 Hao Zhang
Hao Zhang Lu Ren
Lu Ren Rabindra Vishwadev Shivnaraine
Rabindra Vishwadev Shivnaraine
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cardiovasc. Med., 06 October 2022
Sec. General Cardiovascular Medicine
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.1011176
This article is part of the Research TopicReceptors in Cardiovascular Diseases: Mechanisms, diagnosis and treatmentView all 5 articles
Cardiac fibrosis occurs ubiquitously in ischemic heart failure, genetic cardiomyopathies, diabetes mellitus, and aging. It triggers myocardial stiffness, which impairs cardiac function, ultimately progressing to end-stage heart failure and increased mortality. Although several targets for anti-fibrotic therapies have been identified, including TGF-β and receptor tyrosine kinase, there is currently no FDA-approved drug specifically targeting cardiac fibrosis. G protein-coupled receptors (GPCRs) are integral, multipass membrane-bound receptors that exhibit diverse and cell-specific expression, offering novel and unrealized therapeutic targets for cardiac fibrosis. This review highlights the emerging roles of several GPCRs and briefly explores their downstream pathways that are crucial in cardiac fibrosis. We will not only provide an overview of the GPCRs expressed on cardiac fibroblasts that are directly involved in myofibroblast activation but also describe those GPCRs which contribute to cardiac fibrosis via indirect crosstalk mechanisms. We also discuss the challenges of identifying novel effective therapies for cardiac fibrosis and offer strategies to circumvent these challenges.
Cardiovascular diseases (CVD), which affect the heart or vasculature, remain the leading cause of mortality, responsible for ~31% of all deaths worldwide (1). Fibrotic diseases lead to nearly 1 million deaths annually, most of which are due to lung and cardiac fibrosis (2). Different types and consequences of cardiac fibrosis exist, and their pathology depends on the underlying cause. For example, aging, hypertension, and diabetes induce myocardial interstitial fibrosis and decrease ventricular compliance, leading to the pathogenesis of heart failure with diastolic dysfunction (3, 4). However, fibrosis is not invariably detrimental. In myocardial infarction (MI), the loss of a substantial number of cardiomyocytes triggers myofibroblasts activation, representing reparative fibrosis and contributing to scar formation (5). Although the scar lacks contractile ability, it serves an important protective role in keeping cardiac structural integrity and preventing catastrophic mechanical complications, such as cardiac rupture (6).
Cardiac fibroblast is an abundant cell type in the heart (7). In fibrosis diseases, cardiac fibroblasts are activated to myofibroblasts (MyoFB), which become highly proliferative and specialized in the generation of extracellular matrix proteins and contractile genes, such as ACTA2, encoding for smooth muscle α-actin (8, 9). Histologically, cardiac fibrosis is identified as the enlargement of the cardiac interstitium resulting from the deposition of extracellular proteins such as collagen, fibronectin, and elastin (3). This accumulation accompanies most cardiac pathologic conditions, such as myocardial stiffness and diastolic dysfunction, and the extent of cardiac fibrosis is a predictor of adverse outcomes (3).
Although there is no FDA-approved drug specifically targeting cardiac myofibroblast activation, pirfenidone and nintedanib have been approved to treat idiopathic lung fibrosis (10). In addition, pirfenidone has been tested in Phase II clinical trial “PIROUETTE” among patients with heart failure with preserved ejection fraction (HFpEF) and myocardial fibrosis (11). The results show that the administration of pirfenidone for 52 weeks reduced myocardial fibrosis (12), indicating fibrosis shares similar pathways, and antifibrotic therapy can be utilized universally among different organs.
G protein-coupled receptors (GPCRs), located at the cell membrane, possess diverse expression profiles throughout the body and are known to exhibit tissue and cell-specific membrane expressions (13). The basic function of GPCRs is to transduce extracellular stimulus into intracellular signals (14). The “G-proteins” that coupled to GPCRs are composed of a heterotrimer of α, β, and γ subunits—Gα, Gβ, and Gγ, respectively—that is kept in an inactive basal state where αβγ are tightly bound and activated when α dissociates from βγ (14). Since GPCRs consist of more than 800 receptors and account for the most diverse family of proteins in the human genome, the combination of GPCRs at a particular cell type may be substantially heterogeneous and diverse (15). In this review, we will focus on the roles of GPCR and the downstream signaling in cardiac fibrosis regulation and illustrate their utility as promising targets for cardiac fibrosis (Table 1).
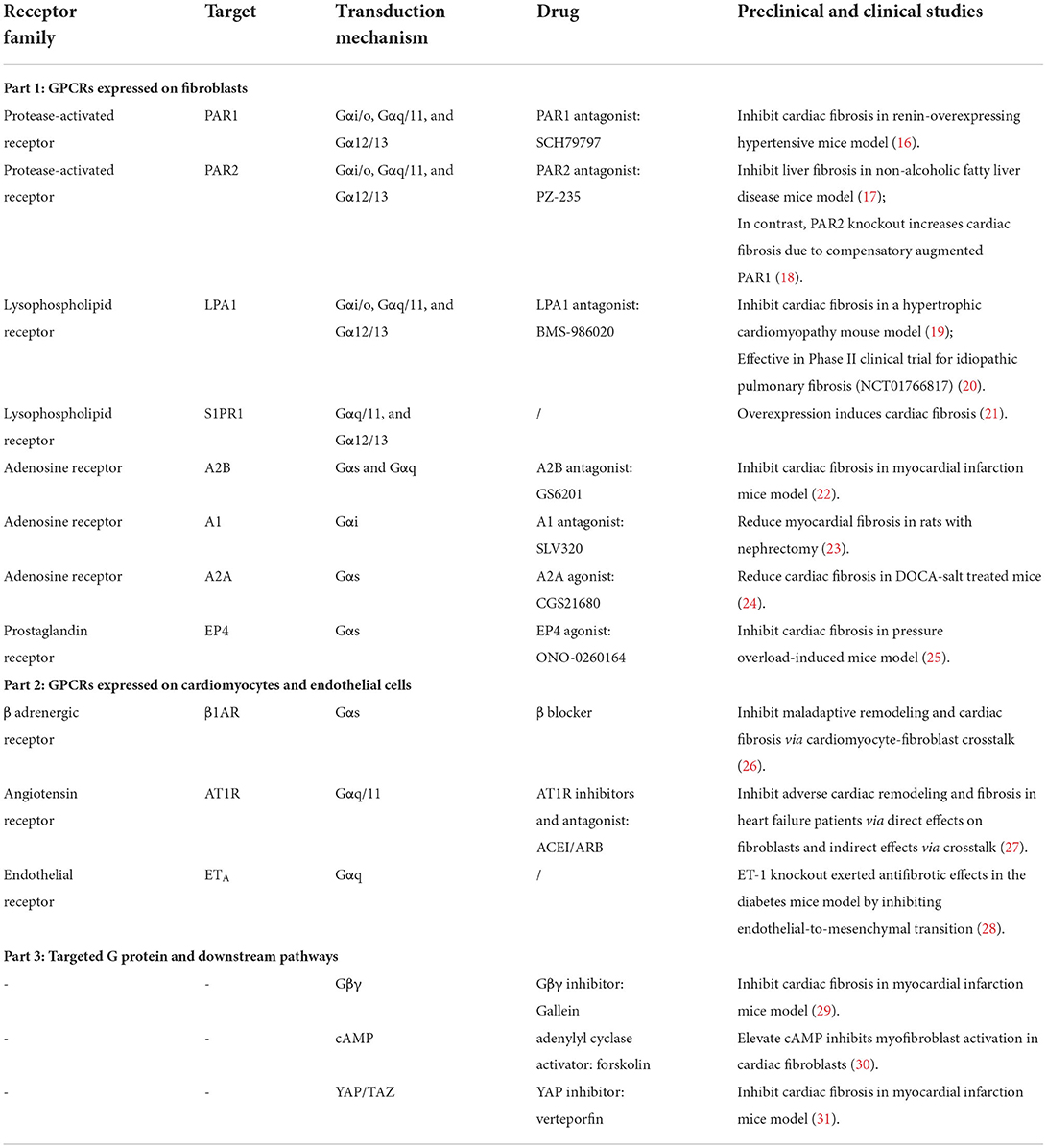
Table 1. Representative GPCRs and downstream pathways involved in fibrosis regulation.
GPCR signaling may also significantly vary within the cell. To date, 21 Gα, 6 Gβ, and 12 Gγ have been discovered (14). Heterotrimeric G proteins are generally identified by their Gα subunits and classified into four major groups: Gα stimulatory (Gαs), Gα inhibitory (Gαi), Gαq, and Gα12/13 (14). Different G-protein subfamilies execute distinct signaling cascades. Gαs stimulates adenylyl cyclase to increase the second messenger cyclic adenosine monophosphate (cAMP), which results in protein kinase A (PKA) activation and subsequent phosphorylation of intracellular proteins (32). Conversely, Gαi exhibits a suppressive effect on adenylyl cyclase, decreasing intracellular cAMP (32). Gαq activates phospholipase C (PLC), resulting in phosphatidylinositol 4,5–bisphosphate (PIP2) cleavage and producing the second messengers inositol 1,4,5–triphosphate (IP3) and diacylglycerol (DAG). IP3 triggers Ca2+ release from the endoplasmic reticulum, along with DAG to activate protein kinase C (PKC) (33). Gα12/13 can activate the small GTPase Rho, which serves as a regulator of a number of intracellular processes, including actin stress fibers formation and cell growth controller (34). GPCRs exist widely and heterogeneously in a cell-specific distribution among the three major cell types found within the heart (i.e., cardiomyocytes, endothelial cells, cardiac fibroblasts) (35, 36).
Besides the G protein signaling cascades, β-arrestins have the capacity to regulate GPCR activity independently (37). Previously, β-arrestins were discovered to mediate receptor internalization and desensitization, exemplified by the role of β-arrestin in β1 adrenergic receptors (β1AR) desensitization in cardiomyocytes (38). Later, other functions of β-arrestins were identified as signal transducers to regulate a variety of intracellular signaling pathways (39), including transforming growth factor-β (TGF-β), as well as downstream kinases such as mitogen-activated protein kinase (MAPK) and phosphoinositide 3-kinase (PI3K) (40), which are highly involved in the regulation of fibrosis.
The GPCR expression level depends on a steady-state transport of receptors between the cell membrane and endosomes (41). Agonists of GPCRs induce internalization that alters the membrane expression level in the short term. Prolonged over-stimulation leads to a downregulation of these receptors at the transcriptional level (41). For example, β1AR and β2AR are the predominant GPCR subtypes expressed in cardiomyocytes and the primary regulators of cardiovascular function (42). Epinephrine stimulates β2AR and initially activates Gs, which increases the beating rate. However, after 10–15 min of stimulation, β2AR signals predominantly through Gi, which decreases the contraction rate (43). Therefore, pathophysiological states may alter not only the expression of these receptors, but also the coupling specificity between the receptors and G proteins (41).
The diverse and cell-specific membrane expression of GPCRs, combined with their important roles in pathophysiology, make them frequent therapeutic targets, leading to ~35% of all FDA-approved drugs (13). A variety of GPCRs have been implicated in the pathogenesis of fibrosis (44, 45). Although several earlier drug discovery campaigns have targeted GPCRs to treat fibrosis, effective treatment is still lacking (44). As fibroblasts among different organs share similar features (46), we will discuss the common and distinct features of those GPCRs involved in cardiac fibrosis compared to fibrosis in other organs.
Protease-activated receptors (PAR) become activated from specific cleavage of the amino-terminal sequence that consequently exposes a new N-terminal sequence functioned as a tethered ligand (47) (Figure 1). Activation of protease-activated receptor-1 (PAR1), which couples to Gq/11, Gi/o, and G12/13, elevates the level of profibrotic gene expression in rat cardiac fibroblasts, leading to myofibroblast activation and increasing collagen synthesis by 60% (48). Conversely, inhibiting PAR1 with SCH79797 attenuates cardiac fibrosis and hypertrophy in the renin-overexpressing induced hypertensive mice model (16). However, PAR1 is highly expressed in platelets, and its antagonist is used for antiplatelet therapy (49). The off-target effect restricts the application of this target for fibrosis treatment. Protease-activated receptor-2 (PAR2), which also couples to Gq/11, Gi/o, and G12/13, is expressed on liver stellate cells that regulate the response to liver cirrhosis (17). PAR2 inhibitor PZ-235 has significantly suppressed liver fibrosis and collagen deposition up to 50–100% (17). In contrast, 1-year-old PAR2-knockout mice suffered from diastolic dysfunction, associated with an increased α-SMA, collagen deposition, lysyl oxidase activity, and collagen cross-linking. The absence of PAR2 contributed to an augmented profibrotic PAR1 and dependent signaling in heart (18), indicating that the same GPCR may exert a distinct role in fibrosis regulation among different organs.
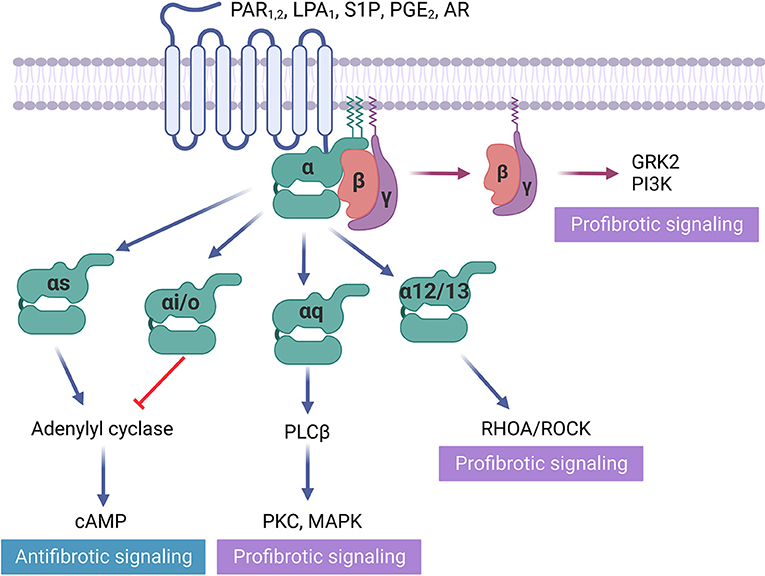
Figure 1. GPCRs in fibrosis regulation. Some GPCRs mediate profibrotic signals, including protease-activated receptors (PAR1), lysophospholipid receptors (LPA1, S1P), and adenosine receptors (AR), that activate receptors coupled to Gαi/o, Gαq/11, Gα12/13. Gβγ activation is also proved to be profibrotic. In contrast, prostaglandin receptor (PGE2), which activates receptors Gαs and direct cAMP activator exert antifibrotic effects.
The lysophospholipid receptor group is a member of the GPCR family of integral membrane proteins essential for lipid signaling (50). Lysophosphatidic acid (LPA) and its G protein-coupled receptor (LPA1) contribute to fibrosis progression (51). LPA1 is highly expressed on human primary fibroblasts, which predominantly couples to Gαq/11, Gαi/o, and Gα12/13 proteins (52). Myocardial fibrosis is a key pathologic feature of hypertrophic cardiomyopathy (HCM). In the HCM mouse model, LPA1 ablation developed significantly less hypertrophy and fibrosis (19). Since LPA1 antagonist BMS-986020 has been tested in Phase II clinical trial for idiopathic pulmonary fibrosis (20), it could potentially be used to treat cardiac fibrosis in the future. Sphingosine 1 phosphate receptor-1 (S1PR1), which also belongs to the lysophospholipid receptor family, couples to Gαq/11, Gαi/o, and Gα12/13. Its overexpression was capable to induces cardiac hypertrophy and fibrosis through angiotensin II and interleukin-6 in S1PR1-transgenic mouse heart (21).
In addition to adenosine and its receptor's crucial role in wound healing (53), they have also been found to promote fibrosis by producing excess matrix in the heart, skin, lungs, and liver (54). The adenosine receptor (AR) family comprises four GPCRs: A1, A2A, A2B, and A3 (55). Previous studies have suggested AR antagonists as an effective fibrosis treatment. For example, caffeine, a non-selective adenosine receptor antagonist, has been shown to alleviate liver fibrosis in animal models and to reduce liver fibrosis in patients with chronic hepatitis C (56, 57). However, the weak affinity of caffeine to ARs requires micromolar concentration in plasma, which prevents its potential for drug development.
A pro-fibrotic role for the A2B, which couples to Gαs and Gαq, has been supported by a study using A2B knockout mice, which exhibited improved diastolic dysfunction and attenuated interstitial fibrosis 8 weeks after MI (22). Selective A2B antagonist, GS-6201, significantly reduced cardiac enlargement and dysfunction compared to vehicle in the mouse MI model (58). Similarly, in the rat model of myocardial ischemia-reperfusion, GS-6201 improved ejection fraction and decreased fibrosis in the non-infarct and border zones (59). In addition, the adenosine A1 receptor (i.e., a Gαi coupled receptor) antagonist SLV320 reduces myocardial fibrosis in rats with nephrectomy (23). In contrast, the activation of A2A (i.e., a Gαs coupled receptor) by CGS21680 reduced cardiomyocyte hypertrophy, cardiac inflammation, and fibrosis in deoxycorticosterone acetate (DOCA) treated mice (24). These interesting findings suggest the effects of ARs on myocardial adaptation are subtype-specific. The distinct role of AR subtypes in cardiac fibrosis regulation needs further investigation.
Prostaglandin E2 (PGE2) receptor, which couples to Gαs, acts via cAMP and is abundantly expressed in fibroblasts (60). Prostaglandin E2 receptor 4 (EP4) stimulation was reported to be cardioprotective. Its agonist ONO-0260164 significantly prevented systolic dysfunction and the progression of myocardial fibrosis in the mouse model of cardiac hypertrophy induced by transverse aortic constriction (25). Conversely, EP4 knockout mice exhibited concentric hypertrophy and myocardial fibrosis in mice fed with high-fat diet (61).
Of particular relevance to cardiac fibrosis compared to fibrosis in other organs are the distinct features of the heart tissue environment (4). In the heart, the crosstalk between fibroblasts, cardiomyocytes, and endothelial cells plays an important role in fibrosis regulation (62). Different types of intercellular crosstalk exist, including direct contact communication via gap junction or nanotubes, indirect cell interaction via paracrine factors, and cell-extracellular matrix (ECM) interaction (63). In particular, several GPCRs and downstream pathways have been involved in cardiac fibrosis regulation via the crosstalk mechanism (Figure 2).
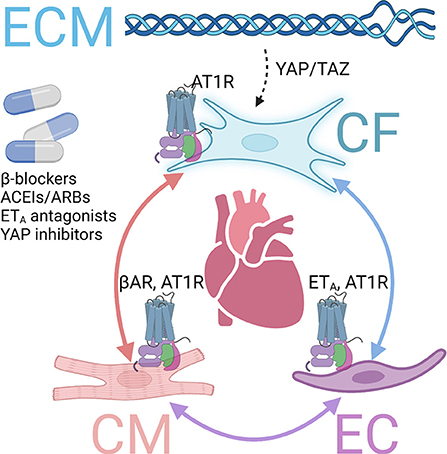
Figure 2. GPCRs involved in fibrosis via crosstalk mechanisms. Several GPCRs contribute to cardiac fibrosis via interactions among cardiomyocytes (CMs), endothelial cells (ECs), cardiac fibroblasts (CFs), and extracellular matrix (ECM). Inhibiting those and inhibiting those GPCRs or downstream pathways exert antifibrotic effects in the heart. βAR, β adrenergic receptor. AT1R, angiotensin receptor type 1. ETA, endothelin receptor A.
The stimulation of β1AR on cardiomyocytes increases contractility through PKA-mediated phosphorylation of specific regulatory proteins to increase intracellular Ca2+ level or sensitivity, such as L-type Ca2+ channels, phospholamban, ryanodine receptor, and troponin I (64). However, long-term stimulation of β1AR leads to cardiac hypertrophy and fibrosis, progressively developing into heart failure (26). Although β-blockers cannot directly prevent MyoFB activation, those drugs have been demonstrated to prevent cardiac fibrosis and improve survival in mice models and clinics via cardiomyocyte-fibroblast communications (26).
Angiotensin receptors, particularly AT1R, play an important role in heart pathophysiology (65). Cardiac AT1R, coupled to Gαq, is upregulated with hypertrophic and ischemia, promoting adverse maladaptive cardiac remodeling, including cardiac fibrosis in chronic heart failure (66). Ang II, the endogenous ligand, is a peptide hormone that regulates several critical physiological processes, representing a principal component of the renin-angiotensin-aldosterone system (66). AT1R is expressed in cardiomyocytes, endothelial cells, and cardiac fibroblasts. Its overexpression lead to cardiac fibrosis and hypertrophy, whereas knockout of AT1R improved cardiac function (67). The mechanisms of profibrotic AT1R are multifold, including direct effects on fibroblasts and, more importantly, via crosstalk between fibroblasts, cardiomyocytes, or endothelial cells (62). Due to its central role in cardiovascular pathophysiology, AT1R inhibitors and antagonists, ACEI and ARB, are essential drugs for heart failure treatment and have been proven to protect against maladaptive remodeling and cardiac fibrosis (27).
Among the endothelial receptors, ETA is the major isoform found in the cardiovascular system (68). Chronic stimulation with ETA agonist ET-1 is associated with adverse effects, including cardiac hypertrophy and fibrosis (4). High levels of ET-1 in plasma have been found in heart failure patients (68). ETA, which couples to Gαq, promotes IP formation and activates MAPK signaling (69). ET1 knockout exerted antifibrotic effects in the diabetes mice model via endothelial-fibroblast interaction (70).
Increased ECM stiffness and tension are known to activate fibroblasts through the yes-associated protein 1 (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) signaling pathway, which mediates the mechanical sensing of cardiac fibroblasts in fibrotic diseases (71). The Hippo-YAP-TAZ pathway is mediated by multiple G proteins, and therefore involved in common GPCR downstream pathways (72). Fibroblast conditional deletion of Yap1 attenuated injury-induced cardiac fibrosis either after acute MI or chronic angiotensin II–phenylephrine stimulation (73).
Multiple profibrotic GPCRs are co-expressed in fibroblasts, indicating the possibility of redundancy of G protein activity in myofibroblast activation mechanisms. These results challenge the effectiveness of antifibrotic therapy by antagonizing one single GPCR. To address the redundancy issue, one strategy is to target the G protein itself or convergent downstream pathways.
GPCRs coupled to Gαs activate adenylyl cyclase to elevate the intracellular cAMP level (14). To examine the consequence of Gαs signaling, forskolin is used as an activator of adenyl cyclase (AC) to increase cAMP. Experiments using forskolin to activate AC have identified that this pathway results in the inhibition of TGF-β-induced fibroblast activation (74). In rat cardiac fibroblasts, elevating cAMP formation by AC overexpression or by forskolin attenuates myofibroblast marker gene expression and collagen synthesis (30).
G protein βγ subunits were identified as G protein components almost 30 years ago (75). Since then, Gβγ signaling has been demonstrated as diverse, regulating a number of downstream pathways depending on the interacting effectors. For example, Gβγ has the capacity to activate G protein-coupled inward rectifier K+ (GIRK) channels, phospholipase A and C, plasma membrane Ca2+ pump, PI3K-AKT, GPCR kinase 2 (GRK2), and guanine exchange factors (GEFs) for small G proteins (76). Moreover, a small molecule Gβγ inhibitor Gallein has been shown to inhibit myofibroblast activation in vitro (77). Gallein treatment a week after ischemic/reperfusion is effective in reducing cardiac fibrosis and preserving ejection fraction in a mouse model of chronic heart failure (29). Since GPCRs are comprised of α and βγ subunits, differentiating the effects of Gas (i.e., antifibrotic) from that of Gβγ (i.e., fibrotic) is challenging and may depend on factors such as cell type of receptor expression.
As previously described, Hippo-YAP is the common downstream pathway of multiple G proteins (72). Specially, activation of lysophospholipid receptors, such as LPA and S1P, coupled simultaneously with Gαi, Gas, and Gα12/13, inhibit the Hippo signaling kinases 1/2 (Lats1/2) (72). The inhibition of Lats1/2 then stimulates the transcription coactivators YAP and TAZ, which are related to cell proliferation and migration (72). YAP and TAZ have recently been identified as promotors for myofibroblast activation in cardiac fibrosis (31). Interestingly, a previous study indicated that the suppression of YAP/TAZ signaling by lovastatin attenuates angiotensin II-induced cardiac fibrosis, both in vitro and in vivo (78).
Given that fibrosis may develop from multiple stimuli, one approach is to explore polypharmacology, which has the capacity to simultaneously target multiple receptors and signaling cascades (79). Polypharmacology has the potential to produce higher efficacy drugs and reduced drug resistance (80). For example, one of the FDA-approved drugs for idiopathic lung fibrosis, nintedanib, simultaneously targets fibroblast growth factor receptors, platelet-derived growth factor receptors, and vascular endothelial growth factor receptors (81). With the advancements in GPCR structural biology and in-silico modeling, compounds can be rationally predicted, modified, and designed to target multiple receptors (41, 82).
Another potential challenge for developing GPCR regulators for antifibrotic therapy is the widespread expression of these receptors among different cell types. Targeting the receptor may exert off-target effects. For example, PAR1 inhibition may not only attenuate myofibroblast activation, but also impair platelet function due to PAR1's high expression on platelets (49). Similarly, AR inhibition by caffeine inhibits liver fibrosis, but may simultaneously alter neurological states because of AR's high expression in neurons (83). Recent published single-cell RNA sequencing (sgRNA-seq) and single-nucleus RNA sequencing (snRNA-seq) datasets from human hearts demonstrated distinct fibrotic responses in various types of cardiomyopathies (28, 84–86). Therefore, multiple signaling pathways and crosstalk can be exploited, as they offer a rich blueprint of receptor distribution across different cell types and organs. By leveraging the single-cell sequencing database, we can identify GPCRs more specifically expressed on fibroblasts at a given tissue and upregulated during fibrosis to reduce potential off-target effects (28, 87).
GPCR targeting approaches have yet to produce FDA-approved drugs for fibrotic diseases. Nevertheless, targeting GPCRs offers a tantalizing and promising therapy for treating cardiac fibrosis, as more GPCR-targeted antifibrotic therapies continue to enter clinical trials. Several challenges remain, and major questions need to be addressed before GPCR-mediated therapies for cardiac fibrosis are realized. The redundancy of profibrotic G protein signals and widespread GPCR expressions must be comprehensively considered to determine and refine our approach for antifibrotic therapy. A few solutions have been proposed to circumvent the challenges. Candidate drugs with polypharmacology or targeting common downstream signaling pathways have the potential to overcome the redundancy challenge.
On the other hand, more specific targets should be identified to minimize off-target effects by single-cell seq datasets of cardiomyopathy. The unique cardiac environment provides opportunities to target GPCRs highly specific expressed in the heart via crosstalk between fibroblasts, cardiomyocytes, endothelial cells, and ECM. Furthermore, the human induced pluripotent stem cell platform can be utilized as a high throughput and more complex human cellular model platform to complement animal models for drug development (88, 89). Collectively, these advances in our knowledge of GPCRs in cardiac fibrosis provide a unique perspective on the challenges and opportunities in exploring GPCRs to treat cardiac fibrosis.
HZ and LR prepared the illustration, organized, and wrote the manuscript. RS wrote and edited the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by American Heart Association 828308 (HZ).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM, et al. Global burden of cardiovascular diseases and risk factors, 1990-2019: update from the GBD 2019 study. J Am Coll Cardiol. (2020) 76:2982–3021. doi: 10.1016/j.jacc.2020.11.010
2. Hinderer S, Schenke-Layland K. Cardiac fibrosis - a short review of causes and therapeutic strategies. Adv Drug Deliv Rev. (2019) 146:77–82. doi: 10.1016/j.addr.2019.05.011
4. Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci. (2014) 71:549–74. doi: 10.1007/s00018-013-1349-6
5. Richardson WJ, Clarke SA, Quinn TA, Holmes JW. Physiological implications of myocardial scar structure. Compr Physiol. (2015) 5:1877–909. doi: 10.1002/cphy.c140067
6. Talman V, Ruskoaho H. Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res. (2016) 365:563–81. doi: 10.1007/s00441-016-2431-9
7. Tallquist MD, Molkentin JD. Redefining the identity of cardiac fibroblasts. Nat Rev Cardiol. (2017) 14:484–91. doi: 10.1038/nrcardio.2017.57
8. Shinde AV, Humeres C, Frangogiannis NG. The role of α-smooth muscle actin in fibroblast-mediated matrix contraction and remodeling. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:298–309. doi: 10.1016/j.bbadis.2016.11.006
9. Santiago JJ, Dangerfield AL, Rattan SG, Bathe KL, Cunnington RH, Raizman JE, et al. Cardiac fibroblast to myofibroblast differentiation in vivo and in vitro: expression of focal adhesion components in neonatal and adult rat ventricular myofibroblasts. Dev Dyn. (2010) 239:1573–84. doi: 10.1002/dvdy.22280
10. Asmani M, Velumani S, Li Y, Wawrzyniak N, Hsia I, Chen Z, et al. Fibrotic microtissue array to predict anti-fibrosis drug efficacy. Nat Commun. (2018) 9:2066. doi: 10.1038/s41467-018-04336-z
11. Lewis GA, Dodd S, Clayton D, Bedson E, Eccleson H, Schelbert EB, et al. Pirfenidone in heart failure with preserved ejection fraction: a randomized phase 2 trial. Nat Med. (2021) 27:1477–82. doi: 10.1038/s41591-021-01452-0
12. Lewis GA, Rosala-Hallas A, Dodd S, Schelbert EB, Williams SG, Cunnington C, et al. Impact of myocardial fibrosis on cardiovascular structure, function and functional status in heart failure with preserved ejection fraction. J Cardiovasc Transl Res. (2022). doi: 10.21203/rs.3.rs-1332963/v1. [Epub ahead of print].
13. Sriram K, Insel PA, G. Protein-coupled receptors as targets for approved drugs: how many targets and how many drugs? Mol Pharmacol. (2018) 93:251–8. doi: 10.1124/mol.117.111062
14. Hauser AS, Attwood MM, Rask-Andersen M, Schiöth HB, Gloriam DE. Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov. (2017) 16:829–42. doi: 10.1038/nrd.2017.178
15. Gacasan SB, Baker DL, Parrill AL, G. protein-coupled receptors: the evolution of structural insight. AIMS Biophys. (2017) 4:491–527. doi: 10.3934/biophy.2017.3.491
16. Yokono Y, Hanada K, Narita M, Tatara Y, Kawamura Y, Miura N, et al. Blockade of PAR-1 signaling attenuates cardiac hypertrophy and fibrosis in renin-overexpressing hypertensive mice. J Am Heart Assoc. (2020) 9:e015616. doi: 10.1161/JAHA.119.015616
17. Shearer AM, Rana R, Austin K, Baleja JD, Nguyen N, Bohm A, et al. Targeting liver fibrosis with a cell-penetrating protease-activated receptor-2 (PAR2) pepducin. J Biol Chem. (2016) 291:23188–98. doi: 10.1074/jbc.M116.732743
18. Friebel J, Weithauser A, Witkowski M, Rauch BH, Savvatis K, Dörner A, et al. Protease-activated receptor 2 deficiency mediates cardiac fibrosis and diastolic dysfunction. Eur Heart J. (2019) 40:3318–32. doi: 10.1093/eurheartj/ehz117
19. Axelsson Raja A, Wakimoto H, DeLaughter DM, Reichart D, Gorham J, Conner DA, et al. Ablation of lysophosphatidic acid receptor 1 attenuates hypertrophic cardiomyopathy in a mouse model. Proc Natl Acad Sci U S A. (2022) 119:e2204174119. doi: 10.1073/pnas.2204174119
20. Decato BE, Leeming DJ, Sand JMB, Fischer A, Du S, Palmer SM, et al. LPA(1) antagonist BMS-986020 changes collagen dynamics and exerts antifibrotic effects in vitro and in patients with idiopathic pulmonary fibrosis. Respir Res. (2022) 23:61. doi: 10.1186/s12931-022-01980-4
21. Ohkura SI, Usui S, Takashima SI, Takuwa N, Yoshioka K, Okamoto Y, et al. Augmented sphingosine 1 phosphate receptor-1 signaling in cardiac fibroblasts induces cardiac hypertrophy and fibrosis through angiotensin II and interleukin-6. PLoS ONE. (2017) 12:e0182329. doi: 10.1371/journal.pone.0182329
22. Vecchio EA, White PJ, May LT. Targeting adenosine receptors for the treatment of cardiac fibrosis. Front Pharmacol. (2017) 8:243. doi: 10.3389/fphar.2017.00243
23. Kalk P, Eggert B, Relle K, Godes M, Heiden S, Sharkovska Y, et al. The adenosine A1 receptor antagonist SLV320 reduces myocardial fibrosis in rats with 5/6 nephrectomy without affecting blood pressure. Br J Pharmacol. (2007) 151:1025–32. doi: 10.1038/sj.bjp.0707319
24. Zhou YP, Ruan CC, Kong LR, Gao PJ. Adenosine A(2A) receptor activation prevents DOCA-salt induced hypertensive cardiac remodeling via iBAT. Biochem Biophys Res Commun. (2020). doi: 10.1016/j.bbrc.2020.02.035. [Epub ahead of print].
25. Wang Q, Oka T, Yamagami K, Lee JK, Akazawa H, Naito AT, et al. An EP4 receptor agonist inhibits cardiac fibrosis through activation of PKA signaling in hypertrophied heart. Int Heart J. (2017) 58:107–14. doi: 10.1536/ihj.16-200
26. Bristow MR. beta-adrenergic receptor blockade in chronic heart failure. Circulation. (2000) 101:558–69. doi: 10.1161/01.CIR.101.5.558
27. Funabiki K, Onishi K, Dohi K, Koji T, Imanaka-Yoshida K, Ito M, et al. Combined angiotensin receptor blocker and ACE inhibitor on myocardial fibrosis and left ventricular stiffness in dogs with heart failure. Am J Physiol Heart Circ Physiol. (2004) 287:H2487–92. doi: 10.1152/ajpheart.00462.2004
28. Tabula Muris Consortium Overall coordination Logistical coordination Organ collection and processing Library preparation and sequencing Computational data analysis. Single-cell transcriptomics of 20 mouse organs creates a Tabula Muris. Nature. (2018) 562:367–72. doi: 10.1038/s41586-018-0590-4
29. Travers JG, Kamal FA, Valiente-Alandi I, Nieman ML, Sargent MA, Lorenz JN, et al. Pharmacological and activated fibroblast targeting of Gβγ-GRK2 after myocardial ischemia attenuates heart failure progression. J Am Coll Cardiol. (2017) 70:958–71. doi: 10.1016/j.jacc.2017.06.049
30. Swaney JS, Roth DM, Olson ER, Naugle JE, Meszaros JG, Insel PA. Inhibition of cardiac myofibroblast formation and collagen synthesis by activation and overexpression of adenylyl cyclase. Proc Natl Acad Sci U S A. (2005) 102:437–42. doi: 10.1073/pnas.0408704102
31. Garoffolo G, Casaburo M, Amadeo F, Salvi M, Bernava G, Piacentini L, et al. Reduction of cardiac fibrosis by interference with YAP-dependent transactivation. Circ Res. (2022) 131:239–57. doi: 10.1161/CIRCRESAHA.121.319373
32. Van Eps N, Altenbach C, Caro LN, Latorraca NR, Hollingsworth SA, Dror RO, et al. G(i)- and G(s)-coupled GPCRs show different modes of G-protein binding. Proc Natl Acad Sci U S A. (2018) 115:2383–8. doi: 10.1073/pnas.1721896115
33. Mizuno N, Itoh H. Functions and regulatory mechanisms of Gq-signaling pathways. Neurosignals. (2009) 17:42–54. doi: 10.1159/000186689
34. Suzuki N, Hajicek N, Kozasa T. Regulation and physiological functions of G12/13-mediated signaling pathways. Neurosignals. (2009) 17:55–70. doi: 10.1159/000186690
35. Salazar NC, Chen J, Rockman HA. Cardiac GPCRs: GPCR signaling in healthy and failing hearts. Biochim Biophys Acta. (2007) 1768:1006–18. doi: 10.1016/j.bbamem.2007.02.010
36. Wang J, Gareri C, Rockman HA. G-Protein-coupled receptors in heart disease. Circ Res. (2018) 123:716–35. doi: 10.1161/CIRCRESAHA.118.311403
37. Jean-Charles PY, Kaur S, Shenoy SK, G. Protein-coupled receptor signaling through β-arrestin-dependent mechanisms. J Cardiovasc Pharmacol. (2017) 70:142–58. doi: 10.1097/FJC.0000000000000482
38. van Gastel J, Hendrickx JO, Leysen H, Santos-Otte P, Luttrell LM, Martin B, et al. β-Arrestin based receptor signaling paradigms: potential therapeutic targets for complex age-related disorders. Front Pharmacol. (2018) 9:1369. doi: 10.3389/fphar.2018.01369
39. Miller WE, Maudsley S, Ahn S, Khan KD, Luttrell LM, Lefkowitz RJ. beta-arrestin1 interacts with the catalytic domain of the tyrosine kinase c-SRC. role of beta-arrestin1-dependent targeting of c-SRC in receptor endocytosis. J Biol Chem. (2000) 275:11312–9. doi: 10.1074/jbc.275.15.11312
40. Gu YJ, Sun WY, Zhang S, Wu JJ, Wei W. The emerging roles of β-arrestins in fibrotic diseases. Acta Pharmacol Sin. (2015) 36:1277–87. doi: 10.1038/aps.2015.74
41. Congreve M, de Graaf C, Swain NA, Tate CG. Impact of GPCR structures on drug discovery. Cell. (2020) 181:81–91. doi: 10.1016/j.cell.2020.03.003
42. Madamanchi A. Beta-adrenergic receptor signaling in cardiac function and heart failure. Mcgill J Med. (2007) 10:99–104. doi: 10.26443/mjm.v10i2.458
43. Strohman MJ, Maeda S, Hilger D, Masureel M, Du Y, Kobilka BK. Local membrane charge regulates β(2) adrenergic receptor coupling to G(i3). Nat Commun. (2019) 10:2234. doi: 10.1038/s41467-019-10108-0
44. Haak AJ, Ducharme MT, Diaz Espinosa AM, Tschumperlin DJ. Targeting GPCR signaling for idiopathic pulmonary fibrosis therapies. Trends Pharmacol Sci. (2020) 41:172–82. doi: 10.1016/j.tips.2019.12.008
45. Kimura T, Singh S, Tanaka N, Umemura T. Role of G protein-coupled receptors in hepatic stellate cells and approaches to anti-fibrotic treatment of non-alcoholic fatty liver disease. Front Endocrinol. (2021) 12:773432. doi: 10.3389/fendo.2021.773432
46. Distler JHW, Györfi AH, Ramanujam M, Whitfield ML, Königshoff M, Lafyatis R. Shared and distinct mechanisms of fibrosis. Nat Rev Rheumatol. (2019) 15:705–30. doi: 10.1038/s41584-019-0322-7
47. Heuberger DM, Schuepbach RA. Protease-activated receptors (PARs): mechanisms of action and potential therapeutic modulators in PAR-driven inflammatory diseases. Thromb J. (2019) 17:4. doi: 10.1186/s12959-019-0194-8
48. Snead AN, Insel PA. Defining the cellular repertoire of GPCRs identifies a profibrotic role for the most highly expressed receptor, protease-activated receptor 1, in cardiac fibroblasts. FASEB J. (2012) 26:4540–7. doi: 10.1096/fj.12-213496
49. Lee S. Discovery of an orally available PAR-1 antagonist as a novel antiplatelet agent. Arch Pharm Res. (2011) 34:515–7. doi: 10.1007/s12272-011-0421-8
50. Riaz A, Huang Y, Johansson S. G-Protein-coupled lysophosphatidic acid receptors and their regulation of AKT signaling. Int J Mol Sci. (2016) 17:215. doi: 10.3390/ijms17020215
51. Taleb SJ, Wei J, Mialki RK, Dong S, Li Y, Zhao J, et al. A blocking peptide stabilizes lysophosphatidic acid receptor 1 and promotes lysophosphatidic acid-induced cellular responses. J Cell Biochem. (2021) 122:827–34. doi: 10.1002/jcb.29919
52. Tager AM, LaCamera P, Shea BS, Campanella GS, Selman M, Zhao Z, et al. The lysophosphatidic acid receptor LPA1 links pulmonary fibrosis to lung injury by mediating fibroblast recruitment and vascular leak. Nat Med. (2008) 14:45–54. doi: 10.1038/nm1685
53. Feoktistov I, Biaggioni I, Cronstein BN. Adenosine receptors in wound healing, fibrosis and angiogenesis. Handb Exp Pharmacol. (2009) 193:383-97. doi: 10.1007/978-3-540-89615-9_13
54. Chan ES, Cronstein BN. Adenosine in fibrosis. Mod Rheumatol. (2010) 20:114–22. doi: 10.3109/s10165-009-0251-4
55. Chen JF, Eltzschig HK, Fredholm BB. Adenosine receptors as drug targets–what are the challenges? Nat Rev Drug Discov. (2013) 12:265–86. doi: 10.1038/nrd3955
56. Modi AA, Feld JJ, Park Y, Kleiner DE, Everhart JE, Liang TJ, et al. Increased caffeine consumption is associated with reduced hepatic fibrosis. Hepatology. (2010) 51:201–9. doi: 10.1002/hep.23279
57. Vitaglione P, Morisco F, Mazzone G, Amoruso DC, Ribecco MT, Romano A, et al. Coffee reduces liver damage in a rat model of steatohepatitis: the underlying mechanisms and the role of polyphenols and melanoidins. Hepatology. (2010) 52:1652–61. doi: 10.1002/hep.23902
58. Toldo S, Zhong H, Mezzaroma E, Van Tassell BW, Kannan H, Zeng D, et al. GS-6201, a selective blocker of the A2B adenosine receptor, attenuates cardiac remodeling after acute myocardial infarction in the mouse. J Pharmacol Exp Ther. (2012) 343:587–95. doi: 10.1124/jpet.111.191288
59. Zhang H, Zhong H, Everett THt, Wilson E, Chang R, Zeng D, et al. Blockade of A2B adenosine receptor reduces left ventricular dysfunction and ventricular arrhythmias 1 week after myocardial infarction in the rat model. Heart Rhythm. (2014) 11:101–9. doi: 10.1016/j.hrthm.2013.10.023
60. Elwakeel E, Brüne B, Weigert A. PGE(2) in fibrosis and cancer: insights into fibroblast activation. Prostaglandins Other Lipid Mediat. (2019) 143:106339. doi: 10.1016/j.prostaglandins.2019.106339
61. Ying F, Liu H, Ching Tang EH, Lakhani I, Liu N, Xia Z, et al. Prostaglandin E receptor subtype 4 protects against diabetic cardiomyopathy by modulating cardiac fatty acid metabolism via FOXO1/CD36 signalling. Biochem Biophys Res Commun. (2021) 548:196–203. doi: 10.1016/j.bbrc.2021.01.038
62. Hall C, Gehmlich K, Denning C, Pavlovic D. Complex relationship between cardiac fibroblasts and cardiomyocytes in health and disease. J Am Heart Assoc. (2021) 10:e019338. doi: 10.1161/JAHA.120.019338
63. Nicin L, Wagner JUG, Luxán G, Dimmeler S. Fibroblast-mediated intercellular crosstalk in the healthy and diseased heart. FEBS Lett. (2022) 596:638–54. doi: 10.1002/1873-3468.14234
64. Lohse MJ, Engelhardt S, Eschenhagen T. What is the role of beta-adrenergic signaling in heart failure? Circ Res. (2003) 93:896–906. doi: 10.1161/01.RES.0000102042.83024.CA
65. De Mello WC, Danser AH. Angiotensin II and the heart: on the intracrine renin-angiotensin system. Hypertension. (2000) 35:1183–8. doi: 10.1161/01.HYP.35.6.1183
66. Kawai T, Forrester SJ, O'Brien S, Baggett A, Rizzo V, Eguchi S. AT1 receptor signaling pathways in the cardiovascular system. Pharmacol Res. (2017) 125:4–13. doi: 10.1016/j.phrs.2017.05.008
67. Dasgupta C, Zhang L. Angiotensin II receptors and drug discovery in cardiovascular disease. Drug Discov Today. (2011) 16:22–34. doi: 10.1016/j.drudis.2010.11.016
68. Leary PJ, Jenny NS, Bluemke DA, Kawut SM, Kronmal RA, Lima JA, et al. Hinckley Stukovsky, Tedford RJ, Endothelin-1, cardiac morphology, heart failure: the MESA angiogenesis study. J Heart Lung Transplant. (2020) 39:45–52. doi: 10.1016/j.healun.2019.07.007
69. Hersch E, Huang J, Grider JR, Murthy KS. Gq/G13 signaling by ET-1 in smooth muscle: MYPT1 phosphorylation via ETA and CPI-17 dephosphorylation via ETB. Am J Physiol Cell Physiol. (2004) 287:C1209–18. doi: 10.1152/ajpcell.00198.2004
70. Widyantoro B, Emoto N, Nakayama K, Anggrahini DW, Adiarto S, Iwasa N, et al. Endothelial cell-derived endothelin-1 promotes cardiac fibrosis in diabetic hearts through stimulation of endothelial-to-mesenchymal transition. Circulation. (2010) 121:2407–18. doi: 10.1161/CIRCULATIONAHA.110.938217
71. Humphrey JD, Dufresne ER, Schwartz MA. Mechanotransduction and extracellular matrix homeostasis. Nat Rev Mol Cell Biol. (2014) 15:802–12. doi: 10.1038/nrm3896
72. Yu FX, Zhao B, Panupinthu N, Jewell JL, Lian I, Wang LH, et al. Regulation of the Hippo-YAP pathway by G-protein-coupled receptor signaling. Cell. (2012) 150:780–91. doi: 10.1016/j.cell.2012.06.037
73. Francisco J, Zhang Y, Jeong JI, Mizushima W, Ikeda S, Ivessa A, et al. Blockade of fibroblast YAP attenuates cardiac fibrosis and dysfunction through MRTF-A inhibition. JACC Basic Transl Sci. (2020) 5:931–45. doi: 10.1016/j.jacbts.2020.07.009
74. Schiller M, Dennler S, Anderegg U, Kokot A, Simon JC, Luger TA, et al. Increased cAMP levels modulate transforming growth factor-beta/Smad-induced expression of extracellular matrix components and other key fibroblast effector functions. J Biol Chem. (2010) 285:409–21. doi: 10.1074/jbc.M109.038620
75. Kramer GM, Jones CD, Xie K, Martemyanov KA. Distinct profiles of functional discrimination among G proteins determine the actions of G protein-coupled receptors. Sci Signal. (2015) 8:ra123. doi: 10.1126/scisignal.aab4068
76. Dupré DJ, Robitaille M, Rebois RV, Hébert TE. The role of Gbetagamma subunits in the organization, assembly, and function of GPCR signaling complexes. Annu Rev Pharmacol Toxicol. (2009) 49:31–56. doi: 10.1146/annurev-pharmtox-061008-103038
77. Kamal FA, Travers JG, Schafer AE, Ma Q, Devarajan P, Blaxall BC, et al. Protein-coupled receptor-G-Protein βγ-subunit signaling mediates renal dysfunction and fibrosis in heart failure. J Am Soc Nephrol. (2017) 28:197–208. doi: 10.1681/ASN.2015080852
78. Wu P, Liu Z, Zhao T, Xia F, Gong L, Zheng Z, et al. Lovastatin attenuates angiotensin II induced cardiovascular fibrosis through the suppression of YAP/TAZ signaling. Biochem Biophys Res Commun. (2019) 512:736–41. doi: 10.1016/j.bbrc.2019.03.158
79. Reddy AS, Zhang S. Polypharmacology: drug discovery for the future. Expert Rev Clin Pharmacol. (2013) 6:41–7. doi: 10.1586/ecp.12.74
80. Masnoon L, Shakib S, Kalisch-Ellett L, Caughey GE. What is polypharmacy? a systematic review of definitions. BMC Geriatr. (2017) 17:230. doi: 10.1186/s12877-017-0621-2
81. Wollin L, Wex E, Pautsch A, Schnapp G, Hostettler KE, Stowasser S, et al. Mode of action of nintedanib in the treatment of idiopathic pulmonary fibrosis. Eur Respir J. (2015) 45:1434–45. doi: 10.1183/09031936.00174914
82. Becker OM, Marantz Y, Shacham S, Inbal B, Heifetz A, Kalid O, et al. G protein-coupled receptors: in silico drug discovery in 3D. Proc Natl Acad Sci U S A. (2004) 101:11304–9. doi: 10.1073/pnas.0401862101
83. Nehlig A, Daval JL, Debry G. Caffeine and the central nervous system: mechanisms of action, biochemical, metabolic and psychostimulant effects. Brain Res Brain Res Rev. (1992) 17:139–70. doi: 10.1016/0165-0173(92)90012-B
84. Reichart D, Lindberg EL, Maatz H, Miranda AMA, Viveiros A, N. Shvetsov, et al. Pathogenic variants damage cell composition and single cell transcription in cardiomyopathies. Science. (2022) 377:eabo1984. doi: 10.1126/science.abo1984
85. Jones RC, Karkanias J, Krasnow MA, Pisco AO, Quake SR, J. Salzman, et al. The Tabula Sapiens: A multiple-organ, single-cell transcriptomic atlas of humans. Science. (2022) 376:eabl4896. doi: 10.1126/science.abl4896
86. Chaffin M, Papangeli I, Simonson B, Akkad AD, Hill MC, Arduini A, et al. Single-nucleus profiling of human dilated and hypertrophic cardiomyopathy. Nature. (2022) 608:174–80. doi: 10.1038/s41586-022-04817-8
87. Karlsson M, Zhang C, Méar L, Zhong W, Digre A, Katona B, et al. A single-cell type transcriptomics map of human tissues. Sci Adv. (2021) 7:eabh2169. doi: 10.1126/sciadv.abh2169
88. Zhang H, Tian L, Shen M, Tu C, Wu H, Gu M, et al. Generation of quiescent cardiac fibroblasts from human induced pluripotent stem cells for in vitro modeling of cardiac fibrosis. Circ Res. (2019) 125:552–66. doi: 10.1161/CIRCRESAHA.119.315491
Keywords: cardiac fibrosis, GPCR, antifibrotic therapy, fibroblast, G protein, crosstalk
Citation: Zhang H, Ren L and Shivnaraine RV (2022) Targeting GPCRs to treat cardiac fibrosis. Front. Cardiovasc. Med. 9:1011176. doi: 10.3389/fcvm.2022.1011176
Received: 03 August 2022; Accepted: 20 September 2022;
Published: 06 October 2022.
Edited by:
Phung N. Thai, University of California, Davis, United StatesReviewed by:
Di Lang, University of California, San Francisco, United StatesCopyright © 2022 Zhang, Ren and Shivnaraine. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hao Zhang, emhhbmdoYW9Ac3RhbmZvcmQuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.