
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med. , 07 November 2022
Sec. Cardiovascular Metabolism
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.1033457
This article is part of the Research Topic Insights in Cardiovascular Metabolism: 2022 View all 5 articles
 Shin-ichi Oka1
Shin-ichi Oka1 Karthi Sreedevi2
Karthi Sreedevi2 Thirupura S. Shankar3
Thirupura S. Shankar3 Shreya Yedla2Sumaita Arowa2Amina James2Kathryn G. Stone2
Shreya Yedla2Sumaita Arowa2Amina James2Kathryn G. Stone2 Katia Olmos2
Katia Olmos2 Amira D. Sabry3
Amira D. Sabry3 Amanda Horiuchi3Keiko M. Cawley3
Amanda Horiuchi3Keiko M. Cawley3 Sean A. O’very3Mingming Tong1
Sean A. O’very3Mingming Tong1 Jaemin Byun1Xiaoyong Xu1Sanchita Kashyap1Youssef Mourad1Omair Vehra1Dallen Calder3Ty Lunde3Tong Liu4Hong Li4
Jaemin Byun1Xiaoyong Xu1Sanchita Kashyap1Youssef Mourad1Omair Vehra1Dallen Calder3Ty Lunde3Tong Liu4Hong Li4 J. Alan Mashchek5
J. Alan Mashchek5 James Cox5,6Yukio Saijoh3Stavros G. Drakos3,7Junco S. Warren2,8,9,10*
James Cox5,6Yukio Saijoh3Stavros G. Drakos3,7Junco S. Warren2,8,9,10*Aims: PERM1 is a striated muscle-specific regulator of mitochondrial bioenergetics. We previously demonstrated that PERM1 is downregulated in the failing heart and that PERM1 positively regulates metabolic genes known as targets of the transcription factor ERRα and its coactivator PGC-1α in cultured cardiomyocytes. The aims of this study were to determine the effect of loss of PERM1 on cardiac function and energetics using newly generated Perm1-knockout (Perm1–/–) mice and to investigate the molecular mechanisms of its transcriptional control.
Methods and results: Echocardiography showed that ejection fraction and fractional shortening were lower in Perm1–/– mice than in wild-type mice (both p < 0.05), and the phosphocreatine-to-ATP ratio was decreased in Perm1–/– hearts (p < 0.05), indicating reduced contractile function and energy reserves of the heart. Integrated proteomic and metabolomic analyses revealed downregulation of oxidative phosphorylation and upregulation of glycolysis and polyol pathways in Perm1–/– hearts. To examine whether PERM1 regulates energy metabolism through ERRα, we performed co-immunoprecipitation assays, which showed that PERM1 bound to ERRα in cardiomyocytes and the mouse heart. DNA binding and reporter gene assays showed that PERM1 was localized to and activated the ERR target promoters partially through ERRα. Mass spectrometry-based screening in cardiomyocytes identified BAG6 and KANK2 as potential PERM1’s binding partners in transcriptional regulation. Mammalian one-hybrid assay, in which PERM1 was fused to Gal4 DNA binding domain, showed that the recruitment of PERM1 to a gene promoter was sufficient to activate transcription, which was blunted by silencing of either PGC-1α, BAG6, or KANK2.
Conclusion: This study demonstrates that PERM1 is an essential regulator of cardiac energetics and function and that PERM1 is a novel transcriptional coactivator in the ERRα/PGC-1α axis that functionally interacts with BAG6 and KANK2.
The heart is the muscle which never rests and consumes more energy than any other organ in the body. The healthy human heart hydrolyses greater than 6 kg of ATP per day, which is mainly utilized by myosin ATPase to generate force for contractility (1). Aberrations in energy production occur in chronic heart failure. Metabolic remodeling in the failing heart includes downregulation of fatty acid metabolism and mitochondrial dysfunction, which further aggravates cardiac contractile dysfunction in the failing heart (2).
Energy metabolism is controlled and fine-tuned by a complex plethora of transcription factors and their coactivators targeting expression of metabolic enzymes and transporters (3). Prominent among those is the Estrogen-Related Receptor Alpha (ERRα), a key transcription factor that coordinates the expression of genes related to oxidative phosphorylation, fatty acid oxidation, and muscle contraction in the heart (4). ERRα belongs to ERR subfamily that includes three isoforms: ERRα (Esrra), β (Esrrb), and γ (Esrrg). ERR is known as an “orphan” nuclear receptor, having no known ligands and requiring the interaction with co-activators for transactivation (5). ERRα and ERRγ are highly expressed in tissues with high energy needs including the heart. Both ERRα and ERRγ bind to an ERR-alpha response element (ERRE) containing a single consensus half-site, 5′-TNAAGGTCA-3′. ERRα and ERRγ are partly redundant in their regulatory function (6), but may play distinct roles in response to pathological stress in the heart (4).
The peroxisome proliferator-activated regulator gamma coactivator-1 (PGC-1) family of transcriptional coactivators plays a critical role in the regulation of mitochondrial biogenesis and energy metabolism in the heart (7, 8). Two members of this family, PGC-1α and PGC-1β, are highly expressed in cardiac muscle and play mostly redundant roles as co-activators of ERRs (9). There is large body of evidence supporting functional co-dependency between PGC-1 coactivators and ERR family, which have led to a notion of “ERR/PGC-1 axis” of metabolic regulation in a variety of biological contexts (3, 10).
The past few years have witnessed an interesting refinement to the concept of “ERR/PGC-1 axis” in the form of organ- or tissue- specific modulators of this regulatory network (11, 12). In particular, we found that the histone methyltransferase SMYD1, expressed predominantly in skeletal and cardiac muscle, positively regulates the expression of PGC-1α (13) as well as that of PERM1 (PGC- 1-, ERR-induced regulator, muscle 1) (14). PERM1 was initially identified as a new downstream target of PGC-1 and ERRs that regulates mitochondrial bioenergetics in skeletal muscle through an undefined mechanism (15). Similar to SMYD1, PERM1 is predominantly expressed in cardiac and skeletal muscle (15). Thus, it seems that both SMYD1 and PERM1 modulate the ERR/PGC-1 regulatory axis to meet the unique energy demands in organs translating energy to mechanical work. Downregulation of the ERR/PGC-1 regulatory axis is an important component of the pathobiology of heart failure (3). As the first indication of the relevance of PERM1 to this highly prevalent disease, we have showed that PERM1 is downregulated in the human and mouse failing hearts, and that silencing of PERM1 in cultured cardiomyocyte leads to reduced mitochondrial respiration capacity (14).
Here we embarked on a comprehensive analysis of the specific role of PERM1 in regulation of cardiac metabolism, and the molecular mechanism of this regulation. The results presented here reveal broad and complex influence of PERM1 on substrate metabolism which appears to be essential for maintenance of full pumping capacity of the heart. Furthermore, we demonstrate that PERM1 acts as a novel transcriptional coactivator forming transcriptional complex with ERRα and PGC-1α, as well as transcription regulators BAG6 and KANK2 revealed as PERM1 binding partners through unbiased mass-spectrometry analysis (12).
All animal experiments were approved by the University of Utah and the Rutgers New Jersey Medical School Institutional Animal Care and Use Committee and conducted according to the Guide for the Care and Use of Laboratory Animals (The National Academy Press (16) 8th edition, 2010).
All animal experiments were approved by the University of Utah and the Rutgers New Jersey Medical School Institutional Animal Care and Use Committee and conducted according to the Guide for the Care and Use of Laboratory Animals (The National Academy Press (16) 8th edition, 2011). Perm1-KO mice were generated by Crispr/Cas9 system with gRNA targeting at the exon 2 in Sadoshima lab at the Rutgers New Jersey Medical School. The Perm1-KO allele lacks 1,157 bp in exon 2 and induces frame shift and an artificial stop codon. Genotyping of WT and mutant mice was performed using the primers, which target the Crispr target region (see Supplementary Methods for primer sequence). The heterozygous and homozygous mice and their littermates at the age of 10–12 weeks were used in this study. Echocardiography was performed on a Vevo 2100 (Visual Sonics) to determine cardiac parameters in live mice as previously described (14). Tissue samples from ventricles were immediately frozen in liquid nitrogen and stored at −80°C until they were used for analysis.
The heterozygous and homozygous mice and their littermates at the age of 9–15 weeks were used in this study. Mice were anesthetized (2% isoflurane mixed with 0.5 L/min 100% O2) in the induction chamber prior to the imaging studies. All echocardiographic analysis of those mice were performed using a Vivo 2100 (Visual Sonics) at the University of Utah as previously described (17).
Constriction of the transverse thoracic aorta (TAC) was performed on 3-month-old WT male mice at the Rutgers New Jersey Medical School, as previously described (14, 18). Thirty minutes before surgery a single dose of ZooPharm Buprenorphine SR (sustained release) (0.15 mg/kg; 72 h efficacy) was administered subcutaneously. Mice were, then, anesthetized with isoflurane (2% in O2) through nosecone, which was delivered from a VetFlo Vaporizer. Lidocaine (7 mg/kg) was introduced under the skin at the incision site. During recovery, if the animal was showing signs of distress, buprenorphine (regular release) was administered at 8-h intervals for up to 48 h as needed. In addition to buprenorphine SR, the NSAID carprofen (5 mg/kg) was given subcutaneously after induction of anesthesia and redosed once every 24 h for 72 h post-surgery. Mice were sacrificed 1 week after TAC surgery to collect cardiac tissue. Unbanded WT mice (sham) were used as controls.
Whole hearts were rapidly excised from WT and Perm1-KO mice at the age of 10 weeks and were fixed in 4% paraformaldehyde and then embedded in paraffin. Hearts were then sectioned at 4 μm and placed on slides. Masson’s trichrome (Sigma) staining was performed according to the manufacturer’s protocols. Tissue sections were visualized with imaged on Nikon eclipse TE2000-U microscope with SPOT software (Diagnostic Instruments).
Primary cultures of ventricular cardiac myocytes were prepared from 1-day-Crl:(WI) BR-Wistar rats (Harlan) as previously described in our publication (14). Briefly, a cardiac myocyte- and fibroblast-rich fraction was obtained by centrifugation through a discontinuous Percoll gradient. Cells were cultured in complete medium containing Dulbecco’s modified Eagles’s medium/F12 supplemented with 5% horse serum, 4 μg/ml transferrin, 0.7 ng/ml sodium selenite, 2 g/l bovine serum albumin (fraction V), 3 mM pyruvate, 15 mM Hepes pH 7.1, 100 μM ascorbate, 100 mg/l ampicillin, 5 mg/l linoleic acid, and 100 μM 5-bromo-2′-deoxyuridine (Sigma). Culture dish were coated with 0.3% gelatin. For silencing of genes, small interfering RNA (siRNA) were used, which were obtained from Qiagen.
Mammalian expression vector for Perm1 (pDC316-Perm1) and luciferase reporter gene driven by 3 repeats of ERRE (3xERRE-luc) were described previously (14). C-terminal Flag tagged PERM1 were generated with pDC316 (pDC316-Perm1-Flag). Gal4 DNA binding domain fused Perm1 (pFA-Perm1) was generated with insertion of Perm1 cDNA into pFA-CMV (Agilent).
Adenoviruses harboring Perm1-Flag were made using the AdMax system (Microbix) with the shuttle vectors pDC316- Perm1-Flag.
ATP, ADP, AMP, PCr, creatine, and NAD+ levels were determined using Shimadzu HPLC system as described in our previous work (19, 20). The HPLC system was composed of CMB-20Lite, LC-20D HPLC pump, micro mixer MR100, SPD-20 UV detector, DGU-403 3-channel degasser, and RT-7725I manual injector with a fixed loop of a 20 μl capability. For the preparation of samples, approximately 10 mg of frozen tissue was homogenized in 120 μl of 0.4 M perchloric acid using a ceramic bead tube kit (MO BIO Laboratories, Inc., Carlsbad, CA, USA) and Bead Ruptor (Omini International, Kennesaw, GA, USA), which provides ultra-rapid shaking. After precipitation with 1 M KOH, the extracts were centrifuged for 3 min at 4°C two times (13,000 × g) Twenty microliters of the extracted sample was manually injected to the Shimadzu HPLC system.
The chromatographic separation of high-energy phosphates and relative metabolites were performed using a C18 reverse-phase column YMC-Pack Pro C18 (5 μm, 120 Å, 150 mm × 4.6 mm I.D.). The mobile phase was composed of 215 mM potassium dihydrogen phosphate, 2.3 mM TBAHS, 4% acetonitrile, 0.4% potassium hydroxide (1M), and the flow rate was set at 1 ml/min. The sample injection volume was 20 μl and the components were monitored at 210 nm (for creatine and PCr) and 254 nm (adenine nucleotides) during isocratic acquisition. All instruments and the columns were operated at room temperature (22–25°C). Standard stock solutions were freshly prepared on the day of experiments (ATP, ADP, AMP, PCr, creatine, and NAD+, all from Sigma, St Louis, MO, USA), and dilution was made by adding mobile phase immediately before each assay, which was used as references for peaks quantification. The cocktail of these standards was run before and after actual samples to make sure that the system performance did not decline.
Proteomic analysis of ventricular tissue collected from Perm1–/– mice and WT littermates at the age of 10 weeks were conducted using label-free quantitative LC-MS/MS, as described in our previous publication (21), with some modifications.
Ventricular samples from WT and Perm1–/– hearts were analyzed using two metabolomic platforms [GC-TOF MS and hydrophilic interaction chromatography (HILIC)-quad-time of flight (QTOF)/MS] for untargeted metabolomic screening, as previously performed (13).
Agilent 6490 triple quadrupole (QqQ) mass spectrometry was used to perform lipidomic profiling of WT and Perm1–/– hearts, as described in our previous publication (22).
Principal component analysis (PCA), and the analyses of heart maps, fold-change, and volcano plots were carried out using the online software Metaboanalyst (23). Enrichment analyses and the generation of network maps of proteome were carried out using the online software STRING database.
Perm1-Flag was expressed in primary cultured cardiomyocytes with adenovirus vector (10 cm dish), followed by immunoprecipitation with anti-Flag antibody (Sigma, A2220). The immune-complex was subjected to SDS-PAGE followed by mass spectrometric analysis.
Heart tissue was cross-linked with formaldehyde. The nuclear fraction was isolated and sonicated to generate a chromatin solution that was then used for immunoprecipitation. Primers used for investigating mouse genome are as follows: (5′ to 3′). Cpt2–CCCTGTGGGCGGAGTTGAACT and ATTTTGTCCGTGACCTTCGCGC; Mcad–ATCTAGCCCAGAATTTGTTGTTCCAGTG and GCGGTGGCTGAGGGAGTTCC; Idh3a–TGATGGATGGCGGAGGCGAGC and TGGGGACACCCGGAGCAGTAC; Sdhd–CCTTCCGGTTCACGCTTCAGGT and TCGCTTTCGAGGGCTCAAGGT; ERRα–CTCGTGTCTCACCTCTGCCTTT and GACACTTGACAGATTTGGTAGATCAG; Ndufv1–GCTCCCCACCCCGTGTGTTT and GGGAGCCAGACGGGTCACCT; and Ndufs1–CTTATATAGAGACGGAGGCGTTTC and GACGACCTGTGGGCCCTTTAGTTA. The corrected chromatin fragment was validated by qPCR with Maxima SYBR Green qPCR master mix (Fermentas).
DNA binding assay using the Idh3a promoter was performed in cultured neonatal rat cardiomyocytes, as described in our previous publication (18) with some modifications. Briefly, biotin-labeled Idh3a promoter (380 bp) was prepared by PCR with a biotin-labeled primer. Endogenous ERRα was knocked down in primary cultured cardiomyocytes by siRNA transfection. After 2–3 days of transfection, the cells were lysed with 300 μl of a nuclear lysis buffer (25 mM Hepes, pH7.9, 0.1 mM EDTA, 10 mM Sodium Butyrate, 10 mM NaF, 1 mM DTT, 1% NP-40, 420 mM KCl, 1 × Proteinase inhibitor cocktail). The 100 μl lysate was diluted with 300 μl of a dilution buffer (10 mM Hepes, pH 7.9, 2.5 mM MgCl2, 5% Glycerol, 1 mM DTT, 0.1% NP-40). The total 400 μl of diluted lysate was incubated with 1 μg biotin-labeled DNA (Idh3a promoter) and streptavidin-beads at 4°C for 2–4 h with rotation. The protein-DNA complex was washed with 1 ml of a washing buffer (10 mM Hepes, pH 7.9, 2.5 mM MgCl 2, 5% Glycerol, and 50 mM KCl 1 mM DTT, 0.1% NP-40) for 3 times. The protein bound to the DNA was analyzed with Western blot analyses.
Luciferase assays were performed in primary cultured rat cardiac myocytes as previously performed (13). Briefly, reporter plasmids (0.3 μg per well), including 3xERRE-luc and UAS-luc, mammalian expression vectors (0.7 μg per well), including pDC316 control vector, pDC316-Perm1 and pFA-Perm1, and siRNA (20 pM) against PGC-1α, ERRα, Ankrd1, Bag6, Kank2, and TIF1β, were transfected into cells plated in 12 well plates using LipofectAmine 2000 (Invitrogen). Total plasmids were kept at 1 μg per well using pDC316 control vector. The total siRNA was kept 20 pM per well using control siRNA. Two μl LipofectAmine 2000 was used per well. Fifty μl OPTI-MEM was used for dilution of plasmid vectors and of LipofectAmine 2000, respectively. Luciferase assays were performed 2 days after transduction with a luciferase assay system (Promega).
Conventional electron microscopic analyses were performed with heart tissue from WT and Perm1–/– mice. Morphometric analysis of mitochondrial cristae and quantification of density and disorganized mitochondrial cristae were performed as previously demonstrated (24).
Total protein lysates (10–30 μg) from mouse left ventricle tissue were incubated with SDS sample buffer at 95°C for 5–20 min. These denatured protein samples were transferred to either polyvinylidene difluoride or nitrocellulose membranes and probed with antibodies against Perm1 (Sigma HPA032711), ERRα (Millipore ERR46Y), PGC-1α (Millipore Ab3242), Ankrd1 (Novus NBP2-15397), BAG6 (Cell Signaling Technology 8523), Kank2 (Novus NBP3-04645), TIF1b (Cell Signaling Technology 4123), and Trx1 (Cell Signaling Technology 2429). The complete methods are in Supplementary material online.
PERM1 and ERRα were immunoprecipitated from mouse heart lysate with anti-PERM1 (Sigma HPA032711) and anti-ERRα antibody (PPMX PP-H5844-00). Protein A/G agarose (Santa Cruz sc-2003) was used.
Real-time PCR was performed with either Taqman method or the SYBR green method on a CFX96 Real-Time PCR system (Bio-Rad) as described previously (13, 22). Briefly, RNA was extracted using TRizol (Life Technologies), followed by ethanol extraction. Reverse transcription was performed from 1 μg of RNA using QuantiTect Reverse Transcription Kit according to the manufacture’s instruction (Qiagen). Relative expression was calculated using the ΔΔCt method with normalization to either Tbp or β-actin. The complete methods and a list of primers that were used in this study are in Supplementary material online.
For comparisons between WT, Perm1± and Perm1–/–, one-way ANOVA with Fisher’s LSD post-hoc test was applied using GraphPad Prism version 9. Student’s T-test was used to compare WT vs. Perm1–/– mice. A value of p < 0.05 was considered statistically significant. Data are given as mean ± SEM.
Our previous study showed that PERM1 is downregulated in the human and the mouse failing heart (14). To determine whether downregulation of PERM1 can be associated with cardiac dysfunction, we generated mouse strains deficient in Perm1 (Perm1–/– mice) using the Crispr/Cas9 genome editing technique. We targeted exon 2 to delete 1,157 bp (Figure 1A), leading to a large deletion after amino acid 27 in the Perm1 locus (Figure 1B). Perm1± and Perm1–/– mice were obtained from offspring of heterozygous breeding pairs. Genotypes of heterozygotes and homozygotes were confirmed by hybridization and PCR analysis of genomic DNA (Figure 1C). Consistent with previous studies, we detected two isoforms of PERM1 at 100 kDa and 90 kDa, both of which significantly decreased in Perm1± hearts (44.3 and 20.2% of WT in 100 and 90 kDa, respectively, both p < 0.05, Figure 1D). PERM1 was undetectable in Perm1–/– hearts (Figure 1D). Pups lacking Perm1 were born at the expected Mendelian ratio (20.3, 54.2, and 25.4% in WT, Perm1± and Perm1–/– mice), suggesting that PERM1 is dispensable for embryonic development. Echocardiographic analysis revealed decreased cardiac function in Perm1–/– mice. Ejection fraction (EF) and fractional shortening (FS) were reduced in Perm1± and Perm1–/– mice as compared with WT littermates, proportionally to the protein levels of PERM1 in the heart (EF: 50.1 ± 1.9 vs. 42.8 ± 2.86 vs. 34.7 ± 1.38%, FS: 24.8 ± 1.18 vs. 20.7 ± 1.76 vs. 16.0 ± 0.89% in WT, Perm1± and Perm1–/–, respectively; Figures 1E,F and Supplementary Table 1, n = 15 per group). The differences in EF and FS were significant between Perm1–/– and WT mice (both p < 0.05), but not between Perm1± and WT mice. There were no differences in heart-to-body weight ratios, (right panel in Figure 1F), cell size (Figure 1H), and gross-anatomy of the heart (Figure 1G) between Perm1–/– and WT mice. Interestingly, Perm1–/– mice exhibited a significant increase in the mRNA level of atrial natriuretic factor (ANF) (Figure 1I), which is known to be upregulated in the failing heart (25), but the related biomarkers α-MHC and β-MHC were unchanged.. In summary, our data suggest that the expression level of PERM1 in the heart is correlated to cardiac function. Thus, PERM1 is essential for maintaining the normal cardiac function.
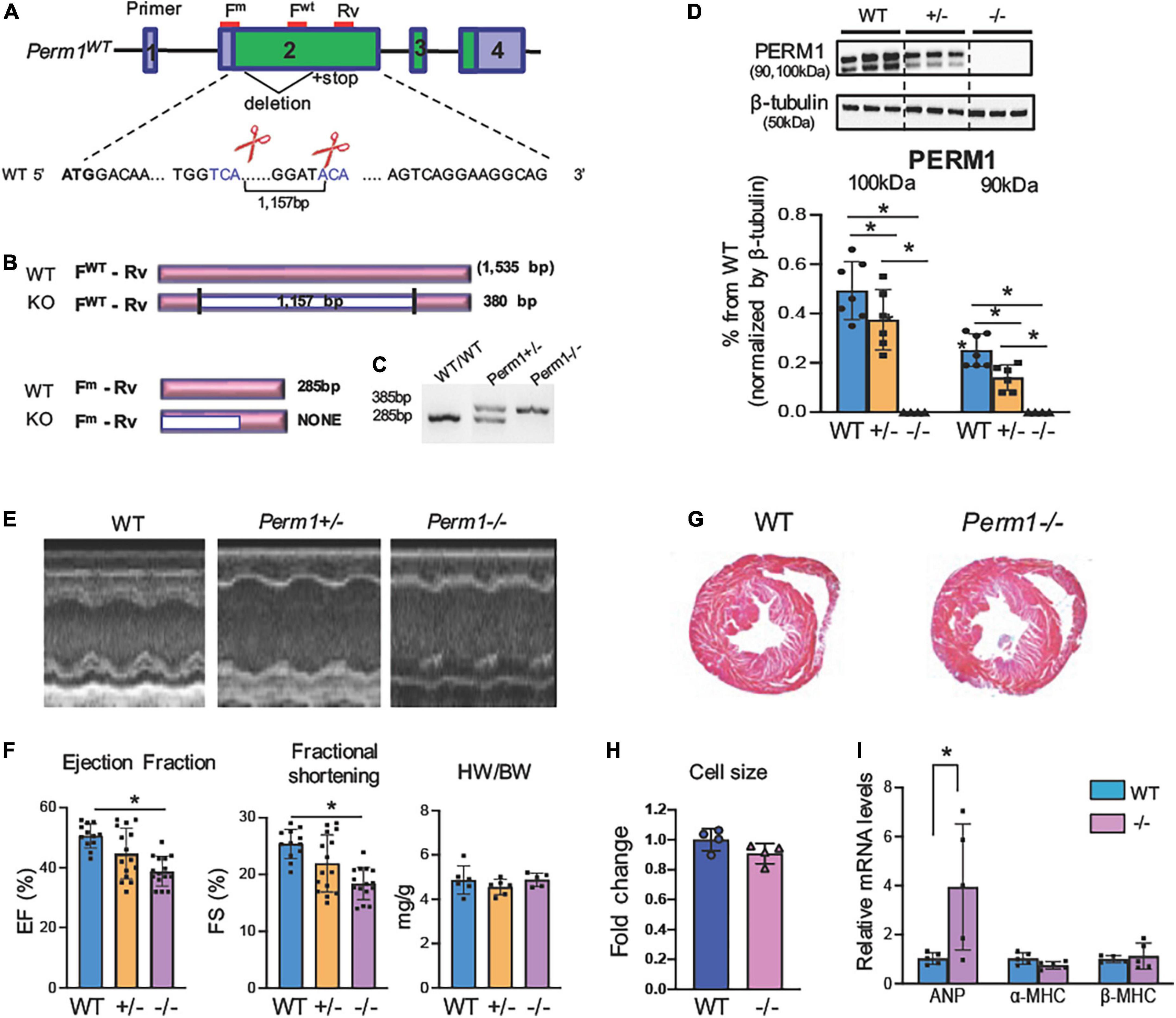
Figure 1. (A) Perm1–/– (KO) mice exhibit reduced cardiac function (age, 3 months). (Schematic of Perm1 gene and the CRISPR/Cas9-mediated gene editing in Perm1 loci in C57BL/6 mice. Exon 2 was targeted, where the primers for genotyping are indicated in red lines (FWT + Rv for WT; Fm + Rv for mutant). (B) Schematic of PCR products from WT and CRISPR/Cas9 mice (KO). PCR with the primer set, FWT + Rv, distinguishes WT (not detected) from either Perm1± or Perm1–/– (360-bp band), while PCR with the primer set, Fm + Rv, distinguishes WT (285 bp) from Perm1± or Perm1–/– mice (not detected). (C) Genotyping of WT, Perm1± and Perm1–/– mice using those primers. (D) Western blotting analysis showing the reduced endogenous levels of PERM1 in Perm1± and Perm1–/– hearts (n = 6/group). (E) Sample 2D echocardiographs from WT, Perm1± and Perm1–/– mice. (F) Ejection fraction (EF), fraction shortening (FS), and the heart weight (mg)–body weight (g) ratios (HW/BW) of WT, Perm1± and Perm1–/– mice (n = 15/group). (G) Low-magnification views of hematoxylin/eosin stains of transverse sections of WT and Perm1–/– hearts. (H) Quantification of cell size in WT and Perm1–/– hearts (n = 5/group). (I) qPCR of ANF, α-MHC, and β-MH in WT and Perm1–/– hearts (n = 5/group). *p < 0.05.
We assessed the levels of high-energy phosphates in myocardium using HPLC (Figure 2). The upper three panels (A–C) show the representative chromatograms in WT, Perm1± and Perm1–/– mice, respectively. The levels of phosphocreatine (PCr) were significantly decreased in Perm1± and Perm1–/– hearts (p < 0.05 in all comparison, Figure 2D). The creatine (Cr) content in Perm1–/– hearts was significantly increased as compared to WT hearts, but there was no change in Perm1± hearts (Figure 2E). The total creatine pool (Cr + PCr) was not statistically different among WT, Perm1± and Perm1–/– hearts (Figure 2F). There was no significant difference in the levels of ATP, ADP, AMP, total adenine nucleotide pool (ATP + ADP + AMP, Figure 2K), and the ADP/ATP ratio among the three groups (Figure 2G). There was a trend that the NAD level was increased in Perm1–/– heart (p = 0.058, Figure 2M). Importantly, the PCr/ATP ratio, a sensitive indicator of the energy state, was significantly decreased in both Perm1± and Perm1–/– hearts (p < 0.05 in all comparisons, Figure 2H). Thus, like cardiac function, the myocardial energy reserve was decreased proportionally to the expression levels of PERM1 in the heart.
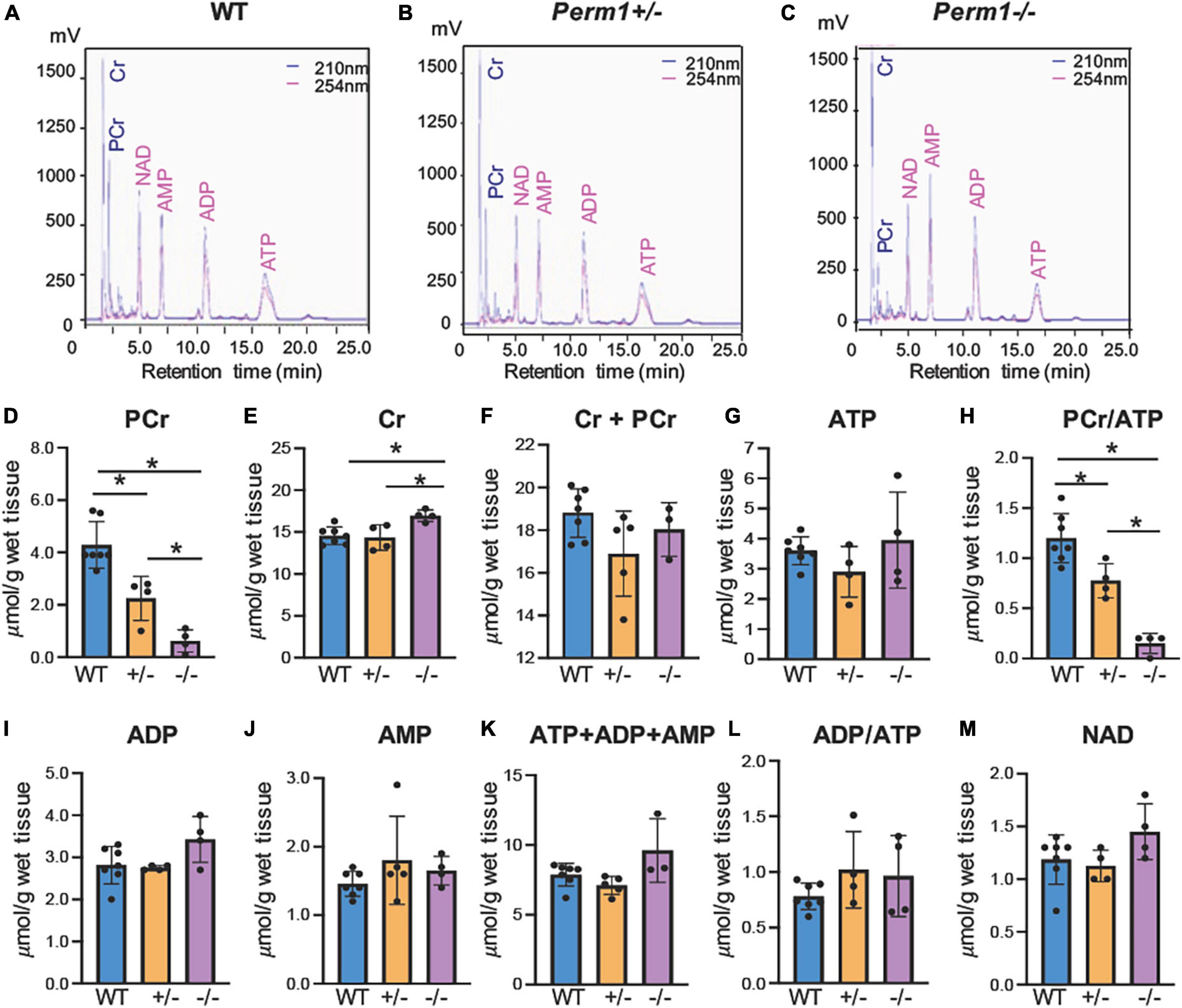
Figure 2. Quantification of high-energy phosphates and related adenine nucleotides. Metabolites were measured by HPLC in ventricular tissue from WT, Perm1± and Perm1–/– mice. Chromatograms of extracts from WT (A), Perm1± (B) and Perm1–/– hearts (C). (D–M) Quantitative analysis of the levels of creatine (Cr), phosphocreatine (PCr), ATP, ADP, AMP, and NAD+, respectively, normalized by wet tissue weight. *p < 0.05.
To investigate biological pathways regulated by PERM1 in the heart, we performed LC-MS/MS-based proteomic analysis of WT and Perm1–/– hearts (n = 4 per group). The PCA of the total of 2,868 detected proteins (Supplementary Dataset 1) separated WT and Perm1–/– groups (Figure 3A), suggesting altered proteomic profile of Perm1–/– hearts. A heat map shows 527 proteins that are differentially expressed in Perm1–/– hearts (207 downregulated, 320 upregulated, p < 0.05, Figure 3B). Using the STRING Database web-based tools (26), the 10 most significant enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways are shown in Figures 3C,D (enrichment of upregulated proteins in Figure 3C, enrichment of downregulated proteins in Figure 3D). We found that the most upregulated metabolic pathways were annotated into the ribosome and glycolysis pathways (Figure 3C, Supplementary Dataset 2). Gene ontology (GO) terms for cellular component showed that many of upregulated proteins Perm1–/– hearts were localized in the cytoplasm (Supplementary Figure 1A and Supplementary Dataset 3). Of note, it has been reported that ribosomal protein expression is increased in response to oxidative stress (27), while upregulation of glycolysis occurs in the progression of heart failure, concomitant with downregulation of OXPHOS (28). The two most enriched KEGG pathways of downregulated proteins in Perm1–/– hearts are the wide, encompassing pathway terms of metabolic pathways and OXPHOS (Figure 3D, Supplementary Dataset 4), and at the top of the list for cellular component terms of downregulated proteins is mitochondria (Supplementary Figure 1B and Supplementary Dataset 5). To get the insights into the downregulation of metabolic pathway in the enrichment analysis, the interaction network view of the 82 significantly altered proteins within the metabolic pathways KEGG term was generated (Figure 3E). Note that highly interconnected clusters include proteins involved in OXPHOS (pink), TCA cycle (green), pyruvate metabolism (light blue), branched-chain amino acid (BCAA) metabolism (red), ubiquinone (blue), and propanoate metabolism (yellow). The comparative analysis of the 35 downregulated OXPHOS proteins in the enrichment analysis of downregulated proteins (Figure 3D) is depicted in heat maps (Figure 3F), displaying the protein names and fold changes.
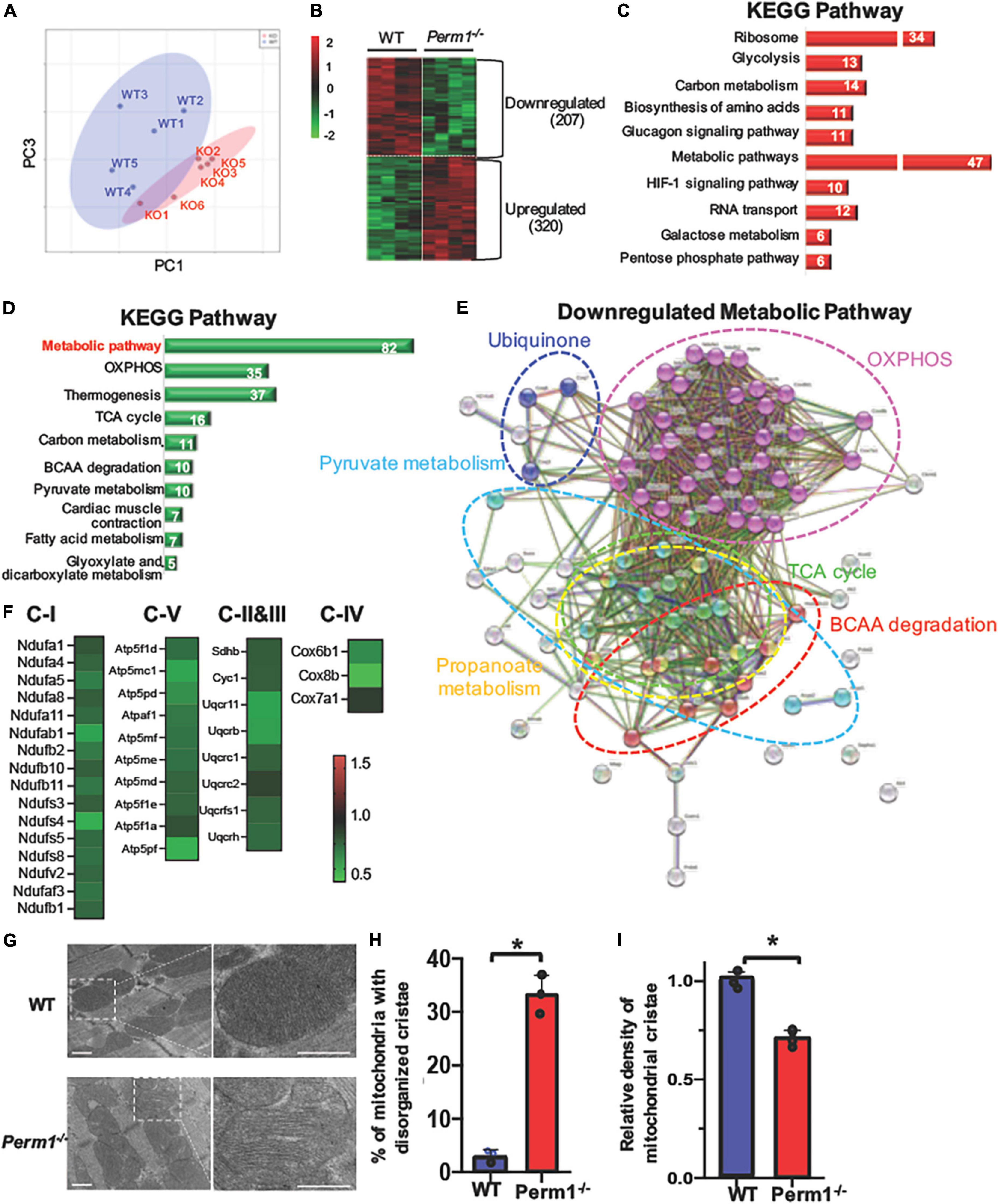
Figure 3. Proteomic analysis of left ventricle tissue from Perm1–/– mice and WT mice (n = 5/group). Among 2,868 detected proteins, 207 proteins were significantly downregulated and 320 proteins were significantly upregulated in Perm1–/– hearts (p < 0.05 by t-test). (A) PCA of all detected proteins from Perm1–/– (red) and WT hearts (blue). (B) Heat map of the proteins that were differentially expressed in Perm1–/– heart. Enrichment analyses of upregulate (C) and downregulated proteins (D) for KEGG pathways. The white numbers in bar graphs indicate the number of proteins that were detected in each pathway. (E) Network map of the 82 significantly changed proteins which belong to the collective KEGG term metabolic pathways in panel (D). (F) Heat maps of fold changes from the 35 proteins comprising the network map presented in panel (E). Analysis of cardiac ultrastructure with representative electron microscopy images (G), quantification of changes in mitochondrial cristae morphology (H), and quantification of mitochondrial cristae density (I) of hearts of WT mice (blue bars, n = 3) and Perm1–/– mice (red bars, n = 3). *p < 0.05 (student’s t-test), ± SED.
Electron microscopic analysis of PERM1-null hearts showed that the density of mitochondrial cristae was remarkably reduced, concurrent with increased disorganization as compared to WT hearts (Figures 3G–I). Overall, these results suggest that PERM1 deletion leads to downregulation of OXPHOS proteins, upregulation of glycolysis, and altered mitochondrial morphology, all of which resembles metabolic remodeling in the failing heart (8, 29).
To examine if the changes in protein expression (Figure 3) alter myocardial metabolic contents, we performed GCMS-based metabolomics and LC/MS-based lipidomics in the hearts from WT and Perm1–/– mice. Metabolomics (n = 6 per group) detected 115 metabolites (Supplementary Dataset 6). The PCA plot of metabolome clearly separates the WT and Perm1–/– groups (Figure 4A). The volcano plot in Figure 4B, where the x-axis indicates fold changes in log-2 scale and the y-axis indicates p-values in -log10 scale, highlights the increased levels of the intermediates of glycolysis and polyol pathways (sorbitol; fructose-6-phosphate; fructose, indicated in red, all p < 0.05) and the decreased levels of free fatty acids (FFAs) and monoglycerides (MGs) (palmitic acid; stearic acid; elaidic acid; 1-stearoylglycerol; 1-palmitoylglycerol, indicated in green, all p < 0.05). Heat maps in Figure 4C summarize the changes in glucose metabolites, FFAs, and MGs in Perm1–/– hearts. Lipidomics (n = 5 per group) detected 394 lipids, including carnitines (CAR), ceramides (Cer), cholesterol ester (CE), diglycerides (DG), phosphatidylcholines (PC), phosphatidylethanolamine (PE), sphingomyelin (SM), and triglycerides (TG) (Supplementary Dataset 7). Similar to metabolome, the PCA plot of these lipids clearly separated WT and Perm1–/– hearts (Figure 4D). The detected lipids were further grouped into the abovementioned classes using the LIPID MAPS classification system (30), and the total levels of lipids in each class were analyzed using box and violin plots (Figure 4E), which revealed global decrease of TG and ceramides (Cer), and an increase of cholesterol ester (CE), in Perm1–/– hearts as compared with WT hearts.
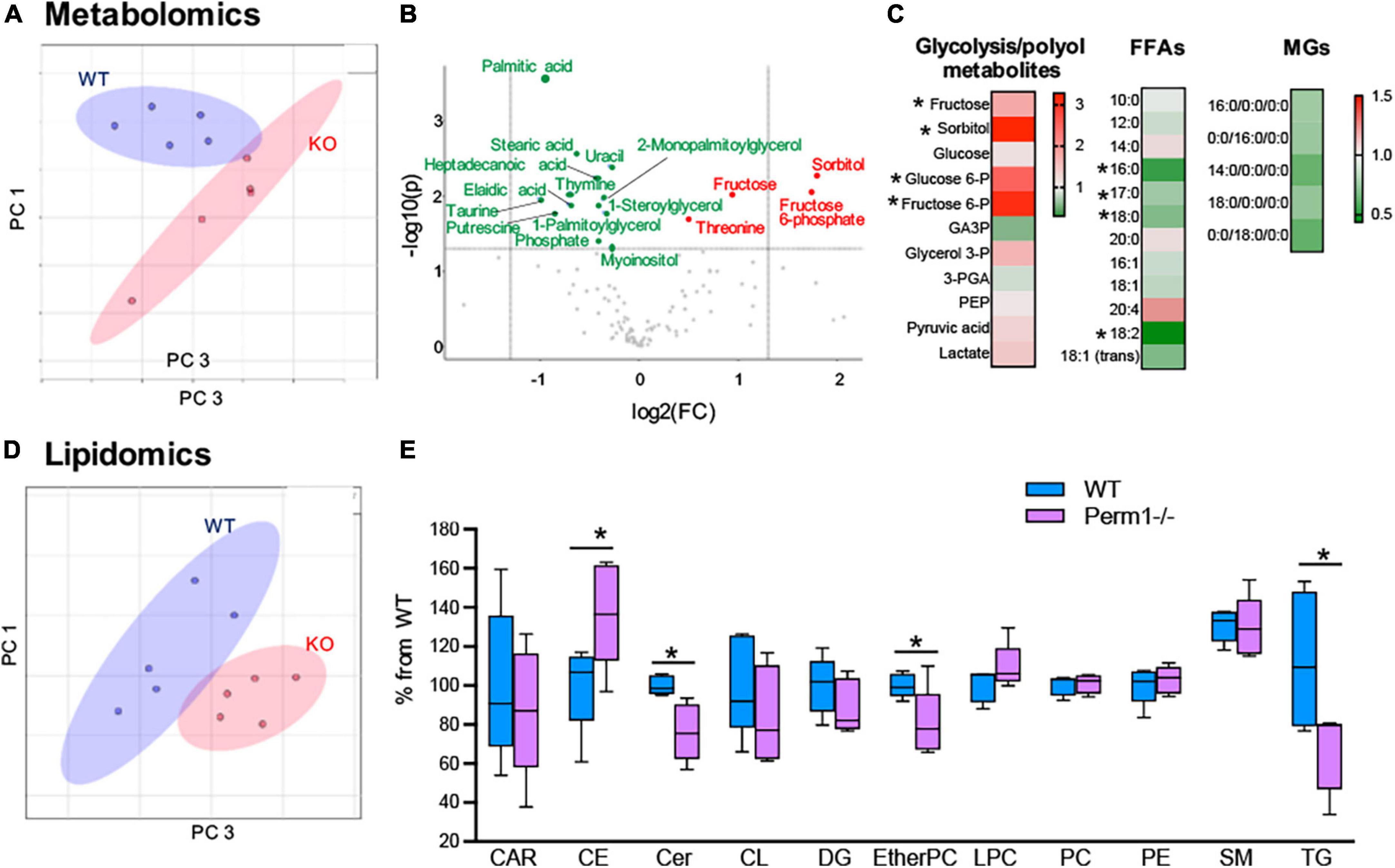
Figure 4. Metabolomic and lipidomic analyses of left ventricle tissue from Perm1–/– mice and WT mice (n = 6/group in metabolomics and n = 5/group in lipidomics). (A–C) Metabolomic profile of Perm1–/– hearts. (A) PCA of all detected metabolites in Perm1-null hearts (red) and WT hearts (blue). (B) Volcano plot contains the significantly increased (red) and decreased (green) metabolites in Perm1–/– hearts compared with those in WT hearts (p < 0.05). X-axis indicates fold change (FC) in log2 scale and y-axis indicates p-value in -log10 scale. (C) Heat maps of metabolites in glycolysis and polyol pathways, and free fatty acids (FFAs) and monoglycerides (MGs). (D,E) Lipidomic profile of Perm1–/– hearts. PCA of all detected lipids in Perm1–/– (red) and WT hearts (blue) (D), and box and violin plot of the total lipid species in each lipid group (E). *p < 0.05.
For a better understanding of the changes in the substrate metabolism due to Perm1 deletion, we created an integrative view combining all metabolomic, lipidomic, proteomic, and gene expression data yielded by this study (Figure 5). As shown in Figure 5A, the accumulation of glycolytic intermediates (glucose-6-P; fructose-6-P, and lactate) was associated with global upregulation of glycolytic enzymes (GPI; PFK1; PGM; PLM, all p < 0.05, Figures 5A,B). qPCR analysis showed that both GLUT1 and GLUT4 were upregulated in Perm1–/– mice (both p < 0.05, Supplementary Figure 2A). Upregulation of lactate dehydrogenase (LDHA, p < 0.05) concomitant with downregulation of the pyruvate dehydrogenase complex (ODHB; PDHX; DLAT; DLD, Figure 5C) and the increased myocardial level of lactate (FC = 1.43, p = 0.088) suggest the increased reliance on anaerobic metabolism in Perm1–/– hearts. We must note that the increased levels of sorbitol and fructose in Perm1–/– hearts (FC = 3.28, p < 0.05) were not associated with a significant change in the protein expression of aldose reductase (AR, AKR1B10) and sorbitol dehydrogenase (SDH, SORD), which catalyze glucose to sorbitol and further to fructose, respectively (FC = 1.01 and 1.21, p = 0.171 and 0.174, respectively, Figure 5A). It is worth noting that one of the most significantly upregulated glycolytic enzymes in Perm1–/– hearts was glycerol-3-P dehydrogenase (GPD1, FC = 1.414, p = 0.01, Figures 5A,B), which serves as a major link between glycolysis and lipid biosynthesis. The initial step of glycerolipid biosynthesis is catalyzed by glycerol-3-P acyltransferase (GPAT), which esterify fatty acid acyl-CoA (acyl-CoA) to glycerol-3-phosphate, yielding lysophosphatidic acid (LPA). Our proteomic data showed that GPAT1 was significantly upregulated in Perm1–/– hearts (Figure 5F), concomitant with upregulation of 1-acylglycerol-3-phosphate acyltransferase (AGPAT) (Figures 5A,F), the latter catalyzing the acylation of LPA to form phosphatidic acid (PA). Taken together, these data suggest that the glycerolipid biosynthesis was promoted in Perm1–/– hearts. However, despite the upregulation of the enzymes responsible for glycerolipid biosynthesis (GPD; GPAD; AGPAT), the myocardial levels of TGs, a major energy storage in cardiomyocytes, were reduced in Perm1–/– hearts (Figures 4E,F, 5A), indicating the imbalanced input and output in the TG pool. We did not observe significant changes in the protein levels of enzymes involved in lipid breakdown from TGs in Perm1–/– hearts (ACS; MGL, Figures 5A,E). Importantly, the key enzymes involved in fatty acid β-oxidation (FAO) were consistently downregulated in Perm1–/– hearts (ACADM; ACAA2; HADH; ECHS1, Figure 5D). qPCR data showed that the mRNA levels of CD36, which facilitates fatty acid uptake into muscle cells, were significantly decreased in Perm1–/– hearts, whereas there was no significant difference in the mRNA levels of CPT1 and CPT2, which are responsible for fatty acid uptake into mitochondria for FAO (FC = 0.84 and 0.96, respectively, both p > 0.05, Supplementary Figure 2B). Of note, qPCR analysis also showed downregulation of the OXPHOS genes Idh3a and Ndufv1 in Perm1–/– mice, which are consistent with our previous study in cardiomyocytes (both p < 0.05, Supplementary Figure 2C).
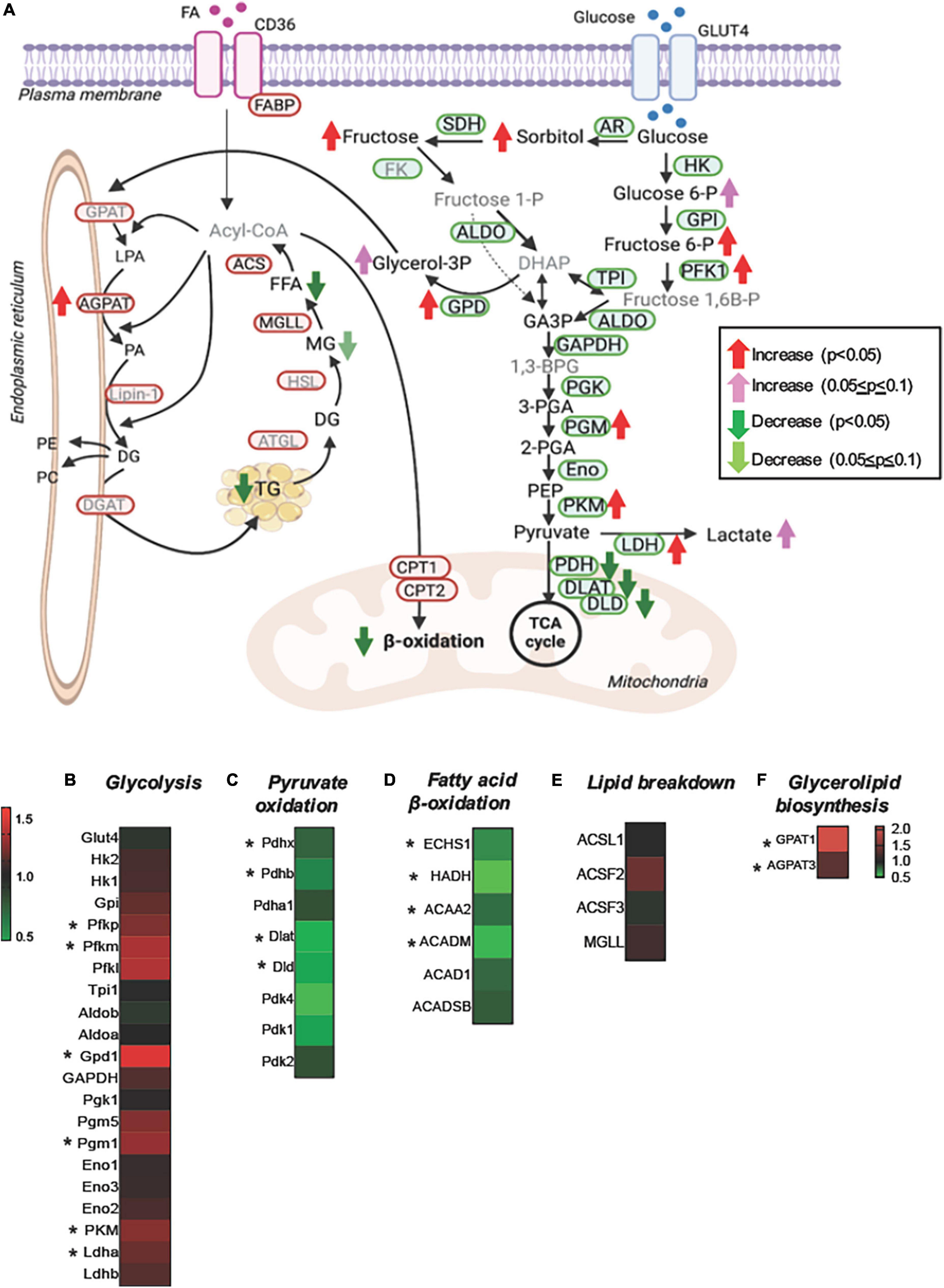
Figure 5. Integrated metabolomic and lipidomic analyses with proteomic data of Perm1–/– hearts. (A) Schematics of glucose and lipid metabolism pathways, in which the altered levels of metabolites, lipids, and proteins are indicated by arrows. Detected metabolites and proteins are indicated in black fonts, and undetected metabolites and proteins are indicated in gray fonts (created using BioRender). Heat maps of the detected proteins involved in glycolysis (B), pyruvate oxidation (C), fatty acid β-oxidation (D), lipid breakdown (E), and glycerolipid biosynthesis (F). Glucose-6-P:glucose-6-phosphate, fructose-6-P:fructose-6-phosphate, fructose-1-P:fructose-1-phosphate, fructose-1,6B-P: fructose-1,6-bisphosphate, DHAP:dihydroxyacetone phosphate, GA3P:glyceraldehyde-3-phosphate, glycerol-3P:glycerol-3-phosphate, 1,3-BPG:1,3-bisphosphoglycerate, 3-PGA:3-phosphoglycerate, 2-PGA:2-phosphoglycerate, PEP, phosphoenolpyruvate; FFA, free fatty acid; MG, monoglyceride; DG, diglyceride; LPA, lysophosphatidate; PA, phosphatidate; PE, phosphatidylethanolamine; PC, phosphatidylcholines. *p < 0.05.
Summarizing, the unique metabolic profile induced by Perm1 deletion combines evidence for reduced lipid utilization and storage, partially upregulated glycerolipid biosynthesis, accumulation of glycolytic and polyol intermediates, and downregulation of pyruvate oxidation.
We previously demonstrated that Perm1 activates transcription through ERR response element (ERRE) (14) where ERRα binds for the transcriptional activation of its downstream target genes. Thus, we hypothesized that PERM1 regulates energy metabolism through ERRα. To test if PERM1 physically interacts with ERRα as part of its transcription complex, we performed co-immunoprecipitation (Co-IP) assays in cardiac tissue from WT mice. ERRα was co-immunoprecipitated with PERM1 using mouse heart lysate (Figure 6A, left), whereas Perm1 was pulled down by anti-ERRα antibody (Figure 6A, right), indicating the binding of Perm1 with ERRα in the heart. Furthermore, we performed Co-IP assay using subcellular fraction of WT mouse cardiac tissue. As shown in Figure 6B, PERM1 was pulled down by anti-ERRα in the nucleus fraction, suggesting that PERM1 could be a transcription partner of ERRα in the heart. These data suggest that PERM1 forms transcriptional complex with both ERRα and PGC-1α. To test if the recruitment of PERM1 to the ERRE depends on ERRα, we performed in vitro DNA binding assay using biotin-labeled DNA comprising 380 bp of the promoter of Idh3a, an ERRα target gene, containing ERREs and a transcription start site (TSS) (Figure 6C). The promoter was incubated with cell lysate from cardiomyocytes, which were transfected either with scrambled-siRNA (control, “siERRα-“) or siRNA for ERRα (siERRα, ERR knockdown, “siERRα + “). PERM1 was recruited to the promoter of Idh3a, which was partly inhibited by ERRα knockdown, suggesting that PERM1 was localized to the promoter of the ERR target gene partly through ERRα. Next, we examined if PERM1 activates the transcription of the ERRE in an ERRα-dependent manner. A reporter gene assay was performed using the cardiomyocytes that were treated with either scrambled-siRNA or siERRα, followed by transfection with a construct that contains a luciferase reporter gene driven by a minimal promoter having upstream 3 repeats of ERR response element (ERRE), 3xERRE-luc. Consistent with our previous observation (14), the reporter activity of 3xERRE-luc was significantly increased by overexpression of PERM1, as compared with control vector, which was partially, yet, significantly decreased by ERRα knockdown (siERRα) (white bar vs. black bar in “ERRα,” p > 0.05; “scrambled” vs. “ERRα” in black bars, p < 0.05 in Figure 6D), suggesting that PERM1 activates the ERRE transcription, in part, through ERRα. To test whether PERM1 is localized to the ERR target gene promoters in the heart, ChIP assays were performed using ventricular tissue from WT mice that were subjected to either sham or TAC surgery, the latter of which induces pressure overload in the heart. As shown in Figure 6E, PERM1 was localized to the proximal region of ERRE that are present in the promoters of ERRα and its target genes (Cpt2; Mcad; Sdhd; Ndufv1; Ndufs1; Idh3a), while the occupancy of PERM1 on the ERRE of ERRα and those ERR target gene promoters was reduced in the TAC mouse heart, suggesting that the transcriptional regulation of ERR target genes by PERM1 was decreased during pressure overload (Figure 6E). These results are consistent with downregulation of PERM1 in the mouse TAC hearts, which we previously shown (14). Taken together, these results suggest that PERM1 positively regulates the transcription of ERR target genes by localizing to and activating the ERRE, likely through interacting with ERRα.
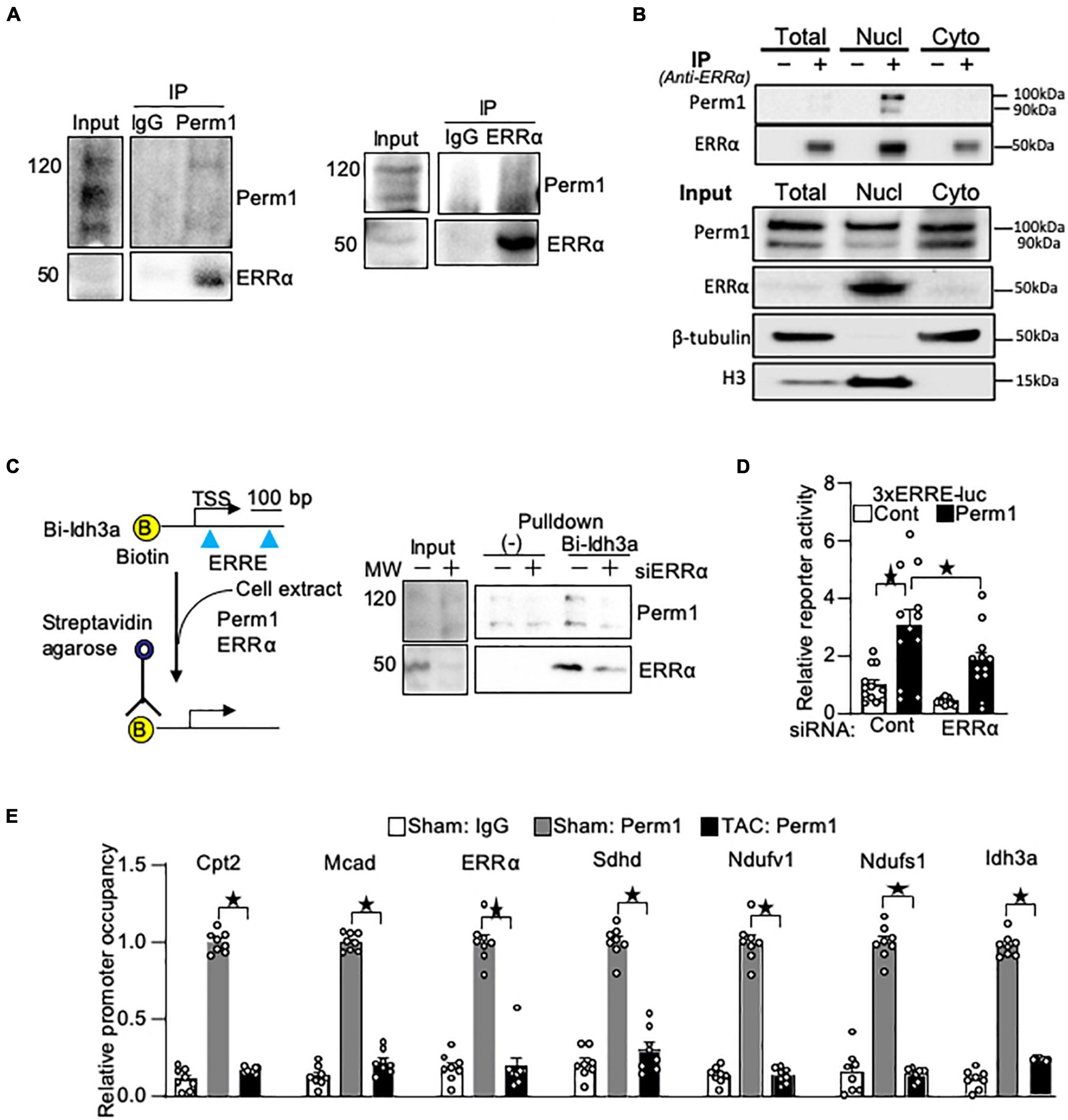
Figure 6. PERM1 interacts with ERRα and activates ERRE in an ERRα-dependent manner. (A) PERM1 binds to ERRα in the adult mouse heart. (B) Co-IP assay of subcellular fractionation from the mouse adult heart shows that PERM1 binds to ERRα in the nucleus. (C) In vitro DNA binding assay using biotin-labeled DNA comprising the promoter of Idh3a (ERR target gene) and cardiomyocyte lysate shows that PERM1 bound to the ERRE of the ERR target gene, which was partly inhibited by silencing of ERRα (siERRα). (D) Reporter gene assays were performed in cardiomyocytes using a luciferase reporter gene driven by 3 repeats of EER response element (3xERRE-luc) (n = 12/group). Adenovirus-mediated overexpression of PERM1 increased the ERRE activity (white bar vs. black bar, scrambled-siRNA), which was partially inhibited by silencing of ERRα (siERRα). Cont: control (adenovirus-null), PERM1: adenovirus-PERM1 (overexpression) (E) ChIP-PCR was performed with anti-PERM1 antibody in the mouse heart that was subjected to either sham or 4 weeks of transverse aortic constriction (TAC) surgery (n = 8/group). PERM1 was localized to the flanking promoter region (ERRE) of the well-known ERR target genes (Ctp2; Mcad; ERRα; Sdhd; Ndufv1; Ndufs1; Idh3a), which was abolished by pressure overload. *p < 0.05 (one-way ANOVA), ± SED.
To gain the insights into the mechanism by which PERM1 promotes transcription, we performed MS-based unbiased screening of PERM1-bound proteins. Flag-tagged PERM1 was expressed in cultured cardiomyocytes, followed by immunoprecipitation with anti-Flag antibody. PERM1-bound proteins of whole lysates were identified using LC-MS/MS. The total of 545 proteins (Supplementary Dataset 8) were analyzed for GO-oriented biological process, which identified 278 pathways that were significantly recognized as biological process (false discovery rate, FDR < 0.05, Supplementary Dataset 9). Among those pathways, we focused on “regulation of gene expression” (Supplementary Dataset 9, FDR = 0.0029), which includes ankyrin repeat domain 1 (ANKRD1), ANKYRIN2, ANKYRIN3, KN motif and ankyrin repeat domains 1 (KANK1), KANK2, BLC2-associated anthanogene-6 (BAG6), transcriptional intermediary factor 1β (TIF1β) and thioredoxin 1 (TRX1). To verify their binding to PERM1, Co-IP assays were performed. Consistent with our result shown in Figures 6A,B and a previous study (31), ERRα and PGC-1α were pulled down by anti-Flag antibody in cardiomyocytes (Figure 7A). In addition, we confirmed the interaction of PERM1 with ANKRD1, BAG6, KANK2, and TIF1β, however, the binding of PERM1 with TRX1 was not observed. To test whether those transcriptional regulators are required for the transcription activation of ERRE by PERM1, luciferase reporter gene assays were performed in cardiomyocytes that were treated with either scramble-siRNA, PGC-1α, ANKRD1, BAG6, KANK2, or TIF1β. As shown in Figure 7B, PERM1-induced transcriptional activation via ERRE was inhibited by knockdown of PGC-1α, BAG6, and KANK2, while silencing of Ankrd1 did not affect the ERRE activation by PERM1. Unexpectedly, knockdown of TIF1β promoted transcription even without PERM1 overexpression. These results suggest that the transcriptional regulators PGC-1α, BAG6, and KANK2 are required for PERM1-induced transcriptional activation via ERRE to some extent. To test if the recruitment of PERM1 to gene promoters has a regulatory role in transcription, we used mammalian one-hybrid system with Gal4-DNA binding domain fused with PERM1 and UAS-luc. Artificial recruitment of PERM1 to a gene promoter activated the transcription of UAS-luc, which was blunted by silencing of either PGC-1α, BAG6, or KANK2 (Figure 7C). Whether BAG6 or KANK2 regulates ERR target gene expression is unknown. We examined if silencing BAG6 or KANK2 alters ERR target gene expression. Primary cultured cardiomyocytes were treated with either scrambled siRNA (siControl), siRNA-Bag6 (siBag6), or siRNA-Kank2 (siKank2) and gene expression analysis was performed. As shown in Figure 7D, siRNA-mediated BAG6 and KANK2 significantly decreased the mRNA levels of ERR target genes (Mcad; Sdha; Sdhd; Fh1; Cycs; Ndufv1; Idh3a, all p < 0.05), suggesting that BAG6 and KANK2 are involved in transcriptional control of ERR target genes. Of note, we previously demonstrated that Perm1 directly regulates Ndufv1 expression (14). Knockdown of PGC-1α, ANKRD1, BAG6, KANK2, and TIF1β by siRNA in cardiomyocytes was verified by western blotting (Figure 7E). Taken together with the results shown in Figure 6B, in which PERM1 interacts with ERRα in the nucleus, these results suggest that PERM1 acts as a transcription coactivator of ERRα that functionally interacts with PGC-1α, BAG6 and KANK2 (Figure 7E).
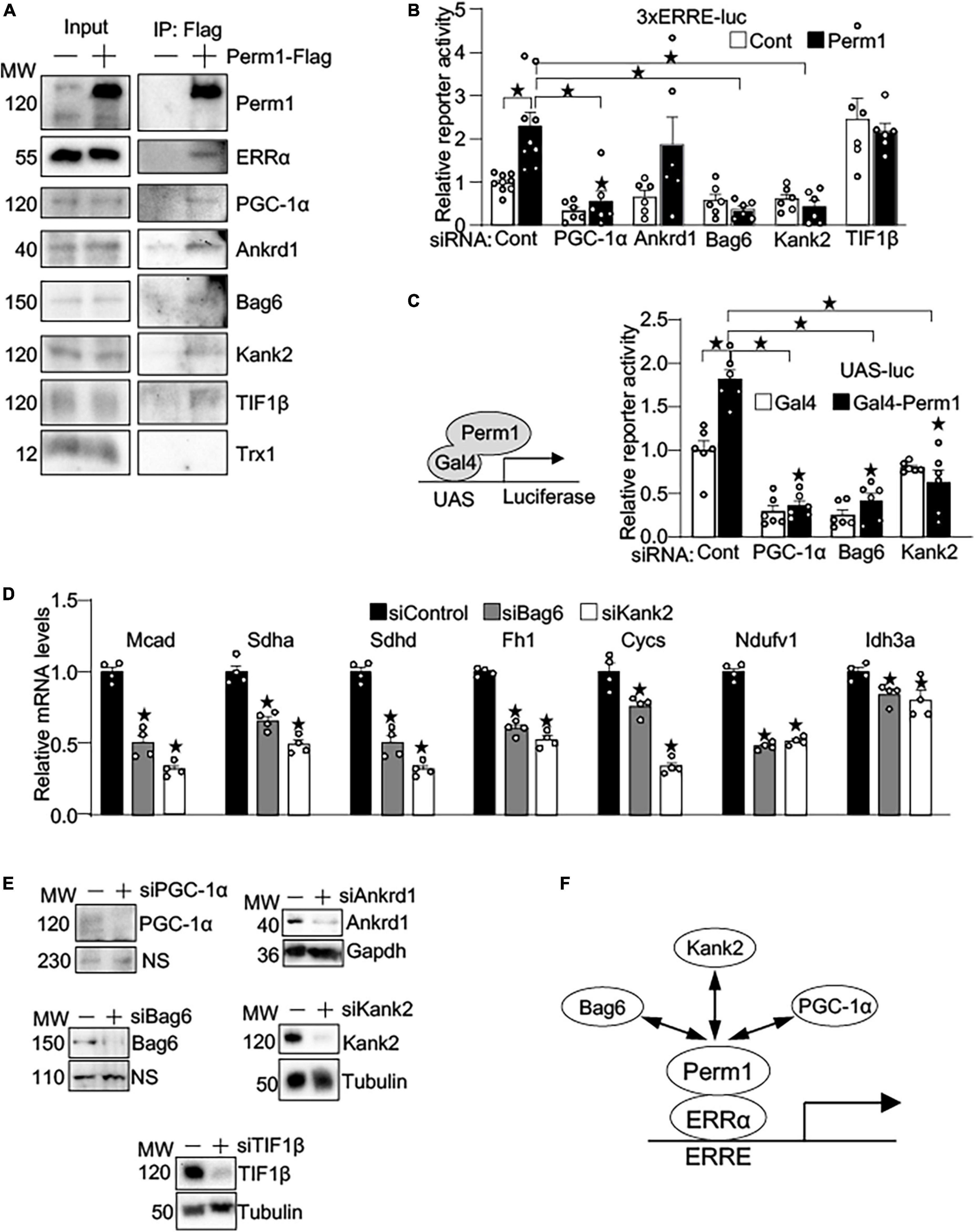
Figure 7. PERM1 is a novel transcription coactivator that functionally interacts with PGC-1α, BAG6, and KANK2. (A) Immunoprecipitation assays show that PERM1 binds to the transcriptional regulators ERRα, PGC-1α, ANKRD1, BAG6, KANK2, and TIF1β. Adenovirus Flag-tagged Perm1 (PERM1-Flag) was transduced to cardiomyocytes, and PERM1-bound proteins were pulled down by anti-Flag antibody. Immunoblotting confirmed the interaction of PERM1 with ERRα, PGC-1α, ANKRD1, BAG6, KANK2, and TIF1β. In contrast, TRX1, which was also identified through MS-based screening as a potential binding partner in transcription regulation, did not bind to PERM1. (B) Luciferase reporter gene assays show that the PERM1-induced transcriptional activation of the ERRE requires PGC-1α, BAG6, and KANK2 (n = 7-9/group). Cardiomyocytes were transduced with 3xERRE-luc, followed by transfecting with either scrambled-siRNA (scr), siPGC-1α, siANKRD1, siBAG6, siKANK2, or TIF1β. (C) In vitro Gal4 assay shows the recruitment of PERM1 to a gene promoter induces transcriptional activation (n = 6/group). Gal4-fused Perm1 (Gal4-Perm1) was expressed with UAS-luc, a reporter gene driven by Gal4 binding sequence, in cardiomyocytes. In control (scrambled-siRNA, “scr”), the reporter activity was significantly increased by Gal4-Perm1, indicating that PERM1 can act as a transcription coactivator. Silencing of PGC-1α, BAG6, KANK2 inhibited the transcription activation by PERM1. (D) qPCR shows downregulation of ERR target genes by silencing BAG6 and KANK2 (siBag6 and siKank2) in cultured cardiomyocytes (n = 4/group). (E) siRNA-mediated knockdown of PGC-1α, ANKRD1, BAG6, KANK2, or TIF1β were verified by western blotting analyses. (F) Hypothetical model of transcriptional regulation by PERM1 on the ERRE. PERM1 is localized to and activates the ERRE in ERR target genes through interacting with ERRα and the other transcriptional regulators BAG6, KANK2, and PGC-1α (*p < 0.05).
This study has revealed that PERM1 ablation in mice leads to a reduction of cardiac function and energy reserve in association with downregulation of OXPHOS proteins and broad changes in substrate metabolism. Furthermore, we show for the first time that in cardiomyocytes PERM1 binds to ERRα, a key transcription factor that regulates genes important for energy homeostasis. The recruitment of PERM1 to a gene promoter led to the activation of transcription (Figure 7C), indicating the role of PERM1 as a new transcription coactivator in the ERRα/PGC-1α axis in the heart. These findings position PERM1 as an important muscle-specific transcriptional regulator that coordinates cardiac function and energy metabolism, presumably via the ERRα/PGC-1α axis.
Perm1 deletion caused extensive alterations in substrate metabolism. Reduced energy reserve in association with downregulation of OXPHOS and upregulation of glycolysis pathway in Perm–/– hearts recapitulates the metabolic phenotype of the failing heart (4, 8, 29). However, the reduced myocardial TG contents in Perm1–/– mice contrasts the phenotype produced by pressure overload-induced heart failure in mice, where accumulation of TGs, DGs and MGs occurs, in part, due to the reduced fatty acid oxidation, in part, through downregulation of CPT1/2 (32). Of note, neither CTP1b nor CPT2 were downregulated in PERM1-null hearts (Supplementary Figure 2). The relative depletion of the global TG pool could explain decreased content of MGs and FFAs (Figure 4), leading to a relative fatty acid “starvation” in Perm–/– mice. Given downregulation of CD36, which is localized to the plasma membrane and facilitates fatty acid uptake into muscle cells (Supplementary Figure 2B), it is plausible that cardiac fatty acid intake was persistently decreased in Perm1 deficient mice.
In parallel to alterations in lipid metabolism, we observed accumulation of intermediates in the proximal part of glycolytic pathway (G6-P and F6-P, Figure 5). Despite upregulation of several glycolytic enzymes (OFK; BPGM; PGM, Figure 5), there is a significant downregulation of PDH, limiting the input to the TCA cycle. The glycolytic flux has to be diverted to bypass the bottleneck at the level of PDH. The diversion apparently occurs via the polyol pathway leading to remarkable increases in sorbitol and fructose content. Presumably, this flux is further directed to glycerol-3-phosphate (G-3P) synthesis (1.64-fold increase, p = 0.1, Figure 5) facilitated by upregulation on the cytoplasmic isoform of G-3P dehydrogenase (GPD1). The fact that the TG pool is decreased suggests, however, that the excess of G-3P is not utilized for de novo TG synthesis.
The most prominent change in metabolome of Perm–/– hearts is the remarkable increase of sorbitol and fructose contents (Figure 4B). Increase in myocardial sorbitol and fructose is a signature of a diabetic heart, and it was recently shown that myocardial sorbitol and fructose content are directly correlated to the degree of diastolic dysfunction in diabetic patients (33). However, the causative link between the elevated levels of polyol pathway intermediates and contractile dysfunction is not well understood. Polyol pathway-mediated oxidative stress has been implicated in dysregulation of myocardial Ca2+ cycling, leading to contractile dysfunction (34). Fructose accumulation can also lead to increased formation of methylglyoxal, which reacts with arginine and lysine residues to form irreversible carbonyl adducts. Glycation of proteins by methylglyoxal was implicated in mitochondrial dysfunction and oxidative stress (35). Finally, channeling fructose excess into de novo TG synthesis may lead to enhanced cardiac lipid droplet formation (steatosis), which has been shown to be an independent predictor of diastolic dysfunction in type 2 diabetic patients (36). An interesting metabolic signature of Perm–/– mice, which sets it apart from the diabetic heart, is a large accumulation of polyol pathway intermediates amid a global decrease in myocardial TG pool. It would be interesting to address whether Perm–/– hearts are tolerant to high-fat diet through limiting lipid storage and oxidation. Future studies should address a possible role of PERM1 in pathological metabolic remodeling and related contractile dysfunction in the diabetic heart.
The ratio of myocardial PCr to ATP is a sensitive index of the energetic state of the heart. PCr serves as a fast-exchange reserve of high-energy phosphates, the energy currency with the highest liquidity in the heart. Whenever energy demand exceeds energy supply, PCr levels decline first, and ATP decreases only when PCr is substantially depleted (37). PCr/ATP ratio is reduced during and after acute hypoxia (38) and in patients suffering from type 2 diabetes and various forms of heart failure (39). Perm1–/– mice exhibit a significant decrease in PCr/ATP ratio as compared to WT mice (Figure 2), indicating a relative “energy starvation.” It should be noted that this observation may be confounded by the procedure of the heart harvesting necessary to perform high-energy phosphate profiling. With the best effort made, it took within 30 sec to transfer the tissue sample from the state of full oxygenation to the state of the full arrest of metabolic activity. This time may be sufficient to create a hypoxic challenge and contribute to PCr depletion. However, the conditions of tissue harvesting were exactly the same in Perm1–/– and WT groups. Hence, whereas a potential hypoxic event cannot be excluded, the relative energy starvation in Perm–/– mice would still be indicative of an energetic vulnerability specific to Perm1 deletion. This raises a possibility that Perm1–/– mice exhibit a reduced tolerance to acute hypoxia and/or increase in cardiac workload, which should be tested in future studies. The mechanism of the relative energy starvation in Perm1–/– heart cannot be exactly defined at this point and may be a result of multiple factors. A computational study addressing possible factors contributing to a reduction in PCr/ATP ratio highlighted the 3 most important factors controlling this ratio: the activity of adenine nucleotide translocase, substrate dehydrogenation, and the activity of cytochrome oxidase (Complex IV). In this model, substrate dehydrogenation is a collective term of all processes supplying NADH to the electron-transport chain (ETC). In the heart, the main supplier of NADH to ETC is FAO. Of interest, cytochrome oxidase becomes the strongest controlling factor under conditions of hypoxia (40). Our proteomics analysis detected total of 17 proteins related to Complex IV. Out of those, four (COX6B1, COX7A1, COX8B, CMC1) were significantly downregulated, and others were unchanged. Whether such selective downregulation is sufficient to reduce the overall Complex IV activity, remains to be investigated. Nevertheless, the relative energy starvation in Perm1–/– mice may be due to downregulation of FAO and/or downregulation of proteins involved in Complex IV.
The total ATP contents remain relatively stable and preserved in the stage of compensated hypertrophy (41), and it becomes significantly reduced only in advanced stages of heart failure (2, 42). Together with the moderate downregulation of OXPHOS proteins and upregulation of glycolysis (Figure 3), our data suggest that metabolic profile in the Perm1 deficit heart mimics the metabolic remodeling at the early stage of heart failure. If this is true, maintaining PERM1 expression during pressure overload might prevent energy imbalance and cardiac dysfunction. This will be investigated in our future study.
The PGC-1/ERR transcriptional module has been established as a center of metabolism regulation in the heart. PERM1 was discovered by Kralli’s group as a muscle-specific protein that is induced by ERRs and PGC-1α/β in C2C12 myoblasts (15). It was initially reported that PERM1 regulates the expression of selective PGC-1/ERR target genes through unknown mechanisms (15). Consistent with conserved putative nuclear localization signals in the C-terminus of PERM1 (15), we found that PERM1 is localized to the nucleus in cultured cardiomyocytes (14). In this study, we show that PERM1 interacts with ERRα in the nucleus of the mouse heart (Figure 6), suggesting that PERM1 is a transcription partner of ERRα. Recent study by Cho et al. showed that PERM1 interacts with PGC-1α in the mouse heart and U2OS cells (cell lines from human bone osteosarcoma epithelial cells) (31). We confirmed the interaction of PERM1 with PGC-1α in cardiomyocytes (Figure 7). Transactivation of the ERRE by PERM1 through ERRα and PGC-1α (Figure 7) further suggests that PERM1 forms the functional transcription complex with ERRα and PGC-1α. Given that the predicted sequence of PERM1 contains ∅XXLL-type motif in the N-terminus that is often involved in protein-protein interaction but there is no other obvious protein motifs suggestive of a molecular function (15), it is plausible that PERM1 plays a role as an adaptor protein that promotes recruitment of transcription regulators to the ERRE to initiate transcription. It is interesting to note that the promoters of both Perm1 and ERRs contain the ERRE (15, 43). We speculate that the PERM1/ERRα/PGC-1α transcriptional complex acts as a feedforward mechanism to dramatically boost transcription of ERRα and its target genes involved in OXPHOS in response to increased energy demands in the heart. This needs to be investigated in our future study.
MS-based unbiased screening of PERM1-bound proteins in cardiomyocytes identified several candidates that may be involved in the transcription regulation by PERM1 (Figure 7). Among them, we found that BAG6 (also known as BAT3) and KANK2 (also known as SIP) are required for PEMR1 to transactivate the ERRE (Figure 7). BAG6 is a nuclear protein, and recent study suggests that BAG6 may act as a component of some chromatin regulator complex that regulates histone 3 lysine 4 dimethylation (H3K4me2) (44), which generally leads to gene activation. Given that PERM1 requires BAG6 to activate the ERRE (Figure 7), it is plausible that PERM1 also recruits epigenetic modulator(s), presumably for chromatin remodeling of ERR/PGC-1 target genes. A cardioprotective effect of KANK2 against myocardial infarction through decreasing collagen deposition and apoptosis has been reported (45). KANK2 is also known to be involved in transcription regulation and interacts with steroid receptor co-activator 1 (SRC-1) in the human heart (46), which facilitates the transcription initiation mediated by nuclear receptors. However, the role of KANK2 in transcriptional control through the ERR/PGC-1 axis needs to be further investigated.
The current study was aimed at elucidating the role of PERM1 as a transcriptional regulator. Bock et al. recently demonstrated that PERM1 is localized in mitochondrial outer membrane that interacts with the MICOS (mitochondrial contact site and cristae organizing system)-MIB (mitochondrial intermembrane space bridging) complex and connects the mitochondria and sarcolemma (47). Our current study and the other studies also confirmed the localization of PERM1 in the mitochondria of cardiac tissue as well as in the nucleus and cytosol (31) (Supplementary Figure 3). Consistently, our LC-MS/MS-based screening of PERM1-bound proteins in cardiomyocytes (whole lysates) detected the components of MICOS-MIB complex, such as MIC60 and SAMM50 (Supplementary Dataset 8). Overall, our current study and the other studies (31, 47) suggest that PERM1 can regulate mitochondrial function through both transcription dependent and independent mechanisms.
This study demonstrates that Perm1 functionally interacts with PGC1α and ERRα. Impaired ERRα/PGC-1α pathways are associated with metabolic dysfunction in the failing heart. Downregulated genes important for energy metabolism in the failing heart are mostly recognized as the ERR/PGC-1 target genes (4, 8, 48). Genetic deletion of either PGC-1α or ERRα leads to global downregulation of OXPHOS genes in the heart (4, 7, 18, 43). However, neither ERRα-KO nor PGC-1α-KO mice (systemic, constitutive) exhibit any change in cardiac function (4, 7). Of note, cardiac-specific PGC-1α-KO mice develop cardiac dysfunction at the age of 17 weeks (-29% in EF), in association of downregulation of FAO proteins (CPT1b, FATP) as well as glycolysis enzymes (GLUT4; PFKm) (49). Downregulation of ERRα occurs early in response to pressure overload in mice (7 days of TAC), whereas there is no change in the expression of ERRγ (4). As discussed in our review (12), a variability of outcomes in PGC-1α expression has been reported in the human and mouse failing hearts: downregulation (8, 50–54); unchanged expression (4, 18, 55, 56); and upregulation (10). In addition, impaired mitochondrial biogenesis was observed in the human and mouse heart failure even when PGC-1α expression was unchanged (29). Thus, the changes in the expression and the activity of PGC-1α alone cannot completely explain metabolic reprogramming in the diseased heart. More importantly, maintaining PGC-1α expression during pressure overload in mice failed to rescue contractile function in this setting. (56, 57) To date, it remains unknown whether maintenance or overexpression of ERRα has any beneficial effect for cardiac function under conditions of hemodynamic stress. Our study demonstrated that both mRNA and proteins levels of Perm1 were significantly reduced in the human and mouse failing heart (14). More importantly, the initial response to phenylephrine-induced hypertrophy in cardiomyocytes, which mimics metabolic changes in pressure overload-induced heart failure, occurred in the reduced mRNA level of Perm1, followed by downregulation of ERRα an PGC-1α (14). Thus, it is possible that downregulation of Perm1 is an early precursor of metabolic remodeling under pathological stress. If so, maintaining Perm1 expression might attenuate the impairment of the ERRα/PGC-1α axis during pressure overload and prevent metabolic perturbations and the progression of heart failure at the early stage. This needs to be determined in our future study.
In conclusion, this study establishes the role of PERM1 as a new transcriptional regulator in the heart that maintains energy reserve and contractile function through interacting with ERRα and PGC-1α. The therapeutic potential of PERM1 in heart failure and cardiac metabolic syndromes should be tested in future studies.
The original contributions presented in this study are included in the article/Supplementary material, further inquiries can be directed to the corresponding author.
This animal study was reviewed and approved by University of Utah and the Rutgers New Jersey Medical School Institutional Animal Care and Use Committee.
JW and SO contributed to the initial design of study and manuscript preparation. TS performed echocardiography. SA, SY, AJ, KGS, KO, AS, and AH performed gene expression study. KS, KC, and SAO performed western blotting analysis. MT performed EM analysis. SO, JB, XX, SK, and YM prepared primary cultured neonatal cardiomyocytes and performed western blotting analysis, reporter gene assays, and DNA binding assays. DC and TOL contributed to the echocardiography analysis. TOL and HL performed LC/MS-MS analyses. JC performed GCMS analysis. JM performed lipidomic analysis. YS contributed to the mouse study. SD supervised echocardiography analysis and edited the manuscript. All authors contributed to the article and approved the submitted version.
This study was supported by the National Heart, Lung, and Blood Institute (NHLBI) 7R01HL156667 (JW and SD), HL135121-01 and HL132067-01A1 (SD); the Harold S. Geneen Charitable Trust Awards Program for Coronary Heart Disease Research (JW); the Nora Eccles Treadwell Foundation (JW and SD); the American Heart Association (AHA) Transformational Project Award 19TPA34850170 (SO) and Heart Failure Strategically Focused Research Network 16SFRN29020000 (SD); Rutgers Office of Research Core Facility utilization grant (SO); and United States Department of Veterans Affairs I01 CX002291 (SD).
We thank Alexey Zaitsev for their critical reading of the manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2022.1033457/full#supplementary-material
1. Ardehali H, Sabbah HN, Burke MA, Sarma S, Liu PP, Cleland JG, et al. Targeting myocardial substrate metabolism in heart failure: potential for new therapies. Eur J Heart Fail. (2012) 14:120–9. doi: 10.1093/eurjhf/hfr173
2. Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure: implications beyond atp production. Circ Res. (2013) 113:709–24. doi: 10.1161/CIRCRESAHA.113.300376
3. Huss JM, Kelly DP. Nuclear receptor signaling and cardiac energetics. Circ Res. (2004) 95:568–78. doi: 10.1161/01.RES.0000141774.29937.e3
4. Huss JM, Imahashi K, Dufour CR, Weinheimer CJ, Courtois M, Kovacs A, et al. The nuclear receptor erralpha is required for the bioenergetic and functional adaptation to cardiac pressure overload. Cell Metab. (2007) 6:25–37. doi: 10.1016/j.cmet.2007.06.005
5. Villena JA, Kralli A. Erralpha: a metabolic function for the oldest orphan. Trends Endocrinol Metab. (2008) 19:269–76. doi: 10.1016/j.tem.2008.07.005
6. Huss JM, Garbacz WG, Xie W. Constitutive activities of estrogen-related receptors: transcriptional regulation of metabolism by the err pathways in health and disease. Biochim Biophys Acta. (2015) 1852:1912–27. doi: 10.1016/j.bbadis.2015.06.016
7. Arany Z, He H, Lin J, Hoyer K, Handschin C, Toka O, et al. Transcriptional coactivator Pgc-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. (2005) 1:259–71. doi: 10.1016/j.cmet.2005.03.002
8. Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM. Transverse aortic constriction leads to accelerated heart failure in mice lacking ppar-gamma coactivator 1alpha. Proc Natl Acad Sci USA. (2006) 103:10086–91. doi: 10.1073/pnas.0603615103
9. Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. (2000) 106:847–56. doi: 10.1172/jci10268
10. Sihag S, Cresci S, Li AY, Sucharov CC, Lehman JJ. Pgc-1alpha and erralpha target gene downregulation is a signature of the failing human heart. J Mol Cell Cardiol. (2009) 46:201–12. doi: 10.1016/j.yjmcc.2008.10.025
11. Finck BN, Kelly DP. Pgc-1 coactivators: inducible regulators of energy metabolism in health and disease. J Clin Invest. (2006) 116:615–22. doi: 10.1172/JCI27794
12. Oka S, Sabry AD, Cawley KM, Warren JS. Multiple levels of pgc-1alpha dysregulation in heart failure. Front Cardiovasc Med. (2020):7:2. doi: 10.3389/fcvm.2020.00002
13. Warren JS, Tracy CM, Miller MR, Makaju A, Szulik MW, Oka SI, et al. Histone methyltransferase smyd1 regulates mitochondrial energetics in the heart. Proc Natl Acad Sci USA. (2018) 115:E7871–80. doi: 10.1073/pnas.1800680115
14. Oka SI, Sabry AD, Horiuchi AK, Cawley KM, O’Very SA, Zaitsev MA, et al. Perm1 regulates cardiac energetics as a downstream target of the histone methyltransferase Smyd1. PLoS One. (2020) 15:e0234913. doi: 10.1371/journal.pone.0234913
15. Cho Y, Hazen BC, Russell AP, Kralli A. Peroxisome proliferator-activated receptor gamma coactivator 1 (Pgc-1)- and estrogen-related receptor (Err)-induced regulator in muscle 1 (Perm1) is a tissue-specific regulator of oxidative capacity in skeletal muscle cells. J Biol Chem. (2013) 288:25207–18. doi: 10.1074/jbc.M113.489674
16. National Research Council Committee for the Update of the Guide for the, Care Use of Laboratory, Animals. The national academies collection: reports funded by national institutes of health. In: Guide for the Care and Use of Laboratory Animals. Washington, DC: National Academies Press (2011).
17. Shankar TS, Ramadurai DKA, Steinhorst K, Sommakia S, Badolia R, Thodou Krokidi A, et al. Cardiac-specific deletion of voltage dependent anion channel 2 leads to dilated cardiomyopathy by altering calcium homeostasis. Nat Commun. (2021) 12:4583. doi: 10.1038/s41467-021-24869-0
18. Bhat S, Chin A, Shirakabe A, Ikeda Y, Ikeda S, Zhai P, et al. Recruitment of Rna polymerase II to metabolic gene promoters is inhibited in the failing heart possibly through pgc-1alpha (peroxisome proliferator-activated receptor-gamma coactivator-1alpha) dysregulation. Circ Heart Fail. (2019) 12:e005529. doi: 10.1161/circheartfailure.118.005529
19. Shibayama J, Taylor TG, Venable PW, Rhodes NL, Gil RB, Warren M, et al. Metabolic determinants of electrical failure in ex-vivo canine model of cardiac arrest: evidence for the protective role of inorganic pyrophosphate. PLoS One. (2013) 8:e57821. doi: 10.1371/journal.pone.0057821
20. Shibayama J, Yuzyuk TN, Cox J, Makaju A, Miller M, Lichter J, et al. Metabolic remodeling in moderate synchronous versus dyssynchronous pacing-induced heart failure: integrated metabolomics and proteomics study. PLoS One. (2015) 10:e0118974. doi: 10.1371/journal.pone.0118974
21. Nakamura M, Liu T, Husain S, Zhai P, Warren JS, Hsu CP, et al. Glycogen synthase kinase-3α promotes fatty acid uptake and lipotoxic cardiomyopathy. Cell Metab. (2019) 29:1119–34.e12. doi: 10.1016/j.cmet.2019.01.005
22. Oka SI, Byun J, Huang CY, Imai N, Ralda G, Zhai P, et al. Nampt potentiates antioxidant defense in diabetic cardiomyopathy. Circ Res. (2021) 129:114–30. doi: 10.1161/circresaha.120.317943
23. Xia J, Sinelnikov IV, Han B, Wishart DS. Metaboanalyst 3.0–making metabolomics more meaningful. Nucleic Acids Res. (2015) 43:W251–7. doi: 10.1093/nar/gkv380
24. Tong M, Saito T, Zhai P, Oka SI, Mizushima W, Nakamura M, et al. Alternative mitophagy protects the heart against obesity-associated cardiomyopathy. Circ Res. (2021) 129:1105–21. doi: 10.1161/circresaha.121.319377
25. Liu CF, Tang WHW. Epigenetics in cardiac hypertrophy and heart failure. JACC Basic Transl Sci. (2019) 4:976–93. doi: 10.1016/j.jacbts.2019.05.011
26. Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, et al. String V9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. (2013) 41:D808–15. doi: 10.1093/nar/gks1094
27. Huang Y, Li Z, Lin E, He P, Ru G. Oxidative damage-induced hyperactive ribosome biogenesis participates in tumorigenesis of offspring by cross-interacting with the wnt and Tgf-B 1 pathways in Ivf embryos. Exp Mol Med. (2021) 53:1792–806. doi: 10.1038/s12276-021-00700-0
28. Stanley WC, Chandler MP. Energy metabolism in the normal and failing heart: potential for therapeutic interventions. Heart Fail Rev. (2002) 7:115–30.
29. Karamanlidis G, Bautista-Hernandez V, Fynn-Thompson F, Del Nido P, Tian R. Impaired mitochondrial biogenesis precedes heart failure in right ventricular hypertrophy in congenital heart disease. Circ Heart Fail. (2011) 4:707–13. doi: 10.1161/circheartfailure.111.961474
30. Liebisch G, Fahy E, Aoki J, Dennis EA, Durand T, Ejsing CS, et al. Update on lipid maps classification, nomenclature, and shorthand notation for ms-derived lipid structures. J Lipid Res. (2020) 61:1539–55. doi: 10.1194/jlr.S120001025
31. Cho Y, Tachibana S, Lam K, Arita Y, Khosrowjerdi S, Zhang O, et al. Perm1 promotes cardiomyocyte mitochondrial biogenesis and protects against hypoxia/reoxygenation-induced damage in mice. J Biol Chem. (2021) 297:100825. doi: 10.1016/j.jbc.2021.100825
32. Lai L, Leone TC, Keller MP, Martin OJ, Broman AT, Nigro J, et al. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: a multisystems approach. Circ Heart Fail. (2014) 7:1022–31. doi: 10.1161/CIRCHEARTFAILURE.114.001469
33. Daniels LJ, Annandale M, Koutsifeli P, Li X, Bussey CT, van Hout I, et al. Elevated myocardial fructose and sorbitol levels are associated with diastolic dysfunction in diabetic patients, and cardiomyocyte lipid inclusions in vitro. Nutr Diabetes. (2021) 11:8. doi: 10.1038/s41387-021-00150-7
34. Tang WH, Cheng WT, Kravtsov GM, Tong XY, Hou XY, Chung SK, et al. Cardiac contractile dysfunction during acute hyperglycemia due to impairment of serca by polyol pathway-mediated oxidative stress. Am J Physiol Cell Physiol. (2010) 299:C643–53. doi: 10.1152/ajpcell.00137.2010
35. Rabbani N, Thornalley PJ. Dicarbonyls linked to damage in the powerhouse: glycation of mitochondrial proteins and oxidative stress. Biochem Soc Trans. (2008) 36:1045–50. doi: 10.1042/bst0361045
36. Rijzewijk LJ, van der Meer RW, Smit JW, Diamant M, Bax JJ, Hammer S, et al. Myocardial steatosis is an independent predictor of diastolic dysfunction in type 2 diabetes mellitus. J Am Coll Cardiol. (2008) 52:1793–9. doi: 10.1016/j.jacc.2008.07.062
37. Neubauer S. The failing heart–an engine out of fuel. N Engl J Med. (2007) 356:1140–51. doi: 10.1056/NEJMra063052
38. Holloway C, Cochlin L, Codreanu I, Bloch E, Fatemian M, Szmigielski C, et al. Normobaric hypoxia impairs human cardiac energetics. FASEB J. (2011) 25:3130–5. doi: 10.1096/fj.11-183426
39. Neubauer S, Horn M, Cramer M, Harre K, Newell JB, Peters W, et al. Myocardial phosphocreatine-to-Atp ratio is a predictor of mortality in patients with dilated cardiomyopathy. Circulation. (1997) 96:2190–6. doi: 10.1161/01.cir.96.7.2190
40. Edwards LM, Ashrafian H, Korzeniewski B. In silico studies on the sensitivity of myocardial Pcr/Atp to changes in mitochondrial enzyme activity and oxygen concentration. Mol Biosyst. (2011) 7:3335–42. doi: 10.1039/c1mb05310h
41. Doenst T, Pytel G, Schrepper A, Amorim P, Farber G, Shingu Y, et al. Decreased rates of substrate oxidation ex vivo predict the onset of heart failure and contractile dysfunction in rats with pressure overload. Cardiovasc Res. (2010) 86:461–70. doi: 10.1093/cvr/cvp414
42. Gupta A, Chacko VP, Weiss RG. Abnormal energetics and Atp depletion in pressure-overload mouse hearts: in vivo high-energy phosphate concentration measures by noninvasive magnetic resonance. Am J Physiol Heart Circ Physiol. (2009) 297:H59–64. doi: 10.1152/ajpheart.00178.2009
43. Dufour CR, Wilson BJ, Huss JM, Kelly DP, Alaynick WA, Downes M, et al. Genome-wide orchestration of cardiac functions by the orphan nuclear receptors erralpha and gamma. Cell Metab. (2007) 5:345–56. doi: 10.1016/j.cmet.2007.03.007
44. Nguyen P, Bar-Sela G, Sun L, Bisht KS, Cui H, Kohn E, et al. Bat3 and set1a form a complex with Ctcfl/Boris to modulate H3k4 histone dimethylation and gene expression. Mol Cell Biol. (2008) 28:6720–9. doi: 10.1128/mcb.00568-08
45. Li L, Li ZB, Jia M, Chu HT. Therapeutic effects of Kank2 in myocardial infarction rats might be associated with the Nf-K b P65 inhibition. Int Immunopharmacol. (2020) 86:106687. doi: 10.1016/j.intimp.2020.106687
46. Zhang Y, Zhang H, Liang J, Yu W, Shang Y. Sip, a novel ankyrin repeat containing protein, sequesters steroid receptor coactivators in the cytoplasm. EMBO J. (2007) 26:2645–57. doi: 10.1038/sj.emboj.7601710
47. Bock T, Turk C, Aravamudhan S, Keufgens L, Bloch W, Rozsivalova DH, et al. Perm1 interacts with the micos-mib complex to connect the mitochondria and sarcolemma via ankyrin B. Nat Commun. (2021) 12:4900. doi: 10.1038/s41467-021-25185-3
48. Karamanlidis G, Nascimben L, Couper GS, Shekar PS, del Monte F, Tian R. Defective DNA replication impairs mitochondrial biogenesis in human failing hearts. Circ Res. (2010) 106:1541–8. doi: 10.1161/circresaha.109.212753
49. Karkkainen O, Tuomainen T, Mutikainen M, Lehtonen M, Ruas JL, Hanhineva K, et al. Heart specific Pgc-1alpha deletion identifies metabolome of cardiac restricted metabolic heart failure. Cardiovasc Res. (2019) 115:107–18. doi: 10.1093/cvr/cvy155
50. Sebastiani M, Giordano C, Nediani C, Travaglini C, Borchi E, Zani M, et al. Induction of mitochondrial biogenesis is a maladaptive mechanism in mitochondrial cardiomyopathies. J Am Coll Cardiol. (2007) 50:1362–9. doi: 10.1016/j.jacc.2007.06.035
51. Piquereau J, Moulin M, Zurlo G, Mateo P, Gressette M, Paul JL, et al. Cobalamin and folate protect mitochondrial and contractile functions in a murine model of cardiac pressure overload. J Mol Cell Cardiol. (2016) 102:34–44. doi: 10.1016/j.yjmcc.2016.11.010
52. Watanabe S, Horie T, Nagao K, Kuwabara Y, Baba O, Nishi H, et al. Cardiac-specific inhibition of kinase activity in calcium/calmodulin-dependent protein kinase kinase-beta leads to accelerated left ventricular remodeling and heart failure after transverse aortic constriction in mice. PLoS One. (2014) 9:e108201. doi: 10.1371/journal.pone.0108201
53. Faerber G, Barreto-Perreia F, Schoepe M, Gilsbach R, Schrepper A, Schwarzer M, et al. Induction of heart failure by minimally invasive aortic constriction in mice: reduced peroxisome proliferator-activated receptor gamma coactivator levels and mitochondrial dysfunction. J Thorac Cardiovasc Surg. (2011) 141:492–500.e1. doi: 10.1016/j.jtcvs.2010.03.029
54. Garnier A, Zoll J, Fortin D, N’Guessan B, Lefebvre F, Geny B, et al. Control by circulating factors of mitochondrial function and transcription cascade in heart failure: a role for endothelin-1 and angiotensin II. Circ Heart Fail. (2009) 2:342–50. doi: 10.1161/circheartfailure.108.812099
55. Oka S, Zhai P, Yamamoto T, Ikeda Y, Byun J, Hsu CP, et al. Peroxisome proliferator activated receptor-alpha association with silent information regulator 1 suppresses cardiac fatty acid metabolism in the failing heart. Circ Heart Fail. (2015) 8:1123–32. doi: 10.1161/circheartfailure.115.002216
56. Karamanlidis G, Garcia-Menendez L, Kolwicz SC Jr, Lee CF, Tian R. Promoting Pgc-1alpha-driven mitochondrial biogenesis is detrimental in pressure-overloaded mouse hearts. Am J Physiol Heart Circ Physiol. (2014) 307:H1307–16. doi: 10.1152/ajpheart.00280.2014
Keywords: PERM1, heart, ERRα, metabolomics, proteomics, metabolism, transcription control
Citation: Oka S-i, Sreedevi K, Shankar TS, Yedla S, Arowa S, James A, Stone KG, Olmos K, Sabry AD, Horiuchi A, Cawley KM, O’very SA, Tong M, Byun J, Xu X, Kashyap S, Mourad Y, Vehra O, Calder D, Lunde T, Liu T, Li H, Mashchek JA, Cox J, Saijoh Y, Drakos SG and Warren JS (2022) PERM1 regulates energy metabolism in the heart via ERRα/PGC−1α axis. Front. Cardiovasc. Med. 9:1033457. doi: 10.3389/fcvm.2022.1033457
Received: 31 August 2022; Accepted: 11 October 2022;
Published: 07 November 2022.
Edited by:
Ippei Shimizu, Juntendo University, JapanReviewed by:
Wataru Kimura, RIKEN Center for Biosystems Dynamics Research (BDR), JapanCopyright © 2022 Oka, Sreedevi, Shankar, Yedla, Arowa, James, Stone, Olmos, Sabry, Horiuchi, Cawley, O’very, Tong, Byun, Xu, Kashyap, Mourad, Vehra, Calder, Lunde, Liu, Li, Mashchek, Cox, Saijoh, Drakos and Warren. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Junco S. Warren, anVuY293YXJyZW5AdnRjLnZ0LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.