 Anuj Narwal
Anuj Narwal Claire S. Whyte
Claire S. Whyte Nicola J. Mutch
Nicola J. Mutch
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cardiovasc. Med., 18 January 2023
Sec. Thrombosis and Haemostasis
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.1070502
This article is part of the Research TopicUnraveling Clot Composition and Structure in Vitro, in Vivo and in SilicoView all 4 articles
Thrombi are heterogenous in nature with composition and structure being dictated by the site of formation, initiating stimuli, shear stress, and cellular influences. Arterial thrombi are historically associated with high platelet content and more tightly packed fibrin, reflecting the shear stress in these vessels. In contrast, venous thrombi are generally erythrocyte and fibrin-rich with reduced platelet contribution. However, these conventional views on the composition of thrombi in divergent vascular beds have shifted in recent years, largely due to recent advances in thromboectomy and high-resolution imaging. Interestingly, the distribution of fibrinolytic proteins within thrombi is directly influenced by the cellular composition and vascular bed. This in turn influences the susceptibility of thrombi to proteolytic degradation. Our current knowledge of thrombus composition and its impact on resistance to thrombolytic therapy and success of thrombectomy is advancing, but nonetheless in its infancy. We require a deeper understanding of thrombus architecture and the downstream influence on fibrinolytic susceptibility. Ultimately, this will aid in a stratified and targeted approach to tailored antithrombotic strategies in patients with various thromboembolic diseases.
Thrombosis is the underlying pathology of major cardiovascular diseases, including myocardial infarction, ischemic stroke, and venous thromboembolism (VTE) which encompasses deep vein thrombosis and pulmonary embolism. Despite advances in diagnostics and novel antithrombotic drugs the mortality rate remains at 1 in 4 worldwide, creating a considerable burden on healthcare and society (1). In addition, thrombosis is a major cause of mortality in other disease, such as cancer, pathogenic infections, and autoimmune diseases (2–4). Hemodynamic forces and anatomic location significantly impact the formation, structure and stability of a thrombus within the vasculature. The resulting thrombi are heterogenous in nature, comprised of varying degrees of fibrin, platelets, erythrocytes, leukocytes, and neutrophil extracellular traps (NETs) (5). Thrombotic structures also vary within the arterial, venous, and microcirculation, at areas of turbulent flow, arising from atherosclerotic lesions, prosthetic devices or irregular vessel geometries and in different anatomic sites, such as the lung or ventricles of the heart (6). Analysis of thrombi has been hampered by availability of fresh samples, however, advancements in thrombectomy to remove thrombi from human blood vessels provided opportunities to examine the structure and composition of a thrombus (7). In addition, various ex vivo and in vivo models of thrombus formation and thrombolysis have provided useful tools to understand thrombus initiation in different vascular beds, the composition of various thrombus components, impact of shear and their downstream impact on fibrinolysis (8–12). Developing an understanding of thrombus composition, localization and abundance of fibrinolytic proteins in specific settings is crucial to personalize antithrombotic treatment strategies and develop novel drugs to target thrombosis.
The trigger for thrombosis depends largely on the vascular bed (Figure 1). Nonetheless, an initial step is adherence of platelets to the vessel wall via various receptors, including the GPIb-IX-V/GPVI adheso-signaling complex thereby initiating platelet activation and aggregation (13). Activated platelets provide a catalytic aminophospholipid surface to assemble the prothrombinase complex, thereby catalyzing conversion of prothrombin to thrombin. These events elicit a conformation change in integrin αIIbβ3, allowing interaction with fibrinogen, which permits tethering of platelets to the forming fibrin network. Fibrinogen binding initiates outside-in signaling and promotes clot retraction, a process whereby activated platelets transduce contractile forces to the fibrin network augmenting clot density and decreasing clot size. Clot retraction is important for clot stability and maintaining blood vessel patency. Interestingly, a recent study found a direct link between endogenous fibrinolysis and clot retraction, suggesting that these processes are inextricably linked in vivo (14). Additional platelet receptors for fibrin have been proposed, including GPVI (15), which can directly instigate platelet activation and drive thrombus propagation (16).
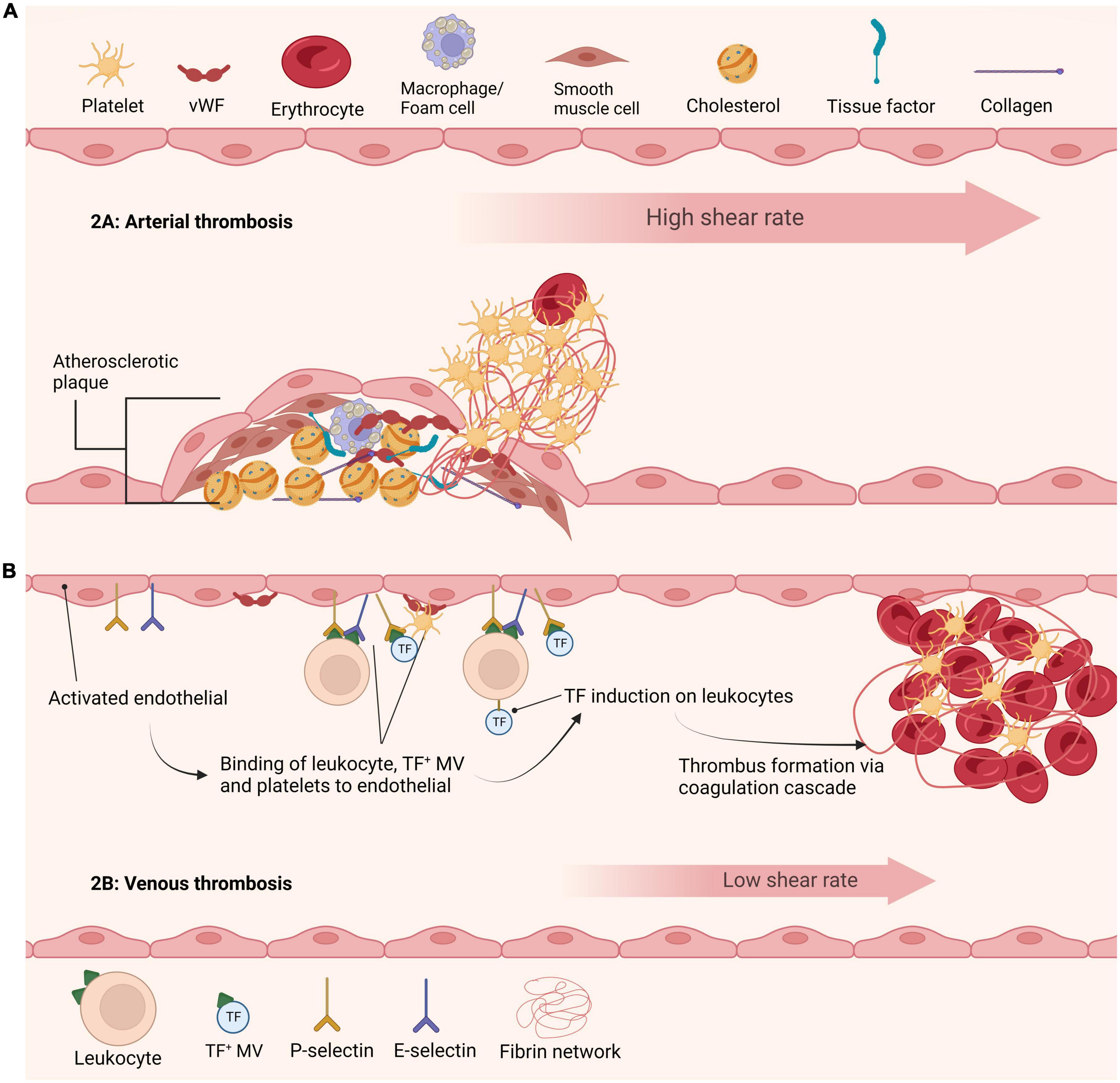
Figure 1. Current understanding of arterial and venous thrombi initiation. (A) Initiation of arterial thrombus formation is triggered by atherosclerotic plaque rupture. Exposure of collagen and tissue factor leads to recruitment and activation of platelets at the site of injury. Thrombi in arteries are formed under high shear stress and are rich in platelets. (B) Formation of venous thrombi is currently understood to be triggered by various mechanisms. The activated endothelium leads to recruitment and binding of cells and factors including leukocytes, tissue factor positive microvesicles and platelets. These agents further promote tissue factor recruitment ultimately leading to formation of a venous thrombus.
Differential agonist distribution in the evolving platelet mass results in phenotypically different subpopulations of platelets (17–20) within the microenvironment of the thrombus. Phosphatidylserine (PS)-negative (aggregating platelets) have a spread morphology, avidly bind fibrinogen, via activated αIIbβ3, and generate fibrin on their surface (21). PS-exposing platelets (procoagulant) bind the prothrombinase complex and exhibit a characteristic balloon shape with prolonged spikes in cytosolic Ca2+; these platelets lack activation of integrin αIIbβ3 (20–22). Work from our group (10, 23, 24) and others (25–28) has shown that PS is concentrated in the “cap” or “body” of these platelets along with key hemostatic proteins.
Thrombin generation on the activated platelets amplifies fibrin formation, thereby providing structural support, mechanical stability and integrity to the thrombus. Fibrin structure is altered by multiple parameters, and is largely dictated by fibrinogen and thrombin concentration (29). Low thrombin concentrations generate thick fibrin fibers, loosely woven, and permeable fibrin clots leading to hyperfibrinolysis. In contrast, high thrombin concentrations give rise to clots comprising of a dense network of thin fibrin fibers associated with hypofibrinolysis (30). Various studies have shown that compact fibrin structures, with highly branched networks are associated with pathophysiological diseases such as, coronary artery disease, ischemic stroke and pulmonary embolism [reviewed by Undas and Ariens (31)]. Fibrin fibers orient in the direction of flow, with increased alignment as shear stress is magnified (32). Interestingly, recent studies indicate that fibrin forms a protective biofilm over the external area of a thrombus, as a protection against invading pathogens (33).
Arterial thrombosis is typically triggered by rupture of an atherosclerotic plaque, permitting contact of highly prothrombotic material, rich in tissue factor and lipids, with plasma thereby prompting platelet activation and coagulation (Figure 1A). Structural analysis of coronary arterial thrombi revealed they are comprised of fibrin (43% of thrombus volume) and platelets (31%) (34). Fibrin was tightly packed, perhaps not surprising given the high shear stress (1,000–1,500 s–1), and arranged in bundles, possibly reflecting the lateral association of fibers due to increased compression exerted by platelet contraction (35). Interestingly, a small number of compressed erythrocytes, termed polyhedrocytes, were evident and the remainder of thrombus volume was occupied by microvesicles and leukocytes (34). Thrombi from ST-segment-elevation myocardial infarction (STEMI) patients, were again largely composed of fibrin with increased erythrocyte to platelet ratio than reported in coronary artery thrombi (36). Intriguingly, in primary coronary intervention fibrin content correlated with plasminogen activator inhibitor-1 (PAI-1) and P-selectin, indicative of a role for platelets in driving fibrin formation (36).
Interestingly, thromboemboli retrieved from the middle cerebral artery or intracranial carotid artery of patients with acute ischemic stroke revealed significant heterogeneity, but again platelet-fibrin areas were dominant, interspersed with areas of nucleated cells and erythrocytes (37). More recent studies unveil heterogenous areas, comprised of erythrocyte-rich and fibrin poor areas and platelet- and fibrin-rich areas (38). A recent elegant study of thrombi from acute ischemic stroke, using scanning electron microscopy and immunohistochemistry, revealed an outer shell, comprised of densely packed fibrin, von Willebrand factor, and aggregated platelets (39). Parameters affecting thrombus growth shift at the point of occlusion when shear stress decreases due to diversion of the blood. Nonetheless, microfluidic modeling of occlusive thrombus formation that permits pressure release demonstrated that despite the variations in shear stress fibrin accumulation under arterial rates was still reduced in comparison to the venous circulation (40).
The mechanisms underpinning development of venous thrombosis are still debatable, with a call for action and prioritization of funding in this area (41). The concepts of Virchow’s triangle, including changes in blood composition, reduction in blood flow, and changes to the vascular endothelium are considered key drivers, but further work is required to tease out precise mechanisms. Genetic and acquired risk factors augment the risk of venous thrombosis [reviewed by Wolberg et al. (42)]. A pivotal study by von-Bruhl et al. (43) demonstrated that initiating events of venous thrombosis in vivo involve crosstalk between platelets, monocytes and neutrophils (Figure 1B). They elegantly demonstrated that neutropenia, genetic ablation of FXII, or disintegration of NETs individually confer protection against deep vein thrombosis (DVT) in vivo (43).
Erythrocytes comprise nearly 60% of the volume of venous thrombi with fibrin fibers accounting for about 30% (34). Polyhedrocytes were also found in venous thrombi, with around 5% of thrombus volume composed of echinocytes (34). These “thorny” erythrocytes are indicative of oxidative stress and perhaps cellular aging within the thrombus environment. Leukocytes and microvesicles were detected but were less abundant (34). The endothelial contribution in venous thrombosis is vital, as it captures leukocytes, tissue factor-positive microvesicles and platelets (Figure 1B). The composition of pulmonary emboli (PE) largely mirrored that of venous thrombi, with polyhedrocytes accounting for majority of the thrombus volume (34). A recent report indicates that PE thrombi are generally “earlier” stage in terms of composition with a higher erythrocyte component (44). Intriguingly, within venous thrombi fibrin fibers were largely evident as individual fibers rather than bundles, perhaps reflecting a decrease in mechanical stability, and accounting for their tendency to readily embolize.
Severe COVID-19 disease is associated with an increased risk of thrombosis (45), both systemically and locally within the pulmonary vasculature (46). Studies indicate that PE derived from critically ill COVID-19 patients differ significantly from non-COVID PE (47). Thrombi were located directly within opacitated lung segments, indicative of in situ thrombogenesis (48). An increased rate of in situ PE in COVID-19 may suggest that leukocytes drive thrombogenesis. Indeed, an in vivo model of DVT has revealed significant fibrin deposition in rats with normal neutrophil counts which is attenuated in neutropenia (49). Further research is required to directly compare the structural composition of PE formed in situ vs. those that embolize to the pulmonary vasculature which will aid understanding of underlying mechanisms and personalize diagnosis and care.
The mechanistic contributions of various circulating cells, including erythrocytes and inflammatory cells, to thrombosis is currently the subject of intense scientific scrutiny. Many avenues of interplay between hemostatic factors and cells or cell-cell interactions have and continue to be uncovered, some of these are highlighted below.
Erythrocytes were long considered to be innocent bystanders in thrombi but are now considered to play a more significant role than previously thought [reviewed by Byrnes and Wolberg (50)]. Erythrocytes express the Fas ligand, FasL and the death receptor, FasR (51). Activation of FasR induces loss of asymmetry and integrity of the phospholipid bilayer thus exposing aminophospholipids. This provides an “eat-me” signal to remove older erythrocytes from the circulation, however, these aminophospholids can also assemble the prothrombinase complex leading to thrombin generation. ADP-activated platelets express FasL on their membrane which interacts with FasR on erythrocytes augmenting aminophospholipid exposure (52). To date this unique cell-cell interaction has only been demonstrated in vitro, however, it provides a novel mechanism in which erythrocytes can promote thrombus formation.
Erythrocyte aggregation influences blood flow and is a cardiovascular risk factor. It was hypothesized that fibrinogen and other plasma proteins induced erythrocyte aggregation via non-specific binding. However, Carvalho et al. (53) demonstrated a unique interaction between fibrinogen and an unknown receptor on erythrocytes using atomic force microscopy. A patient with Glanzmann thrombastenia, a hereditary bleeding disorder caused by deficiency of integrin αIIbβ3, showed defective binding of fibrinogen to erythrocytes. Similarly, the αIIbβ3 inhibitor, eftifibatide, attenuated binding of fibrinogen to erythrocytes, albeit to a lesser degree than on platelets. Interestingly, mice carrying a homozygous mutation for γ390-396 in fibrinogen showed a 50% reduction in thrombus weight, due to reduced erythrocyte volume (54). This effect was mediated via factor XIII activation and crosslinking (54). The group later showed this was dependent on the presence of plasma FXIII (55) and that retention of erythrocytes in clots is mediated via fibrin α-chain cross-linking (56). As discussed, erythrocytes accrued within the clot are frequently observed as polyhedrocytes rather than their native bioconcave state (34, 57). It is the process of clot contraction, mediated by platelets that generates the necessary force to compress and alter the rigidity of erythrocytes into these tightly packed arrays of polyhedrocytes (58, 59). These lines of evidence indicate that erythrocytes play an active role in the size and structural integrity of pathophysiological thrombi.
The intrinsic link between the innate immune system and coagulation is now firmly established (60). Fibrin(ogen) binds to the integrin αMβ2 which is crucial for leukocyte function and innate immunity in vivo (61). Platelet-leukocyte aggregates, mediated via interaction of platelet P-selectin and GPIbα with neutrophil P-selectin glycoprotein ligand-1 and αMβ2 integrin, respectively, are common features in thromboinflammatory disorders [reviewed by Swystun and Liaw (62)]. This cell-cell interaction induces a hypercoaguable state inciting platelet activation, binding of coagulation factors and adhesive proteins such as von Willebrand factor (vWF).
Monocytes harbor the largest circulating pool of tissue factor, which is principally in a quiescent state but can be exposed and decrypted in response to various inflammatory stimuli. Activated monocytes shed microvesicles that carry tissue factor, expose PS, and other procoagulant factors (63). It is well known that monocytes house a large pool of intracellular factor XIII-A (64). Our laboratory has recently shown that monocytes externalize factor XIII-A in response to inflammatory stimuli which stabilizes thrombi in a transglutaminase-dependent manner (65). Monocytes are also the largest circulating pool of the fibrinolytic inhibitor, PAI-2 which is upregulated in response to thrombin and LPS stimulus (66). Interestingly, this serpin is considered to be largely intracellular in nature, but can down-regulate uPA and is cross-linked to fibrin (67). PAI-2 is also found in extracts of human arterial and venous thrombi suggesting secretion from monocytes in response to various stimuli (67). Mice deficient in PAI-2 exhibit superior venous thrombus resolution due to inflammatory and uPA-mediated mechanism (68). Conversely, reports indicate that monocytes recruitment into the thrombi is important for resolution, which is largely uPA-mediated (69). Clearly there is a strong need to understand the nuances by which immune cells function to explain existing controversies in the literature and their role in governing thrombus stability.
Neutrophils accumulate at sites of injury acting to limit invading pathogens. Brinkmann et al. described the extrusion of neutrophil nuclear and cytoplasmic content forming NETs in the cell death process of NETosis (70). These web-like structures are formed in response to inflammatory stimuli, microbial invasion and are composed of histones, DNA strands and granular proteins including neutrophil elastase (70). NETs have been detected in both venous (71) and arterial thrombi (72–74). NETs contribute to thrombus formation through multiple mechanisms, including the release of neutrophil elastase and cathepsin G, as well as externalization of nucleosomes (75). NETs expose tissue factor and protein disulfide isomerase, an enzyme responsible for activating blood cell derived tissue factor (43, 76, 77) thereby driving coagulation. NETosis is promoted under high shear conditions (78) independent of thrombin and fibrin generation (79). Indeed, fibrin limits NET formation and tPA facilitates shear-induced NET formation (78). NETs promote platelet adhesion, activation and aggregation (80) and citrullinated histone H3 (CitH3) are detected in close proximity to vWF within fibrin-rich areas of thrombi (81). Conversely, platelets contribute to the formation of NETs through lipopolysaccharide binding of Toll like receptor 4 (TLR4) (82).
The fibrinolytic system is nature’s endogenous system programmed to dissolve intravascular clots and counteract the opposing coagulation system (Figure 2). Plasmin, the key proteolytic enzyme, is formed via cleavage of circulating plasminogen through the action of plasminogen activators, primarily tissue-type PA (tPA) and uPA. Endothelial cells (83), neurons (84) and hepatocytes (85) express and secrete tPA, with recent evidence suggested that hepatocyte-derived tPA contributes to basal circulating levels of tPA. In contrast, uPA is largely expressed by migratory and inflammatory cells (86). The system is governed by several inhibitors, including α2-antiplasmin and PAI-1 and PAI-2. Activated thrombin activatable fibrinolysis inhibitor (TAFIa; CPB2) down-regulates fibrinolysis, via removal of C-terminal lysine residues from partially degraded fibrin, thereby attenuating binding of plasminogen and tPA. In thrombosis the fibrinolytic balance is disturbed, favoring fibrin formation and persistence, which can be partially attributed to the cellular composition of thrombi and their relative contributions to the system.
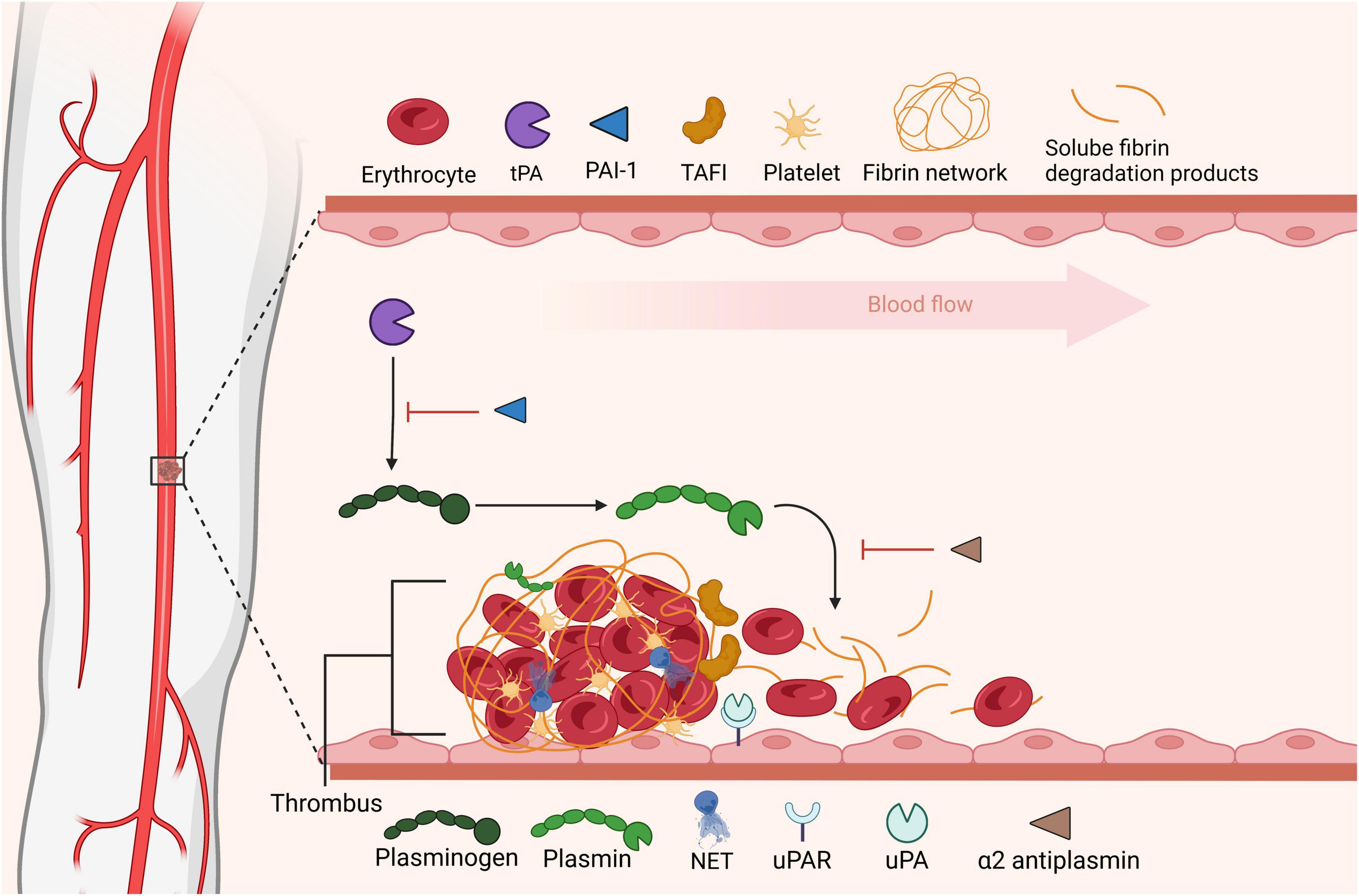
Figure 2. Fibrinolytic dissolution of thrombi. Fibrinolysis is initiated by plasminogen activators that convert circulating plasminogen to its active form plasmin promoting degradation of fibrin. Soluble fibrin degradation products can be cleared from the circulation. Fibrinolysis is regulated at the level of plasminogen activation, via plasminogen activator inhibitor-1 (PAI-1) or by direct inhibition of plasmin by α2-antiplasmin (α2AP). Thrombin activable fibrinolysis inhibitor (TAFI) impedes fibrinolysis by removing C-terminal residues from fibrin, these lysine residues are vital for plasminogen binding to fibrin. In this figure was adapted from “Tissue Plasminogen Activator Activity at Ischemic Region in the Brain,” by BioRender.com (2022). Retrieved from https://app.biorender.com/biorender-templates.
Accumulation of tPA and plasminogen is observed in the head of Chandler model thrombi, directly aligning with localization of fibrinolytic activity (12). This observation is unexpected given the knowledge that the head is rich in platelets and leukocytes, while the tail is fibrin-rich (8). Our previous work had established that the cellular-rich head is rich in active uPA, which is largely leukocyte in origin (9). Within this microenvironment proteolytic activity of the plasminogen activators is largely protected from inhibition by PAI-1 (12), contrasting the situation in plasma where tPA is largely found in complex with PAI-1 (87). Elevation in endogenous tPA during thrombus formation increases retention within thrombi (12), indicating that thrombus resolution is dictated by the levels of activators present during formation. However, there is evidence of infiltration of monocyte/macrophages and neutrophils into forming thrombi (43, 69, 81, 88). Accumulation of plasminogen in thrombi, formed under shear stress, has been depicted by our laboratory (10), plasminogen was demonstrated to be primarily localized in the thrombus core directly on fibrin and on the surface of PS-exposing platelets, via both fibrin dependent and independent processes (10). Consistent with this, plasminogen accumulated in the thrombus core with PS-exposing platelets in an in vivo laser-injury model and this process was enhanced by endogenous plasmin activity (89). Similarly, plasminogen accumulates in fibrin-rich areas on preformed thrombi formed under high shear rates (90). Model thrombi formed using non-anticoagulated blood under high shear rates show elevated levels of PAI-1, whilst both tPA and plasminogen were reduced, resulting in slower rates of fibrinolysis compared to that in thrombi formed at low shear (12). Indeed, cells that are incorporated into thrombi harbor many receptors for plasminogen, largely utilizing C-terminal lysines (91). Plg-RKT was identified in 2010 as the first receptor for plasminogen to be synthesized with a C-terminal lysine (92). Plg-RKT demonstrates affinity for tPA and is known to co-localize with uPAR on monocytes and macrophages (92). We have subsequently identified Plg-RKT on platelets and found that it is directly responsible for anchoring plasminogen to the activated platelet membrane (93). Interestingly, while platelets do not express uPAR, we have shown that the platelet membrane stimulates reciprocal activation of scuPA and plasminogen to their active forms (94), thereby highlighting the importance of cellular surfaces in regulating profibrinolytic activity. Intriguingly, plasminogen bound to fibrin, platelets or extracellular matrix proteins can be proteolytically activated by uPA adhered to monocytes or microvesicles (95). Binding of soluble Glu-plasminogen to cell surfaces enhances its activation [reviewed in Miles and Parmer (96)] induces a conformational change distinct from that of Lys-plasminogen (97). These studies and others highlight the importance of the cell membrane in supplying fibrinolytic proteins and catalyzing plasminogen activation.
Platelets are the major pool of circulating PAI-1 (98). Degranulation following platelet activation gives rise to release of platelet-derived PAI-1 into the local milieu (99, 100). Our laboratory has recently showing that functional PAI-1 is retained on the activated platelet membrane and on associated fibrin (101), providing a local pool of serpin within the thrombus. Platelets also contain an abundance of other fibrinolytic inhibitors such as, alpha2-antiplasmin (102, 103) protease nexin I (PN-1) (104–106), C1-inhibitor (107, 108) and TAFI (109). These inhibitors are also secreted following activation and contribute to antifibrinolytic capacity. PN-1 from platelets downregulates the plasmin generating ability of fibrin-bound tPA and the activity of fibrin-bound plasmin and inhibits uPA (106, 110). However, as noted tPA, uPA and plasminogen are largely protected from inhibition if fibrin- or cell-bound. Activated TAFI (TAFIa) has been shown to limit plasminogen and tPA accumulation on the platelet surface and movement within plasma clots (111, 112). Platelets are also a rich-source of factor XIII-A (113–115) which is known to be externalized upon activation and can participate in thrombus stabilization via crosslinking of a2antiplasmin into the forming thrombus (24). Targeting of activated platelets is therefore an attractive therapeutic strategy. Single-chain antibodies to the platelet integrin αIIbβ3 fused to scuPA have shown promise in a mouse ischemic stroke model (116).
Platelet-mediated clot retraction is reportedly resistant to external fibrinolysis, however, is vulnerable to endogenous fibrinolysis (14). Interestingly, the internal rate of fibrinolysis is enhanced by clot retraction, whilst the external rate of fibrinolysis is impeded thereby suggesting differences in the fibrin susceptibility due to structural rearrangements during this process (117). Thrombi containing erythrocytes formed in vitro are more resistant to plasmin-mediated fibrinolysis despite the thrombi being composed of thinner fibers and a less dense fibrin network (118). However, thrombi obtained from stroke patients by endovascular thrombectomy that were more responsive to intravenous thrombolysis were found to be more erythrocyte-rich (119). Higher erythrocyte count has been associated with shorter intervention times, lower thrombolysis resistance and incidences of embolism and successful recanalization (120–122). The presence of higher white blood cell counts, NETs and vWF have been linked to reduced rates of recanalization (123). Clearly, there is a need for deeper research in this area to deepen our understanding of this area and iron out discrepancies in the current literature.
In addition to “conventional” fibrinolytic factors additional modifiers of thrombus stability have been identified. The impact of NETs, and specifically the DNA composition of thrombi, in limiting fibrinolysis has recently garnered attention. Histones alter fibrin fiber thickness and are crosslinked via factor XIIIa into the network which downregulates fibrinolysis (124). There is significant interest in inclusion of a DNase enzyme, as an adjunct to Alteplase (Actilyse®) in thrombolytic therapy. DNase1, an endonuclease that facilitates chromatin breakdown, has been shown to reduce NET formation and considerably limit DVT growth in mice (43). Additionally, DNase accelerates the rate of ex vivo thrombolysis of coronary and acute ischemic stroke thrombi (72, 73). The presence of large vWF multimers formed under high shear conditions also confer thrombolytic resistance, due to resistance to ADAMTS13, which cleaves vWF and tPA (125). Thrombotic thrombocytopenic purpura (TTP) is caused by ADAMTS13 deficiency leading to ultra-large vWF multimers. Targeted plasmin-mediated degradation of vWF polymers using fusion of a nanobody targeting vWF with the protease domain of uPA has recently shown promise as a treatment for TTP (126).
The recent advances in novel ex vivo models combined with in vivo animal models and developments in thrombectomy have significantly improved our understanding of the complex thrombus environment. This in turn gives significant insight into the susceptibility of thrombi to lysis and the factors which govern these processes. Understanding the impact of location, shear stress and vessel geometries on the cellular content and fibrin network is essential for the development of targeted and personalized approaches to treat thrombotic complications.
AN wrote the manuscript and designed the figures. CW and NM conceptualized, wrote, edited and reviewed the manuscript and figures. All authors contributed to the article and approved the submitted version.
Figures were created with BioRender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Wendelboe A, Raskob G. Global burden of thrombosis: Epidemiologic aspects. Circ Res. (2016) 118:1340–7. doi: 10.1161/CIRCRESAHA.115.306841
2. Fernandes C, Morinaga L, Alves J Jr., Castro M, Calderaro D, Jardim C, et al. Cancer-associated thrombosis: the when, how and why. Eur Respir Rev. (2019) 28:180119. doi: 10.1183/16000617.0119-2018
3. Silvestri E, Scalera A, Emmi G, Squatrito D, Ciucciarelli L, Cenci C, et al. Thrombosis in autoimmune diseases: A role for immunosuppressive treatments? Semin Thromb Hemost. (2016) 42:650–61. doi: 10.1055/s-0036-1579642
4. Beristain-Covarrubias N, Perez-Toledo M, Thomas M, Henderson I, Watson S, Cunningham A. Understanding infection-induced thrombosis: Lessons learned from animal models. Front Immunol. (2019) 10:2569. doi: 10.3389/fimmu.2019.02569
5. Hathcock J. Flow effects on coagulation and thrombosis. Arterioscler Thromb Vasc Biol. (2006) 26:1729–37. doi: 10.1161/01.ATV.0000229658.76797.30
6. Alkarithi G, Duval C, Shi Y, Macrae F, Ariens R. Thrombus structural composition in cardiovascular disease. Arterioscler Thromb Vasc Biol. (2021) 41:2370–83. doi: 10.1161/ATVBAHA.120.315754
7. Staessens S, Francois O, Brinjikji W, Doyle K, Vanacker P, Andersson T, et al. Studying stroke thrombus composition after thrombectomy: What can we learn? Stroke. (2021) 52:3718–27. doi: 10.1161/STROKEAHA.121.034289
8. Robbie L, Young S, Bennett B, Booth N. Thrombi formed in a Chandler loop mimic human arterial thrombi in structure and PAI-1 content and distribution. Thromb Haemost. (1997) 77:510–5. doi: 10.1055/s-0038-1655998
9. Mutch N, Moir E, Robbie L, Berry S, Bennett B, Booth N. Localization and identification of thrombin and plasminogen activator activities in model human thrombi by in situ zymography. Thromb Haemost. (2002) 88:996–1002. doi: 10.1055/s-0037-1613346
10. Whyte C, Swieringa F, Mastenbroek T, Lionikiene A, Lance M, van der Meijden P, et al. Plasminogen associates with phosphatidylserine-exposing platelets and contributes to thrombus lysis under flow. Blood. (2015) 125:2568–78. doi: 10.1182/blood-2014-09-599480
11. Jagadeeswaran P, Cooley B, Gross P, Mackman N. Animal models of thrombosis from Zebrafish to nonhuman primates: Use in the elucidation of new pathologic pathways and the development of antithrombotic drugs. Circ Res. (2016) 118:1363–79. doi: 10.1161/CIRCRESAHA.115.306823
12. Whyte C, Mostefai H, Baeten K, Lucking A, Newby D, Booth N, et al. Role of shear stress and tPA concentration in the fibrinolytic potential of thrombi. Int J Mol Sci. (2021) 22:2115. doi: 10.3390/ijms22042115
13. Gardiner E, Andrews R. Structure and function of platelet receptors initiating blood clotting. Adv Exp Med Biol. (2014) 844:263–75. doi: 10.1007/978-1-4939-2095-2_13
14. Samson A, Alwis I, Maclean J, Priyananda P, Hawkett B, Schoenwaelder S, et al. Endogenous fibrinolysis facilitates clot retraction in vivo. Blood. (2017) 130:2453–62. doi: 10.1182/blood-2017-06-789032
15. Alshehri O, Hughes C, Montague S, Watson S, Frampton J, Bender M, et al. Fibrin activates GPVI in human and mouse platelets. Blood. (2015) 126:1601–8. doi: 10.1182/blood-2015-04-641654
16. Bender M, Hagedorn I, Nieswandt B. Genetic and antibody-induced glycoprotein VI deficiency equally protects mice from mechanically and FeCl(3) -induced thrombosis. J Thromb Haemost. (2011) 9:1423–6. doi: 10.1111/j.1538-7836.2011.04328.x
17. Alberio L, Safa O, Clemetson K, Esmon C, Dale G. Surface expression and functional characterization of alpha-granule factor V in human platelets: effects of ionophore A23187, thrombin, collagen, and convulxin. Blood. (2000) 95:1694–702. doi: 10.1182/blood.V95.5.1694.005k24_1694_1702
18. Kulkarni S, Jackson S. Platelet factor XIII and calpain negatively regulate integrin alphaIIbbeta3 adhesive function and thrombus growth. J Biol Chem. (2004) 279:30697–706. doi: 10.1074/jbc.M403559200
19. Kempton C, Hoffman M, Roberts H, Monroe D. Platelet heterogeneity: variation in coagulation complexes on platelet subpopulations. Arterioscler Thromb Vasc Biol. (2005) 25:861–6. doi: 10.1161/01.ATV.0000155987.26583.9b
20. Heemskerk J, Vuist W, Feijge M, Reutelingsperger C, Lindhout T. Collagen but not fibrinogen surfaces induce bleb formation, exposure of phosphatidylserine, and procoagulant activity of adherent platelets: evidence for regulation by protein tyrosine kinase-dependent Ca2+ responses. Blood. (1997) 90:2615–25. doi: 10.1182/blood.V90.7.2615
21. Munnix I, Kuijpers M, Auger J, Thomassen C, Panizzi P, van Zandvoort M, et al. Segregation of platelet aggregatory and procoagulant microdomains in thrombus formation: regulation by transient integrin activation. Arterioscler Thromb Vasc Biol. (2007) 27:2484–90. doi: 10.1161/ATVBAHA.107.151100
22. Berny M, Munnix I, Auger J, Schols S, Cosemans J, Panizzi P, et al. Spatial distribution of factor Xa, thrombin, and fibrin(ogen) on thrombi at venous shear. PLoS One. (2010) 5:e10415. doi: 10.1371/journal.pone.0010415
23. Mitchell J, Lionikiene A, Georgiev G, Klemmer A, Brain C, Kim P, et al. Polyphosphate colocalizes with factor XII on platelet-bound fibrin and augments its plasminogen activator activity. Blood. (2016) 128:2834–45. doi: 10.1182/blood-2015-10-673285
24. Mitchell J, Lionikiene A, Fraser S, Whyte C, Booth N, Mutch N. Functional factor XIII-A is exposed on the stimulated platelet surface. Blood. (2014) 124:3982–90. doi: 10.1182/blood-2014-06-583070
25. Abaeva A, Canault M, Kotova Y, Obydennyy S, Yakimenko A, Podoplelova N, et al. Procoagulant platelets form an alpha-granule protein-covered “cap” on their surface that promotes their attachment to aggregates. J Biol Chem. (2013) 288:29621–32. doi: 10.1074/jbc.M113.474163
26. Podoplelova N, Sveshnikova A, Kotova Y, Eckly A, Receveur N, Nechipurenko D, et al. Coagulation factors bound to procoagulant platelets concentrate in cap structures to promote clotting. Blood. (2016) 128:1745–55. doi: 10.1182/blood-2016-02-696898
27. Agbani E, Hers I, Poole A. Temporal contribution of the platelet body and balloon to thrombin generation. Haematologica. (2017) 102:e379–81. doi: 10.3324/haematol.2017.166819
28. Agbani E, Poole A. Procoagulant platelets: generation, function, and therapeutic targeting in thrombosis. Blood. (2017) 130:2171–9. doi: 10.1182/blood-2017-05-787259
29. Wolberg A. Thrombin generation and fibrin clot structure. Blood Rev. (2007) 21:131–42. doi: 10.1016/j.blre.2006.11.001
30. Collet J, Park D, Lesty C, Soria J, Soria C, Montalescot G, et al. Influence of fibrin network conformation and fibrin fiber diameter on fibrinolysis speed: dynamic and structural approaches by confocal microscopy. Arterioscler Thromb Vasc Biol. (2000) 20:1354–61. doi: 10.1161/01.ATV.20.5.1354
31. Undas A, Ariens R. Fibrin clot structure and function: a role in the pathophysiology of arterial and venous thromboembolic diseases. Arterioscler Thromb Vasc Biol. (2011) 31:e88–99. doi: 10.1161/ATVBAHA.111.230631
32. Gersh K, Edmondson K, Weisel J. Flow rate and fibrin fiber alignment. J Thromb Haemost. (2010) 8:2826–8. doi: 10.1111/j.1538-7836.2010.04118.x
33. Macrae F, Duval C, Papareddy P, Baker S, Yuldasheva N, Kearney K, et al. A fibrin biofilm covers blood clots and protects from microbial invasion. J Clin Invest. (2018) 128:3356–68. doi: 10.1172/JCI98734
34. Chernysh I, Nagaswami C, Kosolapova S, Peshkova A, Cuker A, Cines D, et al. The distinctive structure and composition of arterial and venous thrombi and pulmonary emboli. Sci Rep. (2020) 10:5112. doi: 10.1038/s41598-020-59526-x
35. Kim O, Litvinov R, Chen J, Chen D, Weisel J, Alber M. Compression-induced structural and mechanical changes of fibrin-collagen composites. Matrix Biol. (2017) 60-61:141–56. doi: 10.1016/j.matbio.2016.10.007
36. Sadowski M, Zabczyk M, Undas A. Coronary thrombus composition: links with inflammation, platelet and endothelial markers. Atherosclerosis. (2014) 237:555–61. doi: 10.1016/j.atherosclerosis.2014.10.020
37. Marder V, Chute D, Starkman S, Abolian A, Kidwell C, Liebeskind D, et al. Analysis of thrombi retrieved from cerebral arteries of patients with acute ischemic stroke. Stroke. (2006) 37:2086–93. doi: 10.1161/01.STR.0000230307.03438.94
38. Staessens S, Denorme F, Francois O, Desender L, Dewaele T, Vanacker P, et al. Structural analysis of ischemic stroke thrombi: histological indications for therapy resistance. Haematologica. (2020) 105:498–507. doi: 10.3324/haematol.2019.219881
39. Di Meglio L, Desilles J, Ollivier V, Nomenjanahary M, Di Meglio S, Deschildre C, et al. Acute ischemic stroke thrombi have an outer shell that impairs fibrinolysis. Neurology. (2019) 93:e1686–98. doi: 10.1212/WNL.0000000000008395
40. Colace T, Muthard R, Diamond S. Thrombus growth and embolism on tissue factor-bearing collagen surfaces under flow: role of thrombin with and without fibrin. Arterioscler Thromb Vasc Biol. (2012) 32:1466–76. doi: 10.1161/ATVBAHA.112.249789
41. Cushman M, Barnes G, Creager M, Diaz J, Henke P, Machlus K, et al. Venous thromboembolism research priorities: A scientific statement from the american heart association and the international society on thrombosis and haemostasis. Circulation. (2020) 142:e85–94. doi: 10.1161/CIR.0000000000000818
42. Wolberg A, Rosendaal F, Weitz J, Jaffer I, Agnelli G, Baglin T, et al. Venous thrombosis. Nat Rev Dis Primers. (2015) 1:15006. doi: 10.1038/nrdp.2015.6
43. von Bruhl M, Stark K, Steinhart A, Chandraratne S, Konrad I, Lorenz M, et al. Monocytes, neutrophils, and platelets cooperate to initiate and propagate venous thrombosis in mice in vivo. J Exp Med. (2012) 209:819–35. doi: 10.1084/jem.20112322
44. Silver M, Kawakami R, Jolly M, Huff C, Phillips J, Sakamoto A, et al. Histopathologic analysis of extracted thrombi from deep venous thrombosis and pulmonary embolism: Mechanisms and timing. Catheter Cardiovasc Interv. (2021) 97:1422–9. doi: 10.1002/ccd.29500
45. Klok F, Kruip M, van der Meer N, Arbous M, Gommers D, Kant K, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. (2020) 191:145–7. doi: 10.1016/j.thromres.2020.04.013
46. Loo J, Spittle D, Newnham M. COVID-19, immunothrombosis and venous thromboembolism: biological mechanisms. Thorax. (2021) 76:412–20. doi: 10.1136/thoraxjnl-2020-216243
47. van Dam L, Kroft L, van der Wal L, Cannegieter S, Eikenboom J, de Jonge E, et al. Clinical and computed tomography characteristics of COVID-19 associated acute pulmonary embolism: A different phenotype of thrombotic disease? Thromb Res. (2020) 193:86–9. doi: 10.1016/j.thromres.2020.06.010
48. Mueller-Peltzer K, Krauss T, Benndorf M, Lang C, Bamberg F, Bode C, et al. Pulmonary artery thrombi are co-located with opacifications in SARS-CoV2 induced ARDS. Respir Med. (2020) 172:106135. doi: 10.1016/j.rmed.2020.106135
49. Porembskaya O, Zinserling V, Tomson V, Toropova Y, Starikova E, Maslei V, et al. Neutrophils mediate pulmonary artery thrombosis in situ. Int J Mol Sci. (2022) 23:5829. doi: 10.3390/ijms23105829
50. Byrnes J, Wolberg A. Red blood cells in thrombosis. Blood. (2017) 130:1795–9. doi: 10.1182/blood-2017-03-745349
51. Mandal D, Mazumder A, Das P, Kundu M, Basu J. Fas-, caspase 8-, and caspase 3-dependent signaling regulates the activity of the aminophospholipid translocase and phosphatidylserine externalization in human erythrocytes. J Biol Chem. (2005) 280:39460–7. doi: 10.1074/jbc.M506928200
52. Klatt C, Kruger I, Zey S, Krott K, Spelleken M, Gowert N, et al. Platelet-RBC interaction mediated by FasL/FasR induces procoagulant activity important for thrombosis. J Clin Invest. (2018) 128:3906–25. doi: 10.1172/JCI92077
53. Carvalho F, Connell S, Miltenberger-Miltenyi G, Pereira S, Tavares A, Ariens R, et al. Atomic force microscopy-based molecular recognition of a fibrinogen receptor on human erythrocytes. ACS Nano. (2010) 4:4609–20. doi: 10.1021/nn1009648
54. Aleman M, Byrnes J, Wang J, Tran R, Lam W, Di Paola J, et al. Factor XIII activity mediates red blood cell retention in venous thrombi. J Clin Invest. (2014) 124:3590–600. doi: 10.1172/JCI75386
55. Kattula S, Byrnes J, Martin S, Holle L, Cooley B, Flick M, et al. Factor XIII in plasma, but not in platelets, mediates red blood cell retention in clots and venous thrombus size in mice. Blood Adv. (2018) 2:25–35. doi: 10.1182/bloodadvances.2017011890
56. Byrnes J, Duval C, Wang Y, Hansen C, Ahn B, Mooberry M, et al. Factor XIIIa-dependent retention of red blood cells in clots is mediated by fibrin alpha-chain crosslinking. Blood. (2015) 126:1940–8. doi: 10.1182/blood-2015-06-652263
57. Tutwiler V, Mukhitov A, Peshkova A, Le Minh G, Khismatullin R, Vicksman J, et al. Shape changes of erythrocytes during blood clot contraction and the structure of polyhedrocytes. Sci Rep. (2018) 8:17907. doi: 10.1038/s41598-018-35849-8
58. Cines D, Lebedeva T, Nagaswami C, Hayes V, Massefski W, Litvinov R, et al. Clot contraction: compression of erythrocytes into tightly packed polyhedra and redistribution of platelets and fibrin. Blood. (2014) 123:1596–603. doi: 10.1182/blood-2013-08-523860
59. Tutwiler V, Litvinov R, Protopopova A, Nagaswami C, Villa C, Woods E, et al. Pathologically stiff erythrocytes impede contraction of blood clots. J Thromb Haemost. (2021) 19:1990–2001. doi: 10.1111/jth.15407
60. Engelmann B, Massberg S. Thrombosis as an intravascular effector of innate immunity. Nat Rev Immunol. (2013) 13:34–45. doi: 10.1038/nri3345
61. Flick M, Du X, Witte D, Jirouskova M, Soloviev D, Busuttil S, et al. Leukocyte engagement of fibrin(ogen) via the integrin receptor alphaMbeta2/Mac-1 is critical for host inflammatory response in vivo. J Clin Invest. (2004) 113:1596–606. doi: 10.1172/JCI20741
62. Swystun L, Liaw P. The role of leukocytes in thrombosis. Blood. (2016) 128:753–62. doi: 10.1182/blood-2016-05-718114
63. Angelillo-Scherrer A. Leukocyte-derived microparticles in vascular homeostasis. Circ Res. (2012) 110:356–69. doi: 10.1161/CIRCRESAHA.110.233403
64. Muszbek L, Adany R, Szegedi G, Polgar J, Kavai M. Factor XIII of blood coagulation in human monocytes. Thromb Res. (1985) 37:401–10. doi: 10.1016/0049-3848(85)90069-6
65. Alshehri F, Whyte C, Tuncay A, Williams M, Wilson H, Mutch N. Monocytes expose factor XIII-A and stabilize thrombi against fibrinolytic degradation. Int J Mol Sci. (2021) 22:6591. doi: 10.3390/ijms22126591
66. Ritchie H, Jamieson A, Booth N. Regulation, location and activity of plasminogen activator inhibitor 2 (PAI-2) in peripheral blood monocytes, macrophages and foam cells. Thromb Haemost. (1997) 77:1168–73. doi: 10.1055/s-0038-1656132
67. Ritchie H, Robbie L, Kinghorn S, Exley R, Booth N. Monocyte plasminogen activator inhibitor 2 (PAI-2) inhibits u-PA-mediated fibrin clot lysis and is cross-linked to fibrin. Thromb Haemost. (1999) 81:96–103. doi: 10.1055/s-0037-1614425
68. Siefert S, Chabasse C, Mukhopadhyay S, Hoofnagle M, Strickland D, Sarkar R, et al. Enhanced venous thrombus resolution in plasminogen activator inhibitor type-2 deficient mice. J Thromb Haemost. (2014) 12:1706–16. doi: 10.1111/jth.12657
69. Singh I, Burnand K, Collins M, Luttun A, Collen D, Boelhouwer B, et al. Failure of thrombus to resolve in urokinase-type plasminogen activator gene-knockout mice: rescue by normal bone marrow-derived cells. Circulation. (2003) 107:869–75. doi: 10.1161/01.CIR.0000050149.22928.39
70. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss D, et al. Neutrophil extracellular traps kill bacteria. Science. (2004) 303:1532–5. doi: 10.1126/science.1092385
71. Savchenko A, Martinod K, Seidman M, Wong S, Borissoff J, Piazza G, et al. Neutrophil extracellular traps form predominantly during the organizing stage of human venous thromboembolism development. J Thromb Haemost. (2014) 12:860–70. doi: 10.1111/jth.12571
72. Mangold A, Alias S, Scherz T, Hofbauer M, Jakowitsch J, Panzenbock A, et al. Coronary neutrophil extracellular trap burden and deoxyribonuclease activity in ST-elevation acute coronary syndrome are predictors of ST-segment resolution and infarct size. Circ Res. (2015) 116:1182–92. doi: 10.1161/CIRCRESAHA.116.304944
73. Ducroux C, Di Meglio L, Loyau S, Delbosc S, Boisseau W, Deschildre C, et al. Thrombus neutrophil extracellular traps content impair tPA-Induced thrombolysis in acute ischemic stroke. Stroke. (2018) 49:754–7. doi: 10.1161/STROKEAHA.117.019896
74. Farkas A, Farkas V, Gubucz I, Szabo L, Balint K, Tenekedjiev K, et al. Neutrophil extracellular traps in thrombi retrieved during interventional treatment of ischemic arterial diseases. Thromb Res. (2019) 175:46–52. doi: 10.1016/j.thromres.2019.01.006
75. Massberg S, Grahl L, von Bruehl M, Manukyan D, Pfeiler S, Goosmann C, et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat Med. (2010) 16:887–96. doi: 10.1038/nm.2184
76. Kambas K, Mitroulis I, Apostolidou E, Girod A, Chrysanthopoulou A, Pneumatikos I, et al. Autophagy mediates the delivery of thrombogenic tissue factor to neutrophil extracellular traps in human sepsis. PLoS One. (2012) 7:e45427. doi: 10.1371/journal.pone.0045427
77. Skendros P, Mitsios A, Chrysanthopoulou A, Mastellos D, Metallidis S, Rafailidis P, et al. Complement and tissue factor-enriched neutrophil extracellular traps are key drivers in COVID-19 immunothrombosis. J Clin Invest. (2020) 130:6151–7. doi: 10.1172/JCI141374
78. Yu X, Diamond S. Fibrin modulates Shear-Induced NETosis in sterile occlusive thrombi formed under haemodynamic flow. Thromb Haemost. (2019) 119:586–93. doi: 10.1055/s-0039-1678529
79. Yu X, Tan J, Diamond S. Hemodynamic force triggers rapid NETosis within sterile thrombotic occlusions. J Thromb Haemost. (2018) 16:316–29. doi: 10.1111/jth.13907
80. Fuchs T, Brill A, Duerschmied D, Schatzberg D, Monestier M, Myers D Jr., et al. Extracellular DNA traps promote thrombosis. Proc Natl Acad Sci USA. (2010) 107:15880–5. doi: 10.1073/pnas.1005743107
81. Brill A, Fuchs T, Savchenko A, Thomas G, Martinod K, De Meyer S, et al. Neutrophil extracellular traps promote deep vein thrombosis in mice. J Thromb Haemost. (2012) 10:136–44. doi: 10.1111/j.1538-7836.2011.04544.x
82. Clark S, Ma A, Tavener S, McDonald B, Goodarzi Z, Kelly M, et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. (2007) 13:463–9. doi: 10.1038/nm1565
83. Angles-Cano E, Balaton A, Le Bonniec B, Genot E, Elion J, Sultan Y. Production of monoclonal antibodies to the high fibrin-affinity, tissue-type plasminogen activator of human plasma. Demonstration of its endothelial origin by immunolocalization. Blood. (1985) 66:913–20. doi: 10.1182/blood.V66.4.913.913
84. Sappino A, Madani R, Huarte J, Belin D, Kiss J, Wohlwend A, et al. Extracellular proteolysis in the adult murine brain. J Clin Invest. (1993) 92:679–85. doi: 10.1172/JCI116637
85. Zhang L, Takahara T, Yata Y, Furui K, Jin B, Kawada N, et al. Increased expression of plasminogen activator and plasminogen activator inhibitor during liver fibrogenesis of rats: role of stellate cells. J Hepatol. (1999) 31:703–11. doi: 10.1016/S0168-8278(99)80351-1
86. Plow E, Herren T, Redlitz A, Miles L, Hoover-Plow J. The cell biology of the plasminogen system. FASEB J. (1995) 9:939–45. doi: 10.1096/fasebj.9.10.7615163
87. Kruithof E, Tran-Thang C, Ransijn A, Bachmann F. Demonstration of a fast-acting inhibitor of plasminogen activators in human plasma. Blood. (1984) 64:907–13. doi: 10.1182/blood.V64.4.907.bloodjournal644907
88. Schuhmann M, Gunreben I, Kleinschnitz C, Kraft P. Immunohistochemical analysis of cerebral thrombi retrieved by mechanical thrombectomy from patients with acute ischemic stroke. Int J Mol Sci. (2016) 17:298. doi: 10.3390/ijms17030298
89. Brzoska T, Tanaka-Murakami A, Suzuki Y, Sano H, Kanayama N, Urano T. Endogenously generated plasmin at the vascular wall injury site amplifies lysine binding site-dependent plasminogen accumulation in microthrombi. PLoS One. (2015) 10:e0122196. doi: 10.1371/journal.pone.0122196
90. Loyau S, Ho-Tin-Noe B, Bourrienne M, Boulaftali Y, Jandrot-Perrus M. Microfluidic modeling of thrombolysis. Arterioscler Thromb Vasc Biol. (2018) 38:2626–37. doi: 10.1161/ATVBAHA.118.311178
91. Plow E, Doeuvre L, Das R. So many plasminogen receptors: why? J Biomed Biotechnol. (2012) 2012:141806. doi: 10.1155/2012/141806
92. Andronicos N, Chen E, Baik N, Bai H, Parmer C, Kiosses W, et al. Proteomics-based discovery of a novel, structurally unique, and developmentally regulated plasminogen receptor, Plg-RKT, a major regulator of cell surface plasminogen activation. Blood. (2010) 115:1319–30. doi: 10.1182/blood-2008-11-188938
93. Whyte C, Morrow G, Baik N, Booth N, Jalal M, Parmer R, et al. Exposure of plasminogen and a novel plasminogen receptor, Plg-RKT, on activated human and murine platelets. Blood. (2021) 137:248–57. doi: 10.1182/blood.2020007263
94. Baeten K, Richard M, Kanse S, Mutch N, Degen J, Booth N. Activation of single-chain urokinase-type plasminogen activator by platelet-associated plasminogen: a mechanism for stimulation of fibrinolysis by platelets. J Thromb Haemost. (2010) 8:1313–22. doi: 10.1111/j.1538-7836.2010.03813.x
95. Dejouvencel T, Doeuvre L, Lacroix R, Plawinski L, Dignat-George F, Lijnen H, et al. Fibrinolytic cross-talk: a new mechanism for plasmin formation. Blood. (2010) 115:2048–56. doi: 10.1182/blood-2009-06-228817
96. Miles L, Parmer R. Plasminogen receptors: the first quarter century. Semin Thromb Hemost. (2013) 39:329–37. doi: 10.1055/s-0033-1334483
97. Han J, Baik N, Kim K, Yang J, Han G, Gong Y, et al. Monoclonal antibodies detect receptor-induced binding sites in Glu-plasminogen. Blood. (2011) 118:1653–62. doi: 10.1182/blood-2010-11-316943
98. Booth N, Robbie L, Croll A, Bennett B. Lysis of platelet-rich thrombi: the role of PAI-1. Ann N Y Acad Sci. (1992) 667:70–80. doi: 10.1111/j.1749-6632.1992.tb51599.x
99. Huebner B, Moore E, Moore H, Stettler G, Nunns G, Lawson P, et al. Thrombin Provokes Degranulation of Platelet alpha-Granules Leading to the Release of Active Plasminogen Activator Inhibitor-1 (PAI-1). Shock. (2018) 50:671–6. doi: 10.1097/SHK.0000000000001089
100. Booth N, Anderson J, Bennett B. Platelet release protein which inhibits plasminogen activators. J Clin Pathol. (1985) 38:825–30. doi: 10.1136/jcp.38.7.825
101. Morrow G, Whyte C, Mutch N. Functional plasminogen activator inhibitor 1 is retained on the activated platelet membrane following platelet activation. Haematologica. (2020) 105:2824–33. doi: 10.3324/haematol.2019.230367
102. Plow E, Collen D. The presence and release of alpha 2-antiplasmin from human platelets. Blood. (1981) 58:1069–74. doi: 10.1182/blood.V58.6.1069.1069
103. Gogstad G, Stormorken H, Solum N. Platelet alpha 2-antiplasmin is located in the platelet alpha-granules. Thromb Res. (1983) 31:387–90. doi: 10.1016/0049-3848(83)90339-0
104. Gronke R, Bergman B, Baker J. Thrombin interaction with platelets. Influence of a platelet protease nexin. J Biol Chem. (1987) 262:3030–6. doi: 10.1016/S0021-9258(18)61464-4
105. Gronke R, Knauer D, Veeraraghavan S, Baker JB. A form of protease nexin I is expressed on the platelet surface during platelet activation. Blood. (1989) 73:472–8. doi: 10.1182/blood.V73.2.472.bloodjournal732472
106. Boulaftali Y, Ho-Tin-Noe B, Pena A, Loyau S, Venisse L, Francois D, et al. Platelet protease nexin-1, a serpin that strongly influences fibrinolysis and thrombolysis. Circulation. (2011) 123:1326–34. doi: 10.1161/CIRCULATIONAHA.110.000885
107. Schmaier A, Smith P, Colman R. Platelet C1- inhibitor. A secreted alpha-granule protein. J Clin Invest. (1985) 75:242–50. doi: 10.1172/JCI111680
108. Schmaier A, Amenta S, Xiong T, Heda G, Gewirtz A. Expression of platelet C1 inhibitor. Blood. (1993) 82:465–74. doi: 10.1182/blood.V82.2.465.465
109. Mosnier L, Buijtenhuijs P, Marx P, Meijers J, Bouma B. Identification of thrombin activatable fibrinolysis inhibitor (TAFI) in human platelets. Blood. (2003) 101:4844–6. doi: 10.1182/blood-2002-09-2944
110. Boulaftali Y, Adam F, Venisse L, Ollivier V, Richard B, Taieb S, et al. Anticoagulant and antithrombotic properties of platelet protease nexin-1. Blood. (2010) 115:97–106. doi: 10.1182/blood-2009-04-217240
111. Suzuki Y, Sano H, Mochizuki L, Honkura N, Urano T. Activated platelet-based inhibition of fibrinolysis via thrombin-activatable fibrinolysis inhibitor activation system. Blood Adv. (2020) 4:5501–11. doi: 10.1182/bloodadvances.2020002923
112. Ni R, Neves M, Wu C, Cerroni S, Flick M, Ni H, et al. Activated thrombin-activatable fibrinolysis inhibitor (TAFIa) attenuates fibrin-dependent plasmin generation on thrombin-activated platelets. J Thromb Haemost. (2020) 18:2364–76. doi: 10.1111/jth.14950
113. Luscher E. [Fibrin-stabilizing factor from thrombocytes]. Schweiz Med Wochenschr. (1957) 87:1220–1.
114. Kiesselbach T, Wagner R. Fibrin-stabilizing factor: a thrombin-labile platelet protein. Am J Physiol. (1966) 211:1472–6. doi: 10.1152/ajplegacy.1966.211.6.1472
115. Katona E, Ajzner E, Toth K, Karpati L, Muszbek L. Enzyme-linked immunosorbent assay for the determination of blood coagulation factor XIII A-subunit in plasma and in cell lysates. J Immunol Methods. (2001) 258:127–35. doi: 10.1016/S0022-1759(01)00479-3
116. Palazzolo, J, Ale A, Ho H, Jagdale S, Broughton B, Medcalf R, et al. Platelet-targeted thrombolysis for treatment of acute ischemic stroke. Blood Adv. (2022). doi: 10.1182/bloodadvances.2021006691
117. Tutwiler V, Peshkova A, Le Minh G, Zaitsev S, Litvinov R, Cines D, et al. Blood clot contraction differentially modulates internal and external fibrinolysis. J Thromb Haemost. (2019) 17:361–70. doi: 10.1111/jth.14370
118. Wohner N, Sotonyi P, Machovich R, Szabo L, Tenekedjiev K, Silva M, et al. Lytic resistance of fibrin containing red blood cells. Arterioscler Thromb Vasc Biol. (2011) 31:2306–13. doi: 10.1161/ATVBAHA.111.229088
119. Choi M, Park G, Lee J, Lee S, Lee S, Kim J, et al. Erythrocyte fraction within retrieved thrombi contributes to thrombolytic response in acute ischemic stroke. Stroke. (2018) 49:652–9. doi: 10.1161/STROKEAHA.117.019138
120. Sporns P, Hanning U, Schwindt W, Velasco A, Buerke B, Cnyrim C, et al. Ischemic stroke: Histological thrombus composition and pre-interventional ct attenuation are associated with intervention time and rate of secondary embolism. Cerebrovasc Dis. (2017) 44:344–50. doi: 10.1159/000481578
121. Shin J, Jeong H, Kwon H, Song K, Kim J. High red blood cell composition in clots is associated with successful recanalization during intra-arterial thrombectomy. PLoS One. (2018) 13:e0197492. doi: 10.1371/journal.pone.0197492
122. Yuki I, Kan I, Vinters H, Kim R, Golshan A, Vinuela F, et al. The impact of thromboemboli histology on the performance of a mechanical thrombectomy device. AJNR Am J Neuroradiol. (2012) 33:643–8. doi: 10.3174/ajnr.A2842
123. Abbasi M, Arturo Larco J, Mereuta M, Liu Y, Fitzgerald S, Dai D, et al. Diverse thrombus composition in thrombectomy stroke patients with longer time to recanalization. Thromb Res. (2022) 209:99–104. doi: 10.1016/j.thromres.2021.11.018
124. Locke M, Longstaff C. Extracellular histones inhibit fibrinolysis through noncovalent and covalent interactions with fibrin. Thromb Haemost. (2021) 121:464–76. doi: 10.1055/s-0040-1718760
125. Herbig B, Diamond S. Pathological von Willebrand factor fibers resist tissue plasminogen activator and ADAMTS13 while promoting the contact pathway and shear-induced platelet activation. J Thromb Haemost. (2015) 13:1699–708. doi: 10.1111/jth.13044
Keywords: thrombus, fibrinolysis, platelets, plasminogen activators, fibrin
Citation: Narwal A, Whyte CS and Mutch NJ (2023) Location, location, location: Fibrin, cells, and fibrinolytic factors in thrombi. Front. Cardiovasc. Med. 9:1070502. doi: 10.3389/fcvm.2022.1070502
Received: 14 October 2022; Accepted: 16 December 2022;
Published: 18 January 2023.
Edited by:
Anastasia N. Sveshnikova, Lomonosov Moscow State University, RussiaReviewed by:
Alexey Shibeko, Center for Theoretical Problems of Physicochemical Pharmacology (RAS), RussiaCopyright © 2023 Narwal, Whyte and Mutch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Nicola J. Mutch,  bi5qLm11dGNoQGFiZG4uYWMudWs=
bi5qLm11dGNoQGFiZG4uYWMudWs=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.