 Haiying Wang1
Haiying Wang1 Yijun Han2Shenwei Li3Yunan Chen3
Yijun Han2Shenwei Li3Yunan Chen3 Yafen Chen3Jing Wang4Yuqing Zhang3Yawen Zhang3Jingsuo Wang3
Yafen Chen3Jing Wang4Yuqing Zhang3Yawen Zhang3Jingsuo Wang3 Yong Xia5*
Yong Xia5* Jinxiang Yuan6*
Jinxiang Yuan6*- 1Department of Physiology, Institute of Basic Medical College, Jining Medical University, Jining, China
- 2Clinical Medical College, Jining Medical University, Jining, China
- 3Institute of Basic Medical College, Jining Medical University, Jining, China
- 4Dongying Fifth People's Hospital, Dongying, China
- 5Key Laboratory of Precision Oncology of Shandong Higher Education, Institute of Precision Medicine, Jining Medical University, Jining, China
- 6The Collaborative Innovation Center, Jining Medical University, Jining, China
Mitochondria is a ubiquitous, energy-supplying (ATP-based) organelle found in nearly all eukaryotes. It acts as a “power plant” by producing ATP through oxidative phosphorylation, providing energy for the cell. The bioenergetic functions of mitochondria are regulated by nuclear genes (nDNA). Mitochondrial DNA (mtDNA) and respiratory enzymes lose normal structure and function when nuclear genes encoding the related mitochondrial factors are impaired, resulting in deficiency in energy production. Massive generation of reactive oxygen species and calcium overload are common causes of mitochondrial diseases. The mitochondrial depletion syndrome (MDS) is associated with the mutations of mitochondrial genes in the nucleus. It is a heterogeneous group of progressive disorders characterized by the low mtDNA copy number. TK2, FBXL4, TYPM, and AGK are genes known to be related to MDS. More recent studies identified new mutation loci associated with this disease. Herein, we first summarize the structure and function of mitochondria, and then discuss the characteristics of various types of MDS and its association with cardiac diseases.
Structure and Function of Mitochondria
Mitochondrial Structure
The mitochondrion is a double membraned organelle commonly found in eukaryotic cells. It consists of four compartments: an outer mitochondrial membrane (OMM), an inner mitochondrial membrane (IMM), a mitochondrial intermembrane space (IMS), and membrane matrix. The OMM is smooth and contains plenty of pore-forming components. The IMM curves inward and folds into a crest containing a large number of enzymes. It is the site of biological oxidation, a process involving respiratory chain complex (CI-IV) and ATP synthase (CV) (1). IMS, located between the OMM and IMM, contains cytochrome c (Cyt-c), ADP/ATP carrier (AAC), and biotic factors and enzyme proteins including apoptosis-inducing factor (AIF) and pro-caspase 2, 3, 9. Mitochondrial permeability transition pores (MPTP) exist at the OMM-IMM contact site. The opening of MPTPs is related to mitochondrial membrane permeability and is subjected to regulation by lipometabolism, mitochondrial membrane potential change, oxidative stress, and other factors (2). Membrane matrix is a single continuous space containing reactive enzymes, mitochondrial genome, mitochondrial DNA (mtDNA), ribosomal RNA (rRNA), and transfer RNA (tRNA), which have functional roles in biotransformation and synthesis. Mitochondria are semi-autonomous organelles that are strongly dependent on the biological function of nuclear genes. In mammals, mtDNA consists of 37 genes, including 22 tRNA genes, 13 genes encoding mitochondrial respiratory chain and oxidative phosphorylation-related protein subunits that assemble respiratory chain complex I-IV(CI-CIV), and 2 rRNA genes (12S and 16S) (3). The majority of mitochondrial proteins are encoded by nucleic genes which first synthesize precursor proteins, and then process in the mitochondria to form mature proteins (4). Mitochondria are the “power workshops” of cells. They provide energy for cellular activities and perform biological functions such as signal transduction and biological oxidation.
Mitochondrial Function
ATP Production by Oxidative Phosphorylation
Mitochondria is the metabolic center of cellular activities. ATP is often called “energy currency” of the cell, and the breakage of high energy phosphate bonds by hydrolysis can provide energy for cellular activities. ATP synthesis is indispensable of oxidative phosphorylation (OXPHOS). OXPHOS is closely related to material energy metabolism (5), electron transfer of the mitochondrial respiratory chain and the enzymatic reaction of a series of protease complexes. OXPHOS contributes to ninety percent of the energy production in eukaryotic cells. On the lipid double layers embedded in IMM, electrons are transmitted to O2 by compound I-IV on the respiratory chain, while simultaneously releasing energy for ATP production in coupling with F0/F1-ATP synthase (complex V) (6) (Figure 1). ATP synthesis is impaired when mitochondrial phosphorylation function becomes dysregulated.
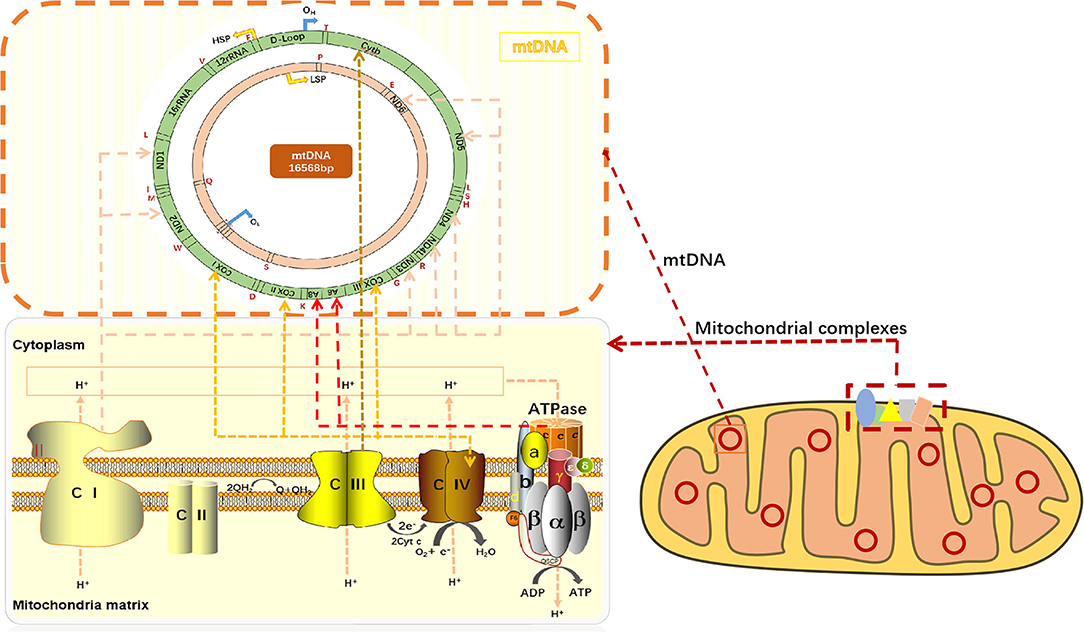
Figure 1. mtDNA encodes respiratory chain complexes for electron transport. Mitochondrial respiratory chain complex I (NADH oxidase) and mitochondrial respiratory chain complex II (succinate dehydrogenase) are the major components of electron entry into mitochondrial electron transport chain (ETC). Complexes I and II catalyze NADH oxidation and succinic acid oxidation to fumaric acid, respectively. Next, coenzyme Q (Q) forms coenzyme (QH2), leading to a decrease in oxygen (the final electron receptor). Mitochondrial respiratory chain complex III (cytochrome c oxidoreductase) is the gatekeeper of the mitochondrial respiratory chain and the main source of reactive oxygen species III, which transfers electrons from panthepanol (reduced coenzyme Q or CoQ) to cytochrome c (cyt c) through a catalytic mechanism of the Q cycle. The mitochondrial respiratory chain complex IV (cytochrome c oxidase) is the final electron acceptor of the mitochondrial ETC, which converts oxygen into water through the oxidation of cyt c, a process associated with ATP synthesis in the mitochondrial membrane. Mitochondrial respiratory chain complex V, also known as F1F0-ATPASE or ATP synthase, works with the four complexes to complete oxidative phosphorylation for ATP production. CI or NADH: Ubiquinone oxidoreductase or NADH encoded by ND1, ND2, ND3, ND4, ND4L, ND5, and ND6 on mtDNA, respectively; CII: dehydrogenase succinic acid, ubiquinone oxidoreductase; CIII: Ubiquinone and ferricytochrome C reducing enzyme encoded by Cytb part on mtDNA, respectively; CIV: CIV, cytochrome c oxidoreductase encoded by COXI, COXII, and COXIII on mtDNA, respectively; The red letters T, F, V, L, I, M, W, D, K, G, R, H, S, L, A, N, C, Y, S, E, P, and Q all indicate tRNA.
Changes in the Mitochondrial Membrane Potential (Δψ) via Electron Transfer in the Mitochondrial Respiratory Chain
The transmission of mitochondrial respiratory chain is indispensable of electronic transfer. Therefore, the process is known as the electron transport chain (ETC). Mitochondria pass through complexes I (CI or NADH: ubiquinone oxidoreductase or NADH dehydrogenase), II (CII, succinic acid: ubiquinone oxidoreductase), III (CIII, ubiquinone: Ferric cytochrome C reductase) and IV (CIV, cytochrome C oxidoreductase), transfer electrons to O2, and release energy which couples with complex V (CV, ATP synthase) to form ATP (7). In mammals, CI is the largest mitochondrial complex composed of 45 subunits, of which 7 are encoded by mitochondrial DNA, namely ND1, ND2, ND3, ND4, ND4L, ND5, and ND6, and the remaining 38 subunits are encoded by nuclear genes. NADH: ubiquinone oxidoreductase subunit AB1 (NDUFAB1), or known as mitochondrial acyl carrier protein, is an important subunit of CI, which can mediate cardioprotective mechanisms and improve mitochondrial bioenergy (8). CIV consists of 13 subunits, of which the largest three subunits (COX I, COX II and COX III) are encoded by mtDNA and can form the core of the cascade reaction, while the remaining 10 subunits (COIV, Va, Vb, VIa-c, VIIa-c, and VIII) are encoded by nDNA. Moreover, the activity of CIV is highly relevant to muscle OXPHOS, followed by CII. CIIIis the most critical mitochondrial respiratory chain enzyme, consisting of 11 different subunits, of which only Cytb is encoded by mtDNA, and Cytb forms a dimer with Cyt C1 and iron-sulfur protein (Fe-S) (9). CIII transfers electrons from panthenol (reductive coenzyme Q or CoQ) to cytochrome C (Cyt C) through a catalytic mechanism, while protons are transferred from the mitochondrial matrix to the IMS (10) (Figure 1). In the process of electron transfer, the complexes interact with each other to form an advanced supramolecular structure known as the supercomplex (SCs) and respiratory corules (11). SCs can improve the efficiency of electron flux through substrate channels or enhanced catalysis, thus promoting electron transport, isolating reaction intermediates and preventing reactive oxygen species (ROS) generation (12).
Mitochondrial and Reactive Oxygen Species
During ATP generation, ETC, and Krebs cycle inevitably lead to electron leakage, causing incomplete reduction of O2 and formation of free radicals in a form of superoxide anion (O2−). At this point, if the muscle remains in a long-term contraction state, the content of ATP generated by oxidative phosphorylation of respiratory chain increases, and electrons overflow into ETC to generate ROS (13). Furthermore, a low level of ROS can act as signaling molecules that maintain normal cell signal transduction. In contrast, a high level of ROS produced more than endogenous scavenging capacity can trigger cascade reactions that damage cellular macromolecules, such as DNA and proteins. Excessive ROS can also reduce the Ca2+sensitivity of myofibril and damage the contractile function of muscle, possibly through enzyme complexes i and iii (Ci, Ciii)-dependent and/or reduced nicotinamide adenine dinucleotide (NADPH) oxidase-dependent pathways. Alternatively, α-ketoglutaric acid in succinyl-coenzyme A can be oxidized indirectly through mitochondria dehydrogenase to generate NADH, which is responsible for ROS overproduction (14). It has been shown that ROS produced by ETC can damage mtDNA. In the absence of proper repair, DNA damage accumulates, causing a series of mitochondrial-related diseases (15).
Role of Oxygen in Mitochondrial Function
Oxygen is key to the functionality of mitochondria. Oxygen affects normal structure and function of the mitochondria. In the third stage of aerobic respiration, mitochondria use NADH, a product of glycolysis and Tricarboxylic Acid Cycle (TCA), reduced flavin adenosine dinucleotide, FADH2 and other high-energy molecules to reduce oxygen, thereby releasing energy to synthesize ATP. The content of oxygen in the environment is closely related to normal oxidative phosphorylation function of mitochondria (16, 17). In mammals, hypoxic signaling protein (HIF) determines the cellular response to hypoxia. Stable HIF protein controls the expression of glycolysis-related target genes at an appropriate level and maintains ROS production at a low, physiological level by changing the structure of CI, II and IV ETC complexes and reducing the enzyme activity of the complex (18). The morphology of fetal cardiac mitochondria at birth is associated with hypoxia signaling. Downregulation of mitochondrial HIF pathway after birth leads to increased respiratory capacity and oxidative phosphorylation. Neary et al. found that HIF signal transduction was maintained under normoxic conditions in mouse heart. The α-subunit proline residues of HIF-α are continuously hydroxylated by Prolyl Hydroxylase (PHD) under normoxic conditions. Hypoxia inhibits PHD, which stabilizes HIF-α and promotes its migration to the nucleus where it dimers with the β subunit and binds to the promoters or enhancers through the hypoxia response elements (HRE) to initiate gene transcription (19, 20). Mitochondria are the main oxygen consumers in the heart. When cardiomyocytes are hypoxic, anaerobic glycolysis increases whereas both mitochondrial oxygen consumption and respiration rate decrease. Ambrose et al. have established a cell model of cardiac hypoxia by using HL-1 cardiomyocytes cells highly related to adult cardiac metabolism. The investigators found that the number of mitochondria increased with a simultaneous decrease in CIV activity after 24 h of hypoxia, suggesting that mitochondria keep their total content fairly consistent during remodeling (21).
Mitochondria and Cell Metabolism
Mitochondria achieve metabolic functions mainly by oxidative phosphorylation, ATP production, NADH oxidation, glycolysis and FADH2 production, TCA cycle, and β–oxidation of fatty acids (22). Meanwhile, mitochondria regulate their fusion, fission, biosynthesis, and degradation through metabolism.
Metabolism-Related Biochemical Reactions
Metabolism is fundamental to cellular activities. It represents distinct processes by which human body converts the nutrients, including sugars, lipids and proteins, into small molecules for absorption and consumption. Fatty acid β -oxidation (FAO) is a major process of long-chain fatty acid metabolism. It produces acetyl coenzyme A (acetyl-CoA), NADH, and FADH2 (23). The TCA, also known as the Krebs cycle, is the hub that links the metabolism of three nutrients. Acetyl-CoA is a common end product of nutrient metabolism, which eventually enters TCA in the form of citrate for thorough oxidation (24). Acetyl CoA is decomposed into CO2 and H. H is delivered to flavin adenine dinucleotide (FAD) and oxidized nicotinamide adenine dinucleotide (NAD+) to generate FADH2 and NADH (25). Metabolism and energy homeostasis are maintained through the activities of various enzymes including AMP-activated protein kinase (AMPK), a conserved protein present in all eukaryotes. AMPK is involved in the metabolism of lipids and carbohydrates. AMPK is activated when the intracellular AMP concentration increases or the skeletal muscle contracts. The activity of AMPK is also regulated by hormones, cytokines, and ROS (26).
Metabolic Regulation of Mitochondrial Dynamics
Mitochondria are highly dynamic organelles. Mitochondrial dynamics is a physiological process in which mitochondria undergo continuous cycles of fusion and division, resulting in changes in the shape, size and position of mitochondria. Mitochondrial dynamics is often determined by the dynamic balances between fission and fusion as well as between biogenesis and degradation such as mitochondrial phagocytosis (27). Mitochondrial fission and fusion are of great importance in the growth and redistribution of mitochondria and the maintenance of a healthy mitochondrial network (28, 29). Through continuous fusion and fission, mitochondria can exchange the damaged mitochondrial DNA and protein lipids with normal counterparts, thus maintaining the stability of mitochondrial structure and function (30). During this process, the core proteins that mediate mitochondrial division and fusion are membrane remodeling mechanochemical enzymes, such as dynein-related protein-like 1 (Drp1/DLP1), optic nerve atrophy 1 (OPA1), and mitochondrial protein (Mfn). Drp1 regulate fission, while OPA1 and Mfn modulate the fusion of IMM and OMM, respectively (31). It is likely that the processes of mitochondrial fission and fusion are asymmetric in the formation of daughter mitochondria. It has been found that the dynamic changes in mitochondria and CR structure formed by endointimal folding have a significant impact on the integrity of mtDNA. CR distortion often leads to mtDNA loss (32). Genetic loss of mitochondrial fusion can result in inadequate mitochondrial mixing, which links to great heterogeneity of mitochondrial proteins, DNA, and membrane potentials. A function of fission is to identify dysfunctional offspring mitochondria, which are subsequently removed by mitochondrial phagocytosis and coordinate for inhibition of mitochondrial fusion (33). Normal mitochondrial biogenesis depends not only on the action of nDNA and mtDNA, but also on the coordination of mitochondrial DNA replication and fusion with division. Environmental factors, such as oxidative stress, exercise, temperature stress (cold or heat), cell division, regeneration and differentiation, can promote mitochondrial biogenesis. Inhibition of mitochondrial dynamics has some effects on the elimination of irreparable mtDNA damage and transmission of mtDNA mutations (34).
Ca2+ Storage
Accumulation of Ca2+ in the mitochondrial matrix regulates aerobic metabolism by activating enzymes in the metabolic reactions. Under physiological conditions, Ca2+ actively regulates the key dehydrogenase in the TCA response, which is conducive to the regulation of Ca2+-dependent functions in the cytoplasm. Under pathological conditions, high Ca2+ concentration can promote the opening of mitochondrial permeability transition pore (MPTP) and induce apoptotic death (35, 36). Excessive deposition of Ca2+ often leads to decreased ATP synthesis and increased ROS production, thus aggravating oxidative stress responses. Ca2+ overload is often associated with intracellular Ca2+ leakage. Sarcoplasmic reticulum (SR) Ca2+ leakage mainly occurs through type 2 Ryanodine receptor (RyR2). It plays a fundamental role in Ca2+ pathophysiology. RyR2 regulates the level of Ca2+ by influencing cardiac excitation-contraction (E-C) coupling (37). Ca2+ concentration in mitochondria is also regulated by genes encoding Ca2+ unidirectional complexes, including mitochondrial calcium uniporter (MCU) and its regulator Ca2+ uptake protein 1 located in the IMM (38). Ca2+ absorption is dependent on the energy transfer of the respiratory chain. Due to the restriction of IMM on MCU, a high level of Ca2+ is required for MCU to enter the mitochondrial matrix, thus, Ca2+ absorption is relatively easy in the vicinity of Ca2+ release reservoirs, such as the endoplasmic reticulum (ER). Mitochondria-associated endoplasmic reticulum membrane (MAM) has been found to transport Ca2+. Ca2+ can be directly transferred to mitochondria via ER membrane where it controls the relevant mitochondrial functions (39).
Mitochondrial Genome Regulation of Its Function
The mitochondrial genome consists of 37 genes, including 22 tRNA genes, 13 genes that encode the subunits of mitochondrial respiratory chain and oxidative phosphorylation-related proteins snd 2 rRNA genes (12S and 16S). Nuclear genes encode about 1,500 mitochondrial proteins, which comprise the majority of mitochondrial respiratory chain polypeptides (40). Additionally, mtDNA also encodes 13 core catalytic peptides, including oxidative phosphorylation complexes i, iii, and iv (41).
It appears that mtDNA depends on mitochondrial nDNA to complete its replication, transcription, translation as well as other processes. Therefore, both nDNA and mtDNA have the regulatory effects on mitochondria, and they jointly ensure the normal function of the mitochondria (42). Given that mtDNA encodes subunits of the respiratory chain complex, mutations in mtDNA can damage the enzymatic activity of oxidative phosphorylation reaction complex, in particular CI, affecting mitochondrial biogenesis (43). The level of mtDNA correlates to mitochondrial fusion. In skeletal muscle, the association between fusion and enzymatic activity in OXPHOS may limit mtDNA defects to the local regions of muscle fibers, which are often associated with mitochondrial myopathy (44). Altogether, mitochondria regulate cell metabolism, growth, senescence, and apoptosis through the coordinated actions of nDNA and mtDNA.
Mitochondrial DNA Depletion Syndrome
As an autosomal recessive disease, mitochondrial DNA depletion syndrome (MDS) is characterized by severe reduction in mtDNA content in the affected tissues and organs (45). Because mtDNA is controlled by nDNA, the copy number of mitochondria may be reduced when the nuclear gene mutations destroy deoxyribonucleotide metabolism in the affected tissues. The production of key subunits of mitochondrial respiratory chain complex and energy production require a sufficient amount of mtDNA. Deletion of mtDNA can lead to organ dysfunction as a result of insufficient synthesis of respiratory chain components (46–48). Commonly mutated genes include TK2, FBXL4, TYPM, AGK, and others. MDS is classified as four forms, which are myopathic form, encephalomyopathic form, hepatocerebral form, and neurogastrointestinal form (49). MDS is usually involved in multiple systems with muscle-related symptoms, such as dystonia and muscle atrophy (50, 51).
Myopathic Mitochondrial DNA Depletion Syndrome
Myopathic MDS, also known as mitochondrial DNA depletion syndrome 2 (MTDPS2) or TK2 deficiency, is characterized by the reduction of mtDNA content in the affected tissues. It is often associated with severe progressive muscle weakness. Dysphagia mainly involves the respiratory system with other clinical manifestations (52). Some patients have clinical symptoms of hypertrophic cardiomyopathy (53). This type of MDS is usually caused by mutation of TK2 gene. It occurs in early childhood and presents in the form of myopathy, mostly accompanied by mtDNA deletion/depletion and increased serum level of creatine kinase (CK) (54, 55). TK2, located on chromosome 16Q21, encodes a mitochondrial enzyme, thymidine kinase 2 (TK2). TK2 is a rate-limiting enzyme which mediates deoxypyrimidine phosphorylation (56). In non-replicating cells, TK2 is also involved in the maintenance of dNTP (57). In clinical and molecular genetics, myopathic MDS can be further divided into three different subtypes, namely, paroxysmal myopathy in infants with severe depletion of mtDNA, paroxysmal myopathy in childhood, and delayed myopathy, among which delayed myopathy is the rarest. In addition to muscle weakness, delayed myopathy can also progress to respiratory insufficiency. Different from the previous two, in addition to muscle weakness, respiratory insufficiency can also be developed at the onset and after 12 years old. Muscle mtDNA can show myopathy with deletion, multiple deletion, or both (58).
Encephalomyopathic Mitochondrial DNA Depletion Syndrome
Encephalomyopathic is a relatively common form of MDS, in particular among adults who have progressive external eye muscle paralysis (adPEO). adPEO manifests by ptosis and severe limitation of eye movement. It may also lead to ataxia and cataract (59). Mitochondrial DNA depletion syndrome 13 (MTDPS 13), also known as FBXL4-related encephalopathy, is a form of MDS manifested by early lactic acidosis, hypotonia, developmental delay, and feeding difficulty (60). In some cases, encephalopathic MDS accompanies liver disease to form hepatocerebral MDS (59). Mitochondrial DNA depletion syndrome 9 (MTDPS 9) can also occur in patients with cerebromyopathic MDS. Lu et al. reported a case of infantile encephalomyopathic MDS associated with SUCLG 1 (succinic acid A ligase α subunit) gene in 2017. The case is manifested by neuromyopathy-like symptoms including muscular hypotonia, psychomotor retardation, brain atrophy, developmental delay, and increased levels of methylmalonic acid and its metabolite (61). Encephalomyopathy can be caused by single gene or polygenic mutations with similar effects. These genes may have synergistic effect on the development of MDS (45).
Hepatocerebral Mitochondrial DNA Depletion Syndrome
Hepatocerebral MDS is one of the most common MDS diseases, and at least 50 kinds of related gene mutations have been discovered (62). This disease may occur in newborns within 6 months after birth, as characterized by vomiting, developmental delay, serious progressive liver failure, hypotonia, hyperreflexia, irritability, and hypoglycemia. Patients often die within a year after the onset of symptoms (63). Hyperlactatemia and 3-methylglutaric aciduria are the well-known characteristics of patients with hepatic encephalopathy, while some patients are accompanied by nephrolithiasis or mild ventricular hypertrophy (64). The liver biopsy can show varying degrees of steatosis, bile duct hyperplasia, fibrosis, lobular collapse, and other symptoms. Studies have shown that most patients presented a pattern of fibrosis with neovascularization following diffuse necrosis (65). Reduction of cytochrome-C oxidase (COX) and combined defects of mitochondrial respiratory chain complex may also be detected in infants' livers (66). Clinically, the identified liver-brain type mutations include POLG1, DGUOK, C10orf2, and MPV17, among which MPV17 is relatively rare (67).
Neurogastrointestinal Mitochondrial DNA Depletion Syndrome
Mitochondrial neurogastrointestinal mitochondrial DNA depletion disease (MNGIE) is a rare metabolic disease caused by TYMP gene mutation encodes thymidine phosphorylase (TP). Patients with inherited, autosomal recessive TYMP mutations show clinical symptoms between 10 and 50 years old, most of which appear before 20 years old (68). In MNGIE, a loss of TP activity leads to the accumulation of deoxyuridine (dUrd) and thymidine (Thd), which infiltrate through the mitochondrial pyrimidine remedy pathway, leading to an imbalance of deoxyuridine triphosphate pool (dNTP). This damages mtDNA, resulting in mitochondrial depletion and ultimately cell death (69). The clinical manifestations of MNGIE include gastrointestinal motility disorders, cachexia, ptosis, ophthalmoplegia, peripheral neuropathy, and related encephalopathy, which often lead to death in adulthood (70). The detection of hypoxia inducible factor-1α (HIF-1α) and vascular endothelial growth factor (VEGF) protein expression proved that MNGIE patients were accompanied by gastrointestinal vascular diseases (71). Currently, hemodialysis can temporarily eliminate the toxic effects of Thd and dUrd in patients. However, permanent TP replacement therapy with AHSCT is required for more effective treatments of MNGIE (69). The correlation between MNGIE vascular changes and neuromuscular abnormalities provides a new strategy for the treatment of this disease (72). Carrier erythrocyte entrapped thymidine phosphorylase therapy and allogeneic hematopoietic stem cell transplantation are currently available clinical therapeutic options for MNGIE (71).
MDS and Its Association With Cardiac Disease
Highly energy-consuming organs including heart requires a proper function of mitochondria. Cardiac energy is primarily provided through mitochondrial OXPHOS (73). Cardiomyocytes are prone to hypoxia. There are two mitochondrial populations in cardiomyocytes, namely, interfibrous mitochondria and submyomembrane mitochondria. The interstitial mitochondria are arranged longitudinally and in parallel. The cristae are mainly arranged in tubular shape. Subsarcolemmal mitochondria gather beneath the sarcolemma in the direction of lamellar ridge (74). Mitochondria are vital for the development and maintenance of heart muscle, the largest energy user in the human body. Mitochondria are not only a source of ATP energy, but also generators of ROS, which can both regulate physiological processes, such as the transition from hyperplastic growth to hypertrophic growth after birth, and cause oxidative damage. Excessive ROS production and oxidative damage are associated with cardiac pathology (75). In addition to lowering bioenergy efficiency, damaged mitochondria also produce more reactive oxygen species, resulting in harmful functional and structural consequences in the cardiovascular system (76). The increased production of ROS leads to vascular inflammation, endothelial cell damage and deposition of oxidized low density protein (oxLDL) in the arterial wall (77). In cardiomyocytes, two mitochondrial populations are electrically coupled to each other, providing electrical conduction from one to the other (78). Dysfunctional mitochondria can impair electronic transfer chain activity, cause ATP depletion, increase oxidative stress, and activate intrinsic apoptosis, leading to myocardial cell death (79). Metabolism is not only crucial for regulating the pump function of the heart, but also for maintaining the functional balance and cellular senescence of cardiomyocytes (80). The important functions of mitochondria in redox balance, energy metabolism and calcium balance determine its pathogenic role in heart failure (HF) and cardiovascular disease (CVD) (81–83). There is no effective treatment at present (84). Accumulation of evidence on mitochondrial therapy may help to provides new targets for the treatment of MDS-associated heart disease (85).
Mitochondria, as an important energy “power plant” of the human body, are closely related to the function of the heart. The changes in mitochondrial structure and physiological function play important roles in heart development, heart remodeling, heart failure, and ischemia-reperfusion injury (86). Some cardiac diseases, such as cardiomyopathy, are mostly related to mitochondrial dysfunction. Pulmonary hypertension may lead to enlargement of the right ventricle. Persistent pulmonary hypertension of neonatal apical hypertrophic cardiomyopathy has been proven to be a rare manifestation related to mitochondrial dysfunction (87). When mitochondria are dysfunctional, mtDNA is damaged due to an increase in ROS production (88). Studies have found that MDS involves not only the brain, liver, skeletal muscle and other organs, but also the heart. The impaired mitochondrial DNA replication may result in mtDNA depletion, and ultimately leads to heart failure. Accordingly, the number of the protein encoded by mtDNA is also reduced (73). Santorelli et al. found a tissue-specific deletion of cardiac mtDNA in an infant patient with hypertrophic cardiomyopathy and mitochondrial exhaustion syndrome. There is increasing evidence that MDS can cause cardiomyopathy (89). In addition to the increased levels of blood lactic acid and pyruvate, liver injury and abnormal myocardial enzymes are also common in laboratory examination (90). Two out of twelve MDS children were reported to have elevated myocardial enzymes (91).
FBXL4, ANT1, AGK, and SLC25A4 have been confirmed to be associated with cardiomyopathy. Wang et al. reported two cases of AGK mutation as a serious manifestation in infants with heart failure. Their skeletal muscle biopsy showed severe mtDNA depletion, along with muscle weakness, growth retardation, and other manifestations of muscle disorders. The cases were diagnosed as Sengers syndrome, thus providing strong evidence for the relationship between mitochondrial depletion syndrome and cardiomyopathy (92). Mitochondrial DNA mutation is considered to change the adaptation between bioenergy systems and dietary patterns, affect the cellular response to stimuli, and increase the susceptibility of mutant individuals to cardiometabolic diseases (93–96).
MDS-Related Genes and Their Defects
TK2
TK2 is a 16q21 gene located on chromosome 16, which encodes for thymidine kinase 2. Many TK2 gene mutations are associated with MDS. Yan et al. demonstrated a mitochondrial DNA depletion syndrome-2 patient caused by TK2 compound heterozygous mutation. The TK2 mutation is considered a recessive genetic mutation type (97). In a case series of 18 patients, Cristina Domínguez-González et al. (99) found that nearly 80% of the patients developed MDS before the age of 12, and only 19% of the patients developed after the age of 12. Clinically, TK2-related MDS patients share common symptoms, including progressive myopathy, skeletal muscle involvement (e.g., facial and axial neck flexor weakness), respiratory system involvement, and chronic progressive external ophthalmoplegia (CPEO) (98). A previous study found that mtDNA depletion mostly affected the brain and heart tissues of TK2 H126N knock-in mice (99). Measurements of mitochondrial TK2 protein by immunoprecipitation found high levels of TK2 in heart, liver, and spleen. The level of thymidylate synthase (TS) protein was also high in the heart tissue (100). Thymidine Triphosphate (TTP) is synthesized mainly by TK2 in the heart. Kamath et al. found that thymidine monophosphate (TMP) could promote the synthesis of TTP independent of TK2 in the cardiac mitochondria of rats in vitro, suggesting a potential therapeutic strategy for TK2 deficiency-related diseases (101).
SUCLG1
SUCLG1 gene encodes the α-subunit of succinate COA ligases (SUCL, also known as succinyl CoA synthase). SUCLG1 subunit is widely expressed in human tissues and abundant in the brain, heart, liver and kidney. A double allele mutation in SUCLG1 gene can promote the deficiency of succinate synthase or ligase activity, resulting in the reduction of mtDNA synthesis and mtDNA depletion (102–104). Several studies have shown that SUCLG1 mutations may be related to mitochondrial hepatic encephalopathy. Hove et al. reported that a neonatal patient had elevated transaminase and liver failure symptoms within 2 months after birth. The mtDNA content of the patient was also decreased, accompanied by progressive myopathies. Gene sequence analysis demonstrated a homozygous mutation [c.40A>T (p.M14L)] in SUCLG1 gene (105). Clinical data showed that succinate ligase deficiency was associated with mtDNA depletion. Liu and co-workers reported five new SUCLG1 mutations, including exon 5 c.550G >A (p.G184S), exon 7 C.751C>T (p.G251S), exon 7 c.809A>C (p.L270W), exon 8 c.961C>G (p.A321P), and exon 9 826-2A>G (splicing), in three Chinese patients with succinate-CoA ligase deficiency (103). The latter two mutations are speculated to be destructive. There are only few case reports linking SUCLG1 mutation to heart diseases (106). Therefore, it is worth of tracking and evaluating the potential relationship between SUCLG1 mutation and impairment of cardiac function due to the abundance of SUCLG1 in the heart (Figure 2).
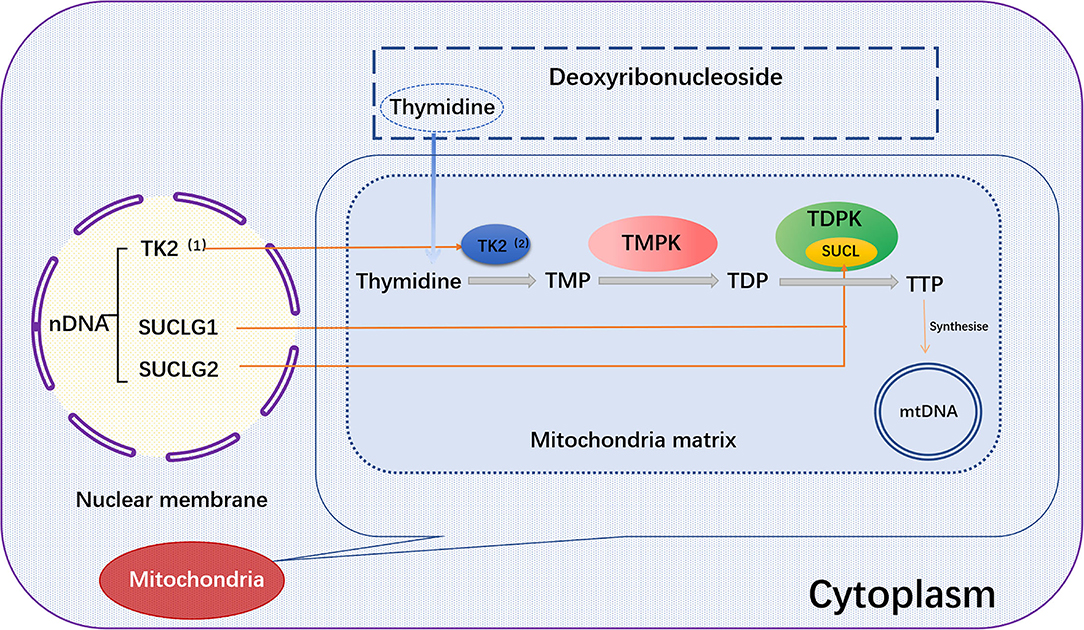
Figure 2. TK2, SUCLG1, or SUCLG2 regulates mtDNA synthesis. In the salvage pathway, the pre-formed deoxyribothymidine enters the mitochondria. The deoxyribonucleoside is then catalyzed by the thymokinases TMPK and TDPK to phosphorylate the deoxyribonucleoside into the triphosphate form in the subsequent three phosphorylation steps, and further synthesize mtDNA. Among them, TK2 and TDPK are encoded by the nuclear genes TK2 and SUCLG1/SUCLG2, respectively. TK2, (1) Thymidine Kinase 2, (2) Thymidine kinase; SUCLG1, succinic acid A ligase α subunit; SUCLG2, succinate-CoA ligase GDP-forming β subunit; Thymidine, deoxyribosylthymine; TMPK, Thymidylate kinase; TDPK, Thiamine diphosphokinase; SUCL, Succinyl CoA ligase; TMP, Thymidine monophosphate; TDP, Thymidine diphosphate; TTP, Thymidine 5'-triphosphate.
SLC25A4
SLC25A4 gene is located on chromosome 4 and encodes for adenine nucleotide translocase 1 (ANT1), which is widely expressed in the heart and skeletal muscles. ANT1 translocates ADP/ATP across the IMM, provides ADP to the mitochondrial ATPase (Complex V), and plays an important role in ATP/ADP exchange between the cytosol and mitochondrial compartment (107, 108). Many studies have shown that SLC25A4 is related to mtDNA depletion sign 12. Thompson et al. found that significant indigenous depletion of mtDNA caused severe mitochondrial respiratory chain deficiencies (109). They identified 2 pathogenic mutation sites: c.703C>G (p.Arg235Gly) and c.239G>A (p.Arg80His). SLC25A4 c.239G>A was found in a case of mitochondrial exhaustion syndrome (MTDPS12) with epileptic encephalopathy. The patient was admitted to hospital due to continuous facial and limb spasm, accompanied by mild respiratory failure (110). A frame-shift null mutation (c.523delC, p.Q175RfsX38) of SLC25A4 resulted in autosomal recessive myopathy and cardiomyopathy. Ten patients with homozygous mutation (adenine nucleotide transposon −1−/−) were followed up for 6 years. These patients displayed continuous adrenergic activation, exercise intolerance, lactic acidosis, hyperalaninemia, and progressive myocardial thickening. Moreover, ECG, echocardiography, and velocity vector imaging analyses indicated the dysregulated left ventricular relaxation, abnormal myocardial repolarization, and abnormal systolic mechanics. Extensive degeneration of cardiomyocytes and structural abnormalities of mitochondria were also evidence in these patients (111). The characteristics of myopathy and cardiomyopathy were also observed in mice lacking the heart/muscle isoform of ANT1 (112).
AGK
Sengers syndrome is an autosomal recessive genetic disease caused by AGK mutations. AGK, a mitochondrial membrane kinase, is involved in protein biogenesis (112). Besides, intracellular LPA is generated by the phosphorylation of monoacylglycerol by AGK (113). Vukotic et al. found that AGK belonged to a subunit of the TIM22 complex that facilitates the transport of metabolite carriers into the IMM. ANT1 levels were reduced in the cells of Sengers syndrome patients, suggesting that AGK is needed for the efficient insertion of metabolite carriers (e.g., SLC25A24 and ANT1) (114). The main clinical features of Sengers syndrome are hypertrophic cardiomyopathy, bi-sided cataracts, myopathy, and lactic acidosis. Hypertrophic cardiomyopathy can eventually contribute to the development of heart failure (91).
MPV17
MPV17 is a nuclear-encoded IMM protein expressed in the brain, kidneys, liver, spleen, and heart in humans (115). A large number of studies have demonstrated that the deficiency of MPV17 may cause abnormalities in mitochondrial ROS production flux, ETC complex activity, mitochondrial membrane potential, and pH value (116, 117). Judith R. Alonzo showed that Mpv17 could transfer dTMP from cytoplasm to mitochondria for mtDNA synthesis (118). Mpv17 deficiency may cause a depletion of dTMP pool in the mitochondria (119). Loss of MPV17 protein leads to a notable reduction in liver mtDNA and ultimately liver dysfunction (120). Mutations in MPV17 gene can result in neurological problems and failure to thrive in pediatric patients (121). The absence of MPV17 protein does not appear to significantly affect mtDNA quantity and overall function of the heart in normal adult mice. However, cardiac mitochondria from MPV17 mutant mice displayed severe cristae damage after ischemia/reperfusion (I/R) injury. MPV17 mutation also disrupted its interactions with ATP synthase, MICOS, Cyclophilin D, and glucose-regulated protein 75 (GRP75). Mitochondrial sensitivity to Ca2+ overload was enhanced in I/R mice, thereby promoting mPTP opening and leading to subsequent cardiomyocyte death (117) (Figure 3).
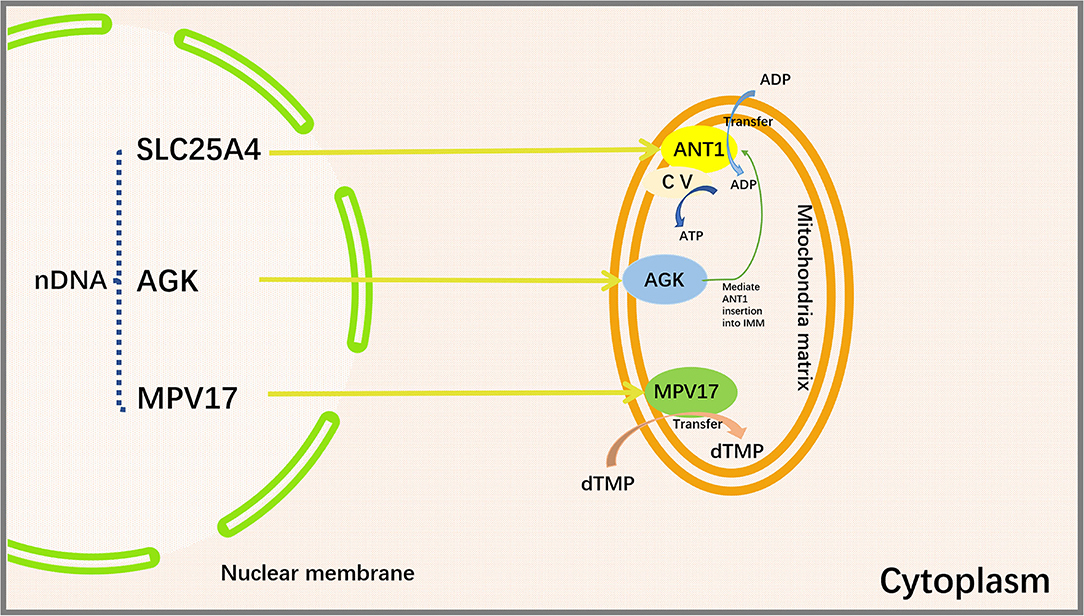
Figure 3. Proteins encoded by SLC25A4, AGK, and MPV17 genes and their functions. SLC25A4, AGK and MPV are important mitochondrial transport proteins. Adenine nucleotide transporter (ANT1) is an ADP/ATP translocator encoded by SLC25A4 and located on the inner mitochondrial membrane. It mediates the transmembrane exchange of ADP and ATP and provides ADP for mitochondrial ATP synthase. Acylglycerol kinase is a mitochondrial lipid kinase encoded by AGK gene. The absence of AGK reduces the content of ANT1 in the inner membrane of muscle mitochondria by affecting lipid metabolism. MPV17 protein is encoded by MPV17 gene, which is involved in the maintenance of nucleotide homeostasis and transfers dTMP from the cytoplasm to the mitochondria to maintain mtDNA synthesis. SLC25A4, Solute Carrier Family 25 Member 4; AGK, Acylglycerol Kinase; MPV17, Mitochondrial Inner Membrane Protein MPV17; ANT1, Adenine nucleotide translocase 1; CV, Complex V, also known as ATP synthase, is one of the mitochondrial respiratory chain electron transport complexes; ADP, Adenosine diphosphate; ATP, Adenosine triphosphate; dTMP, deoxythymidine monophosphate.
POLG
POLG is the only DNA polymerase γ (POLγ) in the mitochondria. This enzyme is a heterologous trimer consisting of three subunits, which are encoded by POLG and POLG2 genes on chromosome 15. POLG gene is a common pathogenic gene of MDS, in which hundreds of mutations have been identified (122). When this gene is mutated, mitochrondria lose the ability to maintain the genome, leading to mtDNA depletion. A large number of mutation sites in POLG have been reported, including the homozygous mutation Chr17:62492543 G>A of Chr17 located in POLG2 gene, which leads to the replacement of R182W in P55. The clinical manifestations of the patient include the reduction in mtDNA content and liver failure, indicative of hepatocerebral MDS. In addition, Hoff et al. showed that the Chr17:62492543 G>A mutation in POLG2 and R182W p55 severely damaged the stability of the auxiliary subunit, and was associated with the disease phenotype (123). Recent studies identified a new homozygous mutation [c.2391G>T (p.M797I)] in the C-terminal subdomain of POLG in a close family with MNGIE syndrome (124). Heart-specific overexpression of POLγY955C mutant caused dysregulation mtDNA biogenesis, resulting in cardiac dysfunction and eventually premature death. Utilizing transgenic mice expressing human POLγY955C in the heart, William Lewis et al. demonstrated that the mice had defective enzyme machinery for mtDNA replication, which resulted in the phenotype of mtDNA depletion. In addition, the mice also showed the phenotypes of increased mitochondrial oxidative stress, histopathological changes, and abnormal mitochondrial ultrastructure in cardiomyocytes. Moreover, cardiac insufficiency is accompanied by cardiac enlargement, increased ventricular volume, and heart mass (125).
RRM2B
RRM2B (ribonucleotide reductase regulatory TP53 inducible subunit M2B), is located on chromosome 8q22.3, and encodes for ribonucleotide diphosphate reductase subunit M2B and p53-inducible ribonucleotide reductase (RNR) small subunit (p53R2). RNR is responsible for catalyzing free radicals and reducing ribonucleotides, in order to dNTPs to maintain the dNTP library at an appropriate level for DNA synthesis and repair. When this gene is defective, it often leads to the occurrence of mitochondrial diseases. Changes in the structural properties of RRM2B protein can also cause mtDNA damage and affect the nervous system (126, 127). Stojanović et al. has reported a missense mutation c.707G>A, p.Cys236Tyr in RRM2B gene in the blood cells of a white male. This mutation appears to be associated with congenital deafness and exercise intolerance due to the impairment of nerve conduction (128). Mutations in RRM2B gene also cause MNGIE. Two RRM2B pathogenic mutations, namely, c.362G>A (p.R121H) and c.329G>A (p.R110H), were identified to be associated with severe mtDNA depletion in a 42-year-old woman. The mutations changed the docking interface of ribonucleoside reductase (RIR2B) homodimeric, thereby affecting the activity of the enzyme (118). In addition, RRM2B mutations can also cause cardiovascular disease. In the case report by Albert Z Lim et al. defects in mtDNA maintenance were found in patients with left ventricular hypertrophy, cardiomyopathy and ventricular septal defects (129).
TWNK
Twinkle (TWNK, previously designated as C10orf2), is responsible for encoding Twinkle proteins, maintaining mtDNA integrity, and displaying 5′ → 3′ DNA helicase activity when unwinding mitochondrial DNA. Mutations in this gene are associated with mitochondrial DNA depletion syndrome 7 (MTDPS7). Li et al. found that the proband carried two TWNK compound heterozygous mutations, of which c.1186C>T (p.Pro396Ser) in exon 1 was inherited from the father, and c.1844G>C (p.Gly615Ala) in exon 5 was inherited from the mother (130). Dominant mutations in TWNK cause progressive ophthalmoplegia, mtDNA deletion, and autosomal dominant inheritance 3, while recessive mutations often lead to MTDPS 7 and Perrault syndrome 5. Among them, MTDPS7 is usually manifested by infantile-onset spinocerebellar ataxia (IOSCA). In a report by Kume et al. TWNK homozygous mutations could lead to cerebellar ataxia in middle-aged patients (131, 132). Twinkle protein has three main domains: primary enzyme domain, junctional domain, and helical enzyme domain. The entire primary enzyme and connector domains as well as a partial helix enzyme domain are located in exon 1. Exons 2–4 encode the remaining helix enzyme domain. Kume et al. (131) found the amino acid at codon 292 changed from Pro to Thr in IOSCA patients. In addition to homozygous mutations in the helix domain, homozygous mutations in the primary enzyme domain 9 and compound heterozygous mutations in both primary enzyme and helix domain 10 were reported in IOSCA patients (133). Pohjoismäki et al. showed that Twinkle protein encoded by TWNK gene may play a direct role in the maintenance of human heart (133). The copy number of myocardial mitochondrial DNA decreased when heart failure occurred, especially after myocardial infarction. Overexpression of Twinkle helicase could greatly increase the copy number of mtDNA, thereby protecting the heart (134). In addition, the overexpression of TWNK also decreased the accumulation of ROS-induced mtDNA mutations, thus benefiting cardiomyopathy in Sod2 +/– mice (135).
FBXL4
FBXL4 (F-box and leucine-rich repeat protein 4), located on chromosome 6, is a nDNA that encodes 621-amino-acid F-box protein. FBXL4 plays vital roles in maintaining mtDNA integrity and stability. It is the first protein family member reported to form Skp1-Cullin-F-box (SCF) (136). In the fibroblasts of patients with FBXL4 double allele mutations, the dysregulated mitochondrial network, decreased mitochondrial energy metabolism-related enzyme activity and reduced consumption of mtDNA were observed. The mutations can lead to mitochondrial exhaustion syndrome 13 (MTDPS13) of cerebral muscular disease type, which is often manifested as decreased muscle tone, difficulty in eating, delayed nerve development, brain atrophy and other symptoms, accompanied by increased blood lactic acid level (137, 138). Wang et al. found a novel FBXL4 frameshift mutation (c.993dupA) in a Han Chinese patient, leading to the premature termination of protein synthesis and defects at the protein level (110). Ballout et al. reported the first case of MTDPS13 in Lebanon. Full-exon group sequencing showed that the female patient had a homozygous non-sense mutation c. 1303C>T (p.Arg435*). In addition to the typical clinical manifestations such as encephalopathy and muscle tone reduction, anemia and normal blood ammonia also appeared (139). Emperador et al. reported new FBXL4 mutations in two patients, which were composite heterozygous mutations c.[858 + 5G>C]; [1510T>C] and homozygous mutation c.[851delC]; [851delC]. Besides, skeletal muscle biopsy showed severe mtDNA depletion (140). In a case reported by Huemer et al. (137) cardiac disease was diagnosed in seven patients with FBXL4 mutations at an initial visit. Tissue examination and cell staining indicated joint defects in the respiratory chain, reduced production of enzymes related to mitochondrial energy metabolism, and reduced mitochondrial DNA content (136). Antoun et al. reported that FBXL4 deficiency could result in severe multi-system diseases, including lactic acidosis, cystic white matter disease, cardiomyopathy, arrhythmia, and immunodeficiency (141). A patient with prenatal diagnosis of polyhydramnios and cerebellar atrophy showed mitochondrial encephalomyopathy. This patient suffered from myocardial hypertrophy with ventricular septal defect. Seven of the nine patients (77%) with FBXL4 deficiency showed abnormal cardiac manifestations, including fallot syndrome (TOF), supraventricular tachycardia (SVT), ventricular septal defect (ASD), and left or right ventricular hypertrophy. These findings imply that cardiac involvement may be one of the most common clinical features of FBXL4 deficiency (142). It has been reported that two siblings harbor homozygous variants in FBXL4 gene (c.1750 T>C; p.Cys584Arg). They had symptoms of encephalomyopathy, lactic acidosis and myocardial hypertrophy, which were consistent with the characteristics of mitochondrial injury myopathy caused by FBXL4 gene mutation. Dichloroacetic acid (DCA) treatment effectively alleviated metabolic acidosis and reversed myocardial hypertrophy in the younger sister (143).
TFAM
The mitochondrial transcription factor A (TFAM) gene, located on nuclear chromosome 10q21, is composed of six introns and seven exons. TFAM is a member of the HMG box protein family. It contains two HMG box domains, an N-terminal domain and a C-terminal structure. TFAM has the ability to maintain mtDNA and initiate mtDNA transcription (144, 145). TFAM is a key factor in promoter selection during the initiation of mitochondrial transcription sites. It recognizes the promoter of POLRMT and initiates transcription by binding to mtDNA without sequence specificity (146). Stile et al. idenfied a TFAM homozygous missense variant (c.533C>T; p. Pro178Leu) among the close relatives of Basque lineage in Colombia. Two siblings showed IUGR, elevated transaminase, preterm infant with hypoglycemia and hyperbilirubinemia progressed to liver failure and death (147). In addition, TFAM and mtDNA copy numbers are essential for the control of mitochondrial gene expression; their reduction can lead to poor heart function and aging (148, 149). Heart-specific knockout of TFAM induced apoptosis in the heart of a transgenic mouse model. Disruption of TFAM in the heart resulted in mtDNA depletion, reduced mitochondrial ATP production rate, and impaired respiratory chain enzyme activities. These defects aggravate heart disease and eventually lead to heart failure (146, 150).
MGME1
Mitochondrial Genome Maintenance Exonuclease 1 (MGME1 or DDK1) belongs to PD-(D/E)XK nuclease superfamily. It is a highly conserved mitochondrial-specific DNA enzyme encoded by C20orf72 gene. Mutation of C20orf72 is associated with exhaustion and rearrangement of mtDNA. Patients with C20orf72 mutations may have CPEO, myasthenia, respiratory distress and gastrointestinal diseases (151). Mouse studies by Matic et al. suggest that MGME1 gene product may be a part of the regulatory switch at the end of the D-loop region since it regulates the replication of mtDNA and the termination of H-chain transcription. Lack of MGME1 may lead to the deletion or depletion of mtDNA in mouse tissue cells (152). MGME1 may also contribute to cerebellar ataxia since mutation of this gene leads to the occurrence of mtDNA depletion syndrome 11, with a series of clinical manifestations of cerebellar diseases. Recently, Hebbar et al. identified a new code-shift mutation c.359del (p.Pro120Leufs*2) in exon 2 in a child with early progressive cerebellar ataxia. The investigator determined that the mutation was the cause of MTDPS11 (153). A high-throughput mutation screening of 16 patients with dilated or hypertrophic cardiomyopathy indicated that MGME1 variants are highly associated with the pathogenesis of heart disease (154).
QIL1/MIC13(C19orf70)
MICOS13, also known as C19orf70, MIC13, or QIL1 (155, 156), is a component of mitochondrial contact site and cristae tissue system (MICOS) complex. It plays vital roles in maintaining the characteristics of mitochondrial cristae and cellular energy metabolism. MICOS13 deficiency leads to morphological defects of cristae and cristae junctions and severely impairs the IMM architecture and mitochondrial respiratory function (157). Patients from a consanguineous family were reported to have MICOS p.(Gly15Glufs*75) variant, thus resulting in mitochondrial hepato-encephalopathy. Homozygous individuals displayed syndromes correlated with mitochondrial dysfunction. In addition, multiple organs with high energy demands, including heart, liver and the central nervous system, were damaged (155, 158). It has been reported that two siblings with QIL1 deficiency exhibited mitochondrial encephalopathy symptom. Loss of QIL1 in this case showed abnormalities in multiple organs (e.g., liver and muscle), while heart ultrasound indicated mild heart hypertrophy as early as 2 years old (159). Furthermore, QIL1 point mutation also resulted in cardiac arrhythmia but not cardiac hypertrophy (160).
Summary and Outlook
MDS is a mitochondrial disease characterized by the reduced amount and/or depletion of mtDNA content. They are caused by mutations in nDNA and can involve two or more organs. In this paper, while discussing the impact of different types of MDS on muscle, brain and nervous system, we explored the potential association between different gene mutations and MDS-associated cardiac diseases, in order to provide guidance for early screening and prevention of these diseases.
Author Contributions
JY and YX: conceptualization and resources. HW, YX, and JY: original draft preparation and resources. YH, SL, YuC, YaC, JingW, and YuZ: writing. YH, SL, YuC, YaC, YuZ, YaZ, JingsW, HW, and JY: review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
The authors would like to acknowledge funding from the Research Start-up Fund of Jining Medical University (Reference: 600791001), the Foundation of Medical Health Science and Technology Development Program of Shandong Province (Reference: 202002061311), the Research Support Fund for Teachers of Jining Medical University (Reference: JYFC2019KJ013), and the Jining Medical College Student Innovation and Entrepreneurship Training Program Project (Reference: cx2021053).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
The authors would like to express their gratitude to EditSprings (https://www.editsprings.com/) for the expert linguistic services. We would also like to acknowledge the support of the research team at Jining Medical University working on the molecular mechanisms and intervention of myocardial infarction.
References
1. El-Hattab AW, Craigen WJ, Scaglia F. Mitochondrial DNA maintenance defects. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:1539–55. doi: 10.1016/j.bbadis.2017.02.017
2. Kai L, Bingzhen C. Progress on cell apoptosis of mitochondrial regulation. Biomed Eng Clin. (2014) 18:501–5. doi: 10.13339/j.cnki.sglc.2014.05.049
3. Thorburn DR, Rahman J, Rahman S. Mitochondrial DNA-Associated Leigh Syndrome and NARP. Seattle, WA: University of Washington (1993).
4. Zhou ZD, Saw WT, Tan EK. Mitochondrial CHCHD-Containing proteins: physiologic functions and link with neurodegenerative diseases. Mol Neurobiol. (2017) 54:5534–46. doi: 10.1007/s12035-016-0099-5
5. Dogan SA, Cerutti R, Benincá C, Brea-Calvo G, Jacobs HT, Zeviani M, et al. Perturbed redox signaling exacerbates a mitochondrial myopathy. Cell Metab. (2018) 28:764–75. doi: 10.1016/j.cmet.2018.07.012
6. Flores-Herrera O, Olvera-Sánchez S, Esparza-Perusquía M, Pardo JP, Rendón JL, Mendoza-Hernández G, et al. Membrane potential regulates mitochondrial ATP-diphosphohydrolase activity but is not involved in progesterone biosynthesis in human syncytiotrophoblast cells. Biochim Biophys Acta. (2015) 1847:143–52. doi: 10.1016/j.bbabio.2014.10.002
7. Fernández-Vizarra E, Tiranti V, Zeviani M. Assembly of the oxidative phosphorylation system in humans: what we have learned by studying its defects. Biochim Biophys Acta. (2009) 1793:200–11. doi: 10.1016/j.bbamcr.2008.05.028
8. Hou T, Zhang R, Jian C, Wan QD, Wang YR, Shu KL, et al. NDUFAB1 confers cardio-protection by enhancing mitochondrial bioenergetics through coordination of respiratory complex and supercomplex assembly. Cell Res. (2019) 29:754–66. doi: 10.1038/s41422-019-0208-x
9. Larsen S, Nielsen J, Hansen CN, Nielesen LB, Wirbrand F, Stride Net, et al. Biomarkers of mitochondrial content in skeletal muscle of healthy young human subjects. J Physiol. (2012) 590:3349–60. doi: 10.1113/jphysiol.2012.230185
10. Protasoni M, Pérez-Pérez R, Lobo-Jarne T, Harbour ME, Ding SJ, Peñas A, et al. Respiratory supercomplexes act as a platform for complex III-mediated maturation of human mitochondrial complexes I and IV. EMBO J. (2020) 39:e102817. doi: 10.15252/embj.2019102817
11. Teodoro JS, Palmeira CM, Rolo AP. Mitochondrial membrane potential (ΔΨ) fluctuations associated with the metabolic states of mitochondria. Methods Mol Biol. (2018) 1782:109–19. doi: 10.1007/978-1-4939-7831-1_6
12. Acin-Perez R, Enriquez JA. The function of the respiratory supercomplexes: the plasticity model. Biochim Biophys Acta. (2014) 1837:444–50. doi: 10.1016/j.bbabio.2013.12.009
13. Ji LL. Redox signaling in skeletal muscle: role of aging and exercise. Adv Physiol Educ. (2015) 39:352–9. doi: 10.1152/advan.00106.2014
14. Bouchez C, Devin A. mitochondrial biogenesis and mitochondrial reactive oxygen species (ROS): a complex relationship regulated by the cAMP/PKA signaling pathway. Cells. (2019) 8:287. doi: 10.3390/cells8040287
15. Saki M, Prakash A. DNA damage related crosstalk between the nucleus and mitochondria. Free Radic Biol Med. (2017) 107:216–27. doi: 10.1016/j.freeradbiomed.2016.11.050
16. Deshpande OA, Mohiuddin SS. Biochemistry, Oxidative Phosphorylation. California, CA: StatPearls (2020).
17. Benard G, Bellance N, Jose C, Melser S, Nouette-Gaulain K, Rossignol R. Multi-site control and regulation of mitochondrial energy production. Biochim Biophys Acta. (2010) 1797:698–709. doi: 10.1016/j.bbabio.2010.02.030
18. Thomas LW, Staples O, Turmaine M, Ashcroft M. CHCHD4 regulates intracellular oxygenation and perinuclear distribution of mitochondria. Front Oncol. (2017) 7:71. doi: 10.3389/fonc.2017.00071
19. Fuhrmann DC, Brüne B. Mitochondrial composition and function under the control of hypoxia. Redox Biol. (2017) 12:208–15. doi: 10.1016/j.redox.2017.02.012
20. Neary MT, Ng KE, Ludtmann MH, Hall AR, Piotrowska I, Wang XB, et al. Hypoxia signaling controls postnatal changes in cardiac mitochondrial morphology and function. J Mol Cell Cardiol. (2014) 74:340–52. doi: 10.1016/j.yjmcc.2014.06.013
21. Ambrose LJ, Abd-Jamil AH, Gomes RS, Carter EE, Carr CA, Clarke K, et al. Investigating mitochondrial metabolism in contracting HL-1 cardiomyocytes following hypoxia and pharmacological HIF activation identifies HIF-dependent and independent mechanisms of regulation. J Cardiovasc Pharmacol Ther. (2014) 19:574–85. doi: 10.1177/1074248414524480
22. Bhatti JS, Bhatti GK, Reddy PH. Mitochondrial dysfunction and oxidative stress in metabolic disorders - a step towards mitochondria based therapeutic strategies. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:1066–77. doi: 10.1016/j.bbadis.2016.11.010
23. Houten SM, Violante S, Ventura FV, Wanders RJA. The biochemistry and physiology of mitochondrial fatty acid β-oxidation and its genetic disorders. Annu Rev Physiol. (2016) 78:23–44. doi: 10.1146/annurev-physiol-021115-105045
24. Prasun P. Disorders of pyruvate metabolism and tricarboxylic acid cycle. Mitochondrial Med. (2019) 2019:83–95. doi: 10.1016/B978-0-12-817006-9.00015-0
25. Martínez-Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun. (2020) 11:102. doi: 10.1038/s41467-019-13668-3
26. Dziewulska A, Dobrzyń P, Dobrzyń A. The role of AMP-activated protein kinase in regulation of skeletal muscle metabolism. Postepy Hig Med Dosw. (2010) 64:513–21. doi: 10.1677/joe.0.0340273
27. Chen H, Chomyn A, Chan DC. Disruption of fusion results in mitochondrial heterogeneity and dysfunction. J Biol Chem. (2005) 280:26185–92. doi: 10.1074/jbc.M503062200
28. Weir HJ, Yao P, Huynh K, Escoubas CC, Goncalves RL, Burkewitz K, et al. Dietary restriction and AMPK increase lifespan via mitochondrial network and peroxisome remodeling. Cell Metab. (2017) 26:884–96. doi: 10.1016/j.cmet.2017.09.024
29. Gómez-Valadés AG, Gonzalez-Franquesa A, Gama-Perez P, Claret M, Garcia-Roves PM. Emerging concepts in diabetes: mitochondrial dynamics and glucose homeostasis. Curr Diabetes Rev. (2017) 13:370–85. doi: 10.2174/1573399812666151012115229
30. Otera H, Ishihara N, Mihara K. New insights into the function and regulation of mitochondrial fission. Biochim Biophys Acta. (2013) 1833:1256–68. doi: 10.1016/j.bbamcr.2013.02.002
31. Lee H, Yoon Y. Mitochondrial fission and fusion. Biochem Soc Trans. (2016) 44:1725–35. doi: 10.1042/BST20160129
32. Kondadi AK, Anand R, Reichert AS. Functional interplay between cristae biogenesis, mitochondrial dynamics and mitochondrial DNA integrity. Int J Mol Sci. (2019) 20:4311. doi: 10.3390/ijms20174311
33. Meyer JN, Leuthner TC, Luz AL. Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology. (2017) 391:42–53. doi: 10.1016/j.tox.2017.07.019
34. Sas K, Szabó E, Vécsei L. Mitochondria, oxidative stress and the kynurenine system, with a focus on ageing and neuroprotection. Molecules. (2018) 23:191. doi: 10.3390/molecules23010191
35. Briston T, Roberts M, Lewis S, Powney B, Staddon JM, Szabadkai G, et al. Mitochondrial permeability transition pore: sensitivity to opening and mechanistic dependence on substrate availability. Sci Rep. (2017) 7:10492. doi: 10.1038/s41598-017-10673-8
36. Gherardi G, Nogara L, Ciciliot S, Fadini GP, Blaauw B, Braghetta P, et al. Loss of mitochondrial calcium uniporter rewires skeletal muscle metabolism and substrate preference. Cell Death Differ. (2019) 26:362–81. doi: 10.1038/s41418-018-0191-7
37. Gherardi G, Monticelli H, Rizzuto R, Mammucari C. The mitochondrial Ca2+ uptake and the fine-tuning of aerobic metabolism. Front Physiol. (2020) 11:554904. doi: 10.3389/fphys.2020.554904
38. Nemani N, Dong Z, Daw CC, Madaris TR, Ramachandran K, Enslow BT, et al. Mitochondrial pyruvate and fatty acid flux modulate MICU1-dependent control of MCU activity. Sci Signal. (2020) 13:eaaz6206. doi: 10.1126/scisignal.aaz6206
39. Gao P, Yan Z, Zhu Z. Mitochondria-Associated endoplasmic reticulum membranes in cardiovascular diseases. Front Cell Dev Biol. (2020) 8:604240. doi: 10.3389/fcell.2020.604240
40. Zapico SC, Ubelaker DH. mtDNA mutations and their role in aging, diseases and forensic sciences. Aging Dis. (2013) 4:364–80. doi: 10.14336/AD.2013.0400364
41. Kraja AT, Liu C, Fetterman JL, Graff M, Have CT, Gu C, et al. Associations of mitochondrial and nuclear mitochondrial variants and genes with seven metabolic traits. Am J Hum Genet. (2019) 104:112–38. doi: 10.1016/j.ajhg.2018.12.001
42. Chaturvedi S, Bala K, Thakur R, Suri V. Mitochondrial encephalomyopathies: advances in understanding. Med Sci Monit. (2005) 11:A238–46. doi: 10.1016/j.lfs.2005.02.003
43. Kopinski PK, Janssen KA, Schaefer PM, Trefely S, Perry CE, Potluri P, et al. Regulation of nuclear epigenome by mitochondrial DNA heteroplasmy. Proc Natl Acad Sci USA. (2019) 116:16028–35. doi: 10.1073/pnas.1906896116
44. Mishra P, Chan DC. Metabolic regulation of mitochondrial dynamics. J Cell Biol. (2016) 212:379–87. doi: 10.1083/jcb.201511036
45. El-Hattab AW, Scaglia F. Mitochondrial DNA depletion syndromes: review and updates of genetic basis, manifestations, and therapeutic options. Neurotherapeutics. (2013) 10:186–98. doi: 10.1007/s13311-013-0177-6
46. Moraes CT, Shanske S, Tritschler HJ, Andreetta F, Bonilla E, et al. MtDNA depletion with variable tissue expression: a novel genetic abnormality in mitochondrial diseases. Am J Hum Genet. (1991) 48:492–501.
47. Sarzi E, Bourdon A, Chrétien D, Corcos J, Slama A, et al. Mitochondrial DNA depletion is a prevalent cause of multiple respiratory chain deficiency in childhood. J Pediatr. (2007) 150:531–4. doi: 10.1016/j.jpeds.2007.01.044
48. Spinazzola A, Invernizzi F, Carrara F, Lamantea E, Donati A, Dirocco M, et al. Clinical and molecular features of mitochondrial DNA depletion syndromes. J Inherit Metab Dis. (2009) 32:143–58. doi: 10.1007/s10545-008-1038-z
49. Lin GZ, Qiu JW, Deng M, Lin WX, Guo L, Song YZ. DGUOK-related mitochondrial DNA depletion syndrome: a case report and literature review. Zhong Guo Dang Dai Er Ke Za Zhi. (2020) 22:274–9. doi: 10.7499/j.issn.1008-8830.2020.03.017
50. El-Hattab AW, Scaglia F. Hattab AW, Scaglia F. SUCLG1-Related Mitochondrial DNA Depletion Syndrome, Encephalomyopathic Form With Methylmalonic Aciduria. GeneReviews® (1993–2021).
51. Wei H, Liu G. Understanding, characteristics and treatment of mitochondrial exhaustion syndrome. J Pract Diabetol. (2018) 14:9–10.
52. Blázquez-Bermejo C, Molina-Granada D, Vila-Julià F, Jiménez-Heis D, Zhou X, Torres-Torronteras J, et al. Age-related metabolic changes limit efficacy of deoxynucleoside-based therapy in thymidine kinase 2-deficient mice. EBio Med. (2019) 46:342–55. doi: 10.1016/j.ebiom.2019.07.042
53. Wang J, El-Hattab AW, Wong LJC. TK2-Related Mitochondrial DNA Maintenance Defect, Myopathic Form. GeneReviews® (1993–2021).
54. Papadimas GK, Vargiami E, Dragoumi P, Van Coster R, Smet J, Seneca S, et al. Mild myopathic phenotype in a patient with homozygous c.416C>T mutation in TK2 gene. Acta Myol. (2020) 39:94–7. doi: 10.36185/2532-1900-012
55. Cámara Y, Carreño-Gago L, Martín MA, Melià MJ, Blázquez A, Delmiro A, et al. Severe TK2 enzyme activity deficiency in patients with mild forms of myopathy. Neurology. (2015) 84:2286–8. doi: 10.1212/WNL.0000000000001644
56. Domínguez-González C, Madruga-Garrido M, Mavillard F, Garone C, Aguirre-Rodríguez FJ, Donati MA, et al. Deoxynucleoside therapy for thymidine kinase 2-deficient myopathy. Ann Neurol. (2019) 86:293–303. doi: 10.1002/ana.25506
57. Chanprasert S, Wang J, Weng SW, Enns GM, Boué DR, Wong BL, et al. Molecular and clinical characterization of the myopathic form of mitochondrial DNA depletion syndrome caused by mutations in the thymidine kinase (TK2) gene. Mol Genet Metab. (2013) 110:153–61. doi: 10.1016/j.ymgme.2013.07.009
58. Garone C, Taylor R W, Nascimento A, Poulton J, Fratter C, Domínguez-González C, et al. Retrospective natural history of thymidine kinase 2 deficiency. J Med Genet. (2018) 55:515–21. doi: 10.1136/jmedgenet-2017-105012
59. Lamperti C, Zeviani M. Encephalomyopathies caused by abnormal nuclear-mitochondrial intergenomic cross-talk. Acta Myol. (2009) 28:2–11.
60. Köse E, Köse M, Edizer S, Akişin Z, Yilmaz ZB, Sahin A, et al. Different clinical presentation in a patient with two novel pathogenic variants of the FBXL4 gene. Turk J Pediatr. (2020) 62:652–6. doi: 10.24953/turkjped.2020.04.016
61. Lu X, Li D, Duan F, Li H, Yao X, Ma B, et al. A case of SUCLG1-related infantile encephalomyopathy mitochondrial DNA depletion syndrome. J Clin Pediatr. (2017) 35:810–4.
62. Mahjoub G, Habibzadeh P, Dastsooz H, Mirzaei M, Kavosi A, Jamali L. Clinical and molecular characterization of three patients with hepatocerebral form of mitochondrial DNA depletion syndrome: a case series. BMC Med Genet. (2019) 20:167. doi: 10.1186/s12881-019-0893-9
63. Tadiboyina VT, Rupar A, Atkison P, Feigenbaum A, Kronick J, Wang J et al. Novel mutation in DGUOK in hepatocerebral mitochondrial DNA depletion syndrome associated with cystathioninuria. Am J Med Genet A. (2005) 135:289–91. doi: 10.1002/ajmg.a.30748
64. Kishita Y, Shimura M, Kohda M, Akita M, Imai-Okazaki A, Yatsuka Y, et al. A novel homozygous variant in MICOS13/QIL1 causes hepato-encephalopathy with mitochondrial DNA depletion syndrome. Mol Genet Genomic Med. (2020) 8:e1427. doi: 10.1002/mgg3.1427
65. Pronicki M, Piekutowska-Abramczuk D, Rokicki D, Iwanicka-Pronicka K, Grajkowska W. Histopathological liver findings in patients with hepatocerebral mitochondrial depletion syndrome with defined molecular basis. Pol J Pathol. (2018) 69:292–8. doi: 10.5114/pjp.2018.79549
66. Nogueira C, Almeida LS, Nesti C, Pezzini I, Videira A, Vilarinho L, et al. Syndromes associated with mitochondrial DNA depletion. Ital J Pediatr. (2014) 40:34. doi: 10.1186/1824-7288-40-34
67. Kim J, Kang E, Kim Y, Kim JM, Lee BH, Murayama K, et al. MPV17 mutations in patients with hepatocerebral mitochondrial DNA depletion syndrome. Mol Genet Metab Rep. (2016) 8:74–6. doi: 10.1016/j.ymgmr.2016.06.006
68. Yadak R, Breur M, Bugiani M. Gastrointestinal dysmotility in MNGIE: from thymidine phosphorylase enzyme deficiency to altered interstitial cells of cajal. Orphanet J Rare Dis. (2019) 14:33. doi: 10.1186/s13023-019-1016-6
69. Hirano M, Garone C, Quinzii C M. CoQ(10) deficiencies and MNGIE: two treatable mitochondrial disorders. Biochim Biophys Acta. (2012) 1820:625–31. doi: 10.1016/j.bbagen.2012.01.006
70. Scarpelli M, Todeschini A, Volonghi I, Padovani A, Filosto M. Mitochondrial diseases: advances and issues. Appl Clin Genet. (2017) 10:21–6. doi: 10.2147/TACG.S94267
71. Filosto M, Cotti Piccinelli S, Caria F, Gallo Cassarino S, Baldelli E, et al. Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE-MTDPS1). J Clin Med. (2018) 7:389. doi: 10.3390/jcm7110389
72. Boschetti E, D'Angelo R, Tardio ML, Costa R, Giordano C, Accarino A, et al. Evidence of enteric angiopathy and neuromuscular hypoxia in patients with mitochondrial neurogastrointestinal encephalomyopathy. Am J Physiol Gastrointest Liver Physiol. (2021) 320:G768–G779. doi: 10.1152/ajpgi.00047.2021
73. Chistiakov DA, Shkurat TP, Melnichenko AA, Grechko AV, Orekhov AN. The role of mitochondrial dysfunction in cardiovascular disease: a brief review. Ann Med. (2018) 50:121–7. doi: 10.1080/07853890.2017.1417631
74. Riva A, Tandler B, Loffredo F, Vazquez E, Hoppel C. Structural differences in two biochemically defined populations of cardiac mitochondria. Am J Physiol Heart Circ Physiol. (2005) 289:H868–72. doi: 10.1152/ajpheart.00866.2004
75. Pohjoismäki JL, Goffart S. The role of mitochondria in cardiac development and protection. Free Radic Biol Med. (2017) 106:345–54. doi: 10.1016/j.freeradbiomed.2017.02.032
76. Marzetti E, Csiszar A, Dutta D, Balagopal G, Calvani R, Leeuwenburgh C. Role of mitochondrial dysfunction and altered autophagy in cardiovascular aging and disease: from mechanisms to therapeutics. Am J Physiol Heart Circ Physiol. (2013) 305:H459–76. doi: 10.1152/ajpheart.00936.2012
77. Victor VM, Apostolova N, Herance R, Hernandez-Mijares A, Rocha M. Oxidative stress and mitochondrial dysfunction in atherosclerosis: mitochondria-targeted antioxidants as potential therapy. Curr Med Chem. (2009) 16:4654–67. doi: 10.2174/092986709789878265
78. Amchenkova AA, Bakeeva LE, Chentsov YS, Skulachev VP, Zorov DB. Coupling membranes as energy-transmitting cables. I. Filamentous mitochondria in fibroblasts and mitochondrial clusters in cardiomyocytes. J Cell Biol. (1988) 107:481–95. doi: 10.1083/jcb.107.2.481
79. Green DR, Reed JC. Mitochondria and apoptosis. Science. (1998) 281:1309–12. doi: 10.1126/science.281.5381.1309
80. Tang X, Li PH, Chen HZ. Cardiomyocyte senescence and cellular communications within myocardial microenvironments. Front Endocrinol. (2020) 11:280. doi: 10.3389/fendo.2020.00280
81. Dey S, DeMazumder D, Sidor A, Foster DB, O'Rourke B. Mitochondrial ROS drive sudden cardiac death and chronic proteome remodeling in heart failure. Circ Res. (2018) 123:356–71. doi: 10.1161/CIRCRESAHA.118.312708
82. Dorn GW II, Vega RB, Kelly DP. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev. (2015) 29:1981–91. doi: 10.1101/gad.269894.115
83. Lee CF, Chavez JD, Garcia-Menendez L, Choi Y, Roe ND, Chiao YA, et al. Normalization of NAD+ redox balance as a therapy for heart failure. Circulation. (2016) 134:883–94. doi: 10.1161/CIRCULATIONAHA.116.022495
84. Nakayama H, Chen X, Baines CP, Klevitsky R, Zhang X, Zhang H, et al. Ca2+ and mitochondrial-dependent cardiomyocyte necrosis as a primary mediator of heart failure. J Clin Invest. (2007) 117:2431–44. doi: 10.1172/JCI31060
85. Wang W, Karamanlidis G, Tian R. Novel targets for mitochondrial medicine. Sci Transl Med. (2016) 8:326rv3. doi: 10.1126/scitranslmed.aac7410
86. Dominic EA, Ramezani A, Anker SD, Verma M, Mehta N, Rao M. Mitochondrial cytopathies and cardiovascular disease. Heart. (2014) 100:611–8. doi: 10.1136/heartjnl-2013-304657
87. Behjati M, Sabri MR, Etemadi Far M, Nejati M. Cardiac complications in inherited mitochondrial diseases. Heart Fail Rev. (2021) 26:391–403. doi: 10.1007/s10741-020-10009-1
88. Yan Y, Li S, Dong W, Liu C, Wang P, Hou W, et al. Correlation of mitochondrial transcription factor A with coronary heart disease. J Pract Med. (2018) 34:2892–5. doi: 10.3969/j.issn.1006-5725.2018.17.018
89. Santorelli FM, Gagliardi MG, Dionisi-Vici C, Parisi F, Tessa A, Carrozzo R, et al. Hypertrophic cardiomyopathy and mtDNA depletion. Successful treatment with heart transplantation. Neuromuscul Disord. (2002) 12:56–9. doi: 10.1016/S0960-8966(01)00248-6
90. Fangdai L, Fang F, Liu Z, Shen D, Ding C, Li J, et al. Clinical phenotype and genotypic characteristics of 12 cases of mitochondrial DNA depletion syndrome. Chin J Pediatr. (2019) 57:211–6. doi: 10.3760/cma.j.issn.0578-1310.2019.03.011
91. Wang B, Du Z, Shan G, Yan C, Zhang VW, Li Z. Case report: two Chinese infants of sengers syndrome caused by mutations in AGK gene. Front Pediatr. (2021) 9:639687. doi: 10.3389/fped.2021.639687
92. Dunham-Snary KJ, Ballinger SW. Mitochondrial genetics and obesity: evolutionary adaptation and contemporary disease susceptibility. Free Radic Biol Med. (2013) 65:1229–37. doi: 10.1016/j.freeradbiomed.2013.09.007
93. Krzywanski DM, Moellering DR, Fetterman JL, Dunham-Snary KJ, Sammy MJ, Ballinger SW. The mitochondrial paradigm for cardiovascular disease susceptibility and cellular function: a complementary concept to Mendelian genetics. Lab Invest. (2011) 91:1122–35. doi: 10.1038/labinvest.2011.95
94. Ruiz-Pesini E, Mishmar D, Brandon M, Procaccio V, Wallace DC. Effects of purifying and adaptive selection on regional variation in human mtDNA. Science. (2004) 303:223–6. doi: 10.1126/science.1088434
95. Tian R, Colucci WS, Arany Z, Bachschmid MM, Ballinger SW, Boudina S, et al. Unlocking the secrets of mitochondria in the cardiovascular system: path to a cure in heart failure—a report from the 2018 national heart, lung, and blood institute workshop. Circulation. (2019) 140:1205–16. doi: 10.1161/CIRCULATIONAHA.119.040551
96. Yan W, Donghai L, Xingxing Z. Thymidine kinase 2 gene compound heterozygous mutation leads to mitochondrial DNA depletion syndrome-2: a case report. Chin J Pediatr. (2018) 56:381–2. doi: 10.3760/cma.j.issn.0578-1310.2018.05.016
97. Domínguez-González C, Hernández-Laín A, Rivas E, Hernández-Voth A, Sayas Catalán J, Fernández-Torrón R, et al. Late-onset thymidine kinase 2 deficiency: a review of 18 cases. Orphanet J Rare Dis. (2019) 14:100. doi: 10.1186/s13023-019-1071-z
98. Dorado B, Area E, Akman HO, Hirano M. Onset and organ specificity of Tk2 deficiency depends on Tk1 down-regulation and transcriptional compensation. Hum Mol Genet. (2011) 20:155–64. doi: 10.1093/hmg/ddq453
99. Wang L, Sun R, Eriksson S. Basic biochemical characterization of cytosolic enzymes in thymidine nucleotide synthesis in adult rat tissues: implications for tissue specific mitochondrial DNA depletion and deoxynucleoside-based therapy for TK2-deficiency. BMC Mol Cell Biol. (2020) 21:33. doi: 10.1186/s12860-020-00272-3
100. Kamath VG, Hsiung CH, Lizenby ZJ, McKee EE. Heart mitochondrial TTP synthesis and the compartmentalization of TMP. J Biol Chem. (2015) 290:2034–41. doi: 10.1074/jbc.M114.624213
101. Landsverk ML, Zhang VW, Wong LC, Andersson HC. A SUCLG1 mutation in a patient with mitochondrial DNA depletion and congenital anomalies. Mol Genet Metab Rep. (2014) 1:451–4. doi: 10.1016/j.ymgmr.2014.09.007
102. Demirbas D, Harris DJ, Arn PH, Huang X, Waisbren SE, Anselm I, et al. Phenotypic variability in deficiency of the α subunit of succinate-CoA ligase. JIMD Rep. (2019) 46:63–9. doi: 10.1002/jmd2.12018
103. Rivera H, Merinero B, Martinez-Pardo M, Arroyo I, Ruiz-Sala P, Bornstein B, et al. Marked mitochondrial DNA depletion associated with a novel SUCLG1 gene mutation resulting in lethal neonatal acidosis, multi-organ failure, and interrupted aortic arch. Mitochondrion. (2010) 10:362–8. doi: 10.1016/j.mito.2010.03.003
104. Van Hove JL, Saenz MS, Thomas JA, Gallagher RC, Lovell MA, Fenton LZ, et al. Succinyl-CoA ligase deficiency: a mitochondrial hepatoencephalomyopathy. Pediatr Res. (2010) 68:159–64. doi: 10.1203/PDR.0b013e3181e5c3a4
105. Liu Y, Li X, Wang Q, Ding Y, Song J, Yang Y. Five novel SUCLG1 mutations in three Chinese patients with succinate-CoA ligase deficiency noticed by mild methylmalonic aciduria. Brain Dev. (2016) 38:61–7. doi: 10.1016/j.braindev.2015.05.002
106. Körver-Keularts IM, de Visser M, Bakker HD, Wanders RJ, Vansenne F, Scholte HR, et al. Two novel mutations in the SLC25A4 gene in a patient with mitochondrial myopathy. JIMD Rep. (2015) 22:39–45. doi: 10.1007/8904_2015_409
107. Hoshino A, Wang WJ, Wada S, McDermott-Roe C, Evans CS, Gosis B, et al. The ADP/ATP translocase drives mitophagy independent of nucleotide exchange. Nature. (2019) 575:375–9. doi: 10.1038/s41586-019-1667-4
108. Thompson K, Majd H, Dallabona C, Reinson K, King MS, Alston CL, et al. Recurrent de novo dominant mutations in SLC25A4 cause severe early-onset mitochondrial disease and loss of mitochondrial DNA copy number. Am J Hum Genet. (2016) 99:860–76. doi: 10.1016/j.ajhg.2016.08.014
109. Kashiki T, Kido J, Momosaki K, Kusunoki S, Ozasa S, Nomura K, et al. Mitochondrial DNA depletion syndrome with a mutation in SLC25A4 developing epileptic encephalopathy: a case report. Brain Dev. (2022) 44:56–62. doi: 10.1016/j.braindev.2021.08.005
110. Wang S, Lin L, Wang Y, Wang A, Liu Z, Wu S, et al. Novel homozygous mutation in the FBXL4 gene is associated with mitochondria DNA depletion syndrome-13. J Neurol Sci. (2020) 416:116948. doi: 10.1016/j.jns.2020.116948
111. Guo W, Liu W, Chen Z, Gu Y, Peng S, Shen L, et al. Tyrosine phosphatase SHP2 negatively regulates NLRP3 inflammasome activation via ANT1-dependent mitochondrial homeostasis. Nat Commun. (2017) 8:2168. doi: 10.1038/s41467-017-02351-0
112. Jackson TD, Hock DH, Fujihara KM, Palmer CS, Frazier AE, Low YC, et al. The TIM22 complex mediates the import of sideroflexins and is required for efficient mitochondrial one-carbon metabolism. Mol Biol Cell. (2021) 32:475–91. doi: 10.1091/mbc.E20-06-0390
113. Kalari S, Zhao Y, Spannhake EW, Berdyshev EV, Natarajan V. Role of acylglycerol kinase in LPA-induced IL-8 secretion and transactivation of epidermal growth factor-receptor in human bronchial epithelial cells. Am J Physiol Lung Cell Mol Physiol. (2009) 296:L328–36. doi: 10.1152/ajplung.90431.2008
114. Vukotic M, Nolte H, König T, Saita S, Ananjew M, Krüger M, et al. Acylglycerol kinase mutated in sengers syndrome is a subunit of the TIM22 protein translocase in mitochondria. Mol Cell. (2017) 67:471–83.e7. doi: 10.1016/j.molcel.2017.06.013
115. Weiher H, Noda T, Gray DA, Sharpe AH Jaenisch R. Transgenic mouse model of kidney disease: insertional inactivation of ubiquitously expressed gene leads to nephrotic syndrome. Cell. (1990) 62:425–34. doi: 10.1016/0092-8674(90)90008-3
116. Shvetsova AN, Mennerich D, Keratar JM, Hiltunen JK Kietzmann T. Non-electron transfer chain mitochondrial defects differently regulate HIF-1alpha degradation and transcription. Redox Biol. (2017) 12:1052–61. doi: 10.1016/j.redox.2017.05.003
117. Antonenkov VD, Isomursu A, Mennerich D, Vapola MH, Weiher H, Kietzmann T Hiltunen JK. The human mitochondrial DNA depletion syndrome gene MPV17 encodes a non-selective channel that modulates membrane potential. J Biol Chem. (2015) 290:13840–61. doi: 10.1074/jbc.M114.608083
118. Shaibani A, Shchelochkov OA, Zhang S, Katsonis P, Lichtarge O, Wong LJ, et al. Mitochondrial neurogastrointestinal encephalopathy due to mutations in RRM2B. Arch Neurol. (2009) 66:1028–32. doi: 10.1001/archneurol.2009.139
119. Alonzo JR, Venkataraman C, Field MS, Stover PJ. The mitochondrial inner membrane protein MPV17 prevents uracil accumulation in mitochondrial DNA. J Biol Chem. (2018) 293:20285–94. doi: 10.1074/jbc.RA118.004788
120. Madungwe NB, Feng Y, Imam Aliagan A, Tombo N, Kaya F, Bopassa JC. Inner mitochondrial membrane protein MPV17 mutant mice display increased myocardial injury after ischemia/reperfusion. Am J Transl Res. (2020) 12:3412–28.
121. El-Hattab AW, Wang J, Dai H, Almannai M, Staufner C, Alfadhel M, et al. MPV17-relatedmitochondrial DNA maintenance defect: new cases and review of clinical, biochemical, andmolecular aspects. Hum Mutat. (2018) 39:461–70. doi: 10.1002/humu.23387
122. Anagnostou ME, Ng YS, Taylor RW, McFarland R. Epilepsy due to mutations in the mitochondrial polymerase gamma (POLG) gene: a clinical and molecular genetic review. Epilepsia. (2016) 57:1531–45. doi: 10.1111/epi.13508
123. Hoff KE, DeBalsi KL, Sanchez-Quintero MJ, Longley MJ, Hirano M, Naini AB, et al. Characterization of the human homozygous R182W POLG2 mutation in mitochondrial DNA depletion syndrome. PLoS ONE. (2018) 13:e0203198. doi: 10.1371/journal.pone.0203198
124. Felhi R, Sfaihi L, Charif M, Desquiret-Dumas V, Bris C, Goudenège D, et al. Next generation sequencing in family with MNGIE syndrome associated to optic atrophy: Novel homozygous POLG mutation in the C-terminal sub-domain leading to mtDNA depletion. Clin Chim Acta. (2019) 488:104–10. doi: 10.1016/j.cca.2018.11.003
125. Lewis W, Day BJ, Kohler JJ, Hosseini SH, Chan SS, Green EC, et al. Decreased mtDNA, oxidative stress, cardiomyopathy, and death from transgenic cardiac targeted human mutant polymerase gamma. Lab Invest. (2007) 87:326–35. doi: 10.1038/labinvest.3700523
126. Ait El Cadi C, Krami AM, Charoute H, Elkarhat Z, Sifeddine N, Lakhiari H, et al. Prediction of the impact of deleterious nonsynonymous single nucleotide polymorphisms on the human RRM2B gene: a molecular modeling study. Biomed Res Int. (2020) 2020:7614634. doi: 10.1155/2020/7614634
127. Tseng YT, Li SW, HuangFu WC, Yen Y, Liu IH. The single nucleotide variant at c.662A>G in human RRM2B is a loss-of-function mutation. Mol Genet Genomic Med. (2020) 8:e1497. doi: 10.1002/mgg3.1497
128. Stojanovic V, Mayr JA, Sperl W, Barišić N, Doronjski A, Milak G. Infantile peripheral neuropathy, deafness, and proximal tubulopathy associated with a novel mutation of the RRM2B gene: case study. Croat Med J. (2013) 54:579–84. doi: 10.3325/cmj.2013.54.579
129. Lim AZ, McFarland R, Taylor R W, Gorman GS. RRM2B Mitochondrial DNA Maintenance Defects. GeneReviews® (1993–2021).
130. Li X, Li L, Sun Y, Lv F, Zhang G, Liu W, et al. Whole exome sequencing reveals two novel compound heterozygous mutations in TWNK as a cause of the hepatocerebral form of mitochondrial DNA depletion syndrome: a case report. BMC Med Genet. (2019) 20:146. doi: 10.1186/s12881-019-0875-y
131. Kume K, Morino H, Miyamoto R, Matsuda Y, Ohsawa R, Kanaya Y, et al. Middle-age-onset cerebellar ataxia caused by a homozygous TWNK variant: a case report. BMC Med Genet. (2020) 21:68. doi: 10.1186/s12881-020-01002-4
132. Jamali F, Ghaedi H, Tafakhori A, Alehabib E, Chapi M, Daftarian N, et al. Homozygous mutation in TWNK cases ataxia, sensorineural hearing loss and optic nerve atrophy. Arch Iran Med. (2019) 22:728–30.
133. Pohjoismäki JL, Goffart S, Taylor RW, Turnbull DM, Suomalainen A, Jacobs HT, et al. Developmental and pathological changes in the human cardiac muscle mitochondrial DNA organization, replication and copy number. PLoS ONE. (2010) 5:e10426. doi: 10.1371/journal.pone.0010426
134. Tanaka A, Ide T, Fujino T, Onitsuka K, Ikeda M, Takehara T, et al. The overexpression of Twinkle helicase ameliorates the progression of cardiac fibrosis and heart failure in pressure overload model in mice. PLoS One. (2013) 8:e67. doi: 10.1371/journal.pone.0067642
135. Pohjoismäki JL, Williams SL, Boettger T, Goffart S, Kim J, Suomalainen A, et al. Overexpression of twinkle-helicase protects cardiomyocytes from genotoxic stress caused by reactive oxygen species. Proc Natl Acad Sci USA. (2013) 110:19408–13. doi: 10.1073/pnas.1303046110
136. Strauss KA, DuBiner L, Simon M, Zaragoza M, Sengupta PP, Li P, et al. Severity of cardiomyopathy associated with adenine nucleotide translocator-1 deficiency correlates with mtDNA haplogroup. Proc Natl Acad Sci USA. (2013) 110:3453–8. doi: 10.1073/pnas.1300690110
137. Huemer M, Karall D, Schossig A, Abdenur JE, Al Jasmi F, Biagosch C, et al. Clinical, morphological, biochemical, imaging and outcome parameters in 21 individuals with mitochondrial maintenance defect related to FBXL4 mutations. J Inherit Metab Dis. (2015) 38:905–14. doi: 10.1007/s10545-015-9836-6
138. Barøy T, Pedurupillay CR, Bliksrud YT, Rasmussen M, Holmgren A, Vigeland MD, et al. A novel mutation in FBXL4 in a Norwegian child with encephalomyopathic mitochondrial DNA depletion syndrome 13. Eur J Med Genet. (2016) 59:342–6. doi: 10.1016/j.ejmg.2016.05.005
139. Ballout RA, Al Alam C, Bonnen PE, Huemer M, El-Hattab AW, Shbarou R. FBXL4-related mitochondrial DNA depletion syndrome 13 (MTDPS13): a case report with a comprehensive mutation review. Front Genet. (2019) 10:39. doi: 10.3389/fgene.2019.00039
140. Emperador S, Garrido-Pérez N, Amezcua-Gil J, Gaudó P, Andrés-Sanz JA, Yubero D, et al. Molecular characterization of new FBXL4 mutations in patients with mtDNA depletion syndrome. Front Genet. (2019) 10:1300. doi: 10.3389/fgene.2019.01300
141. Antoun G, McBride S, Vanstone JR, Naas T, Michaud J, Redpath S, et al. Detailed biochemical and bioenergetic characterization of FBXL4-related encephalomyopathic mitochondrial DNA depletion. JIMD Rep. (2016) 27:1–9. doi: 10.1007/8904_2015_491
142. Dai H, Zhang VW, El-Hattab AW, Ficicioglu C, Shinawi M, Lines M, et al. FBXL4 defects are common in patients with congenital lactic acidemia and encephalomyopathic mitochondrial DNA depletion syndrome. Clin Genet. (2017) 91:634–9. doi: 10.1111/cge.12894
143. Sabouny R, Wong R, Lee-Glover L, Greenway SC, Sinasac DS, Care4Rare Canada, et al. Characterization of the C584R variant in the mtDNA depletion syndrome gene FBXL4, reveals a novel role for FBXL4 as a regulator of mitochondrial fusion. Biochim Biophys Acta Mol Basis Dis. (2019) 1865:165536. doi: 10.1016/j.bbadis.2019.165536
144. de Oliveira VC, Gomes Mariano Junior C, Belizário JE, Krieger JE, Fernandes Bressan F, Roballo KCS, et al. Characterization of post-edited cells modified in the TFAM gene by CRISPR/Cas9 technology in the bovine model. PLoS ONE. (2020) 15:e235856. doi: 10.1371/journal.pone.0235856
145. Zhang J. SLC26A4, TFAM Gene Polymorphism and Late-Onset Alzheimer's Disease in a Han Chinese Population. Qingdao: China Ocean University (2013).
146. Bouda E, Stapon A, Garcia-Diaz M. Mechanisms of mammalian mitochondrial transcription. Protein Sci. (2019) 28:1594–05. doi: 10.1002/pro.3688
147. Stiles AR, Simon MT, Stover A, Eftekharian S, Khanlou N, Wang HL, et al. Mutations in TFAM, encoding mitochondrial transcription factor A, cause neonatal liver failure associated with mtDNA depletion. Mol Genet Metab. (2016) 119:91–9. doi: 10.1016/j.ymgme.2016.07.001
148. Bender A, Krishnan KJ, Morris CM, Taylor GA, Reeve AK, Perry RH, et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet. (2006) 38:515–7. doi: 10.1038/ng1769
149. Hansson A, Hance N, Dufour E, Rantanen A, Hultenby K, Clayton DA, et al. A switch in metabolism precedes increased mitochondrial biogenesis in respiratory chain-deficient mouse hearts. Proc Natl Acad Sci USA. (2004) 101:3136–41. doi: 10.1073/pnas.0308710100
150. Li H, Wang J, Wilhelmsson H, Hansson A, Thoren P, Duffy J, et al. Genetic modification of survival in tissue-specific knockout mice with mitochondrial cardiomyopathy. Proc Natl Acad Sci USA. (2000) 97:3467–72. doi: 10.1073/pnas.97.7.3467
151. Yang C, Wu R, Liu H, Chen Y, Gao Y, Chen X, et al. Structural insights into DNA degradation by human mitochondrial nuclease MGME1. Nucleic Acids Res. (2018) 46:11075–88. doi: 10.1093/nar/gky855
152. Matic S, Jiang M, Nicholls TJ, Uhler JP, Dirksen-Schwanenland C, Polosa PL, et al. Mice lacking the mitochondrial exonuclease MGME1 accumulate mtDNA deletions without developing progeria. Nat Commun. (2018) 9:1202. doi: 10.1038/s41467-018-03552-x
153. Hebbar M, Girisha KM, Srivastava A, Bielas S, Shukla A. Homozygous c.359del variant in MGME1 is associated with early onset cerebellar ataxia. Eur J Med Genet. (2017) 60:533–5. doi: 10.1016/j.ejmg.2017.07.010
154. Szabadosova V, Boronova I, Ferenc P, Tothova I, Bernasovska J, Zigova M, et al. Analysis of selected genes associated with cardiomyopathy by next-generation sequencing. J Clin Lab Anal. (2018) 32:22254. doi: 10.1002/jcla.22254
155. Zeharia A, Friedman JR, Tobar A, Saada A, Konen O, Fellig Y, et al. Mitochondrial hepato-encephalopathy due to deficiency of QIL1/MIC13 (C19orf70), a MICOS complex subunit. Eur J Hum Genet. (2016) 24:1778–82. doi: 10.1038/ejhg.2016.83
156. Zerbes RM, Höß P, Pfanner N, van der Laan M, Bohnert M. Distinct roles of Mic12 and Mic27 in the mitochondrial contact site and cristae organizing system. Mol Biol. (2016) 428:1485–92. doi: 10.1016/j.jmb.2016.02.031
157. Guarani V, McNeill EM, Paulo JA, Huttlin EL, Fröhlich F, Gygi SP, et al. QIL1 is a novel mitochondrial protein required for MICOS complex stability and cristae morphology. Elife. (2015) 4:e06265. doi: 10.7554/eLife.06265
158. Gödiker J, Grüneberg M, DuChesne I, Reunert J, Rust S, Westermann C, et al. QIL1-dependent assembly of MICOS complex-lethal mutation in C19ORF70 resulting in liver disease and severe neurological retardation. J Hum Genet. (2018) 63:707–16. doi: 10.1038/s10038-018-0442-y
159. Guarani V, Jardel C, Chrétien D, Lombès A, Bénit P, Labasse C, et al. QIL1 mutation causes MICOS disassembly and early onset fatal mitochondrial encephalopathy with liver disease. Elife. (2016) 5:e17163. doi: 10.7554/eLife.17163
160. Meurs KM, Friedenberg SG, Olby NJ, Condit J, Weidman J, Rosenthal S, et al. A QIL1 variant associated with ventricular arrhythmias and sudden cardiac death in the juvenile rhodesian ridgeback dog. Genes. (2019) 10:168. doi: 10.3390/genes10020168
Glossary
Keywords: mitochondrial DNA depletion syndrome, nuclear gene mutation, mtDNA, cardiac disease, ATP
Citation: Wang H, Han Y, Li S, Chen Y, Chen Y, Wang J, Zhang Y, Zhang Y, Wang J, Xia Y and Yuan J (2022) Mitochondrial DNA Depletion Syndrome and Its Associated Cardiac Disease. Front. Cardiovasc. Med. 8:808115. doi: 10.3389/fcvm.2021.808115
Received: 03 November 2021; Accepted: 23 December 2021;
Published: 14 February 2022.
Edited by:
Jian Wu, Fudan University, ChinaCopyright © 2022 Wang, Han, Li, Chen, Chen, Wang, Zhang, Zhang, Wang, Xia and Yuan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jinxiang Yuan, eXVhbmppbnhpYW5nMThAMTYzLmNvbQ==; Yong Xia, eGlheW9uZ0BtYWlsLmpubWMuZWR1LmNu