 Claire S. Whyte
Claire S. Whyte Nicola J. Mutch
Nicola J. Mutch- Aberdeen Cardiovascular and Diabetes Centre, Institute of Medical Sciences, School of Medicine, Medical Sciences and Nutrition, University of Aberdeen, Aberdeen, United Kingdom
The formation of thrombi is shaped by intravascular shear stress, influencing both fibrin architecture and the cellular composition which has downstream implications in terms of stability against mechanical and fibrinolytic forces. There have been many advancements in the development of models that incorporate flow rates akin to those found in vivo. Both thrombus formation and breakdown are simultaneous processes, the balance of which dictates the size, persistence and resolution of thrombi. Therefore, there is a requirement to have models which mimic the physiological shear experienced within the vasculature which in turn influences the fibrinolytic degradation of the thrombus. Here, we discuss various assays for fibrinolysis and importantly the development of novel models that incorporate physiological shear rates. These models are essential tools to untangle the molecular and cellular processes which govern fibrinolysis and can recreate the conditions within normal and diseased vessels to determine how these processes become perturbed in a pathophysiological setting. They also have utility to assess novel drug targets and antithrombotic drugs that influence thrombus stability.
Introduction
Fibrinolysis is the process by which fibrin clots are degraded and cleared from the circulation. The central enzyme in this pathway is the serine protease plasmin, which is formed by cleavage of the zymogen, plasminogen, at Arg561-Val562. The endogenous plasminogen activators are tissue plasminogen activator (tPA) and urokinase plasminogen activator (uPA). The structural homology of tPA and uPA is similar, however, they differ in their mechanism of action. Binding to fibrin enhances tPA-mediated plasminogen activation 1250-fold compared to the solution phase (1). In contrast, uPA can efficiently activate plasminogen in the absence of fibrin, either in solution or associated with its cellular receptor, urokinase plasminogen activator receptor (uPAR) (2). Plasmin sequentially cleaves fibrin into fibrin degradation products (FDPs). Measurement of the plasma level of fragment D-dimer, which consists of two D domains from adjacent fibrin monomers, is commonly used as marker of both ongoing thrombosis and fibrinolysis. Several inhibitors regulate fibrinolysis. The serpin, plasminogen activator inhibitor-1 (PAI-1), acts by rapidly forming a 1:1 complex to inhibit tPA and uPA (3, 4). Alpha2-antiplasmin (α2AP) is a fast-acting inhibitor of plasmin present at high circulating concentration (1 μM). Complexes of plasmin/α2AP (PAP) are cleared with a t1/2 of ~0.5 days and elevated levels are frequently used as a marker of pronounced fibrinolytic activity (5). Activated thrombin activatable fibrinolysis inhibitor (TAFIa) is a carboxypeptidase which exerts its anti-fibrinolytic function by cleaving c-terminal lysine residues from fibrin thereby downregulating the binding of tPA and plasminogen and subsequently reducing plasmin generation (6).
Dysregulation of the fibrinolytic system can lead to inefficient resolution of thrombi leading to thrombotic complications and indeed impaired endogenous fibrinolysis is an independent risk factor of cardiovascular events (7, 8). On the other hand, hyperfibrinolysis is associated with bleeding events such as trauma induced coagulopathy [Reviewed in (9)]. Current thrombolytic therapies carry an inherent risk of bleeding events and there is a pressing need to improve our understanding of the fibrinolytic system in order to tailor novel therapeutic strategies.
Classical models of fibrinolysis
Classically, fibrinolysis was studied in static models, most commonly by measuring a change in absorbance over time which increases as the clot forms and decreases as it degrades (Figure 1). This technique can only be used with either platelet-poor or platelet-rich plasma, and not in whole blood due to interference of red blood cells. An adapted Euglobulin clot lysis time (ECLT) assay has been shown to be advantageous when investigating hyperfibrinolysis (Figure 1) (10). This assay uses the euglobulin fraction which allows for fibrinolytic activity to be detected with the endogenous tPA (10). The Halo assay is a plate-based assay where blood/plasma is plated in a ‘halo' around the periphery of the well (Figure 1) (11). This was developed to be a high-throughput thrombolysis assay using small sample volumes of citrated whole blood. Clotting is initiated using tissue factor and once the ‘halo' clot is formed it is overlayed with a plasminogen activator. The change in absorbance is monitored as the clot degrades and red blood cells are released over the center of the well. These assays are advantageous as they are easily accessible, adaptable and relatively high throughput as they are suited to a 96-well plate format. Clot lysis assays are also routinely performed on stored plasma material, rather than necessitating fresh whole blood. These static fibrinolytic assays are useful tools in understanding basic mechanisms of fibrinolysis but lack standardization resulting in inter-laboratory variation and a lack of healthy control ranges. Viscoelastic assays such as the TEG® (Haemonetics Corporation), ROTEM® (Werfen) and more recently the Quantra® (Hemosonics LLC) allow for rapid measurement of clot strength and susceptibility to lysis in whole blood as point of care assays (Figure 1). TEG® and ROTEM® can both be run using either anticoagulated or non-anticoagulated blood which requires the samples to be processed immediately. These viscoelastic assays can detect hyperfibrinolysis but are insensitive to subtle changes in normal fibrinolytic activity. Optimized incorporation of tPA in these viscoelastic tests has allowed for rapid assessment of fibrinolysis in acute clinical settings such as trauma induced coagulopathy (12).
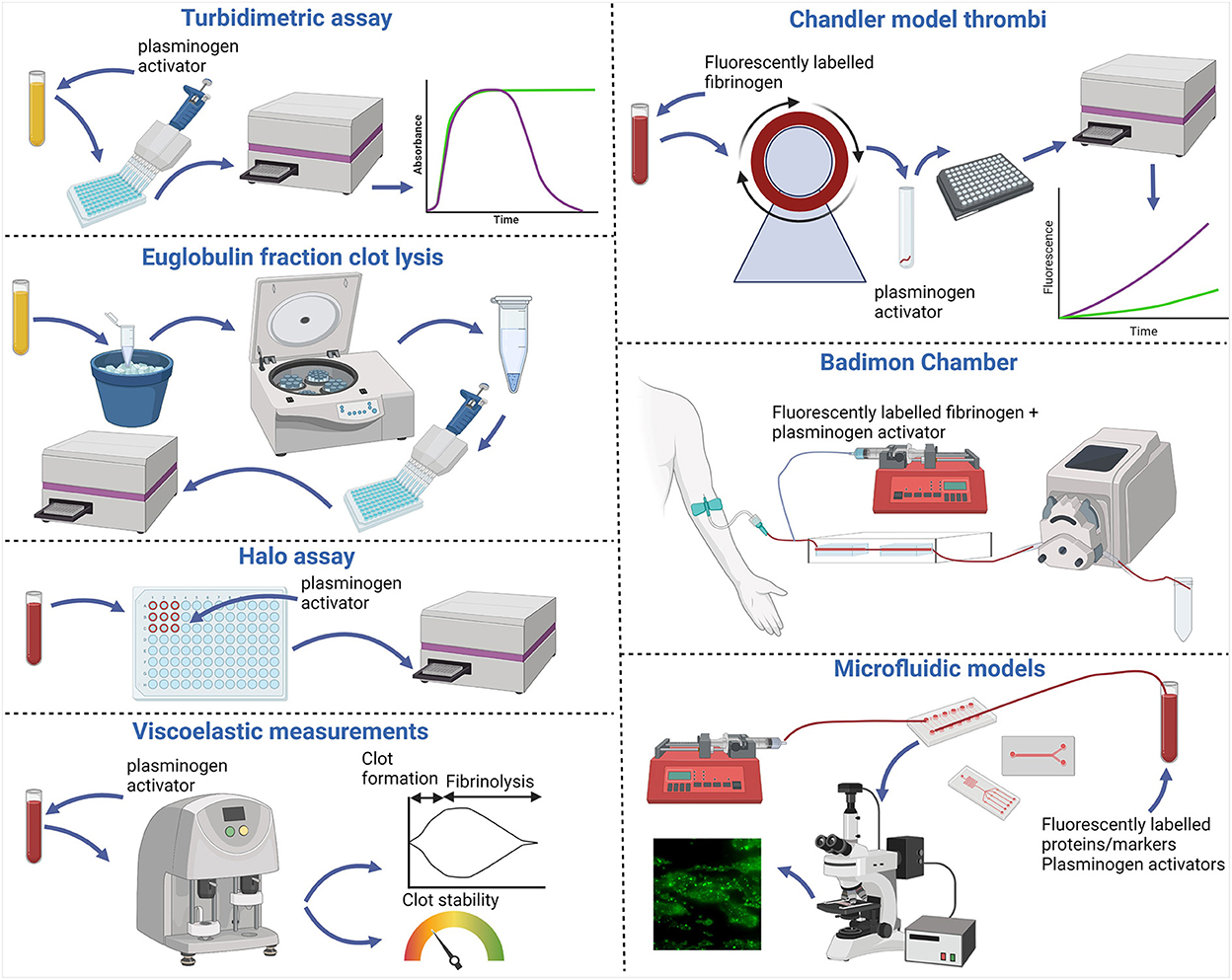
Figure 1. Fibrinolytic assays and models. Turbidimetric clot lysis assays involve clotting plasma in 96 well plates with the addition of calcium and thrombin in the presence of plasminogen activators. Change in absorbance is then measured using a spectrophotometer, increasing as clots form and decreasing during fibrinolysis. The Euglobulin fraction containing plasminogen, plasminogen activators and fibrinogen can be isolated by precipitation on ice cold acetic acid and collected via centrifugation. The pellet is then washed and solubilized before clotting with thrombin and the fibrinolytic activity then measured by change in absorbance. The Halo assay requires whole blood which is clotted with tissue factor in a “Halo” around the outside of a well of a 96 well plate. Plasminogen activators are then added and the absorbance increases as the clot lyses and is released into the well. Viscoelastic measurements are made in whole blood with added plasminogen activator using an analyzer. Data is outputted as traces [TEG® (Haemonetics Corporation) and ROTEM® (Werfen)] or a dial output [Quantra® (Hemosonics LLC)]. Chandler model thrombi are formed after recalcifying whole blood or plasma with added fluorescently labeled fibrinogen and enclosing in tubing under rotation. Thrombi are then removed and bathed in plasminogen activator and samples are taken at regular intervals and change in fluorescence release is measured as a marker of fibrin degradation. The Badimon chamber uses non-anticoagulated blood taken directly from the donor. Plasminogen activators and fluorescently labeled fibrinogen can be mixed with the blood ex vivo which then flows over thrombogenic tissue within the specialized perfusion chambers at predefined shear stresses. Markers of fibrinolysis can be measured from collected effluent. Additionally, thrombi formed on the thrombogenic surface can be removed and fibrinolysis monitored as with the Chandler model. Whole blood microfluidic models use syringe pumps to either pull or push blood over microfluidic biochips. These can be commercially sourced or prepared in house and can have different channel sizes and conformations. Thrombus formation is initiated on thrombogenic coatings within the biochips and viewed via fluorescence microscopy. Various markers and fluorescently labeled proteins can be incorporated pre or post thrombus formation. Fibrinolysis can be monitored as change in fluorescence signal over time and effluents can be collected to monitor markers of fibrinolysis. Figure created with BioRender.com.
However, all of these assays discussed have the disadvantage that they do not consider the influence of shear stress on the structure of the fibrin network and the cellular composition and interactions. Furthermore, the dilutional effect and convectional removal of thrombolytics and fibrinolytic proteins is not replicated in the absence of flow.
The shear stress and the site of formation greatly influences the structure and composition of thrombi (13, 14). Thrombi formed under high shear are more concentrated in platelets than those that are formed under low shear which are more fibrin- and red blood cell-rich (13). However, within a thrombus, there is heterogeneity with areas that are rich in red blood cells ensnared in fibrin and those that are more complex consisting of packed platelets associated with von Willebrand factor, leukocytes and dense fibrin (15). Platelets are reported to account for 31% of thrombus volume in arterial thrombi obtained after thrombectomy whilst they account for only 0.4% in venous thrombi (16). Platelet-rich thrombi display enhanced resistance to lysis when compared to red blood cell-rich thrombi (17) which likely contributes to the failure of thrombolytic therapy in recanalizing some patients (18). Thrombi formed under shear stress tend to have a surface of tightly packed fibers that align in the direction of flow and an interior consisting of fibers that are more heterogenous and porous in nature (19, 20). These structural differences in turn affect the susceptibility to fibrinolytic degradation (21) with clots that consist of dense, tightly packed, thin fibrin networks being less susceptible (22, 23). Therefore, models incorporating physiological flow rates are advantageous and are increasingly utilized in studies of fibrinolysis to mimic the situation in vivo more accurately.
Physiological flow models
Early flow models
The Chandler model was developed in the 1950s (24) and involves the formation of whole blood or plasma thrombi under flow (Figure 1). This simple but effective system forms thrombi in closed loops of polyvinyl tubing sealed using a short piece of tubing of a greater diameter and placed on a rotor at a constant speed. Whole blood or platelet-rich plasma thrombi formed under arterial shear rates mimic those found in vivo with a platelet dense head and fibrin rich tail (13) (Figure 2A). Incorporating fluorescently labeled fibrinogen during formation acts as a tracer to subsequently monitor release of FDPs. Allowing thrombi to proceed to full degradation confirms the sensitivity of this method in detecting differences in release of the degradation products (25). After thrombi have formed under flow, they can be lysed in a bathing solution alone, to monitor spontaneous lysis (26, 27), or with plasminogen activators (3, 27, 28). Release of fluorescent fibrin degradation products over time is directly proportional to the rate of lysis. Plasminogen activators at endogenous concentrations (29) can also be incorporated during thrombus formation under flow, however, the formation time must be decreased to account for simultaneous ongoing fibrinolysis (30). This system is also sensitive to detecting the influence of pharmacological agents on the susceptibility to lysis (31).
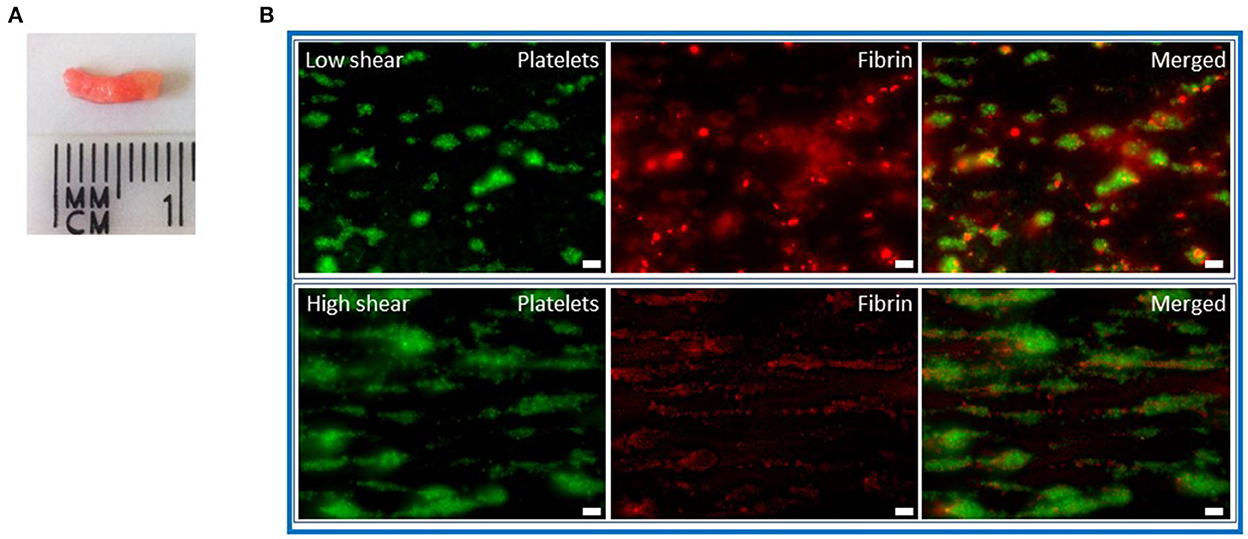
Figure 2. Whole blood thrombi under flow. (A) Chandler model thrombi formed from citrated whole blood that was recalcified (10.9 mM CaCl2). Thrombi were formed under rotation at a constant speed of 30 rpm for 90 min before being removed and washed in 0.9% saline. The platelet-rich white head is visible. (B) Recalcified whole blood thrombi were formed on a collagen (100 ng) and tissue factor (300 pM) surface in Cellix biochips. Thrombi formed under high shear (1,000 s−1) are more platelet-rich with less fibrin than those formed at low shear (250 s−1). DiOC6 (0.5 μg/ml) was included to label platelets (green) and fibrinogen-AF546 (75 μg/ml) added (red). Scale bars represent 10 μm.
Activated Factor XIII (FXIIIa) crosslinks the fibrinolytic inhibitors TAFIa (32), PAI-2 (33) and α2AP (34) to fibrin. Using the Chandler model, we have demonstrated that it is crosslinking of α2AP that is primarily responsible for the antifibrinolytic function of FXIIIa (35, 36). Interestingly, we showed that the antifibrinolytic effect of FXIIIa cannot be visualized in static conditions, emphasizing the importance of incorporation of shear stress. This was subsequently shown by Rijken et al. in a compaction-based model of fibrinolysis (37). The Chandler model can be adapted to model physiological fibrinolytic challenges. For example manipulating the blood by hemodilution to model trauma induced coagulopathy permits us to visualize the efficacy of restoring fibrinogen, via supplementation with fibrinogen concentrates or cryoprecipitate, in stabilizing thrombi against degradation (38).
The Badimon Chamber uses ex vivo blood which is flowed over thrombogenic tissue, such as deendothelialized aorta or Type I collagen bundles (Figure 1) (39). This model has the unique feature that native venous blood is pumped through two cylindrical perfusion chambers with predefined shear stress arranged in series (40). The influence of antithrombotic therapy on thrombus formation can be monitored as thrombus size and release of D-dimer, as a marker of fibrinolysis (41, 42). The nature of this model also allows the direct study of endogenous tPA release from the endothelium after bradykinin infusion (29). Alternatively, fluorescently labeled fibrinogen and exogenous tPA can be administrated via a calibrated syringe-driver into the extracorporeal circuit. Fibrinolysis can be quantified as fluorescence release after removing the porcine aorta strips, with thrombi attached, and placing in tPA bathing fluid and/or as the concentration of fibrinolytic proteins quantified in effluents (29, 30).
Microfluidic models of fibrinolysis
The use of microfluidic devices is a well-established technique for the study of thrombus formation under shear stress in normal and stenotic venous and arterial circulations. The experimental configuration varies greatly from one laboratory to another. The microfluidic device can be comprised of parallel-plate flow chambers (43) commercially available biochips or customizable biochips developed in-house using templates and molded polydimethylsiloxane (PDMS) plasma bonded (44, 45) or vacuum sealed (46) to glass slides or coverslips.
Coagulation is initiated as blood is flowed over surfaces coated with thrombogenic substances including tissue factor, collagen (most commonly of equine source), thrombin and von Willebrand factor and even hydrolyzed atheromatous material. The pattern of the coating varies from micro-spots to whole channel. Platelet-fibrin thrombi are visualized by staining platelets with lipophilic fluorescent dyes, most commonly 3,3′-dihexyloxacarbocyanine iodide (DiOC6), and incorporating fluorescently conjugated fibrinogen. The resulting 3D fibrin-platelet thrombi are visualized by fluorescence and brightfield microscopy with images acquired by a camera. Spatial and temporal analysis of changes in fluorescence can be quantified using imaging software.
The aforementioned flow models permit thrombus formation under flow conditions, yet fibrinolysis is primarily monitored under static conditions. We developed the first physiological microfluidic flow model of fibrinolysis to recapitulate the hemodynamic conditions under which fibrin is both formed and degraded (47). We adapted a well-established parallel-plate perfusion chamber (48) which is capable of forming thrombi with different hierarchal structures (49). Thrombi form with fibrin emanating from aggregated platelets surrounded by phosphatidylserine positive platelets (50). To tailor this model to the study of fibrinolysis we used a similar initial set up to the established model and perfused citrated whole blood (1,000 s−1) over a glass coverslip coated with micro-spots of fibrillar type I collagen in the presence or absence of recombinant human tissue factor. Platelets were labeled with DiOC6 or AlexaFluor labeled Annexin V to detect those that were phosphatidylserine positive and fluorescently labeled fibrinogen or plasminogen were added to the blood in the presence or absence of tPA or uPA. After 7 mins of thrombus formation a Hepes buffer, pH 7.45 was perfused. Fibrin degradation was measured as spatiotemporal changes in fluorescent fibrinogen signal. Dose-dependent fibrinolysis was observed in platelet-fibrin thrombi as change in fluorescence of fibrin(ogen). Fluorescently labeled plasma plasminogen primarily localized to platelet-associated fibrin(ogen) in the thrombus core (47). These findings are akin to an in vivo model examining plasminogen localization after laser injury (51). This model was sensitive to detecting differences in the susceptibility to lysis with a dose-dependent effect of tPA and uPA and demonstrated the balance of ongoing fibrin deposition and fibrinolysis. A higher molar concentration of uPA was required to observe lysis, consistent with the known fibrin specificity of tPA. Similarly, Loyau et al. (52) demonstrated the binding of both plasminogen and tPA in pre-formed platelet-fibrin thrombi and subsequent lysis using supraphysiological concentrations of the plasminogen activator in commercially available biochips (Vena8 Fluoro+ Biochips, Cellix Ltd.) (52). Thrombi formed in microfluidic devices display differences in platelet and fibrin deposition depending on the shear rates applied (Figure 2B). Fibrinolysis can be further quantified by measurements of plasmin activity, D-dimer, FDP and PAP in the effluents from these devices.
Microfluidic devices facilitate the modeling of surgical and pathophysiological conditions. Trauma induced coagulopathy was modeled by addition of exogenous tPA to mimic hyperfibrinolysis (53). This study used an eight-channel device which pulls the blood from a single outlet permitting simultaneous study of multiple conditions. These models are excellent tools for monitoring the influence of pharmacological manipulation of blood samples and have previously been used to monitor platelet deposition in response to P2Y1 and P2Y12 inhibitors and recombinant FVIII (54, 55). The experimental conditions can be manipulated to tease out the contribution of platelets/fibrin, for example using PPACK or hirudin to inhibit thrombin and subsequent fibrin formation. Inhibiting fibrin formation attenuates binding of both plasminogen (47) and tPA (52). Neutralization of the intrinsic pathway can be achieved by incorporation of corn trypsin inhibitor (CTI) (53). Importantly these devices can be used to study the efficacy of novel thrombolytic therapies. Using the Cellix Vena8 Fluoro+ chips, Huang et al. have demonstrated the platelet selectivity of fibrinogen-mimicking multi-arm nanovesicles as a vehicle for targeting thrombolytic therapy (56). This study combined experimental data generated in microfluidic devices with computational modeling further enhancing the power of this technique in developing our understanding of the dynamics of fibrinolysis and predicting kinetics of thrombolytic therapies (56).
Custom designed biochips allow for tailoring to specific experimental questions. An elegant study using PDMS chips bonded to a glass slide with an H-shape channel demonstrated the enhanced ability of tPA-conjugated colloidal micro-wheels in dissolving thrombi (44). The unique chip design represents vascular and extravascular compartments. The tPA micro-wheels were perfused into one vertical channel and, after applying a magnetic field to induce a corkscrew motion, they were visualized to rapidly promote fibrin dissolution in the horizontal “injury channel” (44). This method of combining mechanical and bulk dissolution has been further refined to incorporate plasminogen thereby overcoming rate limiting depletion of this zymogen (57). Although a constant flow rate is applied, the occlusion of the vessel wall leads to increasing shear stress at the site of thrombus formation (58). However, in vivo, blood can be diverted into other branches therefore releasing pressure. This effect is not recapitulated in single channel microfluidics leading to the development of bifurcating biochips which are designed to allow for diversion of blood (46, 58). In a microfluidic model of sterile thrombus occlusion, high interstitial hemodynamic forces promote the formation of neutrophil extracellular traps (NETs) (59). NETs are formed from expulsion of the nuclear and cytoplasmic contents from activated neutrophils resulting in extrusions consisting of chromatin adorned with DNA and proteins. NETs have been shown to exert antifibrinolytic effects (60–62). In this sophisticated model, NETosis was triggered under arterial shear conditions, yet was largely absent under venous conditions (59). Fibrin was observed to repress the formation of NETs while addition of exogenous tPA under arterial shear rates facilitated NET formation (63).
Point of care devices
There is a distinct requirement for rapid and consistent measurements of thrombus formation and thrombolysis. Balancing this with incorporating the impact of shear on thrombus formation is a challenge for the field. Recently there have been a number of point-of-care (POC) devices that aim to tackle this. The Global Thrombosis Test (GTT) (Thromboquest Ltd., UK) uses native non-anticoagulated blood to form thrombi under high shear with blood passing through a ball bearing situated within the chamber. This POC device has recently highlighted an association between clot architecture and platelet reactivity with endogenous fibrinolysis in patients suffering a STEMI (64, 65). Impaired endogenous fibrinolysis has now been proposed as a marker of cardiovascular risk (66, 67).
The Total-Thrombus-formation Analysis System (T-TAS) (Fujimori Kogyo Co. Ltd., Japan) is an integrated automated microchip flow chamber system designed to monitor thrombus formation. This system does not offer customization, as it uses chips with predefined shear stresses and procoagulant precoated surfaces. Nonetheless, the T-TAS analyzer has demonstrated the efficacy of tPA added to blood prior to thrombus formation at high and low shear. Inhibition of PAI-1 by the small molecule inhibitor (PAI-039) had a moderate effect in enhancing the fibrinolytic potential of tPA in thrombi formed in the T-TAS under arterial shear rates (68).
Limitations of flow models
Whilst flow models can recapitulate physiological dynamics of thrombus formation and can be used for screening of novel antithrombotic or thrombolytic therapies, they of course do not consider ongoing pharmacodynamics that may happen in vivo. For whole blood flow models there is a requirement for freshly obtained anticoagulated blood samples that should be used within 4 h of sampling but not within the first 15 to 20 mins (69). This puts a time restriction on the experimental conditions that can be explored and is particularly of note when studying fibrinolysis, as time for both thrombus formation and degradation must be accounted for. The Global thrombosis test uses non-anticoagulated blood and therefore must be processed immediately.
Assays such as the Chandler model thrombus system allow for comparison of multiple conditions simultaneously and generate a thrombus with structural features that resemble in vivo thrombi. However, this is a closed system and therefore does not mimic removal of components as occurs in the circulation and monitoring of the fibrinolytic arm is achieved under static conditions which is sub-optimal. POC devices provide ease of use and standardized methods of obtaining quantitative data which are attractive for clinical assessment of fibrinolytic potential. However, their lack of adaptability does not lend itself to mechanistic studies.
Visualization of thrombus formation and resolution in microfluidic devices requires fluorescence microscopes and cameras capable of appropriate acquisition times and magnifications. The purchase of commercial biochips and associated pumps mean that these models can be very costly, particularly during initial set-up. In-house preparation of biochips using PDMS can offer more affordable and adaptable biochips, but this requires the production of the initial mold, which can be costly, and necessitates design expertise. These assays are difficult to standardize due to the vast range of variables including types of biochips, coating substances, channel conformation and methods of detection. There has been a drive by the International Society on Thrombosis and Haemostasis Scientific Standardization Committee on Biorheology to standardize models of thrombus formation and platelet function in flow-based assays (69). These recommendations are applicable to fibrinolysis models, alongside additional complexities to consider including choice of plasminogen activator, its concentrations and whether it is included pre- or post-thrombus formation in addition to the various methods of monitoring fibrinolysis. Of course, donor variability further confounds these studies which leads to differences in platelet response and surface coverage (70), inter-individual variations in fibrin structure and deposition of hemostatic proteins.
The microvasculature expresses the adhesive glycoprotein thrombomodulin (TM) which drives the anticoagulant function of the endothelium by facilitating the activation of the anticoagulant, protein C. However, TM also participates indirectly in fibrinolysis by promoting thrombin-mediated activation of TAFI. Vascular endothelial cell expression of these fibrinolytic factors, including tPA, uPA is altered in response to shear stress (71, 72). PAI-1 is secreted from endothelial cells (73) and is upregulated by dysfunction of the endothelium due to elevated proinflammatory cytokines (74, 75) and C-reactive protein (76, 77). Endothelial regulation of fibrinolysis is an important component in dictating the overall fibrinolytic response within the vessel wall. Therefore, there is a current drive to develop endothelialized models that can more accurately recapitulate the vasculature not only in shear stress but also contribution of the endothelium.
Conclusions
The thrombus architecture is a dynamic environment and the shear forces that thrombi are exposed to shape the resulting fibrin network and cellular composition. Models that incorporate hemodynamic forces are an excellent tool to unpick the molecular and cellular interactions that dictate thrombus formation, structure, stability and resistance to lysis. The ability to use human blood ex vivo under physiological flow rates mitigates some of the issues of species differences experienced when using animal thrombosis models. Furthermore, this is in line with initiatives to reduce or replace the use of animals in scientific research such as that of the National Center for Replacement, Refinement and Reduction of animals in research. There have been considerable advancements in microfluidic models within the last decade to monitor fibrinolysis, a tool that has been seriously lacking thereby prohibiting our understanding of this complex system. These novel models offer adaptability to mimic shear rates experienced in different anatomical locations and vascular beds and provide important mechanistic insights into the regulation of thrombus stability. Furthermore, they are an excellent tool to facilitate the development and testing of novel thrombolytic therapies and/or drugs that may influence the responsiveness of thrombi to antithrombotic drugs.
Author contributions
CSW and NJM conceived the ideas, researched, and wrote and edited the manuscript. All authors contributed to the article and approved the submitted version.
Funding
CSW and NJM are supported by the British Heart Foundation (PG/20/17/35050) and a NC3Rs-British Heart Foundation Studentship (NC/W001810/1).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Ranby M, Bergsdorf N, Nilsson T. Enzymatic properties of the one- and two-chain form of tissue plasminogen activator. Thromb Res. (1982) 27:175–83. doi: 10.1016/0049-3848(82)90197-9
2. Stoppelli MP, Corti A, Soffientini A, Cassani G, Blasi F, Assoian RK. Differentiation-enhanced binding of the amino-terminal fragment of human urokinase plasminogen activator to a specific receptor on U937 monocytes. Proc Natl Acad Sci USA. (1985) 82:4939–43. doi: 10.1073/pnas.82.15.4939
3. Mutch NJ, Thomas L, Moore NR, Lisiak KM, Booth NA. TAFIa, PAI-1 and alpha-antiplasmin: complementary roles in regulating lysis of thrombi and plasma clots. J Thromb Haemost. (2007) 5:812–7. doi: 10.1111/j.1538-7836.2007.02430.x
4. Booth NA, Anderson JA, Bennett B. Plasminogen activators in alcoholic cirrhosis: demonstration of increased tissue type and urokinase type activator. J Clin Pathol. (1984) 37:772–7. doi: 10.1136/jcp.37.7.772
5. Mast AE, Enghild JJ, Pizzo SV, Salvesen G. Analysis of the plasma elimination kinetics and conformational stabilities of native, proteinase-complexed, and reactive site cleaved serpins: comparison of alpha 1-proteinase inhibitor, alpha 1-antichymotrypsin, antithrombin III, alpha 2-antiplasmin, angiotensinogen, and ovalbumin. Biochemistry. (1991) 30:1723–30. doi: 10.1021/bi00220a039
6. Plug T, Meijers JC. Structure-function relationships in thrombin-activatable fibrinolysis inhibitor. J Thromb Haemost. (2016) 14:633–44. doi: 10.1111/jth.13261
7. Sinnaeve PR, Van de Werf F. Endogenous fibrinolysis in STEMI: important before and after primary PCI. Eur Heart J. (2019) 40:306–8. doi: 10.1093/eurheartj/ehy760
8. Farag M, Spinthakis N, Gue YX, Srinivasan M, Sullivan K, Wellsted D, et al. Impaired endogenous fibrinolysis in ST-segment elevation myocardial infarction patients undergoing primary percutaneous coronary intervention is a predictor of recurrent cardiovascular events: the RISK PPCI study. Eur Heart J. (2019) 40:295–305. doi: 10.1093/eurheartj/ehy656
9. Moore EE, Moore HB, Kornblith LZ, Neal MD, Hoffman M, Mutch NJ, et al. Trauma-induced coagulopathy. Nat Rev Dis Primers. (2021) 7:30. doi: 10.1038/s41572-021-00264-3
10. Ilich A, Noubouossie DF, Henderson M, Ellsworth P, Molitor KF, Campello E, et al. Development and application of global assays of hyper- and hypofibrinolysis. Res Pract Thromb Haemost. (2020) 4:46–53. doi: 10.1002/rth2.12275
11. Bonnard T, Law LS, Tennant Z, Hagemeyer CE. Development and validation of a high throughput whole blood thrombolysis plate assay. Sci Rep. (2017) 7:2346. doi: 10.1038/s41598-017-02498-2
12. Harr JN, Moore EE, Chin TL, Chapman MP, Ghasabyan A, Stringham JR, et al. Viscoelastic hemostatic fibrinogen assays detect fibrinolysis early. Eur J Trauma Emerg Surg. (2015) 41:49–56. doi: 10.1007/s00068-014-0400-0
13. Robbie LA, Young SP, Bennett B, Booth NA. Thrombi formed in a Chandler loop mimic human arterial thrombi in structure and RAI-1 content and distribution. Thromb Haemost. (1997) 77:510–5. doi: 10.1055/s-0038-1655998
14. Falati S, Gross P, Merrill-Skoloff G, Furie BC, Furie B. Real-time in vivo imaging of platelets, tissue factor and fibrin during arterial thrombus formation in the mouse. Nat Med. (2002) 8:1175–81. doi: 10.1038/nm782
15. Staessens S, Denorme F, Francois O, Desender L, Dewaele T, Vanacker P, et al. Structural analysis of ischemic stroke thrombi: histological indications for therapy resistance. Haematologica. (2020) 105:498–507. doi: 10.3324/haematol.2019.219881
16. Chernysh IN, Nagaswami C, Kosolapova S, Peshkova AD, Cuker A, Cines DB, et al. The distinctive structure and composition of arterial and venous thrombi and pulmonary emboli. Sci Rep. (2020) 10:5112. doi: 10.1038/s41598-020-59526-x
17. Jang IK, Gold HK, Ziskind AA, Fallon JT, Holt RE, Leinbach RC, et al. Differential sensitivity of erythrocyte-rich and platelet-rich arterial thrombi to lysis with recombinant tissue-type plasminogen activator. A possible explanation for resistance to coronary thrombolysis. Circulation. (1989) 79:920–8. doi: 10.1161/01.CIR.79.4.920
18. Tomkins AJ, Schleicher N, Murtha L, Kaps M, Levi CR, Nedelmann M, et al. Platelet rich clots are resistant to lysis by thrombolytic therapy in a rat model of embolic stroke. Exp Transl Stroke Med. (2015) 7:2. doi: 10.1186/s13231-014-0014-y
19. Brown AE, Litvinov RI, Discher DE, Purohit PK, Weisel JW. Multiscale mechanics of fibrin polymer: gel stretching with protein unfolding and loss of water. Science. (2009) 325:741–4. doi: 10.1126/science.1172484
20. Varjú I, Sótonyi P, Machovich R, Szabó L, Tenekedjiev K, Silva MM, et al. Hindered dissolution of fibrin formed under mechanical stress. J Thromb Haemost. (2011) 9:979–86. doi: 10.1111/j.1538-7836.2011.04203.x
21. Weisel JW, Litvinov RI. The biochemical and physical process of fibrinolysis and effects of clot structure and stability on the lysis rate. Cardiovasc Hematol Agents Med Chem. (2008) 6:161–80. doi: 10.2174/187152508784871963
22. Collet JP, Park D, Lesty C, Soria J, Soria C, Montalescot G, et al. Influence of fibrin network conformation and fibrin fiber diameter on fibrinolysis speed: dynamic and structural approaches by confocal microscopy. Arterioscler Thromb Vasc Biol. (2000) 20:1354–61. doi: 10.1161/01.ATV.20.5.1354
23. Collet JP, Allali Y, Lesty C, Tanguy ML, Silvain J, Ankri A, et al. Altered fibrin architecture is associated with hypofibrinolysis and premature coronary atherothrombosis. Arterioscler Thromb Vasc Biol. (2006) 26:2567–73. doi: 10.1161/01.ATV.0000241589.52950.4c
24. Chandler AB. In vitro thrombotic coagulation of the blood; a method for producing a thrombus. Lab Invest. (1958) 7:110–4.
25. Hethershaw EL, Cilia La Corte AL, Duval C, Ali M, Grant PJ, Ariëns RA, et al. The effect of blood coagulation factor XIII on fibrin clot structure and fibrinolysis. J Thromb Haemost. (2014) 12:197–205. doi: 10.1111/jth.12455
26. Moir E, Booth NA, Bennett B, Robbie LA. Polymorphonuclear leucocytes mediate endogenous thrombus lysis via a u-PA-dependent mechanism. Br J Haematol. (2001) 113:72–80. doi: 10.1046/j.1365-2141.2001.02696.x
27. Mutch NJ, Moore NR, Wang E, Booth NA. Thrombus lysis by uPA, scuPA and tPA is regulated by plasma TAFI. J Thromb Haemost. (2003) 1:2000–7. doi: 10.1046/j.1538-7836.2003.00383.x
28. Mitchell JL, Lionikiene AS, Fraser SR, Whyte CS, Booth NA, Mutch NJ. Functional factor XIII-A is exposed on the stimulated platelet surface. Blood. (2014) 124:3982–90. doi: 10.1182/blood-2014-06-583070
29. Lucking AJ, Gibson KR, Paterson EE, Faratian D, Ludlam CA, Boon NA, et al. Endogenous tissue plasminogen activator enhances fibrinolysis and limits thrombus formation in a clinical model of thrombosis. Arterioscler Thromb Vasc Biol. (2013) 33:1105–11. doi: 10.1161/ATVBAHA.112.300395
30. Whyte CS, Mostefai HA, Baeten KM, Lucking AJ, Newby DE, Booth NA, et al. Role of shear stress and tPA concentration in the fibrinolytic potential of thrombi. Int J Mol Sci. (2021) 22, 2115. doi: 10.3390/ijms22042115
31. Mutch NJ, Moore NR, Mattsson C, Jonasson H, Green AR, Booth NA. The use of the Chandler loop to examine the interaction potential of NXY-059 on the thrombolytic properties of rtPA on human thrombi in vitro. Br J Pharmacol. (2008) 153:124–31. doi: 10.1038/sj.bjp.0707543
32. Valnickova Z, Enghild JJ. Human procarboxypeptidase U, or thrombin-activable fibrinolysis inhibitor, is a substrate for transglutaminases. Evidence for transglutaminase-catalyzed cross-linking to fibrin. J Biol Chem. (1998) 273:27220–4. doi: 10.1074/jbc.273.42.27220
33. Ritchie H, Robbie LA, Kinghorn S, Exley R, Booth NA. Monocyte plasminogen activator inhibitor 2 (PAI-2) inhibits u-PA-mediated fibrin clot lysis and is cross-linked to fibrin. Thromb Haemost. (1999) 81:96–103. doi: 10.1055/s-0037-1614425
34. Sakata Y, Aoki N. Significance of cross-linking of alpha 2-plasmin inhibitor to fibrin in inhibition of fibrinolysis and in hemostasis. J Clin Invest. (1982) 69:536–42. doi: 10.1172/JCI110479
35. Mutch NJ, Koikkalainen JS, Fraser SR, Duthie KM, Griffin M, Mitchell J, et al. Model thrombi formed under flow reveal the role of factor XIII-mediated cross-linking in resistance to fibrinolysis. J Thromb Haemost. (2010) 8:2017–24. doi: 10.1111/j.1538-7836.2010.03963.x
36. Fraser SR, Booth NA, Mutch NJ. The antifibrinolytic function of factor XIII is exclusively expressed through alpha(2)-antiplasmin cross-linking. Blood. (2011) 117:6371–4. doi: 10.1182/blood-2011-02-333203
37. Rijken DC, Abdul S, Malfliet JJ, Leebeek FW, Uitte de Willige S. Compaction of fibrin clots reveals the antifibrinolytic effect of factor XIII. J Thromb Haemost. (2016) 14:1453–61. doi: 10.1111/jth.13354
38. Whyte CS, Rastogi A, Ferguson E, Donnarumma M, Mutch NJ. The efficacy of fibrinogen concentrates in relation to cryoprecipitate in restoring clot integrity and stability against lysis. Int J Mol Sci. (2022) 23, e2944. doi: 10.3390/ijms23062944
39. Badimon L, Badimon JJ, Galvez A, Chesebro JH, Fuster V. Influence of arterial damage and wall shear rate on platelet deposition. Ex vivo study in a swine model. Arteriosclerosis. (1986) 6:312–20. doi: 10.1161/01.ATV.6.3.312
40. Lucking AJ, Chelliah R, Trotman AD, Connolly TM, Feuerstein GZ, Fox KA, et al. Characterisation and reproducibility of a human ex vivo model of thrombosis. Thromb Res. (2010) 126:431–5. doi: 10.1016/j.thromres.2010.06.030
41. Wolzt M, Gouya G, Kapiotis S, Becka M, Mueck W, Kubitza D. Open-label, randomized study of the effect of rivaroxaban with or without acetylsalicylic acid on thrombus formation in a perfusion chamber. Thromb Res. (2013) 132:240–7. doi: 10.1016/j.thromres.2013.05.019
42. Gouya G, Palkovits S, Kapiotis S, Madl C, Locker G, Stella A, et al. Bioactivity of enoxaparin in critically ill patients with normal renal function. Br J Clin Pharmacol. (2012) 74:806–14. doi: 10.1111/j.1365-2125.2012.04285.x
43. Van Kruchten R, Cosemans JM, Heemskerk JW. Measurement of whole blood thrombus formation using parallel-plate flow chambers—a practical guide. Platelets. (2012) 23:229–42. doi: 10.3109/09537104.2011.630848
44. Tasci TO, Disharoon D, Schoeman RM, Rana K, Herson PS, Marr DW, et al. Enhanced fibrinolysis with magnetically powered colloidal microwheels. Small. (2017) 13, 1700954. doi: 10.1002/smll.201700954
45. Schoeman RM, Rana K, Danes N, Lehmann M, Di Paola JA, Fogelson AL, et al. A microfluidic model of hemostasis sensitive to platelet function and coagulation. Cell Mol Bioeng. (2017) 10:3–15. doi: 10.1007/s12195-016-0469-0
46. Berry J, Peaudecerf FJ, Masters NA, Neeves KB, Goldstein RE, Harper MT. An “occlusive thrombosis-on-a-chip” microfluidic device for investigating the effect of anti-thrombotic drugs. Lab Chip. (2021) 21:4104–17. doi: 10.1039/D1LC00347J
47. Whyte CS, Swieringa F, Mastenbroek TG, Lionikiene AS, Lancé MD, van der Meijden PE, et al. Plasminogen associates with phosphatidylserine-exposing platelets and contributes to thrombus lysis under flow. Blood. (2015) 125:2568–78. doi: 10.1182/blood-2014-09-599480
48. Munnix IC, Strehl A, Kuijpers MJ, Auger JM, van der Meijden PE, van Zandvoort MA, et al. The glycoprotein VI-phospholipase Cgamma2 signaling pathway controls thrombus formation induced by collagen and tissue factor in vitro and in vivo. Arterioscler Thromb Vasc Biol. (2005) 25:2673–8. doi: 10.1161/01.ATV.0000193568.71980.4a
49. de Witt SM, Swieringa F, Cavill R, Lamers MM, Van Kruchten R, Mastenbroek T, et al. Identification of platelet function defects by multi-parameter assessment of thrombus formation. Nat Commun. (2014) 5:4257. doi: 10.1038/ncomms5257
50. Munnix IC, Kuijpers MJ, Auger J, Thomassen CM, Panizzi P, van Zandvoort MA, et al. Segregation of platelet aggregatory and procoagulant microdomains in thrombus formation: regulation by transient integrin activation. Arterioscler Thromb Vasc Biol. (2007) 27:2484–90. doi: 10.1161/ATVBAHA.107.151100
51. Brzoska T, Tanaka-Murakami A, Suzuki Y, Sano H, Kanayama N, Urano T. Endogenously generated plasmin at the vascular wall injury site amplifies lysine binding site-dependent plasminogen accumulation in microthrombi. PLoS ONE. (2015) 10:e0122196. doi: 10.1371/journal.pone.0122196
52. Loyau S, Ho-Tin-Noe B, Bourrienne MC, Boulaftali Y, Jandrot-Perrus M. Microfluidic modeling of thrombolysis. Arterioscler Thromb Vasc Biol. (2018) 38:2626–37. doi: 10.1161/ATVBAHA.118.311178
53. Li R, Elmongy H, Sims C, Diamond SL. Ex vivo recapitulation of trauma-induced coagulopathy and preliminary assessment of trauma patient platelet function under flow using microfluidic technology. J Trauma Acute Care Surg. (2016) 80:440–9. doi: 10.1097/TA.0000000000000915
54. Maloney SF, Brass LF, Diamond SL. P2Y12 or P2Y1 inhibitors reduce platelet deposition in a microfluidic model of thrombosis while apyrase lacks efficacy under flow conditions. Integr Biol (Camb). (2010) 2:183–92. doi: 10.1039/b919728a
55. Li R, Panckeri KA, Fogarty PF, Diamond SL. Recombinant factor VIIa enhances platelet deposition from flowing haemophilic blood but requires the contact pathway to promote fibrin deposition. Haemophilia. (2015) 21:266–74. doi: 10.1111/hae.12558
56. Huang Y, Gu B, Salles-Crawley II, Taylor KA, Yu L, Ren J, et al. Fibrinogen-mimicking, multiarm nanovesicles for human thrombus-specific delivery of tissue plasminogen activator and targeted thrombolytic therapy. Sci Adv. (2021) 7: eabf9033. doi: 10.1126/sciadv.abf9033
57. Disharoon D, Trewyn BG, Herson PS, Marr DWM, Neeves KB. Breaking the fibrinolytic speed limit with microwheel co-delivery of tissue plasminogen activator and plasminogen. J Thromb Haemost. (2022) 20:486–97. doi: 10.1111/jth.15617
58. Colace TV, Muthard RW, Diamond SL. Thrombus growth and embolism on tissue factor-bearing collagen surfaces under flow: role of thrombin with and without fibrin. Arterioscler Thromb Vasc Biol. (2012) 32:1466–76. doi: 10.1161/ATVBAHA.112.249789
59. Yu X, Tan J, Diamond SL. Hemodynamic force triggers rapid NETosis within sterile thrombotic occlusions. J Thromb Haemost. (2018) 16:316–29. doi: 10.1111/jth.13907
60. Longstaff C, Varjú I, Sótonyi P, Szabó L, Krumrey M, Hoell A, et al. Mechanical stability and fibrinolytic resistance of clots containing fibrin, DNA, and histones. J Biol Chem. (2013) 288:6946–56. doi: 10.1074/jbc.M112.404301
61. Varjú I, Longstaff C, Szabó L, Farkas ÁZ, Varga-Szabó VJ, Tanka-Salamon A, et al. DNA, histones and neutrophil extracellular traps exert anti-fibrinolytic effects in a plasma environment. Thromb Haemost. (2015) 113:1289–98. doi: 10.1160/TH14-08-0669
62. Ducroux C, Di Meglio L, Loyau S, Delbosc S, Boisseau W, Deschildre C, et al. Thrombus neutrophil extracellular traps content impair tpa-induced thrombolysis in acute ischemic stroke. Stroke. (2018) 49:754–7. doi: 10.1161/STROKEAHA.117.019896
63. Yu X, Diamond SL. Fibrin modulates shear-induced NETosis in sterile occlusive thrombi formed under haemodynamic flow. Thromb Haemost. (2019) 119:586–93. doi: 10.1055/s-0039-1678529
64. Spinthakis N, Gue Y, Farag M, Ren G, Srinivasan M, Baydoun A, et al. Impaired endogenous fibrinolysis at high shear using a point-of-care test in STEMI is associated with alterations in clot architecture. J Thromb Thrombolysis. (2019) 47:392–5. doi: 10.1007/s11239-018-01799-1
65. Kanji R, Gue YX, Farag MF, Spencer NH, Mutch NJ, Gorog DA. Determinants of endogenous fibrinolysis in whole blood under high shear in patients with myocardial infarction. J Am Coll Cardiol: Basic Transl Sci. (2022) 7:1069–1082. doi: 10.1016/j.jacbts.2022.05.007
66. Gorog DA, Lip GYH. Impaired spontaneous/endogenous fibrinolytic status as new cardiovascular risk factor? JACC Review Topic of the Week J Am Coll Cardiol. (2019) 74:1366–75. doi: 10.1016/j.jacc.2019.07.030
67. Okafor ON, Gorog DA. Endogenous fibrinolysis: an important mediator of thrombus formation and cardiovascular risk. J Am Coll Cardiol. (2015) 65:1683–99. doi: 10.1016/j.jacc.2015.02.040
68. Hosokawa K, Ohnishi-Wada T, Sameshima-Kaneko H, Nagasato T, Miura N, Kikuchi K, et al. Plasminogen activator inhibitor type 1 in platelets induces thrombogenicity by increasing thrombolysis resistance under shear stress in an in-vitro flow chamber model. Thromb Res. (2016) 146:69–75. doi: 10.1016/j.thromres.2016.09.002
69. Mangin PH, Gardiner EE, Nesbitt WS, Kerrigan SW, Korin N, Lam WA, et al. In vitro flow based systems to study platelet function and thrombus formation: recommendations for standardization: Communication from the SSC on Biorheology of the ISTH. J Thromb Haemost. (2020) 18:748–52. doi: 10.1111/jth.14717
70. Neeves KB, Onasoga AA, Hansen RR, Lilly JJ, Venckunaite D, Sumner MB, et al. Sources of variability in platelet accumulation on type 1 fibrillar collagen in microfluidic flow assays. PLoS ONE. (2013) 8:e54680. doi: 10.1371/journal.pone.0054680
71. Dolan JM, Sim FJ, Meng H, Kolega J. Endothelial cells express a unique transcriptional profile under very high wall shear stress known to induce expansive arterial remodeling. Am J Physiol Cell Physiol. (2012) 302:C1109–1118. doi: 10.1152/ajpcell.00369.2011
72. Diamond SL, Eskin SG, McIntire LV. Fluid flow stimulates tissue plasminogen activator secretion by cultured human endothelial cells. Science. (1989) 243:1483–5. doi: 10.1126/science.2467379
73. van Mourik JA, Lawrence DA, Loskutoff DJ. Purification of an inhibitor of plasminogen activator (antiactivator) synthesized by endothelial cells. J Biol Chem. (1984) 259:14914–21. doi: 10.1016/S0021-9258(17)42691-3
74. Kang S, Tanaka T, Inoue H, Ono C, Hashimoto S, Kioi Y, et al. IL-6 trans-signaling induces plasminogen activator inhibitor-1 from vascular endothelial cells in cytokine release syndrome. Proc Natl Acad Sci USA. (2020) 117:22351–6. doi: 10.1073/pnas.2010229117
75. Swiatkowska M, Szemraj J, Cierniewski CS. Induction of PAI-1 expression by tumor necrosis factor alpha in endothelial cells is mediated by its responsive element located in the 4G/5G site. FEBS J. (2005) 272:5821–31. doi: 10.1111/j.1742-4658.2005.04979.x
76. Chen C, Nan B, Lin P, Yao Q. C-reactive protein increases plasminogen activator inhibitor-1 expression in human endothelial cells. Thromb Res. (2008) 122:125–33. doi: 10.1016/j.thromres.2007.09.006
Keywords: fibrinolysis, plasminogen, shear, fibrin, platelets, thrombus, flow
Citation: Whyte CS and Mutch NJ (2022) “Going with the flow” in modeling fibrinolysis. Front. Cardiovasc. Med. 9:1054541. doi: 10.3389/fcvm.2022.1054541
Received: 26 September 2022; Accepted: 18 November 2022;
Published: 02 December 2022.
Edited by:
Henri Spronk, Maastricht University, NetherlandsReviewed by:
Diana Gorog, Imperial College London, United KingdomYvonne Henskens, Maastricht University Medical Centre, Netherlands
Moniek De Maat, Erasmus University Rotterdam, Netherlands
Copyright © 2022 Whyte and Mutch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Claire S. Whyte, Yy5zLndoeXRlQGFiZG4uYWMudWs=