 Meili Wang
Meili Wang Shuanglei Li
Shuanglei Li Hongshan Liu1
Hongshan Liu1 Haixia Huang
Haixia Huang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med. , 09 December 2022
Sec. Atherosclerosis and Vascular Medicine
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.1062695
Background: Vascular smooth muscle cells (VSMCs) phenotype switching is very important during the pathogenesis and progression of vascular diseases. However, it is not well understood how normal VSMCs maintain the differentiated state. The large-conductance Ca2+-activated K+ (BKCa) channels are widely expressed in VSMCs and regulate vascular tone. Nevertheless, there is limited understanding of the role of the BKCa channel in modulation of the VSMC phenotype.
Methods and results: We assessed BKCa channel expression levels in normal and injured carotid arteries from rats of the balloon-injury model. A strong decrease of BKCa-β1 was seen in the injured carotid arteries, accompanied by a parallel decrease of the VSMC contractile markers. BKCa-β1 in primary rat aortic VSMCs was decreased with the increase of passage numbers and the stimulation of platelet-derived growth factor (PDGF)-BB. Conversely, transforming growth factor β upregulated BKCa-β1. Meanwhile, the BKCa-β1 level was positively associated with the levels of VSMC contractile proteins. Intravenous injection of PDGF-BB induced downregulation of BKCa-β1 expression in the carotid arteries. Knockdown of BKCa-β1 favored VSMC dedifferentiation, characterized by altered morphology, abnormal actin fiber organization, decreased contractile proteins expression and reduced contractile ability. Furthermore, the resultant VSMC dedifferentiated phenotype rendered increased proliferation, migration, enhanced inflammatory factors levels, and matrix metalloproteinases activity. Studies using primary cultured aortic VSMCs from human recapitulated key findings. Finally, protein level of BKCa-β1 was reduced in human atherosclerotic arteries.
Conclusion: BKCa-β1 is important in the maintenance of the contractile phenotype of VSMCs. As a novel endogenous defender that prevents pathological VSMC phenotype switching, BKCa-β1 may serve as a potential therapeutic target for treating vascular diseases including post-injury restenosis and atherosclerosis.
Vascular smooth muscle cells (VSMCs) retain plasticity of the arterial wall. Meanwhile, VSMCs can undergo reversible phenotype switching (1). Under physiological conditions, the vessels-resident VSMCs are differentiated, expressing a series of contractile proteins including smooth muscle myosin heavy chain, α-smooth muscle actin (α-SMA), smooth muscle 22α (SM22α), and calponin. However, when exposed to certain pathological factors, VSMCs can exhibit an adaptive reaction and lose the contractile phenotype. This process is called VSMC phenotype switching (2). The dedifferentiated phenotype is characterized by reduced expression of the VSMCs marker proteins. Concomitantly, dedifferentiated VSMCs show high proliferation rate, migration ability, enhanced synthesis of extracellular matrix and inflammatory factors, while they lose the regulating capability of vascular diameter and blood flow (3). Phenotypic modulation of VSMCs is an early event and critical pathological process of many cardiovascular diseases, including atherosclerosis (3), hypertension (2), aneurysm (4), and post-angioplasty restenosis (5). Thus, strategies blocking the VSMC phenotype switching may prevent the progression of these diseases (6–9). Various environmental stimuli, including mechanical injury, reactive oxidative species, growth factors/cytokines, and extracellular matrix instability, have been demonstrated to influence VSMC behavior and induce phenotype switching (2, 10, 11). Besides, it has been shown that transcriptional and epigenetic regulators can induce VSMC phenotypic transition (2). Nevertheless, how normal VSMCs maintain the contractile phenotype and confer vascular homeostasis remains largely unknown.
The differentiated VSMCs express a series of ion channels necessary for their unique contractile properties. VSMCs predominantly express large-conductance Ca2+-activated K+ (BKCa) channels, which dampen depolarization-dependent activation of Ca2+ channels by hyperpolarizing membrane potential. Therefore, BKCa channels have a critical role in VSMC relaxation (12). In VSMCs, BKCa channel is a protein complex composed of two subunits, the pore-forming α-subunit (BKCa-α) and the regulatory β1-subunit (BKCa-β1) (13). We previously showed that BKCa channel activation exerts a beneficial effect in VSMC phenotypic transition by counteracting harmful stimulus (14). Moreover, studies have found that BKCa channel inhibition aggravates VSMC calcification (15). During calcification, VSMCs dedifferentiate to an osteochondrogenic phenotype. However, much more subtle phenotypic changes will also occur in VSMCs. Indeed, VSMC phenotype changes according to the local environmental factors as well as the different stages of development. In this study, we discovered that during the early stage of disease development, BKCa-β1 negatively regulated VSMC dedifferentiation and maintained VSMCs in a quiescent contractile phenotype.
An antibody against BKCa-α (ab99046) used for immunohistochemical staining and western blot was purchased from abcam (Cambridge, UK). An antibody against BKCa-β1 (APC-036) used for immunohistochemical staining and western blot was purchased from Alomone labs (Jerusalem, Israel). Antibodies against α-SMA (ab5694), SM22α (ab10135), and eIF5 (ab228874) used for western blot were purchased from abcam (Cambridge, UK).
All animal studies were approved by the Institutional Animal Care and Use Committee and Ethics Committee of Capital Medical University (Beijing, China) and were in strict accordance with the recommendation in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. Healthy male Sprague-Dawley (SD) rats, aged 8–10 weeks (150–180 g), were obtained from the Animal Center of Capital Medical University and housed in an environment with standard conditions of humidity and room temperature and a light/dark cycle of 12/12 h.
The balloon-injured rat model was established as previously described (16). Briefly, male SD rats were anesthetized by intraperitoneal injection of pentobarbital sodium (40 mg/kg). A balloon catheter of 1.5-mm diameter (Medtronic, Minneapolis, MN, USA) was then inserted into the left external carotid artery lumen and advanced proximally to the common carotid artery lumen. The balloon was inflated to expand the carotid artery to 1.5 times its diameter and then gently pulled back with the rotational movement to the bifurcation. This pulling-back procedure was repeated twice. A similar operation that was performed on right carotid arteries, but without injury, served as a sham control. Two weeks later, the bilateral carotid arteries were harvested.
Tissues embedded in paraffin were cut into slices (7-μm in thickness) and subjected to immunohistochemical staining. Briefly, sections were incubated with corresponding antibodies at 4°C overnight and then HRP-labeled secondary antibodies at room temperature for 1 h. The nuclei were counterstained with hematoxylin.
For F-actin staining, VSMCs were fixed with 4% paraformaldehyde for 15 min and then permeabilized with 0.25% Triton X-100 in PBS for 5 min. Next, VSMCs were incubated with rhodamine phalloidin for 1 h at room temperature.
Primary rat VSMCs were isolated using the modified explant method (17). The thoracic aortas were used for the isolation. VSMCs were cultured in low-glucose Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (Gibco BRL, Grand Island, NY, USA) at 37°C in a humidified atmosphere containing 5% CO2. VSMCs passaged 3–6 were used for the experiments. To evaluate the effects of platelet-derived growth factor (PDGF)-BB or TGF-β, VSMCs at 80–90% confluence were subjected to serum starvation for 24 h followed by stimulation with the indicated concentration of PDGF-BB (25 μg/L) and TGF-β (2.5 μg/L) for 48 h.
Small interfering RNA- (siRNA-) targeting KCNMB1 was designed and synthesized by OBiO (OBiO, China). Sequences corresponding to the small interfering RNA (siRNA) against KCNMB1 were sense, 5′-CCUUGGUUGAUGUGAAGAATT-3′, and antisense, 5′-UUCUUCACAUCAACCAAGGTT-3′. siRNA transfection (50 nM) was performed using the Lipofectamine®3000 Transfection Reagent (Invitrogen, CA, USA), following the manufacturer’s instructions.
A wound-healing assay was used for the assessment of VSMC migration. Briefly, VSMCs were seeded in a 6-cm culture dish. Then the confluent cell monolayers were scratched with a sterile 200 μL pipet tip. The cells were gently rinsed twice with PBS to remove floating cells and incubated with standard culture medium. The migration distance was monitored at 0, 6, 12, and 24 h of incubation. The longest distance of migration from the wound edge was measured (average of five independent microscope fields in each of the independent experiments).
Rat carotid tissue and VSMC extracts that contained equal amounts of total protein were separated by 12% SDS-PAGE gels and subsequently transferred onto PVDF membrane (Millipore, Billerica, MA, USA). The membranes were blocked with 5% milk (in TBST) for 1 h, followed by incubation with the corresponding primary antibodies at 4°C overnight. Following 1 h of incubation with HRP-conjugated secondary antibodies, the membranes were developed using an ECL reagent.
Total RNA was extracted from cultured VSMCs with the TRIzol Reagent (Invitrogen, CA, USA). Equal amounts (1000 ng) were subjected to reverse transcription into cDNA using the ReverTra Ace® qPCR RT kit (TOYOBO, Japan). Quantitation of all gene transcripts was done by qPCR using SYBR® Green Realtime PCR Master Mix (TOYOBO, Japan) and ABI Step One Plus (Applied Biosystems, USA). The primer sequences used are as follows:
BKCa-α: Forward: 5′-AGCGCGGTTAGTGGAAGAAA-3′
Reverse: 5′- ACTCTGGCAAGATCGTGTGG-3′
BKCa-β1: Forward: 5′-CTATGGGCCCCAAATCCTCC-3′
Reverse: 5′-GAGCTGCCAAGACAGAGAGG-3′
PDGF-B: Forward: 5′-TCCGCTCCTTTGATGACCTT-3′
Reverse: 5′-TCCGACTCGACTCCAGAATGT-3′
TGF-β: Forward: 5′-ATACGCCTGAGTGGCTGTCTTT-3′
Reverse: 5′-AAAGCCCTGTATTCCGTCTCCT-3′
MCP-1: Forward: 5′-CAATGAGTCGGCTGGAGAAC-3′
Reverse: 5′-AGTGCTTGAGGTGGTTGTGG-3′
IL-6: Forward: 5′-GCTCTGGTCTTCTGGAGTTCC-3′
Reverse: 5′-GAGTTGGATGGTCTTGGTCCT-3′
IL-1α: Forward: 5′-ATCAGCACCTCACAGCTTCC-3′
Reverse: 5′-TCTCCTCCCGATGAGTAGGC-3′
TNF-α: Forward: 5′-CCAGGTTCTCTTCAAGGGACA-3′
Reverse: 5′-GTACTTGGGCAGGTTGACCTC-3′
GAPDH: Forward: 5′-TGTGAACGGATTTGGCCGTA-3′
Reverse: 5′-TGAACTTGCCGTGGGTAGAG-3′.
Vascular smooth muscle cells treated with BKCa-β1 siRNA or scramble siRNA transfection were incubated in culture medium for additional 48 h. After that, the medium supernatant was separated by 10% SDS-PAGE gels containing 1 mg/ml gelatin (Sigma-Aldrich, MO, USA). The gel was washed with 2.5% Triton X-100 twice and then incubated in zymography buffer (50 mM Tris–HCl, 150 mM NaCl, 10 mM CaCl2, pH 7.5) for about 48 h. Following 6–8 h of incubation with Coomassie brilliant blue R250, the gels were imaged using the imaging instrument.
Vascular smooth muscle cells were digested using trypsin and resuspended in culture medium at a density of 5 × 105 cells/mL. Then the cells were mixed with 10xPBS, 0.1 M NaOH and type I collagen to prepare a collagen lattice. A total of 0.5 mL of the mixture were added to a 24-well plate and incubated for 1.5 h in the cell incubator. After that, each well was added with 0.5 mL of culture medium and the culture plate was incubated for another 24 h before taking pictures.
All human tissue samples were obtained from patients at the Chinese PLA General Hospital (Beijing, China). This study was conducted following the principles outlined in the Declaration of Helsinki and approved by the Medical Ethics Committee of Chinese PLA General Hospital (S2021-406-01). Informed consent was obtained from each participating patient. Internal mammary arteries (IMA) were obtained from patients undergoing coronary artery bypass surgery as control samples, and atherosclerotic arteries were collected from patients undergoing carotid endarterectomy (CEA).
Primary cultures of aortic medial VSMCs were isolated from the thoracic aortas of human who underwent aortic valve replacement or aortic arch surgery using the modified explant method. The aortic specimen was rinsed in PBS to remove blood clots. After the adventitia with peripheral connective tissue was removed, the vessel wall was cut longitudinally and the intima was scraped gently. Then, the tunica media were cut into 1-mm2 explants and inoculated in a 25-cm2 flask. A total of 4–6 h later, culture medium was added carefully into the flask. The flask kept stationary in the incubator for 10 days until the cells grew around the explants. During this period, the culture media were refreshed every 3 days. The purity of VSMCs was tested by immunofluorescence staining for α-SMA and calponin. Isolated VSMCs were maintained in Kaighn’s Modification of Ham’s F-12 Medium (ATCC® 30-2004™) and supplemented with 20% FBS (Gibco BRL, Grand Island, NY, USA) and 10% smooth muscle cell growth supplement (ScienCell).
All the data were expressed as the means ± SEM. Protein band density was normalized to the corresponding control group and then to the mean of the corresponding control. Student’s t-test was used to analyze the differences between two groups. For a pairwise comparison of three or more groups, One-way ANOVA followed by the Student-Newman-Keuls test was used. The Chi-square test was used in the comparison of percentages between two groups. GraphPad Prism 9.0 (GraphPad Software, Inc., San Diego, CA, USA) was used for statistical analysis; P-value < 0.05 was considered statistically significant.
To assess the involvement of BKCa-β1 in VSMC phenotype switching, we established a rat balloon-injury model. H&E staining showed that at 14 days post-injury, moderate to severe intimal hyperplasia developed in the injured carotid arteries (Figure 1A). No significant change was observed in the mRNA or protein level of BKCa-α. However, the expression of BKCa-β1 was downregulated, and the VSMC contractile proteins were reduced in injured vessels compared to sham-operated vessels (Figure 1). These results indicated that BKCa-β1 downregulation was correlated with the changes of VSMC contractile phenotype markers (α-SMA and SM22α).
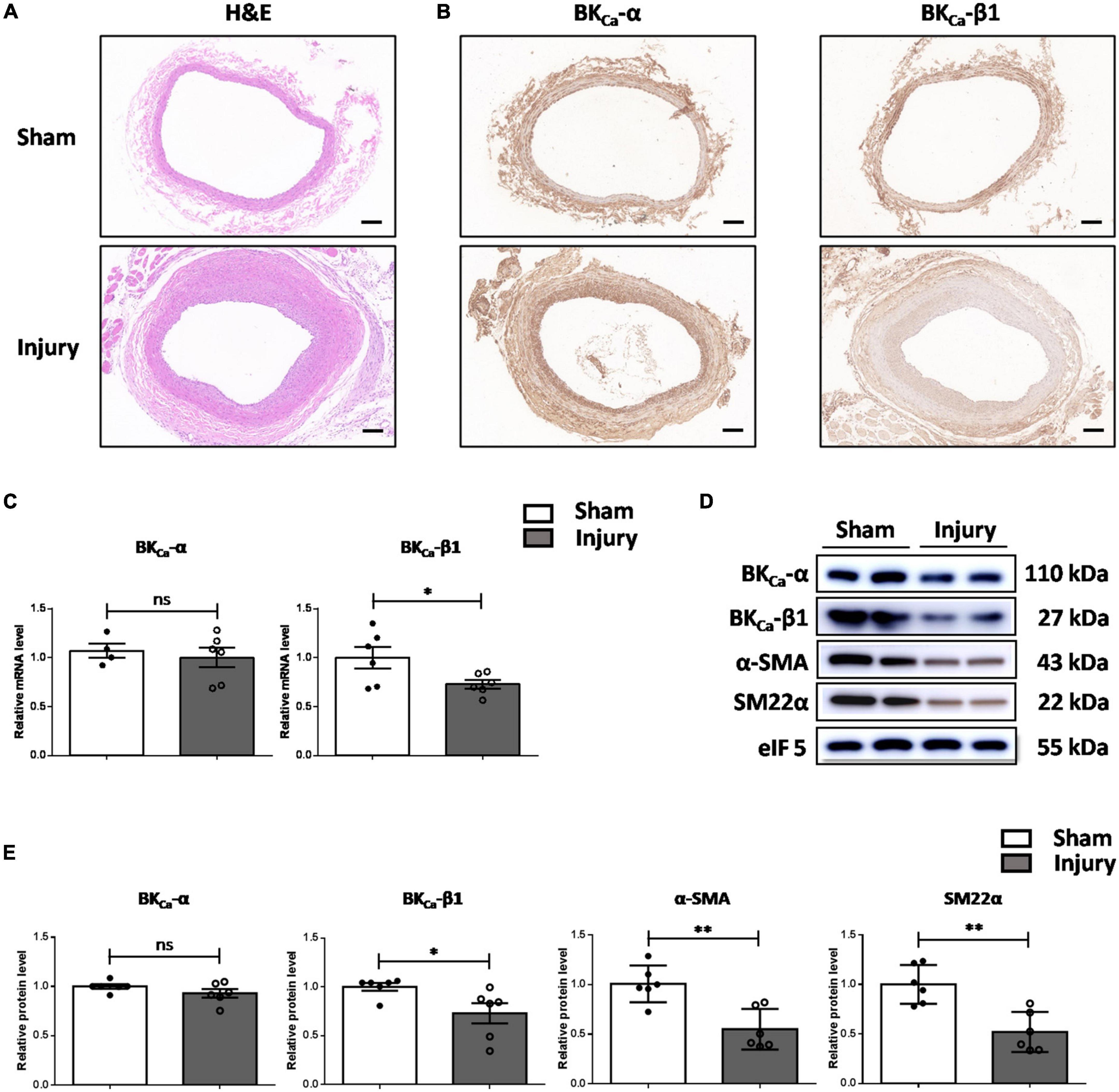
Figure 1. BKCa-β1 expression is reduced in the carotid arteries of rat balloon-injured model. Representative images of H&E (A) and immunohistochemical staining of BKCa−α and BKca−β1 (B) in injured and sham-operated carotid arteries of rat balloon-injured model at 14 days post injury. Scale bar = 100 μm. (C) Quantified mRNA levels of BKca−α and BKca−β1 analyzed by qRT-PCR. Western blot images (D) and quantification (E) for indicated proteins. Eukaryotic initiation factor 5 (eIF5) was used as an internal control. Data are presented as means ± SEM (n = 4∼6 for each group, ns, not significant, *P < 0.05, **P < 0.01 vs. sham).
We then explored the association of BKCa-β1 and the VSMC phenotype. As the cell passage increased, cultured primary rat VSMCs showed greatly reduced expression of differentiation markers, indicating the transformation from differentiated to dedifferentiated phenotype. In addition, compared with early passage VSMCs (passage 1, P1), late-passage VSMCs (passage 6, P6) showed markedly reduced expression of BKCa-β1 (Figures 2A,B).
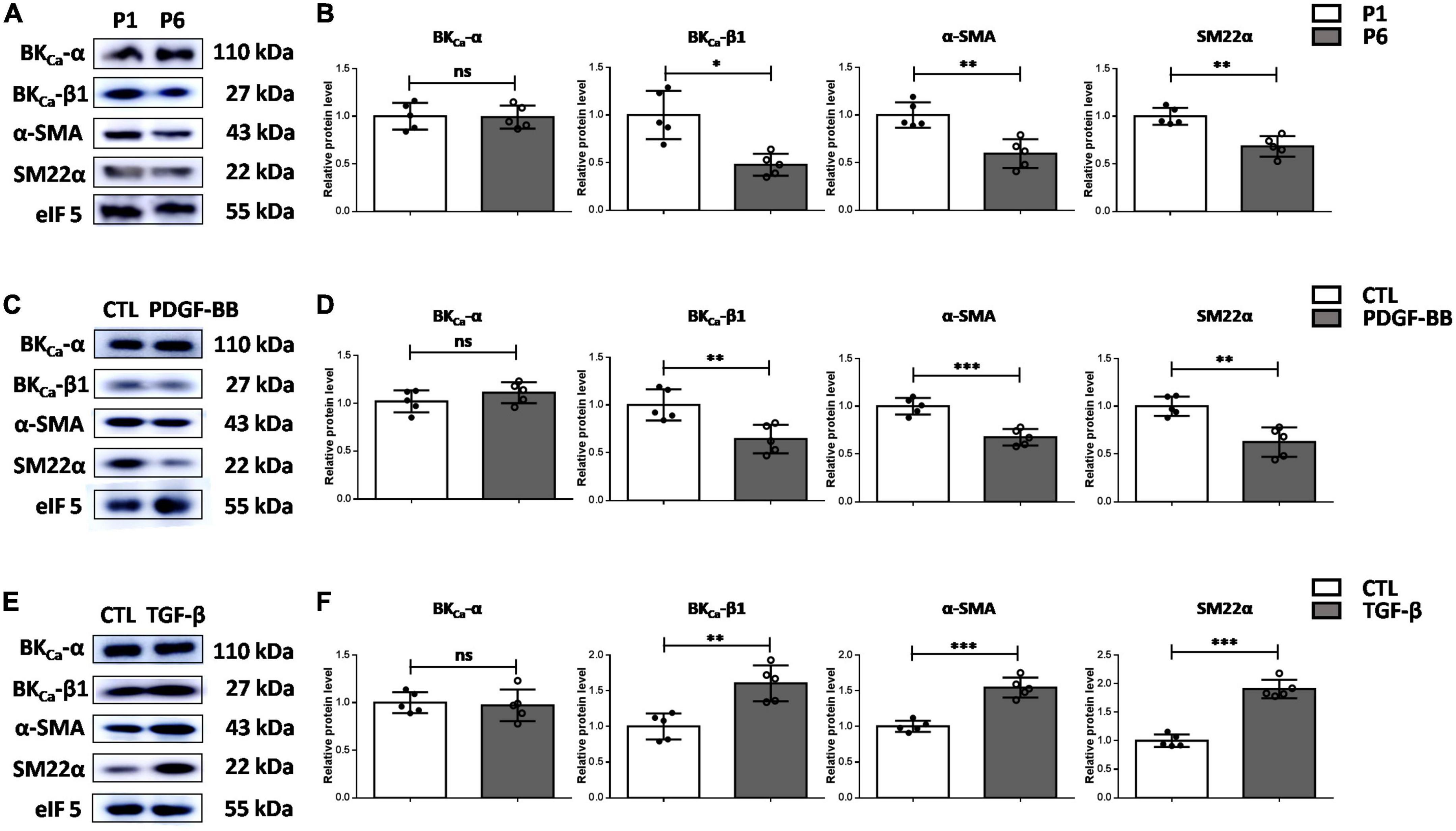
Figure 2. BKCa-β1 expression is positively associated with VSMC differentiation markers in vitro. Western blot images (A) and quantification (B) of BKca−α, BKca−β1, and VSMC differentiation markers expression in VSMCs of passage 1 and passage 6. Western blot images (C) and quantification (D) of BKca−α, BKca−β1, and VSMC differentiation markers expression in VSMCs after PDGF-BB treatment (25 μg/L). Western blot images (E) and quantification (F) of BKca−α, BKca−β1, and VSMC differentiation markers expression in VSMCs treated with TGF-β (2.5 μg/L). VSMCs were subjected to serum-starvation for 24 h and then treated with the indicated stimulation for 48 h. Passages 3-5 of VSMCs were used in (C) through (F). Eukaryotic initiation factor 5 (eIF5) was used as an internal control. Data are presented as means ± SEM (n = 5 for each group, ns, not significant, *P < 0.05, **P < 0.01, and ***P < 0.001 vs. control).
We then examined the response of BKCa-β1 to vascular injury-associated stimuli. Plenty of studies have shown that PDGF-BB can stimulate proliferation and migration of VSMCs and induce the inhibition of VSMC markers expression (18, 19). Moreover, transforming growth factor β (TGF-β) is an effective VSMC differentiation factor in vitro, which transcriptionally regulates genes involved in proliferation and growth (20, 21). Our data indicated that BKCa-β1 expression was reduced in cultured VSMCs due to PDGF-BB stimulation (Figures 2C,D). Inversely, TGF-β stimulation induced an opposite effect (Figures 2E,F). In addition, changes in BKCa-β1 expression occurred in parallel with VSMC trans-differentiation states, as demonstrated by the changes in α-SMA and SM22α. Altogether, these data showed that BKCa-β1 expression was closely related to the changes of VSMC contractile phenotype markers (α-SMA and SM22α).
We further explored the mechanism of the downregulation of BKCa-β1 expression in vivo. It has been reported that PDGF-BB and TGF-β both play important roles as endogenous growth regulatory factors during progressive intimal thickening after balloon angioplasty (22, 23). As Figure 3A shows, the expression of PDGF-B and TGF-β was both significantly increased after carotid injury. It is noteworthy that the change of PDGF-B is consistent with its effect on BKCa-β1 expression in vitro. This result implied that PDGF-BB might be particularly important in BKCa-β1 downregulation. Consistently, BKCa-β1 expression was reduced in the carotid arteries of mice injected with PDGF-BB via tail vein (Figures 3B,C).
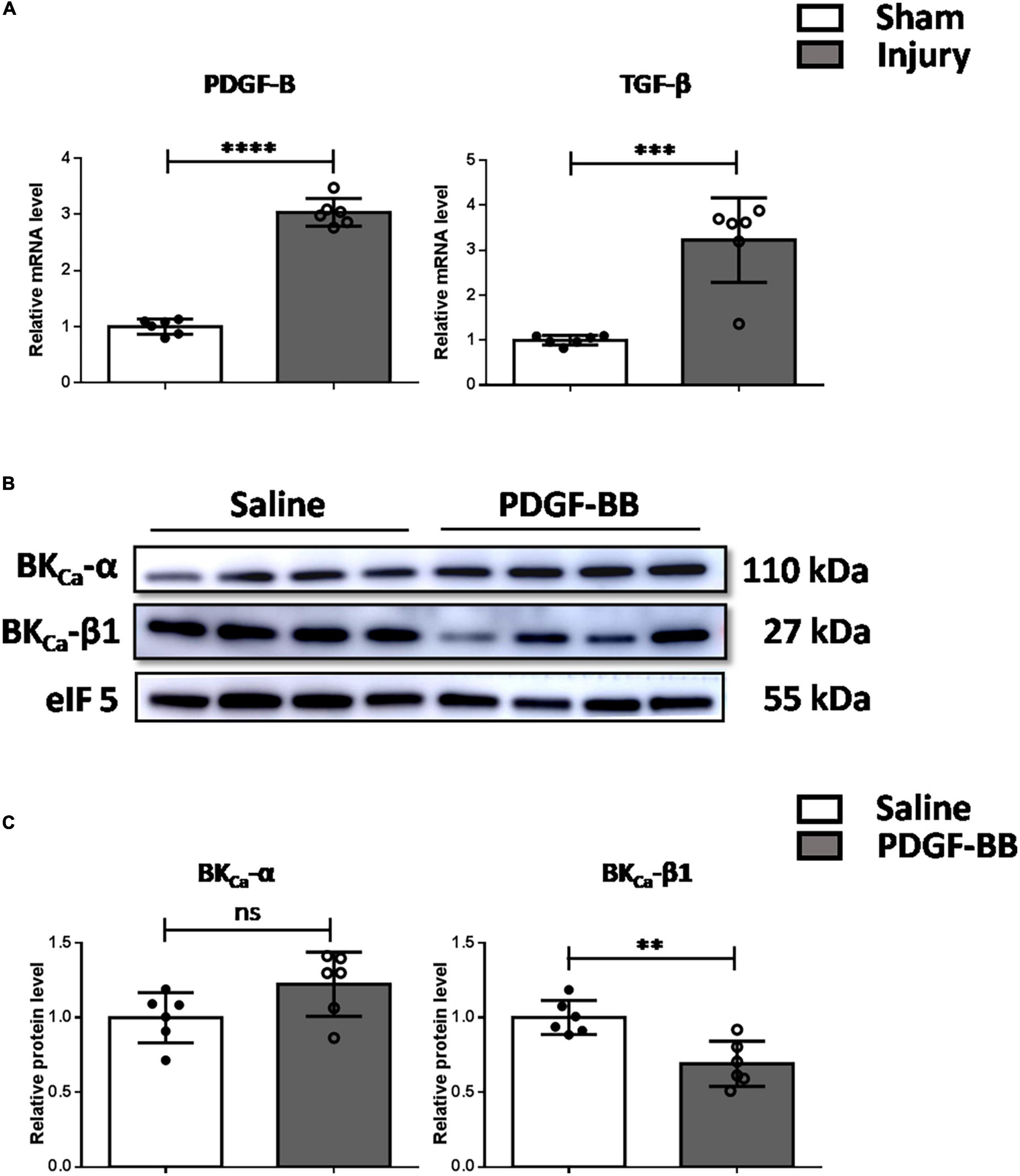
Figure 3. Decreased BKca−β1 expression is mediated by elevated PDGF-BB in vivo. (A) Quantified mRNA levels of PDGF-B and TGF-β analyzed by qRT-PCR in injured and sham-operated carotid arteries. Western blot images (B) and quantification (C) of BKca−α and BKca−β1 in the carotid arteries of mice injected with saline or PDGF-BB via tail vein. Data are presented as means ± SEM (n = 6 for each group, ns, not significant, **P < 0.01, ***P < 0.001, and ****P < 0.0001 vs. control).
To obtain direct evidence supporting the role of BKCa-β1 in VSMC phenotype switching, we used siRNA for the BKCa-β1 (siRNAKCNMB1). Morphologically, scramble siRNA-treated VSMCs were spindle-like, while BKCa-β1-targeting siRNA-treated VSMCs were polygonal (Figure 4A). Dedifferentiated VSMCs show impaired actin filament formation (24). Phalloidin staining showed that BKCa-β1 knockdown disintegrated actin fibers into short and disorganized fibers, resulting in polygonal-shaped VSMCs (Figures 4B,C). Besides, the protein levels of α-SMA and SM22α decreased after siRNA treatment (Figures 4D,E), indicating that BKCa-β1 was required for differentiation markers expression. To assess the contractile function of the VSMCs, the collagen gel contraction assay was performed. In accordance, VSMCs with BKCa-β1 silencing exhibited reduced contractility (Figures 4F,G). These data suggested that BKCa-β1 was necessary for the maintenance of the VSMC differentiated phenotype.
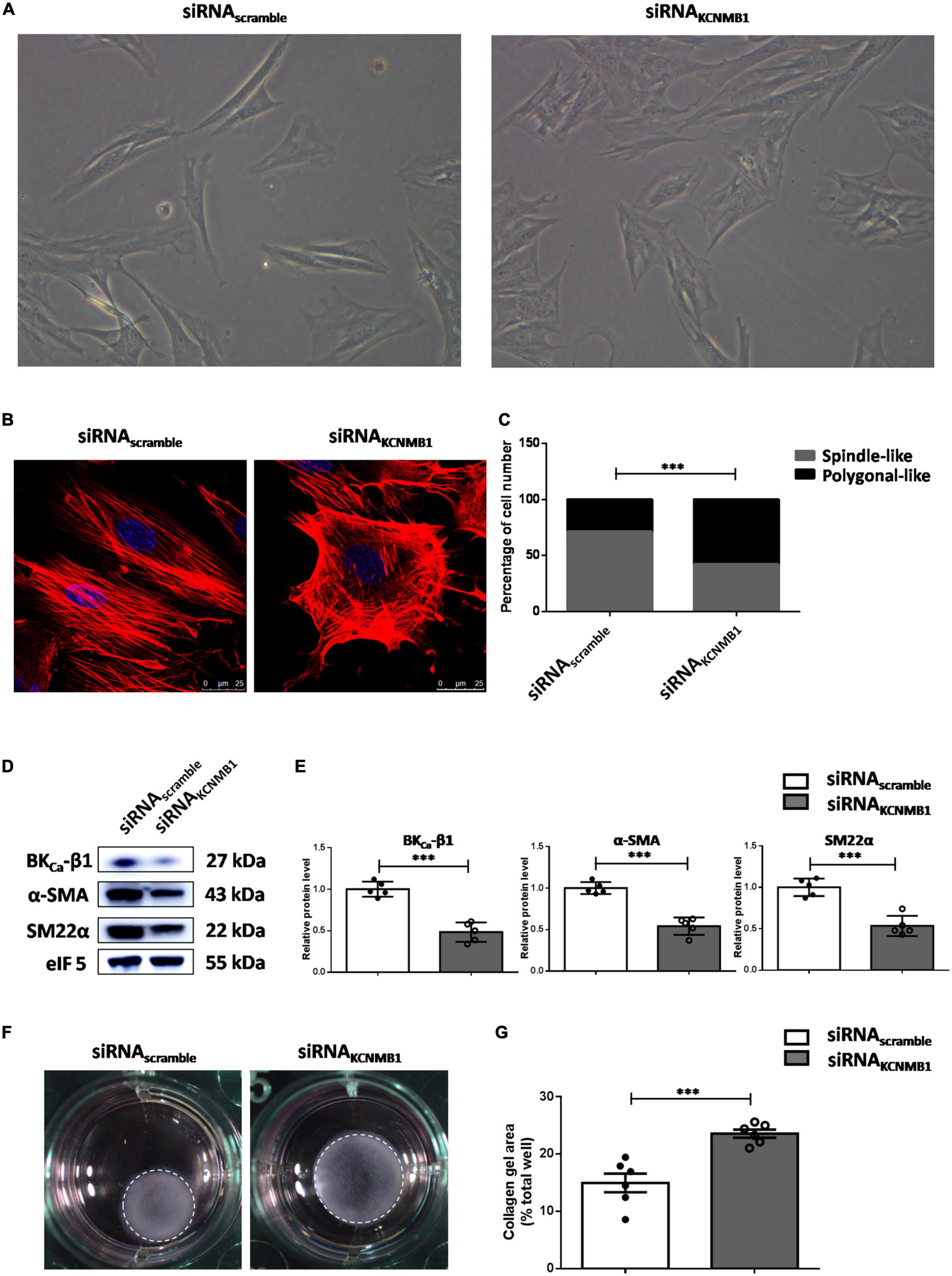
Figure 4. BKCa-β1 is essential for VSMCs to maintain a differentiated phenotype. (A) Representative micrographs of VSMCs treated with BKca−β1 siRNA or scramble siRNA transfection. (B) Representative immunofluorescent images of F-actin of VSMCs stained with phalloidin (red). Nuclei were stained with DAPI (blue). Scale bar = 25 μm. (C) The percentage of spindle-like or polygonal-like cells in each group. Western blot images (D) and quantification (E) of BKca−β1, α-SMA, and SM22α. Representative images (F) and quantification (G) of the areas of collagen gel in each group. The size of the collagen gel in each well was calculated compared to the total area of the well. Data are presented as means ± SEM (n = 5∼6 for each group, ***P < 0.001 vs. scramble).
Dedifferentiated VSMCs display an enhanced proliferative and migratory capacity. As shown in Figure 5A, BKCa-β1 silencing VSMCs showed an increased capacity to proliferate. Scratch-wound assays showed higher migration ability of VSMCs with BKCa-β1 knockdown compared to control cells (Figures 5B,C). Another hallmark of the dedifferentiated VSMCs is the enhanced proinflammatory response and greater proteolytic activity. As Figure 5D shows, the mRNA levels of MCP-1, IL-6, IL-1α, and TNF-α were markedly increased after BKCa-β1 siRNA treatment. In addition, increased activity of matrix metalloproteinases (MMPs, including MMP-9, and MMP-2) was observed using the method of gelatin zymography (Figures 5E,F). Collectively, the dedifferentiated phenotype secondary to BKCa-β1 knockdown exhibited an enhanced proliferative, migratory and synthetic response.
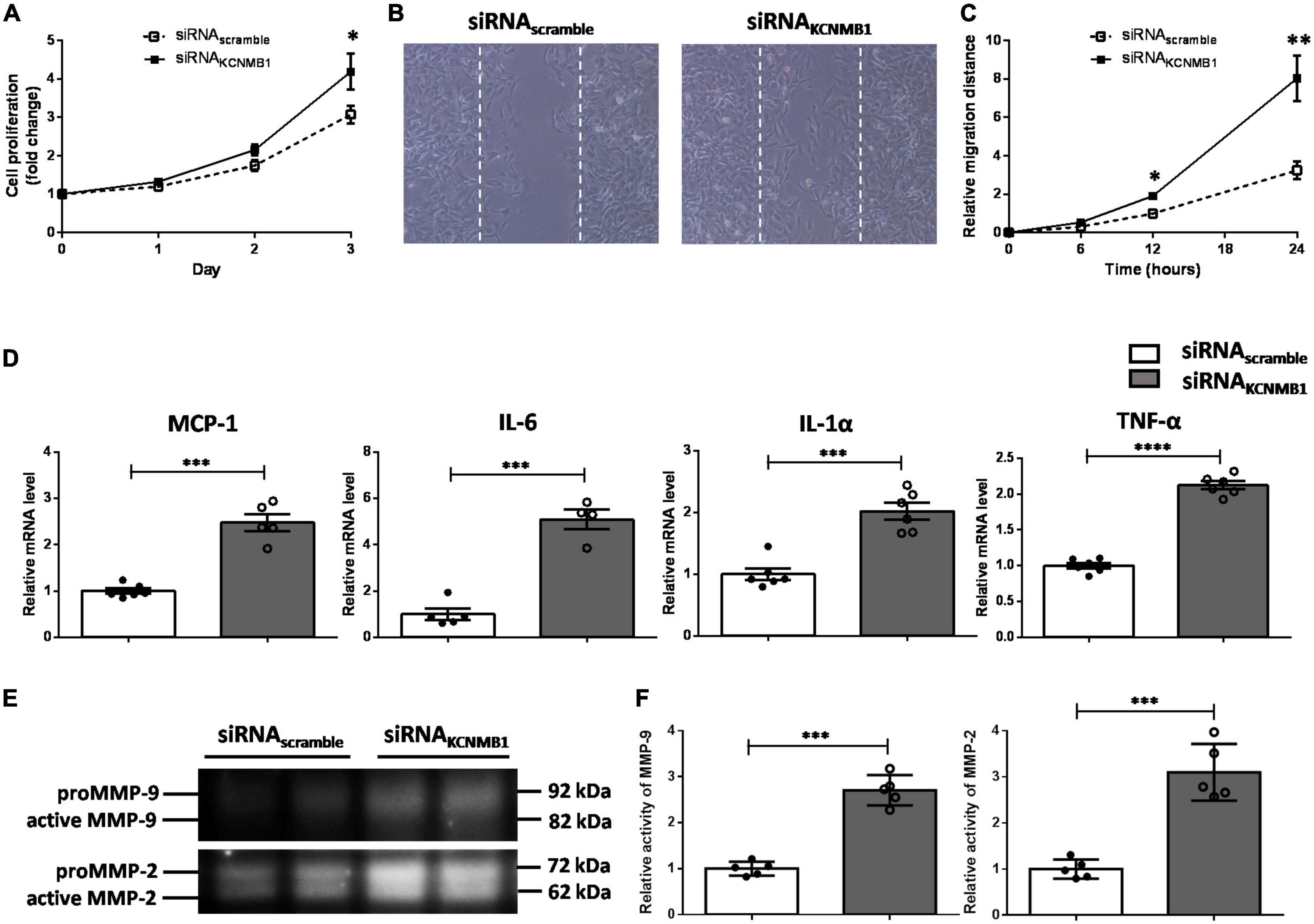
Figure 5. BKCa-β1 knockdown induces proliferative, migratory, and synthetic phenotype of VSMCs. (A) Proliferation curves of VSMCs transfected with BKca−β1 or scrambled siRNA. Representative images (B) and quantification (C) of migration assay. (D) Quantified mRNA levels of inflammatory factors analyzed by qRT-PCR. Representative images (E) and quantification (F) of activities of MMP-9 and MMP-2 tested using gelatin zymography. Data are presented as means ± SEM (n = 5∼6 for each group, *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001 vs. scramble).
To verify the role of BKCa-β1 in the VSMC identity in human, we isolated and cultured primary aortic VSMCs from patients who underwent aortic valve replacement or aortic arch surgery (Supplementary Figure 1). As the results showed, PDGF-BB markedly decreased, whereas TGF-β stimulation significantly increased BKCa-β1 expression, which paralleled the VSMC trans-differentiation state (Figures 6A–D). We further collected atherosclerotic tissue samples from human. Clinical information of human subjects recruited for this study is shown in Table 1. Notably, protein level of BKCa-β1 was significantly reduced in the arteries of patients who underwent CEA compared with control IMA used for coronary artery bypass graft surgery (Figures 6E,F). Together, these results indicated that reduced BKCa-β1 expression was correlated with VSMC dedifferentiation in human.
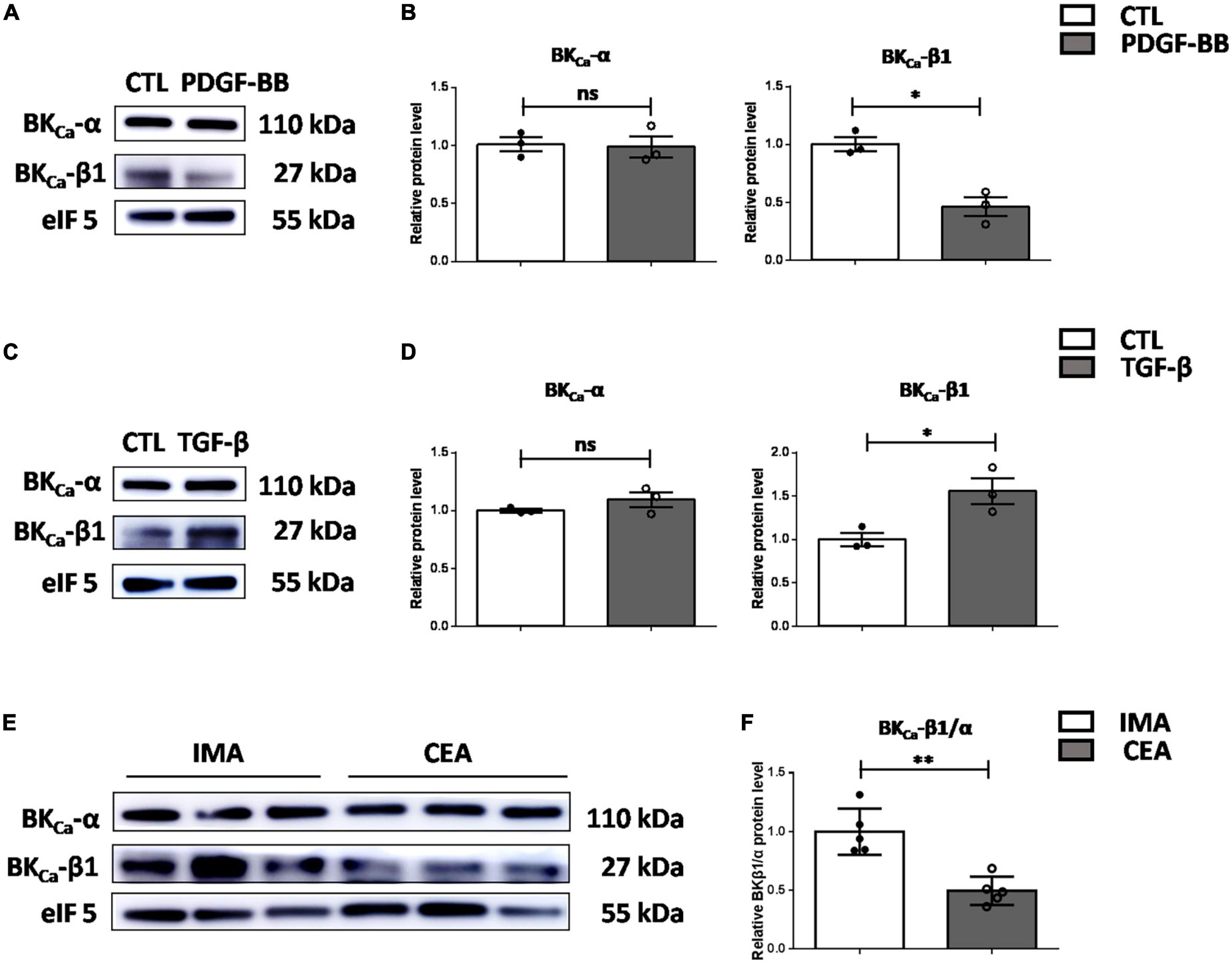
Figure 6. Decreased BKca−β1 expression is correlated with VSMC dedifferentiation and atherosclerosis in human. Western blot images (A) and quantification (B) of BKca−α and BKca−β1 expression in human primary aortic VSMCs after PDGF-BB treatment (25 μg/L). Western blot images (C) and quantification (D) of BKca−α and BKca−β1 expression in human primary aortic VSMCs treated with TGF-β (2.5 μg/L). VSMCs were subjected to serum-starvation for 24 h and then treated with the indicated stimulation for 48 h. Passages 3–5 of VSMCs were used. (E) Representative western blot images of the protein levels of BKca−α and BKca−β1 in lysates of human carotid endarterectomy artery (CEA) and control internal mammary arteries (IMA). (F) Quantification was performed by calculating the ratio between the levels of BKca−β1 and BKca−α. Eukaryotic initiation factor 5 (eIF5) was used as an internal control. Data are presented as means ± SEM (n = 3∼5 for each group, ns, not significant, *P < 0.05, **P < 0.01 vs. control).
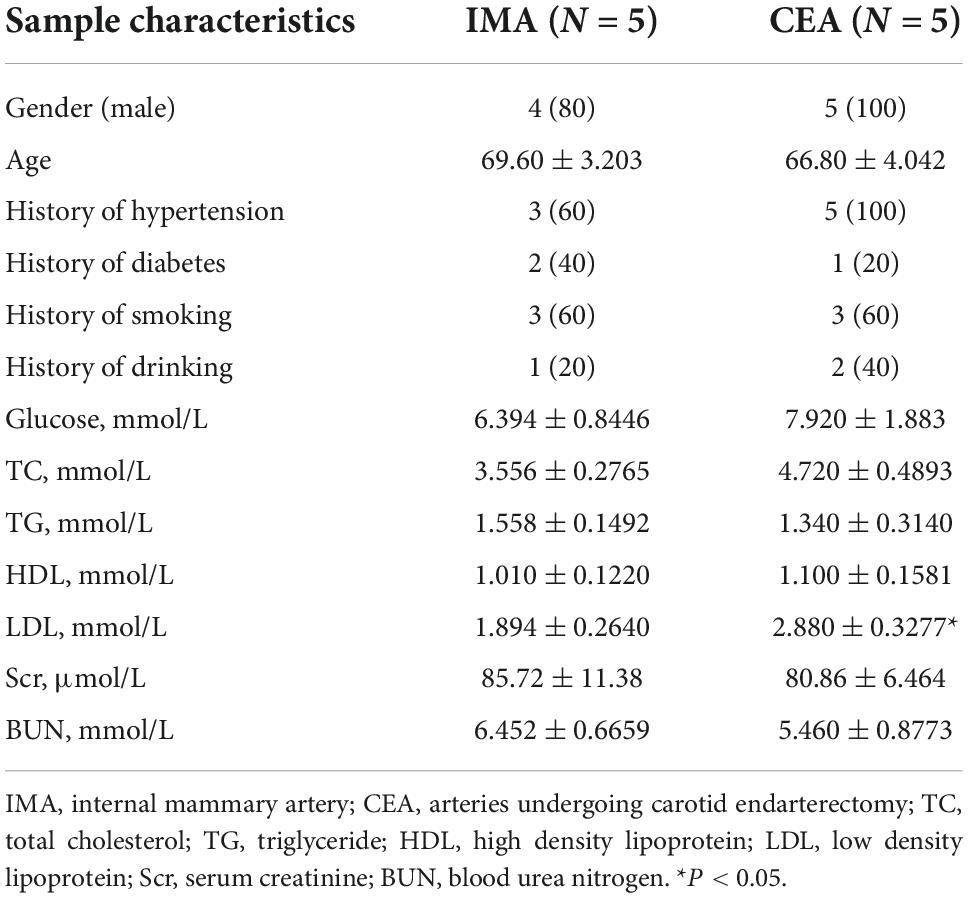
Table 1. Clinical characteristics of the patients.
Differentiated VSMCs share definite contractile characteristics, whereas dedifferentiated VSMCs can show different functional states or phenotypes. According to the functional characteristics, these VSMCs can be osteochondrogenic, proliferative, migratory, synthetic, and inflammatory (2). Although not depicted, there may be distinct modulation for VSMC phenotypes with changing environmental cues. In the early stages of vasculopathy, VSMCs proliferate rapidly and show enhanced migratory capacity. As the disease progresses, VSMCs synthesize a large number of hazardous substances including extracellular matrix protein, inflammatory factors and MMPs. It is generally believed that BKCa channels play a central role in controlling smooth muscle cell contraction. Recent studies have indicated that BKCa channel activation ameliorates VSMCs calcification, functionally related to the α-subunit (15). It is worth noting that during calcification, VSMCs transform into osteoblast/chondrocyte-like cells. However, the possible involvement of the BKCa channel in the VSMC phenotype switching toward other pathological phenotypes has not yet been explored.
Restenosis often occurs after by-pass surgery, angioplasty interventions and vascular injury, during which VSMCs are characterized by pathological proliferation and migration (25). During restenosis, VSMCs can undergo a transient modification of their phenotype, which is originally prepared to repair the vascular injury. The rat model of vascular injury is often used to describe the initial VSMC phenotypic modulation associated with neointimal hyperplasia and can closely mimic restenosis in humans. With the use of the balloon catheter injury model, it has been demonstrated that neointimal formation after angioplasty is associated with downregulated BKCa expression (26). During this process, the mature contractile VSMCs transform into proliferating neointimal VSMCs. Herein, we further proved that the decreased expression of BKCa-β1 was earlier and more obvious (Figure 1). At the same time, reduced expression of VSMCs contractile proteins was observed. During neointima formation, complex interactions between growth-stimulating molecules can promote the proliferation and migration of VSMCs (27). Therefore, reduced BKCa-β1 expression in the neointima might be mediated by many mitotic factors. As reported, TGF-β plays different roles in vitro and in vivo. In vitro, TGF-β can promote SMCs to transform into contractile phenotype and inhibit proliferation (24). Nevertheless, in vivo, TGF-β can promote the formation of neointima and aggravate the restenosis of blood vessels. Besides, purified, recombinant TGF-β stimulated neointimal, but not medial, SMC proliferation in vivo (22). This suggests that functional differences between neointimal and medial SMCs may extend to the level of growth control. In addition, TGF-β can stimulate or inhibit VSMC proliferation depending on cell density (28–32). All these discrepancies above suggest that the phenotype and biological behavior of SMCs are affected by the surrounding microenvironment and there are complex regulations in the body. Anyhow, our results indicated that BKCa-β1 is consistently and positively associated with the contractile phenotype of VSMCs in vitro and in vivo.
Numerous studies have confirmed that PDGF-BB can significantly promote the proliferation and migration of VSMCs after arterial injury (18, 33). In vitro, PDGF-BB stimulation significantly reduced the expression of SM α-actin, SM MHC, and SM22α in the cultured VSMCs (34–37). Our research also proves this (Figures 2C,D). Research has demonstrated that PDGF-BB suppresses VSMC contractile genes expression through the ERK1/2-MAPK pathway, which leads to the dissociation and nucleation of myocardin and SRF. Consistently, previous report has shown that BKCa-β1 expression was also driven by myocardin and SRF (38). Thus, we speculated that the ERK1/2-MAPK pathway would also mediate the downregulation of BKCa-β1 expression. Whether other mechanisms are involved needs further exploration.
Decreased expression or increased degradation of BKCa-β1 has been related to increased vascular tension and hypertension (39). BKCa-β1 expression is decreased in VSMCs from hypertensive (39, 40), aging (41), diabetic (42), and hypoxic rodent models (43). It has also been shown that vascular BKCa channel function is impaired in Type 1 diabetic mice and Type 2 diabetic patients, mainly due to the marked decrease of BKCa-β1 (44, 45). As direct causal evidence, BKCa-β1 deletion in mice leads to increased arterial tension and elevated blood pressure (46, 47). In high-fat-diet mice, BKCa-β1 deficiency exacerbates vascular remodeling and fibrosis (48). Most importantly, loss-of-function mutations in the KCNMB1 lead to hypertension and renal diseases in humans (49). Inversely, a gain-of-function mutation in the KCNMB1 (E65K) is associated with low incidence of diastolic hypertension (50–52). All of the above findings suggest that decreased BKCa-β1 expression can promote the occurrence and development of vascular diseases. To date, there are no reports on the role of BKCa-β1 in the modulation of the VSMC phenotype. Herein, for the first time, we discovered that BKCa-β1 was significantly correlated with the phenotype switching of VSMCs. In addition, we first provided direct evidence that BKCa-β1 knockdown can drive VSMC dedifferentiation. More importantly, we confirmed that decreased BKCa-β1 expression was closely related to the dedifferentiation of VSMCs and atherosclerosis in human. Yet, the exact mechanisms underlying BKCa-β1 deficiency-induced phenotype switching remain to be studied.
BKCa channel is composed of BKCa-α and BKCa-β. In the vascular system, BKCa-β1, encoded by KCNMB1, is the major subtype in VSMCs. BKCa-α forms a functional structure and displays the essential properties of native BKCa channels: voltage and Ca2+ sensitivity, K+ selectivity and large conductance (53). As for B KCa-β1, it confers the BKCa channel with higher Ca2+ and voltage sensitivity, making this channel more efficient in VSMC functions (54). Moreover, BKCa-β1 regulates BKCa-α expression on the membrane via regulating endocytic trafficking signaling (55). Our previous studies identified the mechanosensitivity of the STREX-lacking BKCa channel in the colonic smooth muscle (56) and further verified that BKCa-β1 is involved in the regulation (13). Studies have revealed that BKCa channel activity is significantly affected by the expression of BKCa-β1, and changes in cellular function impaired by BKCa-β1 seem to be more significant than the functional differences among variations in BKCa-α. In accordance with this, many hormones and curative medicines are found to enhance the BKCa channel activity by acting on or interacting with BKCa-β1, and BKCa-β1 targeted compounds prove to be more favorable for VSMCs disorders where its expression is restricted (57). Notably, our present study showed that BKCa-β1 deficiency is specifically involved in the dedifferentiation of VSMCs. In addition, downregulation of BKCa-β1 was characteristic for proliferative and migratory phenotypes of VSMCs.
Taken together, this study demonstrated that BKCa-β1 is an important regulator of VSMC identity by preventing its phenotype switching. These findings reveal a self-protective mechanism of VSMCs against harmful environmental stimuli and support a protective role of BKCa channel activation in various vascular diseases in humans. Targeting BKCa-β1 to precisely modulate BKCa activity may provide novel therapeutic strategy for post-injury restenosis and atherosclerosis.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The studies involving human participants were reviewed and approved by the Medical Ethics Committee of Chinese PLA General Hospital. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by the Institutional Animal Care and Use Committee and Ethics Committee of Capital Medical University (Beijing, China). Written informed consent was obtained from the individual(s) for the publication of any potentially identifiable images or data included in this article.
MW, SL, CX, and HH designed the study. MW and SL performed the experiments and analyzed the data. HL, ML, JZ, and YW analyzed the data. MW wrote the manuscript. HH finalized the manuscript. All authors critically revised the manuscript and approved the submitted version.
This work was supported by funding from the National Natural Science Foundation of China (grant numbers 81900415 and 82000429).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2022.1062695/full#supplementary-material
1. Rzucidlo EM, Martin KA, Powell RJ. Regulation of vascular smooth muscle cell differentiation. J Vasc Surg. (2007) 45(Suppl. A):A25–32. doi: 10.1016/j.jvs.2007.03.001
2. Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. (2004) 84:767–801. doi: 10.1152/physrev.00041.2003
3. Orr AW, Hastings NE, Blackman BR, Wamhoff BR. Complex regulation and function of the inflammatory smooth muscle cell phenotype in atherosclerosis. J Vasc Res. (2010) 47:168–80. doi: 10.1159/000250095
4. Ailawadi G, Moehle CW, Pei H, Walton SP, Yang Z, Kron IL, et al. Smooth muscle phenotypic modulation is an early event in aortic aneurysms. J Thorac Cardiovasc Surg. (2009) 138:1392–9. doi: 10.1016/j.jtcvs.2009.07.075
5. Sai S, Iwata A, Thomas R, Allen MD. Vascular cell adhesion molecule-1 up-regulation and phenotypic modulation of vascular smooth muscle cells predate mononuclear infiltration in transplant arteriopathy. J Thorac Cardiovasc Surg. (2001) 122:508–17. doi: 10.1067/mtc.2001.113601
6. Itoh S, Umemoto S, Hiromoto M, Toma Y, Tomochika Y, Aoyagi S, et al. Importance of NAD(P)H oxidase-mediated oxidative stress and contractile type smooth muscle myosin heavy chain SM2 at the early stage of atherosclerosis. Circulation. (2002) 105:2288–95. doi: 10.1161/01.cir.0000015607.33345.1f
7. Pidkovka NA, Cherepanova OA, Yoshida T, Alexander MR, Deaton RA, Thomas JA, et al. Oxidized phospholipids induce phenotypic switching of vascular smooth muscle cells in vivo and in vitro. Circ Res. (2007) 101:792–801. doi: 10.1161/CIRCRESAHA.107.152736
8. Kumar B, Dreja K, Shah SS, Cheong A, Xu SZ, Sukumar P, et al. Upregulated TRPC1 channel in vascular injury in vivo and its role in human neointimal hyperplasia. Circ Res. (2006) 98:557–63. doi: 10.1161/01.RES.0000204724.29685.db
9. Cheong A, Li J, Sukumar P, Kumar B, Zeng F, Riches K, et al. Potent suppression of vascular smooth muscle cell migration and human neointimal hyperplasia by KV1.3 channel blockers. Cardiovasc Res. (2011) 89:282–9. doi: 10.1093/cvr/cvq305
10. Clempus RE, Sorescu D, Dikalova AE, Pounkova L, Jo P, Sorescu GP, et al. Nox4 is required for maintenance of the differentiated vascular smooth muscle cell phenotype. Arterioscler Thromb Vasc Biol. (2007) 27:42–8. doi: 10.1161/01.ATV.0000251500.94478.18
11. Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol. (2014) 15:786–801. doi: 10.1038/nrm3904
12. Dong DL, Bai YL, Cai BZ. Calcium-activated potassium channels: potential target for cardiovascular diseases. Adv Protein Chem Struct Biol. (2016) 104:233–61. doi: 10.1016/bs.apcsb.2015.11.007
13. Xin F, Cheng Y, Ren J, Zhang S, Liu P, Zhao H, et al. The extracellular loop of the auxiliary beta1-subunit is involved in the regulation of BKCa channel mechanosensitivity. Am J Physiol Cell Physiol. (2018) 315:C485–93. doi: 10.1152/ajpcell.00037.2018
14. Wang M, Yin X, Li S, Zhang X, Yi M, He C, et al. Large-conductance calcium-activated potassium channel opener, NS1619, protects against mesenteric artery remodeling induced by agonistic autoantibodies against the angiotensin II type 1 receptor. J Am Heart Assoc. (2022) 11:e024046. doi: 10.1161/JAHA.121.024046
15. Ning FL, Tao J, Li DD, Tian LL, Wang ML, Reilly S, et al. Activating BK channels ameliorates vascular smooth muscle calcification through Akt signaling. Acta Pharmacol Sin. (2021) 43:1–10. doi: 10.1038/s41401-021-00704-6
16. Zhang W, Trebak M. Vascular balloon injury and intraluminal administration in rat carotid artery. J Vis Exp. (2014) 94:52045. doi: 10.3791/52045
17. Jovinge S, Hultgardh-Nilsson A, Regnstrom J, Nilsson J. Tumor necrosis factor-alpha activates smooth muscle cell migration in culture and is expressed in the balloon-injured rat aorta. Arterioscler Thromb Vasc Biol. (1997) 17:490–7. doi: 10.1161/01.atv.17.3.490
18. Jawien A, Bowen-Pope DF, Lindner V, Schwartz SM, Clowes AW. Platelet-derived growth factor promotes smooth muscle migration and intimal thickening in a rat model of balloon angioplasty. J Clin Invest. (1992) 89:507–11. doi: 10.1172/JCI115613
19. Fu M, Zhu X, Wang Q, Zhang J, Song Q, Zheng H, et al. Platelet-derived growth factor promotes the expression of peroxisome proliferator-activated receptor gamma in vascular smooth muscle cells by a phosphatidylinositol 3-kinase/Akt signaling pathway. Circ Res. (2001) 89:1058–64. doi: 10.1161/hh2301.099642
20. Mikhail N, Fukuda N, Tremblay J, Hamet P. Platelets, growth factors, and vascular smooth-muscle cells in hypertension and diabetes. J Cardiovasc Pharmacol. (1993) 22(Suppl. 6):S64–74.
21. Hadrava V, Kruppa U, Russo RC, Lacourciere Y, Tremblay J, Hamet P. Vascular smooth muscle cell proliferation and its therapeutic modulation in hypertension. Am Heart J. (1991) 122:1198–203. doi: 10.1016/0002-8703(91)90939-f
22. Majesky MW, Lindner V, Twardzik DR, Schwartz SM, Reidy MA. Production of transforming growth factor beta 1 during repair of arterial injury. J Clin Invest. (1991) 88:904–10. doi: 10.1172/JCI115393
23. Huang BF, Wang W, Fu YC, Zhou XH, Wang X. The effect of quercetin on neointima formation in a rat artery balloon injury model. Pathol Res Pract. (2009) 205:515–23. doi: 10.1016/j.prp.2009.01.007
24. Wang L, Zheng J, Du Y, Huang Y, Li J, Liu B, et al. Cartilage oligomeric matrix protein maintains the contractile phenotype of vascular smooth muscle cells by interacting with alpha(7)beta(1) integrin. Circ Res. (2010) 106:514–25. doi: 10.1161/CIRCRESAHA.109.202762
25. Zargham R. Preventing restenosis after angioplasty: a multistage approach. Clin Sci. (2008) 114:257–64. doi: 10.1042/CS20070228
26. Kohler R, Wulff H, Eichler I, Kneifel M, Neumann D, Knorr A, et al. Blockade of the intermediate-conductance calcium-activated potassium channel as a new therapeutic strategy for restenosis. Circulation. (2003) 108:1119–25. doi: 10.1161/01.CIR.0000086464.04719.DD
27. Newby AC, Zaltsman AB. Molecular mechanisms in intimal hyperplasia. J Pathol. (2000) 190:300–9. doi: 10.1002/(SICI)1096-9896(200002)190:33.0.CO;2-I
28. Assoian RK, Sporn MB. Type beta transforming growth factor in human platelets: release during platelet degranulation and action on vascular smooth muscle cells. J Cell Biol. (1986) 102:1217–23. doi: 10.1083/jcb.102.4.1217
29. Majack RA, Majesky MW, Goodman LV. Role of PDGF-A expression in the control of vascular smooth muscle cell growth by transforming growth factor-beta. J Cell Biol. (1990) 111:239–47. doi: 10.1083/jcb.111.1.239
30. Battegay EJ, Raines EW, Seifert RA, Bowen-Pope DF, Ross R. TGF-beta induces bimodal proliferation of connective tissue cells via complex control of an autocrine PDGF loop. Cell. (1990) 63:515–24. doi: 10.1016/0092-8674(90)90448-n
31. Majack RA. Beta-type transforming growth factor specifies organizational behavior in vascular smooth muscle cell cultures. J Cell Biol. (1987) 105:465–71. doi: 10.1083/jcb.105.1.465
32. Owens GK, Geisterfer AA, Yang YW, Komoriya A. Transforming growth factor-beta-induced growth inhibition and cellular hypertrophy in cultured vascular smooth muscle cells. J Cell Biol. (1988) 107:771–80. doi: 10.1083/jcb.107.2.771
33. Ferns GA, Raines EW, Sprugel KH, Motani AS, Reidy MA, Ross R. Inhibition of neointimal smooth muscle accumulation after angioplasty by an antibody to PDGF. Science. (1991) 253:1129–32. doi: 10.1126/science.1653454
34. Blank RS, Owens GK. Platelet-derived growth factor regulates actin isoform expression and growth state in cultured rat aortic smooth muscle cells. J Cell Physiol. (1990) 142:635–42. doi: 10.1002/jcp.1041420325
35. Corjay MH, Thompson MM, Lynch KR, Owens GK. Differential effect of platelet-derived growth factor- versus serum-induced growth on smooth muscle alpha-actin and nonmuscle beta-actin mRNA expression in cultured rat aortic smooth muscle cells. J Biol Chem. (1989) 264:10501–6.
36. Holycross BJ, Blank RS, Thompson MM, Peach MJ, Owens GK. Platelet-derived growth factor-BB-induced suppression of smooth muscle cell differentiation. Circ Res. (1992) 71:1525–32. doi: 10.1161/01.res.71.6.1525
37. Bornfeldt KE, Arnqvist HJ, Norstedt G. Regulation of insulin-like growth factor-I gene expression by growth factors in cultured vascular smooth muscle cells. J Endocrinol. (1990) 125:381–6. doi: 10.1677/joe.0.1250381
38. Long X, Tharp DL, Georger MA, Slivano OJ, Lee MY, Wamhoff BR, et al. The smooth muscle cell-restricted KCNMB1 ion channel subunit is a direct transcriptional target of serum response factor and myocardin. J Biol Chem. (2009) 284:33671–82. doi: 10.1074/jbc.M109.050419
39. Amberg GC, Santana LF. Downregulation of the BK channel beta1 subunit in genetic hypertension. Circ Res. (2003) 93:965–71. doi: 10.1161/01.RES.0000100068.43006.36
40. Amberg GC, Bonev AD, Rossow CF, Nelson MT, Santana LF. Modulation of the molecular composition of large conductance, Ca(2+) activated K(+) channels in vascular smooth muscle during hypertension. J Clin Invest. (2003) 112:717–24. doi: 10.1172/JCI18684
41. Zhang Y, Liao J, Zhang L, Li S, Wu Y, Shi L. BKCa channel activity and vascular contractility alterations with hypertension and aging via beta1 subunit promoter methylation in mesenteric arteries. Hypertens Res. (2018) 41:96–103. doi: 10.1038/hr.2017.96
42. Dong L, Zheng YM, Van Riper D, Rathore R, Liu QH, Singer HA, et al. Functional and molecular evidence for impairment of calcium-activated potassium channels in type-1 diabetic cerebral artery smooth muscle cells. J Cereb Blood Flow Metab. (2008) 28:377–86. doi: 10.1038/sj.jcbfm.9600536
43. Navarro-Antolin J, Levitsky KL, Calderon E, Ordonez A, Lopez-Barneo J. Decreased expression of maxi-K+ channel beta1-subunit and altered vasoregulation in hypoxia. Circulation. (2005) 112:1309–15. doi: 10.1161/CIRCULATIONAHA.104.529404
44. Yi F, Wang H, Chai Q, Wang X, Shen WK, Willis MS, et al. Regulation of large conductance Ca2+-activated K+ (BK) channel beta1 subunit expression by muscle RING finger protein 1 in diabetic vessels. J Biol Chem. (2014) 289:10853–64. doi: 10.1074/jbc.M113.520940
45. Lu T, Chai Q, Jiao G, Wang XL, Sun X, Furuseth JD, et al. Downregulation of BK channel function and protein expression in coronary arteriolar smooth muscle cells of type 2 diabetic patients. Cardiovasc Res. (2019) 115:145–53. doi: 10.1093/cvr/cvy137
46. Sachse G, Faulhaber J, Seniuk A, Ehmke H, Pongs O. Smooth muscle BK channel activity influences blood pressure independent of vascular tone in mice. J Physiol. (2014) 592:2563–74. doi: 10.1113/jphysiol.2014.272880
47. Pluger S, Faulhaber J, Furstenau M, Lohn M, Waldschutz R, Gollasch M, et al. Mice with disrupted BK channel beta1 subunit gene feature abnormal Ca(2+) spark/STOC coupling and elevated blood pressure. Circ Res. (2000) 87:E53–60. doi: 10.1161/01.res.87.11.e53
48. Xu H, Garver H, Fernandes R, Phelps JT, Harkema JJ, Galligan JJ, et al. BK channel beta1-subunit deficiency exacerbates vascular fibrosis and remodelling but does not promote hypertension in high-fat fed obesity in mice. J Hypertens. (2015) 33:1611–23. doi: 10.1097/HJH.0000000000000590
49. Chen Y, Salem RM, Rao F, Fung MM, Bhatnagar V, Pandey B, et al. Common charge-shift mutation Glu65Lys in K+ channel beta(1)-Subunit KCNMB1: pleiotropic consequences for glomerular filtration rate and progressive renal disease. Am J Nephrol. (2010) 32:414–24. doi: 10.1159/000320131
50. Fernandez-Fernandez JM, Tomas M, Vazquez E, Orio P, Latorre R, Senti M, et al. Gain-of-function mutation in the KCNMB1 potassium channel subunit is associated with low prevalence of diastolic hypertension. J Clin Invest. (2004) 113:1032–9. doi: 10.1172/JCI20347
51. Kelley-Hedgepeth A, Peter I, Kip K, Montefusco M, Kogan S, Cox D, et al. The protective effect of KCNMB1 E65K against hypertension is restricted to blood pressure treatment with beta-blockade. J Hum Hypertens. (2008) 22:512–5. doi: 10.1038/jhh.2008.23
52. Nielsen T, Burgdorf KS, Grarup N, Borch-Johnsen K, Hansen T, Jorgensen T, et al. The KCNMB1 Glu65Lys polymorphism associates with reduced systolic and diastolic blood pressure in the Inter99 study of 5729 Danes. J Hypertens. (2008) 26:2142–6. doi: 10.1097/HJH.0b013e32830b894a
53. Cox DH, Aldrich RW. Role of the beta1 subunit in large-conductance Ca(2+)-activated K(+) channel gating energetics. Mechanisms of enhanced Ca(2+) sensitivity. J Gen Physiol. (2000) 116:411–32. doi: 10.1085/jgp.116.3.411
54. Latorre R, Castillo K, Carrasquel-Ursulaez W, Sepulveda RV, Gonzalez-Nilo F, Gonzalez C, et al. Molecular determinants of BK channel functional diversity and functioning. Physiol Rev. (2017) 97:39–87. doi: 10.1152/physrev.00001.2016
55. Toro B, Cox N, Wilson RJ, Garrido-Sanabria E, Stefani E, Toro L, et al. KCNMB1 regulates surface expression of a voltage and Ca2+-activated K+ channel via endocytic trafficking signals. Neuroscience. (2006) 142:661–9. doi: 10.1016/j.neuroscience.2006.06.061
56. Wang W, Huang H, Hou D, Liu P, Wei H, Fu X, et al. Mechanosensitivity of STREX-lacking BKCa channels in the colonic smooth muscle of the mouse. Am J Physiol Gastrointest Liver Physiol. (2010) 299:G1231–40. doi: 10.1152/ajpgi.00268.2010
Keywords: vascular smooth muscle cells, phenotype switching, BKCa channel, atherosclerosis, post-injury restenosis
Citation: Wang M, Li S, Liu H, Liu M, Zhang J, Wu Y, Xiao C and Huang H (2022) Large-conductance Ca2 +-activated K+ channel β1-subunit maintains the contractile phenotype of vascular smooth muscle cells. Front. Cardiovasc. Med. 9:1062695. doi: 10.3389/fcvm.2022.1062695
Received: 06 October 2022; Accepted: 24 November 2022;
Published: 09 December 2022.
Edited by:
Masanori Aikawa, Brigham and Women’s Hospital and Harvard Medical School, United StatesReviewed by:
Jing Wu, University of Rochester Medical Center, United StatesCopyright © 2022 Wang, Li, Liu, Liu, Zhang, Wu, Xiao and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Haixia Huang, aGFpeGlhaEBjY211LmVkdS5jbg==; Cangsong Xiao, eGNzMzAxQHFxLmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.