
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
CASE REPORT article
Front. Cardiovasc. Med., 28 October 2022
Sec. Cardiac Rhythmology
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.1005952
This article is part of the Research TopicCase Reports in Cardiac Rhythmology: 2022View all 19 articles
 Hafiz Omer1,2*†
Hafiz Omer1,2*† Mohamed H. Omer3†
Mohamed H. Omer3† Abdulmohsen R. Alyousef1
Abdulmohsen R. Alyousef1 Ali M. Alzammam4
Ali M. Alzammam4 Omar Ahmad5
Omar Ahmad5 Haitham A. Alanazi1,2,6
Haitham A. Alanazi1,2,6Brugada syndrome is an inherited cardiac channelopathy arising from mutations in voltage-gated cardiac sodium channels. Idiopathic epilepsy portrays a coalescent underlying pathophysiological mechanism pertaining to the premature excitation of neuronal voltage-gated ion channels resulting in the disruption of presynaptic neurons and the unregulated release of excitatory neurotransmitters. The coexistence of epilepsy and Brugada syndrome may be explained by mutations in voltage-gated ion channels, which are coexpressed in cardiac and neural tissue. Moreover, the incidence of sudden unexpected death in epilepsy has been associated with malignant cardiac arrhythmias in the presence of mutations in voltage-gated ion channels. Lamotrigine is an antiepileptic drug that inhibits neuronal voltage-gated sodium channels, thus stabilizing neural impulse propagation and controlling seizure activity in the brain. However, lamotrigine has been shown to inhibit cardiac voltage-gated sodium channels resulting in a potential arrhythmogenic effect and the ability to unmask Brugada syndrome in genetically susceptible individuals. We are reporting a case of a 27-year-old male patient with a background of presumed idiopathic epilepsy who was initiated on lamotrigine therapy resulting in the unmasking of Brugada syndrome and the onset of syncopal episodes. This case provides further evidence for the arrhythmogenic capacity of lamotrigine and highlights the relationship between epilepsy and Brugada syndrome. In this report, we aim to review the current literature regarding the associations between epilepsy and Brugada syndrome and the impact of lamotrigine therapy on such patients.
Brugada syndrome (BrS) is an autosomal dominant channelopathy associated with mutations in voltage-gated ion channels within cardiac myocytes (1). Several genes have been implicated in the development of BrS; however, mutations in the SCN5A gene, which codes for the expression of voltage-gated sodium channels, have been correlated with most cases (2). The clinical features of BrS express wide heterogeneity and can range from complete lack of symptoms to malignant ventricular arrhythmias predisposing to sudden cardiac death (SCD) (1). BrS manifests with a characteristic electrocardiogram (EKG) pattern with coved-type ST-segment elevation in the right precordial leads (3). The malignant nature of BrS arises from the presence of arrhythmogenic epicardial substrate in the right ventricular outflow tract, leading to its association with approximately 20% of cases of SCD in individuals with structurally normal hearts (4, 5).
Idiopathic epilepsy portrays a similar pathophysiological mechanism to BrS, with mutations in the neuronal voltage-gated sodium channels implicated in the condition’s development (6, 7). Moreover, some studies have reported the coexistence of idiopathic epilepsy amongst patients with BrS (7). Interestingly, with the conduction of genome-wide association studies amongst patients with idiopathic epilepsy and BrS, some gene mutations coexpressed in both conditions have been identified (8, 9). A recent emerging hypothesis explaining the coexistence of both conditions suggests that the coexpressed genes code for voltage-gated ion channels expressed within cardiac myocytes and neuronal cells (2, 7, 10).
Lamotrigine is an antiepileptic agent that inhibits neuronal voltage-gated sodium channels, stabilizing presynaptic membranes and hindering excitatory neurotransmitter release (11). However, in vitro studies have demonstrated that lamotrigine may exhibit class IB antiarrhythmic activity by inhibiting cardiac sodium channels (12, 13). There have been six reports in the literature of lamotrigine contributing to the unmasking of Brugada syndrome, which further supports the theory that lamotrigine may exert a sodium channel-blocking effect within cardiac myocytes (14–19).
In this paper, we aim to report a case of a patient with pre-existing epilepsy who was initiated on lamotrigine therapy resulting in the unmasking of a Brugada pattern on the electrocardiogram. As there have only been six reported cases of lamotrigine unmasking BrS, we aim to contribute to the pre-existing literature on the subject. Moreover, the case also highlights the rare coexistence of BrS with idiopathic epilepsy. Furthermore, we carried out an exhaustive literature review using the Medline database of all reports of lamotrigine-induced BrS as well as reports of the coexistence of epilepsy and BrS.
A 27-year-old Saudi-Arabian male with a 4-year history of idiopathic epilepsy was referred to the cardiology department at our hospital for an investigation of his recurrent syncopal episodes and an abnormal electrocardiogram. The patient’s most recent admission was due to a generalized tonic-clonic seizure with lateral tongue biting lasting for 10 min. Seizure activity was rapidly terminated after four milligrams of intravenous lorazepam was administered. The patient recovered immediately with no evidence of confusion or weakness. However, an hour after seizure onset, the patient developed shortness of breath, which lasted for 10 min. A baseline electrocardiogram in the emergency department demonstrated a greater than 2 mm ST segment elevation in leads V1 and V2 with a prominent J wave followed by negative T waves (Figure 1). This was consistent with a type 1 Brugada pattern, facilitating the patient’s referral to our department.
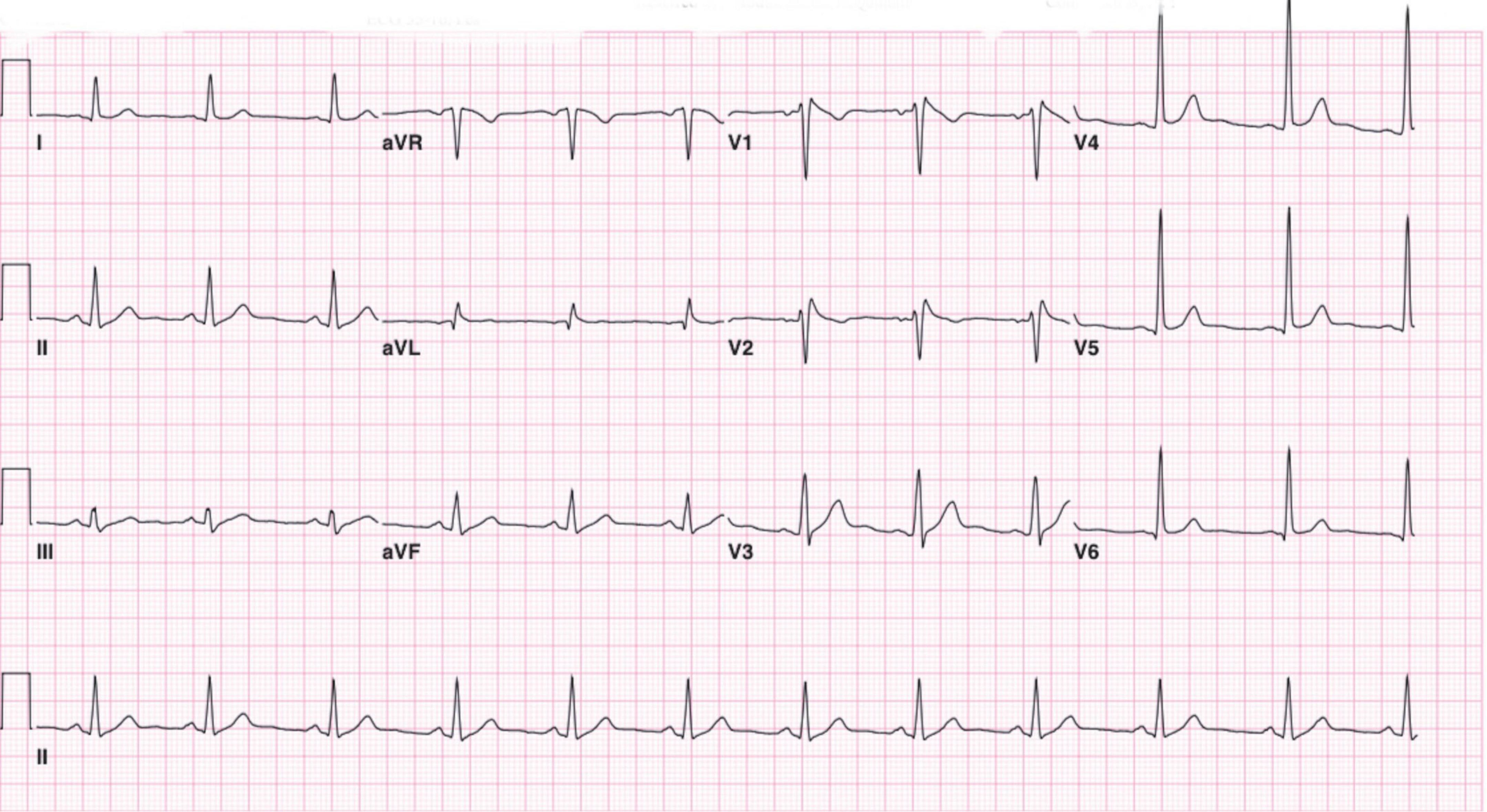
Figure 1. Twelve-lead electrocardiogram demonstrating 2 mm ST segment elevation in leads V1 and V2 with a prominent J wave followed by negative T waves. Suggestive of a type 1 Brugada pattern.
The patient described that he has been experiencing intermittent incidents of transient loss of consciousness preceded by light-headedness and palpitations. Moreover, he reported that this was unusual as he previously experienced one or two episodes annually of generalized tonic-clonic seizures with lateral tongue-biting and urinary incontinence. However, his recent syncopal episodes presented differently as described above and have occurred more frequently. A thorough evaluation of the patient’s family history was undertaken, which did not reveal any history of cardiac disorders, epilepsy, or sudden death. Moreover, the patient described that the incidence of his syncopal episodes began after the initiation of lamotrigine therapy. The patient had previously been prescribed 1,000 milligrams of levetiracetam twice daily; however, 100 milligrams of lamotrigine twice daily was introduced to establish adequate control of seizure activity. Previous electrocardiograms before the initiation of lamotrigine therapy demonstrated normal sinus rhythm with no evidence of a type 1 Brugada pattern.
The patient underwent a transthoracic echocardiogram which revealed no evidence of structural heart disease. Further, electrocardiograms were repeated and were consistent with a type 1 Brugada pattern. Moreover, a CT scan of the head revealed no brain abnormalities. An electroencephalogram (EEG) reported during the awake stage revealed well-organized activity with no spike-and-wave discharges or lateralizing abnormalities. Subsequent phonic stimulation did not produce any abnormalities. An electrophysiological study was performed, which resulted in the induction of polymorphic ventricular tachycardia after programmed electrical stimulation of the right ventricular outflow tract (Figure 2). The ventricular tachycardia was terminated through cardioversion, and the patient remained comfortable throughout the study with no hemodynamic compromise.

Figure 2. Electrophysiological study demonstrating the induction and termination of ventricular fibrillation. (A) Twelve -lead electrocardiogram demonstrating the induction of ventricular fibrillation through triple extrastimuli at the right ventricular outflow tract. (B) Twelve-lead electrocardiogram demonstrating sustained ventricular fibrillation. (C) Twelve-lead electrocardiogram demonstrating the termination of ventricular fibrillation through a 360-J transthoracic direct current shock and subsequent return to sinus rhythm.
Due to the presence of a type 1 Brugada pattern on electrocardiography along with a positive electrophysiological study and the presence of syncope, a recommendation for the placement of an implantable cardiac defibrillator (ICD) was made to the patient in accordance with current guidelines (1, 3). A thorough discussion regarding the risk of sudden cardiac death and the utility of ICD implantation was initiated; however, the patient refused further treatment. Moreover, the patient refused to undergo genetic testing to identify deleterious genetic variants in genes coding for voltage-gated sodium channels. A recommendation was made to the neurology team to discontinue lamotrigine therapy due to its potential arrhythmogenic effects.
The patient is currently receiving 1,000 milligrams of levetiracetam twice daily and has discontinued lamotrigine therapy after his initial admission. A follow-up assessment 3 months after the patient’s initial admission revealed no further incidences of seizures or syncopal episodes. In addition, further electrocardiograms obtained after the discontinuation of lamotrigine therapy display normal sinus rhythm with near complete resolution of the J-point elevation and the type 1 Brugada pattern (Figure 3).
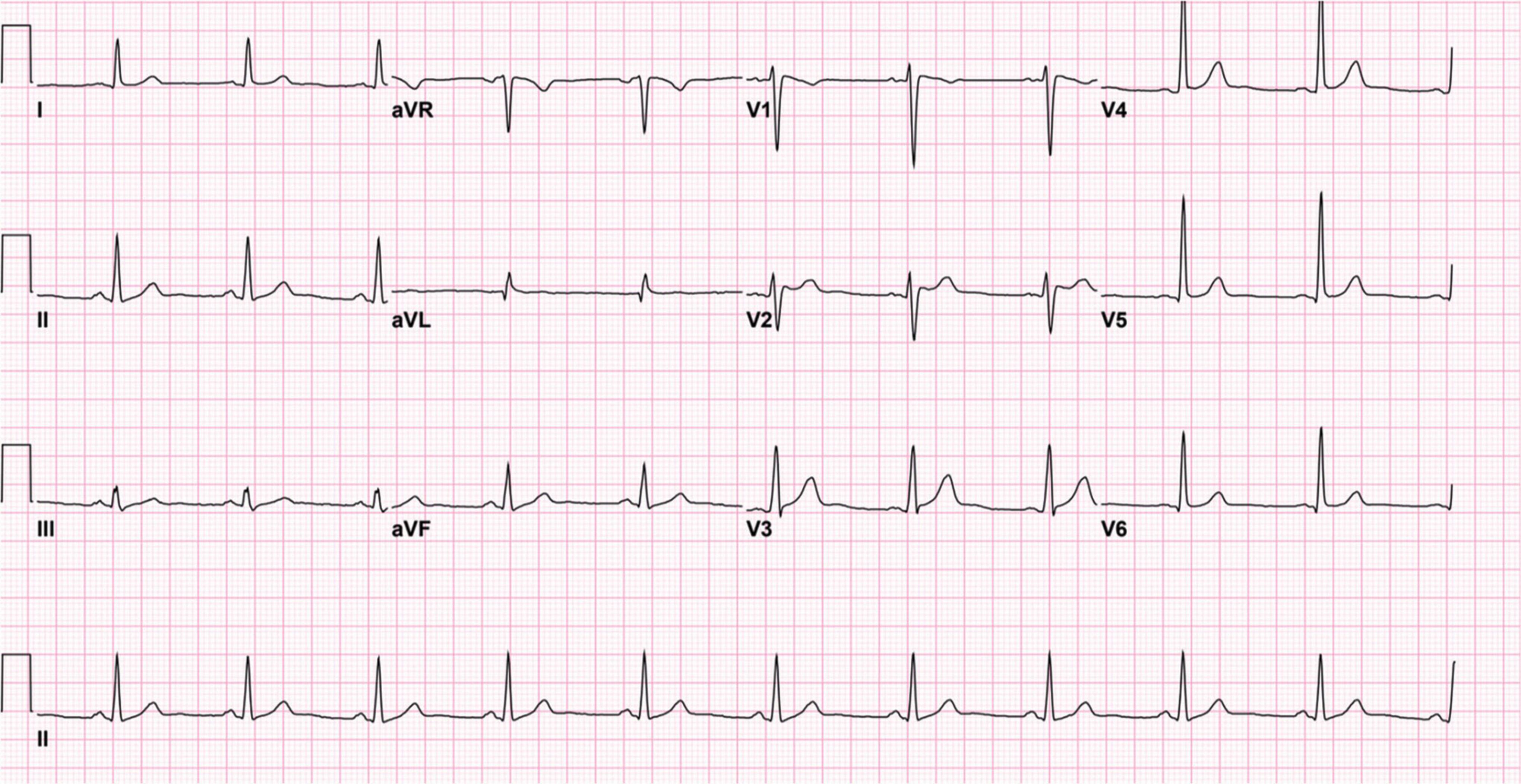
Figure 3. Twelve-lead electrocardiogram after the discontinuation of lamotrigine. The electrocardiogram demonstrates normal sinus rhythm with near resolution of the J-point elevation and type 1 Brugada pattern.
We are reporting a case of lamotrigine-induced unmasking of BrS in a patient with idiopathic epilepsy. This case highlights two exceedingly rare associations, with one being lamotrigine’s unmasking of BrS, while the other association encompasses the coexistence of epilepsy and BrS amongst specific individuals.
The pathophysiological mechanisms underlying BrS and idiopathic epilepsy share a coalescent theme pertaining to the dysregulation of cardiac and neuronal voltage-gated ion channels resulting in the premature excitability of cells, which manifests as cardiac arrhythmias and seizure-like activity, respectively (1, 6, 7). To identify reports highlighting the coexistence of epilepsy and BrS, we performed a Medline search using the keywords’ Brugada syndrome’, “Epilepsy,” and “Seizure.” Our initial search identified seventy-nine studies, and after abstract screening to identify relevant papers, we identified seventeen publications reporting cases of BrS in association with epilepsy (14–16, 19–32). The essential characteristic features of each paper, including electrocardiogram findings (EKG), electroencephalogram (EEG) findings, features of epilepsy, and the type of antiepileptic therapy, are highlighted in Table 1.
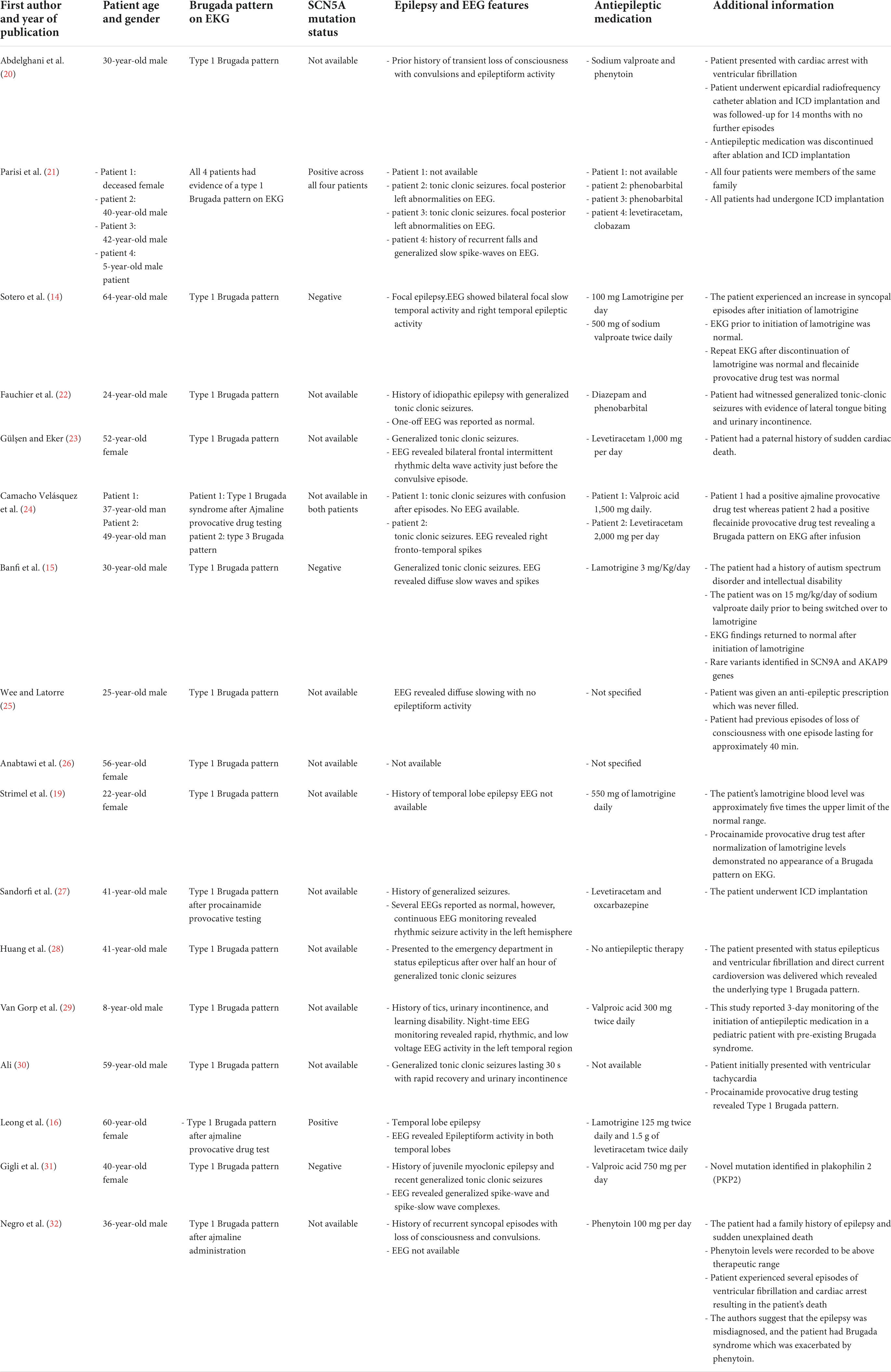
Table 1. Characteristic features of seventeen publications reporting the association between epilepsy and Brugada syndrome.
Mutations in voltage-gated sodium ion channels underlie the repolarization and depolarization abnormalities observed in BrS (1, 2). Over twenty individual genes have been associated with the development of BrS (2). Notably, loss of function mutations in the SCN5A gene, which codes for the alpha-subunit of the NaV1.5 cardiac voltage-gated sodium channels, have been implicated in over 80% of identifiable mutations in BrS (2–33). Parisi et al. provide a report of a family with a mutation in SCN5A with co-expression of BrS and epilepsy along with characteristic EKG and EEG findings which can be found in Table 1 (21). The authors of this paper suggest that individuals who possess the SCN5A mutation may have an age-dependent phenotypic expression of this mutation, with cerebral features presenting earlier in life and cardiac manifestations occurring in the later decades. This hypothesis is further supported by patients with gastrointestinal disease harboring the SCN5A mutation expressing variable symptoms within each age group (34). Furthermore, Leong et al. highlight a case of a patient harboring an SCN5A mutation with temporal lobe epilepsy and BrS (16). Animal studies in rats demonstrate the expression of SCN5A genes within the limbic system, suggesting that it can alter neuronal action potential propagation (35). More recently, the expression of SCN5A genes within the human brain has been described, with studies suggesting an increased expression within astrocytes and astrocytoma tumors (36–38). Hence, co-expression of SCN5A genes within the brain and cardiac tissue may underpin the concurrence of BrS and epilepsy amongst genetically susceptible individuals harboring the SCN5A mutation.
Recently, mutations in the SCN10A gene, which codes for the voltage-gated sodium channels subunit NaV1.8, were implicated in a large percentage of BrS cases and were associated with an increased phenotypic expression of symptoms (39). Interestingly, despite the NaV1.8 being mainly expressed in the peripheral nervous system, in certain pathologies the NaV1.8 has been found within the central nervous system (40). Moreover, a recent genetic analysis has demonstrated that variants in the SCN10A gene were linked to certain epilepsy-related phenotypes (41). Banfi et al. highlight a case of epilepsy associated with novel mutations identified in the voltage-gated sodium channel SCN9A gene, and the AKAP9 gene, which have previously been implicated in cases of BrS (14, 42). The SCN9A gene is also expressed in the brain, with mutations in this gene being associated with the development of certain subtypes of epilepsy (43, 44). It is worth noting that BrS is not the only cardiac channelopathy that has been associated with epilepsy. Namely, the KCNQ1 gene, which codes for voltage-gated potassium channels, has been correlated with epilepsy and long QT syndrome (LQTS) in a subgroup of patients (45).
Sudden unexplained death in epilepsy (SUDEP) is a phenomenon that carries tremendous morbidity among patients diagnosed with epilepsy. Approximately 18% of patients with epilepsy die due to SUDEP (8). One of the leading theories behind the pathophysiological mechanisms of SUDEP involves the onset of fatal cardiac arrhythmias (42). This theory is supported by retrospective cohort studies which suggest that patients with epileptic seizures have an increased incidence of abnormal electrocardiogram signs (46–48). It is also worth noting that SUDEP and sudden cardiac death have been shown to share similar risk factors, including sex and age (49). Furthermore, some gene mutations in genes that code for cardiac voltage-gated ion channels have been identified in patients with SUDEP. In particular, a few studies have reported mutations of the SCN5A and SCN10A genes amongst patients with SUDEP (50, 51).
Interestingly, mutations in the SCN8A gene, which codes for the alpha subunit type 8 of the voltage-gated sodium channel (NaV1.6), have been implicated in cases of epilepsy and SUDEP (52). The NaV1.6 voltage-gated sodium channel is predominantly expressed in the brain; hence mutations in the SCN8A gene are associated with a subtype of epilepsy termed early infantile-epileptic encephalopathy-13 (43). Individuals with SCN8A mutations possess up to a 10% higher risk of SUDEP (43). A possible explanation for this increased risk relates to the expression of NaV1.6 in cardiac tissue, with mutations in this gene having been associated with cardiac arrhythmias in animal models (43, 53). Moreover, it is worth noting that the mere presence of the NaV1.6 channel, under pathological conditions, may contribute to cardiac arrythmias. For instance, an in vivo study utilizing mouse models examined the effects of sodium-channel blockade on the induction of catecholaminergic polymorphic ventricular tachycardia (54). The authors of this study noted that blockade of the NaV1.6 channel reduced the incidence of cardiac arrythmias (54). In addition, the authors hypothesize that NaV1.6 blockade may interfere with sodium and calcium signaling within cardiac myocytes, thus, specific blockade of NaV1.6 may provide a novel approach for the treatment of cardiac arrythmias (54). Munger et al. also examined the impact of NaV1.6 on calcium and sodium channel dysregulation on the induction of atrial fibrillation (55). The authors of this paper suggest that blockade of sodium-channel isoforms such as the NaV1.6 provides a novel approach for the treatment of atrial fibrillation through modulation of calcium release within myocytes (55). Moreover, the SCN1A gene which codes for the NaV1.1 voltage-gated sodium channel, has also been associated with epilepsy, SUDEP, and cardiac conduction abnormalities across in vivo and in vitro studies. Mutations in the SCN1A lead to the development of Dravet syndrome (a form of early onset severe myoclonic epilepsy) in over 80% of cases (56). The incidence of SUDEP amongst patients with Dravet syndrome is the highest SUDEP rate reported amongst epilepsy subtypes, hence, examining the mechanisms which may underpin SUDEP in Dravet syndrome is essential to further the understanding of the role of sodium channel isoforms in cardiac arrhythmogenesis and epilepsy (57). Auerbach et al. explored the mechanisms behind the altered cardiac electrophysiology and SUDEP in a mouse model with Dravet syndrome (58). The authors of this study noted that the mice with Dravet syndrome exhibited abnormalities in transient and persistent sodium current density within cardiac myocytes (58). The authors also noted the presence of increased cardiac excitability and arrhythmogenic electrocardiogram changes within the Dravet mice (58). Kalume et al. also examined the mechanisms behind SUDEP in a Dravet mouse model (59). The findings of this study suggest that SUDEP in association with SCN1A mutations is triggered by parasympathetic overdrive resulting in fatal bradycardia and cardiac dysregulation (59). Experimental models examining the effects of SCN1A mutations in epilepsy have described the overexpression of the NaV1.1 channels within cardiac myocytes and subsequent cardiac hyper-excitability resulting in a predisposition to cardiac arrhythmogenesis (60).
The coexistence of epilepsy and BrS poses a tremendous diagnostic challenge due to the overlap in presentations amongst both diseases. Differentiating true epileptic seizures from arrhythmia-induced syncope amongst patients with BrS is challenging. Seizure-like activity amongst patients with BrS may be a byproduct of cardiac arrhythmias resulting in cerebral hypoperfusion and subsequent syncope (61, 62). There have been four reports of BrS masquerading as a seizure disorder resulting in the misdiagnosis of patients (20, 25, 26, 32). One of the reports by Wee and Latorre was that of a 25-year-old male who was presumed to have epilepsy and presented to the emergency department with an anoxic brain injury resulting in his subsequent death (25). A type 1 BrS pattern was missed on the patient’s previous electrocardiograms; thereby, this report illustrates the significant magnitude associated with a misdiagnosis of epilepsy amongst BrS patients. Moreover, clinical features characteristically associated with epilepsy, such as tongue-biting and generalized tonic-clonic convulsions, may be observed in patients with arrhythmia-induced syncopal episodes, making establishing a final diagnosis a tremendous hurdle (63). The BrS EKG pattern may also not be visible on the initial EKG and may require further assessment through sodium-channel blocker provocative testing. In addition, a multicenter prospective observational study amongst patients with drug-resistant epilepsy found that syncope and epilepsy were coexistent amongst 20% of subjects (64). The discovery of mutations in voltage-gated cardiac ion channels amongst patients with SUDEP may be explained by the existence of potentially undiagnosed cardiac channelopathies and arrhythmias. Misdiagnosis of cardiac channelopathies as epilepsy, is also common with LQTS. A study by MacCormick et al. found that patients diagnosed with epilepsy experienced a delay of approximately 12 years before a correct diagnosis of LQTS was made (65).
Lamotrigine is an antiepileptic drug that inhibits neuronal voltage-gated sodium channels, stabilizing presynaptic neurons and suppressing unregulated glutamate release (11). Recently in 2021, the United States Food and Drug Administration (FDA) issued a warning recommending against using lamotrigine in individuals with structural heart disease and conduction disorders due to its potential arrhythmogenic effect (12). The FDA’s warning was based on in vitro studies suggesting that lamotrigine might inhibit cardiac voltage-gated sodium channels with similar pharmacodynamics to class IB antiarrhythmic agents (12, 13). It is well-established that sodium-channel blockers may induce malignant ventricular arrhythmias in patients with BrS (66). Moreover, sodium-channel blocking antiarrhythmic drugs such as flecainide and ajmaline are used to artificially induce ventricular arrhythmias in patients with BrS as part of the diagnostic process (64). An animal study by Goto et al. investigated the effects of lamotrigine therapy on EKG parameters (67). A notable finding from this study included the elevation of the J wave in over half of the animals (67). The J wave is a distinctive feature of BrS, thereby suggesting that lamotrigine may possess the ability to unmask a BrS pattern in genetically susceptible individuals. Lamotrigine exhibits blockade of the brain’s most abundantly expressed voltage-gated sodium channels, including the NaV1.1, NaV1.2, and NaV1.6 (68). However, lamotrigine has also been shown to impact the cardiac voltage-gated sodium channel isoform NaV1.5 in several in vitro studies. In a paper by Ingleby-Talecki et al., lamotrigine was shown to block NaV1.5 current even at therapeutic dosages (69). Lamotrigine demonstrated a half maximal inhibitory concentration (IC50) of 280.2 and 28.8 μM at holding voltages of –120 and –95, respectively (69). Moreover, other in vitro studies have reported similar potencies regarding lamotrigine’s NaV1.5 blockade (13, 70). In vitro studies have also shown that lamotrigine demonstrates voltage-dependent blockade of the NaV1.5 channels with rapid kinetics, closely resembling the class IB anti-arrhythmic agent mexiletine (69). The findings of the aforementioned in vitro studies suggest that lamotrigine may be capable of blocking NaV1.5 channels at plasma concentrations observed within its therapeutic dosage range. Moreover, a human study by Dixon et al. analyzed the effects of lamotrigine on the PR interval in healthy subjects (71). The study demonstrates that lamotrigine may exhibit a dose-dependent prolongation of the PR interval (71). The PR interval may be prolonged due to NaV1.5 blockade, however, other factors such as heart rate and autonomic stimulation may contribute to PR prolongation (72). The findings of the in vitro studies coupled with lamotrigine’s effects on electrocardiogram markers such as the J wave and PR interval in in vivo studies suggest that at therapeutic concentrations, lamotrigine may possess the ability to inhibit NaV1.5 channels and thus contribute to the Brugada phenotype in genetically susceptible individuals.
To explore the association between lamotrigine therapy and the unmasking of BrS, we performed a Medline search using the terms “Lamotrigine” and “Brugada Syndrome.” A total of 11 publications were identified, and abstract screening resulted in the selection of six reports (14–19). The features of each report, including patient characteristics, EKG findings, and lamotrigine dosage, are summarized in Table 2.
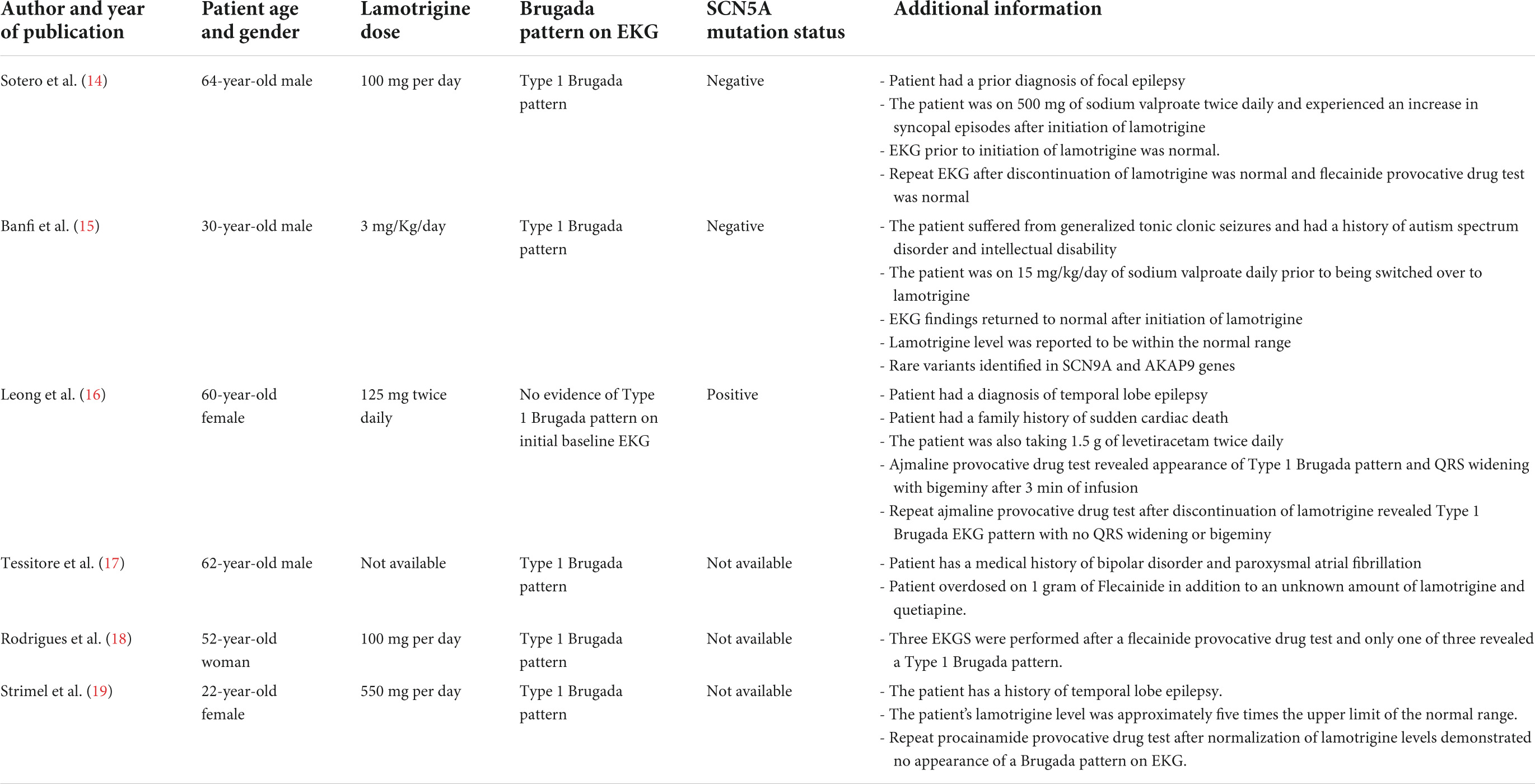
Table 2. Characteristic features of six publications reporting the association between lamotrigine therapy and Brugada syndrome.
Four of the six patients have been treated with lamotrigine for pre-existing epilepsy (14–16, 19). In addition, all patients demonstrated a type 1 BrS pattern on EKG. Interestingly, five patients had a previously normal EKG prior to the initiation of lamotrigine therapy, suggesting that lamotrigine may contribute to unmasking the BrS pattern in susceptible individuals (14–19). Furthermore, the blood levels of lamotrigine were reported to be elevated in a paper by Strimel et al., whereas Banfi et al. reported a lamotrigine level within the normal range (15, 19). The lamotrigine blood level was unavailable within other reports. However, the discrepancy in the lamotrigine blood levels between the two papers highlights that lamotrigine may exhibit an inhibitory effect on cardiac sodium channels at both therapeutic and supra-therapeutic levels. It has previously been hypothesized that lamotrigine may unmask BrS at higher drug concentrations due to loss of specificity for neuronal voltage-gated sodium channels resulting in downstream effects on cardiac voltage-gated channels (19). However, the unmasking of BrS at normal physiological blood levels of lamotrigine contradicts this theory. We hypothesize that individual genetics may interfere with lamotrigine metabolism resulting in significant disparities relating to its pharmacokinetic metabolism amongst different individuals. Lamotrigine exhibits first-order pharmacokinetics and has an excellent oral bioavailability resulting in its rapid absorption with maximal plasma concentrations of the drug occurring within 1–3 h (11). Moreover, the metabolism of lamotrigine is facilitated by different UDP−glucuronosyltransferase enzymes in the liver (11). Individuals express a wide range of heterogeneity amongst genes coding for UDP−glucuronosyltransferases, and multiple polymorphisms can be detected amongst the population (73). The variable pharmacokinetic profile of lamotrigine was investigated in a study of 100 epilepsy patients by Milosheska et al. (74). The study found that multiple factors influenced the clearance of lamotrigine, including genetic polymorphisms in UDP−glucuronosyltransferases, body weight, and renal function (74). Therefore, the authors of this paper suggested that the variability in lamotrigine’s pharmacokinetics governs the need for more precise and individualized drug monitoring and dosage adjustments (74).
As mentioned previously, many patients with cardiac channelopathies have a prior diagnosis of epilepsy requiring antiepileptic drug treatment. The role of antiepileptic drugs in the management of true epileptic seizures is well-established. For instance, the therapeutic interventions used in our patient’s case to manage his epilepsy diagnosis included levetiracetam and lamotrigine, which are supported by clinical and pharmacogenomic studies (75). However, using antiepileptic drugs in patients with non-epileptic seizures and concurrent cardiac channelopathies may result in an arrhythmogenic effect. For instance, a paper by Bardai et al. found an increase in the risk of SCD in association with both epilepsy and the use of antiepileptic drugs (76). Moreover, Ishizue et al. found that polytherapy with multiple sodium channel blocking antiepileptic drugs was associated with arrhythmogenic EKG abnormalities (77). The unmasking of a BrS pattern on EKG has also been observed with other antiepileptic drugs, such as carbamazepine and phenytoin (78, 79).
Nonetheless, the evidence for lamotrigine’s arrhythmogenic effects remains unclear as the FDA’s current recommendation is based solely on the findings of in vitro studies. The findings of the aforementioned in vitro studies regarding lamotrigine’s impact on cardiac activity have yet to translate clinically. For instance, a recent systematic review by Restrepo et al. explored cardiac risk in patients undergoing lamotrigine therapy and found no clear evidence to support an increase in cardiac risk (80). Therefore, as it currently stands, lamotrigine and other sodium-channel blocking antiepileptic drugs are not contraindicated in patients with BrS.
As described previously, the coexistence of epilepsy and BrS has been associated with mutations in genes coding for voltage-gated ion channels, which are coexpressed in cardiac and neural tissue (7). However, it is worth noting that the relationship between genotypes and clinical phenotypes is often not linear; hence mutations in genes co-expressed in cardiac and neuronal tissue may independently induce epilepsy and cardiac arrhythmias. Sudden unexplained death in epilepsy has been associated with malignant cardiac arrhythmias due to the presence of mutations in genes coding for voltage-gated cardiac ion channels (7). Consequently, the identification of potentially pathogenic genetic variants which may underlie the pathogenesis of epilepsy, cardiac conduction disorders, and SUDEP requires the use of targeted next-generation sequencing technology and the execution of genome-wide association studies to further our understanding of the genetic basis of the aforementioned conditions (81). Moreover, the coexistence of epilepsy and BrS poses a diagnostic challenge due to the similar nature of clinical presentations. Therefore, we recommend a thorough neurological and cardiovascular evaluation of patients presenting with seizures and syncope to identify any underlying disorders.
Using sodium-channel blocking antiepileptic drugs such as lamotrigine in patients with coinciding epilepsy and BrS may promote arrhythmogenic effects in cardiac myocytes (12). Furthermore, individual genetic polymorphisms may influence the pharmacokinetic profile of lamotrigine, resulting in impaired metabolism and subsequent supratherapeutic blood levels (72). Accordingly, we propose the need for more research to understand the arrhythmogenic capacity of lamotrigine and other antiepileptic drugs. In addition, we recommend the need for precise and individualized therapeutic monitoring and dosage adjustments amongst patients receiving lamotrigine therapy.
The original contributions presented in this study are included in the article/supplementary material, further inquiries can be directed to the corresponding author.
The studies involving human participants were reviewed and approved by the King Abdullah International Medical Research Center (KAIMRC) Institutional Review Board. Written informed consent was obtained from the participant/s for the publication of this case report.
MO and AMA wrote the initial manuscript and carried out the literature review process. HO and HA contributed to the acquisition of clinical data and the clinical design of the manuscript. OA contributed to the review of the present literature. AA contributed to the collation of clinical data. All authors reviewed the manuscript and authorized its submission for publication.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
BrS, Brugada syndrome; SCD, sudden cardiac death; SUDEP, sudden unexplained death in epilepsy; LQTS, long QT syndrome EKG, electrocardiogram; EEG, electroencephalogram; SCN5A, Sodium Voltage-Gated Channel Alpha Subunit 5; SCN10A, Sodium Voltage-Gated Channel Alpha Subunit 10; SCN9A, Sodium Voltage-Gated Channel Alpha Subunit 9; SCN8A, Sodium Voltage-Gated Channel Alpha Subunit 8; AKAP9, A-Kinase Anchoring Protein 9; KCNQ1, Potassium Voltage-Gated Channel Subfamily Q Member 1; FDA, Food and Drug Administration; UDP − glucuronosyltransferases, Uridine 5 ′′ -diphospho-glucuronosyltransferase.
1. Li K, Lee S, Yin C, Liu T, Ngarmukos T, Conte G, et al. Brugada syndrome: a comprehensive review of pathophysiological mechanisms and risk stratification strategies. Int J Cardiol Heart Vasc. (2020) 26:100468. doi: 10.1016/j.ijcha.2020.100468
3. Antzelevitch C, Yan G. J-wave syndromes: Brugada and early repolarization syndromes. Heart Rhythm. (2015) 12:1852–66.
4. Pappone C, Brugada J, Vicedomini G, Ciconte G, Manguso F, Saviano M, et al. Electrical Substrate elimination in 135 consecutive patients with Brugada syndrome. Circ Arrhythm Electrophysiol. (2017) 10:e005053. doi: 10.1161/CIRCEP.117.005053
5. Brugada R, Campuzano O, Sarquella-Brugada G, Brugada J, Brugada P. Brugada Syndrome. Methodist Debakey Cardiovasc J. (2014) 10:25.
6. Devinsky O, Vezzani A, O’Brien T, Jette N, Scheffer I, de Curtis M, et al. Epilepsy. Nat Rev Dis Primers. (2018) 4:18024.
7. D’Imperio S, Monasky M, Micaglio E, Ciconte G, Anastasia L, Pappone C. Brugada syndrome: warning of a systemic condition? Front Cardiovasc Med. (2021) 8:771349. doi: 10.3389/fcvm.2021.771349
8. Chahal C, Salloum M, Alahdab F, Gottwald J, Tester D, Anwer L, et al. Systematic review of the genetics of sudden unexpected death in epilepsy: potential overlap with sudden cardiac death and arrhythmia−related genes. J Am Heart Assoc. (2020) 9:e012264. doi: 10.1161/JAHA.119.012264
9. Ricci M, Menegon S, Vatrano S, Mandrile G, Cerrato N, Carvalho P, et al. SCN1B gene variants in Brugada syndrome: a study of 145 SCN5A-negative patients. Sci Rep. (2014) 4:6470. doi: 10.1038/srep06470
10. Costagliola G, Orsini A, Coll M, Brugada R, Parisi P, Striano P. The brain–heart interaction in epilepsy: implications for diagnosis, therapy, and SUDEP prevention. Ann Clin Transl Neurol. (2021) 8:1557–68. doi: 10.1002/acn3.51382
12. French J, Perucca E, Sander J, Bergfeldt L, Baulac M, Auerbach D, et al. FDA safety warning on the cardiac effects of lamotrigine: an advisory from the Ad Hoc ILAE/AES Task Force. Epilepsia Open. (2021) 6:45–8.
13. Harmer A, Valentin J, Pollard C. On the relationship between block of the cardiac Na+channel and drug-induced prolongation of the QRS complex. Br J Pharmacol. (2011) 164:260–73. doi: 10.1111/j.1476-5381.2011.01415.x
14. Sotero F, Silva C, Cunha N, Franco A, Pimentel J. Lamotrigine-induced Brugada syndrome: a rare adverse event. Seizure. (2022) 94:7–9. doi: 10.1016/j.seizure.2021.11.001
15. Banfi P, Coll M, Oliva A, Alcalde M, Striano P, Mauri M, et al. Lamotrigine induced Brugada-pattern in a patient with genetic epilepsy associated with a novel variant in SCN9A. Gene. (2020) 754:144847. doi: 10.1016/j.gene.2020.144847
16. Leong K, Seligman H, Varnava A. Proarrhythmogenic effects of lamotrigine during ajmaline testing for Brugada syndrome. HeartRhythm Case Rep. (2017) 3:167–71. doi: 10.1016/j.hrcr.2016.11.006
17. Tessitore E, Ramlawi M, Tobler O, Sunthorn H. Brugada pattern caused by a flecainide overdose. J Emerg Med. (2017) 52:e95–7. doi: 10.1016/j.jemermed.2016.10.045
18. Rodrigues R, Amador P, Rassi L, Seixo F, Parreira L, Fonseca N, et al. Brugada pattern in a patient medicated with lamotrigine. Rev Port Cardiol. (2013) 32:807–10. doi: 10.1016/j.repc.2013.01.009
19. Strimel W, Woodruff A, Cheung P, Kirmani B, Stephen Huang S. Brugada-like electrocardiographic pattern induced by lamotrigine toxicity. Clin Neuropharmacol. (2010) 33:265–7.
20. Abdelghani M, Chapra A, Asaad N, Hayat S. Epilepsy and brugada syndrome: association or uncommon presentation? Heart Views. (2020) 21:114.
21. Parisi P, Oliva A, Coll Vidal M, Partemi S, Campuzano O, Iglesias A, et al. Coexistence of epilepsy and Brugada syndrome in a family with SCN5A mutation. Epilepsy Res. (2013) 105:415–8. doi: 10.1016/j.eplepsyres.2013.02.024
22. Fauchier L, Babuty D, Cosnay P. Epilepsy, Brugada syndrome and the risk of sudden unexpected death. J Neurol. (2000) 247:643–4.
23. Gülşen K, Eker A. PP-205 Brugada syndrome and epilepsy in same patient: Na channel defect in both myocytes and neurons. Am J Cardiol. (2014) 113:S84.
24. Camacho Velásquez J, Rivero Sanz E, Velazquez Benito A, Mauri Llerda J. Epilepsy and Brugada syndrome. Neurología. (2017) 32:58–60.
25. Wee C, Latorre J. A fatal case of mistaken identity: Brugada syndrome masquerading as seizure disorder (P2.264). Am Acad Neurol. (2018). Available online at: https://n.neurology.org/content/90/15_Supplement/P2.264 (accessed July 19, 2022).
26. Anabtawi A, Anabtawi A, Kim A. A case of brugada syndrome presenting as seizure disorder. J Am Coll Cardiol. (2018) 71:A2547.
27. Sandorfi G, Clemens B, Csanadi Z. Electrical storm in the brain and in the heart: epilepsy and Brugada syndrome. Mayo Clin Proc. (2013) 88:1167–73. doi: 10.1016/j.mayocp.2013.06.019
28. Huang C, Chen T, Lin F, Huang M. Status epilepticus as an initial presentation of brugada syndrome: a case report. Kaohsiung J Med Sci. (2005) 21:387–91. doi: 10.1016/S1607-551X(09)70139-1
29. Van Gorp V, Danschutter D, Huyghens L, Hachimi-Idrissi S, Sarkozy A, Chierchia G, et al. Monitoring the safety of antiepileptic medication in a child with Brugada syndrome. Int J Cardiol. (2010) 145:e64–7. doi: 10.1016/j.ijcard.2008.12.156
30. Ali A. Brugada Syndrome in a Black Man with Seizures and Urinary Incontinence. Houston, TX: Texas Heart institute (2006).
31. Gigli L, Bertero G, Vidal M, Iglesias A, Campuzano O, Striano P, et al. Juvenile myoclonic epilepsy and Brugada type 1 ECG pattern associated with (a novel) plakophillin 2 mutation. J Neurol. (2017) 264:792–5. doi: 10.1007/s00415-017-8414-2
32. Negro G, Ciconte G, Borrelli V, Rondine R, Maiolo V, Pappone C. Sudden death of a patient with epilepsy: when Brugada syndrome mimicry can be fatal. HeartRhythm Case Rep. (2022) 8:205–8. doi: 10.1016/j.hrcr.2021.12.008
33. Li W, Yin L, Shen C, Hu K, Ge J, Sun A. SCN5A variants: association with cardiac disorders. Front Physiol. (2018) 9:1372. doi: 10.3389/fphys.2018.01372
34. Locke G, Ackerman M, Zinsmeister A, Thapa P, Farrugia G. Gastrointestinal symptoms in families of patients with an SCN5A-encoded cardiac channelopathy: evidence of an intestinal channelopathy. Am J Gastroenterol. (2006) 101:1299–304. doi: 10.1111/j.1572-0241.2006.00507.x
35. Wang J, Ou S, Wang Y, Kameyama M, Kameyama A, Zong Z. Analysis of four novel variants of Nav1.5/SCN5A cloned from the brain. Neurosci Res. (2009) 64:339–47. doi: 10.1016/j.neures.2009.04.003
36. Wang J, Ou S, Bai Y, Wang Y, Xu Z, Luan G. Multiple Nav1.5 isoforms are functionally expressed in the brain and present distinct expression patterns compared with cardiac Nav1.5. Mol Med Rep. (2017) 16:719–29. doi: 10.3892/mmr.2017.6654
37. Xing D, Wang J, Ou S, Wang Y, Qiu B, Ding D, et al. Expression of neonatal Nav1.5 in human brain astrocytoma and its effect on proliferation, invasion and apoptosis of astrocytoma cells. Oncol Rep. (2014) 31:2692–700. doi: 10.3892/or.2014.3143
38. Johnson J, Hofman N, Haglund C, Cascino G, Wilde A, Ackerman M. Identification of a possible pathogenic link between congenital long QT syndrome and epilepsy. Neurology. (2008) 72:224–31. doi: 10.1212/01.wnl.0000335760.02995.ca
39. Hu D, Barajas-Martínez H, Pfeiffer R, Dezi F, Pfeiffer J, Buch T, et al. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J Am Coll Cardiol. (2014) 64:66–79. doi: 10.1016/j.jacc.2014.04.032
41. Kambouris M, Thevenon J, Soldatos A, Cox A, Stephen J, Ben-Omran T, et al. BiallelicSCN10Amutations in neuromuscular disease and epileptic encephalopathy. Ann Clin Transl Neurol. (2016) 4:26–35.
42. Wang M, Tu X. The genetics and epigenetics of ventricular arrhythmias in patients without structural heart disease. Front Cardiovasc Med. (2022) 9:891399. doi: 10.3389/fcvm.2022.891399
43. Menezes L, Sabiá Júnior E, Tibery D, Carneiro L, Schwartz E. Epilepsy-related voltage-gated sodium channelopathies: a review. Front Pharmacol. (2020) 11:1276. doi: 10.3389/fphar.2020.01276
44. Zhang S, Zhang Z, Shen Y, Zhu Y, Du K, Guo J, et al. SCN9A epileptic encephalopathy mutations display a gain-of-function phenotype and distinct sensitivity to oxcarbazepine. Neurosci Bull. (2019) 36:11–24. doi: 10.1007/s12264-019-00413-5
45. Goldman A, Glasscock E, Yoo J, Chen T, Klassen T, Noebels J. Arrhythmia in heart and brain: KCNQ1 mutations link epilepsy and sudden unexplained death. Sci Transl Med. (2009) 1:2ra6. doi: 10.1126/scitranslmed.3000289
46. Opherk C, Coromilas J, Hirsch L. Heart rate and EKG changes in 102 seizures: analysis of influencing factors. Epilepsy Res. (2002) 52:117–27. doi: 10.1016/s0920-1211(02)00215-2
47. Zare M, Salari M, Tajmirriahi M. Electrocardiographic changes in patients with refractory epilepsy. J Res Med Sci. (2013) 18(Suppl. 1):S32–4.
48. Zijlmans M, Flanagan D, Gotman J. Heart rate changes and ECG Abnormalities during epileptic seizures: prevalence and definition of an objective clinical sign. Epilepsia. (2002) 43:847–54. doi: 10.1046/j.1528-1157.2002.37801.x
49. Velagapudi P, Turagam M, Laurence T, Kocheril A. Cardiac arrhythmias and sudden unexpected death in epilepsy (SUDEP). Pacing Clin Electrophysiol. (2011) 35:363–70.
50. Ravindran K, Powell K, Todaro M, O’Brien T. The pathophysiology of cardiac dysfunction in epilepsy. Epilepsy Res. (2016) 127:19–29.
51. Coll M, Allegue C, Partemi S, Mates J, Del Olmo B, Campuzano O, et al. Genetic investigation of sudden unexpected death in epilepsy cohort by panel target resequencing. Int J Legal Med. (2015) 130:331–9. doi: 10.1007/s00414-015-1269-0
52. Gardella E, Møller R. Phenotypic and genetic spectrum of SCN 8A −related disorders, treatment options, and outcomes. Epilepsia. (2019) 60Suppl. 3:S77–85. doi: 10.1111/epi.16319
53. Frasier C, Wagnon J, Bao Y, McVeigh L, Lopez-Santiago L, Meisler M, et al. Cardiac arrhythmia in a mouse model of sodium channel SCN8A epileptic encephalopathy. Proc Natl Acad Sci U.S.A. (2016) 113:12838–43. doi: 10.1073/pnas.1612746113
54. Radwański P, Ho H, Veeraraghavan R, Brunello L, Liu B, Belevych A, et al. Neuronal Na+ channels are integral components of pro-arrhythmic Na+/Ca2+ signaling nanodomain that promotes cardiac arrhythmias during β-adrenergic stimulation. JACC Basic Transl Sci. (2016) 1:251–66. doi: 10.1016/j.jacbts.2016.04.004
55. Munger M, Olðar Y, Koleske M, Struckman H, Mandrioli J, Lou Q, et al. Tetrodotoxin−sensitive neuronal−type Na + channels: a novel and druggable target for prevention of atrial fibrillation. J Am Heart Assoc. (2020) 9:e015119. doi: 10.1161/JAHA.119.015119
56. Rosander C, Hallböök T. Dravet syndrome in Sweden: a population-based study. Dev Med Child Neurol. (2015) 57:628–33.
57. Cooper M, Mcintosh A, Crompton D, McMahon J, Schneider A, Farrell K, et al. Mortality in Dravet syndrome. Epilepsy Res. (2016) 128:43–7.
58. Auerbach D, Jones J, Clawson B, Offord J, Lenk G, Ogiwara I, et al. Altered cardiac electrophysiology and SUDEP in a model of dravet syndrome. PLoS One. (2013) 8:e77843. doi: 10.1371/journal.pone.0077843
59. Kalume F, Westenbroek R, Cheah C, Yu F, Oakley J, Scheuer T, et al. Sudden unexpected death in a mouse model of Dravet syndrome. J Clin Investig. (2013) 123:1798–808. doi: 10.1172/JCI66220
60. Biet M, Morin N, Lessard-Beaudoin M, Graham R, Duss S, Gagné J, et al. Prolongation of action potential duration and qt interval during epilepsy linked to increased contribution of neuronal sodium channels to cardiac late Na + current. Circ Arrhythm Electrophysiol. (2015) 8:912–20. doi: 10.1161/CIRCEP.114.002693
61. Petkar S, Hamid T, Iddon P, Clifford A, Rice N, Claire R, et al. Prolonged implantable electrocardiographic monitoring indicates a high rate of misdiagnosis of epilepsy–REVISE study. Europace. (2012) 14:1653–60. doi: 10.1093/europace/eus185
63. Akor F, Liu N, Besag F, Ahmed M. Value of tongue biting in differentiating between epileptic seizures and syncope. Seizure. (2013) 22:328.
64. Ungar A, Ceccofiglio A, Pescini F, Mussi C, Tava G, Rafanelli M, et al. Syncope and epilepsy coexist in ‘possible’ and ‘drug-resistant’ epilepsy (overlap between epilepsy and syncope study - OESYS). BMC Neurol. (2017) 17:45. doi: 10.1186/s12883-017-0822-5
65. MacCormick J, McAlister H, Crawford J, French J, Crozier I, Shelling A, et al. Misdiagnosis of long QT syndrome as epilepsy at first presentation. Ann Emerg Med. (2009) 54:26–32.
66. Postema P, Wolpert C, Amin A, Probst V, Borggrefe M, Roden D, et al. Drugs and Brugada syndrome patients: review of the literature, recommendations, and an up-to-date website (www.brugadadrugs.org). Heart Rhythm. (2009) 6:1335–41. doi: 10.1016/j.hrthm.2009.07.002
67. Goto A, Hagiwara-Nagasawa M, Kambayashi R, Nunoi Y, Izumi-Nakaseko H, Kawai S, et al. Reverse translational analysis of clinically reported, lamotrigine-induced cardiovascular adverse events using the halothane-anesthetized dogs. Heart Vessels. (2020) 36:424–9. doi: 10.1007/s00380-020-01716-8
68. Qiao X, Sun G, Clare J, Werkman T, Wadman W. Properties of human brain sodium channel α-subunits expressed in HEK293 cells and their modulation by carbamazepine, phenytoin and lamotrigine. Br J Pharmacol. (2014) 171:1054–67. doi: 10.1111/bph.12534
69. Ingleby−Talecki L, van Dijkman S, Oosterholt S, Della Pasqua O, Winter C, Cunnington M, et al. Cardiac sodium channel inhibition by lamotrigine: in vitro characterization and clinical implications. Clin Transl Sci. (2022) 15:1978–89. doi: 10.1111/cts.13311
70. Guo D, Jenkinson S. Simultaneous assessment of compound activity on cardiac Nav1.5 peak and late currents in an automated patch clamp platform. J Pharmacol Toxicol Methods. (2019) 99:106575. doi: 10.1016/j.vascn.2019.04.001
71. Dixon R, Alexander S, Brickel N. Effect of lamotrigine on the PR interval in healthy subjects. Br J Clin Pharmacol. (2011) 71:961–2.
72. Erdemli G, Kim A, Ju H, Springer C, Penland R, Hoffmann P. Cardiac safety implications of hNav1.5 blockade and a framework for pre-clinical evaluation. Front Pharmacol. (2012) 3:6. doi: 10.3389/fphar.2012.00006
73. Miners J, McKinnon R, Mackenzie P. Genetic polymorphisms of UDP-glucuronosyltransferases and their functional significance. Toxicology. (2002) 18:453–6.
74. Milosheska D, Lorber B, Vovk T, Kastelic M, Dolžan V, Grabnar I. Pharmacokinetics of lamotrigine and its metabolite N-2-glucuronide: influence of polymorphism of UDP-glucuronosyltransferases and drug transporters. Br J Clin Pharmacol. (2016) 82:399–411. doi: 10.1111/bcp.12984
75. Wolking S, Moreau C, Nies A, Schaeffeler E, McCormack M, Auce P, et al. Testing association of rare genetic variants with resistance to three common antiseizure medications. Epilepsia. (2020) 61:657–66. doi: 10.1111/epi.16467
76. Bardai A, Blom M, van Noord C, Verhamme K, Sturkenboom M, Tan H. Sudden cardiac death is associated both with epilepsy and with use of antiepileptic medications. Heart. (2014) 101:17–22.
77. Ishizue N, Niwano S, Saito M, Fukaya H, Nakamura H, Igarashi T, et al. Polytherapy with sodium channel-blocking antiepileptic drugs are associated with arrhythmogenic ST-T abnormality in patients with epilepsy. Seizure. (2016) 40:81–7. doi: 10.1016/j.seizure.2016.06.004
78. Ota H, Kawamura Y, Sato N, Hasebe NA. Carbamazepine-induced Brugada-type electrocardiographic pattern in a patient with schizophrenia. Int Med. (2017) 56:3047–50. doi: 10.2169/internalmedicine.8875-17
79. Swe T, Bhattarai B, Dufresne A. Type 1 Brugada pattern ECG due to supra-therapeutic phenytoin level. BMJ Case Rep. (2016) 2016:bcr2016214899.
80. Restrepo J, MacLean R, Celano C, Huffman J, Januzzi J, Beach S. The assessment of cardiac risk in patients taking lamotrigine; a systematic review. Gen Hosp Psychiatry. (2022) 78:14–27.
Keywords: Brugada syndrome, lamotrigine, epilepsy, syncope, sudden unexpected death in epilepsy (SUDEP), sudden cardiac death (SCD), ion channels
Citation: Omer H, Omer MH, Alyousef AR, Alzammam AM, Ahmad O and Alanazi HA (2022) Unmasking of Brugada syndrome by lamotrigine in a patient with pre-existing epilepsy: A case report with review of the literature. Front. Cardiovasc. Med. 9:1005952. doi: 10.3389/fcvm.2022.1005952
Received: 28 July 2022; Accepted: 10 October 2022;
Published: 28 October 2022.
Edited by:
Matteo Anselmino, University of Turin, ItalyReviewed by:
Pasquale Striano, Giannina Gaslini Institute (IRCCS), ItalyCopyright © 2022 Omer, Omer, Alyousef, Alzammam, Ahmad and Alanazi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hafiz Omer, b21lcmhAbmdoYS5tZWQuc2E=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.