
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cardiovasc. Med., 21 December 2022
Sec. General Cardiovascular Medicine
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.1070935
This article is part of the Research TopicMolecular Mechanism of Vascular Remodelling and Target Organ DamageView all 14 articles
 Andrea Saavedra-Alvarez1Katherine V. Pereyra1Camilo Toledo1Rodrigo Iturriaga1,2,3
Andrea Saavedra-Alvarez1Katherine V. Pereyra1Camilo Toledo1Rodrigo Iturriaga1,2,3 Rodrigo Del Rio1,2,3*
Rodrigo Del Rio1,2,3*Heart failure with preserved ejection fraction (HFpEF) is a complex, heterogeneous disease characterized by autonomic imbalance, cardiac remodeling, and diastolic dysfunction. One feature that has recently been linked to the pathology is the presence of macrovascular and microvascular dysfunction. Indeed, vascular dysfunction directly affects the functionality of cardiomyocytes, leading to decreased dilatation capacity and increased cell rigidity, which are the outcomes of the progressive decline in myocardial function. The presence of an inflammatory condition in HFpEF produced by an increase in proinflammatory molecules and activation of immune cells (i.e., chronic low-grade inflammation) has been proposed to play a pivotal role in vascular remodeling and endothelial cell death, which may ultimately lead to increased arterial elastance, decreased myocardium perfusion, and decreased oxygen supply to the tissue. Despite this, the precise mechanism linking low-grade inflammation to vascular alterations in the setting of HFpEF is not completely known. However, the enhanced sympathetic vasomotor tone in HFpEF, which may result from inflammatory activation of the sympathetic nervous system, could contribute to orchestrate vascular dysfunction in the setting of HFpEF due to the exquisite sympathetic innervation of both the macro and microvasculature. Accordingly, the present brief review aims to discuss the main mechanisms that may be involved in the macro- and microvascular function impairment in HFpEF and the potential role of the sympathetic nervous system in vascular dysfunction.
Heart failure (HF) is a pathological condition affecting mainly the elderly population. A subcategory of this disease is HF with preserved ejection fraction (HFpEF), whose incidence has increased notably in recent years, particularly in the last two decades, from 48 to 57% compared with systolic HF (or reduced ejection fraction HF). Furthermore, HFpEF accounts for the death of 1 in 8 people over 65 years (1). Patients with HFpEF have a poor quality of life, high medical costs, and early death (2). Then, understanding the pathophysiology of HFpEF is relevant for future therapeutic strategies to improve HFpEF outcomes.
Patients with HFpEF display several comorbidities associated with cardiac and vascular disturbances, including but not limited to diabetes mellitus, obesity, pulmonary hypertension, coronary artery disease, chronic renal failure, and systemic inflammation (1), all of which contribute to endothelial dysfunction, cardiomyocyte hypertrophy, and cardiac fibrosis (2, 3). Furthermore, it has been described that autonomic imbalance, a hallmark of HF independent of its etiology (i.e., reduced or preserved EF), plays a key role in disease progression (4). Indeed, patients with HF showing sustained elevations in systemic circulating levels of catecholamines (i.e., norepinephrine) show higher mortality rates (5). Importantly, evidence indicates that the sympathetic nervous system (SNS) is critically influenced, at the central and peripheral levels, by the most relevant factors regulating vascular function, such as nitric oxide (NO), reactive oxygen species (ROS), endothelin 1 (ET-1), and the renin-angiotensin system (RAS). Then, a bidirectional and maladaptive relationship between endothelial function and hyperactivity of the SNS could play a role in short- and long-term vascular dysfunction in HFpEF. Indeed, autonomic imbalance in HFpEF increases sympathetic vasomotor tone (6). The latter results in increased excitatory sympathetic activity to blood vessels changing the balance between vasodilator and vasoconstrictor molecules that regulate endothelial cell function and therefore, cardiovascular integrity (7). In this review, we will focus on the main factors that may contribute to the development/maintenance of vascular cell dysfunction and their potential link to enhanced sympatho-vasomotor tone in the setting of HFpEF.
Endothelium-dependent coronary microvascular dysfunction is present in approximately 30% of patients with HFpEF (8). In addition, more than 30% of patients with HFpEF display endothelium-independent dysfunction, reflected in significant reductions in coronary flow reserve (CFR) (8). Indeed, patients with HFpEF present vascular-ventricular uncoupling and stiffness, which is associated with decreased exercise capacity (9). Accordingly, acute increases in cardiac afterload, in the setting of arterial-ventricular stiffness, lead to increases in arterial blood pressure that impairs diastolic relaxation and increases filling pressures during exercise (10). The specific mechanisms associated with the changes in arterial elastance during HFpEF are not fully elucidated, but they have been associated with blood vessels alterations in the bioavailability and responses to vasoactive molecules such as ET-1 and NO (11, 12).
In addition to systemic functional alterations in the vasculature, a reduction in myocardial microvascular density, called microvascular rarefaction, is observed in patients with HFpEF (13). Microvascular rarefaction contributes to cardiac perfusion failure by decreasing myocardial oxygen delivery in patients with HFpEF (14). Therefore, rarefaction of resistance vessels, including small arteries and arterioles, increases coronary microvascular resistance, resulting in reduced cardiac perfusion (15), which has been proposed as a pathogenic mechanism involved in the progressive decline in cardiac function in HFpEF (15). The precise mechanism(s) underpinning vascular rarefaction in HFpEF is still not completely known; however, due to the exquisite sympathetic regulation of blood vessels, and the fact that sympathoexcitation occurs in HFpEF, it is plausible that enhanced sympatho-vasomotor tone may play a role in vascular rarefaction by changing the vasoconstrictor to vasodilator balance in the vessel microenvironment (Figure 1).
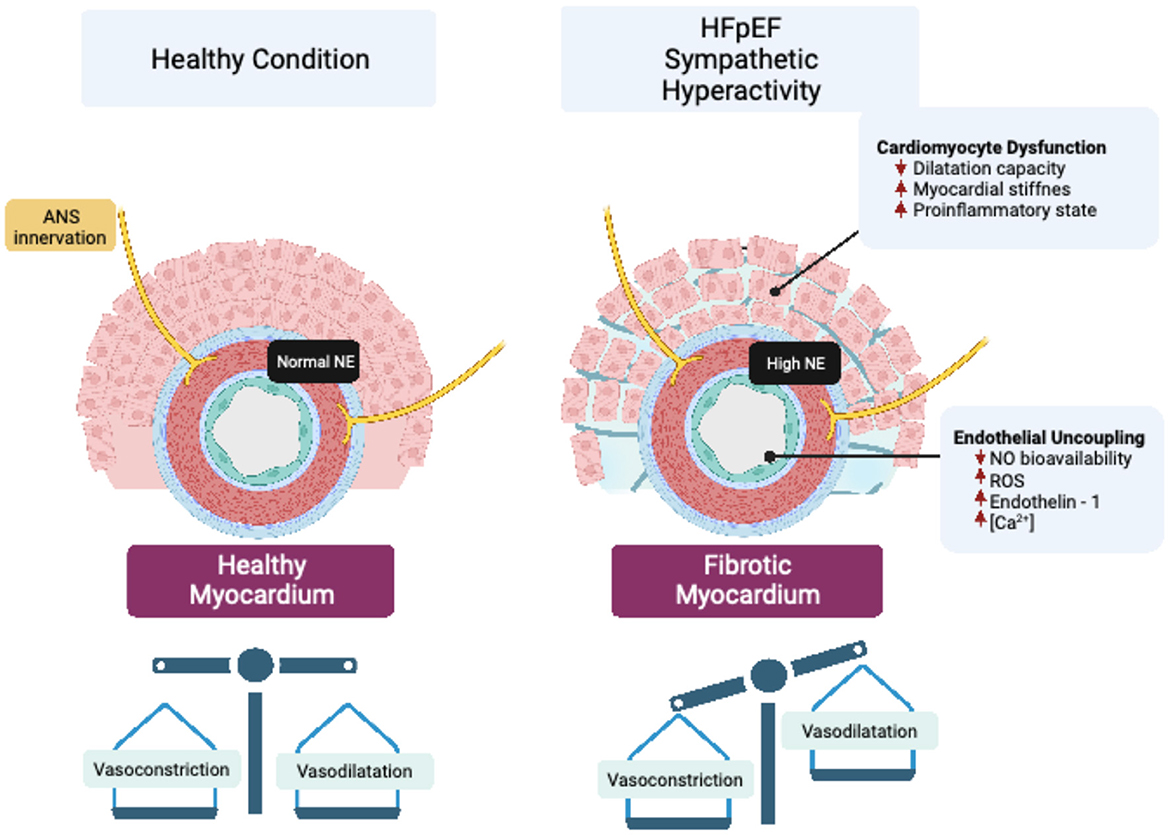
Figure 1. Vascular sympathetic neurotransmission and endothelial dysfunction in HFpEF. Enhanced sympathetic outflow led to increased release of norepinephrine (NE), which impair cardiovascular endothelial function by modifying peptides and signaling molecules that regulate perfusion to vascular beds. Endothelial uncoupling, in turn, can generate cardiomyocyte dysfunction that affects the structure and function of the heart through mechanisms associated with impaired myocardial dilatation capacity, stiffness, and inflammation.
To the best of our knowledge, there is no comprehensive literature providing mechanistic insights into macrovasculature changes in HFpEF. Macrovascular arterial stiffness results in an increase in pulse pressure and wave velocity, which impairs normal microvascular function (16). The latter is particularly relevant for the coronary and renal microvasculature since pathological alterations in pulse pressure and blood flow result in damage to the capillary network of these vascular territories (17). Indeed, coronary artery disease is considered an indicative sign of vascular dysfunction in patients with HFpEF (18). Arterial rarefaction and inadequate angiogenesis that take place during microvascular/macrovascular dysfunction may contribute to a decrease in oxygen supply to the myocardium (19). Accordingly, it has been proposed that left ventricular diastolic dysfunction in patients with HFpEF results from vascular alterations, with aortic stiffness and altered vascular endothelial function being fundamental characteristics of this process (20). Indeed, stiffness at the macrovasculature level is associated with ventricular decreases in elastance, leading to abnormal left arterio-ventricular crowning (21). Notably, ventricular stiffness occurs regardless of several comorbidities presented by patients with HFpEF (22). Besides the changes in vascular stiffness, studies in HFpEF also showed a decrease in brachial flow-mediated dilatation (FMD) and hyperemia, suggesting the presence of endothelial dysfunction at macrovascular/microvascular circulation. Lee et al. (23), proposed that macrovascular dysfunction is indeed a consequence of primary alterations at the microvascular level (23). This is in line with a previous report showing the presence of endothelial dysfunction at the microvasculature with no overt signs of vascular dysfunction in conductance vessels in experimental HFpEF models (24). Together, current evidence supports the role of microvascular/macrovascular alterations in the progression of heart disease. Whether changes/adaptations in the microvasculature/macrovasculature are a cause or consequence to support the failing heart (in the setting of heart failure) remains to be determined.
The endothelium is a highly dynamic layer that works as a barrier that separates the blood from the extravascular tissue and interacts with other cell types contributing to the physiological and homeostatic regulation of blood vessel function (25). In addition, the endothelium prevents the aggregation and adhesion of platelets and leukocytes, inhibits the proliferation of smooth muscle cells (SMC), regulates vascular tone, and plays a protective role against mechanical stimuli such as pressure or frictional stress, through the release of vasoactive substances. This is critical for the maintenance of adequate organ/tissue perfusion (26, 27). While endothelial cells (EC) are located in the most internal layer of blood vessels, SMCs are located in the medial layer and constitute the contractile elements of blood vessels, contributing to the regulation of blood vessel tone, blood pressure, and circulation (28). Then, the correct function of SMC and EC is important for vascular health since both manage vasomotor tone and vasculature integrity.
Both arms of the autonomic nervous system (ANS) (i.e., sympathetic and parasympathetic) innervate blood vessel walls and regulate wall tension (29–31). SMCs at the muscular layer of blood vessel walls receive adrenergic and cholinergic nerve projections from sympathetic and parasympathetic innervation, while ECs do not present a direct neural innervation from the ANS (29, 31). The vascular SMC layer encompasses several ANS nerve terminals. Indeed, SMC constitutively expresses β-adrenergic receptors, which modulate vasodilatation, and α1/α2-adrenergic receptors, which modulate vasoconstriction (29, 31). In addition, parasympathetic stimulation of muscarinic receptors within SMCs also results in blood vessel contraction. Despite not being directly innervated by the sympathetic-adrenergic system, ECs also constitutively express both β-adrenoreceptors and α2-adrenoreceptors. While the effects of β-adrenoreceptors stimulation on EC function remain unknown, the activation of α2-adrenoreceptors leads to the release of vasoactive molecules such as nitric oxide (NO), which acting at SMC induces cell relaxation resulting in blood vessel vasodilation (29, 31). Besides the fine regulation of vascular function by the ANS, how autonomic imbalance could affect vasculature integrity by modulating mechanisms associated with vasoconstriction/relaxation and the vasculature environment is not completely understood, and much less is known about these mechanisms in the pathological setting of HFpEF. In this review, we discussed the potential mechanism of vascular dysfunction in HFpEF and its relation to autonomic imbalance.
The role of vascular NO is essential for vasodilation, inhibition of platelet aggregation, and protection of the integrity of the endothelial layer given its anti-inflammatory, proangiogenic, anti-apoptotic, and anti-fibrotic properties, reducing vascular inflammation and atherosclerosis (32, 33). At the major circulation, NO diffuses into platelets and SMC from EC, which stimulates soluble guanylate cyclase (sGC) and activates the cyclic GMP (cGMP) pathway to induce calcium release from the sarcoplasmic reticulum (SCR) in SMC, preventing platelet aggregation and producing vasodilation, respectively. At the level of cardiac microcirculation, NO can diffuse into cardiomyocytes from adjacent coronary vasculature, modulating cardiac function (7). In addition, NO signaling is involved in tissue repair by mediating the mobilization of stem and progenitor cells (34). In HFpEF, endothelial dysfunction has been linked to decreased production of cGMP and reduced activity of protein kinase G (PKG) and the L-arginine-NO synthetic pathway. Therefore, mechanisms for vasodilation are likely to be impaired in patients with HFpEF. Interestingly, vascular endothelial dysfunction in the heart shared similar mechanisms compared to those found in the systemic circulation, being alterations in sGC-cGMP signaling a common pathway affected at both levels during the progression HFpEF. More importantly, alterations in the sGC-cGMP-PKG pathway in HFpEF promote functional impairment in cardiomyocytes, as evidenced by delayed myocardial relaxation, increased myocardial stiffness, cardiac hypertrophy, and interstitial fibrosis (35). Therefore, direct interventions targeting the NO/cGMP/PKG pathway have been proposed as novel therapeutics to improve both vascular and cardiac function in HFpEF (36, 37).
How autonomic imbalance, a hallmark pathophysiological condition found in experimental and human HFpEF, affects vascular NO production is still not known. Endothelial β2-adrenergic receptors stimulate NO synthesis by the activation of endothelial nitric oxide synthase isoform (eNOS) (32). Interestingly, overexpression or chronic activation of eNOS could be maladaptive since marked increases in intracellular oxidative stress have been reported following eNOS overexpression (38, 39). Furthermore, chronic β-adrenoreceptor activation exacerbates eNOS activity and upregulates eNOS gene expression, favoring superoxide anion generation and vascular dysfunction through reductions in NO bioavailability (38, 40). Indeed, oxygen free radicals rapidly react with NO to form reactive nitrogen species, which are known to promote a prothrombotic and proinflammatory niche within blood vessels (12, 41). Notably, the relevance of reduced NO bioavailability and increased oxidative stress to promote HFpEF pathophysiology has been demonstrated in experimental HF in which concomitant metabolic and vascular stress in mice (high-fat diet and constitutive NOS inhibition using N(omega)-nitro-L-arginine methyl ester) recapitulated the cardiovascular features of human HFpEF (12, 26). Therefore, it is plausible that hyperactivation of the sympathetic nervous system in HFpEF may lead to decreases in NO bioavailability by promoting the formation of reactive nitrogen species within blood vessels. Further investigation is needed to fully determine the contribution of enhanced sympathetic activity on NO and vascular alterations in HFpEF. In addition, HFpEF increases ROS levels and/or antioxidant enzyme suppression, leading to cardiac and endothelial dysfunction. The different risk factors for HFpEF stimulate the production of ROS (42–44). Oxidative stress by their side increases levels of hydrogen peroxide and reactive oxidative metabolites, uncoupled endothelial nitric oxide synthase, endothelial NADPH oxidase 2 (NOX2) expression, and reduced NO levels indicate the presence of myocardial oxidative stress in patients with HFpEF (45). Beyond oxidation, inhibition of NO production can reduce NO bioavailability, for example, through AGE-induced elevation of asymmetric levels of ADMA (dimethyl L-arginine), an inhibitor of eNOS (endothelial NOS), which contributes to endothelium-dependent dysfunction associated with poorer HFpEF prognosis (46). Also, autonomic dysfunction characterized by chronic activation of the SNS might contribute to oxidative stress at the EC level. Previous reports showed high contractile activity in β2-adrenoreceptor deficient mice, and this loss of function can trigger ROS-mediated NO impairment (47). Thus, a lack of β2 receptors increases oxidative stress in the β2-KO mice arteries, and this change the vasoconstrictor response to phenylephrine. In addition, the above evidence suggests a crucial link between adrenergic pathways, oxidative stress, and NO bioavailability in the vasculature (47, 48). Interestingly, patients with HFpEF display not only impaired catecholamine sensitivity and β-adrenoreceptor density at the cardiac level (49, 50) but also display impaired chronotropic and vasodilatation response to exercise (51), suggesting possible desensitization of adrenergic signaling at the cardiac and vascular level. Overall, heightened SNS activity in the setting of HFpEF might contribute to creating a vicious cycle that promotes and maintains vascular dysfunction.
Risk factors in HF, such as diabetes mellitus, aging, and hypertension, among others, trigger systemic low-grade inflammation, characterized by chronic elevations in circulating immune cells, proinflammatory cytokines, and increased expression of endothelial adhesion molecules, such as vascular and intercellular cell adhesion molecules-1 (ICAM-1 and VCAM-1), and the corresponding ligands of circulating leukocytes, increasing myocardial infiltration of CD45+ and CD3+ T-lymphocytes (52). The latter further promotes the infiltration of leukocytes, especially monocytes, into the myocardial tissue, increasing the release of transforming growth factor beta (TGF-β), which ultimately leads to extracellular matrix remodeling and fibrosis (41, 43). Importantly, it has been reported that flow-mediated dilation (FMD) and reactive hyperemic index (RH) are reduced in patients with HFpEF (45), which is closely associated with elevations in inflammatory markers, such as CRP, IL-6, TNF-α, IL-1β, and NFG15 (53, 54). The increase in the inflammatory status leads to coronary microvascular endothelial dysfunction and further increases in inflammatory cytokines (55) partially mediated by the activation of the nuclear factor-kappa B (NFkB) signaling pathway (43). Thus, microvascular dysfunction is proposed to be the central mediator connecting systemic low-grade inflammation with myocardial dysfunction and remodeling in the setting of HFpEF (35).
Chronic elevation of catecholamines in HF, such as epinephrine and norepinephrine, is a hallmark and strong predictor of mortality in patients with HF (56, 57). Catecholamines activate the adenyl cyclase (AC)-cAMP-PKA pathway, leading to IP3R1 activation and in consequence IP3 signal to increased Ca2+ release and vascular tone in VSMCs during HF (58). Also, it has been found that BK potassium channels, which contribute to VSMC hyperpolarization, are downregulated in HF, promoting vasoconstriction, and synergizing with IP3R1 for elevations in cytosolic [Ca2+] (59). Since mRNA and protein levels of inositol 1,4,5 phosphate receptor 1 (IP3R1) are upregulated in HF and increased receptor phosphorylation in HF, it has been suggested that IP3R1 may play an important role in Ca2+ regulation in VSMC (60, 61). However, little is known about the contribution of intracellular calcium (Ca2+) mishandling in the vasculature and subsequent acceleration of cardiac remodeling and progression of HFpEF (58). Nevertheless, alterations in the expression and function of proteins that handle Ca2+ and a maladaptive redistribution of intracellular calcium have been described in HF (62). Some of these proteins are RyR2, Serca2a, Na+-Ca2+ exchanger (NCX), and transient receptor potential cation channels (TRPC) (63). For RyR2, there is evidence of PKA-dependent hyperphosphorylation (in S2808), causing channel dissociation, increasing Ca2+ leakage from the SR, decreasing Ca2+ transients, changing spontaneous Ca2+ release events, and altering cytosolic Ca2+ management (64). In addition, Serca2a is downregulated in HFpEF, then Ca2+ reuptake toward the SR affecting both active and passive cardiovascular functions (65). In addition, increased activity of NCX in HFpEF has also been described (66). Finally, the TRPC channels that participate in the entry of Ca2+ from the extracellular medium that allows the increase of Ca2+ reservoirs into the SR are increased in HFpEF, possibly as an adaptive mechanism due to a decrease in Ca2+ reserves in the SR (67). In addition, increased myosin heavy chain phosphorylation has also been found in the arteries of patients with HF and mice (68). The latter has been linked to VSMC remodeling and has been associated with alterations in VSMC Ca2+ handling (69). Therefore, alterations in the management of intracellular Ca2+ in the vasculature in HF may play an important role not only in vascular cell function but also in the adverse remodeling of several vascular compartments.
Little is known about the role of macro- and microvascular alterations during the onset, development, and progression of HFpEF. However, it is highly likely that vascular rarefaction takes place during the onset, maintenance and/or progression of HFpEF resulting in increases in microvascular resistance, reductions in tissue perfusion, and activation of vasomotor sympathetic fibers that ultimately create a feed-forward mechanism that promotes the further deterioration of vascular function by shifting the balance between vasoconstriction and vasodilation. On the contrary, proinflammatory and pro-oxidative molecules have been associated with the etiology of the disease. At the microvascular level, the decrease in the bioavailability of NO, alterations in the sGC-cGMP-PKG pathway, accumulation of ROS, and chronic low-grade inflammation are the main actions involved in the alteration of vascular function both at the systemic circulation and in the coronary territory, promoting a functional decrease in cardiomyocytes, evidenced by delayed myocardial relaxation, increased myocardial stiffness, cardiac hypertrophy, and interstitial fibrosis. The latter may have fundamental implications for the progressive decline in cardiac function during HFpEF.
To date, there are only preventive and palliative actions to deal with HFpEF, such as exercise and a healthy lifestyle, which do not imply a remission of the disease. In this article, several molecular candidates rise as potential therapeutic targets to improve both vascular and cardiac functions in HFpEF, including but not limited to NO metabolic pathway, IP3R signaling, adrenergic pathways, and reduction of oxidative stress and vascular inflammation.
AS-A wrote the first draft. KP, CT, RI, and RDR contributed to manuscript formulation and revision. All authors have read and approved the final manuscript.
This study was supported by Fondo de Desarrollo Científico y Tecnológico Fondecyt (1220950) and the Basal Center of Excellence in Aging and Regeneration (AFB 170005; ACE 210009).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Tsao CW, Lyass A, Enserro D, Larson MG, Ho JE, Kizer JR, et al. Temporal trends in the incidence of and mortality associated with heart failure with preserved and reduced ejection fraction. JACC Heart Failure. (2018) 6:678–85. doi: 10.1016/j.jchf.2018.03.006
2. Nair N. Epidemiology and pathogenesis of heart failure with preserved ejection fraction. Rev Cardiovasc Med. (2020) 21:531–40. doi: 10.31083/j.rcm.2020.04.154
3. Alem M. Endothelial dysfunction in chronic heart failure: assessment, findings, significance, and potential therapeutic targets. Int J Mol Sci. (2019) 20:3198. doi: 10.3390/ijms20133198
4. Triposkiadis F, Karayannis G, Giamouzis G, Skoularigis J, Louridas G, Butler J, et al. The sympathetic nervous system in heart failure physiology, pathophysiology, and clinical implications. J Am Coll Cardiol. (2009) 54:1747–62. doi: 10.1016/j.jacc.2009.05.015
5. Cohn JN. Physiologic basis of vasodilator therapy for heart failure. Am J Md. (1981) 71:135–9. doi: 10.1016/0002-9343(81)90276-X
6. Heusser K, Wittkoepper J, Bara C, Haverich A, Diedrich A, Levine B, et al. Sympathetic vasoconstrictor activity before and after left ventricular assist device implantation in patients with end-stage heart failure. Eur J Heart Fail. (2021) 23:1955–9. doi: 10.1002/ejhf.2344
7. Wang P, Wei M, Zhu X, Liu Y, Yoshimura K, Zheng M, et al. Nitric oxide down-regulates voltage-gated Na +channel in cardiomyocytes possibly through S-nitrosylation-mediated signaling. Sci Rep. (2021) 11:11273. doi: 10.1038/s41598-021-90840-0
8. Yang JH, Obokata M, Reddy YNV, Redfield MM, Lerman A, Borlaug BA, et al. Endothelium-dependent and independent coronary microvascular dysfunction in patients with heart failure with preserved ejection fraction. Eur J Heart Fail. (2020) 22:432–41. doi: 10.1002/ejhf.1671
9. Hundley WG, Kitzman DW, Morgan TM, Hamilton CA, Darty SN, Stewart KP, et al. Cardiac cycle-dependent changes in aortic area and distensibility are reduced in older patients with isolated diastolic heart failure and correlate with exercise intolerance. J Am Coll Cardiol. (2001) 38:796–802. doi: 10.1016/S0735-1097(01)01447-4
10. Mohammed SF, Borlaug BA, McNulty S, Lewis GD, Lin G, Zakeri R, et al. Resting ventricular–vascular function and exercise capacity in heart failure with preserved ejection fraction: a relax trial ancillary study. Circ Heart Fail. (2014) 7:580–9. doi: 10.1161/CIRCHEARTFAILURE.114.001192
11. Chowdhury MA, Moukarbel GV, Gupta R, Frank SM, Anderson AM, Liu LC, et al. Endothelin 1 is associated with heart failure hospitalization and long-term mortality in patients with heart failure with preserved ejection fraction and pulmonary hypertension. Cardiology. (2019) 143:124–33. doi: 10.1159/000501100
12. Schiattarella G, Altamirano F, Tong D, French K, Villalobos E, Kim S, et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature. (2019) 568:351–6. doi: 10.1038/s41586-019-1100-z
13. Mohammed SF, Hussain S, Mirzoyev SA, Edwards WD, Maleszewski JJ, Redfield MM, et al. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation. (2015) 131:550–9. doi: 10.1161/CIRCULATIONAHA.114.009625
14. Van Empel VP, Mariani J, Borlaug BA, Kaye DM. Impaired myocardial oxygen availability contributes to abnormal exercise hemodynamics in heart failure with preserved ejection fraction. J Am Heart Assoc. (2014) 3:e001293. doi: 10.1161/JAHA.114.001293
15. Van de Wouw J, Sorop O, van Drie RWA, van Duin RWB, Nguyen ITN, Joles JA, et al. Perturbations in myocardial perfusion and oxygen balance in swine with multiple risk factors: a novel model of ischemia and no obstructive coronary artery disease. Basic Res Cardiol. (2020) 115:21. doi: 10.1007/s00395-020-0778-2
16. Mitchell GF. Effects of central arterial aging on the structure and function of the peripheral vasculature: implications for end-organ damage. J Appl Physiol. (2008) 105:1652–60. doi: 10.1152/japplphysiol.90549.2008
17. Van de Wouw J, Broekhuizen M, Sorop O, Joles JA, Verhaar MC, Duncker DJ, et al. Chronic kidney disease as a risk factor for heart failure with preserved ejection fraction: a focus on microcirculatory factors and therapeutic targets. Front Physiol. (2019) 4:1108. doi: 10.3389/fphys.2019.01108
18. Gori M, Lam CS, Gupta DK, Santos AB, Cheng S, Shah AM, et al. Sex-specific cardiovascular structure and function in heart failure with preserved ejection fraction. Eur J Heart Fail. (2014) 16:535–42. doi: 10.1002/ejhf.67
19. Gladden JD, Linke WA, Redfield MM. Heart failure with preserved ejection fraction. Pflugers Arch. (2014) 466:1037–53. s00424-014-1480-8. doi: 10.1007/s00424-014-1480-8
20. Maréchaux S, Samson R, Van Belle E, Breyne J, De Monte J, Dédrie C, et al. Vascular and microvascular endothelial function in heart failure with preserved ejection fraction. J Card Fail. (2016) 22:3–11. doi: 10.1016/j.cardfail.2015.09.003
21. Teo LY, Chan LL, Lam CS. Heart Failure with preserved ejection fraction in hypertension. Curr Opin Cardiol. (2016) 31:410–6. doi: 10.1097/HCO.0000000000000292
22. Mohammed SF, Borlaug BA, Roger VL, Mirzoyev SA, Rodeheffer RJ, Chirinos JA, et al. Comorbidity and ventricular and vascular structure and function in heart failure with preserved ejection fraction. A community-based study. Circ Heart Fail. (2012) 5:710–19. doi: 10.1161/CIRCHEARTFAILURE.112.968594
23. Lee J. F., Barrett-O'Keefe Z., Garten R. S., Nelson A., Ryan J., Nativi J., Richardson R., Wray D. Evidence of microvascular dysfunction in heart failure with preserved ejection fraction. Heart. (2016) 102:278–84. doi: 10.1136/heartjnl-2015-308403
24. Haykowsky MJ, Herrington DM, Brukaber PH, Morgan TM, Hundley WG, Kitzman DW, et al. Relationship of flow-mediated arterial dilation and exercise capacity in older patients with heart failure and preserved ejection fraction. J Gerontol A Biol Sci Med Sci. (2013) 68:161–7. doi: 10.1093/gerona/gls099
25. Pinto A. R., Ilinykh A., Ivey M. J., Kuwabara J. T., D'Antoni M. L., Debuque R., Chandran A., Wang L., Arora K., Rosenthal N. A., Tallquist M. D. (2016). Revisiting cardiac cellular composition. Circ Res. 118, 400–409. doi: 10.1161/CIRCRESAHA.115.307778
26. Riad A, Westermann D, Van Linthout S, Mohr Z, Uyulmaz S, Becher PM, et al. Enhancement of endothelial nitric oxide synthase production reverses vascular dysfunction and inflammation in the hindlimbs of a rat model of diabetes. Diabetologia. (2008) 51:2325–32. doi: 10.1007/s00125-008-1159-9
27. Hayashida R, Kondo K, Morita S, Unno K, Shintani S, Shimizu Y, et al. Diallyl trisulfide augments ischemia-induced angiogenesis via an endothelial nitric oxide synthase-dependent mechanism. Circ J. (2017) 81:870–8. doi: 10.1253/circj.CJ-16-1097
28. Michel J, Li Z, Lacolley P. Smooth muscle cells and vascular diseases. Cardiovasc Res. (2012) 95:135–7. doi: 10.1093/cvr/cvs172
29. Amiya E, Watanabe M, Komuro I. The relationship between vascular function and the autonomic nervous system. Ann Vasc Dis. (2014) 7:109–19. doi: 10.3400/avd.ra.14-00048
30. Harris KF, Matthews KA. Interactions between autonomic nervous system activity and endothelial function: a model for the development of cardiovascular disease. Psychosom Med. (2004). 66:153–64. doi: 10.1097/01.psy.0000116719.95524.e2
31. Sheng Y, Zhu L. The crosstalk between autonomic nervous system and blood vessels. Int J Physiol Pathophysiol Pharmacol. (2018) 10:17–28.
32. Cyr AR, Huckaby LV, Shiva SS, Zuckerbraun BS. Nitric oxide and endothelial dysfunction. Crit Care Clin. (2019) 36:307–21. doi: 10.1016/j.ccc.2019.12.009
33. Clapp BR, Hingorani AD, Kharbanda RK, Mohamed-Ali V, Stephens JW, Vallance P, et al. Inflammation-induced endothelial dysfunction involves reduced nitric oxide bioavailability and increased oxidant stress. Cardiovasc Res. (2004) 64:20. doi: 10.1016/j.cardiores.2004.06.020
34. Emanueli C, Van Linthout S, Bonaria Salis M, Monopoli A, Del Soldato P, Ongini E, et al. Nitric oxide-releasing aspirin derivative, NCX 4016, promotes reparative angiogenesis and prevents apoptosis and oxidative stress in a mouse model of peripheral ischemia. Arterioscler Thromb Vasc Biol. (2004) 24:2082–7. doi: 10.1161/01.ATV.0000144030.39087.3b
35. Chirinos JA, Akers SR, Trieu L, Ischiropoulos H, Doulias PT, Tariq A, et al. Heart failure, left ventricular remodeling, and circulating nitric oxide metabolites. J Am Heart Assoc. (2016) 5:e004133. doi: 10.1161/JAHA.116.004133
36. Greene SJ, Gheorghiade M, Borlaug BA, Pieske B, Vaduganathan M, Burnett JC, et al. The cGMP signaling pathway as a therapeutic target in heart failure with preserved ejection fraction. J Am Heart Assoc. (2013) 2:e000536. doi: 10.1161/JAHA.113.000536
37. Wilck N, Marko L, Balogh A, Kraeker K, Herse F, Bartolomaeus H, et al. Nitric oxide-sensitive guanylyl cyclase stimulation improves experimental heart failure with preserved ejection fraction. JCI Insight. (2018) 3:e96006. doi: 10.1172/jci.insight.96006
38. Toblli JE, DiGennaro F, Giani JF, Dominici FP. Nebivolol: impact on cardiac and endothelial function and clinical utility. Vasc Health Risk Manag. (2012) 8:151–60. doi: 10.2147/VHRM.S20669
39. Suvorava T, Nagy N, Pick S, Lieven O, Rüther U, Dao VT, et al. Impact of eNOS-dependent oxidative stress on endothelial function and neointima formation. Antioxidants Redox Signal. (2015) 23:711–23. doi: 10.1089/ars.2014.6059
40. Ferro A, Queen LR, Priest R, Xu B, Ritter JM, Poston L, et al. Activation of nitric oxide synthase by beta 2-adrenoceptors in human umbilical vein endothelium in vitro. Br J Pharmocol. (1999) 126:1872–80. doi: 10.1038/sj.bjp.0702512
41. Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. (2013) 62:263–71. doi: 10.1016/j.jacc.2013.02.092
42. Zhazykbayeva S, Pabel S, Mügge A, Sossalla S, Hamdani N. The molecular mechanisms associated with the physiological responses to inflammation and oxidative stress in cardiovascular diseases. Biophys Rev. (2020) 12:947–680. doi: 10.1007/s12551-020-00742-0
43. Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res. (2018) 122:877–902. doi: 10.1161/CIRCRESAHA.117.311401
44. Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C dependent activation of NADPH oxidase in cultured vascular cells. Diabetes. (2000) 49:1939–45. doi: 10.2337/diabetes.49.11.1939
45. Franssen C, Chen S, Unger A, Korkmaz HI, De Keulenaer GW, Tschöpe C, et al. Myocardial microvascular inflammatory endothelial activation in heart failure with preserved ejection fraction. JACC Heart Fail. (2016) 4:312–24. doi: 10.1016/j.jchf.2015.10.007
46. Babes E, Babes V. ADMA and prognosis in patients with heart failure and preserved ejection fraction. Esc Heart Fail. (2013) 7:1491. doi: 10.1093/eurheartj/eht308.P1491
47. Davel AP, Ceravolo GS, Wenceslau CF, Carvalho MH, Brum PC, Rossoni LV, et al. Increased vascular contractility and oxidative stress in β2-adrenoceptor knockout mice: the role of NADPH oxidase. J Vasc Res. (2012) 49:342–52. doi: 10.1159/000337486
48. Conti V, Russomanno G, Corbi G, Izzo V, Vecchione C, Filippelli A, et al. Adrenoreceptors and nitric oxide in the cardiovascular system. Front Physiol. (2013) 4:321. doi: 10.3389/fphys.2013.00321
49. Bristow MR, Mann DL. Cardiac adrenergic activation in heart failure with preserved ejection fraction. JACC Basic Transl Sci. (2022) 7:128–30. doi: 10.1016/j.jacbts.2022.01.003
50. van Veldhuisen DJ, Cohen-Solal A, Böhm M, Anker SD, Babalis D, Roughton M, et al. Beta-blockade with nebivolol in elderly heart failure patients with impaired and preserved left ventricular ejection fraction: data from seniors (study of effects of nebivolol intervention on outcomes and rehospitalization in seniors with heart failure). J Am Coll Cardiol. (2009) 53:2150–8. doi: 10.1016/j.jacc.2009.02.046
51. Borlaug BA, Melenovský V, Russell SD, Kessler K, Pacak K, Becker L, et al. Impaired chronotropic and vasodilator reserves limit exercise capacity in patients with heart failure and a preserved ejection fraction. Circulation. (2006) 114:2138–47. doi: 10.1161/CIRCULATIONAHA.106.632745
52. Westermann D, Lindner D, Kasner M, Zietsch C, Savvatis K, Escher F, et al. Cardiac inflammation contributes to changes in the extracellular matrix in patients with heart failure and normal ejection fraction. Circ Heart Fail. (2011) 4:44–52. doi: 10.1161/CIRCHEARTFAILURE.109.931451
53. Collier P, Watson CJ, Voon V, Phelan D, Jan A, Mak G, et al. Can emerging biomarkers of myocardial remodeling identify asymptomatic hypertensive patients at risk for diastolic dysfunction and diastolic heart failure. Eur J Heart Fail. (2011) 13:1087–95. doi: 10.1093/eurjhf/hfr079
54. DuBrock HM, AbouEzzeddine OF, Redfield MM. High-sensitivity C-reactive protein in heart failure with preserved ejection fraction. PLoS ONE. (2018) 13:e0201836. doi: 10.1371/journal.pone.0201836
55. Bishu K, Deswal A, Chen HH, LeWinter MM, Lewis GD, Semigran MJ, et al. Biomarkers in acutely decompensated heart failure with preserved or reduced ejection fraction. Am Heart J. (2012) 164:763–70. doi: 10.1016/j.ahj.2012.08.014
56. Lymperopoulos A, Rengo G, Koch WJ. Adrenergic nervous system in heart failure: pathophysiology and therapy. Circ Res. (2013) 113:739–53. doi: 10.1161/CIRCRESAHA.113.300308
57. Santulli G, Iaccarino G. Adrenergic signaling in heart failure and cardiovascular aging. Maturitas. (2016) 93:65–72. doi: 10.1016/j.maturitas.2016.03.022
58. Dridi H, Santulli G, Gambardella J, Jankauskas S, Yuan Q, Yang Q, et al. IP3 receptor orchestrates maladaptive vascular responses in heart failure. J Clin Invest. (2022) 132:e152859. doi: 10.1172/JCI152859
59. Jackson WF. Potassium channels in regulation of vascular smooth muscle contraction and growth. Adv Pharmacol. (2016) 78:89–144. doi: 10.1016/bs.apha.2016.07.001
60. Lin Q, Zhao G, Fang X, Peng X, Tang H, Wang H, et al. IP3 receptors regulate vascular smooth muscle contractility and hypertension. JCI Insight. (2016) 1:e89402. doi: 10.1172/jci.insight.89402
61. Gambardella J, Lombardi A, Morelli MB, Ferrara J, Santulli G. Inositol 1,4,5-trisphosphate receptors in human disease: a comprehensive update. J Clin Med. (2020) 9:1096. doi: 10.3390/jcm9041096
62. Guo A, Zhang C, Wei S, Chen B, Sheng Song L. Emerging mechanisms of T-tubule remodelling in heart failure. Cardiovasc Res. (2013) 98:204–15. doi: 10.1093/cvr/cvt020
63. Zhao J, Xu T, Zhou Y, Zhou Y, Xia Y, Li D, et al. B-type natriuretic peptide and its role in altering Ca2+-regulatory proteins in heat failure-mechanistic insights. Heart Fail Rev. (2020) 25:861–71. doi: 10.1007/s10741-019-09883-1
64. Bovo E, Huke S, Blatter LA, Zima AV. The effect of PKA-mediated phosphorylation of ryanodine receptor on SR Ca2+ leak in ventricular myocytes. J Mol Cell Cardiol. (2017) 104:9–16. doi: 10.1016/j.yjmcc.2017.01.015
65. Li L, Louch W, Niederer S, Aronsen J, Christensen G, Sejersted O, et al. Sodium accumulation in SERCA Knockout-induced heart failure. Biophys J. (2012) 102:2039–48. doi: 10.1016/j.bpj.2012.03.045
66. Bers DM. Cardiac sarcoplasmic reticulum calcium leak: basis and roles in cardiac dysfunction. Annu Rev Physiol. (2014) 76:107–27. doi: 10.1146/annurev-physiol-020911-153308
67. Eder P, Molkentin JD. TRPC channels as effectors of cardiac hypertrophy. Circ Res. (2011) 108:265–72. doi: 10.1161/CIRCRESAHA.110.225888
68. He WQ, Peng YJ, Zhang WC, Lv N, Tang J, Chen C, et al. Myosin light chain kinase is central to smooth muscle contraction and required for gastrointestinal motility in mice. Gastroenterology. (2008) 135:610–20. doi: 10.1053/j.gastro.2008.05.032
Keywords: inflammation, oxidative stress, heart failure, vascular dysfunction, preserved ejection fraction heart failure (HFpEF)
Citation: Saavedra-Alvarez A, Pereyra KV, Toledo C, Iturriaga R and Del Rio R (2022) Vascular dysfunction in HFpEF: Potential role in the development, maintenance, and progression of the disease. Front. Cardiovasc. Med. 9:1070935. doi: 10.3389/fcvm.2022.1070935
Received: 15 October 2022; Accepted: 17 November 2022;
Published: 21 December 2022.
Edited by:
Jintao Wang, National Heart, Lung, and Blood Institute (NIH), United StatesReviewed by:
Yunhui Du, Beijing Institute of Heart, Capital Medical University, ChinaCopyright © 2022 Saavedra-Alvarez, Pereyra, Toledo, Iturriaga and Del Rio. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rodrigo Del Rio, cmRlbHJpb0BiaW8ucHVjLmNs
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.