 Carina Mauersberger1,2
Carina Mauersberger1,2 Julia Hinterdobler1,2
Julia Hinterdobler1,2 Heribert Schunkert1,2
Heribert Schunkert1,2 Thorsten Kessler1,2
Thorsten Kessler1,2 Hendrik B. Sager1,2*
Hendrik B. Sager1,2*- 1Department of Cardiology, German Heart Center Munich, Technical University Munich, Munich, Germany
- 2DZHK (German Centre for Cardiovascular Research), Partner Site Munich Heart Alliance, Munich, Germany
Atherosclerosis is the leading cause of death worldwide and leukocyte recruitment is a key element of this phenomenon, thus allowing immune cells to enter the arterial wall. There, in concert with accumulating lipids, the invading leukocytes trigger a plethora of inflammatory responses which promote the influx of additional leukocytes and lead to the continued growth of atherosclerotic plaques. The recruitment process follows a precise scheme of tethering, rolling, firm arrest, crawling and transmigration and involves multiple cellular and subcellular players. This review aims to provide a comprehensive up-to-date insight into the process of leukocyte recruitment relevant to atherosclerosis, each from the perspective of endothelial cells, monocytes and macrophages, neutrophils, T lymphocytes and platelets. In addition, therapeutic options targeting leukocyte recruitment into atherosclerotic lesions—or potentially arising from the growing body of insights into its precise mechanisms—are highlighted.
Introduction
Atherosclerosis is a chronic disease characterized by the accumulation of lipoprotein particles and inflammatory cells inside the arterial vessel wall of large- and medium-sized arteries. Atherosclerotic plaques may destabilize during the progression of the disease leading to plaque rupture/erosion ultimately resulting in partial or complete vessel obstruction which may cause cardiovascular events such as myocardial infarction (MI) or stroke (1). Altogether, cardiovascular diseases represent the leading cause of death worldwide (2). Mechanistically, atherosclerosis has long been considered a solely metabolic-driven disease; a result of high plasma lipid levels and passive uptake of cholesterol into the vessel wall at atherosclerosis-prone regions marked by disturbed blood flow patterns (3, 4). Over the past decades, however, evidence accumulated highlighting the contribution of immune cells in the etiology of atherosclerosis (4). Leukocytes, the effector cells of the immune system, contribute to all stages of the disease. Specifically, monocyte-derived macrophages, neutrophils and T lymphocytes are involved in inflammatory processes inside the vessel wall during lesion initiation, progression and rupture (5, 6). Thus, leukocyte recruitment to the vessel represents an essential early step in initiation and progression of atherosclerosis preceding local actions of intimal leukocytes. In this review, we aim to summarize basic concepts of leukocyte recruitment and highlight novel findings in the context of atherosclerosis.
Basic Concepts of Leukocyte Recruitment in Vessels
Leukocyte recruitment from blood to distinct tissues is essential in both non-sterile and sterile inflammation but also under steady-state conditions (7). For simplicity, this review will focus on leukocyte recruitment to diseased vessels in the context of atherosclerosis. Classically, the major players in the recruitment process are the endothelium and different leukocyte subsets including monocytes, neutrophils and lymphocytes. To achieve selective recruitment, all players interact in a strictly orchestrated manner (8). The traditional model of the leukocyte recruitment cascade describes three major steps following tissue/endothelial cell (EC) activation: rolling, activation, and arrest (9). However, recent experimental evidence has expanded our knowledge on the leukocyte recruitment process, suggested additional steps, and refined the molecular principles underlying different stages (10). Within the next paragraph, we aim to outline basic and novel concepts of leukocyte recruitment from the circulation.
Inflammatory Tissue Activation
Inflammatory tissue activation is the initial step in the leukocyte recruitment cascade in both non-sterile (infectious) and sterile (non-infectious) diseases. It occurs as a physiological response of the immune system to various stimuli including tissue damage and cell death, pathogens, or toxic compounds (11). While some responses are shared between non-sterile and sterile diseases, some are specific to certain pathologies. In this context, atherosclerosis-specific mechanisms will be further outlined in later sections.
Classically, acute inflammation is triggered by conserved pathogen-associated molecular patterns (PAMPs) and endogenous stress signals [damage-associated molecular patterns (DAMPs)] that are recognized by respective receptors on tissue-resident immune cells and non-immune cells (12, 13). Activation of these receptors results in the release of pro-inflammatory cytokines and chemokines (14–16). ECs are one of the main targets of pro-inflammatory cytokines (17, 18). As a result, they upregulate adhesion molecule and chemokine expression. Some cytokines such as histamine activate ECs by binding to G-protein-coupled receptors (GPCRs; = type I activation) and induce intracellular signaling cascades that lead to rapid translocation of preformed molecules (19). In contrast, type II activation of ECs is slower but longer lasting (20). It can be triggered by various inflammatory cytokines, such as tumor necrosis factor alpha (TNFα) and interleukin (IL)1-β, and leads to de novo synthesis of adhesion molecules and chemokines (20, 21). The expression and extracellular secretion of various chemokines such as C-C motif chemokine ligand (CCL)2 and C-X-C motif chemokine ligand (CXCL)1 leads to—among other functions—attraction of leukocytes (=chemotaxis) (22–24). Moreover, activated tissue-resident leukocytes, specifically macrophages, can also secrete chemotactic molecules such as CCL3 (23). In addition, activated platelets can deposit chemokines such as CCL5 and CXCL4 on ECs contributing to the chemotaxis of leukocytes to sites of inflammation (25, 26). Of note, differential recruitment of leukocyte subsets is favored by the specificity of certain chemotactic molecules and their respective receptors on leukocytes (27, 28).
In summary, inflammatory tissue activation precedes actual leukocyte recruitment by priming ECs and inducing leukocyte chemotaxis.
Leukocyte Tethering and (Slow) Rolling
Leukocyte tethering (=capture) and subsequent rolling is the first interaction step between ECs and leukocytes. It is mainly mediated by platelet (P)-selectin, endothelial (E)-selectin and leukocyte (L)-selectin (29). Although first described in platelets, P-selectin is also expressed on activated ECs. Selectins consist of an extracellular N-terminal lectin domain, an epidermal growth factor-like domain, a series of repetitive complement control proteins, a transmembrane domain and a C-terminal intracellular domain (20). With their N-terminal lectin domain, they are able to bind to glycosylated ligands in a calcium-dependent manner on the cell surface of opposite cells (30). Of note, selectin binding is highly dependent on correct glycosylation involving modifications by several enzymes that link various types of saccharide molecules (10, 31).
In vessels, leukocyte rolling is predominantly achieved by the interaction of endothelial E- and P-selectin with P-selectin glycoprotein ligand-1 (PSGL-1) and other glycosylated ligands [e.g., CD44 and ESL-1 (E-selectin ligand 1, specifically binding to E-selectin)] on leukocytes (32–36). In ECs, P-selectin is prestored in vesicles called “Weibel-Palade bodies” and translocated to the luminal membrane as a response to inflammatory stimuli, while E-selectin is synthesized de novo upon cell activation (19). L-selectin also interacts with PSGL-1 but is mainly expressed on leukocytes and thus important specifically in secondary leukocyte capture (37, 38). L-selectin on leukocytes was also shown to interact with glycosylated ligands on the endothelial membrane (39–41). However, these experiments were mainly performed in the context of lymph node homing. Still, PSGL-1 is expressed on both leukocytes and vascular ECs (42), which suggests a relevant contribution of L-selectin-mediated rolling in vessels. Additional to leukocyte-leukocyte interactions, platelet-leukocyte interactions are involved in secondary leukocyte capture processes (43, 44). Platelet P-selectin can engage with both endothelial and leukocyte PSGL-1, thereby acting as a bridge between the two cell types. Recent studies have identified further relevant players, such as the interaction of leukocyte macrophage receptor 1 (Mac-1) with platelet CD147 (45) in platelet-mediated leukocyte recruitment. In addition to membrane-bound selectins, extracellular matrix proteins, such as galectins, are likely to be involved in the leukocyte adhesion cascade including rolling (46). Here, extracellular galectins might bind glycosylated ligands on both ECs and leukocytes thus facilitating further interactions. Indeed, slow rolling was impaired in Galectin-3 knockout mice (47). Taken together, leukocyte rolling on ECs is mediated by the interplay of molecules on both cell types. Regarding the important role of endothelial P- and E-selectin, prior EC activation and subsequent selectin expression is key to initiate the leukocyte rolling process.
Recent experimental evidence indicated so-called “integrin-mediated rolling” and “slow rolling” as intermediate steps between rolling and firm arrest (10, 48). Integrin-mediated rolling is achieved by a transient interaction of leukocyte integrins in an intermediate conformational state with their respective adhesion molecules on ECs (10). To some extent, integrin-mediated rolling is also selectin-dependent (49, 50). Although selectins do not bind to leukocyte integrins directly, they can induce integrin activation via an intracellular signaling cascade, for example via leukocyte PSGL-1 (51–53). As a consequence, leukocyte integrins undergo conformational changes that result in increased binding to respective endothelial adhesion molecules (54). Of note, also soluble selectins, known biomarkers for inflammation (55, 56), can induce integrin activation (57). Slow rolling can be viewed as a specific type of integrin-mediated rolling: It is induced by pro-inflammatory cytokine exposure, and mediated mainly by the subsequent upregulation of endothelial E-selectin which induces integrin activation (10, 58).
As described above, myeloid cells, namely monocytes/macrophages and neutrophils, exert key functions in atherosclerosis. Recent evidence suggests that neutrophils are among the first cells recruited to inflamed tissues, thereby facilitating subsequent monocyte uptake (59, 60). This suggests underlying differences in the recruitment cascade of neutrophils and monocytes. Indeed, differences in integrin-mediated rolling and slow rolling have been described. In monocytes, integrin-mediated rolling is mainly conveyed via ß1-integrins such as very late antigen 4 (VLA-4) (10, 61). In contrast, ß2-integrins such as lymphocyte function-associated antigen 1 (LFA-1) and Mac-1 seem to be crucial for integrin-mediated rolling and slow rolling in neutrophils (49, 58, 62).
Taken together, initial leukocyte tethering and subsequent rolling and slow rolling is essential to enable leukocyte contact with ECs.
Leukocyte Activation and Arrest
Following leukocyte rolling, leukocytes need to firmly adhere to the endothelium for further transmigration. This is achieved by stable interactions between leukocyte integrins and endothelial adhesion molecules (19). However, this process requires prior activation of leukocyte integrins (63). Integrin activation is mainly mediated via so-called “inside-out signaling”; that means the activation of intracellular signaling cascades in response to chemokine binding to dedicated receptors on leukocytes (19). Concretely, secreted chemokines that are present in the extracellular glycocalyx bind to GPCRs on leukocytes, which results in conformational (=affinity) and expression density (=avidity/valency) changes (8, 64). Traditionally, chemokines were supposed to be presented by EC-bound glycosaminoglycans (65, 66). This is contrasted by new experimental studies indicating only transient interactions between chemokines and glycosaminoglycans, which allows retention of chemokines in the glycocalyx space close to the endothelium. However, most likely mainly free chemokines have the ability to bind to GPCRs on leukocytes (67). Chemokines inducing integrin activation are often the same molecules relevant for chemotaxis but quantitative differences in receptor expression and ligand binding may explain the differences in chemoattractant and pro-adhesive response (68).
Upon chemokine binding to GPCRs, complex intracellular signaling cascades get activated which are reviewed in detail elsewhere and are still not deciphered completely (8, 69). One major downstream effect, as mentioned above, are conformational changes in the extracellular domains of leukocyte integrins (from a bent, low-affinity conformation to an extended, high-affinity conformation) (69). Recently, talin and kindlin-3 have been identified as two intracellular molecules that bind to the cytoplasmic tail of integrins and, independent of each other, increase integrin affinity via conformational changes (8, 70–73).
Following activation, heterodimeric leukocyte integrins engage with their counterreceptors on ECs and consequently, leukocytes firmly adhere to the endothelium. Classical integrin-adhesion molecule combinations include interactions between VLA-4 with vascular cell adhesion molecule 1 (VCAM-1), LFA-1 with intercellular adhesion molecule (ICAM)-1, ICAM-2 and ICAM-3, and Mac-1—which is specifically described in human neutrophil arrest (74)—with ICAM-1 (75–79). Additionally, Mac-1 binding to CD40 ligand (CD40L) has been recently identified to contribute to the leukocyte arrest process (80–82).
In contrast to “inside-out signaling,” “outside-in signaling” describes the process in which signals are transduced into leukocytes upon integrin engagement (83). Several studies suggest that this mechanism is specifically important in post-arrest adhesion strengthening and further leukocyte activation (83–85).
Firm arrest is crucial for leukocyte emigration into inflamed tissues as it paves the way for final transmigration through the vascular endothelium.
Crawling and Leukocyte Transmigration
Compared to the preceding steps of the leukocyte recruitment cascade, detailed understanding of the final step, leukocyte transmigration, has been achieved fairly recently. Intraluminal crawling is essential to later leukocyte transmigration as cells can thereby migrate to preferred sites of transmigration (86). Leukocyte Mac-1 and LFA-1 binding to endothelial ICAM-1 play a key role in the process of crawling (87–90). Crawling and subsequent leukocyte transmigration is promoted by various stimuli (10). In particular, binding of leukocyte integrins to adhesion molecules was shown to induce EC activation (91), comparable to the outside-in-signaling as observed during firm arrest in leukocytes. Concretely, integrin-adhesion molecule interactions result in the clustering of adhesion molecules in specific EC regions yielding ICAM-1- and VCAM-1-rich domains (10, 92, 93).
Leukocyte transmigration can happen via two different routes: the classical, paracellular route (migration between two EC bodies) or a transcellular route (migration through thin parts of an EC body) (89). If migration happens via the paracellular route, EC junctions need to be modified transiently to reduce contact between adjacent ECs. This is achieved, for example, by active squeezing of leukocyte nuclei to disassemble endothelial actin filaments (94), by EC contraction mediated by cytoplasmic structural proteins and by reduced expression of EC adherens junctions such as vascular endothelial cadherin (VE-cadherin) (51, 95). Mechanistically, this is supposed to be triggered by intracellular signaling cascades as a response to LFA-1 and VLA-4 binding and subsequent ICAM-1 and VCAM-1 clustering on EC, which results in increased cytosolic calcium levels leading to enhanced myosin light chain kinase activity, dissociation of vascular endothelial protein tyrosine phosphatase (VE-PTP) from VE-Cadherin and phosphorylation as well as dephosphorylation of distinct tyrosine residues on VE-cadherin (96–98). As a result, VE-cadherin, which is linked to the actin-cytoskeleton via catenins, dissociates from this connection and is internalized, thus contributing to loosening of endothelial tight junctions (99). By contrast, other adherens junction molecules, such as platelet/endothelial cell-adhesion molecule 1 (PECAM-1/CD31) and CD99, but also tight junction molecules, such as junctional adhesion molecule (JAM)-A, are actively transported to the site of diapedesis in so-called lateral border recycling compartments (LBRC) (8, 100). By both homophilic (leukocytes express identical molecules) and heterophilic (leukocytes express non-identical ligands) interactions, endothelial adhesion and junctional proteins achieve shuffling of the migrating leukocyte through the lateral border (91, 101, 102).
A second route using transcellular migration has been described, specifically at thin parts of ECs (93, 103). In this case, tight junctions between ECs remain intact (103). Instead, leukocytes are transported through the EC body by caveolin 1-rich and adhesion molecule-rich (specifically ICAM-1) transcellular pores (95).
However, despite our increasing knowledge on the molecular processes of para- and transcellular leukocyte migration, the mechanisms that decide on the actual migration route have not yet been deciphered completely. The decision is most likely based on a combination of factors including vessel type (macrovascular vs. microvascular), leukocyte subset, tight junctional organization, inflammatory activation and other, yet unidentified aspects (89, 104).
Leukocyte Recruitment in Atherosclerosis
Whereas the molecular mechanisms of leukocyte recruitment are being elucidated in increasing detail, our knowledge of the specific processes that drive leukocyte migration into atherosclerotic plaques is still rather incomplete. It is likely that there are many shared steps, although some players appear to be more important in atherosclerosis than in recruitment cascades in other tissues. While the involvement of different leukocyte populations in atherosclerosis is known since long on an observational level from histological specimens, most of our current understanding in this field is derived from experiments with induced atherosclerosis in genetically altered mice. Important models for this are mouse lines lacking the genes for apolipoprotein E (Apoe) or low density lipoprotein receptor (Ldlr) fed a cholesterol-enriched diet (in the following referred to as Apoe−/− and Ldlr−/− mice, respectively).
Increasingly, these findings may also be supported by the results of genetic or proteomic association studies in humans: Single nucleotide polymorphisms (SNPs), for example, are used to compare the individual genetic profile of coronary artery disease (CAD) patients and healthy controls, and the enrichment of certain SNPs in the patient cohort can accordingly be linked to CAD in so-called genome-wide association studies (GWAS). Thus, with increasing numbers of SNPs and individuals included, a large number of variants associated with CAD have already been identified (105, 106). Although many of these SNPs reside in areas of unknown function in the genome, some of the associated genes have already been linked to leukocyte recruitment processes (107). Bringing together knowledge derived from these different approaches, we aim to summarize and discuss the currently known important players in immune cell recruitment into atherosclerotic plaques below (Figure 1).
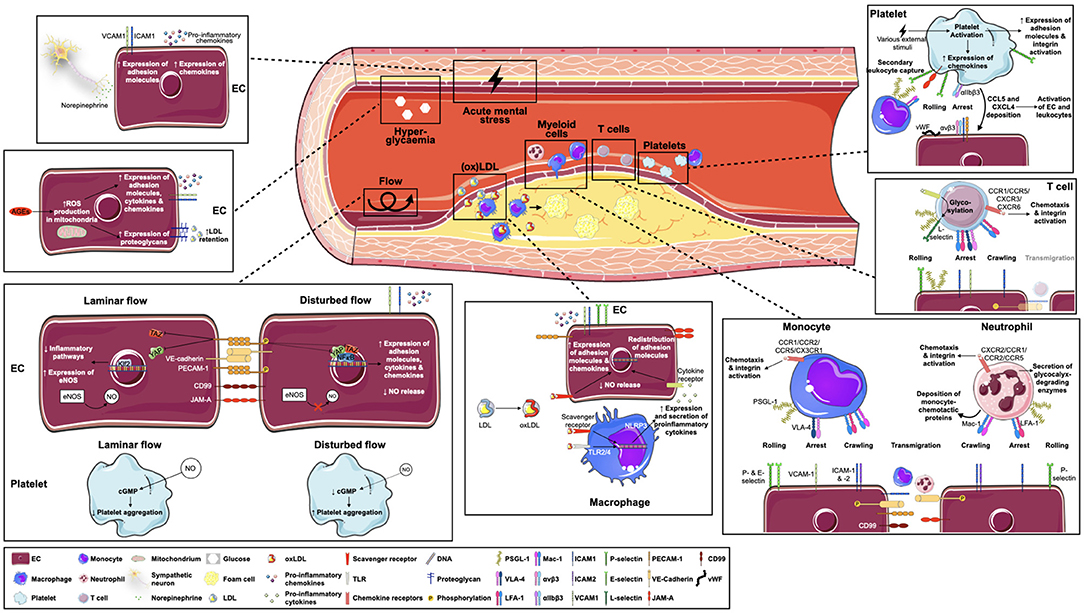
Figure 1. Key factors in atherosclerosis-specific leukocyte recruitment. Leukocyte recruitment into atherosclerotic plaques is multifaceted and involves several players. Endothelial activation through disturbed flow patterns and oxidized lipoproteins, but possibly also via hyperglycemia or local sympathetic innervation, forms the basis for subsequent leukocyte-endothelial interactions. Of note, specific leukocyte populations respond differently to atherogenic triggers and use unique molecules and receptors to achieve leukocyte rolling, arrest, crawling and transmigration. Additionally, platelets strongly contribute to leukocyte recruitment by secondary leukocyte capture and activation of endothelial cells and leukocytes. AEG = advanced glycosylation end products; CCL, C-C motif chemokine ligand; CCR, C-C motif chemokine receptor; CD99, cluster of differentiation 99; cGMP, cyclic guanosine monophosphate; CXCL, C-X-C motif chemokine ligand; CX(3)CR, C-X(3)-C motif chemokine receptor; EC, endothelial cell; eNOS, endothelial nitric oxide synthase; E-selectin, endothelial selectin; ICAM-1/2, intracellular adhesion molecule 1/2; Klf2, Krüppel-like factor 2; NLRP3, NOD-, LRR- and pyrin domain-containing protein 3; L-selectin, leukocyte selectin; Mac-1, macrophage receptor 1; NO, nitric oxide; (ox)LDL, (oxidized) lipoprotein; PECAM-1, platelet endothelial cell adhesion molecule 1; P-selectin, platelet selectin; PSGL-1, P-selectin glycoprotein ligand 1; ROS, reactive oxygen species; TLR, toll-like receptor; VCAM-1, vascular adhesion molecule 1; VE-cadherin, vascular endothelial cadherin; VLA-4, very late antigen 4; vWF, von Willebrand factor, YAP, yes-associated protein.
Endothelial Cell Priming
Keep It Flowing
The endothelium is permanently exposed to blood flow-induced shear stress and adapts to changes in flow by several immediate responses, e.g., conformational remodeling of the glycocalyx, opening of ion channels, and activation of different membrane receptors such as GPCRs and integrins (108). A central role in these mechanotransduced responses can be attributed to the mechanosensory PECAM-1, VE-cadherin and vascular endothelial growth factor receptor (VEGFR) complex: Acute onset of laminar flow promotes PECAM-1 phosphorylation followed by Src-dependent phosphorylation of VEGFR-2 and−3, proteins which are both linked to PECAM-1 via VE-cadherin and subsequently activate multiple intracellular pathways (109–111). Many downstream functions of this complex are transmitted via phosphatidylinositol 3-kinase/Akt signaling, e.g., leading to global activation of β1-integrins and the small GTPase RhoA which finally triggers promotion of focal adhesions, cytoskeletal adaption and alignment of the cell in the direction of flow (112, 113), or activation of endothelial nitric oxide (NO) synthase (eNOS) leading to NO-mediated vasodilation (114–116). Lastly, these processes stimulate the activation of transcription factors such as Krüppel-like factor 2 (Klf2) (117) and inhibit pro-inflammatory action of the Hippo pathway effectors yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ), which have been identified as key mechanotransducers in response to disturbed flow (118). In summary, initial pro-inflammatory activity triggered by flow onset is followed by alignment of the cell in the direction of flow, enhanced NO production and inhibition of inflammatory pathways, maintaining an anti-inflammatory status of the endothelium in response to laminar shear.
In contrast, disturbed flow, which preferentially occurs in branches, bifurcations and curvatures of the vessel—regions, where atherosclerotic plaques are mainly found—has been shown to lead to different responses: impaired NO release (119, 120), reactive oxygen species (ROS) production (121), deposition of fibronectin and fibrinogen to the subendothelial basement membrane (122, 123), and activation of pro-inflammatory transcription factors such as nuclear factor (NF)-κB and the aforementioned transcription cofactors YAP/TAZ, inducing the expression of several pro-inflammatory proteins such as VCAM-1, ICAM-1, CCL2, IL-6, and CXCL8 (111, 118, 124). Additionally, shear stress response regulatory elements have been found in the promoter of NOS3—the gene encoding eNOS—downregulating its expression in response to flow (125). Apart from that, evidence also suggests a flow-dependent expression of Toll-like receptor (TLR) 2 on EC (126, 127). Therefore, permanent changes in shear stress hinder the cell from adapting to the direction of flow and from overcoming the initial pro-inflammatory response phase, rendering these areas prone to leukocyte influx. This is also mirrored by the role of PECAM-1: While loss of PECAM1 is associated with reduced NO production in EC exposed to laminar flow (115, 128) and was shown to accelerate the onset of collagen-induced arthritis in mice (129, 130), in regions of disturbed flow, PECAM-1 elicits pro-inflammatory effects in the vasculature via increased NF-κB activation and VCAM-1 expression as shown in Pecam1-knockout mouse models (111, 131). Accordingly, more recent findings led to the conclusion that PECAM-1 exerts both pro- and anti-atherosclerotic properties in EC depending on the type of flow (132, 133). Moreover, common variants assigned to PECAM1 in humans have been associated with CAD in a large GWAS (134), highlighting the role of PECAM-1 in atherosclerosis beyond its known effects in cell culture and mouse models.
Another striking role in the abovementioned process of flow-induced EC priming can be attributed to NO, a gas produced mainly by eNOS that diffuses across cell membranes and, via soluble guanylyl cyclase (sGC), promotes the formation of the second messenger cyclic guanosine monophosphate (cGMP) in various neighboring cells. Among its several important downstream functions are smooth muscle cell relaxation, thus regulating the vascular tone, and platelet aggregation (135). Recently, it was shown that not only NO production in EC but also cGMP formation in one of its effector cells, namely platelets, is strongly shear-dependent (136). Insufficient NO availability, as it may be caused by impaired blood flow, is the major cause of endothelial dysfunction and leads to several pro-atherogenic responses in the vasculature by directly impacting leukocyte recruitment, e.g., increasing NF-κB activation and enhancing endothelial expression of VCAM-1, E-Selectin, and ICAM-1 (137). Furthermore, common polymorphisms in NOS3, but strikingly also several other genes of this pathway—among them GUCY1A1 encoding for the α1-subunit of sGC and PDE5A for a cGMP degrading enzyme—have been linked to coronary artery disease susceptibility in GWAS (138–140) and once again highlight the role of this pathway in atherogenesis.
oxLDL and Other Evildoers
The second important pillar for atherosclerotic EC priming is attributed to inflammatory mediators. While the role of PAMPs in atherogenesis is not investigated to a large extent, although the role of viruses and bacteria on plaque progression or rupture has been increasingly recognized (141), most research refers to the sterile character of atherosclerotic inflammation. In this context, modified low density lipoprotein (LDL) plays a fundamental role.
LDL from the circulation can be incorporated by EC either by receptor-mediated endocytosis or caveolae mediated transcytosis (142, 143), whereby the latter one is generally regarded as the more relevant way of atherogenic LDL accumulation (144). In the subendothelial space, LDL is retained by extracellular matrix proteoglycans (145) and, catalyzed by enzymes such as lipoxygenases or myeloperoxidases, metal ions and free radicals, chemically modified to various degrees. This leads to the formation of strongly pro-atherogenic LDL variants such as oxidized LDL (oxLDL) (137). The exact process of LDL oxidation is not fully elucidated but is supposed to be linked to oxidative stress resulting from a disbalance between ROS production and antioxidant defense mechanisms (146), as triggered, for example, by aging (147) or smoking (148). As a consequence, LDL loses its ability to bind the LDL receptor but strongly enhances affinity for scavenger receptors such as CD36 and lectin-like oxidized LDL receptor-1 (LOX-1) on EC, vascular smooth muscle cells, and macrophages (149). Intriguingly, a degradation product of LOX-1 can also be found in plasma [soluble Lox-1 (sLox-1)] and has recently emerged as a potential biomarker for cardiovascular disease incidence (150).
Ultimately, among the myriad pro-atherogenic responses of EC to such modified LDL are inhibition of NO production (151, 152), regulation of microRNAs (153), enhanced expression of E- and P-selectin, VCAM-1, ICAM-1 (154, 155), CCL2, CXCL2, 3 and 8 (156), and redistribution of JAM-A to facilitate transmigration (157)—thus paving the way for leukocyte infiltration. Therefore, oxidation of LDL is without doubt a central aspect of atherogenesis (149). However, while oxLDL is the best-studied form of modified LDL, there are several other modifications of LDL with pro-atherogenic properties, such as desialylation (158) or sphingomyosinase-induced aggregation (159).
Strikingly, these processes appear to begin already very early in life in genetically predisposed individuals (160), highlighting the influence of heritable factors on atherogenesis, most of which remain unexplored (161). Among the known contributors to atherogenic EC priming, however, is also hyperglycaemia. For example, hyperglycaemia driven accumulation of advanced glycosylation end products (AGEs) in vessels promotes ROS formation and adhesion molecule expression in EC, and release of pro-inflammatory cytokines such as IL-1β, IL-6, CCL2 and CXCL8 from leukocytes (162). Moreover, it impairs eNOS function and promotes the expression of proteoglycans, associated with increased LDL retention in the vascular wall (137, 163). Another important driver of atherogenesis is psychological stress (164, 165). Just recently, we showed that acute mental stress promotes atherosclerosis-related recruitment of leukocytes in mice by increasing the expression of endothelial adhesion molecules and the release of chemokines (166), adding to the knowledge of neuroimmune linkages in atherosclerosis.
Stage Free for Leukocytes
In response to upregulated adhesion molecules on EC and an increased chemokine gradient, leukocyte adhesion is initiated. However, this is not a static “one after the other” process. Several inflammatory stimuli emanating from vascular cells, platelets and leukocytes—as a response to the flow and lipid-driven EC priming, but also interlocking from the very beginning—continuously contribute to EC activation. Therefore, this section will focus on the role of the second important player in the atherosclerotic recruitment process: leukocytes.
The Classic: Monocytes
Monocytes and macrophages are the central figures in the history of atherosclerosis research. Once migrated, monocytes differentiate into macrophages, the major leukocyte population within atherosclerotic plaques (167), which strongly engulf modified LDL and fulfill several proatherogenic functions. There are three main subsets of monocyte populations in humans, that is classical CD14++ CD16− and non-classical CD14+ CD16++ monocytes and a small, less investigated group of intermediate CD14+ CD16+ monocytes (168, 169). In mice, C-C motif chemokine receptor (CCR)2+ C-X3-C motif chemokine receptor (CX3CR)1+ lymphocyte antigen 6 complex, locus C (Ly6C)high monocytes are closely related to the classical cells in humans and CCR2− CX3CR1++ Ly6Clow monocytes to the non-classical population (170). Although fate mapping and adoptive transfer experiments suggest that Ly6Clow monocytes are derived from Ly6Chigh cells, hypercholesterolaemia is associated with impaired Ly6Clow formation, despite a strong expansion of the Ly6Chigh population leading to systemic monocytosis (171–173). Importantly, both monocyte subsets fulfill different roles in monocyte recruitment. In steady state, Ly6Clow monocytes are dependent on CX3CL1 (fractalkine) stimulation via CX3CR1 and are constantly patrolling the vessel by communicating with endothelial ICAM-1 and−2 via LFA-1, but scarcely transmigrate (174). Although in the setting of tissue damage it has been suggested that they are among the first cells to extravasate and to promote recruitment of other leukocytes by release of inflammatory cytokines (175), Ly6Clow monocytes were associated with markedly less recruitment to atherosclerotic plaques than Ly6Chigh monocytes (176). However, under atherosclerotic conditions their patrolling behavior is strongly upregulated in a CX3CR1-independent manner, and genetic depletion of Ly6Clow monocytes was associated with pronounced endothelial apoptosis, suggesting an important role for endothelial maintenance in atherosclerosis (177). Nonetheless, in the following, we focus on the recruitment of inflammatory monocytes.
An initial trigger for leukocyte extravasation are local chemotactic gradients. For monocytes, CCL2 is a key chemokine that targets the CCR2 receptor highly expressed on classical/ Ly6Chigh monocytes (28). Both Ccr2 and Ccl2 knockout in atherosclerosis-prone mice (178, 179) as well as Ccr2 targeted siRNA treatment (180) significantly reduced atherosclerotic plaque formation, whereas in contrast, leukocyte-specific overexpression of Ccl2 in Apoe−/− mice promoted atherosclerosis progression (181). In humans, CCL2 levels in atherosclerotic lesions have only recently emerged as a potential indicator of plaque vulnerability (182). Interestingly, individuals with familiar hypercholesterolemia—a strong genetic predisposition to atherosclerosis—were found to have a 3-fold higher CCR2 expression on classical monocytes than healthy subjects, whereas cholesterol lowering therapy with a proprotein convertase subtilisin/kexin type 9 (PCSK9) antibody reduced monocyte CCR2 surface expression by 60% in these patients (183). This is further evidence that lipid-related and inflammatory processes strongly interact. Yet, despite the striking role of CCR2, also CCR5 is crucially involved in Ly6Chigh monocyte chemotaxis. While Ccr5 deficiency resulted in reduced mononuclear cell infiltration and lesion formation as well as decreased neointima formation (184–186), combined inhibition of CCL2, CX3CR1, and CCR5 resulted in as much as a 90 % reduction in atherosclerotic plaque formation in Apoe−/− mice, in spite of persistent hypercholesterolaemia (187). However, rather than having chemotactic functions, the CX3CR1/CX3CL1 interaction may impact cell survival (188).
Following attraction to atherosclerosis-prone endothelium, pro-inflammatory monocytes initiate rolling particularly by interaction of PSGL-1 with P- and E-selectin expressed on activated EC. Apoe−/− mice with a functional knockout of the gene encoding for PSGL-1 were shown to develop smaller atherosclerotic plaques (189) and in a similar way, P-selectin deficiency was associated with less leukocyte recruitment in atherosclerosis (190–192). Plaque leukocyte recruitment and consequently plaque size were also decreased when atherosclerotic mice were treated with EC-avid nanoparticles inducing endothelial silencing of P- and E-selectin, in parallel with ICAM-1, ICAM-2 and VCAM-1 (193). While lack of E-selectin alone reduced the progression of atherosclerotic plaque formation only to a minor extent (194), the combined genetic silencing of P-and E-selectin in Ldlr−/− mice even led to an 80% reduction in lesion formation (195). An inhibitory peptide preventing monocyte binding to selectins was shown to decrease monocyte recruitment and subsequently atherosclerotic lesion size, particularly by inhibiting monocyte activation via NF-κB (196). Moreover, intravital microscopy experiments in inflamed cremasteric veins indicated that E-selectin selectively affects the rolling velocity of inflammatory monocytes, whereas the flux of rolling neutrophils is regulated by P- and L-selectin (197). However, it is open whether this is also true for atherosclerotic arteries. As described above, transition of rolling to slow rolling and eventually to firm arrest requires integrin activation on the surface of leukocytes (102). Intravital microscopy in carotid arteries revealed a crucial function of CCR1 and CCR5 in this step for classical monocytes, but not of CCR2 (198). Although monocytes, in common with neutrophils, express the integrins LFA-1 and Mac-1, monocyte arrest seems to depend particularly on the VLA-4-VCAM-1 interaction (199–201). In line, blocking VLA-4 decreased leukocyte recruitment in atherosclerotic mice (202, 203) and functional downregulation of Vcam1 significantly reduced atherosclerotic lesion formation in mice in a gene-dose dependent manner (204, 205), while similarly, treatment with a VCAM-1 blocking antibody attenuated atherosclerosis in Apoe−/− mice (206). Of note, it has been shown that rolling of monocytes may also be mediated by platelets bound to the extracellular membrane, but rather in a ß2-integrin dependent way (207).
When firmly attached to the endothelium, monocytes engage in crawling behavior in search of ideal sites for extravasation. Thereby, using the intracellular actomyosin machinery for directed movement, they develop integrin-rich protrusions that scan the endothelial lumen for chemotactic directionality (102) mainly by interacting with ICAM-1 or ICAM-2 on EC via both LFA-1 and Mac-1 (208). Experiments in mice could partly confirm the involvement of these adhesion molecules in atherosclerosis: Apoe−/− Icam1−/− mice displayed reduced atherosclerotic lesions (209) and similarly, fatty streak lesion area was smaller in mice deficient for either ICAM-1 or ß2-integrin (the common subunit of LFA-1 and Mac-1), but most distinctly for mice with a double knockout of both encoding genes (210). Also, Icam1−/− Apoe−/− mice were shown to have significantly reduced atherosclerotic lesions, and soluble levels of ICAM-1 paralleled atherosclerosis progression in Apoe−/− mice with significantly elevated plasma concentrations compared to experiment onset (211). However, other studies could not confirm a substantial involvement of ICAM-1 loss on atherogenesis (204, 212). While the participation of LFA-1, on the other hand, was associated with atherosclerosis progression in rats (213), a study in Ldlr−/− mice could not endorse significant influence of Mac-1 on atherosclerotic lesion formation (214). Interestingly, a recent publication proposes a divergent influence of ß2-integrin on different stages of atherosclerosis—being protective in the initial phase, but pro-atherogenic in later stages, which could be partly driven by chronic dyslipidaemia (215).
Transmigration, the final step of the recruitment cascade, is facilitated by the redistribution of adhesion molecules such as JAM-A and VE-cadherin and formation of transmigratory cups characterized by local clustering of ICAM-1 and VCAM-1, actin remodeling and the formation of endothelial protrusions developing around the penetrating leukocyte (86). Thereby, also several effectors of the Rho-GTPase family are activated in EC, such as triple functional domain protein (Trio), Ras-related C3 botulinum toxin substrate 1 (Rac1), RhoG and its exchange factor SH3-containing guanine nucleotide exchange factor (SGEF) (216), stimulating the formation of the cup-like structures during transmigration of leukocytes but also promoting ROS production and subsequent activation of matrix metalloproteinases (MMP). In a later step, also RhoA and its effector Rho-associated protein kinase (ROCK) are activated, enlarging the transmigratory gap through enhanced actin-myosin contractility (217). Indeed, SGEF-deficient mice displayed decreased atherosclerotic plaque formation supposedly via reduced formation of endothelial docking structures (218) and similarly, inhibition of ROCK reduces atherosclerosis in mice (219, 220). Strikingly, the genes encoding for RhoA, Rac1 and SGEF were also associated with CAD by GWAS (139, 221–223).
Further, VE-cadherin plays a crucial role in leukocyte transmigration. It is linked to the actin-cytoskeleton via catenins and constitutively associated with the phosphatase VE-PTP, which stabilizes VE-cadherin junctions both by dephosphorylation and inhibition of Rho GTPase signaling (96, 224, 225). Beyond its previously described effects, oxLDL was also shown to directly promote monocyte transmigration by down-regulating VE-cadherin and upregulating PECAM-1 (226). Besides, PECAM-1 deficiency or blocking antibodies have been shown to specifically inhibit transmigration in vitro and in vivo in various inflammatory disease models in mice (99). Together with CD99, but also JAM-A, PECAM-1 is actively transported to the transmigration site via LBRCs and likely required for leukocyte diapedesis, as blocking of this targeted process resulted in considerably lower monocyte transmigration in vitro (227). In line, impaired JAM-A expression in EC or blocking JAM-A by a peptide antagonist inhibited leukocyte recruitment and atherosclerotic plaque formation in hyperlipidemic mice (228, 229). Remarkably, also the expression and distribution of JAM-A appears to respond to changes in flow (230). Given that CD99 also exerts an important influence on monocyte transmigration (231), in an interesting experiment, vaccination directed against CD99 was shown to reduce leukocyte numbers in atherosclerotic plaques and attenuate atherosclerotic lesion formation in mice (232). However, the role of PECAM-1—although quite obviously promoting leukocyte transmigration in vitro—seems to be more complex in atherosclerosis in general, as it exerts different influences on atherogenesis partly depending on the hemodynamic environment (see section Keep it Flowing).
Following their recruitment into plaques, monocytes massively differentiate into macrophages and, to a lesser extent, presumably also into dendritic cells (233). However, accumulation of macrophages in atherosclerotic lesions is likely a complex interplay of monocyte recruitment and local macrophage proliferation which also involves tissue-resident macrophages (234, 235). Within plaques, macrophages strongly engulf modified LDL particularly via scavenger receptor A1 (SRA1), LOX-1 and CD36, which is supported by TLR2, 4 and 6 signaling and promoting their phenotypic change to cholesterol-rich foam cells (236). Subsequently, foam cells can induce the release of pro-inflammatory cytokines (237) and Vcam1 expression in early aortic fatty streaks in mice (238). Excessive cholesterol accumulation may also lead to the formation of cholesterol crystals which trigger activation of the NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) inflammasome, a huge cytosolic oligomer inducing cleavage and secretion of IL-1ß and IL-18 (239). However, the inflammatory role of foam cells has been challenged by recent findings, which support a more diverse function in atherosclerosis (240) and suggest non-foamy macrophages to be the actual contributors of pro-inflammatory signaling in atherosclerotic plaques (241). As such, monocyte-derived macrophages again strongly promote endothelial activation and subsequent invasion of additional leukocytes, so permitting the plaque to grow and grow under continuous LDL supply (242).
The Newcomer: Neutrophils
Neutrophils are the most abundant leukocyte population in human blood and it has been just in the course of the last decades that they emerged from more or less neglected bystanders in atherogenesis to forerunners of monocyte infiltration (243). In infection, they are among the first cells to invade into inflamed tissues and promptly release cytotoxic ROS and proteases or form neutrophil extracellular traps (NET) targeted to rapidly eliminate pathogens (77). However, such behavior has also been observed in sterile inflammation as in atherosclerosis, making neutrophils furthermore an important contributor to atherosclerosis progression and complications such as stroke or acute coronary syndrome (244). While many steps of neutrophil recruitment into inflamed tissues are shared with monocytes and therefore close to the mechanisms described above, below, we focus on specific differences in the recruitment behavior of neutrophils.
Comparable to atherosclerotic monocytosis, also neutrophil levels in the blood are frequently increased in atherosclerosis (245) and related to future major adverse cardiovascular events in patients with acute coronary syndrome (246). Neutrophils circulating in the bloodstream are highly sensitive to various chemotactic signals. The traditional view that monocyte chemotaxis rather depends on CC-chemokines and neutrophil chemotaxis rather on CXC-chemokines is supported by predominant expression of CXC-chemokine receptors in neutrophils (247, 248) and fueled by a recent study concluding that CCR1, CCR2, CCR3, and CCR5 are not involved in neutrophil recruitment in acute inflammation in mice (28). However, several other studies have also demonstrated an important role for CC-chemokines in neutrophil attraction (249), suggesting that a strict separation of monocyte and neutrophil relevant chemokines does likely not represent the whole picture. As follows, important chemokines to trigger neutrophil activation in mice are CXCL1, CXCL2, and CXCL5 (presumably also representing CXCL8 in humans) as well as CCL5. Evasin-3, a pharmacological inhibitor of CXCL1 and CXCL2, reduced intraplaque neutrophil and MMP9 content (250). Similarly, the nicotinamide phosphoribosyltransferase inhibitor FK866, which was shown to strongly inhibit CXCL1 production in EC in vitro, reduced neutrophil infiltration and MMP-9 content in atherosclerotic lesions (251). Moreover, neutrophil recruitment to large arteries was shown to depend on CCR1, CCR2, CCR5, and C-X-C motif chemokine receptor (CXCR)2 in early stages of atherosclerosis, thus being particularly dependent on platelet-derived CCL5 stimulation (243). Another important role in neutrophil chemotaxis was also shown for CCL3, as leukocyte-specific CCL3 depletion inhibited atherosclerotic lesion formation particularly by affecting neutrophil accumulation (252).
Importantly, neutrophils also contribute to monocyte recruitment by depositing chemotactic proteins on the endothelium (244). One such example is cathelicidin. Mice lacking the corresponding gene developed significantly smaller atherosclerotic lesions with lower numbers of plaque macrophages (253). Similarly, it was reported that neutrophil-derived α-defensin can complex with CCL5 and be presented on EC, causing enhanced monocyte adhesion and vascular inflammation (254); while cathepsin G—also released from granules of neutrophils—is specifically deposited on the arterial endothelium of arteries but not on venule EC, promoting adhesion and extravasation of myeloid cells specifically in atherosclerosis-susceptible areas of vessels (255).
Neutrophils can facilitate their way to the endothelium through the dense network of endothelial glycocalyx by releasing proteolytic proteins, MMPs and ROS, thus locally breaking this physical barrier down (256). At the endothelium, neutrophil tethering and rolling is mainly mediated by PSGL-1 binding to P-selectin and CD44, but not to E-selectin, which is thus assumed to be selectively used by inflammatory monocytes (197). This is an important observation, as preformed P-selectin is available much quicker upon activation of EC than E-selectin which requires de novo synthesis, and which could therefore partly explain the delayed secondary recruitment of inflammatory monocytes compared to neutrophils in inflammation. CD44 has been studied quite extensively for its involvement in atherosclerosis, but depletion in atherosclerotic mice tended to yield conflicting results (257). Depletion of L-selectin, which might be specifically important in secondary capture, was consensually shown to promote atherosclerosis, accompanied by a drop of aortic B cells (258, 259). In vivo, however, the rolling behavior of monocytes and neutrophils on carotid artery bifurcations of Apoe−/− mice appears to be quite different: While the number of rolling neutrophils, in contrast to monocytes, increased during high-fat diet, the rolling rate of monocytes decreased during the same period (260). Another striking observation is that under high shear stress, by cytoskeletal reorganization during rolling neutrophils can form slings out of their membrane which are characterized by surface expression of distinct sticky PSGL-1 clusters and LFA-1, thus facilitating contact with the endothelium (261). During rolling, neutrophils were also shown to secrete S100 calcium-binding protein (S100)A8 and S100A9, calcium-binding proteins constitutively expressed in myeloid cells that account for ~45% of the cytoplasmic proteins in neutrophils (262). Interestingly, apart from their numerous functions in inflammation, these proteins were also associated with leukocyte chemotaxis, inducing VCAM-1 and ICAM-1 expression in EC while upregulating Mac-1 expression in leukocytes, subsequently resulting in increased TLR4-mediated Mac-1/ICAM-1 binding, decelerated leukocyte rolling and enhanced firm adhesion on the endothelium (263, 264). In line, blocking antibodies or genetic depletion of S100A9 reduced leukocyte recruitment in several murine inflammatory disease models (262), making these proteins a promising pharmacological target in inflammation-related diseases.
Transition from slow rolling to neutrophil arrest seems to particularly depend on LFA-1 binding to endothelial ICAM-1, which is in contrast to monocytes requiring VLA-4/VCAM-1 interactions. Hereby, CXCR2 is assumed to be of central importance for “inside-out” activation of LFA-1 (265). And while monocyte crawling is dependent on both Mac-1 and LFA-1, neutrophils seem to exclusively crawl via Mac-1, interacting with ICAM-1 and ICAM-2 (88, 208, 266). Remarkably, when Mac-1 is blocked, neutrophils can also crawl against the direction of flow in vitro by engaging LFA-1, while Mac-1 favors flow-directed crawling behavior (87). During crawling, neutrophils flatten and form protrusions reaching into the endothelial surface similar to monocytes, while on the front of the moving cell, filamentous actin (F-actin) and on the end, the so-called uropod, myosin filaments aid the cell in directed movement on the endothelium (267). Lack of the actin cytoskeleton transcription factor MKL1 in neutrophils almost completely abrogated migration in vitro (268). Importantly, neutrophils were recently found to actively scan for activated platelets via protruding PSGL-1 clusters at the uropod, and such interactions with platelets were crucial for intravascular migration of neutrophils (269). Also EC derived CXCL1 was shown to support neutrophil crawling, while CXCL2, mainly produced by neutrophils themselves, particularly aided in breaching of endothelial junctions together with its atypical chemokine receptor 1 which was found to be enriched in endothelial junctions (270).
Following firm arrest, neutrophil interaction with EC via β2-integrin/ICAM-1 triggers VE-cadherin phosphorylation and subsequent loosening of endothelial adherent junctions to promote the favored paracellular route for diapedesis (97). This is quite similar to the process described for monocytes above, and requires the formation of transmigratory cups which were reported to form around neutrophils specifically in a “dome” shaped manner (271), involving RhoA and leukocyte-specific protein 1 mediated formation of contractile F-actin structures that tightly surround the invading cell in order to prevent vascular leakage during transmigration (272, 273). Strikingly, neutrophils were also observed to return to the circulation by reverse transendothelial migration in the context of low JAM-C expression in mice (274). Mechanistically, local proteolytic cleavage of JAM-C, driven by neutrophil-derived elastase, was shown to promote this behavior (275).
After infiltrating the plaque, neutrophils have a plethora of possibilities to further promote leukocyte recruitment and subsequent progression of atherosclerosis. They can release granula proteins such as cathelicidin, cathepsin G, elastases, MMPs and ROS, or activate NET formation, thus attracting further leukocytes, promoting oxidative stress, LDL modification, EC activation, activation of macrophages and cellular damage (244).
The Player With the Many Faces: T Cells
While myeloid cells form the first line of defense and respond rapidly but uniformly to a broad spectrum of identified threats, cells of the adaptive immune system perform highly specific tasks that are individually tailored to the particular profile of their target, resulting in a delayed but finely matched immune response. Thus, it is not surprising that T cells—a highly abundant population in human atherosclerotic plaques—are divided into multiple different subpopulations. This includes naïve, memory and effector CD4+ and CD8+ T cells, but also regulatory T cells (Treg) (276). CD4+ T cells are generally associated with increased atherosclerotic plaque growth (277–280). Following antigen-presentation, CD4+ T cells are activated and differentiate into T helper (Th)1, Th2, Th17 cells or other subsets and resemble, together with memory T cells, the major proportion of T cells to be found within atherosclerotic plaques (281). However, they can also give rise to Tregs in the periphery (282). Among the CD4+ T cells, Th1 cells are the most abundant T-subtype in human atherosclerotic lesions (283) and can generally be attributed as pro-atherosclerotic (284, 285), while the role of the other T cell subsets is still a matter of debate and discussed in detail elsewhere (286). Tregs, on the other hand, which express the characteristic transcription factor forkhead box protein P3 (FOXP3) and CD25, are clearly associated with an anti-inflammatory role by suppressing the proliferation of pro-inflammatory effector T cells and influencing macrophage function toward an anti-inflammatory phenotype (287, 288). Depletion of Treg cells in mice aggravates atherosclerosis (289–291) and Tregs express IL-10 and transforming growth factor (TGF)-β, both associated with anti-atherosclerotic effects in atherosclerosis (292). Interestingly, however, just recently an autoreactive phenotype in Tregs directed against apolipoprotein B-100 (ApoB-100), the core protein of LDL, in late-stage atherosclerosis was identified (293), questioning the classical view of Tregs as solely beneficial cells. Moreover, a recent study showed that dyslipidaemia reprograms the metabolic footprint of Tregs toward an effector-like migratory phenotype, challenging the classical hypothesis that Treg migration into plaques might be reduced (294). Finally, CD8+ T cells are more frequent in blood of CAD patients than in healthy individuals (295, 296) and generally associated with pro-atherosclerotic effects in preclinical studies (297–299), but, in contrast, inhibiting CD8+ cells in advanced lesions also resulted in less stable lesions (300).
T cells can use both classical myeloid cell like and antigen-dependent patterns for migration into tissues (256). While most of the T cells found within plaques are antigen-experienced T cells (286) and T cells targeting ApoB-100 were shown to circulate in human blood (301), a recent study suggests that naïve T cells can also be primed directly in the vessel wall (302). Of note, this was not related to tertiary lymphoid organs which can be found in later stages of atherosclerosis within the adventitia and promote Treg expansion (303). However, the antigens to which T cells respond in atherosclerosis are mostly unknown which renders it difficult to study antigen-dependent effects in T cell migration. Therefore, the precise mechanism of T cell recruitment to atherosclerotic plaques, albeit of great interest, is still subject of basic research, and many of the subsequent insights are derived from in vitro findings.
To migrate into murine atherosclerosis-prone vessels, circulating T cells roll on endothelial P-selectin using PSGL-1 in vivo (302), with a potential role for its co-factors CD44 and CD43 (304, 305). In contrast to monocytes, however, to be fully active, PSGL-1 in T cells requires prior glycosylation (35). Mac-1 has not been described to participate in T cell recruitment but L-selectin, which is important for lymphocyte trafficking into lymph nodes (256), was shown to play a role in T cell migration into peripheral tissues, especially in recruitment into the adventitia of healthy, non-inflamed arteries in mice (306). This again suggests a role for naïve T cell recruitment, as T cells generally lose L-selectin expression upon antigen-presentation. Notably, in a study using intravital microscopy, rolling of T cells on carotid artery bifurcations could not be observed in early atherosclerosis, but was induced on pronounced atherosclerotic lesions in mice (260), indicating a more pronounced role for T cells in late atherosclerosis.
Induction of firm adhesion requires integrin activation, which is accomplished by chemokines in a similar way as in myeloid cells. Different T cell subsets react to different cytokines, but CCR1 and CCR5 have been shown to be expressed on most atherosclerosis relevant T cells and thus are supposed to play a major role in T cell recruitment in response to CCL5 (307). However, CCR1 and CCR5 appear to exhibit opposing effects, as CCR1 seems to be anti-atherosclerotic in the context of T cell recruitment in murine atherosclerosis (308), whereas CCR5 rather has a pro-atherogenic role (section The Classic: Monocytes). Another chemokine receptor to be found on Th1 cells is CXCR3, which is required for Th1 differentiation (309). It requires binding of CXCL10, which, when inhibited, decreases atherosclerotic lesion size and specifically T cell accumulation in murine atherosclerotic lesions (310). Interestingly, CXCL10 levels were suggested to be higher in obese compared to non-obese subjects, functionally promoting adhesion capacity of leukocytes in vitro (311). Similarly, genetic depletion of Cxcr3 or antagonizing CXCR3 pharmacologically in mice reduced atherosclerosis progression and infiltration of inflammatory T cells, while Treg numbers rised (312, 313). CXCR6 has been described as a marker of polarized Th1 cells (314) important for T cell homing, as absence of CXCR6 inhibited recruitment of T cells, diminished IFNγ production and atherosclerotic lesion formation in Apoe−/− mice (315). CXCL16 is the ligand for CXCR6 and chemoattractive when expressed and deposited on EC, but also functions as a scavenger receptor for oxLDL on monocytes and macrophages (316). In contrast to other oxLDL scavenger receptors, CXCL16 depletion was associated with reduced plaque formation in Ldlr−/− mice (317), thus acting in both pro- and anti-atherosclerotic ways, depending on the context. Last but not least, CCR7, which mediates T cell homing to lymph nodes, and its ligands CCL19 and CCL21 have also been identified within atherosclerotic lesions of humans and mice (318). However, studies on atherosclerosis in Ccr7−/− mice achieved controversial findings (281).
To summarize, several chemokines and respective receptors are involved in T cell attraction and inside-out signaling. However, Th1 cells are also able to bypass extracellular chemokine signals by absorbing chemokines stored intraendothelially in vesicles via dense lymphocyte-endothelial synapses (319). Moreover, EC also seem to be able to act in an APC like manner, presenting antigens specifically to memory T cells via their T cell receptor (TCR) and thereby activating them toward tissue migration (320). Whether these scenarios are also relevant for recruitment into atherosclerotic vessels is thus far not known.
Adhesion of the now activated T lymphocytes occurs presumably via the β2-integrin LFA-1, whereby VLA-4 and CD47 are also thought to play a role (79, 321). Similar to monocytes, crawling T cells interact with ICAM-1 via LFA-1 (322), polarize into a leading edge and tailing uropod and probe the endothelium by invasive protrusions (323) which is controlled by RhoA and continuous actin reorganization (324). The transmigration process of T cells in atherosclerotic arteries to date is mostly unknown, but according to T cell migration into other peripheral inflamed tissues likely similar to other leukocytes, favoring paracellular migration involving ICAM-1-mediated signaling and VE-cadherin dissociation from VE-PTP (325, 326). In contrast, TCR-activated effector memory CD4+ cells can also transmigrate in an alternative way involving CX3CL1 and LBRC adhesion proteins in vitro (327). Again, if this also happens in transmigration through atherosclerotic arteries, is not known.
Platelets: Small but Effective Partners in Crime
Although far from giving a full picture, the role of platelets in atherosclerosis is becoming increasingly clear, revealing that their involvement extends well-beyond thrombus formation. In fact, thrombocytes also contribute to leukocyte recruitment in early atherogenesis, as will be elucidated in the following.
While shear stress can trigger endothelial activation, leading to upregulation of adhesion molecules and chemokines (see section Keep it Flowing), shear stress is also known to directly trigger platelet activation and aggregation in atherosclerotic vessels, thus promoting thrombotic arterial occlusion (328). However, shear stress or shear activated EC can stimulate platelets also in earlier phases of atherogenesis and promote platelet adhesion to the EC surface via enhanced adhesion molecule expression or decreased release of NO and prostacyclin (329), thus shifting the balance between inhibitory and activating pathways in platelets in favor of platelet activation. Of interest, impaired function of the ATP-binding cassette transporter G4 in bone marrow megakaryocyte progenitors giving rise to platelets has been shown to inhibit cholesterol efflux from these cells, thus promoting platelet production and accelerating atherosclerosis (330). Additionally, oxLDL was also shown to directly activate platelets via binding to CD36, thereby impairing cGMP mediated anti-inflammatory effects (331), while the traditional risk factors hyperlipidaemia, hyperglycaemia and hypertension were likewise associated with increased platelet reactivity (332).
Upon activation, platelets undergo shape change and increase surface expression of P-selectin and CD40L, while integrin αIIbβ3 adopts its active conformation (333). Such activated circulating platelets were shown to readily bind to EC and monocytes, deposit the chemokines CCL5 and CXCL4 (also known as PF4) on both EC and monocytes and subsequently promote leukocyte accumulation and atherosclerotic lesion formation in the arterial intima, notably already prior to the development of manifest atherosclerotic lesions (25, 334). Moreover, atherosclerotic plaques also suggest presence of macrophage-platelet aggregates, indicating that platelet-binding to monocytes persists beyond the recruitment process (335). In this interplay, platelets induce a pro-inflammatory phenotype in macrophages characterized by increased production of the cytokines IL-6 and IL-1β and impaired capability to phagocytose dying cells, which in turn results in increased necrotic core area in atherosclerotic plaques of Ldlr−/− mice. In line, the amount of circulating monocyte-platelet aggregates was significantly increased in CAD patients (336, 337). Mechanistically, platelet binding to EC is a two-step process initiated by platelet rolling on the glycoprotein von Willebrand factor (vWF) released and bound by the endothelium, which involves platelet glycoprotein (GP)Ibα and P-selectin. Subsequently, firm adhesion of platelets is mediated via integrin αIIbβ3 which binds to endothelial αvβ3 and ICAM-1 (338), and involves PECAM-1 signaling (339). The interaction between activated platelets and leukocytes is conveyed by P-selectin interaction with leukocyte PSGL-1, which is supported by platelet glycoprotein Ibα (GPIbα), JAM-A, and JAM-C binding to leukocyte Mac-1 (340–342). In this context, the role of JAM-A is striking, as it seems to act as a brake on platelet activation: not only do knockout mice deficient for JAM-A display enlarged thrombi (343) and accelerated early-stage neointima formation (342), but also a hyperreactive phenotype significantly aggravating atherosclerotic lesion formation (344). However, in sharp contrast to these previous findings, a peptide antagonist intended to inhibit JAM-A function was shown to exert beneficial effects in atherosclerotic Apoe−/− mice by inhibiting platelet adhesion to the endothelium (228). Therefore, further studies are warranted to clarify a possible correlation of these findings.
Strikingly, endothelium-adherent platelets were observed to form exceptionally long, flow-induced protrusions (FLIPR) from their membrane under high shear stress. Mediated by P-selectin/PSGL-1-interaction, such FLIPR can deliver platelet microvesicles to rolling monocytes and neutrophils which promotes their activation, as demonstrated by increased CD11b expression and L-selectin shedding (345). Another interesting observation is that, although platelets can adhere to EC at various shear rates in vitro, their ability to capture leukocytes may be limited to regions of disturbed flow (346). Moreover, the absence of platelets in mice markedly suppresses neutrophil crawling, whereas depletion of their neutrophil ligand PSGL-1 significantly alters neutrophil surface distribution of Mac-1 and CXCR2, thereby impairing directed intravascular motility and transmigration (269). On the other hand, platelets were also shown to recruit to atherosclerotic plaques by interacting with previously adhered monocytes and neutrophils in form of secondary capture (260).
Apart from favoring leukocyte recruitment by direct binding, several pro-inflammatory mediators released from activated platelets also promote leukocyte recruitment (332). One such platelet-derived chemokine of pivotal relevance to leukocyte recruitment is CCL5. It is deposited on activated EC in arteries mainly by platelets and significantly involved in monocyte and neutrophil adhesion to EC, as shown by in vitro and in vivo experiments (243, 347). Antagonizing CCL5, in turn, reduced neointima formation, leukocyte infiltration and atherosclerotic plaque formation in mice (348–350). CCL5 was also shown to form complexes with CXCL4 which synergistically enhances the capacity of CCL5 to recruit monocytes (351). Strikingly, this interaction could be selectively disrupted by the peptidic inhibitor CKEY2 and its mouse ortholog MKEY, thus decreasing monocyte recruitment and atherosclerotic plaque formation in mice (352). Another such liaison was found between CCL5 and the neutrophil-derived protein human neutrophil peptide 1 (HNP1), also facilitating monocyte recruitment to sites of inflammation. This could likewise be inhibited by application of the peptide antagonist RRYGTSKYQ (254).
A further important platelet derived chemokine is CXCL4 which is highly abundant in platelet α-granules and plays a critical role in coagulation. Importantly, CXCL4 could be localized in human atherosclerotic lesions and its presence on EC and macrophages positively correlated with clinical parameters for atherosclerosis (353). Moreover, CXCL4 was shown to directly bind oxLDL and to increase its uptake in vascular cells and macrophages (354), which is supported by histological findings in human atherosclerotic lesions (355). Also, upon stimulation with CXCL4, macrophages abolish expression of the atheroprotective scavenger receptor CD163 (356) and, as suggested by a recent study, give rise to a new macrophage phenotype called M4, characterized by the simultaneous expression of MMP7 and S100A8 (357). Additionally, CXCL4 appears to prompt differentiation of monocytes to macrophages (358). In line with these observations, depletion of CXCL4 in mice significantly reduced atherosclerotic lesion formation (359).
Another platelet derived factor is CXCL12. Interestingly, polymorphisms within the gene encoding for CXCL12 in humans have been genome-wide significantly associated with CAD (360, 361). CXCL12 can signal both via CXCR4—relevant for neutrophil retention in the bone marrow—and CXCR7. While CXCR7 is barely detectable on blood leukocytes, it appears to be upregulated during monocyte-to-macrophage differentiation. This was accompanied by a switch in intracellular signaling in response to CXCL12 toward more pro-inflammatory pathways and subsequently enhanced phagocytotic activity of macrophages (362). While CXCL12 promotes monocyte chemotaxis in a CXCR4-dependent manner, monocyte adhesion to platelet-bound CXCL12 is rather mediated by CXCR7 (363). Furthermore, in a paracrine manner, CXCL12 was also shown to regulate platelet activation (364, 365). In line, Cxcl12 overexpression increased, while endothelial-cell specific Cxcl12 depletion reduced atherosclerotic lesion area in mice (366, 367) and platelet surface CXCL12 expression correlated with the risk of adverse cardiac events in symptomatic CAD patients undergoing percutaneous coronary intervention (PCI) (368). To the several other mediators released from platelets upon activation belong CXCL3, CXCL5, CXCL7, CXCL16, CCL3, and macrophage migration inhibitory factor (MIF). Noteworthy, platelets also release several angiogenesis-related proteins such as vascular endothelial growth factor (VEGF) or platelet derived growth factor (PDGF) with important influence on atherosclerosis. While angiogenic factors do not necessarily appear to influence leukocyte recruitment directly, neovascularization within atherosclerotic plaques is a hallmark of progressive atherosclerosis exponentially expanding the area over which leukocytes can penetrate, hence further promoting leukocyte infiltration and plaque destabilization (369).
Lessons Learned: Clinical Implications and Therapeutic Options
Plasma levels of cholesterol, which circulates in the blood via LDL and strongly stimulates atherogenesis, can be successfully lowered by treatment with statins—an approach representing the mainstay of atherosclerosis therapy today. Further pharmacological strategies to prevent cardiovascular events include antihypertensive and antihyperglycemic agents, if applicable (370). However, according to a 2010 meta-analysis, average statin therapy in randomized, controlled trials (RCT) still leaves a mean residual risk of over 75% for major cardiovascular events in these patients (371). Intensified statin treatment and additional use of ezetimibe or PCSK9 inhibitors, which reduce levels of circulating LDL by a different mechanism, further strongly reduces this risk but, again, far from completely abolishing it (372, 373). Therefore, it becomes clear that fighting only the traditional risk factors is not sufficient to eliminate atherosclerosis.
At this point, anti-inflammatory treatment strategies come to play (Figure 2). The IL-1β neutralizing antibody canakinumab was one of the first solely anti-inflammatory drugs shown to reduce the recurrence of cardiovascular events in high-risk CAD patients (374), and as suggested from mouse experiments, particularly does so by reducing leukocyte production in the bone marrow and deactivating EC toward less leukocyte recruitment (375). Moreover, another such highly anti-inflammatory drug, colchicine, was proven to reduce the risk of myocardial infarction, ischemic stroke, or cardiovascular death by over 25%, of note in addition to baseline treatment with lipid-lowering agents in 97% of enrolled patients (376). Colchicine's mechanism of action is not fully understood, but suggested to inhibit inflammasome activation, neutrophil recruitment and leukocyte-platelet interactions (377), thus directly affecting leukocyte migration into atherosclerotic plaques.
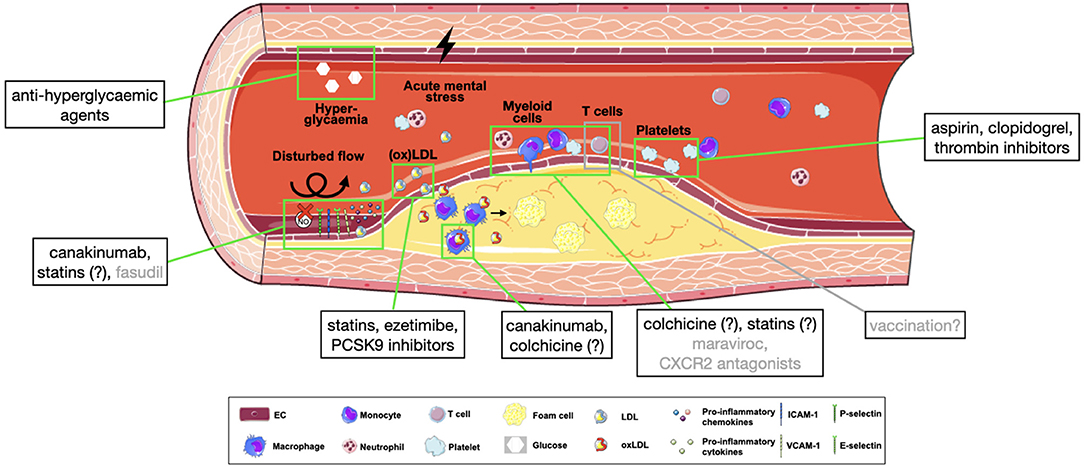
Figure 2. Pharmacological targets in atherosclerotic leukocyte recruitment. Simplified overview of approved (black) or potential (gray) targets in the process of leukocyte recruitment into atherosclerotic plaques. Substances with an incompletely clarified mechanism of action are marked with (?). CXCL2, C-X-C motif chemokine ligand 2; ICAM-1, intracellular adhesion molecule 1; ICAM-2, intracellular adhesion molecule 2; NO, nitric oxide; (ox)LDL, (oxidized) lipoprotein; PCSK9, proprotein convertase subtilisin/kexin type 9; VCAM-1, vascular adhesion molecule 1.
Of great interest, statins exert anti-inflammatory effects beyond their action on LDL (378), which may partly explain their superiority in cardiovascular risk reduction compared with other lipid-lowering agents. Mechanistically, in absence of hypercholesterolaemia, statins were shown to improve endothelial function, particularly by improving NO availability, stability of adherens junctions and reducing ROS formation (379–381), to inhibit neovascularization (382, 383), and, importantly, to selectively block LFA-1 and subsequent lymphocyte adhesion (384). Similarly, antithrombotic agents—a major pillar of secondary prevention—do not only reduce aggregation, but also platelet activation. Aspirin, for example, inhibits GPIIb/IIIa and P-selectin expression and release of chemokines (385, 386), clopidogrel was shown to improve systemic NO bioavailability and reduce soluble CD40L and CCL5 release (387) and thrombin inhibitors reduce formation of platelet-leukocyte aggregates and atherosclerotic plaques in mice (388, 389). Also some anti-hyperglycaemic agents have shown to exert beneficial effects on cardiovascular outcomes, both in diabetic and non-diabetic patients with heart failure (390, 391). Of note, the mechanism of action of some of these agents may affect NLRP3 inflammasome function in macrophages (392).
Up until now, strategies aimed at reducing vascular oxidative stress have not been shown beneficial in atherosclerosis patients (137). Interestingly, however, the Rho kinase inhibitor fasudil was associated with enhanced NO bioavailability, thus improving endothelial function in atherosclerotic patients (393). When regarding chemokine receptors, CXCR2 antagonists have been or are currently investigated in pilot or phase II studies in inflammatory diseases and COPD, while the CoronAry heart DiseAse (CICADA) study is specifically testing the cardiovascular effects of such agent (394). The CCR5 inhibitor maraviroc was shown to reduce aortic plaque size when treating atherosclerotic mice (395) but also to decrease atherosclerosis progression in HIV patients (396) and thus represents another interesting target for further studies.
A major fly in the ointment, however, is that large-scale inhibition of adhesion molecules or chemokines is often a double-edged sword, as several key players in leukocyte recruitment act differently in different tissues or have different, sometimes conflicting effects on atherosclerosis, as is the case with PECAM-1 (397). Another drawback is the importance of leukocyte recruitment for fighting infections. This limits therapeutical benefits of canakinumab, e.g., (374). Therefore, in contrast to targeting LDL, ubiquitous inhibition of adhesion molecules or chemokines is often not feasible. However, chrono-pharmacological treatment is one example for a more targeted approach: In mice, CCL2-dependent myeloid cell recruitment to atherosclerotic plaques peaks in the early morning and could be effectively targeted by time-adjusted treatment—importantly, without affecting cell adhesion in the cremasteric microcirculation at the same time (398). Another nascent concept is vaccination in atherosclerosis, aiming at inducing antigen-specific regulatory T cells to suppress deleterious effector T cell expansion and thus inhibit atherosclerosis. However, it is left open whether this strategy is feasible and beneficial (286).
Thus, it remains exciting to see what new therapeutics or therapeutic concepts will emerge in the future that prove helpful in curbing the inflammatory aspect of atherosclerosis and, in particular, leukocyte recruitment into the vessel walls.
Conclusions
By following leukocytes step by step on their way into atherosclerotic plaques, it became clear that all leukocytes, although differing in their affinity for specific adhesion molecules or chemokines, use the same overall concept of tethering and rolling, adhesion, crawling and transmigration. Rolling is mainly mediated by PSGL-1 and during rolling, chemokine-chemokine receptor interactions activate the high affinity conformation of leukocyte integrins in a process called inside-out signaling, paving the way for firm adhesion via VLA-4 or LFA-1. Many chemokines have different affinities for different leukocytes, e.g., CCL2 rather favors classical monocytes and CXCL1 and 2 neutrophils, which may allow for targeted recruitment of a particular cell type. Other chemokines such as CCL5 are similarly important for monocytes, neutrophils and T cells, and are partially derived from platelets, which also directly contribute to leukocyte migration by binding to ECs and leukocytes. The concept of crawling and transmigration, however, appears to be very similar between leukocytes, involving cup formation and breaking of endothelial junctions. An important part of the recruitment process is EC priming, which enables expression or upregulation of adhesion factors such as P-selectin, ICAM-1 or VCAM-1 on ECs as binding partners for leukocyte ligands. ECs are mainly activated by two mechanisms: Disturbed flow and inflammatory mediators, most importantly oxLDL. Recent research shows important and divergent contributions of the different leukocyte subsets on plaque formation, with neutrophils being among the first cells to invade, paving the way for monocyte migration which, inside the plaque, differentiate to macrophages, ingest oxLDL, and promote further recruitment of leukocytes. T cells, on the other side, might contribute to atherosclerosis by targeting specific—mainly unidentified—antigens within the plaque.
Therapeutically targeting recruitment related processes is mainly drawn back by the multitude of important functions that most involved factors have in the immune system. Nevertheless, canakinumab and colchicine, two anti-inflammatory agents contributing to recruitment-related processes, were already proven beneficial in CAD patients. And surprisingly, statins, the mainstay of current atherosclerosis therapy, appear to not only lower LDL levels but also to inhibit leukocyte migration by affecting endothelial and leukocyte function. The same holds true for antithrombotic agents, which affect leukocyte recruitment in multiple ways. Many ongoing studies are investigating the effect of potential additional treatment strategies that mainly target the inflammatory nature of atherosclerosis. However, several open questions show that there is still a long way to go in basic research and on the road from bench to bedside, before we can control the excess inflammatory recruitment of leukocytes in atherosclerotic plaques in patients.
Author Contributions
CM, JH, and HBS conceptualized the content, reviewed literature, wrote the manuscript, and generated figures. HS and TK discussed and edited the review. All authors contributed to the article and approved the submitted version.
Funding
HBS has received funding from the European Research Council under the European Union's Horizon 2020 Research and Innovation Programme (STRATO, Grant Agreement No. 759272), the Else-Kröner-Fresenius-Stiftung (2020_EKSE.07), Else-Kröner-Forschungskolleg München, Technical University Munich, the Deutsche Herzstiftung (F/28/17), and the Deutsche Forschungsgemeinschaft (DFG) (SA 1668/5-1). TK was funded by the Corona Foundation as part of the Junior Research Group Translational Cardiovascular Genomics (S199/10070/2017) and the German Research Foundation (DFG) as part of the collaborative research center SFB 1123 (B02).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We thank Servier Medical Art (https://smart.servier.com) for providing free illustrations for figures.
Abbreviations
AGE, advanced glycosylation end product; ApoB-100, apolipoprotein B 100; Apoe−/−, apolipoprotein E knockout; CAD, coronary artery disease; CCL, C-C motif chemokine ligand; CCR, C-C motif chemokine receptor; CD, cluster of differentiation; cGMP, cyclic guanosine monophosphate; CICADA, CoronAry heart DiseAse study; COPD, chronic obstructive pulmonary disease; CX3CL1, C-X3-C motif chemokine ligand 1; CX3CR1, C-X3-C motif chemokine receptor 1; CXCL, C-X-C motif chemokine ligand; CXCR, C-X-C motif chemokine receptor; EC, endothelial cells; eNOS, endothelial nitric oxide synthase; ESL-1, endothelial selectin ligand 1; F-actin, filamentous actin; FLIPR, flow-induced protrusions; FOXP3, forkhead box protein P3; GPCR, G-protein-coupled receptor; GPIbα, glycoprotein Ibα; GWAS, genome-wide association study; ICAM-1/-2/-3, intercellular adhesion molecule 1/2/3; IL, interleukin; JAM-A/-C, junctional adhesion molecule; Klf2, Krüppel-like factor 2; LBRC, lateral border recycling compartments; LDL, low density lipoprotein; Ldlr−/−, low density lipoprotein receptor knockout; LFA-1, lymphocyte function-associated antigen 1; LOX-1, lectin-like oxidized LDL receptor 1; Ly6C, lymphocyte antigen 6 complex, locus C; Mac-1, macrophage receptor 1; MMP, matrix metalloproteinase; NET, neutrophil extracellular traps; NF-κB, nuclear factor κB; NLRP3, NOD-, LRR- and pyrin domain-containing protein 3; NO, nitric oxide; NOS3, nitric oxide synthase 3 (= eNOS) gene; Nrp1, neuropilin 1; oxLDL, oxidized low density lipoprotein; PAMP, pathogen-associated molecular patterns; PECAM-1, platelet/endothelial cell-adhesion molecule 1; PCSK9, proprotein convertase subtilisin/kexin type 9; PSGL-1, platelet selectin glycoprotein ligand 1; Rac1, Ras-related C3 botulinum toxin substrate 1; ROCK, Rho-associated protein kinase; ROS, reactive oxygen species; S100A8/9, S100 calcium-binding protein A8; sLox-1, soluble lectin-like oxidized LDL receptor 1; sGC, soluble guanylyl cyclase; SGEF, SH3-containing guanine nucleotide exchange factor; SNP, single nucleotide polymorphism; TAZ, transcriptional coactivator with PDZ-binding motif; TCR, T cell receptor; Th1/2/17, T helper cell subtype 1, 2, 17; TLR, Toll-like receptor; TNFα, tumor necrosis factor alpha; Treg, regulatory T cell; Trio, triple functional domain protein; VCAM-1, vascular cell adhesion molecule 1; VE-cadherin, vascular endothelial cadherin; VEGFR, vascular endothelial growth factor receptor; VE-PTP, vascular endothelial protein tyrosine phosphatase; VLA-4, very late antigen 4; YAP, yes-associated protein.
References
1. Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, et al. Atherosclerosis. Nature Reviews Disease Primers. (2019) 5:56. doi: 10.1038/s41572-019-0106-z
2. Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM, et al. Global burden of cardiovascular diseases and risk factors, 1990-2019: update from the GBD 2019 study. J Am Coll Cardiol. (2020) 76:2982–3021. doi: 10.1016/j.jacc.2020.11.010
3. Borén J, Chapman MJ, Krauss RM, Packard CJ, Bentzon JF, Binder CJ, et al. Low-density lipoproteins cause atherosclerotic cardiovascular disease: pathophysiological, genetic, and therapeutic insights: a consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. (2020) 41:2313–30. doi: 10.1093/eurheartj/ehz962
5. Tabas I, García-Cardeña G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol. (2015) 209:13–22. doi: 10.1083/jcb.201412052
6. Wolf D, Ley K. Immunity and inflammation in atherosclerosis. Circ Res. (2019) 124:315–27. doi: 10.1161/CIRCRESAHA.118.313591
7. Ley K, Reutershan J. Leucocyte-endothelial interactions in health and disease. Handb Exp Pharmacol. (2006) 176:97–133. doi: 10.1007/3-540-36028-X_4
8. Imhof BA, Aurrand-Lions M. Adhesion mechanisms regulating the migration of monocytes. Nat Rev Immunol. (2004) 4:432–44. doi: 10.1038/nri1375
9. Butcher EC. Leukocyte-endothelial cell recognition: three (or more) steps to specificity and diversity. Cell. (1991) 67:1033–6. doi: 10.1016/0092-8674(91)90279-8
10. Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. (2007) 7:678–89. doi: 10.1038/nri2156
11. Furman D, Campisi J, Verdin E, Carrera-Bastos P, Targ S, Franceschi C, et al. Chronic inflammation in the etiology of disease across the life span. Nat Med. (2019) 25:1822–32. doi: 10.1038/s41591-019-0675-0
12. Netea MG, Balkwill F, Chonchol M, Cominelli F, Donath MY, Giamarellos-Bourboulis EJ, et al. A guiding map for inflammation. Nat Immunol. (2017) 18:826–31. doi: 10.1038/ni.3790
13. Shao Y, Saredy J, Yang WY, Sun Y, Lu Y, Saaoud F, et al. Vascular endothelial cells and innate immunity. Arterioscler Thromb Vasc Biol. (2020) 40:e138–52. doi: 10.1161/ATVBAHA.120.314330
14. Gong T, Liu L, Jiang W, Zhou R. DAMP-sensing receptors in sterile inflammation and inflammatory diseases. Nat Rev Immunol. (2020) 20:95–112. doi: 10.1038/s41577-019-0215-7
15. Mogensen TH. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin Microbiol Rev. (2009) 22:240–73. doi: 10.1128/CMR.00046-08
16. Kawasaki T, Kawai T. Toll-like receptor signaling pathways. Front Immunol. (2014) 5:461. doi: 10.3389/fimmu.2014.00461
17. Ait-Oufella H, Taleb S, Mallat Z, Tedgui A. Recent advances on the role of cytokines in atherosclerosis. Arterioscler Thromb Vasc Biol. (2011) 31:969–79. doi: 10.1161/ATVBAHA.110.207415
18. Pober JS, Sessa WC. Evolving functions of endothelial cells in inflammation. Nat Rev Immunol. (2007) 7:803–15. doi: 10.1038/nri2171
19. Muller WA. Getting leukocytes to the site of inflammation. Vet Pathol. (2013) 50:7–22. doi: 10.1177/0300985812469883
20. Patten DA, Shetty S. More than just a removal service: scavenger receptors in leukocyte trafficking. Front Immunol. (2018) 9:2904. doi: 10.3389/fimmu.2018.02904
21. Choi HJ, Kim NE, Kim BM, Seo M, Heo JH. TNF-alpha-induced YAP/TAZ activity mediates leukocyte-endothelial adhesion by regulating VCAM1 expression in endothelial cells. Int J Mol Sci. (2018) 19:3428. doi: 10.3390/ijms19113428
22. Drechsler M, Duchene J, Soehnlein O. Chemokines control mobilization, recruitment, and fate of monocytes in atherosclerosis. Arterioscler Thromb Vasc Biol. (2015) 35:1050–5. doi: 10.1161/ATVBAHA.114.304649
23. Gencer S, Evans BR, van der Vorst EPC, Döring Y, Weber C. Inflammatory chemokines in atherosclerosis. Cells. (2021) 10:226. doi: 10.3390/cells10020226
24. Shi C, Pamer EG. Monocyte recruitment during infection and inflammation. Nat Rev Immunol. (2011) 11:762. doi: 10.1038/nri3070
25. Huo Y, Schober A, Forlow SB, Smith DF, Hyman MC, Jung S, et al. Circulating activated platelets exacerbate atherosclerosis in mice deficient in apolipoprotein E. Nat Med. (2003) 9:61–7. doi: 10.1038/nm810
26. Noels H, Weber C, Koenen RR. Chemokines as therapeutic targets in cardiovascular disease. Arterioscler Thromb Vasc Biol. (2019) 39:583–92. doi: 10.1161/ATVBAHA.118.312037
27. Dyer DP. Understanding the mechanisms that facilitate specificity, not redundancy, of chemokine-mediated leukocyte recruitment. Immunology. (2020) 160:336–44. doi: 10.1111/imm.13200
28. Dyer DP, Medina-Ruiz L, Bartolini R, Schuette F, Hughes CE, Pallas K, et al. Chemokine receptor redundancy and specificity are context dependent. Immunity. (2019) 50:378–89.e5. doi: 10.1016/j.immuni.2019.01.009
29. Kansas G. Selectins and their ligands: current concepts and controversies. Blood. (1996) 88:3259–87. doi: 10.1182/blood.V88.9.3259.bloodjournal8893259
30. Cummings RD, Smith DF. The selectin family of carbohydrate-binding proteins: structure and importance of carbohydrate ligands for cell adhesion. Bioessays. (1992) 14:849–56. doi: 10.1002/bies.950141210
31. Maly P, Thall A, Petryniak B, Rogers CE, Smith PL, Marks RM, et al. The alpha(1,3)fucosyltransferase Fuc-TVII controls leukocyte trafficking through an essential role in L-, E-, and P-selectin ligand biosynthesis. Cell. (1996) 86:643–53. doi: 10.1016/S0092-8674(00)80137-3
32. Levinovitz A, Mühlhoff J, Isenmann S, Vestweber D. Identification of a glycoprotein ligand for E-selectin on mouse myeloid cells. J Cell Biol. (1993) 121:449–59. doi: 10.1083/jcb.121.2.449
33. Somers WS, Tang J, Shaw GD, Camphausen RT. Insights into the molecular basis of leukocyte tethering and rolling revealed by structures of P- and E-selectin bound to SLe(X) and PSGL-1. Cell. (2000) 103:467–79. doi: 10.1016/S0092-8674(00)00138-0
34. Yago T, Shao B, Miner JJ, Yao L, Klopocki AG, Maeda K, et al. E-selectin engages PSGL-1 and CD44 through a common signaling pathway to induce integrin alphaLbeta2-mediated slow leukocyte rolling. Blood. (2010) 116:485–94. doi: 10.1182/blood-2009-12-259556
35. Zarbock A, Ley K, McEver RP, Hidalgo A. Leukocyte ligands for endothelial selectins: specialized glycoconjugates that mediate rolling and signaling under flow. Blood. (2011) 118:6743–51. doi: 10.1182/blood-2011-07-343566
36. Huo Y, Xia L. P-selectin glycoprotein ligand-1 plays a crucial role in the selective recruitment of leukocytes into the atherosclerotic arterial wall. Trends Cardiovasc Med. (2009) 19:140–5. doi: 10.1016/j.tcm.2009.07.006
37. Alon R, Fuhlbrigge RC, Finger EB, Springer TA. Interactions through L-selectin between leukocytes and adherent leukocytes nucleate rolling adhesions on selectins and VCAM-1 in shear flow. J Cell Biol. (1996) 135:849–65. doi: 10.1083/jcb.135.3.849
38. Ivetic A, Hoskins Green HL, Hart SJ. L-selectin: a major regulator of leukocyte adhesion, migration and signaling. Front Immunol. (2019) 10:1068. doi: 10.3389/fimmu.2019.01068
39. Hemmerich S, Butcher EC, Rosen SD. Sulfation-dependent recognition of high endothelial venules (HEV)-ligands by L-selectin and MECA 79, and adhesion-blocking monoclonal antibody. J Exp Med. (1994) 180:2219–26. doi: 10.1084/jem.180.6.2219
40. Kimura N, Mitsuoka C, Kanamori A, Hiraiwa N, Uchimura K, Muramatsu T, et al. Reconstitution of functional L-selectin ligands on a cultured human endothelial cell line by cotransfection of alpha1–>3 fucosyltransferase VII and newly cloned GlcNAcbeta:6-sulfotransferase cDNA. Proc Natl Acad Sci USA. (1999) 96:4530–5. doi: 10.1073/pnas.96.8.4530
41. Uchimura K, Gauguet JM, Singer MS, Tsay D, Kannagi R, Muramatsu T, et al. A major class of L-selectin ligands is eliminated in mice deficient in two sulfotransferases expressed in high endothelial venules. Nat Immunol. (2005) 6:1105–13. doi: 10.1038/ni1258
42. da Costa Martins P, Garcia-Vallejo JJ, van Thienen JV, Fernandez-Borja M, van Gils JM, Beckers C, et al. P-selectin glycoprotein ligand-1 is expressed on endothelial cells and mediates monocyte adhesion to activated endothelium. Arterioscler Thromb Vasc Biol. (2007) 27:1023–9. doi: 10.1161/ATVBAHA.107.140442
43. Rossaint J, Margraf A, Zarbock A. Role of platelets in leukocyte recruitment and resolution of inflammation. Front Immunol. (2018) 9:2712. doi: 10.3389/fimmu.2018.02712
44. Drechsler M, Soehnlein O. The complexity of arterial classical monocyte recruitment. J Innate Immun. (2013) 5:358–66. doi: 10.1159/000348795
45. Heinzmann D, Noethel M, Ungern-Sternberg SV, Mitroulis I, Gawaz M, Chavakis T, et al. CD147 is a novel interaction partner of integrin αMβ2 mediating leukocyte and platelet adhesion. Biomolecules. (2020) 10:541. doi: 10.3390/biom10040541
46. Lightfoot A, McGettrick HM, Iqbal AJ. Vascular endothelial galectins in leukocyte trafficking. Front Immunol. (2021) 12:687711. doi: 10.3389/fimmu.2021.687711
47. Gittens BR, Bodkin JV, Nourshargh S, Perretti M, Cooper D. Galectin-3: a positive regulator of leukocyte recruitment in the inflamed microcirculation. J Immunol. (2017) 198:4458–69. doi: 10.4049/jimmunol.1600709
48. Kunkel EJ, Dunne JL, Ley K. Leukocyte arrest during cytokine-dependent inflammation in vivo. J Immunol. (2000) 164:3301–8. doi: 10.4049/jimmunol.164.6.3301
49. Chesnutt BC, Smith DF, Raffler NA, Smith ML, White EJ, Ley K. Induction of LFA-1-dependent neutrophil rolling on ICAM-1 by engagement of E-selectin. Microcirculation. (2006) 13:99–109. doi: 10.1080/10739680500466376
50. Kunkel EJ, Ley K. Distinct phenotype of E-selectin-deficient mice. E-selectin is required for slow leukocyte rolling in vivo. Circ Res. (1996) 79:1196–204. doi: 10.1161/01.RES.79.6.1196
51. Maas SL, Soehnlein O, Viola JR. Organ-specific mechanisms of transendothelial neutrophil migration in the lung, liver, kidney, and aorta. Front Immunol. (2018) 9:2739. doi: 10.3389/fimmu.2018.02739
52. Yago T, Zhang N, Zhao L, Abrams CS, McEver RP. Selectins and chemokines use shared and distinct signals to activate β2 integrins in neutrophils. Blood Adv. (2018) 2:731–44. doi: 10.1182/bloodadvances.2017015602
53. Mestas J, Ley K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc Med. (2008) 18:228–32. doi: 10.1016/j.tcm.2008.11.004
54. Salas A, Shimaoka M, Kogan AN, Harwood C, von Andrian UH, Springer TA. Rolling adhesion through an extended conformation of integrin alphaLbeta2 and relation to alpha I and beta I-like domain interaction. Immunity. (2004) 20:393–406. doi: 10.1016/S1074-7613(04)00082-2
55. Baidildinova G, Nagy M, Jurk K, Wild PS, ten Cate H, van der Meijden PEJ. Soluble platelet release factors as biomarkers for cardiovascular disease. Front Cardiovasc Med. (2021) 8:684920. doi: 10.3389/fcvm.2021.684920
56. Miwa K, Igawa A, Inoue H. Soluble E-selectin, ICAM-1 and VCAM-1 levels in systemic and coronary circulation in patients with variant angina. Cardiovasc Res. (1997) 36:37–44. doi: 10.1016/S0008-6363(97)00143-0
57. Ruchaud-Sparagano MH, Walker TR, Rossi AG, Haslett C, Dransfield I. Soluble E-selectin acts in synergy with platelet-activating factor to activate neutrophil beta 2-integrins. Role of tyrosine kinases and Ca2+ mobilization. J Biol Chem. (2000) 275:15758–64. doi: 10.1074/jbc.M907390199
58. Dunne JL, Ballantyne CM, Beaudet AL, Ley K. Control of leukocyte rolling velocity in TNF-alpha-induced inflammation by LFA-1 and Mac-1. Blood. (2002) 99:336–41. doi: 10.1182/blood.V99.1.336
59. Soehnlein O. Multiple roles for neutrophils in atherosclerosis. Circ Res. (2012) 110:875–88. doi: 10.1161/CIRCRESAHA.111.257535
60. Soehnlein O, Lindbom L, Weber C. Mechanisms underlying neutrophil-mediated monocyte recruitment. Blood. (2009) 114:4613–23. doi: 10.1182/blood-2009-06-221630
61. Huo Y, Hafezi-Moghadam A, Ley K. Role of vascular cell adhesion molecule-1 and fibronectin connecting segment-1 in monocyte rolling and adhesion on early atherosclerotic lesions. Circ Res. (2000) 87:153–9. doi: 10.1161/01.RES.87.2.153
62. Dunne JL, Collins RG, Beaudet AL, Ballantyne CM, Ley K. Mac-1, but not LFA-1, uses intercellular adhesion molecule-1 to mediate slow leukocyte rolling in TNF-alpha-induced inflammation. J Immunol. (2003) 171:6105–11. doi: 10.4049/jimmunol.171.11.6105
63. Hynes RO. Integrins: versatility, modulation, and signaling in cell adhesion. Cell. (1992) 69:11–25. doi: 10.1016/0092-8674(92)90115-S
64. Arnaout MA, Mahalingam B, Xiong JP. Integrin structure, allostery, bidirectional signaling. Annu Rev Cell Dev Biol. (2005) 21:381–410. doi: 10.1146/annurev.cellbio.21.090704.151217
65. Spillmann D, Witt D, Lindahl U. Defining the interleukin-8-binding domain of heparan sulfate. J Biol Chem. (1998) 273:15487–93. doi: 10.1074/jbc.273.25.15487
66. Tanaka Y, Adams DH, Hubscher S, Hirano H, Siebenlist U, Shaw S. T-cell adhesion induced by proteoglycan-immobilized cytokine MIP-1 beta. Nature. (1993) 361:79–82. doi: 10.1038/361079a0
67. Graham GJ, Handel TM, Proudfoot AEI. Leukocyte adhesion: reconceptualizing chemokine presentation by glycosaminoglycans. Trends Immunol. (2019) 40:472–81. doi: 10.1016/j.it.2019.03.009
68. Campbell JJ, Qin S, Bacon KB, Mackay CR, Butcher EC. Biology of chemokine and classical chemoattractant receptors: differential requirements for adhesion-triggering versus chemotactic responses in lymphoid cells. J Cell Biol. (1996) 134:255–66. doi: 10.1083/jcb.134.1.255
69. Bednarczyk M, Stege H, Grabbe S, Bros M. β2 Integrins-Multi-Functional Leukocyte Receptors in Health and Disease. Int J Mol Sci. (2020) 21:1402. doi: 10.3390/ijms21041402
70. Moser M, Legate KR, Zent R, Fassler R. The tail of integrins, talin, and kindlins. Science. (2009) 324:895–9. doi: 10.1126/science.1163865
71. Sun Z, Costell M, Fassler R. Integrin activation by talin, kindlin and mechanical forces. Nat Cell Biol. (2019) 21:25–31. doi: 10.1038/s41556-018-0234-9
72. Tadokoro S, Shattil SJ, Eto K, Tai V, Liddington RC, de Pereda JM, et al. Talin binding to integrin beta tails: a final common step in integrin activation. Science. (2003) 302:103–6. doi: 10.1126/science.1086652
73. Wen L, Marki A, Roy P, McArdle S, Sun H, Fan Z, et al. Kindlin-3 recruitment to the plasma membrane precedes high-affinity beta2-integrin and neutrophil arrest from rolling. Blood. (2021) 137:29–38. doi: 10.1182/blood.2019003446
74. Fan Z, McArdle S, Marki A, Mikulski Z, Gutierrez E, Engelhardt B, et al. Neutrophil recruitment limited by high-affinity bent β2 integrin binding ligand in cis. Nature Commun. (2016) 7:12658. doi: 10.1038/ncomms12658
75. Berlin C, Bargatze RF, Campbell JJ, von Andrian UH, Szabo MC, Hasslen SR, et al. alpha 4 integrins mediate lymphocyte attachment and rolling under physiologic flow. Cell. (1995) 80:413–22. doi: 10.1016/0092-8674(95)90491-3
76. Sadik CD, Kim ND, Luster AD. Neutrophils cascading their way to inflammation. Trends Immunol. (2011) 32:452–60. doi: 10.1016/j.it.2011.06.008
77. Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. (2013) 13:159–75. doi: 10.1038/nri3399
78. Meerschaert J, Furie MB. The adhesion molecules used by monocytes for migration across endothelium include CD11a/CD18, CD11b/CD18, and VLA-4 on monocytes and ICAM-1, VCAM-1, and other ligands on endothelium. J Immunol. (1995) 154:4099–112.
79. Walling BL, Kim M. LFA-1 in T cell migration and differentiation. Front Immunol. (2018) 9:952. doi: 10.3389/fimmu.2018.00952
80. Wolf D, Anto-Michel N, Blankenbach H, Wiedemann A, Buscher K, Hohmann JD, et al. A ligand-specific blockade of the integrin Mac-1 selectively targets pathologic inflammation while maintaining protective host-defense. Nat Commun. (2018) 9:525. doi: 10.1038/s41467-018-02896-8
81. Wolf D, Hohmann JD, Wiedemann A, Bledzka K, Blankenbach H, Marchini T, et al. Binding of CD40L to Mac-1's I-domain involves the EQLKKSKTL motif and mediates leukocyte recruitment and atherosclerosis–but does not affect immunity and thrombosis in mice. Circ Res. (2011) 109:1269–79. doi: 10.1161/CIRCRESAHA.111.247684
82. Zirlik A, Maier C, Gerdes N, MacFarlane L, Soosairajah J, Bavendiek U, et al. CD40 ligand mediates inflammation independently of CD40 by interaction with Mac-1. Circulation. (2007) 115:1571–80. doi: 10.1161/CIRCULATIONAHA.106.683201
83. Ley K, Zarbock A. Hold on to your endothelium: postarrest steps of the leukocyte adhesion cascade. Immunity. (2006) 25:185–7. doi: 10.1016/j.immuni.2006.08.001
84. Hirahashi J, Mekala D, Van Ziffle J, Xiao L, Saffaripour S, Wagner DD, et al. Mac-1 signaling via Src-family and Syk kinases results in elastase-dependent thrombohemorrhagic vasculopathy. Immunity. (2006) 25:271–83. doi: 10.1016/j.immuni.2006.05.014
85. Zhang H, Schaff UY, Green CE, Chen H, Sarantos MR, Hu Y, et al. Impaired integrin-dependent function in Wiskott-Aldrich syndrome protein-deficient murine and human neutrophils. Immunity. (2006) 25:285–95. doi: 10.1016/j.immuni.2006.06.014
86. Sluiter TJ, van Buul JD, Huveneers S, Quax PHA, de Vries MR. Endothelial barrier function and leukocyte transmigration in atherosclerosis. Biomedicines. (2021) 9:328. doi: 10.3390/biomedicines9040328
87. Buffone A Jr., Anderson NR, Hammer DA. Human neutrophils will crawl upstream on ICAM-1 If Mac-1 is blocked. Biophys J. (2019) 117:1393–404. doi: 10.1016/j.bpj.2019.08.044
88. Phillipson M, Heit B, Colarusso P, Liu L, Ballantyne CM, Kubes P. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med. (2006) 203:2569–75. doi: 10.1084/jem.20060925
89. Salminen AT, Allahyari Z, Gholizadeh S, McCloskey MC, Ajalik R, Cottle RN, et al. In vitro studies of transendothelial migration for biological and drug discovery. Front Med Technol. (2020) 2:616. doi: 10.3389/fmedt.2020.600616
90. Schenkel AR, Mamdouh Z, Muller WA. Locomotion of monocytes on endothelium is a critical step during extravasation. Nat Immunol. (2004) 5:393–400. doi: 10.1038/ni1051
91. Leick M, Azcutia V, Newton G, Luscinskas FW. Leukocyte recruitment in inflammation: basic concepts and new mechanistic insights based on new models and microscopic imaging technologies. Cell Tissue Res. (2014) 355:647–56. doi: 10.1007/s00441-014-1809-9
92. Barreiro O, Yanez-Mo M, Serrador JM, Montoya MC, Vicente-Manzanares M, Tejedor R, et al. Dynamic interaction of VCAM-1 and ICAM-1 with moesin and ezrin in a novel endothelial docking structure for adherent leukocytes. J Cell Biol. (2002) 157:1233–45. doi: 10.1083/jcb.200112126
93. Carman CV, Springer TA. A transmigratory cup in leukocyte diapedesis both through individual vascular endothelial cells and between them. J Cell Biol. (2004) 167:377–88. doi: 10.1083/jcb.200404129
94. Barzilai S, Yadav SK, Morrell S, Roncato F, Klein E, Stoler-Barak L, et al. Leukocytes breach endothelial barriers by insertion of nuclear lobes and disassembly of endothelial actin filaments. Cell Rep. (2017) 18:685–99. doi: 10.1016/j.celrep.2016.12.076
95. Nourshargh S, Hordijk PL, Sixt M. Breaching multiple barriers: leukocyte motility through venular walls and the interstitium. Nat Rev Mol Cell Biol. (2010) 11:366–78. doi: 10.1038/nrm2889
96. Broermann A, Winderlich M, Block H, Frye M, Rossaint J, Zarbock A, et al. Dissociation of VE-PTP from VE-cadherin is required for leukocyte extravasation and for VEGF-induced vascular permeability in vivo. J Exp Med. (2011) 208:2393–401. doi: 10.1084/jem.20110525
97. Vestweber D. How leukocytes cross the vascular endothelium. Nat Rev Immunol. (2015) 15:692–704. doi: 10.1038/nri3908
98. Wessel F, Winderlich M, Holm M, Frye M, Rivera-Galdos R, Vockel M, et al. Leukocyte extravasation and vascular permeability are each controlled in vivo by different tyrosine residues of VE-cadherin. Nat Immunol. (2014) 15:223–30. doi: 10.1038/ni.2824
99. Gerhardt T, Ley K. Monocyte trafficking across the vessel wall. Cardiovasc Res. (2015) 107:321–30. doi: 10.1093/cvr/cvv147
100. Muller WA. Mechanisms of leukocyte transendothelial migration. Annu Rev Pathol. (2011) 6:323–44. doi: 10.1146/annurev-pathol-011110-130224
101. Goswami D, März S, Li Y.-T., Artz A, Schäfer K, et al. Endothelial CD99 supports arrest of mouse neutrophils in venules and binds to neutrophil PILRs. Blood. (2017) 129:1811–22. doi: 10.1182/blood-2016-08-733394
102. Nourshargh S, Alon R. Leukocyte migration into inflamed tissues. Immunity. (2014) 41:694–707. doi: 10.1016/j.immuni.2014.10.008
103. Engelhardt B, Wolburg H. Mini-review: transendothelial migration of leukocytes: through the front door or around the side of the house? Eur J Immunol. (2004) 34:2955–63. doi: 10.1002/eji.200425327
104. Rao RM, Yang L, Garcia-Cardena G, Luscinskas FW. Endothelial-dependent mechanisms of leukocyte recruitment to the vascular wall. Circ Res. (2007) 101:234–47. doi: 10.1161/CIRCRESAHA.107.151860b
105. Chen Z, Schunkert H. Genetics of coronary artery disease in the post-GWAS era. J Intern Med. (2021) 290:980–92. doi: 10.1111/joim.13362
106. Kessler T, Schunkert H. Coronary artery disease genetics enlightened by genome-wide association studies. JACC Basic Transl Sci. (2021) 6:610–23. doi: 10.1016/j.jacbts.2021.04.001
107. Mauersberger C, Schunkert H, Sager HB. Inflammation-related risk loci in genome-wide association studies of coronary artery disease. Cells. (2021) 10:440. doi: 10.3390/cells10020440
108. Yamashiro Y, Yanagisawa H. The molecular mechanism of mechanotransduction in vascular homeostasis and disease. Clin. Sci. (2020) 134:2399–418. doi: 10.1042/CS20190488
109. Conway DE, Breckenridge MT, Hinde E, Gratton E, Chen CS, Schwartz MA. Fluid shear stress on endothelial cells modulates mechanical tension across VE-cadherin and PECAM-1. Curr Biol. (2013) 23:1024–30. doi: 10.1016/j.cub.2013.04.049
110. Coon BG, Baeyens N, Han J, Budatha M, Ross TD, Fang JS, et al. Intramembrane binding of VE-cadherin to VEGFR2 and VEGFR3 assembles the endothelial mechanosensory complex. J Cell Biol. (2015) 208:975–86. doi: 10.1083/jcb.201408103
111. Tzima E, Irani-Tehrani M, Kiosses WB, Dejana E, Schultz DA, Engelhardt B, et al. A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature. (2005) 437:426–31. doi: 10.1038/nature03952
112. Collins C, Guilluy C, Welch C, O'Brien ET, Hahn K, Superfine R, et al. Localized tensional forces on PECAM-1 elicit a global mechanotransduction response via the integrin-RhoA pathway. Curr Biol. (2012) 22:2087–94. doi: 10.1016/j.cub.2012.08.051
113. Orr AW, Ginsberg MH, Shattil SJ, Deckmyn H, Schwartz MA. Matrix-specific suppression of integrin activation in shear stress signaling. Mol Biol Cell. (2006) 17:4686–97. doi: 10.1091/mbc.e06-04-0289
114. Dusserre N, L'Heureux N, Bell KS, Stevens HY, Yeh J, Otte LA, et al. PECAM-1 interacts with nitric oxide synthase in human endothelial cells: implication for flow-induced nitric oxide synthase activation. Arterioscler Thromb Vasc Biol. (2004) 24:1796–802. doi: 10.1161/01.ATV.0000141133.32496.41
115. Fleming I, Fisslthaler B, Dixit M, Busse R. Role of PECAM-1 in the shear-stress-induced activation of Akt and the endothelial nitric oxide synthase (eNOS) in endothelial cells. J Cell Sci. (2005) 118:4103–11. doi: 10.1242/jcs.02541
116. Lakshmikanthan S, Zheng X, Nishijima Y, Sobczak M, Szabo A, Vasquez-Vivar J, et al. Rap1 promotes endothelial mechanosensing complex formation, NO release and normal endothelial function. EMBO Rep. (2015) 16:628–37. doi: 10.15252/embr.201439846
117. Novodvorsky P, Chico TJ. The role of the transcription factor KLF2 in vascular development and disease. Prog Mol Biol Transl Sci. (2014) 124:155–88. doi: 10.1016/B978-0-12-386930-2.00007-0
118. Wang L, Luo JY, Li B, Tian XY, Chen LJ, Huang Y, et al. Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature. (2016) 540:579–82. doi: 10.1038/nature20602
119. Gambillara V, Chambaz C, Montorzi G, Roy S, Stergiopulos N, Silacci P. Plaque-prone hemodynamics impair endothelial function in pig carotid arteries. Am J Physiol Heart Circ Physiol. (2006) 290:H2320–8. doi: 10.1152/ajpheart.00486.2005
120. Lu X, Kassab GS. Nitric oxide is significantly reduced in ex vivo porcine arteries during reverse flow because of increased superoxide production. J Physiol. (2004) 561:575–82. doi: 10.1113/jphysiol.2004.075218
121. Wang C, Baker BM, Chen CS, Schwartz MA. Endothelial cell sensing of flow direction. Arterioscler Thromb Vasc Biol. (2013) 33:2130–6. doi: 10.1161/ATVBAHA.113.301826
122. Feaver RE, Gelfand BD, Wang C, Schwartz MA, Blackman BR. Atheroprone hemodynamics regulate fibronectin deposition to create positive feedback that sustains endothelial inflammation. Circ Res. (2010) 106:1703–11. doi: 10.1161/CIRCRESAHA.109.216283
123. Orr AW, Sanders JM, Bevard M, Coleman E, Sarembock IJ, Schwartz MA. The subendothelial extracellular matrix modulates NF-kappaB activation by flow: a potential role in atherosclerosis. J Cell Biol. (2005) 169:191–202. doi: 10.1083/jcb.200410073
124. Bondareva O, Tsaryk R, Bojovic V, Odenthal-Schnittler M, Siekmann AF, Schnittler HJ. Identification of atheroprone shear stress responsive regulatory elements in endothelial cells. Cardiovasc Res. (2019) 115:1487–99. doi: 10.1093/cvr/cvz027
125. Davis ME, Grumbach IM, Fukai T, Cutchins A, Harrison DG. Shear stress regulates endothelial nitric-oxide synthase promoter activity through nuclear factor kappaB binding. J Biol Chem. (2004) 279:163–8. doi: 10.1074/jbc.M307528200
126. Dunzendorfer S, Lee HK, Tobias PS. Flow-dependent regulation of endothelial Toll-like receptor 2 expression through inhibition of SP1 activity. Circ Res. (2004) 95:684–91. doi: 10.1161/01.RES.0000143900.19798.47
127. Mullick AE, Soldau K, Kiosses WB, Bell TA 3rd, Tobias PS, Curtiss LK. Increased endothelial expression of Toll-like receptor 2 at sites of disturbed blood flow exacerbates early atherogenic events. J Exp Med. (2008) 205:373–83. doi: 10.1084/jem.20071096
128. Xu S, Ha CH, Wang W, Xu X, Yin M, Jin FQ, et al. PECAM1 regulates flow-mediated Gab1 tyrosine phosphorylation and signaling. Cell Signal. (2016) 28:117–24. doi: 10.1016/j.cellsig.2015.12.007
129. Tada Y, Koarada S, Morito F, Ushiyama O, Haruta Y, Kanegae F, et al. Acceleration of the onset of collagen-induced arthritis by a deficiency of platelet endothelial cell adhesion molecule 1. Arthritis Rheum. (2003) 48:3280–90. doi: 10.1002/art.11268
130. Wong MX, Hayball JD, Hogarth PM, Jackson DE. The inhibitory co-receptor, PECAM-1 provides a protective effect in suppression of collagen-induced arthritis. J Clin Immunol. (2005) 25:19–28. doi: 10.1007/s10875-005-0354-7
131. Harry BL, Sanders JM, Feaver RE, Lansey M, Deem TL, Zarbock A, et al. Endothelial cell PECAM-1 promotes atherosclerotic lesions in areas of disturbed flow in ApoE-deficient mice. Arterioscler Thromb Vasc Biol. (2008) 28:2003–8. doi: 10.1161/ATVBAHA.108.164707
132. Goel R, Schrank BR, Arora S, Boylan B, Fleming B, Miura H, et al. Site-specific effects of PECAM-1 on atherosclerosis in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. (2008) 28:1996–2002. doi: 10.1161/ATVBAHA.108.172270
133. Harrison M, Smith E, Ross E, Krams R, Segers D, Buckley CD, et al. The role of platelet-endothelial cell adhesion molecule-1 in atheroma formation varies depending on the site-specific hemodynamic environment. Arterioscler Thromb Vasc Biol. (2013) 33:694–701. doi: 10.1161/ATVBAHA.112.300379
134. Howson JMM, Zhao W, Barnes DR, Ho WK, Young R, Paul DS, et al. Fifteen new risk loci for coronary artery disease highlight arterial-wall-specific mechanisms. Nat Genet. (2017) 49:1113–9. doi: 10.1038/ng.3874
135. Dang TA, Schunkert H, Kessler T. cGMP signaling in cardiovascular diseases: linking genotype and phenotype. J Cardiovasc Pharmacol. (2020) 75:516–25. doi: 10.1097/FJC.0000000000000744
136. Wen L, Feil S, Wolters M, Thunemann M, Regler F, Schmidt K, et al. A shear-dependent NO-cGMP-cGKI cascade in platelets acts as an auto-regulatory brake of thrombosis. Nat Commun. (2018) 9:4301. doi: 10.1038/s41467-018-06638-8
137. Förstermann U, Xia N, Li H. Roles of vascular oxidative stress and nitric oxide in the pathogenesis of atherosclerosis. Circ Res. (2017) 120:713–35. doi: 10.1161/CIRCRESAHA.116.309326
138. CARDIoGRAMplusC4D Consortium, Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. (2013) 45:25–33. doi: 10.1038/ng.2480
139. Nelson CP, Goel A, Butterworth AS, Kanoni S, Webb TR, Marouli E, et al. Association analyses based on false discovery rate implicate new loci for coronary artery disease. Nat Genet. (2017) 49:1385–91. doi: 10.1038/ng.3913
140. Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, et al. A comprehensive 1,000 genomes-based genome-wide association meta-analysis of coronary artery disease. Nat Genet. (2015) 47:1121–30. doi: 10.1038/ng.3396
141. Zimmer S, Grebe A, Latz E. Danger signaling in atherosclerosis. Circ Res. (2015) 116:323–40. doi: 10.1161/CIRCRESAHA.116.301135
142. Huang L, Chambliss KL, Gao X, Yuhanna IS, Behling-Kelly E, Bergaya S, et al. SR-B1 drives endothelial cell LDL transcytosis via DOCK4 to promote atherosclerosis. Nature. (2019) 569:565–9. doi: 10.1038/s41586-019-1140-4
143. Luchetti F, Crinelli R, Nasoni MG, Benedetti S, Palma F, Fraternale A, et al. LDL receptors, caveolae and cholesterol in endothelial dysfunction: oxLDLs accomplices or victims? Br J Pharmacol. (2021) 178:3104–14. doi: 10.1111/bph.15272
144. Barreto J, Karathanasis SK, Remaley A, Sposito AC. Role of LOX-1 (lectin-like oxidized low-density lipoprotein receptor 1) as a cardiovascular risk predictor: mechanistic insight and potential clinical use. Arterioscler Thromb Vasc Biol. (2021) 41:153–66. doi: 10.1161/ATVBAHA.120.315421
145. Kang H, Lu J, Yang J, Fan Y, Deng X. Interaction of arterial proteoglycans with low density lipoproteins (LDLs): from theory to promising therapeutic approaches. Medicine in Novel Technol Devices. (2019) 3:100016. doi: 10.1016/j.medntd.2019.100016
146. Marchio P, Guerra-Ojeda S, Vila JM, Aldasoro M, Victor VM, Mauricio MD. Targeting early atherosclerosis: a focus on oxidative stress and inflammation. Oxid Med Cell Longev. (2019) 2019:8563845. doi: 10.1155/2019/8563845
147. Ungvari Z, Tarantini S, Kiss T, Wren JD, Giles CB, Griffin CT, et al. Endothelial dysfunction and angiogenesis impairment in the ageing vasculature. Nat Rev Cardiol. (2018) 15:555–65. doi: 10.1038/s41569-018-0030-z
148. Münzel T, Hahad O, Kuntic M, Keaney JF, Deanfield JE, Daiber A. Effects of tobacco cigarettes, e-cigarettes, and waterpipe smoking on endothelial function and clinical outcomes. Eur Heart J. (2020) 41:4057–70. doi: 10.1093/eurheartj/ehaa460
149. Poznyak AV, Nikiforov NG, Markin AM, Kashirskikh DA, Myasoedova VA, Gerasimova EV, et al. Overview of OxLDL and its impact on cardiovascular health: focus on atherosclerosis. Front Pharmacol. (2020) 11:613780. doi: 10.3389/fphar.2020.613780
150. Hofmann A, Brunssen C, Wolk S, Reeps C, Morawietz H. Soluble LOX-1: a novel biomarker in patients with coronary artery disease, stroke, and acute aortic dissection? J Am Heart Assoc. (2020) 9:e013803. doi: 10.1161/JAHA.119.013803
151. Blair A, Shaul PW, Yuhanna IS, Conrad PA, Smart EJ. Oxidized low density lipoprotein displaces endothelial nitric-oxide synthase (eNOS) from plasmalemmal caveolae and impairs eNOS activation. J Biol Chem. (1999) 274:32512–9. doi: 10.1074/jbc.274.45.32512
152. Ryoo S, Lemmon CA, Soucy KG, Gupta G, White AR, Nyhan D, et al. Oxidized low-density lipoprotein-dependent endothelial arginase II activation contributes to impaired nitric oxide signaling. Circ Res. (2006) 99:951–60. doi: 10.1161/01.RES.0000247034.24662.b4
153. Dégano IR, Camps-Vilaro A, Subirana I, Garcia-Mateo N, Cidad P, Munoz-Aguayo D, et al. Association of circulating microRNAs with coronary artery disease and usefulness for reclassification of healthy individuals: the REGICOR Study. J Clin Med. (2020) 9:1402. doi: 10.3390/jcm9051402
154. Li D, Chen H, Romeo F, Sawamura T, Saldeen T, Mehta JL. Statins modulate oxidized low-density lipoprotein-mediated adhesion molecule expression in human coronary artery endothelial cells: role of LOX-1. J Pharmacol Exp Ther. (2002) 302:601–5. doi: 10.1124/jpet.102.034959
155. Li D, Mehta JL. Antisense to LOX-1 inhibits oxidized LDL-mediated upregulation of monocyte chemoattractant protein-1 and monocyte adhesion to human coronary artery endothelial cells. Circulation. (2000) 101:2889–95. doi: 10.1161/01.CIR.101.25.2889
156. Mattaliano MD, Huard C, Cao W, Hill AA, Zhong W, Martinez RV, et al. LOX-1-dependent transcriptional regulation in response to oxidized LDL treatment of human aortic endothelial cells. Am J Physiol Cell Physiol. (2009) 296:C1329–37. doi: 10.1152/ajpcell.00513.2008
157. Schmitt MM, Fraemohs L, Hackeng TM, Weber C, Koenen RR. Atherogenic mononuclear cell recruitment is facilitated by oxidized lipoprotein-induced endothelial junctional adhesion molecule-A redistribution. Atherosclerosis. (2014) 234:254–64. doi: 10.1016/j.atherosclerosis.2014.03.014
158. Mezentsev A, Bezsonov E, Kashirskikh D, Baig MS, Eid AH, Orekhov A. Proatherogenic sialidases and desialylated lipoproteins: 35 years of research and current state from bench to bedside. Biomedicines. (2021) 9:600. doi: 10.3390/biomedicines9060600
159. Ruuth M, Nguyen SD, Vihervaara T, Hilvo M, Laajala TD, Kondadi PK, et al. Susceptibility of low-density lipoprotein particles to aggregate depends on particle lipidome, is modifiable, and associates with future cardiovascular deaths. Eur Heart J. (2018) 39:2562–73. doi: 10.1093/eurheartj/ehy319
160. Galkina E, Ley K. Leukocyte influx in atherosclerosis. Curr Drug Targets. (2007) 8:1239–48. doi: 10.2174/138945007783220650
161. Bjorkegren JLM, Kovacic JC, Dudley JT, Schadt EE. Genome-wide significant loci: how important are they? Systems genetics to understand heritability of coronary artery disease and other common complex disorders. J Am Coll Cardiol. (2015) 65:830–45. doi: 10.1016/j.jacc.2014.12.033
162. Pezhman L, Tahrani A, Chimen M. Dysregulation of leukocyte trafficking in type 2 diabetes: mechanisms and potential therapeutic avenues. Front Cell Dev Biol. (2021) 9:624184. doi: 10.3389/fcell.2021.624184
163. Basehore S, Bohlman S, Weber C, Swaminathan S, Zhang Y, Jang C, et al. Laminar flow on endothelial cells suppresses eNOS O-GlcNAcylation to promote eNOS activity. Circ Res. (2021) 129:1054–1066. doi: 10.1161/CIRCRESAHA.121.318982
164. Heidt T, Sager HB, Courties G, Dutta P, Iwamoto Y, Zaltsman A, et al. Chronic variable stress activates hematopoietic stem cells. Nat Med. (2014) 20:754–8. doi: 10.1038/nm.3589
165. Hinterdobler J, Schunkert H, Kessler T, Sager HB. Impact of acute and chronic psychosocial stress on vascular inflammation. Antioxid Redox Signal. (2021) 35:1531–1550. doi: 10.1089/ars.2021.0153
166. Hinterdobler J, Schott S, Jin H, Meesmann A, Steinsiek AL, Zimmermann AS, et al. Acute mental stress drives vascular inflammation promotes plaque destabilization in mouse atherosclerosis. Eur Heart J. (2021) 42:4077–4088. doi: 10.1093/eurheartj/ehab371
167. Zernecke A, Winkels H, Cochain C, Williams JW, Wolf D, Soehnlein O, et al. Meta-analysis of leukocyte diversity in atherosclerotic mouse aortas. Circ Res. (2020) 127:402–26. doi: 10.1161/CIRCRESAHA.120.316903
168. Hamers AAJ, Dinh HQ, Thomas GD, Marcovecchio P, Blatchley A, Nakao CS, et al. Human monocyte heterogeneity as revealed by high-dimensional mass cytometry. Arterioscler Thromb Vasc Biol. (2019) 39:25–36. doi: 10.1161/ATVBAHA.118.311022
169. Ziegler-Heitbrock L, Ancuta P, Crowe S, Dalod M, Grau V, Hart DN, et al. Nomenclature of monocytes and dendritic cells in blood. Blood. (2010) 116:e74–80. doi: 10.1182/blood-2010-02-258558
170. Sager HB, Nahrendorf M. Inflammation: a trigger for acute coronary syndrome. Q J Nucl Med Mol Imaging. (2016) 60:185–93.
171. Rahman MS, Murphy AJ, Woollard KJ. Effects of dyslipidaemia on monocyte production and function in cardiovascular disease. Nat Rev Cardiol. (2017) 14:387–400. doi: 10.1038/nrcardio.2017.34
172. Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R, et al. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. (2007) 117:195–205. doi: 10.1172/JCI29950
173. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. (2013) 38:79–91. doi: 10.1016/j.immuni.2012.12.001
174. Marcovecchio PM, Thomas GD, Mikulski Z, Ehinger E, Mueller KAL, Blatchley A, et al. Scavenger receptor CD36 directs nonclassical monocyte patrolling along the endothelium during early atherogenesis. Arterioscler Thromb Vasc Biol. (2017) 37:2043–52. doi: 10.1161/ATVBAHA.117.309123
175. Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, et al. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. (2007) 317:666–70. doi: 10.1126/science.1142883
176. Tacke F, Alvarez D, Kaplan TJ, Jakubzick C, Spanbroek R, Llodra J, et al. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. (2007) 117:185–94. doi: 10.1172/JCI28549
177. Quintar A, McArdle S, Wolf D, Marki A, Ehinger E, Vassallo M, et al. Endothelial protective monocyte patrolling in large arteries intensified by western diet and atherosclerosis. Circ Res. (2017) 120:1789–99. doi: 10.1161/CIRCRESAHA.117.310739
178. Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2-/- mice reveals a role for chemokines in the initiation of atherosclerosis. Nature. (1998) 394:894–7. doi: 10.1038/29788
179. Gu L, Okada Y, Clinton SK, Gerard C, Sukhova GK, Libby P, et al. Absence of monocyte chemoattractant protein-1 reduces atherosclerosis in low density lipoprotein receptor-deficient mice. Mol Cell. (1998) 2:275–81. doi: 10.1016/S1097-2765(00)80139-2
180. Leuschner F, Dutta P, Gorbatov R, Novobrantseva TI, Donahoe JS, Courties G, et al. Therapeutic siRNA silencing in inflammatory monocytes in mice. Nat Biotechnol. (2011) 29:1005–10. doi: 10.1038/nbt.1989
181. Aiello RJ, Bourassa PA, Lindsey S, Weng W, Natoli E, Rollins BJ, et al. Monocyte chemoattractant protein-1 accelerates atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. (1999) 19:1518–25. doi: 10.1161/01.ATV.19.6.1518
182. Georgakis MK, van der Laan SW, Asare Y, Mekke JM, Haitjema S, Schoneveld AH, et al. Monocyte-chemoattractant protein-1 levels in human atherosclerotic lesions associate with plaque vulnerability. Arterioscler Thromb Vasc Biol. (2021) 41:2038–48. doi: 10.1161/ATVBAHA.121.316091
183. Bernelot Moens SJ, Neele AE, Kroon J, van der Valk FM, Van den Bossche J, Hoeksema MA, et al. PCSK9 monoclonal antibodies reverse the pro-inflammatory profile of monocytes in familial hypercholesterolaemia. Eur Heart J. (2017) 38:1584–93. doi: 10.1093/eurheartj/ehx002
184. Braunersreuther V, Zernecke A, Arnaud C, Liehn EA, Steffens S, Shagdarsuren E, et al. Ccr5 but not Ccr1 deficiency reduces development of diet-induced atherosclerosis in mice. Arterioscler Thromb Vasc Biol. (2007) 27:373–9. doi: 10.1161/01.ATV.0000253886.44609.ae
185. Potteaux S, Combadiere C, Esposito B, Lecureuil C, Ait-Oufella H, Merval R, et al. Role of bone marrow-derived CC-chemokine receptor 5 in the development of atherosclerosis of low-density lipoprotein receptor knockout mice. Arterioscler Thromb Vasc Biol. (2006) 26:1858–63. doi: 10.1161/01.ATV.0000231527.22762.71
186. Zernecke A, Liehn EA, Gao JL, Kuziel WA, Murphy PM, Weber C. Deficiency in CCR5 but not CCR1 protects against neointima formation in atherosclerosis-prone mice: involvement of IL-10. Blood. (2006) 107:4240–3. doi: 10.1182/blood-2005-09-3922
187. Combadière C, Potteaux S, Rodero M, Simon T, Pezard A, Esposito B, et al. Combined inhibition of CCL2, CX3CR1, and CCR5 abrogates Ly6C(hi) and Ly6C(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice. Circulation. (2008) 117:1649–57. doi: 10.1161/CIRCULATIONAHA.107.745091
188. Landsman L, Bar-On L, Zernecke A, Kim KW, Krauthgamer R, Shagdarsuren E, et al. CX3CR1 is required for monocyte homeostasis and atherogenesis by promoting cell survival. Blood. (2009) 113:963–72. doi: 10.1182/blood-2008-07-170787
189. An G, Wang H, Tang R, Yago T, McDaniel JM, McGee S, et al. P-selectin glycoprotein ligand-1 is highly expressed on Ly-6Chi monocytes and a major determinant for Ly-6Chi monocyte recruitment to sites of atherosclerosis in mice. Circulation. (2008) 117:3227–37. doi: 10.1161/CIRCULATIONAHA.108.771048
190. Dong ZM, Brown AA, Wagner DD. Prominent role of P-selectin in the development of advanced atherosclerosis in ApoE-deficient mice. Circulation. (2000) 101:2290–5. doi: 10.1161/01.CIR.101.19.2290
191. Johnson RC, Chapman SM, Dong ZM, Ordovas JM, Mayadas TN, Herz J, et al. Absence of P-selectin delays fatty streak formation in mice. J Clin Invest. (1997) 99:1037–43. doi: 10.1172/JCI119231
192. Mayadas TN, Johnson RC, Rayburn H, Hynes RO, Wagner DD. Leukocyte rolling and extravasation are severely compromised in P selectin-deficient mice. Cell. (1993) 74:541–54. doi: 10.1016/0092-8674(93)80055-J
193. Sager HB, Dutta P, Dahlman JE, Hulsmans M, Courties G, Sun Y, et al. RNAi targeting multiple cell adhesion molecules reduces immune cell recruitment and vascular inflammation after myocardial infarction. Sci Transl Med. (2016) 8:342ra80. doi: 10.1126/scitranslmed.aaf1435
194. Collins RG, Velji R, Guevara NV, Hicks MJ, Chan L, Beaudet al. P-Selectin or intercellular adhesion molecule (ICAM)-1 deficiency substantially protects against atherosclerosis in apolipoprotein E-deficient mice. J Exp Med. (2000) 191:189–94. doi: 10.1084/jem.191.1.189
195. Dong ZM, Chapman SM, Brown AA, Frenette PS, Hynes RO, Wagner DD. The combined role of P- and E-selectins in atherosclerosis. J Clin Invest. (1998) 102:145–52. doi: 10.1172/JCI3001
196. Ye Z, Zhang S, Liu Y, Wang S, Zhang J, Huang R. A peptide analogue of selectin ligands attenuated atherosclerosis by inhibiting monocyte activation. Mediators Inflamm. (2019) 2019:8709583. doi: 10.1155/2019/8709583
197. Zuchtriegel G, Uhl B, Hessenauer ME, Kurz AR, Rehberg M, Lauber K, et al. Spatiotemporal expression dynamics of selectins govern the sequential extravasation of neutrophils and monocytes in the acute inflammatory response. Arterioscler Thromb Vasc Biol. (2015) 35:899–910. doi: 10.1161/ATVBAHA.114.305143
198. Soehnlein O, Drechsler M, Doring Y, Lievens D, Hartwig H, Kemmerich K, et al. Distinct functions of chemokine receptor axes in the atherogenic mobilization and recruitment of classical monocytes. EMBO Mol Med. (2013) 5:471–81. doi: 10.1002/emmm.201201717
199. Henderson RB, Hobbs JA, Mathies M, Hogg N. Rapid recruitment of inflammatory monocytes is independent of neutrophil migration. Blood. (2003) 102:328–35. doi: 10.1182/blood-2002-10-3228
200. Huo Y, Weber C, Forlow SB, Sperandio M, Thatte J, Mack M, et al. The chemokine KC, but not monocyte chemoattractant protein-1, triggers monocyte arrest on early atherosclerotic endothelium. J Clin Invest. (2001) 108:1307–14. doi: 10.1172/JCI12877
201. Hyduk SJ, Cybulsky MI. Role of alpha4beta1 integrins in chemokine-induced monocyte arrest under conditions of shear stress. Microcirculation. (2009) 16:17–30. doi: 10.1080/10739680802425195
202. Barringhaus KG, Phillips JW, Thatte JS, Sanders JM, Czarnik AC, Bennett DK, et al. Alpha4beta1 integrin (VLA-4) blockade attenuates both early and late leukocyte recruitment and neointimal growth following carotid injury in apolipoprotein E (-/-) mice. J Vasc Res. (2004) 41:252–60. doi: 10.1159/000078646
203. Shih PT, Brennan ML, Vora DK, Territo MC, Strahl D, Elices MJ, et al. Blocking very late antigen-4 integrin decreases leukocyte entry and fatty streak formation in mice fed an atherogenic diet. Circ Res. (1999) 84:345–51. doi: 10.1161/01.RES.84.3.345
204. Cybulsky MI, Iiyama K, Li H, Zhu S, Chen M, Iiyama M, et al. A major role for VCAM-1, but not ICAM-1, in early atherosclerosis. J Clin Invest. (2001) 107:1255–62. doi: 10.1172/JCI11871
205. Dansky HM, Barlow CB, Lominska C, Sikes JL, Kao C, Weinsaft J, et al. Adhesion of monocytes to arterial endothelium and initiation of atherosclerosis are critically dependent on vascular cell adhesion molecule-1 gene dosage. Arterioscler Thromb Vasc Biol. (2001) 21:1662–7. doi: 10.1161/hq1001.096625
206. Park JG, Ryu SY, Jung IH, Lee YH, Kang KJ, Lee MR, et al. Evaluation of VCAM-1 antibodies as therapeutic agent for atherosclerosis in apolipoprotein E-deficient mice. Atherosclerosis. (2013) 226:356–63. doi: 10.1016/j.atherosclerosis.2012.11.029
207. Kuijper PH, Gallardo Torres HI, Houben LA, Lammers JW, Zwaginga JJ, Koenderman L. P-selectin and MAC-1 mediate monocyte rolling and adhesion to ECM-bound platelets under flow conditions. J Leukoc Biol. (1998) 64:467–73. doi: 10.1002/jlb.64.4.467
208. Sumagin R, Prizant H, Lomakina E, Waugh RE, Sarelius IH. LFA-1 and Mac-1 define characteristically different intralumenal crawling and emigration patterns for monocytes and neutrophils in situ. J Immunol. (2010) 185:7057–66. doi: 10.4049/jimmunol.1001638
209. Bourdillon MC, Poston RN, Covacho C, Chignier E, Bricca G, McGregor JL. ICAM-1 deficiency reduces atherosclerotic lesions in double-knockout mice (ApoE(-/-)/ICAM-1(-/-)) fed a fat or a chow diet. Arterioscler Thromb Vasc Biol. (2000) 20:2630–5. doi: 10.1161/01.ATV.20.12.2630
210. Nageh MF, Sandberg ET, Marotti KR, Lin AH, Melchior EP, Bullard DC, et al. Deficiency of inflammatory cell adhesion molecules protects against atherosclerosis in mice. Arterioscler Thromb Vasc Biol. (1997) 17:1517–20. doi: 10.1161/01.ATV.17.8.1517
211. Kitagawa K, Matsumoto M, Sasaki T, Hashimoto H, Kuwabara K, Ohtsuki T, et al. Involvement of ICAM-1 in the progression of atherosclerosis in APOE-knockout mice. Atherosclerosis. (2002) 160:305–10. doi: 10.1016/S0021-9150(01)00587-1
212. Manka D, Collins RG, Ley K, Beaudet AL, Sarembock IJ. Absence of p-selectin, but not intercellular adhesion molecule-1, attenuates neointimal growth after arterial injury in apolipoprotein e-deficient mice. Circulation. (2001) 103:1000–5. doi: 10.1161/01.CIR.103.7.1000
213. Nie Q, Fan J, Haraoka S, Shimokama T, Watanabe T. Inhibition of mononuclear cell recruitment in aortic intima by treatment with anti-ICAM-1 and anti-LFA-1 monoclonal antibodies in hypercholesterolemic rats: implications of the ICAM-1 and LFA-1 pathway in atherogenesis. Lab Invest. (1997) 77:469–82.
214. Kubo N, Boisvert WA, Ballantyne CM, Curtiss LK. Leukocyte CD11b expression is not essential for the development of atherosclerosis in mice. J Lipid Res. (2000) 41:1060–6. doi: 10.1016/S0022-2275(20)32010-1
215. Merched A, Tollefson K, Chan L. Beta2 integrins modulate the initiation and progression of atherosclerosis in low-density lipoprotein receptor knockout mice. Cardiovasc Res. (2010) 85:853–63. doi: 10.1093/cvr/cvp347
216. van Buul JD, Allingham MJ, Samson T, Meller J, Boulter E, Garcia-Mata R, et al. RhoG regulates endothelial apical cup assembly downstream from ICAM1 engagement and is involved in leukocyte trans-endothelial migration. J Cell Biol. (2007) 178:1279–93. doi: 10.1083/jcb.200612053
217. Heemskerk N, van Rijssel J, van Buul JD. Rho-GTPase signaling in leukocyte extravasation: an endothelial point of view. Cell Adh Migr. (2014) 8:67–75. doi: 10.4161/cam.28244
218. Samson T, van Buul JD, Kroon J, Welch C, Bakker EN, Matlung HL, et al. The guanine-nucleotide exchange factor SGEF plays a crucial role in the formation of atherosclerosis. PLoS ONE. (2013) 8:e55202. doi: 10.1371/journal.pone.0055202
219. Mallat Z, Gojova A, Sauzeau V, Brun V, Silvestre JS, Esposito B, et al. Rho-associated protein kinase contributes to early atherosclerotic lesion formation in mice. Circ Res. (2003) 93:884–8. doi: 10.1161/01.RES.0000099062.55042.9A
220. Wu DJ, Xu JZ, Wu YJ, Jean-Charles L, Xiao B, Gao PJ, et al. Effects of fasudil on early atherosclerotic plaque formation and established lesion progression in apolipoprotein E-knockout mice. Atherosclerosis. (2009) 207:68–73. doi: 10.1016/j.atherosclerosis.2009.04.025
221. Klarin D, Zhu QM, Emdin CA, Chaffin M, Horner S, McMillan BJ, et al. Genetic analysis in UK Biobank links insulin resistance and transendothelial migration pathways to coronary artery disease. Nat Genet. (2017) 49:1392–7. doi: 10.1038/ng.3914
222. van der Harst P, Verweij N. Identification of 64 novel genetic loci provides an expanded view on the genetic architecture of coronary artery disease. Circ Res. (2018) 122:433–43. doi: 10.1161/CIRCRESAHA.117.312086
223. Verweij N, Eppinga RN, Hagemeijer Y, van der Harst P. Identification of 15 novel risk loci for coronary artery disease and genetic risk of recurrent events, atrial fibrillation and heart failure. Sci Rep. (2017) 7:2761. doi: 10.1038/s41598-017-03062-8
224. Juettner VV, Kruse K, Dan A, Vu VH, Khan Y, Le J, et al. VE-PTP stabilizes VE-cadherin junctions and the endothelial barrier via a phosphatase-independent mechanism. J Cell Biol. (2019) 218:1725–42. doi: 10.1083/jcb.201807210
225. Nottebaum AF, Cagna G, Winderlich M, Gamp AC, Linnepe R, Polaschegg C, et al. VE-PTP maintains the endothelial barrier via plakoglobin and becomes dissociated from VE-cadherin by leukocytes and by VEGF. J Exp Med. (2008) 205:2929–45. doi: 10.1084/jem.20080406
226. Hashimoto K, Kataoka N, Nakamura E, Tsujioka K, Kajiya F. Oxidized LDL specifically promotes the initiation of monocyte invasion during transendothelial migration with upregulated PECAM-1 and downregulated VE-cadherin on endothelial junctions. Atherosclerosis. (2007) 194:e9–17. doi: 10.1016/j.atherosclerosis.2006.11.029
227. Mamdouh Z, Kreitzer GE, Muller WA. Leukocyte transmigration requires kinesin-mediated microtubule-dependent membrane trafficking from the lateral border recycling compartment. J Exp Med. (2008) 205:951–66. doi: 10.1084/jem.20072328
228. Babinska A, Clement CC, Przygodzki T, Talar M, Li Y, Braun M, et al. A peptide antagonist of F11R/JAM-A reduces plaque formation and prolongs survival in an animal model of atherosclerosis. Atherosclerosis. (2019) 284:92–101. doi: 10.1016/j.atherosclerosis.2019.02.014
229. Schmitt MM, Megens RT, Zernecke A, Bidzhekov K, van den Akker NM, Rademakers T, et al. Endothelial junctional adhesion molecule-a guides monocytes into flow-dependent predilection sites of atherosclerosis. Circulation. (2014) 129:66–76. doi: 10.1161/CIRCULATIONAHA.113.004149
230. Curaj A, Wu Z, Rix A, Gresch O, Sternkopf M, Alampour-Rajabi S, et al. Molecular ultrasound imaging of junctional adhesion molecule a depicts acute alterations in blood flow and early endothelial dysregulation. Arterioscler Thromb Vasc Biol. (2018) 38:40–8. doi: 10.1161/ATVBAHA.117.309503
231. Schenkel AR, Mamdouh Z, Chen X, Liebman RM, Muller WA. CD99 plays a major role in the migration of monocytes through endothelial junctions. Nat Immunol. (2002) 3:143–50. doi: 10.1038/ni749
232. van Wanrooij EJ, de Vos P, Bixel MG, Vestweber D, van Berkel TJ, Kuiper J. Vaccination against CD99 inhibits atherogenesis in low-density lipoprotein receptor-deficient mice. Cardiovasc Res. (2008) 78:590–6. doi: 10.1093/cvr/cvn025
233. Swirski FK, Weissleder R, Pittet MJ. Heterogeneous in vivo behavior of monocyte subsets in atherosclerosis. Arterioscler Thromb Vasc Biol. (2009) 29:1424–32. doi: 10.1161/ATVBAHA.108.180521
234. Robbins CS, Hilgendorf I, Weber GF, Theurl I, Iwamoto Y, Figueiredo JL, et al. Local proliferation dominates lesional macrophage accumulation in atherosclerosis. Nat Med. (2013) 19:1166–72. doi: 10.1038/nm.3258
235. Williams JW, Zaitsev K, Kim KW, Ivanov S, Saunders BT, Schrank PR, et al. Limited proliferation capacity of aortic intima resident macrophages requires monocyte recruitment for atherosclerotic plaque progression. Nat Immunol. (2020) 21:1194–204. doi: 10.1038/s41590-020-0768-4
236. Binder CJ, Papac-Milicevic N, Witztum JL. Innate sensing of oxidation-specific epitopes in health and disease. Nat Rev Immunol. (2016) 16:485–97. doi: 10.1038/nri.2016.63
237. Bekkering S, Quintin J, Joosten LA, van der Meer JW, Netea MG, Riksen NP. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler Thromb Vasc Biol. (2014) 34:1731–8. doi: 10.1161/ATVBAHA.114.303887
238. Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. (2016) 354:472–7. doi: 10.1126/science.aaf6659
239. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. (2010). NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 464:1357–61. doi: 10.1038/nature0893
240. Spann NJ, Garmire LX, McDonald JG, Myers DS, Milne SB, Shibata N, et al. Regulated accumulation of desmosterol integrates macrophage lipid metabolism and inflammatory responses. Cell. (2012) 151:138–52. doi: 10.1016/j.cell.2012.06.054
241. Kim K, Shim D, Lee JS, Zaitsev K, Williams JW, Kim KW, et al. Transcriptome analysis reveals nonfoamy rather than foamy plaque macrophages are proinflammatory in atherosclerotic murine models. Circ Res. (2018) 123:1127–42. doi: 10.1161/CIRCRESAHA.118.312804
242. Kim KW, Ivanov S, Williams JW. Monocyte recruitment, specification, and function in atherosclerosis. Cells. (2020) 10:15. doi: 10.3390/cells10010015
243. Drechsler M, Megens RT, van Zandvoort M, Weber C, Soehnlein O. Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation. (2010) 122:1837–45. doi: 10.1161/CIRCULATIONAHA.110.961714
244. Silvestre-Roig C, Braster Q, Ortega-Gomez A, Soehnlein O. Neutrophils as regulators of cardiovascular inflammation. Nat Rev Cardiol. (2020) 17:327–40. doi: 10.1038/s41569-019-0326-7
245. Chistiakov DA, Grechko AV, Myasoedova VA, Melnichenko AA, Orekhov AN. The role of monocytosis and neutrophilia in atherosclerosis. J Cell Mol Med. (2018) 22:1366–82. doi: 10.1111/jcmm.13462
246. Guasti L, Dentali F, Castiglioni L, Maroni L, Marino F, Squizzato A, et al. Neutrophils and clinical outcomes in patients with acute coronary syndromes and/or cardiac revascularisation. A systematic review on more than 34:000 subjects. Thromb Haemost. (2011) 106:591–9. doi: 10.1160/TH11-02-0096
247. Human-Protein-Atlas. Human Protein Atlas. Available from: http://www.proteinatlas.org (accessed November 12, 2021).
248. Thul PJ, Akesson L, Wiking M, Mahdessian D, Geladaki A, Ait Blal H, et al. A subcellular map of the human proteome. Science. (2017) 356:6340. doi: 10.1126/science.aal3321
249. Döring Y, Drechsler M, Soehnlein O, Weber C. Neutrophils in atherosclerosis: from mice to man. Arterioscler Thromb Vasc Biol. (2015) 35:288–95. doi: 10.1161/ATVBAHA.114.303564
250. Copin JC, da Silva RF, Fraga-Silva RA, Capettini L, Quintao S, Lenglet S, et al. Treatment with Evasin-3 reduces atherosclerotic vulnerability for ischemic stroke, but not brain injury in mice. J Cereb Blood Flow Metab. (2013) 33:490–8. doi: 10.1038/jcbfm.2012.198
251. Nencioni A, da Silva RF, Fraga-Silva RA, Steffens S, Fabre M, Bauer I, et al. Nicotinamide phosphoribosyltransferase inhibition reduces intraplaque CXCL1 production and associated neutrophil infiltration in atherosclerotic mice. Thromb Haemost. (2014) 111:308–22. doi: 10.1160/TH13-07-0531
252. de Jager SC, Bot I, Kraaijeveld AO, Korporaal SJ, Bot M, van Santbrink PJ, et al. Leukocyte-specific CCL3 deficiency inhibits atherosclerotic lesion development by affecting neutrophil accumulation. Arterioscler Thromb Vasc Biol. (2013) 33:e75–83. doi: 10.1161/ATVBAHA.112.300857
253. Döring Y, Drechsler M, Wantha S, Kemmerich K, Lievens D, Vijayan S, et al. Lack of neutrophil-derived CRAMP reduces atherosclerosis in mice. Circ Res. (2012) 110:1052–6. doi: 10.1161/CIRCRESAHA.112.265868
254. Alard JE, Ortega-Gomez A, Wichapong K, Bongiovanni D, Horckmans M, Megens RT, et al. Recruitment of classical monocytes can be inhibited by disturbing heteromers of neutrophil HNP1 and platelet CCL5. Sci Transl Med. (2015) 7:317ra196. doi: 10.1126/scitranslmed.aad5330
255. Ortega-Gomez A, Salvermoser M, Rossaint J, Pick R, Brauner J, Lemnitzer P, et al. Cathepsin G controls arterial but not venular myeloid cell recruitment. Circulation. (2016) 134:1176–88. doi: 10.1161/CIRCULATIONAHA.116.024790
256. Schnoor M, Alcaide P, Voisin MB, van Buul JD. Crossing the vascular wall: common and unique mechanisms exploited by different leukocyte subsets during extravasation. Mediat Inflamm. (2015) 2015:946509. doi: 10.1155/2015/946509
257. Krolikoski M, Monslow J, Pure E. The CD44-HA axis and inflammation in atherosclerosis: a temporal perspective. Matrix Biol. (2019) 78–9, 201–18. doi: 10.1016/j.matbio.2018.05.007
258. Gjurich BN, Taghavie-Moghadam PL, Ley K, Galkina EV. L-selectin deficiency decreases aortic B1a and Breg subsets and promotes atherosclerosis. Thromb Haemost. (2014) 112:803–11. doi: 10.1160/TH13-10-0865
259. Rozenberg I, Sluka SH, Mocharla P, Hallenberg A, Rotzius P, Boren J, et al. Deletion of L-selectin increases atherosclerosis development in ApoE-/- mice. PLoS ONE. (2011) 6:e21675. doi: 10.1371/journal.pone.0021675
260. Chevre R, Gonzalez-Granado JM, Megens RT, Sreeramkumar V, Silvestre-Roig C, Molina-Sanchez P, et al. High-resolution imaging of intravascular atherogenic inflammation in live mice. Circ Res. (2014) 114:770–9. doi: 10.1161/CIRCRESAHA.114.302590
261. Sundd P, Gutierrez E, Koltsova EK, Kuwano Y, Fukuda S, Pospieszalska MK, et al. 'Slings' enable neutrophil rolling at high shear. Nature. (2012) 488:399–403. doi: 10.1038/nature11248
262. Wang S, Song R, Wang Z, Jing Z, Wang S, Ma J. S100A8/A9 in inflammation. Front Immunol. (2018) 9:1298. doi: 10.3389/fimmu.2018.01298
263. Pruenster M, Kurz AR, Chung KJ, Cao-Ehlker X, Bieber S, Nussbaum CF, et al. Extracellular MRP8/14 is a regulator of beta2 integrin-dependent neutrophil slow rolling and adhesion. Nat Commun. (2015) 6:6915. doi: 10.1038/ncomms7915
264. Viemann D, Strey A, Janning A, Jurk K, Klimmek K, Vogl T, et al. Myeloid-related proteins 8 and 14 induce a specific inflammatory response in human microvascular endothelial cells. Blood. (2005) 105:2955–62. doi: 10.1182/blood-2004-07-2520
265. Lefort CT, Ley K. Neutrophil arrest by LFA-1 activation. Front Immunol. (2012) 3:157. doi: 10.3389/fimmu.2012.00157
266. Halai K, Whiteford J, Ma B, Nourshargh S, Woodfin A. ICAM-2 facilitates luminal interactions between neutrophils and endothelial cells in vivo. J Cell Sci. (2014) 127:620–9. doi: 10.1242/jcs.137463
267. Filippi MD. Neutrophil transendothelial migration: updates and new perspectives. Blood. (2019) 133:2149–58. doi: 10.1182/blood-2018-12-844605
268. Record J, Malinova D, Zenner HL, Plagnol V, Nowak K, Syed F, et al. Immunodeficiency and severe susceptibility to bacterial infection associated with a loss-of-function homozygous mutation of MKL1. Blood. (2015) 126:1527–35. doi: 10.1182/blood-2014-12-611012
269. Sreeramkumar V, Adrover JM, Ballesteros I, Cuartero MI, Rossaint J, Bilbao I, et al. Neutrophils scan for activated platelets to initiate inflammation. Science. (2014) 346:1234–8. doi: 10.1126/science.1256478
270. Girbl T, Lenn T, Perez L, Rolas L, Barkaway A, Thiriot A, et al. Distinct compartmentalization of the chemokines CXCL1 and CXCL2 and the atypical receptor ACKR1 determine discrete stages of neutrophil diapedesis. Immunity. (2018) 49:1062–76 e6. doi: 10.1016/j.immuni.2018.09.018
271. Phillipson M, Kaur J, Colarusso P, Ballantyne CM, Kubes P. Endothelial domes encapsulate adherent neutrophils and minimize increases in vascular permeability in paracellular and transcellular emigration. PLoS ONE. (2008) 3:e1649. doi: 10.1371/journal.pone.0001649
272. Heemskerk N, Schimmel L, Oort C, van Rijssel J, Yin T, Ma B, et al. F-actin-rich contractile endothelial pores prevent vascular leakage during leukocyte diapedesis through local RhoA signalling. Nat Commun. (2016) 7:10493. doi: 10.1038/ncomms10493
273. Petri B, Kaur J, Long EM, Li H, Parsons SA, Butz S, et al. Endothelial LSP1 is involved in endothelial dome formation, minimizing vascular permeability changes during neutrophil transmigration in vivo. Blood. (2011) 117:942–52. doi: 10.1182/blood-2010-02-270561
274. Woodfin A, Voisin MB, Beyrau M, Colom B, Caille D, Diapouli FM, et al. The junctional adhesion molecule JAM-C regulates polarized transendothelial migration of neutrophils in vivo. Nat Immunol. (2011) 12:761–9. doi: 10.1038/ni.2062
275. Colom B, Bodkin JV, Beyrau M, Woodfin A, Ody C, Rourke C, et al. Leukotriene B4-neutrophil elastase axis drives neutrophil reverse transendothelial cell migration in vivo. Immunity. (2015) 42:1075–86. doi: 10.1016/j.immuni.2015.05.010
276. Kumar BV, Connors TJ, Farber DL. Human T cell development, localization, and function throughout life. Immunity. (2018) 48:202–13. doi: 10.1016/j.immuni.2018.01.007
277. Emeson EE, Shen ML, Bell CG, Qureshi A. Inhibition of atherosclerosis in CD4 T-cell-ablated and nude (nu/nu) C57BL/6 hyperlipidemic mice. Am J Pathol. (1996) 149:675–85.
278. Zhou X, Nicoletti A, Elhage R, Hansson GK. Transfer of CD4(+) T cells aggravates atherosclerosis in immunodeficient apolipoprotein E knockout mice. Circulation. (2000) 102:2919–22. doi: 10.1161/01.CIR.102.24.2919
279. Zhou X, Robertson AK, Hjerpe C, Hansson GK. Adoptive transfer of CD4+ T cells reactive to modified low-density lipoprotein aggravates atherosclerosis. Arterioscler Thromb Vasc Biol. (2006) 26:864–70. doi: 10.1161/01.ATV.0000206122.61591.ff
280. Zhou X, Robertson AK, Rudling M, Parini P, Hansson GK. Lesion development and response to immunization reveal a complex role for CD4 in atherosclerosis. Circ Res. (2005) 96:427–34. doi: 10.1161/01.RES.0000156889.22364.f1
281. Li J, Ley K. Lymphocyte migration into atherosclerotic plaque. Arterioscler Thromb Vasc Biol. (2015) 35:40–9. doi: 10.1161/ATVBAHA.114.303227
282. Zhu J, Paul WE. CD4 T cells: fates, functions, and faults. Blood. (2008) 112:1557–69. doi: 10.1182/blood-2008-05-078154
283. Frostegård J, Ulfgren AK, Nyberg P, Hedin U, Swedenborg J, Andersson U, et al. Cytokine expression in advanced human atherosclerotic plaques: dominance of pro-inflammatory (Th1) and macrophage-stimulating cytokines. Atherosclerosis. (1999) 145:33–43. doi: 10.1016/S0021-9150(99)00011-8
284. Buono C, Binder CJ, Stavrakis G, Witztum JL, Glimcher LH, Lichtman AH. T-bet deficiency reduces atherosclerosis and alters plaque antigen-specific immune responses. Proc Natl Acad Sci USA. (2005) 102:1596–601. doi: 10.1073/pnas.0409015102
285. Laurat E, Poirier B, Tupin E, Caligiuri G, Hansson GK, Bariety J, et al. In vivo downregulation of T helper cell 1 immune responses reduces atherogenesis in apolipoprotein E-knockout mice. Circulation. (2001) 104:197–202. doi: 10.1161/01.CIR.104.2.197
286. Saigusa R, Winkels H, Ley K. T cell subsets and functions in atherosclerosis. Nat Rev Cardiol. (2020) 17:387–401. doi: 10.1038/s41569-020-0352-5
287. Foks AC, Lichtman AH, Kuiper J. Treating atherosclerosis with regulatory T cells. Arterioscler Thromb Vasc Biol. (2015) 35:280–7. doi: 10.1161/ATVBAHA.114.303568
288. Proto JD, Doran AC, Gusarova G, Yurdagul A Jr, Sozen E, Subramanian M, et al. (2018) Regulatory T cells promote macrophage efferocytosis during inflammation resolution. Immunity. 49:666–77 e6. doi: 10.1016/j.immuni.2018.07.015
289. Ait-Oufella H, Salomon BL, Potteaux S, Robertson AK, Gourdy P, Zoll J, et al. Natural regulatory T cells control the development of atherosclerosis in mice. Nat Med. (2006) 12:178–80. doi: 10.1038/nm1343
290. Klingenberg R, Gerdes N, Badeau RM, Gistera A, Strodthoff D, Ketelhuth DF, et al. Depletion of FOXP3+ regulatory T cells promotes hypercholesterolemia and atherosclerosis. J Clin Invest. (2013) 123:1323–34. doi: 10.1172/JCI63891
291. van Es T, van Puijvelde GH, Foks AC, Habets KL, Bot I, Gilboa E, et al. Vaccination against Foxp3(+) regulatory T cells aggravates atherosclerosis. Atherosclerosis. (2010) 209:74–80. doi: 10.1016/j.atherosclerosis.2009.08.041
292. Ley K. Role of the adaptive immune system in atherosclerosis. Biochem Soc Trans. (2020) 48:2273–81. doi: 10.1042/BST20200602
293. Wolf D, Gerhardt T, Winkels H, Michel NA, Pramod AB, Ghosheh Y, et al. Pathogenic autoimmunity in atherosclerosis evolves from initially protective apolipoprotein B100-reactive CD4(+) T-regulatory cells. Circulation. (2020) 142:1279–93. doi: 10.1161/CIRCULATIONAHA.119.042863
294. Amersfoort J, Schaftenaar FH, Douna H, van Santbrink PJ, van Puijvelde GHM, Slütter B, et al. Diet-induced dyslipidemia induces metabolic and migratory adaptations in regulatory T cells. Cardiovasc Res. (2021) 117:1309–24. doi: 10.1093/cvr/cvaa208
295. Bergström I, Backteman K, Lundberg A, Ernerudh J, Jonasson L. Persistent accumulation of interferon-γ-producing CD8+CD56+ T cells in blood from patients with coronary artery disease. Atherosclerosis. (2012) 224:515–20. doi: 10.1016/j.atherosclerosis.2012.07.033
296. Hwang Y, Yu HT, Kim DH, Jang J, Kim HY, Kang I, et al. Expansion of CD8(+) T cells lacking the IL-6 receptor α chain in patients with coronary artery diseases (CAD). Atherosclerosis. (2016) 249:44–51. doi: 10.1016/j.atherosclerosis.2016.03.038
297. Cochain C, Koch M, Chaudhari SM, Busch M, Pelisek J, Boon L, et al. CD8+ T cells regulate monopoiesis and circulating Ly6C-high monocyte levels in atherosclerosis in mice. Circ Res. (2015) 117:244–53. doi: 10.1161/CIRCRESAHA.117.304611
298. Kyaw T, Winship A, Tay C, Kanellakis P, Hosseini H, Cao A, et al. Cytotoxic and proinflammatory CD8+ T lymphocytes promote development of vulnerable atherosclerotic plaques in apoE-deficient mice. Circulation. (2013) 127:1028–39. doi: 10.1161/CIRCULATIONAHA.112.001347
299. Seijkens TTP, Poels K, Meiler S, van Tiel CM, Kusters PJH, Reiche M, et al. Deficiency of the T cell regulator Casitas B-cell lymphoma-B aggravates atherosclerosis by inducing CD8+ T cell-mediated macrophage death. Eur Heart J. (2019) 40:372–82. doi: 10.1093/eurheartj/ehy714
300. van Duijn J, Kritikou E, Benne N, van der Heijden T, van Puijvelde GH, Kroner MJ, et al. CD8+ T-cells contribute to lesion stabilization in advanced atherosclerosis by limiting macrophage content and CD4+ T-cell responses. Cardiovasc Res. (2019) 115:729–38. doi: 10.1093/cvr/cvy261
301. Kimura T, Kobiyama K, Winkels H, Tse K, Miller J, Vassallo M, et al. Regulatory CD4(+) T cells recognize major histocompatibility complex class II molecule-restricted peptide epitopes of apolipoprotein B. Circulation. (2018) 138:1130–43. doi: 10.1161/CIRCULATIONAHA.117.031420
302. MacRitchie N, Grassia G, Noonan J, Cole JE, Hughes CE, Schroeder J, et al. The aorta can act as a site of naive CD4+ T-cell priming. Cardiovasc Res. (2020) 116:306–16. doi: 10.1093/cvr/cvz102
303. Hu D, Mohanta SK, Yin C, Peng L, Ma Z, Srikakulapu P, et al. Artery tertiary lymphoid organs control aorta immunity and protect against atherosclerosis via vascular smooth muscle cell lymphotoxin beta receptors. Immunity. (2015) 42:1100–15. doi: 10.1016/j.immuni.2015.05.015
304. Matsumoto M, Shigeta A, Miyasaka M, Hirata T. CD43 plays both antiadhesive and proadhesive roles in neutrophil rolling in a context-dependent manner. J Immunol. (2008) 181:3628–35. doi: 10.4049/jimmunol.181.5.3628
305. Nacher M, Blazquez AB, Shao B, Matesanz A, Prophete C, Berin MC, et al. Physiological contribution of CD44 as a ligand for E-Selectin during inflammatory T-cell recruitment. Am J Pathol. (2011) 178:2437–46. doi: 10.1016/j.ajpath.2011.01.039
306. Galkina E, Kadl A, Sanders J, Varughese D, Sarembock IJ, Ley K. Lymphocyte recruitment into the aortic wall before and during development of atherosclerosis is partially L-selectin dependent. J Exp Med. (2006) 203:1273–82. doi: 10.1084/jem.20052205
307. Li J, McArdle S, Gholami A, Kimura T, Wolf D, Gerhardt T, et al. CCR5+T-bet+FoxP3+ effector CD4 T cells drive atherosclerosis. Circ Res. (2016) 118:1540–52. doi: 10.1161/CIRCRESAHA.116.308648
308. Potteaux S, Combadiere C, Esposito B, Casanova S, Merval R, Ardouin P, et al. Chemokine receptor CCR1 disruption in bone marrow cells enhances atherosclerotic lesion development and inflammation in mice. Mol Med. (2005) 11:16–20. doi: 10.2119/2005-00028.Potteaux
309. Groom JR, Richmond J, Murooka TT, Sorensen EW, Sung JH, Bankert K, et al. CXCR3 chemokine receptor-ligand interactions in the lymph node optimize CD4+ T helper 1 cell differentiation. Immunity. (2012) 37:1091–103. doi: 10.1016/j.immuni.2012.08.016
310. Heller EA, Liu E, Tager AM, Yuan Q, Lin AY, Ahluwalia N, et al. Chemokine CXCL10 promotes atherogenesis by modulating the local balance of effector and regulatory T cells. Circulation. (2006) 113:2301–12. doi: 10.1161/CIRCULATIONAHA.105.605121
311. Moreno B, Hueso L, Ortega R, Benito E, Martinez-Hervas S, Peiro M, et al. Association of chemokines IP-10/CXCL10 and I-TAC/CXCL11 with insulin resistance and enhance leukocyte endothelial arrest in obesity. Microvasc Res. (2021) 139:104254. doi: 10.1016/j.mvr.2021.104254
312. van Wanrooij EJ, de Jager SC, van Es T, de Vos P, Birch HL, Owen DA, et al. CXCR3 antagonist NBI-74330 attenuates atherosclerotic plaque formation in LDL receptor-deficient mice. Arterioscler Thromb Vasc Biol. (2008) 28:251–7. doi: 10.1161/ATVBAHA.107.147827
313. Veillard NR, Steffens S, Pelli G, Lu B, Kwak BR, Gerard C, et al. Differential influence of chemokine receptors CCR2 and CXCR3 in development of atherosclerosis in vivo. Circulation. (2005) 112:870–8. doi: 10.1161/CIRCULATIONAHA.104.520718
314. Kim CH, Kunkel EJ, Boisvert J, Johnston B, Campbell JJ, Genovese MC, et al. Bonzo/CXCR6 expression defines type 1-polarized T-cell subsets with extralymphoid tissue homing potential. J Clin Invest. (2001) 107:595–601. doi: 10.1172/JCI11902
315. Galkina E, Harry BL, Ludwig A, Liehn EA, Sanders JM, Bruce A, et al. CXCR6 promotes atherosclerosis by supporting T-cell homing, interferon-gamma production, and macrophage accumulation in the aortic wall. Circulation. (2007) 116:1801–11. doi: 10.1161/CIRCULATIONAHA.106.678474
316. Wuttge DM, Zhou X, Sheikine Y, Wågsäter D, Stemme V, Hedin U, et al. CXCL16/SR-PSOX is an interferon-gamma-regulated chemokine and scavenger receptor expressed in atherosclerotic lesions. Arterioscler Thromb Vasc Biol. (2004) 24:750–5. doi: 10.1161/01.ATV.0000124102.11472.36
317. Aslanian AM, Charo IF. Targeted disruption of the scavenger receptor and chemokine CXCL16 accelerates atherosclerosis. Circulation. (2006) 114:583–90. doi: 10.1161/CIRCULATIONAHA.105.540583
318. Damås JK, Smith C, Øie E, Fevang B, Halvorsen B, Waehre T, et al. Enhanced expression of the homeostatic chemokines CCL19 and CCL21 in clinical and experimental atherosclerosis: possible pathogenic role in plaque destabilization. Arterioscler Thromb Vasc Biol. (2007) 27:614–20. doi: 10.1161/01.ATV.0000255581.38523.7c
319. Shulman Z, Cohen SJ, Roediger B, Kalchenko V, Jain R, Grabovsky V, et al. Transendothelial migration of lymphocytes mediated by intraendothelial vesicle stores rather than by extracellular chemokine depots. Nat Immunol. (2012) 13:67–76. doi: 10.1038/ni.2173
320. Pober JS, Merola J, Liu R, Manes TD. Antigen presentation by vascular cells. Front Immunol. (2017) 8:1907. doi: 10.3389/fimmu.2017.01907
321. Azcutia V, Routledge M, Williams MR, Newton G, Frazier WA, Manica A, et al. CD47 plays a critical role in T-cell recruitment by regulation of LFA-1 and VLA-4 integrin adhesive functions. Mol Biol Cell. (2013) 24:3358–68. doi: 10.1091/mbc.e13-01-0063
322. Shulman Z, Shinder V, Klein E, Grabovsky V, Yeger O, Geron E, et al. Lymphocyte crawling and transendothelial migration require chemokine triggering of high-affinity LFA-1 integrin. Immunity. (2009) 30:384–96. doi: 10.1016/j.immuni.2008.12.020
323. Carman CV, Sage PT, Sciuto TE, de la Fuente MA, Geha RS, Ochs HD, et al. Transcellular diapedesis is initiated by invasive podosomes. Immunity. (2007) 26:784–97. doi: 10.1016/j.immuni.2007.04.015
324. Dupré L, Houmadi R, Tang C, Rey-Barroso J. T lymphocyte migration: an action movie starring the actin and associated actors. Front Immunol. (2015) 6:586. doi: 10.3389/fimmu.2015.00586
325. Lee J, Song KH, Kim T, Doh J. Endothelial cell focal adhesion regulates transendothelial migration and subendothelial crawling of T cells. Front Immunol. (2018) 9:48. doi: 10.3389/fimmu.2018.00048
326. Vockel M, Vestweber D. How T cells trigger the dissociation of the endothelial receptor phosphatase VE-PTP from VE-cadherin. Blood. (2013) 122:2512–22. doi: 10.1182/blood-2013-04-499228
327. Manes TD, Pober JS. Antigen presentation by human microvascular endothelial cells triggers ICAM-1-dependent transendothelial protrusion by, and fractalkine-dependent transendothelial migration of, effector memory CD4+ T cells. J Immunol. (2008) 180:8386–92. doi: 10.4049/jimmunol.180.12.8386
328. Rana A, Westein E, Niego B, Hagemeyer CE. Shear-dependent platelet aggregation: mechanisms and therapeutic opportunities. Front Cardiovasc Med. (2019) 6:141. doi: 10.3389/fcvm.2019.00141
329. Wang L, Tang C. Targeting platelet in atherosclerosis plaque formation: current knowledge and future perspectives. Int J Mol Sci. (2020) 21:9760. doi: 10.3390/ijms21249760
330. Murphy AJ, Bijl N, Yvan-Charvet L, Welch CB, Bhagwat N, Reheman A, et al. Cholesterol efflux in megakaryocyte progenitors suppresses platelet production and thrombocytosis. Nat Med. (2013) 19:586–94. doi: 10.1038/nm.3150
331. Magwenzi S, Woodward C, Wraith KS, Aburima A, Raslan Z, Jones H, et al. Oxidized LDL activates blood platelets through CD36/NOX2-mediated inhibition of the cGMP/protein kinase G signaling cascade. Blood. (2015) 125:2693–703. doi: 10.1182/blood-2014-05-574491
332. Bakogiannis C, Sachse M, Stamatelopoulos K, Stellos K. Platelet-derived chemokines in inflammation and atherosclerosis. Cytokine. (2019) 122:154157. doi: 10.1016/j.cyto.2017.09.013
333. Shevchuk O, Begonja AJ, Gambaryan S, Totzeck M, Rassaf T, Huber TB, et al. Proteomics: a tool to study platelet function. Int J Mol Sci. (2021) 22:4776. doi: 10.3390/ijms22094776
334. Massberg S, Brand K, Gruner S, Page S, Muller E, Muller I, et al. A critical role of platelet adhesion in the initiation of atherosclerotic lesion formation. J Exp Med. (2002) 196:887–96. doi: 10.1084/jem.20012044
335. Barrett TJ, Schlegel M, Zhou F, Gorenchtein M, Bolstorff J, Moore KJ, et al. Platelet regulation of myeloid suppressor of cytokine signaling 3 accelerates atherosclerosis. Sci Transl Med. (2019) 11:481. doi: 10.1126/scitranslmed.aax0481
336. Allen N, Barrett TJ, Guo Y, Nardi M, Ramkhelawon B, Rockman CB, et al. Circulating monocyte-platelet aggregates are a robust marker of platelet activity in cardiovascular disease. Atherosclerosis. (2019) 282:11–8. doi: 10.1016/j.atherosclerosis.2018.12.029
337. Czepluch FS, Kuschicke H, Dellas C, Riggert J, Hasenfuss G, Schafer K. Increased proatherogenic monocyte-platelet cross-talk in monocyte subpopulations of patients with stable coronary artery disease. J Intern Med. (2014) 275:144–54. doi: 10.1111/joim.12145
338. Coenen DM, Mastenbroek TG, Cosemans J. Platelet interaction with activated endothelium: mechanistic insights from microfluidics. Blood. (2017) 130:2819–28. doi: 10.1182/blood-2017-04-780825
339. Meza D, Shanmugavelayudam SK, Mendoza A, Sanchez C, Rubenstein DA, Yin W. Platelets modulate endothelial cell response to dynamic shear stress through PECAM-1. Thromb Res. (2017) 150:44–50. doi: 10.1016/j.thromres.2016.12.003
340. Santoso S, Sachs UJ, Kroll H, Linder M, Ruf A, Preissner KT, et al. The junctional adhesion molecule 3 (JAM-3) on human platelets is a counterreceptor for the leukocyte integrin Mac-1. J Exp Med. (2002) 196:679–91. doi: 10.1084/jem.20020267
341. Wang Y, Sakuma M, Chen Z, Ustinov V, Shi C, Croce K, et al. Leukocyte engagement of platelet glycoprotein Ibalpha via the integrin Mac-1 is critical for the biological response to vascular injury. Circulation. (2005) 112:2993–3000. doi: 10.1161/CIRCULATIONAHA.105.571315
342. Zhao Z, Vajen T, Karshovska E, Dickhout A, Schmitt MM, Megens RTA, et al. Deletion of junctional adhesion molecule A from platelets increases early-stage neointima formation after wire injury in hyperlipidemic mice. J Cell Mol Med. (2017) 21:1523–31. doi: 10.1111/jcmm.13083
343. Naik MU, Stalker TJ, Brass LF, Naik UP. JAM-A protects from thrombosis by suppressing integrin alphaIIbbeta3-dependent outside-in signaling in platelets. Blood. (2012) 119:3352–60. doi: 10.1182/blood-2011-12-397398
344. Karshovska E, Zhao Z, Blanchet X, Schmitt MM, Bidzhekov K, Soehnlein O, et al. Hyperreactivity of junctional adhesion molecule A-deficient platelets accelerates atherosclerosis in hyperlipidemic mice. Circ Res. (2015) 116:587–99. doi: 10.1161/CIRCRESAHA.116.304035
345. Tersteeg C, Heijnen HF, Eckly A, Pasterkamp G, Urbanus RT, Maas C, et al. FLow-induced PRotrusions (FLIPRs): a platelet-derived platform for the retrieval of microparticles by monocytes and neutrophils. Circ Res. (2014) 114:780–91. doi: 10.1161/CIRCRESAHA.114.302361
346. Skilbeck CA, Walker PG, David T, Nash GB. Disturbed flow promotes deposition of leucocytes from flowing whole blood in a model of a damaged vessel wall. Br J Haematol. (2004) 126:418–27. doi: 10.1111/j.1365-2141.2004.05057.x
347. von Hundelshausen P, Weber KS, Huo Y, Proudfoot AE, Nelson PJ, Ley K, et al. RANTES deposition by platelets triggers monocyte arrest on inflamed and atherosclerotic endothelium. Circulation. (2001) 103:1772–7. doi: 10.1161/01.CIR.103.13.1772
348. Braunersreuther V, Steffens S, Arnaud C, Pelli G, Burger F, Proudfoot A, et al. A novel RANTES antagonist prevents progression of established atherosclerotic lesions in mice. Arterioscler Thromb Vasc Biol. (2008) 28:1090–6. doi: 10.1161/ATVBAHA.108.165423
349. Schober A, Manka D, von Hundelshausen P, Huo Y, Hanrath P, Sarembock IJ, et al. Deposition of platelet RANTES triggering monocyte recruitment requires P-selectin and is involved in neointima formation after arterial injury. Circulation. (2002) 106:1523–9. doi: 10.1161/01.CIR.0000028590.02477.6F
350. Veillard NR, Kwak B, Pelli G, Mulhaupt F, James RW, Proudfoot AE, et al. Antagonism of RANTES receptors reduces atherosclerotic plaque formation in mice. Circ Res. (2004) 94:253–61. doi: 10.1161/01.RES.0000109793.17591.4E
351. von Hundelshausen P, Koenen RR, Sack M, Mause SF, Adriaens W, Proudfoot AE, et al. Heterophilic interactions of platelet factor 4 and RANTES promote monocyte arrest on endothelium. Blood. (2005) 105:924–30. doi: 10.1182/blood-2004-06-2475
352. Koenen RR, von Hundelshausen P, Nesmelova IV, Zernecke A, Liehn EA, Sarabi A, et al. Disrupting functional interactions between platelet chemokines inhibits atherosclerosis in hyperlipidemic mice. Nat Med. (2009) 15:97–103. doi: 10.1038/nm.1898
353. Pitsilos S, Hunt J, Mohler ER, Prabhakar AM, Poncz M, Dawicki J, et al. Platelet factor 4 localization in carotid atherosclerotic plaques: correlation with clinical parameters. Thromb Haemost. (2003) 90:1112–20. doi: 10.1160/TH03-02-0069
354. Baidzajevas K, Hadadi E, Lee B, Lum J, Shihui F, Sudbery I, et al. Macrophage polarisation associated with atherosclerosis differentially affects their capacity to handle lipids. Atherosclerosis. (2020) 305:10–8. doi: 10.1016/j.atherosclerosis.2020.05.003
355. Nassar T, Sachais BS, Akkawi S, Kowalska MA, Bdeir K, Leitersdorf E, et al. Platelet factor 4 enhances the binding of oxidized low-density lipoprotein to vascular wall cells. J Biol Chem. (2003) 278:6187–93. doi: 10.1074/jbc.M208894200
356. Gleissner CA, Shaked I, Erbel C, Bockler D, Katus HA, Ley K. CXCL4 downregulates the atheroprotective hemoglobin receptor CD163 in human macrophages. Circ Res. (2010) 106:203–11. doi: 10.1161/CIRCRESAHA.109.199505
357. Erbel C, Tyka M, Helmes CM, Akhavanpoor M, Rupp G, Domschke G, et al. CXCL4-induced plaque macrophages can be specifically identified by co-expression of MMP7+S100A8+ in vitro and in vivo. Innate Immun. (2015) 21:255–65. doi: 10.1177/1753425914526461
358. Scheuerer B, Ernst M, Durrbaum-Landmann I, Fleischer J, Grage-Griebenow E, Brandt E, et al. The CXC-chemokine platelet factor 4 promotes monocyte survival and induces monocyte differentiation into macrophages. Blood. (2000) 95:1158–66. doi: 10.1182/blood.V95.4.1158.004k31_1158_1166
359. Sachais BS, Turrentine T, Dawicki McKenna JM, Rux AH, Rader D, Kowalska MA. Elimination of platelet factor 4 (PF4) from platelets reduces atherosclerosis in C57Bl/6 and apoE-/- mice. Thromb Haemost. (2007) 98:1108–13. doi: 10.1160/TH07-04-0271
360. Samani NJ, Erdmann J, Hall AS, Hengstenberg C, Mangino M, Mayer B, et al. Genomewide association analysis of coronary artery disease. N Engl J Med. (2007) 357:443–53. doi: 10.1056/NEJMoa072366
361. Schunkert H, Konig IR, Kathiresan S, Reilly MP, Assimes TL, Holm H, et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat Genet. (2011) 43:333–8. doi: 10.1038/ng.784
362. Ma W, Liu Y, Ellison N, Shen J. Induction of C-X-C chemokine receptor type 7 (CXCR7) switches stromal cell-derived factor-1 (SDF-1) signaling and phagocytic activity in macrophages linked to atherosclerosis. J Biol Chem. (2013) 288:15481–94. doi: 10.1074/jbc.M112.445510
363. Chatterjee M, von Ungern-Sternberg SN, Seizer P, Schlegel F, Buttcher M, Sindhu NA, et al. Platelet-derived CXCL12 regulates monocyte function, survival, differentiation into macrophages and foam cells through differential involvement of CXCR4-CXCR7. Cell Death Dis. (2015) 6:e1989. doi: 10.1038/cddis.2015.233
364. Karim ZA, Alshbool FZ, Vemana HP, Conlon C, Druey KM, Khasawneh FT. CXCL12 regulates platelet activation via the regulator of G-protein signaling 16. Biochim Biophys Acta. (2016) 1863:314–21. doi: 10.1016/j.bbamcr.2015.11.028
365. Walsh TG, Harper MT, Poole AW. SDF-1alpha is a novel autocrine activator of platelets operating through its receptor CXCR4. Cell Signal. (2015) 27:37–46. doi: 10.1016/j.cellsig.2014.09.021
366. Döring Y, van der Vorst EPC, Duchene J, Jansen Y, Gencer S, Bidzhekov K, et al. CXCL12 derived from endothelial cells promotes atherosclerosis to drive coronary artery disease. Circulation. (2019) 139:1338–40. doi: 10.1161/CIRCULATIONAHA.118.037953
367. Gao JH, He LH, Yu XH, Zhao ZW, Wang G, Zou J, et al. CXCL12 promotes atherosclerosis by downregulating ABCA1 expression via the CXCR4/GSK3beta/beta-catenin(T120)/TCF21 pathway. J Lipid Res. (2019) 60:2020–33. doi: 10.1194/jlr.RA119000100
368. Rath D, Chatterjee M, Bongartz A, Muller K, Droppa M, Stimpfle F, et al. Platelet surface expression of SDF-1 is associated with clinical outcomes in the patients with cardiovascular disease. Platelets. (2017) 28:34–9. doi: 10.1080/09537104.2016.1203399
369. Eriksson EE. Intravital microscopy on atherosclerosis in apolipoprotein e-deficient mice establishes microvessels as major entry pathways for leukocytes to advanced lesions. Circulation. (2011) 124:2129–38. doi: 10.1161/CIRCULATIONAHA.111.030627
370. Visseren FLJ, Mach F, Smulders YM, Carballo D, Koskinas KC, Back M, et al. 2021 ESC Guidelines on cardiovascular disease prevention in clinical practice. Eur Heart J. (2021) 42:3227–337. doi: 10.1093/eurheartj/ehab484
371. Baigent C, Blackwell L, Emberson J, Holland LE, Reith C, Bhala N, et al. Efficacy and safety of more intensive lowering of LDL cholesterol: a meta-analysis of data from 170,000 participants in 26 randomised trials. Lancet. (2010) 376:1670–81. doi: 10.1016/S0140-6736(10)61350-5
372. Cannon CP, Blazing MA, Giugliano RP, McCagg A, White JA, Theroux P, et al. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. (2015) 372:2387–97. doi: 10.1056/NEJMoa1410489
373. Wong ND, Shapiro MD. Interpreting the findings from the recent PCSK9 monoclonal antibody cardiovascular outcomes trials. Front Cardiovasc Med. (2019) 6:14. doi: 10.3389/fcvm.2019.00014
374. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
375. Hettwer J, Hinterdobler J, Miritsch B, Deutsch MA, Li X, Mauersberger C, et al. Interleukin-1beta suppression dampens inflammatory leukocyte production and uptake in atherosclerosis. Cardiovasc Res. (2021) doi: 10.1093/cvr/cvab337 [Online ahead of print].
376. Nidorf SM, Fiolet ATL, Mosterd A, Eikelboom JW, Schut A, Opstal TSJ, et al. Colchicine in patients with chronic coronary disease. N Engl J Med. (2020) 383:1838–47. doi: 10.1056/NEJMoa2021372
377. Fiolet ATL, Opstal TSJ, Mosterd A, Eikelboom JW, Jolly SS, Keech AC, et al. Efficacy and safety of low-dose colchicine in patients with coronary disease: a systematic review and meta-analysis of randomized trials. Eur Heart J. (2021) 42:2765–75. doi: 10.1093/eurheartj/ehab115
378. Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr., Kastelein JJ, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med. (2008) 359:2195–207. doi: 10.1056/NEJMoa0807646
379. Brili S, Tousoulis D, Antonopoulos AS, Antoniades C, Hatzis G, Bakogiannis C, et al. Effects of atorvastatin on endothelial function and the expression of proinflammatory cytokines and adhesion molecules in young subjects with successfully repaired coarctation of aorta. Heart. (2012) 98:325–9. doi: 10.1136/heartjnl-2011-300287
380. Huo Z, Kong Y, Meng M, Cao Z, Zhou Q. Atorvastatin enhances endothelial adherens junctions through promoting VE-PTP gene transcription and reducing VE-cadherin-Y731 phosphorylation. Vascul Pharmacol. (2019) 117:7–14. doi: 10.1016/j.vph.2018.06.003
381. Wagner AH, Kohler T, Ruckschloss U, Just I, Hecker M. Improvement of nitric oxide-dependent vasodilatation by HMG-CoA reductase inhibitors through attenuation of endothelial superoxide anion formation. Arterioscler Thromb Vasc Biol. (2000) 20:61–9. doi: 10.1161/01.ATV.20.1.61
382. Baganha F, de Jong RCM, Peters EA, Voorham W, Jukema JW, Delibegovic M, et al. Atorvastatin pleiotropically decreases intraplaque angiogenesis and intraplaque haemorrhage by inhibiting ANGPT2 release and VE-Cadherin internalization. Angiogenesis. (2021) 24:567–81. doi: 10.1007/s10456-021-09767-9
383. Chiang KH, Cheng WL, Shih CM, Lin YW, Tsao NW, Kao YT, et al. Statins, HMG-CoA reductase inhibitors, improve neovascularization by increasing the expression density of CXCR4 in endothelial progenitor cells. PLoS ONE. (2015) 10:e0136405. doi: 10.1371/journal.pone.0136405
384. Weitz-Schmidt G, Welzenbach K, Brinkmann V, Kamata T, Kallen J, Bruns C, et al. Statins selectively inhibit leukocyte function antigen-1 by binding to a novel regulatory integrin site. Nat Med. (2001) 7:687–692. doi: 10.1038/89058
385. Goetzl EJ, Goetzl L, Karliner JS, Tang N, Pulliam L. Human plasma platelet-derived exosomes: effects of aspirin. FASEB J. (2016) 30:2058–63. doi: 10.1096/fj.201500150R
386. McKenzie ME, Malinin AI, Bell CR, Dzhanashvili A, Horowitz ED, Oshrine BR, et al. Aspirin inhibits surface glycoprotein IIb/IIIa, P-selectin, CD63, and CD107a receptor expression on human platelets. Blood Coagul Fibrinolysis. (2003) 14:249–53. doi: 10.1097/01.mbc.0000046182.72384.ab
387. Heitzer T, Rudolph V, Schwedhelm E, Karstens M, Sydow K, Ortak M, et al. Clopidogrel improves systemic endothelial nitric oxide bioavailability in patients with coronary artery disease: evidence for antioxidant and antiinflammatory effects. Arterioscler Thromb Vasc Biol. (2006) 26:1648–52. doi: 10.1161/01.ATV.0000225288.74170.dc
388. Christersson C, Johnell M, Siegbahn A. The influence of direct thrombin inhibitors on the formation of platelet-leukocyte aggregates and tissue factor expression. Thromb Res. (2010) 126:e327–33. doi: 10.1016/j.thromres.2010.03.019
389. Pingel S, Tiyerili V, Mueller J, Werner N, Nickenig G, Mueller C. Thrombin inhibition by dabigatran attenuates atherosclerosis in ApoE deficient mice. Arch Med Sci. (2014) 10:154–60. doi: 10.5114/aoms.2014.40742
390. McMurray JJV, Solomon SD, Inzucchi SE, Køber L, Kosiborod MN, Martinez FA, et al. Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med. (2019) 381:1995–2008. doi: 10.1056/NEJMoa1911303
391. Packer M, Anker SD, Butler J, Filippatos G, Pocock SJ, Carson P, et al. Cardiovascular and renal outcomes with empagliflozin in heart failure. N Engl J Med. (2020) 383:1413–24. doi: 10.1056/NEJMoa2022190
392. Kim SR, Lee SG, Kim SH, Kim JH, Choi E, Cho W, et al. SGLT2 inhibition modulates NLRP3 inflammasome activity via ketones and insulin in diabetes with cardiovascular disease. Nat Commun. (2020) 11:2127. doi: 10.1038/s41467-020-15983-6
393. Nohria A, Grunert ME, Rikitake Y, Noma K, Prsic A, Ganz P, et al. Rho kinase inhibition improves endothelial function in human subjects with coronary artery disease. Circ Res. (2006) 99:1426–32. doi: 10.1161/01.RES.0000251668.39526.c7
394. Joseph JP, Reyes E, Guzman J, O'Doherty J, McConkey H, Arri S, et al. CXCR2 Inhibition - a novel approach to treating CoronAry heart DiseAse (CICADA): study protocol for a randomised controlled trial. Trials. (2017) 18:473. doi: 10.1186/s13063-017-2210-2
395. Cipriani S, Francisci D, Mencarelli A, Renga B, Schiaroli E, D'Amore C, et al. Efficacy of the CCR5 antagonist maraviroc in reducing early, ritonavir-induced atherogenesis and advanced plaque progression in mice. Circulation. (2013) 127:2114–24. doi: 10.1161/CIRCULATIONAHA.113.001278
396. Francisci D, Pirro M, Schiaroli E, Mannarino MR, Cipriani S, Bianconi V, et al. Maraviroc intensification modulates atherosclerotic progression in HIV-suppressed patients at high cardiovascular risk. A randomized, crossover pilot study. Open Forum Infect Dis. (2019) 6:ofz112. doi: 10.1093/ofid/ofz112
397. Caligiuri G. CD31 as a therapeutic target in atherosclerosis. Circ Res. (2020) 126:1178–89. doi: 10.1161/CIRCRESAHA.120.315935
Keywords: atherosclerosis, vascular inflammation, leukocyte recruitment, adhesion molecules, integrin, transendothelial migration
Citation: Mauersberger C, Hinterdobler J, Schunkert H, Kessler T and Sager HB (2022) Where the Action Is—Leukocyte Recruitment in Atherosclerosis. Front. Cardiovasc. Med. 8:813984. doi: 10.3389/fcvm.2021.813984
Received: 12 November 2021; Accepted: 15 December 2021;
Published: 11 January 2022.
Edited by:
Holger Winkels, Universitätsklinikum Köln, GermanyReviewed by:
Jesse W. Williams, University of Minnesota, United StatesKlaus Ley, La Jolla Institute for Immunology (LJI), United States
Anton Gisterå, Karolinska Institutet (KI), Sweden
Copyright © 2022 Mauersberger, Hinterdobler, Schunkert, Kessler and Sager. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hendrik B. Sager, aGVuZHJpay5zYWdlckB0dW0uZGU=