
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med. , 18 January 2022
Sec. Cardiovascular Biologics and Regenerative Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.810241
This article is part of the Research Topic MicroRNAs in Heart Regeneration: Regulatory mechanisms and therapeutic applications View all 6 articles
 Evelyn Gabriela Rusu-Nastase
Evelyn Gabriela Rusu-Nastase Ana-Mihaela Lupan
Ana-Mihaela Lupan Catalina Iolanda Marinescu
Catalina Iolanda Marinescu Carmen Alexandra Neculachi
Carmen Alexandra Neculachi Mihai Bogdan Preda
Mihai Bogdan Preda Alexandrina Burlacu*
Alexandrina Burlacu*Deregulation of microRNA (miRNA) profile has been reportedly linked to the aging process, which is a dominant risk factor for many pathologies. Among the miRNAs with documented roles in aging-related cardiac diseases, miR-18a, -21a, -22, and -29a were mainly associated with hypertrophy and/or fibrosis; however, their relationship to aging was not fully addressed before. The purpose of this paper was to evaluate the variations in the expression levels of these miRNAs in the aging process. To this aim, multiple organs were harvested from young (2–3-months-old), old (16–18-months-old), and very old (24–25-months-old) mice, and the abundance of the miRNAs was evaluated by quantitative real-time (RT)-PCR. Our studies demonstrated that miR-21a, miR-22, and miR-29a were upregulated in the aged heart. Among them, miR-29a was highly expressed in many other organs, i.e., the brain, the skeletal muscle, the pancreas, and the kidney, and its expression was further upregulated during the natural aging process. Western blot, immunofluorescence, and xCELLigence analyses concurrently indicated that overexpression of miR-29a in the muscle cells decreased the collagen levels as well as cell migration and proliferation. Computational prediction analysis and overexpression studies identified SERPINH1, a specific chaperone of procollagens, as a potential miR-29a target. Corroborating to this, significantly downregulated SERPINH1 levels were found in the skeletal muscle, the heart, the brain, the kidney, and the pancreas harvested from very old animals, thereby indicating the role of the miR-29a-SERPINH1 axis in the aging process. In vitro analysis of miR-29a effects on fibroblast and cardiac muscle cells pointed toward a protective role of miR-29a on aging-related fibrosis, by reducing cell migration and proliferation. In conclusion, our study indicates an adaptive increase of miR-29 in the natural aging process and suggests its role as a transcriptional repressor of SERPINH1, with a potential therapeutic value against adverse matrix remodeling and aging-associated tissue fibrosis.
The persistent increase in human longevity has stimulated the interest of scientists in understanding the fundamental mechanisms underlying natural aging, which is the dominant risk factor for many pathologies, including cardiovascular diseases (1). Aging is a progressive process characterized by gradual changes and alterations accumulated in the tissues and organs, which eventually results in the reduced ability of the organism to meet increased demands (2, 3). The alterations associated with the aging process can be grouped into several hallmarks of aging, such as genomic instability, telomere attrition, epigenetic alterations, loss of protein homeostasis, deregulated nutrient sensing, mitochondrial dysfunction, cellular senescence, stem cell exhaustion, and altered intercellular communication (4). Thus, during aging, the accumulation of somatic mutations in the nuclear DNA may affect the essential genes and transcriptional pathways that may compromise tissue homeostasis (4). Besides, natural aging is associated with telomere attrition, which may promote the development of degenerative defects, such as pulmonary fibrosis or dyskeratosis congenita (5, 6). Aging is also associated with increased production of reactive oxygen species, which are known to induce damages in mitochondrial DNA and redox-sensitive mitochondrial proteins, and to promote mitochondrial dysfunction, thus leading to heart failure and other pathologies (7). All these hallmarks are interwoven and promote the gradual loss of physiological integrity and cell function, and eventually lead to the death of the organism.
During aging, the cardiac tissue architecture deteriorates, leading to a gradual decline in the cardiac reserves, a major pathophysiological feature of the heart in old individuals (8). Cardiac fibrosis and hypertrophy are two important hallmarks of an aged heart and they are tightly interwoven and mutually triggered by each other. Thus, the baseline activation of fibrotic pathways in the aged heart is followed by defective scar formation and left ventricular hypertrophy, making the heart prone to adverse dilative remodeling. Consequently, cardiac aging is associated with diastolic dysfunction and heart failure with preserved systolic function, which has significant impacts on the elderly population. Dissecting the mechanisms of interstitial fibrosis and cardiomyocyte hypertrophy in the aging heart is therefore critical in order to inspire new approaches for the prevention of adverse remodeling and heart failure in old patients.
MicroRNAs (miRNAs) are a class of small non-coding RNAs that regulate gene expression at the posttranscriptional level and together with other non-coding RNA molecules, are powerful regulators of a wide range of biological mechanisms in many disorders (9, 10). Notably, numerous basic and clinical studies have connected miRNA to the initiation and/or progression of hypertrophy and fibrosis (11, 12), which make them exciting potential therapeutic targets in these conditions.
One of the most influencing miRNAs in the cardiovascular system is miR-21, which is the major miRNA in cardiac fibroblasts, where it accounts for around 20% of total miRNAs (13). MiR-21 was found increased in the naturally aged murine hearts (14), as well as in plasma samples from centenarians (15). Besides, various animal models of cardiac fibrosis (16), hypertrophic heart (17), and heart failure (18) identified the presence of miR-21 in increased amounts in comparison to control groups, being related to cardiac dysfunction. This miRNA has attracted attention for its diverse effects in cardiac function (19) and was extensively reported to exert both protective and harmful effects, being involved in different cell processes, such as cell growth and proliferation, mitochondrial damage, chemosensitivity, response to stress, and cell invasion (15). In accordance to this, the dysregulation of miR-21 occurred in diverse pathologies, like cancer and metabolic disorders (20, 21).
MiR-17-92 cluster, which consists of six mature miRNAs (miR-17, miR-18a, miR-19a, miR-19b, miR-20a, and miR-92a), has also been involved in cardiac aging. Within this cluster, miR-18a was reported to regulate the expression of several extracellular matrix proteins (22) and its decreased expression in heart failure in the aged individuals was linked to aging-induced heart remodeling. Corroborating to this, downregulation of miR-18a was associated with increased fibrosis and a decline in heart function (23), while over-expression of miR-18a reduced fibrosis, hypertrophy, and the apoptosis of cardiomyocytes in heart failure (24).
Another miRNA involved in cardiac aging is miR-22. Previous studies reported a prohypertrophic effect of miR-22, with a critical role in cardiac remodeling (25–27). This miRNA was found increased in aging hearts and this upregulation was linked to accelerated senescence and increased migration of cardiac fibroblasts (28). Corroborating to this, the inhibition of miR-22 stimulated cardiac autophagy and enhanced cardiac function in older mice with myocardial infarction (29).
Another miRNA with a prominent role in cardiac hypertrophy and fibrosis is miR-29a, which is the most stable and most abundantly expressed member of its family (30). MiR-29 is an essential regulator of extracellular matrix proteins in pathways related to fibrosis (31, 32) and has been reported to be dramatically downregulated in the region of the fibrotic scar after myocardial infarction (33). Recent studies have shown that miR-29a has different roles in cardiac fibroblasts, where it inhibits collagen synthesis, thus attenuating fibrosis (33) and cardiomyocytes, which induces hypertrophy. Overall, the deregulation of miR-29a was associated with conditions, such as myocardial fibrosis, cardiac hypertrophy, congestive heart failure, chronic hepatic injury, hepatitis C virus (HCV) infection and inflammation, hypertensive and diabetic nephropathies, and chronic kidney diseases (30).
Aiming at identifying miRNAs with therapeutic value for the cardiac fibrosis associated with the natural aging process, we explored whether the expression of these four miRNAs changed during the natural aging process. Here, we report that miR-29a increase is a common feature of multiple organs in the natural aging process and it may represent a compensatory mechanism against cardiac fibrosis through SERPINH1 downregulation.
All animal experiments were conducted in accordance with the European Guidelines for Animal Welfare (Directive 2010/63/EU) and approved by the National Sanitary Veterinary and Food Safety Authority (nr 389/22.03.2018). C57Bl/6 mice were purchased from Jackson Laboratory and bred in the ICBP animal facility, under specific pathogen-free conditions, in a controlled environment of 12/12 h light/dark cycle, 21°C, 55–60% humidity, with chow and water ad libitum. Female and male C57BL/6J mouse cohorts used in this study were divided into three age groups: young (2–3 months-old, n = 20), old (16–18 months-old, n = 4), and very old (24–25 months-old, n = 10).
The NIH/3T3 and HL-1 cell lines were purchased from ATCC and Merck Millipore, MA, USA, respectively. NIH/3T3 cells were cultured in high-glucose DMEM (Thermo Fisher Scientific, MA, USA), supplemented with 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, MA, USA) and 1% antibiotic-antimycotic solution (Merck KGaA, Darmstadt, Germany) and passaged two times a week at a density of 3 × 103 cells/cm2. HL-1 cells were cultured in Claycomb medium supplemented with 10% FBS, 1% non-essential amino acids (NEAA), 0.1 mM norepinephrine, and 2mM L-glutamine and passaged every three days by 1:3 splitting onto gelatin/fibronectin-coated tissue flasks.
Primary cardiac fibroblasts were isolated from mouse ventricles as previously described (13). Briefly, the mice were anesthetized with ketamine: xylazine (100:10, mg/kg b.w.); then, the hearts were removed, rinsed with ice-cold phosphate-buffered saline (PBS), and minced into 1- to 2-mm pieces under a sterile laminar flow hood. The fresh tissue was dissociated into a single-cell suspension through 5-min-sequential steps using 5 ml Hanks' balanced salt solution (HBSS) with 0.1% trypsin and 100 μg/ml type I-S collagenase (Merck KGaA, Darmstadt, Germany). Around 5–8 fractions were collected until the whole tissue was digested. Each collected fraction was mixed with 2 ml FBS before centrifugation at 400 × g, 4°C for 5 min. In the end, all cellular pellets were pooled and resuspended in 10 ml of Dulbecco's Modified Eagle's Medium (DMEM)/F12 supplemented with 10% FBS, 1% antibiotic-antimycotic solution, 1 × L-glutamine (Merck KGaA, Darmstadt, Germany), 10 ng/μl fibroblast growth factor (FGF) (R&D Systems, Canada), and 10 μg/ml insulin for plating. Cells were allowed to adhere and proliferate in culture for 5 days before being used in experiments.
Mice were anesthetized with ketamine: xylazine (100/10 mg/kg b.w.). The organs were harvested as follows: the cardiac ventricle, the mediastinal lymph nodes, the thymus, a portion of the right liver lobe, the spleen, the pancreas, the left kidney, the left quadriceps muscle, and the whole brain. The lymphatic organs were harvested only from young and old mice. The organs were immediately flash-frozen in liquid nitrogen in 2-ml RNase-free tubes and stored at −80°C. Organ powder was obtained from each collected organ by grinding in liquid nitrogen using a mortar and pestle and stored at −80°C until use.
The total RNA was extracted from 10 mg frozen organ powder in 1 ml of Trizol® reagent (Thermo Fisher Scientific, MA, USA) with phenol/chloroform, by following the manufacturer's recommendations. For RNA isolation from cardiac fibroblasts, the miRNeasy kit (Qiagen, MD, USA), as per the protocol recommended by the manufacturer, was used. Total RNA was eluted in 30 μl of nuclease-free H2O and stored at −80°C until use.
For quantification of mature miRNA strands, 25 ng of the total RNA were reverse-transcribed to complementary DNA (cDNA) using TaqMan® MiRNA assays and Applied Biosystems® TaqMan® MiRNA Reverse Transcription Kit. The reverse transcription was carried out on a Veriti™ 96-Well Thermal Cycler and the real-time quantitative reverse transcription PCR (RT-qPCR) was carried out using 0.2 μl primers and TaqMan probe, at a final reaction volume of 10 μl, on ViiA™ 7 RT-PCR System. The cycling conditions were as follows: enzyme activation at 95°C for 20 s, denaturation at 95°C for 1 s, and annealing/extension at 60°C for 20 s. Forty cycles of amplification were performed.
The reverse-transcription reaction was made with 500 ng total RNA, using the High-Capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific, MA, USA). Quantification of SERPINH1 was carried out using SYBR™ Select Master Mix (Thermo Fisher Scientific, MA, USA) and ViiA™ 7 Real-Time PCR System. Relative expression was calculated using the comparative CT method and mouse S18 was used for normalization.
Cells were seeded onto a 24-well plate at 70% confluence, one day prior to transfection. Cells were transiently transfected with mature miR-29a-3p mimic (Thermo Fisher Scientific, MA, USA, ID MC12499) or scrambled miRNA control (negative control) (Thermo Fisher Scientific, MA, USA, Cat. 4464058) at a final concentration of 10 nM, using Lipofectamine™ RNAiMAX Transfection Reagent (Thermo Fisher Scientific, MA, USA). The medium was replaced with a complete growth medium after 6 h of transfection, and cells were maintained in culture for another 24 h, before cellular assays.
The miR-29a targets were predicted using three online analysis tools, such as TargetScan (34), DIANA-microT-CDS (35), and miRDB (36). To reduce possible false positives, only the overlapped predicted targets of at least two databases were considered for functional analysis. Biological process enrichment analysis was performed using ClueGO v2.5.7 (37), a plug-in of Cytoscape v3.8.2 (38), and the criteria for grouping the genes for each term were as follows: 24 genes per term, 10% genes per term with a kappa score threshold of 0.4, minimum GO level of 3, and maximum GO level of 8. The enrichment/depletion test for terms was set to two-sided (enrichment/depletion) based on a hypergeometric test, and the corrected method for p-value was set as per the Benjamini-Hochberg method. The adjusted p < 0.01 was considered statistically significant.
For PPI analysis, the target genes were mapped to the STRING v11 database (39) and only the interactions with a combined score higher than 0.4 and FDR stringency less than 1% were considered significant. The network file was loaded in Cytoscape software for visual adjustment. To emphasize the relationships between proteins, the size of nodes was correlated with the number of interactions of each particular protein, and the thickness of edges was correlated with the combined score.
Quantification of total collagen level in the cardiac ventricle was performed using a Hydroxyproline Assay Kit (Merck KGaA, Darmstadt, Germany, MAK008) and following the manufacturer's recommendations. Briefly, 10 mg wet tissue was homogenized in 100 μl of water and hydrolyzed with 100 μl concentrated HCl for 3 h at 120 °C. The mixture was centrifuged and 30 μl supernatant was transferred into a 96-well plate and evaporated to dryness in a 60°C oven. The oxidation reaction was done using Chloramine T for 5 min at room temperature, and then the samples were mixed with DMAB/perchloric acid/isopropanol reagent and incubated for 90 min at 60°C. Hydroxyproline was measured spectrophotometrically at 560 nm using a standard curve.
Cells were fixed in 4% formaldehyde, permeabilized with 0.1 % TritonX100, and incubated with anti-Collagen I (Novus Biologicals, CO, USA, NB600-408-1, dilution 1: 50) or anti-Collagen III (Abcam, Cambridge, UK, ab7778, dilution 1: 100) rabbit polyclonal antibody, overnight at 4 °C. Washed cells were then incubated with anti-rabbit IgG (whole molecule)-FITC secondary antibody (Merck KGaA, Darmstadt, Germany, F9887, dilution 1: 320) for 1 h at room temperature in the dark. Image acquisition was performed using a Leica DMi8 inverted fluorescent microscope (Leica Microsystems GmbH, Wetzlar, Germany) equipped with HC PL APO 20x/0.75 CS2 dry objective. Fluorophores were excited with a multi-LED Spectra-X light source (Lumencor, OR, USA) and images were captured with an sCMOS camera Leica DFC9000 and subsequently processed with Leica LAS X software. Z-stack images (15-μm depth) were acquired with LAS X Navigator module and mosaic merged and then digitally processed for extended depth of field. The fluorescence was quantified with the analysis tool of Las X software by averaging the least four random fields per each experimental condition and the results were expressed as mean fluorescence intensity.
Cells were lysed in 2X Laemmli buffer containing proteases inhibitor cocktail (Merck KGaA, Darmstadt, Germany), and protein concentration was determined using the Amido Black assay. A total of 40 μg of protein was loaded and resolved on 4–15% of precast polyacrylamide gel, followed by a transfer on 0.45 μm nitrocellulose membrane and Ponceau S reversible staining. After blocking 3 h in 5% skim milk in Tris-buffered saline containing 0.1% Tween 20 (TBS-T), the membranes were incubated with anti-collagen III antibody (Abcam, Cambridge, UK, ab7778, 1 μg/ml) overnight at 4°C. Membranes were rinsed in TBS and further incubated with HRP-conjugated secondary antibody (R&D Systems, Canada, HAF008) for 1 h. Protein bands were imaged after enhancement with a chemiluminescence agent (Thermo Fisher Scientific, MA, USA) using an ImageQuant LAS4000 system (Fujifilm, Minato, Japan). The protein expression was quantified by densitometry with TotalLab software and normalized to total protein as quantified by Ponceau S staining.
The effect of miR-29a overexpression on HL-1 cell proliferation was evaluated with the xCELLigence System (Agilent Technologies, CA, USA). This system monitors cellular events in real-time by using the microelectrodes integrated at the bottom of the specially designed E-plates. Briefly, 24 h after transfection initiation, the cells (expressing scramble miRNA sequence or miR-29a mimic) were seeded onto E-plates (15 × 103 cells/well) in a complete medium, and the cell index was recorded at 1-h interval for up to 48 h. The migratory capacity of the transfected cells was evaluated using CIM-plates, which are double-chamber systems (with 8-μm pore membranes) bearing microelectrodes onto the inner face of the membrane. Briefly, the cell suspension prepared in serum-free medium was added to the upper chamber (40 × 103 cells/well) and the cells were allowed to migrate through the membrane toward the lower chamber, containing the complete medium. When cells pass through the membrane in response to serum, they come in contact with the microelectrodes and generate impedance signals.
Cell proliferation was analyzed using Propidium iodide staining and flow cytometry analysis. Briefly, the cell suspension containing 105 cells was fixed with 70% ice-cold ethanol (3 h at −20°C) and then incubated with 20 μg/ml propidium iodide and 100 μg/ml RNAse A. The stained cells were analyzed with a CytoFlex flow cytometer (Beckman Coulter, CA, USA). At least 20,000 events were analyzed per sample. The different phases of the cell cycle (G1, S, and G2/M) were determined based on the fluorescent peak signal, which was proportional to cellular DNA content.
All results were expressed as mean +/– SD. Statistical analysis was performed using GraphPad Prism 9.1.2 (GraphPad Software Inc, CA, USA). P-value has been calculated using unpaired t-test for equal variances and unpaired t-test with Welch's correction for unequal variances. For cell cycle experiments, two-way ANOVA was used. Correlations were done using Pearson's correlation coefficient, for normally distributed data, or Spearman's correlation coefficient, for non-normally distributed data. A p-value less than 0.05 was considered statistically significant.
To address whether the aging process impacts the abundance of these miRNAs in the myocardium, the expression levels were evaluated in the heart ventricle and the skeletal muscle of young (2–3-months-old) and old (16–18-months-old) animals (Figure 1A). The rationale of introducing skeletal muscle samples in the analysis was to help discern the cardiac specificity of miRNA changes. The results showed that miR-21a, -22, and -29a had significantly higher levels in old mice-derived cardiac muscle (Figure 1B). Among them, miR-29a had a tendency of increased expression in the skeletal muscle of old animals as well (Figure 1C), while miR-21a and miR-22 were not modified (Figure 1C), thus suggesting the probability of heart-specific changes for miR-21a and miR-22 during the natural aging process. On the contrary, miR-18a had different age-associated patterns, with constant expression in the old ventricle and decreased expression in the old skeletal muscle.
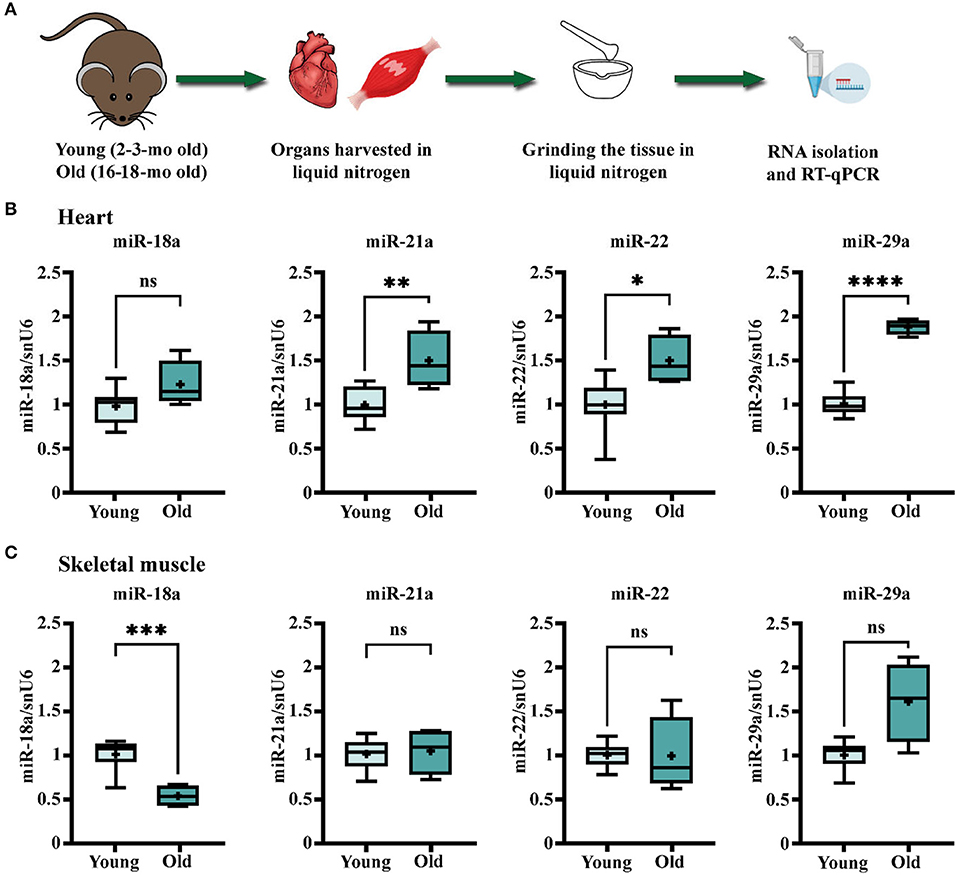
Figure 1. Age-related changes in the expression levels of miR-18a, miR-21a, miR-22, and miR-29a in heart and skeletal muscle of C57Bl/6J mice. (A) Schematic illustration of the experimental design. The heart ventricles and left quadriceps muscle were harvested from young (n = 10) and old (n = 4) mice and processed for total RNA isolation and real-time quantitative reverse transcription PCR (RT-qPCR) analysis. (B) The gene expression level of the four miRNAs in the heart. (C) The gene expression level of the four miRNAs in the skeletal muscle. *p < 0.05, **p < 0.01, ***p < 0.005, and **** p < 0.001.
In an attempt to find out whether the immune cells infiltrating the old heart might be the source of the increased miRNA levels, the expression levels of miR-21a, -22, and -29a were determined in the mediastinal lymph nodes (as the heart draining lymph nodes), the spleen, and the thymus, knowing that the chronic low-grade systemic inflammation is a key pathological characteristic of aged myocardium. The qRT-PCR analysis showed no age-dependent alteration of miR-22 levels in the lymphoid organs in old animals (Figure 2A), and generally, there were very low expression levels of this miRNA in the three organs (data not shown). These results excluded the contribution of the lymphoid organs to the miR-22 increase in the old heart and further suggested that the age-associated miR-22 increase might be confined to the heart. Indeed, gene expression analysis of miR-22 in other organs harvested from different experimental lots including young (2–3-months-old) and very old (24–25-months-old) animals confirmed the steady expression levels in the kidney, the liver, and the brain (Supplementary Figure 1). It is however uncertain which cell type might be the source of miR-22 increase in the cardiac tissue as neither miRNA sequencing (GEO accession number GSE153214) nor qRT-PCR analysis identified any change in the expression level of this miRNA in isolated populations of cardiac fibroblasts and cardiomyocytes (data not shown). Interestingly, both miR-21a and miR-29a had significantly higher expressions in the heart draining lymph nodes in old animals, as well as in the spleen and the thymus (Figures 2B,C), which make them more conceivable in the hypothesis of the infiltrating immune cells into the aged heart as contributors to the increase in these miRNA levels and also advances these modifications as being common features of aging tissues.
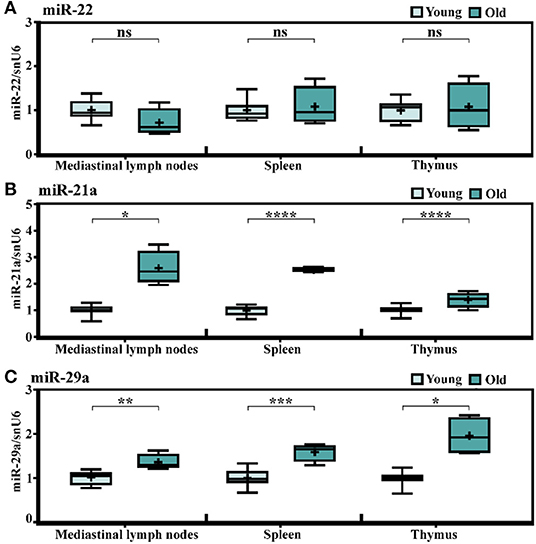
Figure 2. Age-related changes in the expression levels of miR-21a, miR-22, and miR-29a in lymph organs of C57Bl/6 mice. The expressions of (A) miR-22, (B) miR-21a, and (C) miR-29a in the mediastinal lymph nodes, the spleen, and the thymus harvested from the young (n = 10) and old (n = 4) animals. The results are presented as fold changes relative to the young group. *p < 0.05, **p < 0.01, ***p < 0.005, ****p < 0.001.
To further investigate whether the increases in miR-21a and miR-29a expression levels in the natural aging process are general aging features, several other organs have been included in the analysis. Having the steady or not-significantly increased expressions of miR-21a and miR-29a in the skeletal muscle of old animals (16–18-months-old) in comparison to young counterparts (described in Figure 1C), we attempted to investigate the miRNA expressions at even older ages, by establishing another experimental lot comprising young (2–3-months-old) and very old (24–25-months-old) animals. Multiple organs have been included in the analysis i.e., the cardiac ventricle, the skeletal muscle, the brain, the kidney, the pancreas, and the liver. The results confirmed the aging-induced increase of miR-21a expression in the cardiac ventricle and also showed significant increases in all the other analyzed organs, except the liver (Figure 3A). However, apart from the heart, the increases in the other organs were minor and probably without biological significance.
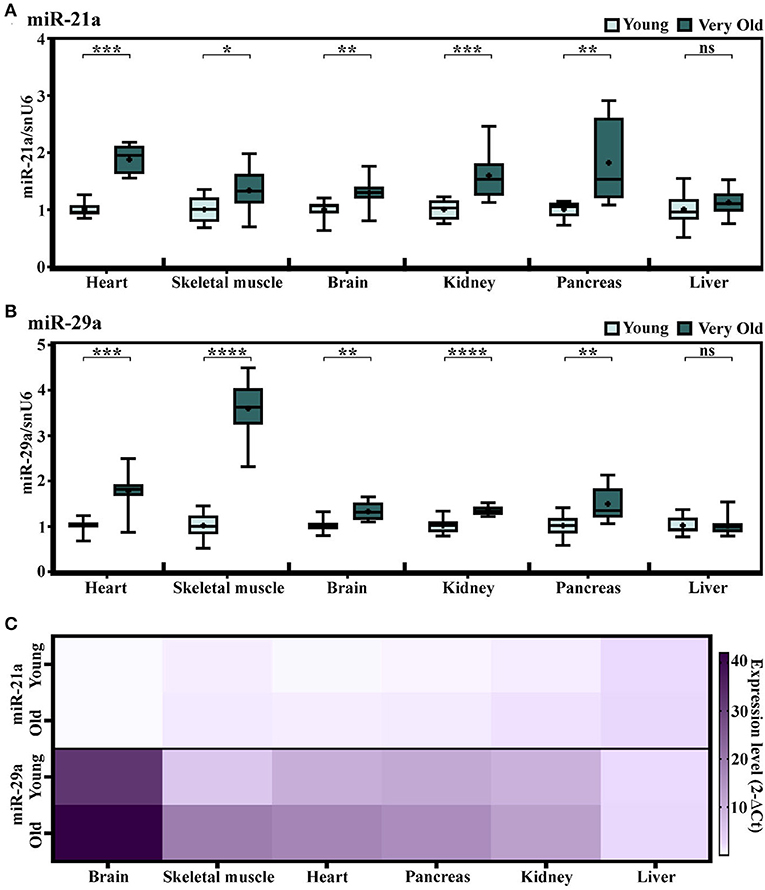
Figure 3. (A,B) The expression levels of miR-21a (A) and miR-29a (B) in multiple organs harvested from young (2–3-months-old) and very old (24–25-months-old) mice. The results are presented as fold changes relative to the young group per each organ (the heart, the skeletal muscle, the brain, the kidney, the pancreas, and the liver); n = 10 animals/group. *p < 0.05, **p < 0.01, ***p < 0.005, and ****p < 0.001. (C) Heat map showing the expression of miR-21 and miR-29a normalized to snoRNA/U6 in each organ. Note the higher expression of miR-29a in comparison to miR-21 in the analyzed organs.
Similarly, miR-29a showed statistically significant increases in the heart, the skeletal muscle, the brain, the kidney, and the pancreas, yet not in the liver of the old animals (Figure 3B). Nevertheless, in contrast to miR-21a, the increase in miR-29a was higher in the skeletal muscle and its expression level had a gradually increasing tendency during aging, which was more obvious than in the cardiac ventricle (Supplementary Figure 2). Supporting this, the miRNA sequencing of cardiac fibroblasts isolated from the young and old mice (13) showed slightly increased levels of miR-29a (Supplementary Figure 3), which could not be further validated by qRT-PCR analysis. It is also worth mentioning that, although highly abundant in the cardiac fibroblasts, miR-21a was not found altered in old cardiac fibroblasts (Supplementary Figure 3) and, furthermore, its levels were lower than those of miR-29a in either the organ investigated from the young or the old individuals (Figure 3C). Together, these data suggest that the aging-related increase in miR-29a, although apparently a feature of multiple organs, is more obvious in muscle tissues, with putative contributors being the infiltrating immune cells, rather than the resident cardiac cell populations.
The potential impact of miR-29a increase during the aging process was investigated through bioinformatic analysis of the targets predicted by three databases. Concisely, the targets predicted by at least two of the three databases (TargetScan v7.2, miRDB, and DIANA v5.0) were used for Gene Ontology (GO)-Biological Processes enrichment analysis (Figure 4A). This analysis highlighted extracellular matrix (ECM) organization as the most significantly enriched GO term. This result was in accordance with the ability of miR-29 to regulate a broad collection of mRNAs with roles in fibrosis and ECM remodeling (33). Other enriched GO terms mostly included biological processes involved in connective tissue homeostasis, e.g., cell-matrix adhesion, metallopeptidase activity, regulation of smooth muscle cell proliferation, etc (Figure 4B).
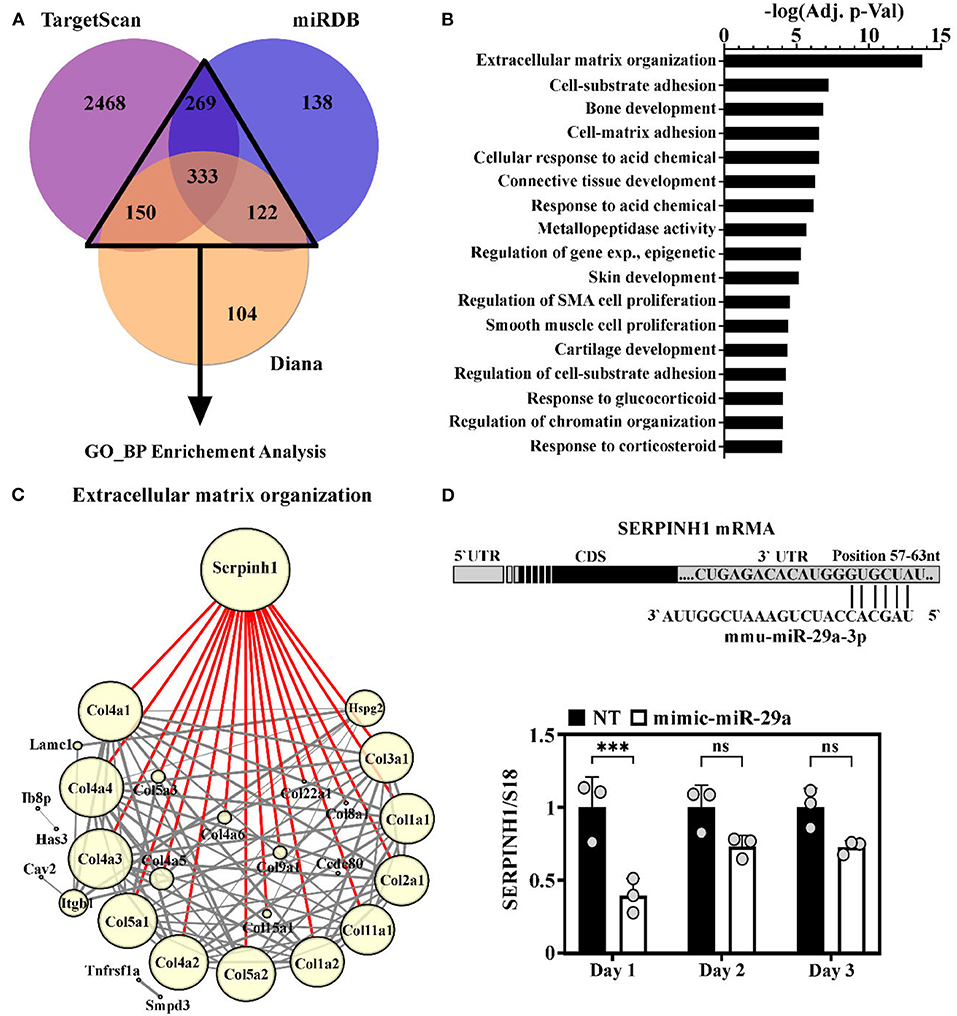
Figure 4. Identification of SERPINH1 as a miR-29a target. (A) Venn diagram showing the predicted targets of the miR-29a, obtained by interrogating three computational algorithms i.e., TargetScan, miRDB, and Diana. The targets predicted by at least 2 databases (with numbers illustrated in a triangle) were used in gene ontology (GO) analysis. (B) Enrichment analysis of predicted miR-29a target genes. Note that extracellular matrix (ECM) organization emerged as the most significantly enriched process involving miR-29a target genes. (C) String analysis of genes involved in ECM organization. Node size directly correlates with the number of interactions of each protein (min = 1; max = 18); Edge width denotes the confidence score calculated by STRING: the thinnest = 0.4 (medium confidence); the thickest ≥ 0.9 (the highest confidence); Highlighted in red are the interaction of SERPINH1. (D) Illustration of the 3′ UTR region of the mouse SERPINH1 gene with the predicted site for miR-29a (below). The expression of SERPINH1 mRNA level in HL-1 cells at one, two, and three days after transient transfection of cells with miR-29a mimic; ***p < 0.005.
To better understand the relationship between the predicted targets implicated in ECM organization, the PPI analysis was done using the STRING database. To visualize this network, the 46 genes included in this term were imported into Cytoscape. The generated network was further processed by eliminating the proteins with no direct interactions among them. The final network, containing 28 nodes and 188 interactions, is illustrated in Figure 4C. The node size correlates with the number of interactions between proteins and the edge thickness correlates with the confidence score. This analysis highlighted Serpinh1 as the most important component in the network. SERPINH1 interacts with high confidence scores (> 0.9) with 18 proteins, all from the collagen family (Figure 4C).
To experimentally validate miR-29a as a modulator for SERPINH1 in the muscle cells, the gain-of-function approach was used in HL-1 cardiac muscle cells by transient transfection with miR-29a mimic, which resulted in a marked increase of the endogenous expression level of miR-29a, peaking at 1 day after oligonucleotide addition (Supplementary Figure 3). The RT-qPCR analysis of SERPINH1 in the transfected cells showed that the overexpression of miR-29a caused a pronounced reduction of SERPINH1 (2.5-fold decrease) at the mRNA level at 1 day after transfection (Figure 4D). Similar results were obtained with the 3T3 fibroblasts cell line (Supplementary Figure 5). This data validates SERPINH1 as a direct target of miR-29a in cardiac muscle cells.
To further validate the biological link between miR-29a and SERPINH1 synthesis in vivo, the transcriptional levels of SERPINH1 were measured in various organs from very old mice (24–25-months-old) relative to their young counterparts. Quantitative RT-PCR analysis revealed that SERPINH1 was decreased in all aged organs, except the liver (Figure 5A). Noteworthy, the expression level of miR-29a inversely correlated with SERPINH1. The strongest correlation was found in the skeletal muscle, whereas the heart, the kidney, and the pancreas only showed moderate associations. Still, no significant association was found in the brain (Figure 5B). Our results are also concurrent with the data from the Tabula Muris Senis registry, which showed progressive decreases of SERPINH1 mRNA levels in the heart, the limb muscle, the kidney, and the pancreas of the mouse, during aging (https://twc-stanford.shinyapps.io/maca/).
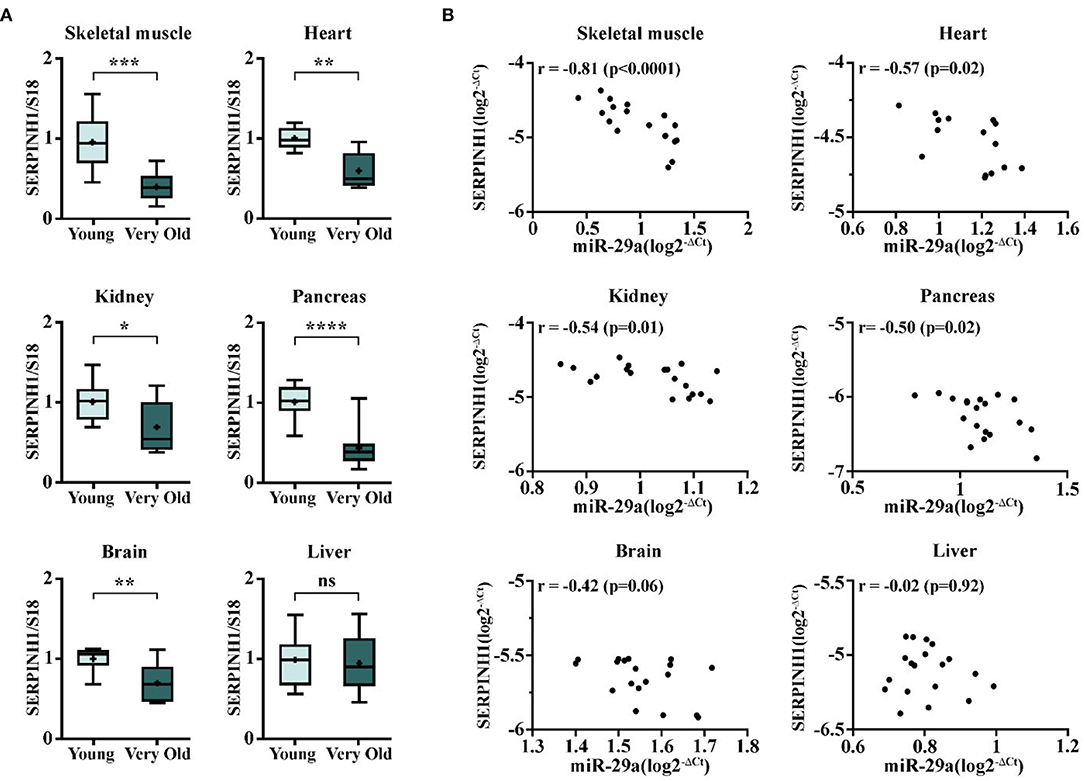
Figure 5. The expression of SERPINH1 and correlation between SERPINH1 and miR-29a expression levels in various organs. (A) The expression of SERPINH1 in the heart, the skeletal muscle, the brain, the kidney, the pancreas, and the liver. For each organ, the expression is presented relative to the young group. N = 5–10 animals/group; *p < 0.05, **p < 0.01, ***p < 0.005, and ****p < 0.001. (B) Pearson's correlations between SERPINH1 and miR-29a expression levels in the six organs mentioned above.
To further explore the function of miR-29a increase in the old heart and the skeletal muscle, the protein levels of collagen I and III, two targets of this miRNA which are both under the direct control of SERPINH1, were determined in miR-29a-overexpressing HL-1 cells. Immunofluorescence staining followed by fluorescence quantification showed decreased levels of collagen III (Figure 6A) and collagen I (Supplementary Figure 6) in the HL-1cells 1 day after transfection. This data was also validated by Western blot analysis (Figure 6B), which showed a reduced level of collagen III in cell lysates obtained from miR-29a-overexpressing cells when compared to miRNA scramble-transfected cells. However, the quantification of hydroxyproline in the cardiac ventricle lysates obtained from the young, old, and very old animals, as a measure to estimate total collagen levels, showed increased levels of collagens in the old animals (Figure 6C), a result that was not intriguingly, being in agreement with the fibrosis development in the aging heart. This data suggests that the increase in the miR-29a level, noticed in multiple organs in the old individuals, is an adaptive response of the organism to the increased fibrosis associated with the aging process.
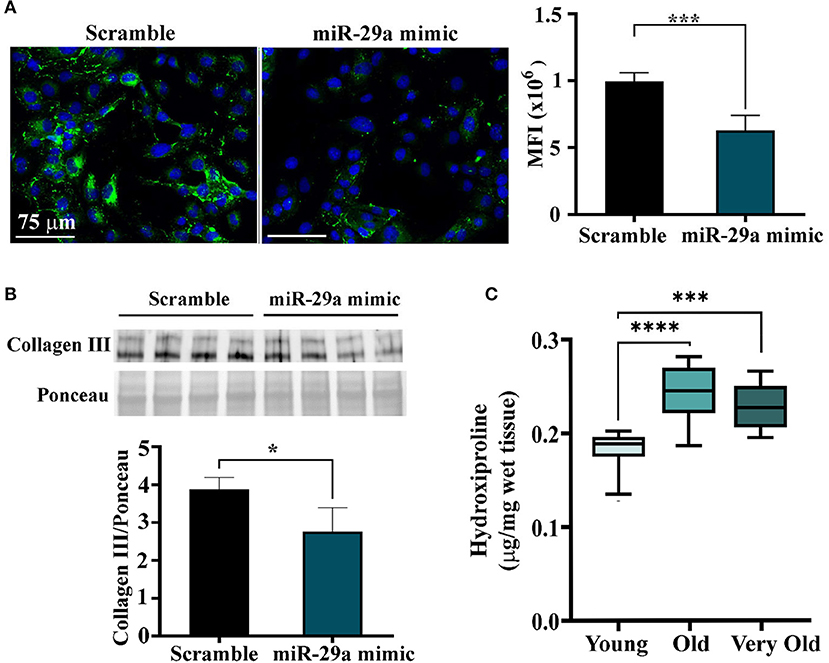
Figure 6. Quantification of collagen proteins in HL-1 cells overexpressing miR-29a and in ventricular lysates obtained from young (2–3-months-old), old (16–18-months-old), and very old (24–25-months-old) animals. (A) Immunofluorescence images of HL-1 cells at 24 h after transfection with mir-29a mimic or scramble miRNA. The histogram shows the quantification of the fluorescence signal produced by collagen III staining, expressed as mean fluorescence intensity. ***p < 0.005. (B) Western blot analysis of the collagen III in miR-29a-overexpressing HL-1 cells. The histogram shows the densitometry quantification normalized to total protein. *p < 0.05. (C) The hydroxyproline levels in tissue lysates of cardiac ventricles were obtained from young, old, and very old animals. ***p < 0.005 and ****p < 0.001.
The effects of miR-29a have been further evaluated in HL-1 cells by examining its effects on cell migration and proliferation. Cell migration was analyzed with the xCELLigence system, by following the capacity of cells to infiltrate the membrane pores of CIM plates in response to the chemoattractant molecules within the serum. The results showed a decreased migration ability of miR-29a overexpressing cells in comparison with the designed control cells (Figure 7A). Moreover, cell proliferation was also affected by miR-29a, as demonstrated by xCELLigence analysis of HL-1 myocytes after transfection (Figure 7B), as well as by cell cycle analysis of cells evaluated at 24 h after transfection initiation (Figure 7C). The results concurred toward a slower proliferation rate of cells overexpressing miR-29a, as illustrated by the decreased cell index and diminished percentage of cells in the G2/M phase, in comparison to scramble-transfected cells. The same effects have been observed on 3T3 fibroblasts cells (data not shown). Considering the activation and proliferation of fibroblasts as common processes accompanying fibrosis development, this data might indicate a protective role of miR-29a upregulation during the natural aging process. However, additional experiments are necessary to confirm the protective role of miR-29a in the aging process, as well as the downregulation of SERPINH1 as a direct target of miR-29a in the old organs.
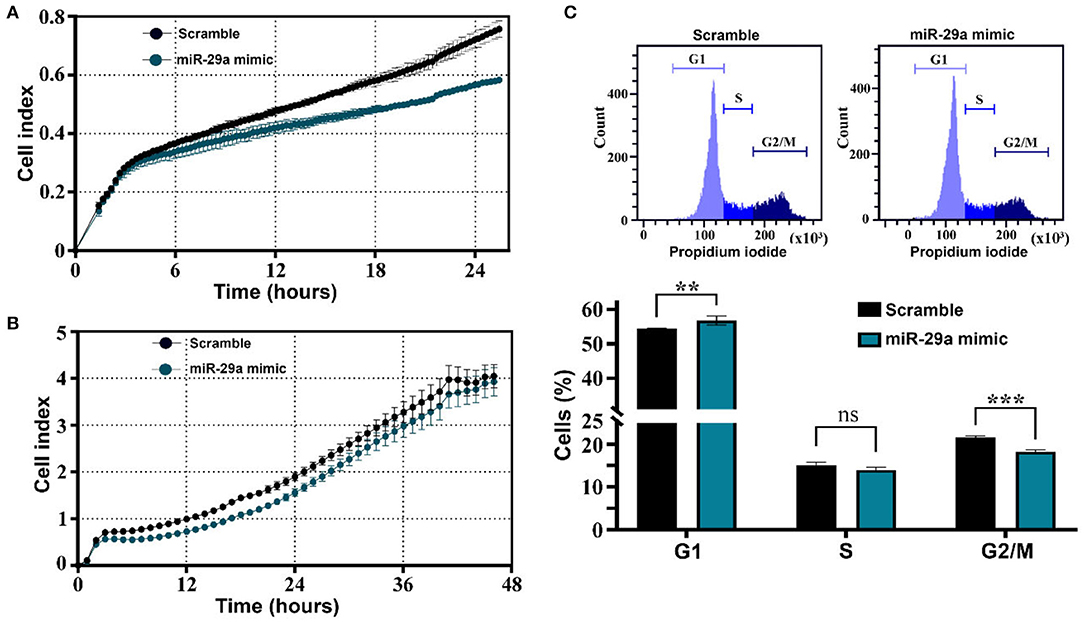
Figure 7. Impact of miR-29a on muscle cell migration and proliferation. (A) Real-time profiling of HL-1 cell migration in CIM plates, after transfection with miR-29a mimic or scrambled miRNA control. The data represent one representative experiment of three experiments performed with similar results. (B) Real-time profiling of HL-1 cell adherence and proliferation in E-plates, after transfection with miR-29a mimic or scrambled miRNA control. The data represent one representative experiment of three experiments performed in quadruplicates with similar results. (C) Cell-cycle distribution and percentage of cells in G0/G1, S, and G2 /M phases. The data represent the mean +/– SD of 3 independent experiments performed. **p < 0.01 and ***p < 0.005.
In conclusion, our data indicate that miR-29a is a potent posttranscriptional repressor of SERPINH1 in multiple organs and its upregulation is a common feature of the aging process, suggesting that it might represent a potential therapeutic target for age-related adverse matrix remodeling and tissue fibrosis.
Deregulation of the miRNA profile in aging has been reported in several studies (40–42), yet only a small number of miRNAs have been directly associated with aging or aging-related cardiac diseases. Fibrosis and hypertrophy are important pathologies in the aged heart and are the primary causes of heart failure (43). Among the miRNAs with documented roles in cardiac diseases, miR-22 was mainly associated with hypertrophy, while miR-18a, miR-21, and miR-29a were associated with fibrosis. The aim of this paper was to evaluate the expression levels of miR-18a, miR-21a, miR-22, and miR-29a in the aging process. Our studies demonstrated that (i) miR-21a, miR-22, and miR-29a are upregulated in the aged heart; (ii) miR-21a and miR-29a are also upregulated in other organs during aging; (iii) miR-29a is highly expressed in many organs and it negatively regulates with the SERPINH1 gene expression level; (iv) a direct negative correlation exists between miR-29a and SERPINH1 expression levels during aging in the cardiac ventricle, the skeletal muscle, the kidney, and the pancreas.
It might be worth mentioning that, although the miR-18a level was not found significantly altered in the heart of old mice (16–18-months-old), its expression slightly, yet significantly, increased in the very old animals (data not shown), indicating that this miRNA gradually changed with aging, but was detected only at very old ages. Similar findings were reported by Schroen's group, which showed an increased expression of miR-18a in the heart of 104-week-old C57Bl/6 mice (22). This increased expression was further associated with the resistance to heart failure, by reducing thrombospondin-1 (THSP-1) and connective tissue growth factor (CTGF), the two direct targets of members of the miR-17–92 cluster (22).
Besides an increase in the size of the old ventricle, the miR-21 and miR-29a were also found in higher levels in other aged organs, including the skeletal muscle and the brain, which are in accordance with other previous studies (44–46). The increase of miR-29 in the skeletal muscle in old rodents resulted in the inhibition of proliferation of muscle progenitor cells, by suppressing the mediators of cell proliferation, IGF-1, p85α, and B-Myb, which in turn led to cellular senescence and contributed to the aging-induced loss of muscle mass and senescence (45). As both miRNAs were found upregulated in the mediastinal lymph nodes, the spleen, and the thymus, we presumed that these changes might be connected to the immune cells infiltrating the organs in advanced age (inflammaging). In addition, this hypothesis was reinforced by the unchanged expression levels of these two miRNAs in cardiac fibroblasts and cardiomyocytes isolated from young and old mice, which made likely the existence of external miRNA sources. According to a recently published comprehensive analysis of aging dynamics, based on RNA-sequencing of 17 organs and plasma proteomics at 10 ages across the mouse lifespan, a widespread immune cell activation became detectable in multiple organs after middle age (47). Therefore, inflammaging may act as both the driver of the whole-organ inflammatory signal, as well as a contributor to the shifts in miRNA levels in aged organs. Interestingly, upregulation of miR-29a in the aged brain was associated with inflammation, by decreasing the expression of two important regulators of microglial activation, such as the IGF-1 and CX3CL1 (44).
Multiple studies documented the increased deposition of ECM proteins in the heart, the pancreas, the kidney, and other organs with advancing age (2, 48, 49), making fibrosis a hallmark of aging. Although both miR-21 and miR-29a have been involved in fibrosis, they appear to have opposite roles. MiR-21 has been associated with the development of fibrosis in patients with pulmonary, hepatic, renal, or cardiac pathologies. Thus, in diabetic cardiomyopathy, an increased level of miR-21 was associated with cardiac interstitial and perivascular fibrosis through NFκB/SMAD7 signaling pathway (8). Other findings showed that miR-21 promoted cardiac fibrosis via the TGF-β/Smad7 signaling pathway after myocardial infarction (50) and via the CADM1/STAT3 pathway in patients with atrial fibrillation (51). In a transgenic mouse model of cardiac failure, the elevated expression level of miR-21 induced fibrosis, hypertrophy, and cardiac dysfunction, by inhibiting SPRY1, which promoted ERK–MAPK signaling and cardiac fibroblast survival (18). MiR-21 was also found upregulated in the lungs of patients with idiopathic pulmonary fibrosis (52), as well as in kidney fibrosis that followed acute kidney injury or chronic allograft dysfunction (53). Moreover, miR-21 upregulation was correlated with hepatic fibrosis in human cirrhotic liver samples (54).
In contrast, the members of the miR-29 family are known for their anti-fibrotic effects in various organs, by targeting genes involved in ECM production and fibrosis (55). Thus, miR-29 was found downregulated in the border zone of the infarcted myocardium and its expression was correlated with the upregulation of multiple ECM proteins, such as collagen I, II, III, and fibronectin 1 (33). In the mouse model of angiotensin II (Ang II)-induced hypertension, the cardiac fibrosis was abrogated by miR-29b overexpression, which was negatively regulated by TGF-β/Smad signaling in fibrosis (56), while in a model of isoproterenol-induced fibrosis, miR-29a attenuated cardiac fibrosis by targeting DNMT3A expression leading to inhibition of the Ras/ERK1/2 signaling pathway (57). The same mechanisms involving TGF-β/Smad3 signaling were described for miR-29b inhibition in bleomycin-induced pulmonary fibrosis (58) and the development of progressive renal fibrosis in obstructive nephropathy (59). Moreover, the miR-29 family was found downregulated in carbon tetrachloride-induced liver fibrosis in mice, as well as in the livers of patients with advanced liver fibrosis (60).
Although there is substantial evidence for the fact that the miR-29 family abrogates the development of fibrosis by inhibiting collagen synthesis in cardiac fibroblasts, its role in cardiac hypertrophy is still controversial. In a mouse model of cardiac pressure overload, miR-29 was shown to promote pathologic hypertrophy of cardiac myocytes and overall cardiac dysfunction (61). As the expression level of miR-29 in cardiomyocytes isolated from hearts with transverse aortic constriction surpassed those in cardiac fibroblasts, its relevance in the progression of cardiac hypertrophy appeared as more meaningful, thus explaining the overall protecting effect of miR-29 inhibition (or its genetic deletion) on the cardiac hypertrophy and fibrosis in this pathology. The authors also reported that the overexpression of miR-29 in primary cardiomyocytes led to the activation of Wnt signaling, which promoted hypertrophy and the sequential release of profibrotic factors (61). Such discrepancies may be explained by the diversity of mechanisms regulating the miR-29 family, with different stimuli activating distinct signaling pathways and leading to various responses.
A comparative analysis of miR-21a and miR-29a expression levels in various organs tilt the balance in favor of miR-29a, whose expression was much stronger in all analyzed organs. This data suggests that the anti-fibrotic effect of miR-29a prevails in old organs, thus conferring an overall protective anti-fibrotic mechanism in aging.
Besides its anti-fibrotic role, miR-29 was also reported to have protective effects in cancer, elicited by various mechanisms (30). Thus, miR-29 was shown to directly target CDK6 (62) and CDC42 (63), proteins with important roles in controlling cell proliferation in several cancer types. In addition, it was reported that the mantle cell lymphoma patients with downregulated miR-29 showed shorter survival rates compared to patients with relatively higher levels of miR-29 (62). MiR-29a was also shown to inhibit autophagy in pancreatic cancer cells, a mechanism by which the cancer cells survive and acquire chemoresistance. MiR-29a was significantly downregulated in pancreatic cancer cells, yet its overexpression led to increased sensitization and inhibition of autophagy in chemo-resistant cell lines (64). Moreover, miR-29b was found downregulated in ovarian cancer and its restoration led to the inhibition of glycolysis and glucose metabolism in cancer cells by targeting AKT2 and AKT3 (65).
Although miR-29 was found to increase in multiple aged organs in our study, its expression was not changed in the liver. Similar results were found in another report, which analyzed the expression of 367 miRNAs in the aged liver, yet no variations in miR-29 were reported (66). However, it would be worth mentioning that miR-29 was found upregulated in the liver of young mice with Hutchinson-Gilford progeria syndrome, but not in the naturally aged mice (67).
Pathway enrichment analysis of target genes of miR-29a revealed SERPINH1 as a major player in connective tissue homeostasis. SERPINH1, also named Hsp47 (Heat-shock protein 47), is a molecular chaperone that belongs to the Serpin family, yet has no serin protease inhibitory effect. SERPINH1 is found primarily in the secretory pathway of collagen-producing cells, where it binds to and stabilizes procollagen, preventing incomplete folding and lateral aggregation prior secretion (68–70). Without the assistance of SERPINH1, the type I collagen is unable to form the rigid, protease-resistant, and triple-helical structure (71). The importance of SERPINH1 in collagen secretion, processing, and fibril assembling, as well as its deposition in the ECM, had been demonstrated in multiple studies (72, 73). In the human breast cancer tissue, SERPINH1 was positively correlated with collagen I, collagen IV, and fibronectin expression (72). Mutation of SERPINH1 was associated with a severe phenotype of Osteogenesis Imperfecta, in which the secreted type I collagen structure was compromised and became sensitive to protease activity (73). Moreover, knockout mice presented abnormal epithelial tissues, ruptured blood vessels, discontinued basement membranes; they did not survive beyond 11.5 days post-coitus (71).
In this paper, we report that the overexpression of miR-29a in HL-1 cells led to SERPINH1 downregulation, thus endorsing SERPINH1 as a direct target of miR-29a. Furthermore, we showed that miR-29a negatively correlated with SERPINH1 expression levels in various organs during aging. To the best of our knowledge, this is the first report showing that SERPINH1 is downregulated in aged organs, likely as a consequence of the increase in miR-29a level. The presence of miR-29a-binding site within SERPINH1- 3' UTR was previously validated via luciferase reporter assay and western blot (74). Besides, the downregulation of miR-29 was found to be associated with the upregulation of SERPINH1 in various cancers (glioma, cervical, breast, and renal), being involved in the growth, proliferation, migration, and invasion of cancer cells. Consequently, the suppression of SERPINH1 by miR-29- overexpression canceled the progression of cancers (72, 74–77). In good agreement with these studies, we reported here that the overexpression of miR-29a inhibited the proliferation and migration of HL-1 cardiac muscle cells and 3T3 fibroblasts cells. Considering that fibroblast activation and proliferation are important processes leading to cardiac fibrosis (78), this data might indicate a protective role of miR-29a upregulation during the natural aging process. However, additional experiments are necessary to confirm the protective role of the miR-29a/SERPINH1 axis in the aging process.
Recent studies showed that SERPINH1 increased in mouse cardiac endothelial cells and this increase was associated with endothelial dysfunction and impaired vascular remodeling (79). Moreover, multiple studies showed that SERPINH1 was overexpressed in fibrosis-related diseases. In Fischer 344 rats, which spontaneously develop kidney fibrosis in aging, the overexpression of SERPINH1 was associated with increased synthesis of collagens I, III, and IV (80). The same results were observed in carbon tetrachloride-induced liver fibrosis and bleomycin-induced pulmonary fibrosis (81, 82). Additionally, cardiac fibrosis after pressure overload injury was also associated with the increased levels of SERPINH1, collagen I, III, and V and, moreover, myofibroblast-specific deletion of SERPINH1 reduced fibrosis and cardiac hypertrophy (83). The SERPINH1 was also associated with cardiac fibrosis following myocardial infarction. Thus, rats with ligated left anterior descendant coronary artery showed upregulated levels of SERPINH1 in the infarcted zone, which were maintained for up to 28 days after the intervention (84). Moreover, administration of SERPINH1 antisense oligonucleotides reduced cardiac remodeling and improved cardiac function in a rat model of myocardial infarction (85). These findings strengthen our hypothesis that upregulation of miR-29a during natural aging represents a protective mechanism against fibrosis by targeting SERPINH1.
Although collagen level is elevated in fibrosis-related pathologies, multiple studies showed that its increased synthesis was not the primary cause of fibrosis in aging. Contrarily, several studies reported that mRNA expression of collagens I and III was even reduced in aged rat myocardium (86, 87). Important sources of collagen accumulation in aging also result in the attenuation of matrix-degrading pathways (88), as well as the accumulation of advanced glycation end products, which caused collagen cross-linking (89). In this paper, we found increased amounts of hydroxyproline in the cardiac ventricles harvested from old and very old mice, in comparison to their young counterparts. Our results are in accordance with other reports showing that cardiac fibrosis was associated with the aging process. A possible explanation for increased collagen levels is the upregulation of miR-29a and subsequent downregulation of SERPINH1. Thus, miR-29a concomitantly regulates the transcriptional levels of collagen proteins, as well as collagen maturation process (by targeting the specific collagen chaperon, SERPINH1). Our results suggest that miR-29a increase in multiple organs in old individuals develops as an adaptive response of the organism to the increased fibrosis associated with the natural aging process. However, further studies are necessary to confirm the protective role of miR-29a in the aging process.
In conclusion, our study demonstrates that miR-29a is highly expressed in many organs and its expression is further upregulated during the natural aging process. This process occurs in association with the downregulation of SERPINH1, which thus might be a potential therapeutic target for adverse matrix remodeling and aging-associated tissue fibrosis.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by National Sanitary Veterinary and Food Safety Authority (nr 389/22.03.2018).
ER-N, A-ML, MP, CM, and CN performed experiments. ER-N and AB designed the work, analyzed and interpreted the data, and wrote the manuscript. AB gave the final approval of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Romanian Ministry of Education, PN-III-P4-ID-PCE-2020-1340-contract 122/2021.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2021.810241/full#supplementary-material
1. Gupta SK, Piccoli MT, Thum T. Non-coding RNAs in cardiovascular ageing. Ageing Res Rev. (2014) 17:79–85. doi: 10.1016/j.arr.2014.01.002
3. Lee S, Choi E, Cha MJ, Park AJ, Yoon C, Hwang KC. Impact of miRNAs on cardiovascular aging. J Geriatr Cardiol. (2015) 12:569–74. doi: 10.11909/j.issn.1671-5411.2015.05.011
4. Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. (2013) 153:1194–217. doi: 10.1016/j.cell.2013.05.039
5. Armanios M, Alder JK, Parry EM, Karim B, Strong MA, Greider CW. Short telomeres are sufficient to cause the degenerative defects associated with aging. Am J Hum Genet. (2009) 85:823–32. doi: 10.1016/j.ajhg.2009.10.028
6. Bilgili H, Bialas AJ, Gorski P, Piotrowski WJ. Telomere abnormalities in the pathobiology of idiopathic pulmonary fibrosis. J Clin Med. (2019) 8:1232. doi: 10.3390/jcm8081232
7. Li H, Hastings MH, Rhee J, Trager LE, Roh JD, Rosenzweig A. Targeting Age-Related Pathways in Heart Failure. Circ Res. (2020) 126:533–51. doi: 10.1161/CIRCRESAHA.119.315889
8. Li Q, Yao Y, Shi S, Zhou M, Zhou Y, Wang M, et al. Inhibition of miR-21 alleviated cardiac perivascular fibrosis via repressing EndMT in T1DM. J Cell Mol Med. (2020) 24:910–20. doi: 10.1111/jcmm.14800
9. Desvignes T, Batzel P, Berezikov E, Eilbeck K, Eppig JT, McAndrews MS, et al. miRNA nomenclature: a view incorporating genetic origins, biosynthetic pathways, and sequence variants. Trends Genet. (2015) 31:613–26. doi: 10.1016/j.tig.2015.09.002
10. Graves P, Zeng Y. Biogenesis of mammalian microRNAs: a global view. Genom Proteomics Bioinform. (2012) 10:239–45. doi: 10.1016/j.gpb.2012.06.004
11. Bernardo BC, Gao XM, Winbanks CE, Boey EJ, Tham YK, Kiriazis H, et al. Therapeutic inhibition of the miR-34 family attenuates pathological cardiac remodeling and improves heart function. Proc Natl Acad Sci U S A. (2012) 109:17615–20. doi: 10.1073/pnas.1206432109
12. Boon RA, Iekushi K, Lechner S, Seeger T, Fischer A, Heydt S, et al. MicroRNA-34a regulates cardiac ageing and function. Nature. (2013) 495:107–10. doi: 10.1038/nature11919
13. Lupan AM, Rusu EG, Preda MB, Marinescu CI, Ivan C, Burlacu A. miRNAs generated from Meg3-Mirg locus are downregulated during aging. Aging (Albany NY). (2021) 13:15875–97. doi: 10.18632/aging.203208
14. Zhang X, Azhar G, Wei JY. The expression of microRNA and microRNA clusters in the aging heart. PLoS One. (2012) 7:e34688. doi: 10.1371/journal.pone.0034688
15. Serna E, Gambini J, Borras C, Abdelaziz KM, Belenguer A, Sanchis P, et al. Centenarians, but not octogenarians, up-regulate the expression of microRNAs. Sci Rep. (2012) 2:961. doi: 10.1038/srep00961
16. Lorenzen JM, Schauerte C, Hubner A, Kolling M, Martino F, Scherf K, et al. Osteopontin is indispensible for AP1-mediated angiotensin II-related miR-21 transcription during cardiac fibrosis. Eur Heart J. (2015) 36:2184–96. doi: 10.1093/eurheartj/ehv109
17. van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, et al. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci U S A. (2006) 103:18255–60. doi: 10.1073/pnas.0608791103
18. Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. (2008) 456:980–4. doi: 10.1038/nature07511
19. Dai B, Wang F, Nie X, Du H, Zhao Y, Yin Z, et al. The cell type-specific functions of miR-21 in cardiovascular diseases. Front Genet. (2020) 11:563166. doi: 10.3389/fgene.2020.563166
20. Abue M, Yokoyama M, Shibuya R, Tamai K, Yamaguchi K, Sato I, et al. Circulating miR-483-3p and miR-21 is highly expressed in plasma of pancreatic cancer. Int J Oncol. (2015) 46:539–47. doi: 10.3892/ijo.2014.2743
21. Calo N, Ramadori P, Sobolewski C, Romero Y, Maeder C, Fournier M, et al. Stress-activated miR-21/miR-21* in hepatocytes promotes lipid and glucose metabolic disorders associated with high-fat diet consumption. Gut. (2016) 65:1871–81. doi: 10.1136/gutjnl-2015-310822
22. van Almen GC, Verhesen W, van Leeuwen RE, van de Vrie M, Eurlings C, Schellings MW, et al. MicroRNA-18 and microRNA-19 regulate CTGF and TSP-1 expression in age-related heart failure. Aging Cell. (2011) 10:769–79. doi: 10.1111/j.1474-9726.2011.00714.x
23. Ovchinnikova ES, Schmitter D, Vegter EL, Ter Maaten JM, Valente MA, Liu LC, et al. Signature of circulating microRNAs in patients with acute heart failure. Eur J Heart Fail. (2016) 18:414–23. doi: 10.1002/ejhf.332
24. Na J, Jin H, Wang X, Huang K, Sun S, Li Q, et al. The crosstalk of HDAC3, microRNA-18a and ADRB3 in the progression of heart failure. Cell Biosci. (2021) 11:31. doi: 10.1186/s13578-020-00523-y
25. Gurha P, Wang T, Larimore AH, Sassi Y, Abreu-Goodger C, Ramirez MO, et al. microRNA-22 promotes heart failure through coordinate suppression of PPAR/ERR-nuclear hormone receptor transcription. PLoS ONE. (2013) 8:e75882. doi: 10.1371/journal.pone.0075882
26. Xu XD, Song XW, Li Q, Wang GK, Jing Q, Qin YW. Attenuation of microRNA-22 derepressed PTEN to effectively protect rat cardiomyocytes from hypertrophy. J Cell Physiol. (2012) 227:1391–8. doi: 10.1002/jcp.22852
27. Huang ZP, Chen J, Seok HY, Zhang Z, Kataoka M, Hu X, et al. MicroRNA-22 regulates cardiac hypertrophy and remodeling in response to stress. Circ Res. (2013) 112:1234–43. doi: 10.1161/CIRCRESAHA.112.300682
28. Jazbutyte V, Fiedler J, Kneitz S, Galuppo P, Just A, Holzmann A, et al. MicroRNA-22 increases senescence and activates cardiac fibroblasts in the aging heart. Age (Dordr). (2013) 35:747–62. doi: 10.1007/s11357-012-9407-9
29. Gupta SK, Foinquinos A, Thum S, Remke J, Zimmer K, Bauters C, et al. Preclinical development of a MicroRNA-based therapy for elderly patients with myocardial infarction. J Am Coll Cardiol. (2016) 68:1557–71. doi: 10.1016/j.jacc.2016.07.739
30. Kwon JJ, Factora TD, Dey S, Kota J. A systematic review of miR-29 in cancer. Mol Ther Oncolytics. (2019) 12:173–94. doi: 10.1016/j.omto.2018.12.011
31. Abonnenc M, Nabeebaccus AA, Mayr U, Barallobre-Barreiro J, Dong X, Cuello F, et al. Extracellular matrix secretion by cardiac fibroblasts: role of microRNA-29b and microRNA-30c. Circ Res. (2013) 113:1138–47. doi: 10.1161/CIRCRESAHA.113.302400
32. Cheng J, Wang Y, Wang D, Wu Y. Identification of collagen 1 as a post-transcriptional target of miR-29b in skin fibroblasts: therapeutic implication for scar reduction. Am J Med Sci. (2013) 346:98–103. doi: 10.1097/MAJ.0b013e318267680d
33. van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, et al. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci U S A. (2008) 105:13027–32. doi: 10.1073/pnas.0805038105
34. Agarwal V, Bell GW, Nam JW, Bartel DP. Predicting effective microRNA target sites in mammalian mRNAs. Elife. (2015) 4:e05005. doi: 10.7554/eLife.05005
35. Paraskevopoulou MD, Georgakilas G, Kostoulas N, Vlachos IS, Vergoulis T, Reczko M, et al. DIANA-microT web server v5.0: service integration into miRNA functional analysis workflows. Nucleic Acids Res. (2013) 41:W169–73 doi: 10.1093/nar/gkt393
36. Chen Y, Wang X. miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res. (2020) 48:D127–D31. doi: 10.1093/nar/gkz757
37. Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, et al. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. (2009) 25:1091–3. doi: 10.1093/bioinformatics/btp101
38. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. (2003) 13:2498–504. doi: 10.1101/gr.1239303
39. Szklarczyk D, Gable AL, Nastou KC, Lyon D, Kirsch R, Pyysalo S, et al. The STRING database in 2021: customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. (2021) 49:D605–D12. doi: 10.1093/nar/gkaa1074
40. Min KW, Zealy RW, Davila S, Fomin M, Cummings JC, Makowsky D, et al. Profiling of m6A RNA modifications identified an age-associated regulation of AGO2 mRNA stability. Aging Cell. (2018) 17:e12753. doi: 10.1111/acel.12753
41. Fehlmann T, Lehallier B, Schaum N, Hahn O, Kahraman M, Li Y, et al. Common diseases alter the physiological age-related blood microRNA profile. Nat Commun. (2020) 11:5958. doi: 10.1038/s41467-020-19665-1
42. Smith-Vikos T, Liu Z, Parsons C, Gorospe M, Ferrucci L, Gill TM, et al. A serum miRNA profile of human longevity: findings from the Baltimore Longitudinal Study of Aging (BLSA). Aging (Albany NY). (2016) 8:2971–87. doi: 10.18632/aging.101106
43. North BJ, Sinclair DA. The intersection between aging and cardiovascular disease. Circ Res. (2012) 110:1097–108. doi: 10.1161/CIRCRESAHA.111.246876
44. Fenn AM, Smith KM, Lovett-Racke AE, Guerau-de-Arellano M, Whitacre CC, Godbout JP. Increased micro-RNA 29b in the aged brain correlates with the reduction of insulin-like growth factor-1 and fractalkine ligand. Neurobiol Aging. (2013) 34:2748–58. doi: 10.1016/j.neurobiolaging.2013.06.007
45. Hu Z, Klein JD, Mitch WE, Zhang L, Martinez I, Wang XH. MicroRNA-29 induces cellular senescence in aging muscle through multiple signaling pathways. Aging (Albany NY). (2014) 6:160–75. doi: 10.18632/aging.100643
46. Heid J, Cencioni C, Ripa R, Baumgart M, Atlante S, Milano G, et al. Age-dependent increase of oxidative stress regulates microRNA-29 family preserving cardiac health. Sci Rep. (2017) 7:16839. doi: 10.1038/s41598-017-16829-w
47. Schaum N, Lehallier B, Hahn O, Palovics R, Hosseinzadeh S, Lee SE, et al. Ageing hallmarks exhibit organ-specific temporal signatures. Nature. (2020) 583:596–602. doi: 10.1530/ey.18.14.11
48. Chung KW, Lee EK, Lee MK, Oh GT, Yu BP, Chung HY. Impairment of PPARalpha and the fatty acid oxidation pathway aggravates renal fibrosis during aging. J Am Soc Nephrol. (2018) 29:1223–37. doi: 10.1681/ASN.2017070802
49. Blokland KEC, Pouwels SD, Schuliga M, Knight DA, Burgess JK. Regulation of cellular senescence by extracellular matrix during chronic fibrotic diseases. Clin Sci (Lond). (2020) 134:2681–706. doi: 10.1042/CS20190893
50. Yuan J, Chen H, Ge D, Xu Y, Xu H, Yang Y, et al. Mir-21 promotes cardiac fibrosis after myocardial infarction via targeting Smad7. Cell Physiol Biochem. (2017) 42:2207–19. doi: 10.1159/000479995
51. Cao W, Shi P, Ge JJ. miR-21 enhances cardiac fibrotic remodeling and fibroblast proliferation via CADM1/STAT3 pathway. BMC Cardiovasc Disord. (2017) 17:88. doi: 10.1186/s12872-017-0520-7
52. Liu G, Friggeri A, Yang Y, Milosevic J, Ding Q, Thannickal VJ, et al. miR-21 mediates fibrogenic activation of pulmonary fibroblasts and lung fibrosis. J Exp Med. (2010) 207:1589–97. doi: 10.1084/jem.20100035
53. Chau BN, Xin C, Hartner J, Ren S, Castano AP, Linn G, et al. MicroRNA-21 promotes fibrosis of the kidney by silencing metabolic pathways. Sci Transl Med. (2012) 4:121ra18 doi: 10.1126/scitranslmed.3003205
54. Zhang Z, Zha Y, Hu W, Huang Z, Gao Z, Zang Y, et al. The autoregulatory feedback loop of microRNA-21/programmed cell death protein 4/activation protein-1 (MiR-21/PDCD4/AP-1) as a driving force for hepatic fibrosis development. J Biol Chem. (2013) 288:37082–93. doi: 10.1074/jbc.M113.517953
55. Lin HY, Wang FS, Yang YL, Huang YH. MicroRNA-29a suppresses CD36 to ameliorate high fat diet-induced steatohepatitis and liver fibrosis in mice. Cells. (2019) 8:1298. doi: 10.3390/cells8101298
56. Zhang Y, Huang XR, Wei LH, Chung AC, Yu CM, Lan HY. miR-29b as a therapeutic agent for angiotensin II-induced cardiac fibrosis by targeting TGF-beta/Smad3 signaling. Mol Ther. (2014) 22:974–85. doi: 10.1038/mt.2014.25
57. Qin RH, Tao H, Ni SH, Shi P, Dai C, Shi KH. microRNA-29a inhibits cardiac fibrosis in Sprague-Dawley rats by downregulating the expression of DNMT3A. Anatol J Cardiol. (2018) 20:198–205. doi: 10.14744/AnatolJCardiol.2018.98511
58. Xiao J, Meng XM, Huang XR, Chung AC, Feng YL, Hui DS, et al. miR-29 inhibits bleomycin-induced pulmonary fibrosis in mice. Mol Ther. (2012) 20:1251–60. doi: 10.1038/mt.2012.36
59. Qin W, Chung AC, Huang XR, Meng XM, Hui DS, Yu CM, et al. TGF-beta/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. J Am Soc Nephrol. (2011) 22:1462–74. doi: 10.1681/ASN.2010121308
60. Roderburg C, Urban GW, Bettermann K, Vucur M, Zimmermann H, Schmidt S, et al. Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology. (2011) 53:209–18. doi: 10.1002/hep.23922
61. Sassi Y, Avramopoulos P, Ramanujam D, Gruter L, Werfel S, Giosele S, et al. Cardiac myocyte miR-29 promotes pathological remodeling of the heart by activating Wnt signaling. Nat Commun. (2017) 8:1614. doi: 10.1038/s41467-017-01737-4
62. Zhao JJ, Lin J, Lwin T, Yang H, Guo J, Kong W, et al. microRNA expression profile and identification of miR-29 as a prognostic marker and pathogenetic factor by targeting CDK6 in mantle cell lymphoma. Blood. (2010) 115:2630–9. doi: 10.1182/blood-2009-09-243147
63. Zhang M, Guo W, Qian J, Wang B. Negative regulation of CDC42 expression and cell cycle progression by miR-29a in breast cancer. Open Med (Wars). (2016) 11:78–82. doi: 10.1515/med-2016-0015
64. Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, et al. Pancreatic cancers require autophagy for tumor growth. Genes Dev. (2011) 25:717–29. doi: 10.1101/gad.2016111
65. Teng Y, Zhang Y, Qu K, Yang X, Fu J, Chen W, et al. MicroRNA-29B (mir-29b) regulates the Warburg effect in ovarian cancer by targeting AKT2 and AKT3. Oncotarget. (2015) 6:40799–814. doi: 10.18632/oncotarget.5695
66. Maes OC, An J, Sarojini H, Wang E. Murine microRNAs implicated in liver functions and aging process. Mech Ageing Dev. (2008) 129:534–41. doi: 10.1016/j.mad.2008.05.004
67. Ugalde AP, Ramsay AJ., de la Rosa J, Varela I, Marino G, Cadinanos J, et al. Aging and chronic DNA damage response activate a regulatory pathway involving miR-29 and p53. EMBO J. (2011) 30:2219–32. doi: 10.1038/emboj.2011.124
68. Ito S, Nagata K. Biology of Hsp47 (Serpin H1), a collagen-specific molecular chaperone. Semin Cell Dev Biol. (2017) 62:142–51. doi: 10.1016/j.semcdb.2016.11.005
69. Kohler A, Morgelin M, Gebauer JM, Ocal S, Imhof T, Koch M, et al. New specific HSP47 functions in collagen subfamily chaperoning. FASEB J. (2020) 34:12040–52. doi: 10.1096/fj.202000570R
70. Sasikumar P, AlOuda KS, Kaiser WJ, Holbrook LM, Kriek N, Unsworth AJ, et al. The chaperone protein HSP47: a platelet collagen binding protein that contributes to thrombosis and hemostasis. J Thromb Haemost. (2018) 16:946–59. doi: 10.1111/jth.13998
71. Nagai N, Hosokawa M, Itohara S, Adachi E, Matsushita T, Hosokawa N, et al. Embryonic lethality of molecular chaperone hsp47 knockout mice is associated with defects in collagen biosynthesis. J Cell Biol. (2000) 150:1499–506. doi: 10.1083/jcb.150.6.1499
72. Zhu J, Xiong G, Fu H, Evers BM, Zhou BP, Xu R. Chaperone Hsp47 drives malignant growth and invasion by modulating an ECM gene network. Cancer Res. (2015) 75:1580–91. doi: 10.1158/0008-5472.CAN-14-1027
73. Christiansen HE, Schwarze U, Pyott SM, AlSwaid A, Al Balwi M, Alrasheed S, et al. Homozygosity for a missense mutation in SERPINH1, which encodes the collagen chaperone protein HSP47, results in severe recessive osteogenesis imperfecta. Am J Hum Genet. (2010) 86:389–98. doi: 10.1016/j.ajhg.2010.01.034
74. Yamamoto N, Kinoshita T, Nohata N, Yoshino H, Itesako T, Fujimura L, et al. Tumor-suppressive microRNA-29a inhibits cancer cell migration and invasion via targeting HSP47 in cervical squamous cell carcinoma. Int J Oncol. (2013) 43:1855–63. doi: 10.3892/ijo.2013.2145
75. Yamada Y, Sugawara S, Arai T, Kojima S, Kato M, Okato A, et al. Molecular pathogenesis of renal cell carcinoma: Impact of the anti-tumor miR-29 family on gene regulation. Int J Urol. (2018) 25:953–65. doi: 10.1111/iju.13783
76. Zhao D, Jiang X, Yao C, Zhang L, Liu H, Xia H, et al. Heat shock protein 47 regulated by miR-29a to enhance glioma tumor growth and invasion. J Neurooncol. (2014) 118:39–47. doi: 10.1007/s11060-014-1412-7
77. Kamran F, Andrade AC, Nella AA, Clokie SJ, Rezvani G, Nilsson O, et al. Evidence that up-regulation of MicroRNA-29 contributes to postnatal body growth deceleration. Mol Endocrinol. (2015) 29:921–32. doi: 10.1210/me.2015-1047
78. Humeres C, Frangogiannis NG. Fibroblasts in the infarcted, remodeling, and failing heart. JACC Basic Transl Sci. (2019) 4:449–67. doi: 10.1016/j.jacbts.2019.02.006
79. Hemanthakumar KA, Fang S, Anisimov A, Mayranpaa MI, Mervaala E, Kivela R. Cardiovascular disease risk factors induce mesenchymal features and senescence in mouse cardiac endothelial cells. Elife. (2021) 10:e62678. doi: 10.7554/eLife.62678
80. Razzaque MS, Shimokawa I, Nazneen A, Higami Y, Taguchi T. Age-related nephropathy in the Fischer 344 rat is associated with overexpression of collagens and collagen-binding heat shock protein 47. Cell Tissue Res. (1998) 293:471–8. doi: 10.1007/s004410051139
81. Masuda H, Fukumoto M, Hirayoshi K, Nagata K. Coexpression of the collagen-binding stress protein HSP47 gene and the alpha 1(I) and alpha 1(III) collagen genes in carbon tetrachloride-induced rat liver fibrosis. J Clin Invest. (1994) 94:2481–8. doi: 10.1172/JCI117617
82. Razzaque MS, Hossain MA, Kohno S, Taguchi T. Bleomycin-induced pulmonary fibrosis in rat is associated with increased expression of collagen-binding heat shock protein (HSP) 47. Virchows Arch. (1998) 432:455–60. doi: 10.1007/s004280050191
83. Khalil H, Kanisicak O, Vagnozzi RJ, Johansen AK, Maliken BD, Prasad V, et al. Cell-specific ablation of Hsp47 defines the collagen-producing cells in the injured heart. JCI Insight. (2019) 4:e128722. doi: 10.1172/jci.insight.128722
84. Takeda K, Kusachi S, Ohnishi H, Nakahama M, Murakami M, Komatsubara I, et al. Greater than normal expression of the collagen-binding stress protein heat-shock protein-47 in the infarct zone in rats after experimentally-induced myocardial infarction. Coron Artery Dis. (2000) 11:57–68. doi: 10.1097/00019501-200002000-00011
85. Hagiwara S, Iwasaka H, Shingu C, Matumoto S, Hasegawa A, Noguchi T. Heat shock protein 47 (HSP47) antisense oligonucleotides reduce cardiac remodeling and improve cardiac function in a rat model of myocardial infarction. Thorac Cardiovasc Surg. (2011) 59:386–92. doi: 10.1055/s-0030-1250658
86. Annoni G, Luvara G, Arosio B, Gagliano N, Fiordaliso F, Santambrogio D, et al. Age-dependent expression of fibrosis-related genes and collagen deposition in the rat myocardium. Mech Ageing Dev. (1998) 101:57–72. doi: 10.1016/S0047-6374(97)00165-6
87. Besse S, Robert V, Assayag P, Delcayre C, Swynghedauw B. Nonsynchronous changes in myocardial collagen mRNA and protein during aging: effect of DOCA-salt hypertension. Am J Physiol. (1994) 267:H2237–44. doi: 10.1152/ajpheart.1994.267.6.H2237
88. Robert V, Besse S, Sabri A, Silvestre JS, Assayag P, Nguyen VT, et al. Differential regulation of matrix metalloproteinases associated with aging and hypertension in the rat heart. Lab Invest. (1997) 76:729–38.
Keywords: aging, miRNA, fibrosis, SERPINH1, prediction analysis, miR-29a
Citation: Rusu-Nastase EG, Lupan A-M, Marinescu CI, Neculachi CA, Preda MB and Burlacu A (2022) MiR-29a Increase in Aging May Function as a Compensatory Mechanism Against Cardiac Fibrosis Through SERPINH1 Downregulation. Front. Cardiovasc. Med. 8:810241. doi: 10.3389/fcvm.2021.810241
Received: 06 November 2021; Accepted: 17 December 2021;
Published: 18 January 2022.
Edited by:
Katherine Athayde Teixeira De Carvalho, Pelé Pequeno Príncipe Research Institute, BrazilReviewed by:
Dayu Sun, Army Medical University, ChinaCopyright © 2022 Rusu-Nastase, Lupan, Marinescu, Neculachi, Preda and Burlacu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Alexandrina Burlacu, c2FuZGEuYnVybGFjdUBpY2JwLnJv
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.