathy. Cardiovasc Res. (2020) 116:1585–99. doi: 10.1093/cvr/cvaa124
30. Ferrantini C, Coppini R, Pioner JM, Gentile F, Tosi B, Mazzoni L, et al. Pathogenesis of hypertrophic cardiomyopathy is mutation rather than disease specific: a comparison of the cardiac troponin T E163R and R92Q mouse models. J Am Heart Assoc. (2017) 6:e005407. doi: 10.1161/JAHA.116.005407
31. Maurizi N, Michels M, Rowin EJ, Semsarian C, Girolami F, Tomberli B, et al. Clinical course and significance of hypertrophic cardiomyopathy without left ventricular hypertrophy. Circulation. (2019) 139:830–3. doi: 10.1161/CIRCULATIONAHA.118.037264
32. Bagnall RD, Weintraub RG, Ingles J, Duflou J, Yeates L, Lam L, et al. A prospective study of sudden cardiac death among children and young adults. N Engl J Med. (2016) 374:2441–52. doi: 10.1056/NEJMoa1510687
33. Mascia G, Crotti L, Groppelli A, Canepa M, Carlo Merlo A, Benenati S, et al. Syncope in hypertrophic cardiomyopathy (part I): an updated systematic review and meta-analysis. Int J Cardiol. (2022) 357:88–94. doi: 10.1016/j.ijcard.2022.03.028
34. Odak M, Douedi S, Mararenko A, Alshami A, Elkherpitawy I, Douedi H, et al. Arrhythmogenic right ventricular cardiomyopathy: the role of genetics in diagnosis, management, and screening. Cardiol Res. (2022) 13:177–84. doi: 10.14740/cr1373
35. Vissing CR, Espersen K, Mills HL, Bartels ED, Jurlander R, Skriver SV, et al. Family screening in dilated cardiomyopathy: prevalence, incidence, and potential for limiting follow-up. JACC Heart Fail. (2022) 10:792–803. doi: 10.1016/j.jchf.2022.07.009
36. Krahn AD, Behr ER, Hamilton R, Probst V, Laksman Z, Han HC. Brugada syndrome. JACC Clin Electrophysiol. (2022) 8:386–405. doi: 10.1016/j.jacep.2021.12.001
37. Zaffalon D, Papatheodorou E, Merghani A, Dhutia H, Moccia E, Malhotra A, et al. Role of the electrocardiogram in differentiating genetically determined dilated cardiomyopathy from athlete’s heart. Eur J Clin Invest. (2022) 52:e13837. doi: 10.1111/eci.13837
38. Efremidis M, Vlachos K, Kyriakopoulou M, Mililis P, Martin CA, Bazoukis G, et al. The RV1-V3 transition ratio: a novel electrocardiographic criterion for the differentiation of right versus left outflow tract premature ventricular complexes. Heart Rhythm O2. (2021) 2:521–8. doi: 10.1016/j.hroo.2021.07.009
Keywords: juvenile sudden cardiac death, ToRSADE registry, molecular autopsy, next generation sequencing, genetics, arrhythmia
Citation: Girolami F, Spinelli V, Maurizi N, Focardi M, Nesi G, Maio V, Grifoni R, Albora G, Bertaccini B, Targetti M, Coppini R, Favilli S, Olivotto I and Cerbai E (2022) Genetic characterization of juvenile sudden cardiac arrest and death in Tuscany: The ToRSADE registry. Front. Cardiovasc. Med. 9:1080608. doi: 10.3389/fcvm.2022.1080608
Received: 26 October 2022; Accepted: 28 November 2022;
Published: 14 December 2022.
Edited by:
Aviram Hochstadt, Tel Aviv Sourasky Medical Center, IsraelReviewed by:
Giuseppe Mascia, University of Genoa, ItalyCopyright © 2022 Girolami, Spinelli, Maurizi, Focardi, Nesi, Maio, Grifoni, Albora, Bertaccini, Targetti, Coppini, Favilli, Olivotto and Cerbai. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesca Girolami, ZnJhbmNlc2NhLmdpcm9sYW1pQG1leWVyLml0
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.