
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 03 November 2022
Sec. Cardiovascular Biologics and Regenerative Medicine
Volume 9 - 2022 | https://doi.org/10.3389/fcvm.2022.1000578
This article is part of the Research Topic Cardiac Regeneration View all 12 articles
 Runran Miao1
Runran Miao1 Libo Wang1,2Zhigang Chen1,3,4Shiqi Ge1Li Li1Kai Zhang1Yingen Chen1Wenjing Guo1Xulei Duan1Mingyang Zhu1
Libo Wang1,2Zhigang Chen1,3,4Shiqi Ge1Li Li1Kai Zhang1Yingen Chen1Wenjing Guo1Xulei Duan1Mingyang Zhu1 Guoan Zhao1,3,4*
Guoan Zhao1,3,4* Fei Lin1,3,4*
Fei Lin1,3,4*Myocardial remodeling is a key pathophysiological basis of heart failure, which seriously threatens human health and causes a severe economic burden worldwide. During chronic stress, the heart undergoes myocardial remodeling, mainly manifested by cardiomyocyte hypertrophy, apoptosis, interstitial fibrosis, chamber enlargement, and cardiac dysfunction. The NADPH oxidase family (NOXs) are multisubunit transmembrane enzyme complexes involved in the generation of redox signals. Studies have shown that NOXs are highly expressed in the heart and are involved in the pathological development process of myocardial remodeling, which influences the development of heart failure. This review summarizes the progress of research on the pathophysiological processes related to the regulation of myocardial remodeling by NOXs, suggesting that NOXs-dependent regulatory mechanisms of myocardial remodeling are promising new therapeutic targets for the treatment of heart failure.
Myocardial remodeling is a fundamental pathophysiological process in heart failure (HF) and is closely related to high morbidity and mortality of cardiovascular diseases (CVD) worldwide (1, 2). The main pathological features of myocardial remodeling are cardiomyocyte hypertrophy and apoptosis, excessive extracellular matrix protein (ECM) deposition, collagen deposition, and perivascular fibrosis, which lead to myocardial stiffness, chamber dilation, cardiac insufficiency, and ultimately heart failure (1, 3). Furthermore, various cardiovascular diseases, such as myocardial infarction, cardiomyopathy, heart valve disease, myocarditis, and hypertension, can lead to myocardial remodeling, myocardial stiffness, and reduced compliance, followed by heart failure and death (4). Therefore, reversing myocardial remodeling and reducing morbidity and mortality of heart failure is the key to clinical treatment of cardiovascular diseases. Accumulating evidence suggests that ROS produced by nicotinamide adenine dinucleotide phosphate (NADPH) oxidases (NOXs) can regulate the level of oxidative stress and participate in the development of myocardial remodeling. This review explores the involvement of NADPH oxidase-related isoforms in the pathophysiological process of myocardial remodeling development through the regulation of complex molecular pathways and provides new ideas and approaches for the treatment of heart failure.
NADPH oxidase, a multi-enzyme complex originally found in neutrophils, is involved in the non-specific host immune defense response against pathogenic microorganisms and is a key regulator of the redox homeostatic response. Seven mammalian isoforms of NOXs have been identified, including NOX1, NOX2, NOX3, NOX4, NOX5, double oxidase 1 (DUX1), and double oxidase 2 (DUOX2) (5, 6). All NOX isoforms contain NADPH-and flavin adenine dinucleotide (FAD)-binding domains. The different isoforms comprise homologs containing the NOX2 gp91Phox subunit but have different localization and regulatory roles (7). NOX1–4 consists of the NOX subunit catalytic subunit and p22phox as the only membrane-bound subunit. NOX5 is a structurally active enzyme body consisting of only the NOX subunit. DUOX 1 and DUOX2 consist of the DUOX-1/2 subunit, DUOXA1/2 subunit, and amino-terminal transmembrane structural domain with the peroxidase-like structural domain (8, 9). Table 1 describes the composition, localization and related functions of NADPH oxidase.
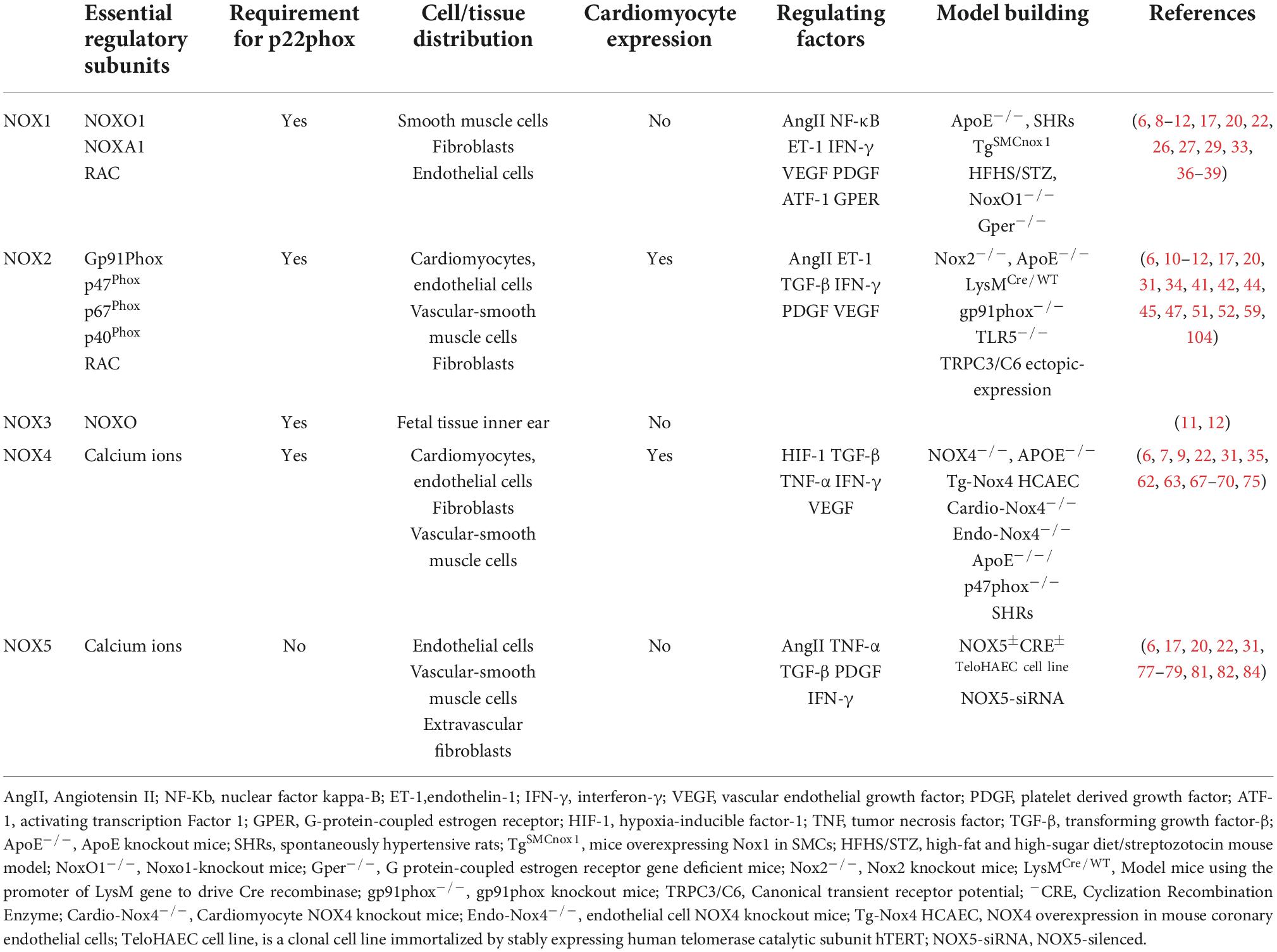
Table 1. Introduction to the composition, localization and related functions of NADPH oxidase.
The highest levels of NOX1, NOX2, NOX4, and NOX5 are found in the cardiovascular system (10), and the main isoforms expressed in the skeletal system are NOX1, NOX2, and NOX4. NOX3 is expressed only in fetal tissues and in the inner ear, especially in the cochlea, sensory epithelium, and spiral ganglia of the vestibule, which are essential for vestibular development and function (11, 12). DUOX1 and DUOX2 are mainly expressed in thyroid tissues (13). NOXs are composed of different regulatory subunits. NOX1 consists of p22Phox, NOXO1, NOXA1, and RAC1/2 regulatory subunits, NOX2 consists of Gp91Phox, p22Phox, p47Phox, p67Phox, p40Phox, and RAC1/2 regulatory subunits, and NOX3 consists of p22Phox and NOXO regulatory subunits. The p22Phox regulatory subunits regulate NOX4, and NOX5, DUOX 1, and DUOX2 are mainly regulated by the calcium-dependent mode of the N-terminal structural domain without p22Phox regulatory subunits (8, 14, 15).
NOXs are one of the main sources of reactive oxygen species (ROS) that are involved in the pathophysiological processes of CVD through redox reactions (11, 16). ROS in the heart is mainly produced through NOX1, NOX2, NOX4, and NOX5 catalysis (17). The main enzymatic sources of ROS are mitochondrial respiratory chain enzymes, xanthine oxidase (XO), uncoupled endothelial nitric oxide synthase (eNOS), and NOXs (18, 19). ROS, known as “second messengers,” play a role in rapidly regulating the activity of signaling molecules and transcription factors. The main types of ROS include superoxide radical anion (O2⋅-), hydrogen peroxide (H2O2), and hydroxyl radicals (⋅OH). NOX1, 2, and 5 produce O2⋅-, while NOX4 mainly produces H2O2. NOX-induced oxidative stress that produces ROS can lead to eNOS dysregulation and endothelial dysfunction, reduce NO bioavailability, and induce myocardial damage (20). Under normal conditions, a small amount of ROS maintains the homeostasis of the cardiovascular system. However, excess ROS can cause redox imbalance, resulting in myocardial cell apoptosis and necrosis, fibroblast proliferation, activation of matrix metalloproteinases (MMPs), collagen deposition, and other pathophysiological processes, activating oxidative stress and causing pathological myocardial remodeling, leading to various diseases, such as myocardial infarction, hypertension, arrhythmia, heart failure, and vascular dysfunction (21–23). Excessive production of ROS also activates a variety of hypertrophic signaling kinases and transcription factors, disrupting the contractile function of excitation-contraction coupling core proteins, causing mitochondrial DNA(mtDNA) damage, lipid and protein peroxidation, abnormal ion channel function, leading to fibroblast proliferation, vascular endothelial dysfunction, interstitial fibrosis, atheromatous plaque formation, thromboembolism formation and inadequate cardiac energy supply, eventually progressing to heart failure (24–28). Recent studies have shown that ROS produced by mitochondria activate the oxidative stress process, causing apoptosis and interstitial fibrosis, resulting in functional and structural impairment, leading to deterioration of right ventricular (RV) function and pressure overload, which in turn leads to right heart failure (29). Studies have shown that NOX-related subtypes can mediate the development of cardiovascular diseases through the production of excess ROS. Figure 1 shows the functional properties and basic activators of the four isoforms involved in myocardial remodeling.
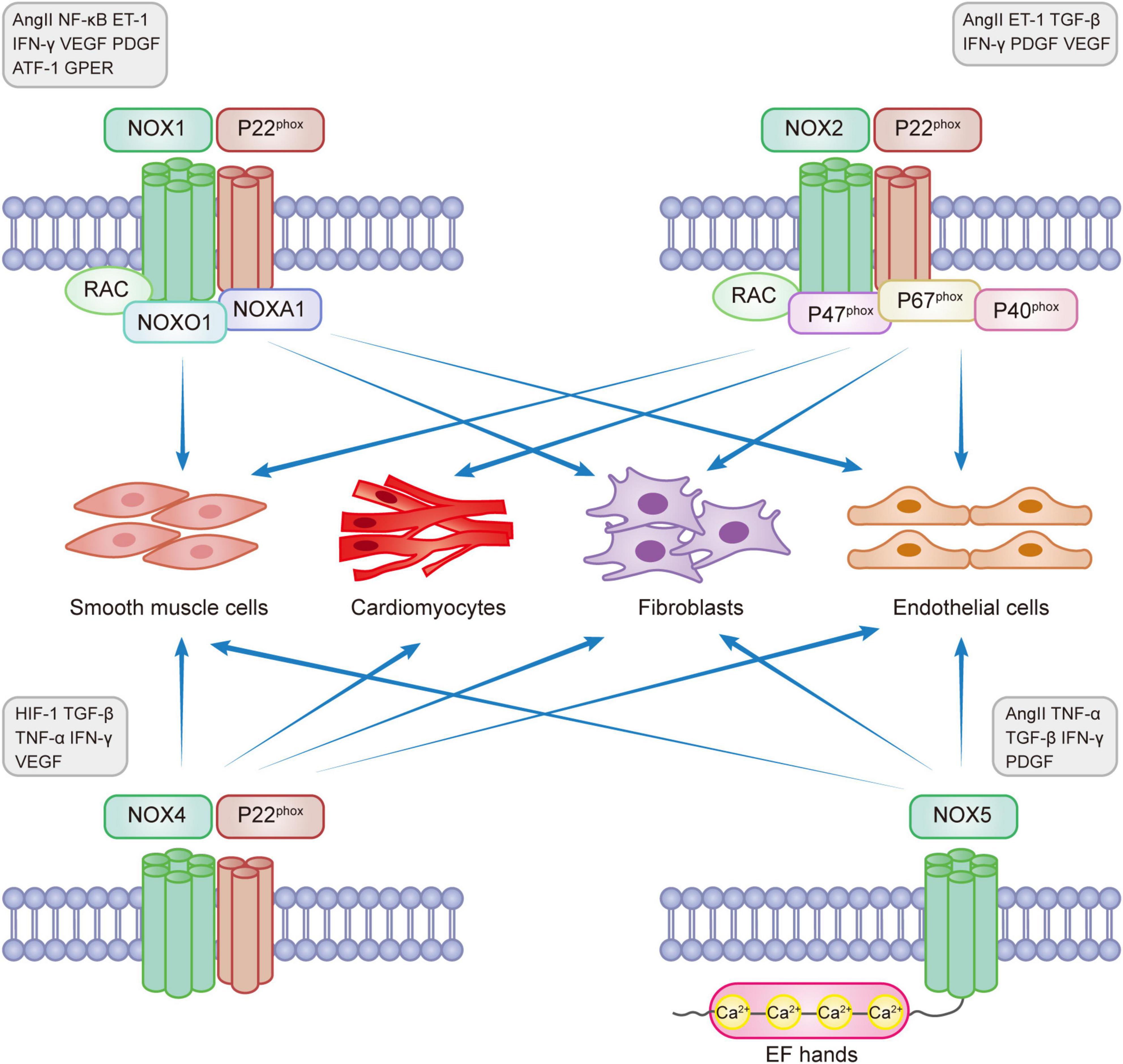
Figure 1. NADPH oxidase expression in myocardial remodeling.
NOX1 was initially found in colon epithelial cells but can also be expressed in smooth muscle cells, fibroblasts, and endothelial cells. NOX1 is involved in the development of many diseases, such as atherosclerotic cardiovascular disease, lung disease, chronic kidney disease, cerebrovascular disease, and cancer (5, 30, 31). NOX1 is expressed on the X chromosome and determines the risk of sex-linked diseases (32). It is the main isoform regulating hypertensive diseases and aortic thickening. NOX1 has a self-regulatory function and is mainly regulated by the expression levels of various proteins such as AngII, NF-κB, endothelin-1 (ET-1), interferon-γ(IFN-γ), vascular endothelial growth factor (VEGF), platelet-derived growth factor (PDGF), activating transcription factor 1 (ATF-1), and G protein-coupled estrogen receptor (GPER), which play pro-proliferative and pro-inflammatory roles (6, 31, 33, 34).
Studies have shown that NOX1 promotes the progression of atherosclerotic disease, hypertensive disease, and myocardial remodeling through multiple signaling targets. Experimental studies showed that NOX1 expression was barely detectable in the left ventricle of adult rats and cultured cardiomyocytes of neonatal rats, but NOX2 and NOX4 could be detected, and the NOX2 expression level was higher than that of NOX4 (35). High NOX1 expression activates macrophage infiltration and promotes systemic inflammatory responses, thereby accelerating atherosclerosis development in female mice (36). Studies have found that NOX1 expression in ApoE–/– mouse models activates endothelial cell and macrophage proliferation and collagen deposition, significantly enlarging the intra-aortic plaque area and promoting atherosclerotic disease progression concurrently (33, 37). Studies have also reported that diabetic APOE–/– mice can increase the NOX1 level by mediating the overexpression of ET-1, increasing the production of ROS in peri-aortic adipose tissue, and promoting atherosclerosis progression (38). Vascular smooth muscle cell phenotype transition is a pathophysiological mechanism of atherosclerotic disease, and protein disulfide isomerase-A1 (PDIA1) is an upstream regulator of smooth muscle cell (SMC) phenotype transition, upregulating the expression levels of NOX1 and NOX4, causing cytoskeleton reconstruction and promoting atherosclerotic disease progression and exacerbating myocardial remodeling (39). AngII directly enhances the binding of NOX1 to angiotensin type 1 (AT1) receptors and stimulates vascular smooth muscle cell activation (5), which has pro-inflammatory and pro-atherosclerotic effects. The epidermal growth factor receptor (EGFR), an important target of NOX1, upregulates ATF-1-mediated NOX1 expression levels and regulates cell proliferation and migration, promoting atherosclerosis progression (33). A study showed that NOX1 and NOX2 are highly expressed in aged spontaneously hypertensive rats (40). By constructing transgenic mice overexpressing NOX1 in vascular smooth muscle cells (TgSMCnox1), releasing excessive ROS from NOX1 could cause vascular endothelial cell dysfunction, leading to vessel wall hypertrophy and hypertension (41). In another study, NOX1-deficient mice (KO) had reduced expression of the surface adhesion molecules VCAM-1 and ICAM-1, reduced levels of myocardial inflammatory markers such as Mac-2, IL-1, and NLRP3, and downregulated myocardial metabolic remodeling in cardiac endothelial cells compared to male wild-type mice (WT) using a high-fat, high-sugar diet (HFHS)/streptozotocin (STZ) mouse model (42). GPER receptors promote ROS production by directly enhancing NOX1 binding to AT1, stimulating the proliferation of cardiomyocytes and vascular smooth muscle cells, affecting myocardial structural and morphological changes (43), and leading to myocardial remodeling.
NOX2 (gp91Phox), present in phagocytes, is the first typical member of the NADPH oxidase family to be identified. NOX2 can be expressed in cardiomyocytes, endothelial cells, vascular smooth muscle cells, fibroblasts, and inflammatory cells and is present in the heart, blood vessels, neural tissue, and kidney (14, 44, 45). NOX2 is a major enzyme involved in the membrane-bound release of ROS and the development of heart failure, dyskinesia, and myotonic diseases (46). Many factors could enhance the expression of NOX2, such as Hyperglycemia, hyperlipidemia, ischemia-reperfusion injury, Ang II, ET-1, transforming growth factor β (TGF-β), IFN-γ, PDGF, and VEGF (6, 7, 31, 34).
NOX2 is involved in the normal heart developmental process but aggravates the progression of diseases such as atherosclerosis, hypertension, arrhythmias, and heart failure. NOX2–/– embryonic hearts develop abnormal endocardial cushion development, which severely affects the endocardial-to-mesenchymal transition (EndMT) of atrial cushion explants, resulting in endocardial cell proliferation, apoptosis, and cardiac malformations (47). When cardiomyocytes are subjected to mechanical distortion by physiological stretch, they activate NOX2 in the sarcoplasmic and t-tubular membranes, induce ROS production through a microtubule-dependent network (X-ROS signaling), activate oxidative stress, sensitize ryanodine receptors (RyRs) around the sarcoplasmic reticulum (SR), induce massive cytoplasmic calcium ion (Ca2 +) release and increase the incidence of arrhythmias and cardiomyopathy (48). Studies have demonstrated that NOX2 expression in ApoE–/– mice reduces NO bioavailability, downregulates oxidative stress, decreases ROS release, and activates vascular smooth muscle cells, which can cause atherosclerosis (49). By constructing a post-infarction heart failure model in the left anterior descending branch of mice, AT1 receptor blockers acted on NOX2+ myeloid cells, inducing inflammatory cell infiltration and activation of oxidative stress, causing vascular endothelial dysfunction and exacerbating heart failure (50). Transverse aortic constriction(TAC) induction model mice showed that the absence of NOX2 interfered with oxidative stress in the heart and produced a sustained protective effect against heart failure (45). By constructing gp91phox–/– mouse models, inhibition of NOX2 expression reduced the release of ROS and MMPs, decreased the activation of vascular endothelial cells owing to hypoxia and inflammatory response, and attenuated vascular remodeling (51). NOX2 and NOX4 in the hypothalamic paraventricular nucleus are key sources of aldosterone, leading to ROS release, causing sympathetic excitation, and contributing to the development of hypertensive disease (52). Activation of the renin-angiotensin system (RAS) promotes the high expression of NOX2 in cardiac endothelial cells, releasing excess superoxide, leading to NO inactivation, mediating inflammatory responses, and enhancing endothelial-mesenchymal transition (EMT), which leads to vasodilator dysfunction and increased cardiac interstitial fibrosis (53). Clinical studies have demonstrated a certain thrombotic risk in COVID-19 patients, correlating with oxidative stress caused by NOX2 activation (54, 55). Therefore, NOX2 is expected to be a novel pharmacological target for the treatment and prevention of COVID-19. This shows that NOX2 can be involved in the progression of cardiovascular diseases by participating in the pathogenesis of inflammatory response, apoptosis, and oxidative stress, providing novel ideas for clinical treatment.
NOX2 is involved in cellular hypertrophy and apoptosis during myocardial remodeling. Studies have shown that hyperglycemia can participate in NOX2-mediated oxidative stress in an AMPK-dependent manner, causing myocardial remodeling by causing apoptosis (56, 57). Ras-related C3 botulinum toxin substrate 1 (RAC1) is an important component of NADPH oxidase that promotes the transfer of cytoplasmic subunits to the membrane and induces NOXs activity. RAC1 is an important regulator that mediates NADPH oxidase activity to produce myocardial remodeling (58). RAC1 is involved in NOX2-induced hypertrophy of cardiomyocytes, causing myocardial remodeling (35). NOX2 expression mediates toll-like receptor 5 (TLR5) in adriamycin (Doxorubicin, DOX) toxicity, exacerbating cardiomyocyte death and interstitial fibrosis, leading to acute myocardial injury (59). Transient receptor potential canonical (TRPC) subfamily proteins are components of calcium channels that mediate calcium signaling and are key mediators involved in the development of myocardial remodeling. TRPC3 mediates pathological myocardial remodeling by forming a stable protein complex with NOX2 and p22phox, releasing ROS, amplifying redox signals, and inducing fibrotic responses in cardiomyocytes and fibroblasts through mechanical stimulation (60). Studies have shown that NOX2 is the main source of superoxide anion production by human atrial myocytes, causing apoptosis and interstitial fibrotic remodeling, which is an important basis for oxidative stress and electrophysiological remodeling in patients with atrial fibrillation (61). Therefore, NOX2 downregulation can act as an oxidative stress inhibitor and reduce ROS formation, providing long-term therapeutic value for the treatment of myocardial remodeling and the development of new drugs.
NOX4 is the most widely expressed protein initially found in the kidney. NOX4 is present in the mitochondria, endoplasmic reticulum, cardiomyocytes, endothelial cells, vascular smooth muscle cells, fibroblasts, blood vessels, and various organs (31, 39). During various stress conditions, NOX4 is an important source of oxidative stress in myocardial mitochondria and is involved in the energy metabolic process of myocardial remodeling (62). TGF-β, TNF-α, and IFN-γ can activate high NOX4 expression (6, 31, 63, 64). NOX4 can be involved in the pathogenesis of various cardiovascular diseases and myocardial remodeling as a central component of endoplasmic reticulum stress, participating in the oxidation of sarcoplasmic reticulum calcium ATPase (SERCA), increasing intracellular calcium ion transport, mediating myocardial electrophysiological activity, activating oxidative stress, and reducing autophagy (65).
NOX4 expression plays a partially protective role in the heart (66). NOX4 serves as a vasodilator through H2O2. NOX4–/– and APOE–/– mice can induce atherosclerotic disease progression by increasing endothelial cell inflammation and oxidative stress levels (67). NOX4 attenuates age-related mitochondrial oxidative stress and vascular inflammation and reduces the incidence of cardiovascular disease (62, 68). NOX4–/– mice (c-NOX4 KO) inhibited cysteine oxidation and nuclear withdrawal of HDAC4, and TAC-induced pathological myocardial hypertrophy was attenuated (69). During myocardial ischemia-reperfusion (I/R), cardiomyocyte NOX4 levels contributed to macrophage proliferation and polarization responses, and volume overload improved ventricular remodeling after myocardial infarction (MI) through NOX4 activation of Akt and an increase in downstream protein synthesis markers (S6 ribosomal protein and eIF4E-BP1), a therapeutic target (70).
High expression of NOX4, an endogenous anti-atherosclerotic enzyme, can cause damage to the heart. Lack of NOX4 in cardiac myocytes is beneficial, while the lack of NOX4 in blood vessels causes a decreased adaptive response, leading to post-ischemic and hypoxic myocardial injury. During hypoxia, NOX4 mediates the release of HIF-1α and vascular endothelial growth factor (VEGF), causing apoptosis and dysfunction of cardiomyocytes (7, 71). Excess ROS produced by NOX4 promotes cardiomyocyte apoptosis and mitochondrial dysfunction by mediating the upregulation of the pro-apoptotic proteins Bax and caspase-3 and the downregulation of the anti-apoptotic protein BCL2 and the survival protein p-AKTser473, resulting in myocardial structural damage and cardiac systolic and diastolic insufficiency (7, 72). Increased NOX4 expression in male Wistar rats treated with dexamethasone caused sympathetic excitation of the heart and blood vessels, upregulated oxidative stress levels, elevated blood pressure, and caused myocardial fibrosis. Simultaneously, the incidence of ventricular arrhythmias was significantly increased when induced by drugs or abrupt pacing (73). Clinically, the novel hypoglycemic GLP-1 agonist liraglutide exerts an anti-inflammatory effect by interfering with Sirtuin-1 (SIRT1)/NOX4/ROS signaling and NLRP3 inflammatory vesicle-dependent cellular scorching, downregulating NOX4 expression, reducing hypoxia-induced oxidative stress, and acting as a myocardial protector (74). JunD is a member of the activator protein 1 (AP-1) family of transcription factors and serves as a key target protein involved in anti-oxidative stress. JunD–/– mice upregulate oxidative stress levels by activating signaling pathways involved in JNK1 (c-Jun N-terminal kinases), increasing NOX2/4 expression, and inducing inflammatory response pathways involved in NF-κB signaling, causing left heart dysfunction (75). NOX1 and NOX4 mediate signaling pathways that promote smooth muscle cell proliferation, differentiation, and migration, causing vascular remodeling and exacerbating the progression of atherosclerosis and hypertension. Dual inhibitors of Nox1/4 increase blood pressure and perivascular macrophage infiltration, exacerbating perivascular inflammation and fibrosis levels in models of spontaneous hypertension (76). Thus, NOX4 exerts a dual regulatory effect on the myocardium and blood vessels to control the development of myocardial remodeling. The potential risk of cardiovascular disease should be considered when using NOX4 inhibitors.
NOX5 is found only in humans and is not expressed in other mammals. It is mainly found in the vascular and renal systems and can be expressed in endothelial cells, vascular smooth muscle cells, and extravascular fibroblasts (77, 78). The NOX5 gene is located on chromosome 15 and includes six different splice isoforms: α, β, γ, δ, ε, and ζ, all of which are expressed in the vascular endothelial and smooth muscle cells. NOX5-α and NOX5-β are the most abundantly expressed and functional proteins for ROS production, and NOX5-ε may be a negative regulator of NOX5 (79). NOX5 does not require regulatory subunit activation and depends on an increased Ca2 + concentration for activation, causing a conformational change in the N-terminal structural domain containing the EF-hand loop (78).
Clinical studies have found that NOX5 is involved in the development of myocardial infarction, atherosclerosis, hypertension, and aortic aneurysm and aggravates myocardial remodeling. Hyperglycemia, TNF, TGF-β, IFN-γ, PDGF and Ang II promote increased intracellular calcium, and NOX5 binds directly to intracellular calcium and is activated for overexpression (6, 31, 79). Protein kinase C (PKC) also promotes NOX5 overexpression, regulates the endothelial cyclooxygenase-2 (COX-2)/prostaglandin-2 (PGE2) axis, causes ROS overproduction, and accelerates the progression of myocardial remodeling by activating the post-myocardial infarction (MI) inflammatory response (80, 81). NF-κB, a transcription factor involved in NOX5-induced adenocarcinoma, is involved in apoptosis through the upregulation of ROS-mediated activation of cyclooxygenase-2 (COX-2) (81). Recent studies have shown that NOX5, a gene newly associated with hypertension (82), ameliorates abnormal VSMC proliferation, inhibits oxidative stress, and attenuates hypertensive complications through the NOX5/ROS/c-Src signaling pathway (redox-sensitive protein) (83). In response to platelet-derived growth factor (PDGF) stimulation, NOX5 produces ROS to activate the JAK/STAT pathway to induce the proliferation of human aortic endothelial cells (84). NOX5 promotes the release of ROS from extracellular vesicles (EVS), induces phenotypic conversion of vascular smooth muscle cells, activates oxidative stress, promotes vascular smooth muscle cell proliferation, and induces myocardial remodeling (85). Therefore, NOX5 may be a new therapeutic target for the clinical treatment of heart failure.
NADPH is involved in oxidative stress-induced myocardial remodeling, mainly in endothelial dysfunction, ROS overproduction, and reduced antioxidant capacity (79, 86). ROS produced by NOXs can regulate the level of oxidative stress and participate in the development of myocardial remodeling, a response produced by the heart to pathological stimuli and consists of two main categories: physiological and pathological (87). Physiological myocardial remodeling is a compensatory and adaptive change of the heart to the body to maintain normal physiological functions of the heart. Pathological myocardial remodeling mainly occurs in structural, morphological, and energy metabolic remodeling, resulting in cardiac scarring, leading to myocardial stiffness, cavity dilation, and ventricular systolic dysfunction, resulting in arrhythmias and heart failure (1). The myocardial remodeling processes involve cardiomyocytes, fibroblasts, endothelial cells, vascular smooth muscle cells (VSMCs), and immune cells. Numerous studies have shown that modulation of NOX levels can improve the progression of myocardial remodeling. In contrast, NOXs produce ROS that act on mitochondrial enzymes, protein kinases, and transcription factors, causing cardiomyocyte hypertrophy, apoptosis, and necrosis, severely impairing cardiomyocyte energy metabolism and mitochondrial function, affecting myocardial excitation-contraction coupling, leading to myocardial hypertrophy and end-stage heart failure. Studies have confirmed that Ang II is important in regulating ROS generation by NOXs through AT1 receptors (86). Ang II inhibition also helps reduce the expression of NOXs and regulate myocardial remodeling.
Myocardial structural remodeling includes cardiomyocyte hypertrophy, apoptosis, and interstitial fibrosis (14, 88). Cardiomyocyte hypertrophy is manifested by increased cardiomyocyte length, width, and mass, but the number of cardiomyocytes remains unchanged. Pathological myocardial hypertrophy is stimulated by cardiomyocyte apoptosis, fibroblasts, endothelial cells, smooth muscle cells and phagocytes activation and differentiation, extracellular matrix protein deposition, and interstitial and perivascular fibrosis (87), causing myocardial stiffness, abnormal cardiac function, and eventually heart failure (Figure 2).
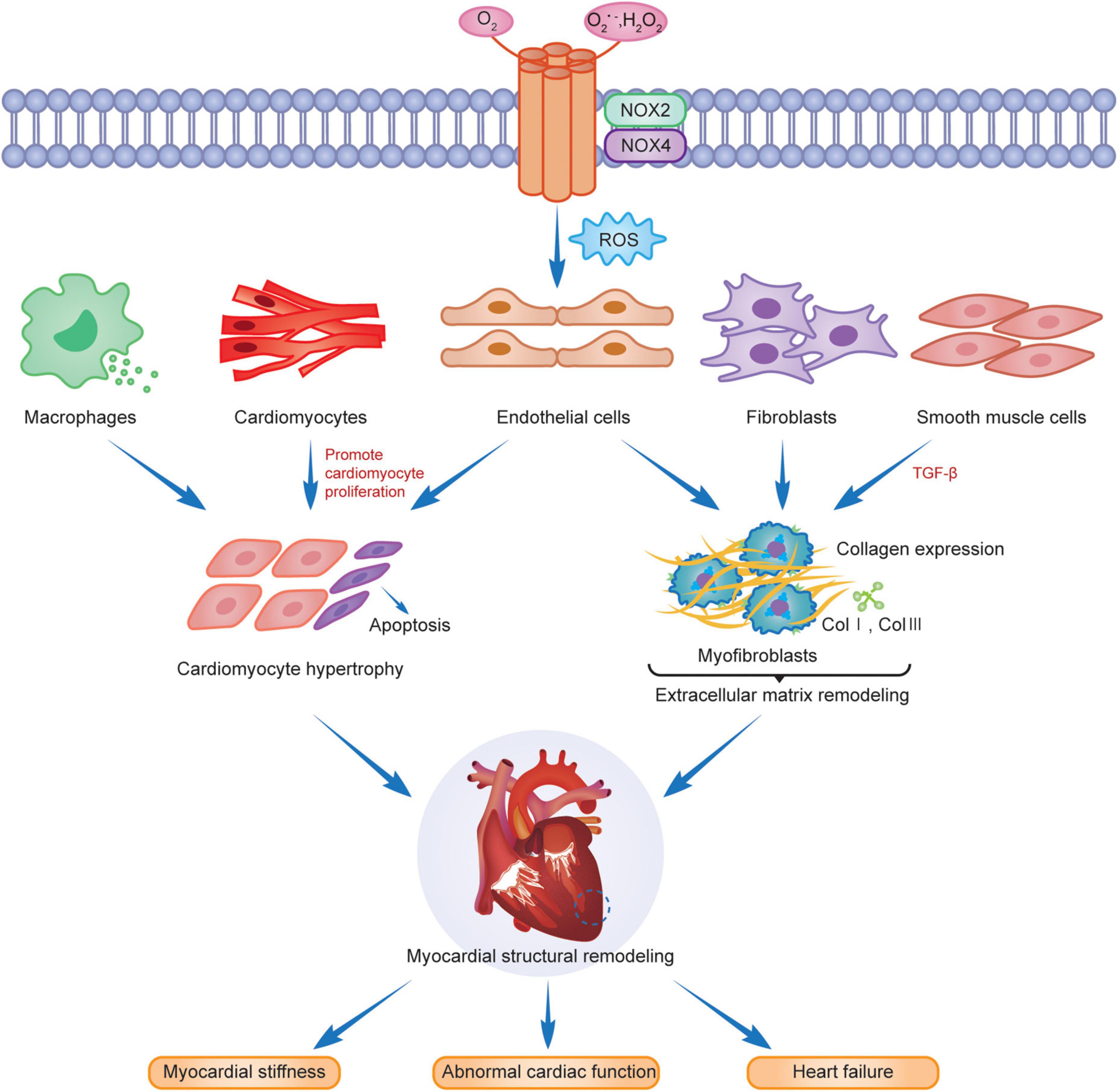
Figure 2. NOX2 and NOX4 are involved in myocardial remodeling.
Cardiomyocyte hypertrophy and apoptosis are the underlying features of myocardial structural remodeling. Studies have demonstrated that the main isoforms in cardiomyocytes are NOX2 and NOX4, with NOX2 mainly localized in the plasma membrane and NOX4 distributed in the nuclear membrane. Both are associated with the myocardial structural remodeling in chronic stress states. Intracellular NOX-mediated ROS, activated via the Ras-MEK1/2-ERK1/2 pathway, are involved in α1-adrenoceptor (α1-AR) mediated cardiomyocyte hypertrophy (22), which leads to myocardial structural and morphological remodeling. RAC1 is an important active regulator of NOXs. Ang II activates RAC1, upregulates the expression of NOXs, promotes cardiomyocyte hypertrophy, fibroblast proliferation, and myofibroblast differentiation, and induces myocardial remodeling (89). Studies have revealed that activation of NOX2 was induced after AngII, pressure overload and myocardial infarction, leading to cardiomyocyte hypertrophy, apoptosis, vascular smooth muscle cell proliferation and extracellular interstitial fibrosis, and aggravating myocardial structural remodeling (90–92). The AMPK/NOX2 pathway attenuates oxidative stress in diabetic hearts with ischemia/reperfusion injury and may reduce myocardial hypertrophic lesions by reducing cardiomyocyte death (93). NOX2, expressed in the heart, not only mediates oxidative stress, apoptosis, and inflammation through multiple target pathway proteins but is also subject to gene-level regulators, such as microRNAs (miRNAs) for its downstream products, to reduce its activity and slow the progression of cardiomyopathy and myocardial remodeling after MI (94). Cardiomyocyte hypertrophy caused by erythromycin (DOX) is associated with the activation of TRPC3/NOX2 signaling targets, which causes left ventricular hypertrophy, resulting in cardiac dysfunction (95). FYN is a tyrosine kinase of the Src family that negatively regulates NOX4 activity, attenuates oxidative stress and cardiomyocyte death, and slows myocardial remodeling through translational modification of the phosphorylated C-terminal Y566 of NOX4 (96). Another study indicated that high NOX4 expression activates the AKT/mTOR/NF-κB signaling pathway, mediates oxidative stress, releases excess ROS, causes cardiomyocyte hypertrophy and interstitial fibrosis, and leads to cardiac remodeling. Myocardial remodeling was significantly reduced after the treatment of mouse hearts with the NOX inhibitor GKT137831 (97).
Interstitial fibrosis is another characteristic change in the structural remodeling of the myocardium. Extracellular matrix protein deposition is an important component of interstitial fibrosis, and the main basic proteins included are type I and type III collagen while altering the myocardial meshwork. The proliferation and migration of cardiac fibroblasts are crucial in the interstitial fibrosis process of myocardial structural remodeling (1), and the two contribute to each other to aggravate myocardial remodeling progression. TGF-βis critical for the conversion of cardiac fibroblasts to a myofibroblast phenotype. Intervention in the TGF-β/Smad signaling pathway attenuates the activation and phenotypic conversion of fibroblasts due to Ang II induced NADPH oxidase activation and attenuates extracellular matrix remodeling due to cascade reactions (98). NOX4 induces TGF-β-induced actin upregulation and phenotypic transition in fibroblasts by stimulating Smads 2/3 phosphorylation and activation (99). ROS production after NOX4 expression upregulation can promote endothelial cell migration and vascular remodeling in an eNOS-dependent manner (100), which are jointly involved in promoting extracellular matrix remodeling. In the case of chronic hemodynamic overload, the NOX4 expression level in cardiomyocytes and endothelial cells increases, enhancing HIF-1 and VEGF signaling, activating oxidative stress, causing myocardial fibrosis and vascular proliferation, increasing myocardial capillary density, and acting as a partial myocardial compensation but leading to the development of myocardial remodeling (66). However, NOX2 is deleterious in oxidative stress, and salusin-β is a bioactive peptide with 20 amino acids that can regulate the proliferative activity of vascular endothelial and smooth muscle cells. Salusin-β inhibits NOX-mediated ROS production, activates eNOS and NO production, causes dysregulation of the balance of contractile and vasodilator factors released from the vascular endothelium (101), improves endothelial dysfunction, cardiovascular remodeling, and cardiac dysfunction, and promotes early depolarization of rabbit cardiomyocytes by activating the calmodulin-dependent protein kinase II (CaMKII) pathway. Studies demonstrated its use as a downstream target of NOX2 to increase the incidence of arrhythmias in AngII-induced mouse models (102, 103), suggesting that NOX2 is involved in cardiac structural remodeling. Myocardial interstitial fibrosis due to pressure overload induced by Ang II injection, aldosterone, and aortic ligation was significantly reduced in the NOX2–/– mouse model (104).
In cardiac energy metabolism, 95% of energy is provided by mitochondrial oxidative phosphorylation in the form of adenosine triphosphate (ATP), and the remaining 5% is provided by glycolysis. Mitochondria provide ATP mainly from the oxidation of fatty acids, approximately 40–60%, and the rest from the oxidation of ketone bodies, amino acids, and pyruvate (derived from glucose and lactate) (105, 106). Therefore, the remodeling of myocardial energy metabolism consists mainly of disorders of cardiac metabolic substrates and intracellular mitochondrial dysfunction (4, 107, 108).
NOX2 and NOX4 are involved in disrupting metabolic substrates for myocardial remodeling. The expression of NOX2 facilitates the polarization of M1 macrophages, promotes the coupling of inflammatory receptors to NOX2, activates the inflammatory cascade response, enhances the transport of glucose transporter proteins to the cytoplasmic membrane, and increases glucose uptake by glycolytic metabolic processes (109). Excitation-contraction coupling and metabolic disorders can cause increased mitochondrial respiratory chain superoxide synthesis, increased mitochondrial ROS release, and an imbalance between myocardial oxygen supply and demand, triggering oxidative stress and affecting various cellular survival rates during myocardial remodeling (110). NOX4 is mainly localized in the mitochondria and is involved in tricarboxylic acid cycle (TCA) and energy metabolism in the electron transport chain through redox reactions, thereby affecting nutrient metabolism (97).
Mitochondria are key to the remodeling of myocardial energy metabolism. Mitochondria are the center of intracellular processes of calcium homeostasis, energy metabolism, and oxidative stress. Intracellular Ca2+ and Na+ imbalance impede mitochondrial calcium overload, leading to the abnormal opening of the mitochondrial permeability transition pore (mPTP), resulting in abnormal mitochondrial energy metabolism, triggering oxidative stress, leading to excessive release of ROS from mitochondria, interfering with the excitation-contraction coupling process in the myocardium, and inducing cardiomyocyte death (111, 112). Studies have shown that NOX4 upregulation mediates mitochondrial dysfunction, cardiomyocyte hypertrophy, and apoptosis and is involved in myocardial structure and energy metabolism (97). AMPK is a physiological inhibitor of NOX and ROS production in endothelial cells. During pathological conditions, AMPK expression is reduced, and mitochondria produce excessive ROS, affecting myocardial energy supply and promoting inflammatory responses, exacerbating myocardial energy metabolic remodeling (113). Thus AMPK can be a therapeutic target for NOX-mediated remodeling of myocardial energy metabolism as a key mediator. AngII induces NADPH oxidase activation and ROS production, leading to mitochondrial microstructure and dysfunction by decreasing mitochondrial membrane potential and affecting mitochondrial morphology. Ang II stimulates NADPH oxidase through a protein kinase C (PKC)-mediated pathway-dependent stimulation, causing increased ROS release, activating mitochondrial Na+-K+- ATP channels, causing mitochondrial membrane potential abnormalities, disrupting the mitochondrial respiratory enzyme complex, resulting in mitochondrial dysfunction and remodeling of myocardial energy metabolism (114). NOX4 is mainly localized in the mitochondria of cardiomyocytes and endothelial cells. Mitochondrial DNA (mtDNA) is the main target of NADPH oxidase-mediated ROS action (25). NOX4 plays an adaptive role by promoting the oxidation of fatty acids and providing the energy required by cardiomyocytes (115). NOX4 and ROS produced by mitochondria are involved in NLRP3 inflammatory vesicles, dynamin-related protein 1 (Drp1) activation, and mitochondrial division induced by dilated cardiomyopathy (DCM) (116). NOX4 is crucial for myocardial energy metabolic remodeling. Therefore, mitochondrial dysfunction is critical in myocardial energy metabolic remodeling during pathological myocardial remodeling and simultaneously promotes myocardial pathological structural remodeling, leading to cardiac hypertrophy and functional decline (87). Figure 3 shows the involvement of NOXs in the remodeling of myocardial energy metabolism through the release of ROS. The intervention of NADPH-activated oxidative stress provides new ideas for the treatment of myocardial remodeling and end-stage heart failure.
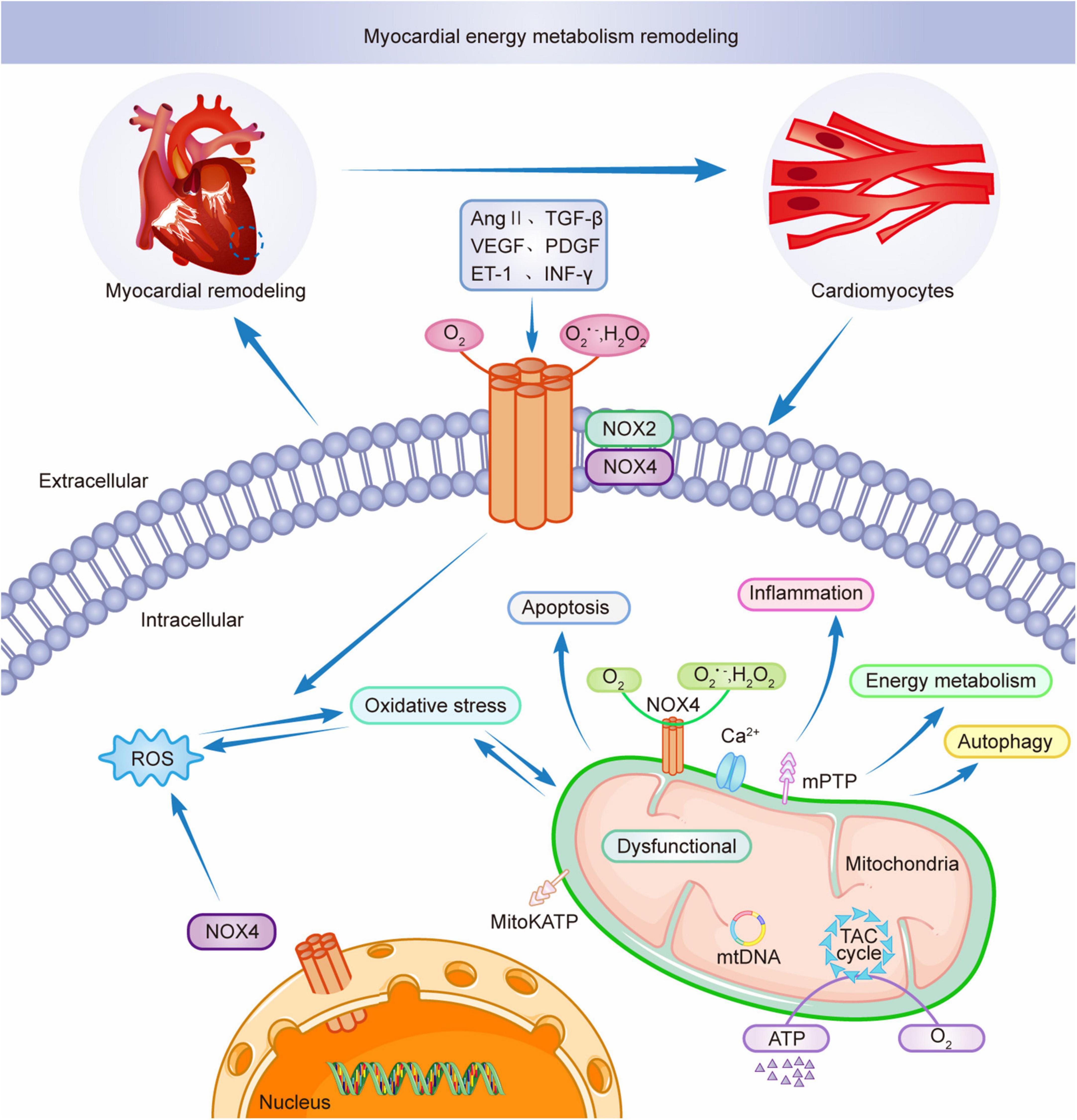
Figure 3. NOX2 and NOX4 participate in myocardial energy metabolism remodeling.
Drugs targeting NOX subtypes and related pathways are well researched and can be useful in the clinical management of cardiac and vascular disease. Numerous studies have shown that clinical agents acting on NOX family targets can slow the progression of myocardial remodeling and heart failure. RAC1 knockdown and NADPH oxidase inhibitors were shown to reduce myocardial apoptosis, inhibit the conversion of fibroblasts into myofibroblasts, provide antihypertrophic and antifibrotic effects, and improve the progression of myocardial remodeling (117). As an NOX inhibitor, celastrol directly binds to signal transduction and transcriptional activator proteins, reduces tyrosine phosphorylation and nuclear translocation, and inhibits AngII involvement in NOX2-mediated myocardial remodeling, presumably leading to a protective effect on the myocardium through anti-oxidative stress (118, 119). Apocynin reduces DNA damage and telomere shortening in cardiomyocytes, inhibits the release of ROS from NOX, reduces oxidative stress levels, improves myocardial remodeling and reduces death in heart failure. Apocynin has been shown to reduce fibroblast activation, cardiomyocyte hypertrophy and apoptosis through anti-oxidative stress, preventing deterioration of cardiac function and pathological remodeling in Diabetic cardiomyopathy (DCM) mouse model (120, 121). Vitamin D deficiency is an important potential risk factor for cardiovascular disease. Vitamin D regulates NO synthesis by mediating the activity of eNOS. Studies have shown that vitamin D enhances oxidative capacity through the activity of antioxidant enzymes and can inhibit NOX production of ROS (122). The key to the development of NOX subtypes of cardiovascular disease and the exploration of NOX-mediated related targets provide potential therapeutic directions for the clinical treatment of cardiovascular disease caused by myocardial remodeling.
Myocardial remodeling is a common pathophysiological process in many cardiovascular diseases, accelerating disease progression and eventually leading to heart failure. There are few clinically effective drugs to reverse myocardial remodeling, and NADPH oxidases play a crucial role in the pathogenesis of cardiovascular diseases. NOX-mediated ROS production is associated with important cellular functions such as cell differentiation, proliferation, migration, apoptosis, and immune response. In addition, it is involved in different signaling pathways mediating the pathogenesis of cardiovascular diseases, such as inflammatory response, autophagy, apoptosis, and oxidative stress. Increased ROS release and oxidative stress-induced cellular damage lead to structural and functional alterations in the heart, leading to cardiac arrest, the major cause of death worldwide (123). Studies have confirmed that NADPH oxidase can induce ROS production and accelerate the morbidity and mortality associated with heart failure through different pathways and molecular mechanisms.
In this review, the basic pathophysiological process of myocardial remodeling was explored regarding the NADPH oxidase family mediating related signaling pathways through multiple signaling targets, upregulating oxidative stress levels, activating increased ROS expression levels, and severely affecting pathological changes such as cardiomyocyte hypertrophy, apoptosis, and interstitial fibrosis, and causing changes in myocardial structural and energy metabolic remodeling, leading to the deterioration of heart failure. However, its role in the signaling cascade and the associated molecular mechanisms involved in different pathological conditions are still unknown. Therefore, exploring the pathogenesis of myocardial remodeling and related therapeutic targets of oxidative stress is crucial. Although there are still some limitations to the clinical understanding of NOX in cardiovascular diseases, exploring relevant NOX inhibitory targets and developing new directions for clinical drugs provide potential applications for reversing myocardial remodeling and delaying the progression of heart failure in the clinical setting.
RM wrote the manuscript by reviewing and summarizing the relevant literature. LW and ZC revised the manuscript. SG, LL, and KZ prepared the figures. YC, WG, XD, and MZ involved in the work. GZ and FL supervised the progress of the entire manuscript to ensure completeness and accuracy and approved the submitted version. All authors contributed to the article and approved the submitted version.
This work was supported by the Key Specialized Research and Development Breakthrough in Henan Province (Nos. 212102310350 and 222102310442), the Research Projects of Higher Education Institutions in Henan Province (No. 22A360017), and the Medical Science and Technology Research Program Jointly Co-constructed Project in Henan Province (Nos. LHGL20200521 and LHGL20210513).
We thank Editage (www.editage.cn) for English language editing.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Li L, Zhao Q, Kong W. Extracellular matrix remodeling and cardiac fibrosis. Matrix Biol. (2018) 68–9:490–506. doi: 10.1016/j.matbio.2018.01.013
2. Scofield SL, Amin P, Singh M, Singh K. Extracellular ubiquitin: role in myocyte apoptosis and myocardial remodeling. Compr Physiol. (2015) 6:527–60. doi: 10.1002/cphy.c150025
3. Gonzalez A, Schelbert EB, Diez J, Butler J. Myocardial interstitial fibrosis in heart failure: biological and translational perspectives. J Am Coll Cardiol. (2018) 71:1696–706. doi: 10.1016/j.jacc.2018.02.021
4. Gibb AA, Hill BG. Metabolic coordination of physiological and pathological cardiac remodeling. Circ Res. (2018) 123:107–28. doi: 10.1161/CIRCRESAHA.118.312017
5. Barton M, Meyer MR, Prossnitz ER. Nox1 downregulators: a new class of therapeutics. Steroids. (2019) 152:108494. doi: 10.1016/j.steroids.2019.108494
6. Lassegue B, San Martin A, Griendling KK. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ Res. (2012) 110:1364–90. doi: 10.1161/CIRCRESAHA.111.243972
7. Waghela BN, Vaidya FU, Agrawal Y, Santra MK, Mishra V, Pathak C. Molecular insights of NADPH oxidases and its pathological consequences. Cell Biochem Funct. (2021) 39:218–34. doi: 10.1002/cbf.3589
8. Lambeth JD. Nox enzymes and the biology of reactive oxygen. Nat Rev Immunol. (2004) 4:181–9. doi: 10.1038/nri1312
9. Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. (2007) 87:245–313. doi: 10.1152/physrev.00044.2005
10. Konior A, Schramm A, Czesnikiewicz-Guzik M, Guzik TJ. NADPH oxidases in vascular pathology. Antioxid Redox Signal. (2014) 20:2794–814. doi: 10.1089/ars.2013.5607
11. Forstermann U, Xia N, Li H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ Res. (2017) 120:713–35. doi: 10.1161/CIRCRESAHA.116.309326
12. Burtenshaw D, Kitching M, Redmond EM, Megson IL, Cahill PA. Reactive oxygen species (ROS), intimal thickening, and subclinical atherosclerotic disease. Front Cardiovasc Med. (2019) 6:89. doi: 10.3389/fcvm.2019.00089
13. De Deken X, Wang D, Many MC, Costagliola S, Libert F, Vassart G, et al. Cloning of two human thyroid CDNAS encoding new members of the NADPH oxidase family. J Biol Chem. (2000) 275:23227–33. doi: 10.1074/jbc.M000916200
14. Nabeebaccus A, Zhang M, Shah AM. NADPH oxidases and cardiac remodelling. Heart Fail Rev. (2011) 16:5–12. doi: 10.1007/s10741-010-9186-2
15. Hordijk PL. Regulation of NADPH oxidases: the role of RAC proteins. Circ Res. (2006) 98:453–62. doi: 10.1161/01.RES.0000204727.46710.5e
16. Cai H. Nad(P)H oxidase-dependent self-propagation of hydrogen peroxide and vascular disease. Circ Res. (2005) 96:818–22. doi: 10.1161/01.RES.0000163631.07205.fb
17. Maejima Y, Kuroda J, Matsushima S, Ago T, Sadoshima J. Regulation of myocardial growth and death by NADPH oxidase. J Mol Cell Cardiol. (2011) 50:408–16. doi: 10.1016/j.yjmcc.2010.12.018
18. Zhang Y, Murugesan P, Huang K, Cai H. NADPH oxidases and oxidase crosstalk in cardiovascular diseases: novel therapeutic targets. Nat Rev Cardiol. (2020) 17:170–94. doi: 10.1038/s41569-019-0260-8
19. Bubb KJ, Drummond GR, Figtree GA. New opportunities for targeting redox dysregulation in cardiovascular disease. Cardiovasc Res. (2020) 116:532–44. doi: 10.1093/cvr/cvz183
20. Meza CA, La Favor JD, Kim DH, Hickner RC. Endothelial dysfunction: is there a hyperglycemia-induced imbalance of NOX and NOS? Int J Mol Sci. (2019) 20:3775. doi: 10.3390/ijms20153775
21. Grieve DJ, Byrne JA, Siva A, Layland J, Johar S, Cave AC, et al. Involvement of the nicotinamide adenosine dinucleotide phosphate oxidase isoform Nox2 in cardiac contractile dysfunction occurring in response to pressure overload. J Am Coll Cardiol. (2006) 47:817–26. doi: 10.1016/j.jacc.2005.09.051
22. Xiao L, Pimentel DR, Wang J, Singh K, Colucci WS, Sawyer DB. Role of reactive oxygen species and Nad(P)H oxidase in alpha-adrenoceptor signaling in adult rat cardiac myocytes. Am J Physiol Cell Physiol. (2002) 282:C926–34. doi: 10.1152/ajpcell.00254.2001
23. Maytin M, Siwik DA, Ito M, Xiao L, Sawyer DB, Liao R, et al. Pressure overload-induced myocardial hypertrophy in mice does not require Gp91phox. Circulation. (2004) 109:1168–71. doi: 10.1161/01.CIR.0000117229.60628.2F
24. Panda P, Verma HK, Lakkakula S, Merchant N, Kadir F, Rahman S, et al. Biomarkers of oxidative stress tethered to cardiovascular diseases. Oxid Med Cell Longev. (2022) 2022:9154295. doi: 10.1155/2022/9154295
25. Tsutsui H, Kinugawa S, Matsushima S. Oxidative stress and heart failure. Am J Physiol Heart Circ Physiol. (2011) 301:H2181–90. doi: 10.1152/ajpheart.00554.2011
26. Nishihara T, Tokitsu T, Sueta D, Oike F, Takae M, Fujisue K, et al. Clinical significance of reactive oxidative metabolites in patients with heart failure with reduced left ventricular ejection fraction. J Card Fail. (2021) 27:57–66. doi: 10.1016/j.cardfail.2020.07.020
27. Krzeminska J, Wronka M, Mlynarska E, Franczyk B, Rysz J. Arterial hypertension-oxidative stress and inflammation. Antioxidants. (2022) 11:172. doi: 10.3390/antiox11010172
28. Shi X, Dorsey A, Qiu H. New progress in the molecular regulations and therapeutic applications in cardiac oxidative damage caused by pressure overload. Antioxidants. (2022) 11:877. doi: 10.3390/antiox11050877
29. Muller M, Bischof C, Kapries T, Wollnitza S, Liechty C, Geissen S, et al. Right heart failure in mice upon pressure overload is promoted by mitochondrial oxidative stress. JACC Basic Transl Sci. (2022) 7:658–77. doi: 10.1016/j.jacbts.2022.02.018
30. Gimenez M, Schickling BM, Lopes LR, Miller FJ Jr. Nox1 in cardiovascular diseases: regulation and pathophysiology. Clin Sci. (2016) 130:151–65. doi: 10.1042/CS20150404
31. Cave AC, Brewer AC, Narayanapanicker A, Ray R, Grieve DJ, Walker S, et al. NADPH oxidases in cardiovascular health and disease. Antioxid Redox Signal. (2006) 8:691–728. doi: 10.1089/ars.2006.8.691
32. AlSiraj Y, Chen X, Thatcher SE, Temel RE, Cai L, Blalock E, et al. XX sex chromosome complement promotes atherosclerosis in mice. Nat Commun. (2019) 10:2631. doi: 10.1038/s41467-019-10462-z
33. Sheehan AL, Carrell S, Johnson B, Stanic B, Banfi B, Miller FJ Jr. Role for NOX1 NADPH oxidase in atherosclerosis. Atherosclerosis. (2011) 216:321–6. doi: 10.1016/j.atherosclerosis.2011.02.028
34. Lassegue B, Clempus RE. Vascular Nad(P)H oxidases: specific features, expression, and regulation. Am J Physiol Regul Integr Comp Physiol. (2003) 285:R277–97. doi: 10.1152/ajpregu.00758.2002
35. Hingtgen SD, Tian X, Yang J, Dunlay SM, Peek AS, Wu Y, et al. Nox2-containing NADPH oxidase and AKT activation play a key role in angiotensin II-induced cardiomyocyte hypertrophy. Physiol Genomics. (2006) 26:180–91. doi: 10.1152/physiolgenomics.00029.2005
36. Buchmann GK, Schurmann C, Warwick T, Schulz MH, Spaeth M, Muller OJ, et al. Deletion of Noxo1 limits atherosclerosis development in female mice. Redox Biol. (2020) 37:101713. doi: 10.1016/j.redox.2020.101713
37. Jandeleit-Dahm K. Endothelin in diabetes-associated atherosclerosis: opportunity ‘NOX’. Cardiovasc Res. (2021) 117:987–9. doi: 10.1093/cvr/cvab018
38. Ouerd S, Idris-Khodja N, Trindade M, Ferreira NS, Berillo O, Coelho SC, et al. Endothelium-restricted endothelin-1 overexpression in type 1 diabetes worsens atherosclerosis and immune cell infiltration via NOX1. Cardiovasc Res. (2021) 117:1144–53. doi: 10.1093/cvr/cvaa168
39. Fernandes DC, Wosniak J Jr, Goncalves RC, Tanaka LY, Fernandes CG, Zanatta DB, et al. PDIA1 acts as master organizer of Nox1/Nox4 balance and phenotype response in vascular smooth muscle. Free Radic Biol Med. (2021) 162:603–14. doi: 10.1016/j.freeradbiomed.2020.11.020
40. Wind S, Beuerlein K, Armitage ME, Taye A, Kumar AH, Janowitz D, et al. Oxidative stress and endothelial dysfunction in aortas of aged spontaneously hypertensive rats by NOX1/2 is reversed by NADPH oxidase inhibition. Hypertension. (2010) 56:490–7. doi: 10.1161/HYPERTENSIONAHA.109.149187
41. Dikalova A, Clempus R, Lassegue B, Cheng G, McCoy J, Dikalov S, et al. Nox1 overexpression potentiates angiotensin II-induced hypertension and vascular smooth muscle hypertrophy in transgenic mice. Circulation. (2005) 112:2668–76. doi: 10.1161/CIRCULATIONAHA.105.538934
42. Xu L, Balzarolo M, Robinson EL, Lorenz V, Verde GD, Joray L, et al. Nox1 mediates metabolic heart disease in mice and is upregulated in monocytes of humans with diastolic dysfunction. Cardiovasc Res. (2021) cvab349. doi: 10.1093/cvr/cvab349
43. Meyer MR, Fredette NC, Sharma G, Barton M, Prossnitz ER. GPER is required for the age-dependent upregulation of the myocardial endothelin system. Life Sci. (2016) 159:61–5. doi: 10.1016/j.lfs.2016.02.041
44. Laddha AP, Kulkarni YA. NADPH oxidase: a membrane-bound enzyme and its inhibitors in diabetic complications. Eur J Pharmacol. (2020) 881:173206. doi: 10.1016/j.ejphar.2020.173206
45. Parajuli N, Patel VB, Wang W, Basu R, Oudit GY. Loss of Nox2 (Gp91phox) prevents oxidative stress and progression to advanced heart failure. Clin Sci. (2014) 127:331–40. doi: 10.1042/CS20130787
46. Ferreira LF, Laitano O. Regulation of NADPH oxidases in skeletal muscle. Free Radic Biol Med. (2016) 98:18–28. doi: 10.1016/j.freeradbiomed.2016.05.011
47. Moazzen H, Wu Y, Engineer A, Lu X, Aulakh S, Feng Q. Nox2 is critical to endocardial to mesenchymal transition and heart development. Oxid Med Cell Longev. (2020) 2020:1679045. doi: 10.1155/2020/1679045
48. Prosser BL, Ward CW, Lederer WJ. X-ROS signaling: rapid mechano-chemo transduction in heart. Science. (2011) 333:1440–5. doi: 10.1126/science.1202768
49. Urner S, Ho F, Jha JC, Ziegler D, Jandeleit-Dahm K. NADPH oxidase inhibition: preclinical and clinical studies in diabetic complications. Antioxid Redox Signal. (2020) 33:415–34. doi: 10.1089/ars.2020.8047
50. Molitor M, Rudi WS, Garlapati V, Finger S, Schuler R, Kossmann S, et al. Nox2+ myeloid cells drive vascular inflammation and endothelial dysfunction in heart failure after myocardial infarction via angiotensin II receptor type 1. Cardiovasc Res. (2021) 117:162–77. doi: 10.1093/cvr/cvaa042
51. Tojo T, Ushio-Fukai M, Yamaoka-Tojo M, Ikeda S, Patrushev N, Alexander RW. Role of Gp91phox (Nox2)-containing Nad(P)H oxidase in angiogenesis in response to hindlimb ischemia. Circulation. (2005) 111:2347–55. doi: 10.1161/01.CIR.0000164261.62586.14
52. Xue B, Beltz TG, Johnson RF, Guo F, Hay M, Johnson AK. PVN adenovirus-sirna injections silencing either Nox2 or Nox4 attenuate aldosterone/Nacl-induced hypertension in mice. Am J Physiol Heart Circ Physiol. (2012) 302:H733–41. doi: 10.1152/ajpheart.00873.2011
53. Murdoch CE, Chaubey S, Zeng L, Yu B, Ivetic A, Walker SJ, et al. Endothelial NADPH oxidase-2 promotes interstitial cardiac fibrosis and diastolic dysfunction through proinflammatory effects and endothelial-mesenchymal transition. J Am Coll Cardiol. (2014) 63:2734–41. doi: 10.1016/j.jacc.2014.02.572
54. Sindona C, Schepici G, Contestabile V, Bramanti P, Mazzon E. Nox2 activation in Covid-19: possible implications for neurodegenerative diseases. Medicina. (2021) 57:604. doi: 10.3390/medicina57060604
55. Violi F, Oliva A, Cangemi R, Ceccarelli G, Pignatelli P, Carnevale R, et al. Nox2 activation in Covid-19. Redox Biol. (2020) 36:101655. doi: 10.1016/j.redox.2020.101655
56. Hafstad AD, Hansen SS, Lund J, Santos CXC, Boardman NT, Shah AM, et al. NADPH oxidase 2 mediates myocardial oxygen wasting in obesity. Antioxidants. (2020) 9:171. doi: 10.3390/antiox9020171
57. Wang C, Zhu L, Yuan W, Sun L, Xia Z, Zhang Z, et al. Diabetes aggravates myocardial ischaemia reperfusion injury via activating Nox2-related programmed cell death in an AMPK-dependent manner. J Cell Mol Med. (2020) 24:6670–9. doi: 10.1111/jcmm.15318
58. Satoh M, Ogita H, Takeshita K, Mukai Y, Kwiatkowski DJ, Liao JK. Requirement of RAC1 in the development of cardiac hypertrophy. Proc Natl Acad Sci U.S.A. (2006) 103:7432–7. doi: 10.1073/pnas.0510444103
59. Ma ZG, Kong CY, Wu HM, Song P, Zhang X, Yuan YP, et al. Toll-like receptor 5 deficiency diminishes doxorubicin-induced acute cardiotoxicity in mice. Theranostics. (2020) 10:11013–25. doi: 10.7150/thno.47516
60. Kitajima N, Numaga-Tomita T, Watanabe M, Kuroda T, Nishimura A, Miyano K, et al. TRPC3 positively regulates reactive oxygen species driving maladaptive cardiac remodeling. Sci Rep. (2016) 6:37001. doi: 10.1038/srep37001
61. Kim YM, Guzik TJ, Zhang YH, Zhang MH, Kattach H, Ratnatunga C, et al. A myocardial Nox2 containing Nad(P)H oxidase contributes to oxidative stress in human atrial fibrillation. Circ Res. (2005) 97:629–36. doi: 10.1161/01.RES.0000183735.09871.61
62. Ago T, Matsushima S, Kuroda J, Zablocki D, Kitazono T, Sadoshima J. The NADPH oxidase NOX4 and aging in the heart. Aging. (2010) 2:1012–6. doi: 10.18632/aging.100261
63. Sturrock A, Cahill B, Norman K, Huecksteadt TP, Hill K, Sanders K, et al. Transforming growth factor-beta1 induces Nox4 Nad(P)H oxidase and reactive oxygen species-dependent proliferation in human pulmonary artery smooth muscle cells. Am J Physiol Lung Cell Mol Physiol. (2006) 290:L661–73. doi: 10.1152/ajplung.00269.2005
64. Yoshida LS, Tsunawaki S. Expression of NADPH oxidases and enhanced HO-generating activity in human coronary artery endothelial cells upon induction with tumor necrosis factor-alpha. Int Immunopharmacol. (2008) 8:1377–85. doi: 10.1016/j.intimp.2008.05.004
65. Ochoa CD, Wu RF, Terada LS. Ros signaling and ER stress in cardiovascular disease. Mol Aspects Med. (2018) 63:18–29. doi: 10.1016/j.mam.2018.03.002
66. Zhang M, Mongue-Din H, Martin D, Catibog N, Smyrnias I, Zhang X, et al. Both cardiomyocyte and endothelial cell Nox4 mediate protection against hemodynamic overload-induced remodelling. Cardiovasc Res. (2018) 114:401–8. doi: 10.1093/cvr/cvx204
67. Schurmann C, Rezende F, Kruse C, Yasar Y, Lowe O, Fork C, et al. The NADPH oxidase Nox4 has anti-atherosclerotic functions. Eur Heart J. (2015) 36:3447–56. doi: 10.1093/eurheartj/ehv460
68. Vendrov AE, Vendrov KC, Smith A, Yuan J, Sumida A, Robidoux J, et al. Nox4 NADPH oxidase-dependent mitochondrial oxidative stress in aging-associated cardiovascular disease. Antioxid Redox Signal. (2015) 23:1389–409. doi: 10.1089/ars.2014.6221
69. Matsushima S, Kuroda J, Ago T, Zhai P, Park JY, Xie LH, et al. Increased oxidative stress in the nucleus caused by NOX4 mediates oxidation of HDAC4 and cardiac hypertrophy. Circ Res. (2013) 112:651–63. doi: 10.1161/CIRCRESAHA.112.279760
70. Mongue-Din H, Patel AS, Looi YH, Grieve DJ, Anilkumar N, Sirker A, et al. NADPH oxidase-4 driven cardiac macrophage polarization protects against myocardial infarction-induced remodeling. JACC Basic Transl Sci. (2017) 2:688–98. doi: 10.1016/j.jacbts.2017.06.006
71. Zhang M, Brewer AC, Schroder K, Santos CX, Grieve DJ, Wang M, et al. NADPH oxidase-4 mediates protection against chronic load-induced stress in mouse hearts by enhancing angiogenesis. Proc Natl Acad Sci U.S.A. (2010) 107:18121–6. doi: 10.1073/pnas.1009700107
72. Wen SY, Tamilselvi S, Shen CY, Day CH, Chun LC, Cheng LY, et al. Protective effect of HDL on NADPH oxidase-derived super oxide anion mediates hypoxia-induced cardiomyocyte apoptosis. PLoS One. (2017) 12:e0179492. doi: 10.1371/journal.pone.0179492
73. Macedo FN, Souza DS, Araujo J, Dantas CO, Miguel-Dos-Santos R, Silva-Filha E, et al. NOX-dependent reactive oxygen species production underlies arrhythmias susceptibility in dexamethasone-treated rats. Free Radic Biol Med. (2020) 152:1–7. doi: 10.1016/j.freeradbiomed.2020.03.005
74. Zhang L, Qi X, Zhang G, Zhang Y, Tian J. Saxagliptin protects against hypoxia-induced damage in H9c2 cells. Chem Biol Interact. (2020) 315:108864. doi: 10.1016/j.cbi.2019.108864
75. Hussain S, Khan AW, Akhmedov A, Suades R, Costantino S, Paneni F, et al. Hyperglycemia induces myocardial dysfunction via epigenetic regulation of jund. Circ Res. (2020) 127:1261–73. doi: 10.1161/CIRCRESAHA.120.317132
76. Nosalski R, Mikolajczyk T, Siedlinski M, Saju B, Koziol J, Maffia P, et al. Nox1/4 inhibition exacerbates age dependent perivascular inflammation and fibrosis in a model of spontaneous hypertension. Pharmacol Res. (2020) 161:105235. doi: 10.1016/j.phrs.2020.105235
77. Zhao GJ, Zhao CL, Ouyang S, Deng KQ, Zhu L, Montezano AC, et al. Ca(2+)-dependent Nox5 (NADPH oxidase 5) exaggerates cardiac hypertrophy through reactive oxygen species production. Hypertension. (2020) 76:827–38. doi: 10.1161/HYPERTENSIONAHA.120.15558
78. BelAiba RS, Djordjevic T, Petry A, Diemer K, Bonello S, Banfi B, et al. Nox5 variants are functionally active in endothelial cells. Free Radic Biol Med. (2007) 42:446–59. doi: 10.1016/j.freeradbiomed.2006.10.054
79. Jha JC, Watson AMD, Mathew G, de Vos LC, Jandeleit-Dahm K. The emerging role of NADPH oxidase NOX5 in vascular disease. Clin Sci. (2017) 131:981–90. doi: 10.1042/CS20160846
80. Pandey D, Patel A, Patel V, Chen F, Qian J, Wang Y, et al. Expression and functional significance of NADPH oxidase 5 (Nox5) and its splice variants in human blood vessels. Am J Physiol Heart Circ Physiol. (2012) 302:H1919–28. doi: 10.1152/ajpheart.00910.2011
81. Marques J, Cortes A, Pejenaute A, Ansorena E, Abizanda G, Prosper F, et al. Induction of cyclooxygenase-2 by overexpression of the human NADPH oxidase 5 (NOX5) gene in aortic endothelial cells. Cells. (2020) 9:637. doi: 10.3390/cells9030637
82. Touyz RM, Anagnostopoulou A, Camargo LL, Rios FJ, Montezano AC. Vascular biology of superoxide-generating NADPH oxidase 5-implications in hypertension and cardiovascular disease. Antioxid Redox Signal. (2019) 30:1027–40. doi: 10.1089/ars.2018.7583
83. Camargo LL, Montezano AC, Hussain M, Wang Y, Zou Z, Rios FJ, et al. Central role of C-Src in Nox5- mediated redox signalling in vascular smooth muscle cells in human hypertension. Cardiovasc Res. (2022) 118:1359–73. doi: 10.1093/cvr/cvab171
84. Jay DB, Papaharalambus CA, Seidel-Rogol B, Dikalova AE, Lassegue B, Griendling KK. Nox5 mediates Pdgf-induced proliferation in human aortic smooth muscle cells. Free Radic Biol Med. (2008) 45:329–35. doi: 10.1016/j.freeradbiomed.2008.04.024
85. Furmanik M, Chatrou M, van Gorp R, Akbulut A, Willems B, Schmidt H, et al. Reactive oxygen-forming NOX5 links vascular smooth muscle cell phenotypic switching and extracellular vesicle-mediated vascular calcification. Circ Res. (2020) 127:911–27. doi: 10.1161/CIRCRESAHA.119.316159
86. Yousefian M, Shakour N, Hosseinzadeh H, Hayes AW, Hadizadeh F, Karimi G. The natural phenolic compounds as modulators of NADPH oxidases in hypertension. Phytomedicine. (2019) 55:200–13. doi: 10.1016/j.phymed.2018.08.002
87. Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. (2018) 15:387–407. doi: 10.1038/s41569-018-0007-y
88. Murdoch CE, Zhang M, Cave AC, Shah AM. NADPH oxidase-dependent redox signalling in cardiac hypertrophy, remodelling and failure. Cardiovasc Res. (2006) 71:208–15. doi: 10.1016/j.cardiores.2006.03.016
89. Lyu L, Chen J, Wang W, Yan T, Lin J, Gao H, et al. Scoparone alleviates ang II-induced pathological myocardial hypertrophy in mice by inhibiting oxidative stress. J Cell Mol Med. (2021) 25:3136–48. doi: 10.1111/jcmm.16304
90. Bendall JK, Cave AC, Heymes C, Gall N, Shah AM. Pivotal role of a Gp91(Phox)-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation. (2002) 105:293–6. doi: 10.1161/hc0302.103712
91. Greenberg HZE, Zhao G, Shah AM, Zhang M. Role of oxidative stress in calcific aortic valve disease and its therapeutic implications. Cardiovasc Res. (2022) 118:1433–51. doi: 10.1093/cvr/cvab142
92. Byrne JA, Grieve DJ, Bendall JK, Li JM, Gove C, Lambeth JD, et al. Contrasting roles of NADPH oxidase isoforms in pressure-overload versus angiotensin II-induced cardiac hypertrophy. Circ Res. (2003) 93:802–5. doi: 10.1161/01.RES.0000099504.30207.F5
93. Marino A, Hausenloy DJ, Andreadou I, Horman S, Bertrand L, Beauloye C. Amp-activated protein kinase: a remarkable contributor to preserve a healthy heart against ROS injury. Free Radic Biol Med. (2021) 166:238–54. doi: 10.1016/j.freeradbiomed.2021.02.047
94. Zhou H, Wang B, Yang YX, Jia QJ, Zhang A, Qi ZW, et al. Long noncoding RNAS in pathological cardiac remodeling: a review of the update literature. Biomed Res Int. (2019) 2019:7159592. doi: 10.1155/2019/7159592
95. Chen DS, Yan J, Yang PZ. Cardiomyocyte atrophy, an underestimated contributor in doxorubicin-induced cardiotoxicity. Front Cardiovasc Med. (2022) 9:812578. doi: 10.3389/fcvm.2022.812578
96. Matsushima S, Kuroda J, Zhai P, Liu T, Ikeda S, Nagarajan N, et al. Tyrosine kinase fyn negatively regulates NOX4 in cardiac remodeling. J Clin Invest. (2016) 126:3403–16. doi: 10.1172/JCI85624
97. Ago T, Kuroda J, Pain J, Fu C, Li H, Sadoshima J. Upregulation of Nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circ Res. (2010) 106:1253–64. doi: 10.1161/CIRCRESAHA.109.213116
98. Fu B, Su Y, Ma X, Mu C, Yu F. Scoparone attenuates angiotensin II-induced extracellular matrix remodeling in cardiac fibroblasts. J Pharmacol Sci. (2018) 137:110–5. doi: 10.1016/j.jphs.2018.05.006
99. Cucoranu I, Clempus R, Dikalova A, Phelan PJ, Ariyan S, Dikalov S, et al. Nad(P)H oxidase 4 mediates transforming growth factor-beta1-induced differentiation of cardiac fibroblasts into myofibroblasts. Circ Res. (2005) 97:900–7. doi: 10.1161/01.RES.0000187457.24338.3D
100. Craige SM, Chen K, Pei Y, Li C, Huang X, Chen C, et al. Nadph oxidase 4 promotes endothelial angiogenesis through endothelial nitric oxide synthase activation. Circulation. (2011) 124:731–40. doi: 10.1161/CIRCULATIONAHA.111.030775
101. Xu Y, Pan Y, Wang X, Chen A, Tang X, Liu X, et al. Knockdown of salusin-beta improves cardiovascular function in myocardial infarction-induced chronic heart failure rats. Oxid Med Cell Longev. (2021) 2021:8896226. doi: 10.1155/2021/8896226
102. Lu S, Liao Z, Lu X, Katschinski DM, Mercola M, Chen J, et al. Hyperglycemia acutely increases cytosolic reactive oxygen species via O-linked glcnacylation and camkii activation in mouse ventricular myocytes. Circ Res. (2020) 126:e80–96. doi: 10.1161/CIRCRESAHA.119.316288
103. Zhang M, Perino A, Ghigo A, Hirsch E, Shah AM. NADPH oxidases in heart failure: poachers or gamekeepers? Antioxid Redox Signal. (2013) 18:1024–41. doi: 10.1089/ars.2012.4550
104. Zhang M, Shah AM. ROS signalling between endothelial cells and cardiac cells. Cardiovasc Res. (2014) 102:249–57. doi: 10.1093/cvr/cvu050
105. Lopaschuk GD, Karwi QG, Tian R, Wende AR, Abel ED. Cardiac energy metabolism in heart failure. Circ Res. (2021) 128:1487–513. doi: 10.1161/CIRCRESAHA.121.318241
106. Wu C, Zhang Z, Zhang W, Liu X. Mitochondrial dysfunction and mitochondrial therapies in heart failure. Pharmacol Res. (2022) 175:106038. doi: 10.1016/j.phrs.2021.106038
107. Bertero E, Maack C. Metabolic remodelling in heart failure. Nat Rev Cardiol. (2018) 15:457–70. doi: 10.1038/s41569-018-0044-6
108. Yang J, Guo Q, Feng X, Liu Y, Zhou Y. Mitochondrial dysfunction in cardiovascular diseases: potential targets for treatment. Front Cell Dev Biol. (2022) 10:841523. doi: 10.3389/fcell.2022.841523
109. Griffiths HR, Gao D, Pararasa C. Redox regulation in metabolic programming and inflammation. Redox Biol. (2017) 12:50–7. doi: 10.1016/j.redox.2017.01.023
110. Vasquez-Trincado C, Garcia-Carvajal I, Pennanen C, Parra V, Hill JA, Rothermel BA, et al. Mitochondrial dynamics, mitophagy and cardiovascular disease. J Physiol. (2016) 594:509–25. doi: 10.1113/JP271301
111. Bertero E, Maack C. Calcium signaling and reactive oxygen species in mitochondria. Circ Res. (2018) 122:1460–78. doi: 10.1161/CIRCRESAHA.118.310082
112. Zhou B, Tian R. Mitochondrial dysfunction in pathophysiology of heart failure. J Clin Invest. (2018) 128:3716–26. doi: 10.1172/JCI120849
113. Wang S, Zhang M, Liang B, Xu J, Xie Z, Liu C, et al. Ampkalpha2 deletion causes aberrant expression and activation of Nad(P)H oxidase and consequent endothelial dysfunction in vivo: role of 26s proteasomes. Circ Res. (2010) 106:1117–28. doi: 10.1161/CIRCRESAHA.109.212530
114. Doughan AK, Harrison DG, Dikalov SI. Molecular mechanisms of angiotensin II-mediated mitochondrial dysfunction: linking mitochondrial oxidative damage and vascular endothelial dysfunction. Circ Res. (2008) 102:488–96. doi: 10.1161/CIRCRESAHA.107.162800
115. Nabeebaccus A, Hafstad A, Eykyn T, Yin X, Brewer A, Zhang M, et al. Cardiac-targeted NADPH oxidase 4 in the adaptive cardiac remodelling of the murine heart. Lancet. (2015) 385(Suppl. 1):S73. doi: 10.1016/S0140-673660388-9
116. Zeng C, Duan F, Hu J, Luo B, Huang B, Lou X, et al. Nlrp3 inflammasome-mediated pyroptosis contributes to the pathogenesis of non-ischemic dilated cardiomyopathy. Redox Biol. (2020) 34:101523. doi: 10.1016/j.redox.2020.101523
117. Li J, Zhu H, Shen E, Wan L, Arnold JM, Peng T. Deficiency of RAC1 blocks NADPH oxidase activation, inhibits endoplasmic reticulum stress, and reduces myocardial remodeling in a mouse model of type 1 diabetes. Diabetes. (2010) 59:2033–42. doi: 10.2337/db09-1800
118. Ye S, Luo W, Khan ZA, Wu G, Xuan L, Shan P, et al. Celastrol attenuates angiotensin II-induced cardiac remodeling by targeting Stat3. Circ Res. (2020) 126:1007–23. doi: 10.1161/CIRCRESAHA.119.315861
119. Jaquet V, Marcoux J, Forest E, Leidal KG, McCormick S, Westermaier Y, et al. NADPH oxidase (NOX) isoforms are inhibited by celastrol with a dual mode of action. Br J Pharmacol. (2011) 164:507–20. doi: 10.1111/j.1476-5381.2011.01439.x
120. Brandt M, Dorschmann H, Khraisat S, Knopp T, Ringen J, Kalinovic S, et al. Telomere shortening in hypertensive heart disease depends on oxidative DNA damage and predicts impaired recovery of cardiac function in heart failure. Hypertension. (2022) 79:2173–84. doi: 10.1161/HYPERTENSIONAHA.121.18935
121. Ding W, Feng H, Li WJ, Liao HH, Zhang N, Zhou ZY, et al. Apocynin attenuates diabetic cardiomyopathy by suppressing Ask1-P38/Jnk signaling. Eur J Pharmacol. (2021) 909:174402. doi: 10.1016/j.ejphar.2021.174402
122. Kim DH, Meza CA, Clarke H, Kim JS, Hickner RC. Vitamin D and endothelial function. Nutrients. (2020) 12:575. doi: 10.3390/nu12020575
Keywords: heart failure, myocardial remodeling, NADPH oxidase, reactive oxygen species, oxidative stress
Citation: Miao R, Wang L, Chen Z, Ge S, Li L, Zhang K, Chen Y, Guo W, Duan X, Zhu M, Zhao G and Lin F (2022) Advances in the study of nicotinamide adenine dinucleotide phosphate oxidase in myocardial remodeling. Front. Cardiovasc. Med. 9:1000578. doi: 10.3389/fcvm.2022.1000578
Received: 22 July 2022; Accepted: 18 October 2022;
Published: 03 November 2022.
Edited by:
Rajika Roy, Temple University, United StatesReviewed by:
Murugesan Velayutham, West Virginia University, United StatesCopyright © 2022 Miao, Wang, Chen, Ge, Li, Zhang, Chen, Guo, Duan, Zhu, Zhao and Lin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Guoan Zhao, Z3VvYW56aGFvQHh4bXUuZWR1LmNu; Fei Lin, bGluZmVpeGl4aUBhbGl5dW4uY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.