 Xiao Jin
Xiao Jin Shengjie Yang
Shengjie Yang Jing Lu2
Jing Lu2 Min Wu
Min Wu
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med., 10 February 2022
Sec. Lipids in Cardiovascular Disease
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.804214
This article is part of the Research TopicInsights in Lipids in Cardiovascular Disease: 2021View all 11 articles
Low-density lipoprotein cholesterol (LDL-C) plays an important role in the formation, incidence, and development of atherosclerosis (AS). Low-density lipoproteins can be divided into two categories: large and light LDL-C and small, dense low-density lipoprotein cholesterol (sdLDL-C). In recent years, an increasing number of studies have shown that sdLDL-C has a strong ability to cause AS because of its unique characteristics, such as having small-sized particles and low density. Therefore, this has become the focus of further research. However, the specific mechanisms regarding the involvement of sdLDL-C in AS have not been fully explained. This paper reviews the possible mechanisms of sdLDL-C in AS by reviewing relevant literature in recent years. It was found that sdLDL-C can increase the atherogenic effect by regulating the activity of gene networks, monocytes, and enzymes. This article also reviews the research progress on the effects of sdLDL-C on endothelial function, lipid metabolism, and inflammation; it also discusses its intervention effect. Diet, exercise, and other non-drug interventions can improve sdLDL-C levels. Further, drug interventions such as statins, fibrates, ezetimibe, and niacin have also been found to improve sdLDL-C levels.
Atherosclerosis (AS) is the formation of fibrofatty lesions within the arterial wall, and it causes widespread morbidity and mortality worldwide together with heart muscle infarction, stroke, and disabling peripheral artery illness (1). AS could be a major condition that seriously harms human health and is understood as the major cause of mortality not only in developed countries, but globally (2). At present, inflammatory reactions (3), lipid metabolism disorders (4), and oxidative stressare the most important and widely recognized pathogenic causes of AS (5). In the field of lipid metabolism, a number of irrefutable pieces of evidence have proven the pathogenic role of low-density lipoprotein cholesterol (LDL-C) in AS, so we have extremely effective tools to reduce LDL-C levels, thus reducing the occurrence of cardiovascular events (2). Some studies have shown the correlation between different low-density lipoprotein (LDL) subgroups and the occurrence of AS, in which small, dense low-density lipoprotein cholesterol (sdLDL-C) is closely related to and has a stronger effect on AS (6). This study aimed to explore the relationship between sdLDL-C levels and AS.
LDL is a lipoprotein with a density between 1.006 and 1.063 g/mL. It is composed of many heterogeneous particles. They can be separated using various experimental methods. Krauss et al. used ultracentrifugation to classify LDL into four types according to density: large and light LDL- I (1.025–1.034 g/mL), intermediate density LDL- II (1.035–1.044 g/mL), low-density LDL- III (1.045–1.060 g/mL), and very low-density LDL-IV (7). Another method widely used to identify low-density lipoproteins is gradient gel electrophoresis (GGE), which separates low-density lipoprotein particles by electrophoretic mobility. In studies using GGE, LDL particles have been separated into four major subfractions, LDL I (large LDL, peak diameter 26.0–28.5 nm), LDL II (intermediate LDL, 25.5–26.4 nm), LDL III A and B (small LDL, 24.2–25.5 nm), and LDL IV A and B (very small LDL, 22.0–24.1 nm) (8). The Lipoprint LDL subcomponent rapid analysis system (Hitachi 7180) is the only diagnostic equipment certified by the United States Food and Drug Administration for the separation and detection of LDL subcomponents. Based on the charge and particle size of LDL-C, they can be divided into seven subcomponents within a short time via polyacrylamide gel electrophoresis, in which components 3–7 were defined as sdLDL-C (Figure 1) (9). In addition, foreign enzyme-linked immunosorbent assay (ELISA, Millipore, St. Louis, MO) kits can quickly detect the concentrations of sdLDL-C but do not rule out the possibility of cross-reactions, so it remains to be verified whether they can be used in clinical settings (10). The homogeneous method (11) can be used to measure sdLDL-C levels by removing lipoproteins other than sdLDL-C using a surfactant and sphingomyelinase, and it is a better technique compared to the traditional detection method of LDL subfraction. Its correlation, accuracy, and stability are high, which provided a cornerstone for the popularization and application of clinical sdLDL-C detection. Other analytical methods to detect sdLDL-C include gel filtration column chromatography, high performance liquid chromatography (HPLC), ion mobility analysis, and dynamic light scattering. In clinical practice, it is important to accurately analyze LDL subclasses thorough analytical methods.
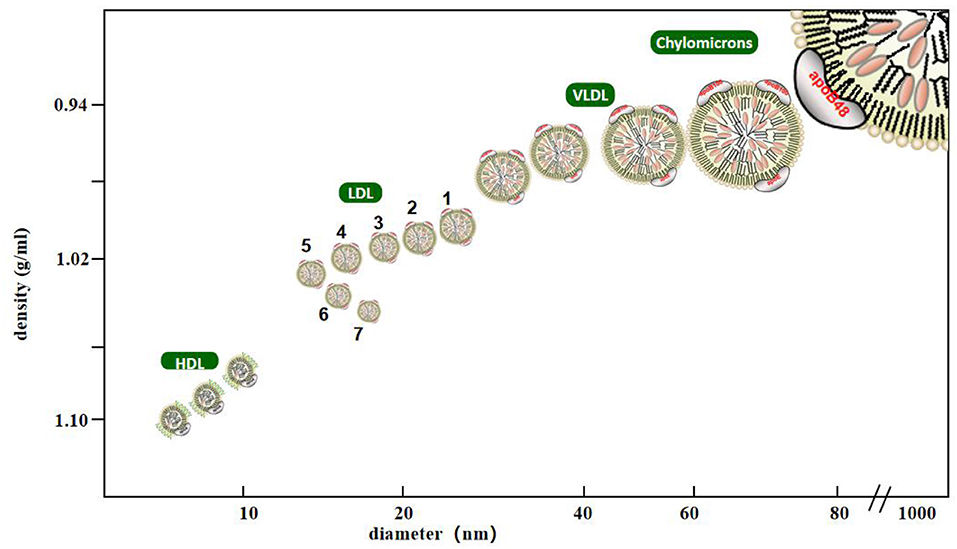
Figure 1. Classification of lipoprotein cholesterol. Lipoprotein with density <0.95 g/ml, diameter of 80–500 nm is chylomicrons, density of 0.95–1.006 g/ml, diameter of 25–80 nm is very low density lipoprotein, density of 1.063–1.21 g/ml, diameter of 8–15 nm is high density lipoprotein, density of 1.006–1.063 g/ml, diameter of 18–28 nm is low density lipoprotein, Among them, low-density lipoprotein is divided into seven subtypes. The third to seventh subtypes are small and dense low-density lipoprotein with density >1,004 g/ml and diameter <25.5 nm.
For the detection of sdLDL-C, Mauree et al. developed a new equation to calculate the content of sdLDL-C {the formula of sd-LDL-C is as follows: ElbLDL-C = 1.43 × LDL-C −[0.14 × (ln (TG) × LDL-C)] − 8.99; EsdLDL-C = LDL-C−ElbLDL-C}. Equations for sdLDL-C was generated with least-squares regression analysis using the direct Denka sdLDL-C assay as reference (n = 20,171). This equation can be widely used for all patients with standard lipid groups without incurring additional laboratory testing costs. The limitation of the study is that the study population is not universal. The equation developed is based on fasting subjects. Therefore, whether this equation can be used in experiments or clinical applications remains to be verified (12).
About the detection method of sdLDL-C (8), ultracentrifugation involves multiple steps and requires specialized equipment and expertise to separate LDL subsets, which is more error-prone, time-consuming, laborious and costly. The resolution of gradient gel electrophoresis is very high, but it requires a lot of manpower, material and financial resources, and the sample flux is low. The polyacrylamide gel tube electrophoresis method is simple to operate and shows a satisfactory coefficient of variation between samples, but the disadvantage is that the equipment is expensive. The homogeneous method can be fully automated and can be used in the high-throughput integration platform, which is helpful for the large-scale testing of sdLDL-C.
At present, it is generally believed that there are two ways to produce sdLDL-C (7, 13). First, when the triglyceride (TG) content in the liver is high, the liver directly secretes VLDL1 (large particles and high TG content) and VLDL2 (low TG content), when the level of TG synthesized by hepatocytes decreases, the liver secretes VLDL1 (small particles and high TG content) and intermediate density lipoprotein2 (low TG content). TG-deficient lipoproteins are the precursors of larger LDL (LDL I and LDL II), while TG-rich lipoproteins are converted into sdLDL (LDL III and LDL IV) after being defatted by lipoprteinlipase (LPL) and hepatic lipase (HL). Second, there is a very active and dynamic lipid exchange between various lipoproteins in the plasma, which is mainly catalyzed by cholesterol lipid transport proteins. The total cholesterol (TC) of LDL is transferred to VLDL and the TG of VLDL is transferred to LDL, but the total amount and synthesis of LDL remains unchanged. When the TG levels in LDL increase to a certain extent, LDL will be hydrolyzed by liver lipase to remove TG, the LDL particles become smaller, then the TC content decreases, thus promoting the formation of sdLDL-C. When the plasma concentration of TG exceeds 1.5 mmol/L, lipid exchange is accelerated; higher TG levels accelerate the lipid interaction between VLDL and LDL, producing more sdLDL-C. It has been reported in the literature that the change in gene locus is also significantly related to sdLDL-C levels. In 2009, Musunuru et al. (14) proved that four genes, CEPT, LIPC, APOA1/A5, and LPL are related to the particle size and distribution of LDL or HDL. Hoogeveen (15) showed that 127 single nucleotide polymorphisms (SNPs) are considerably related to sdLDL-C. These SNPs are distributed in eight different sites on chromosomes 1, 2, 7, 8, 11, and 19, and are distributed in 14 different genes. The genetic variation of these genes is related to lipid metabolism and inflammatory pathways. This study also found that the genetic variation of the new locus PCSK7 was also associated with sdLDL-C levels.
AS mainly affects the intima of the large- and medium-sized arteries, characterized by lipid deposition, focal fibrosis, and the formation of atherosclerotic plaques, resulting in thickening, hardening, and lumen stenosis of the vessel wall, ultimately leading to ischemic changes in the corresponding organs. In recent years, some studies have confirmed that the ability of LDL-C to induce AS varies with different densities, and there is a stronger relationship between sdLDL-C and the stability of AS plaques (16, 17). Ikezaki (18) followed 2,030 men and women [median age 59 years old, no cardiovascular disease (CVD) and not taking cholesterol-lowering drugs] for five years and performed univariate, multivariate regression and least squares analysis to examine the relationship between direct sdLDL-C and other lipoproteins with the progression of carotid intimal medial thickness (cIMT). The plasma levels of direct sdLDL-C and other lipoproteins were measured using a homogeneous detection kit obtained from Denka-Seiken. The results showed that compared with LDL-C, sdLDL-C had a stronger correlation with the progress of cIMT. However, the scale of this line-up study is small and all subjects are Japanese. A larger population of different races is needed to verify this study.
Duran et al. conducted a prospective case cohort study to study the relationship between sdLDL-C and cardiovascular events. The sdLDL-C concentration in this study was directly measured using a two-stage automatic homogenization test also developed by Denka Seiken Co (Niigata, Japan). A total of 27,552 participants provided sufficient blood samples when they entered the group after taking into account the missing key exposure data, the final sample included 480 women with total CVD and 496 women whose age and smoking frequency matched. The study found that the concentration of sdLDL-C in women with myocardial infarction (MI) was much higher than that in the control group, suggested that there was a significant correlation between plasma sdLDL-C concentration and MI (17).
Balling et al. measured sdLDL cholesterol using Denka Seiken's assay in 38,322 individuals participating in the Copenhagen General Population Study from 2013 to 2017. The death and immigration information comes from the Danish civil registration system. Individuals are followed up from baseline to December 2018 for MI and atherosclerotic cardiovascular disease (ASCVD). events, death, immigration, or the end of follow-up, whichever occurs first. Covariates were measured at baseline, including smoking, lipid-lowering therapy, blood pressure, body mass index, diabetes, blood sample analysis, total cholesterol, high-density lipoprotein cholesterol, triglycerides and apolipoprotein B. The Cox regression restricted cubic spline model with multivariate adjustment was also used to examine the association between sdLDL cholesterol and the risk of MI. The median follow-up time was 3.1 years. In the multivariate adjusted spline curve, as the concentration of sdLDL cholesterol increased, the risk of myocardial infarction was observed to increase (6). A large amount of experimental data (9, 15, 19–25) showed that sdLDL-C is closely related to the occurrence of cardiovascular events, so it is necessary to monitor clinically and reduce the concentration of sdLDL-C to reduce the occurrence of cardiovascular events (Table 1).
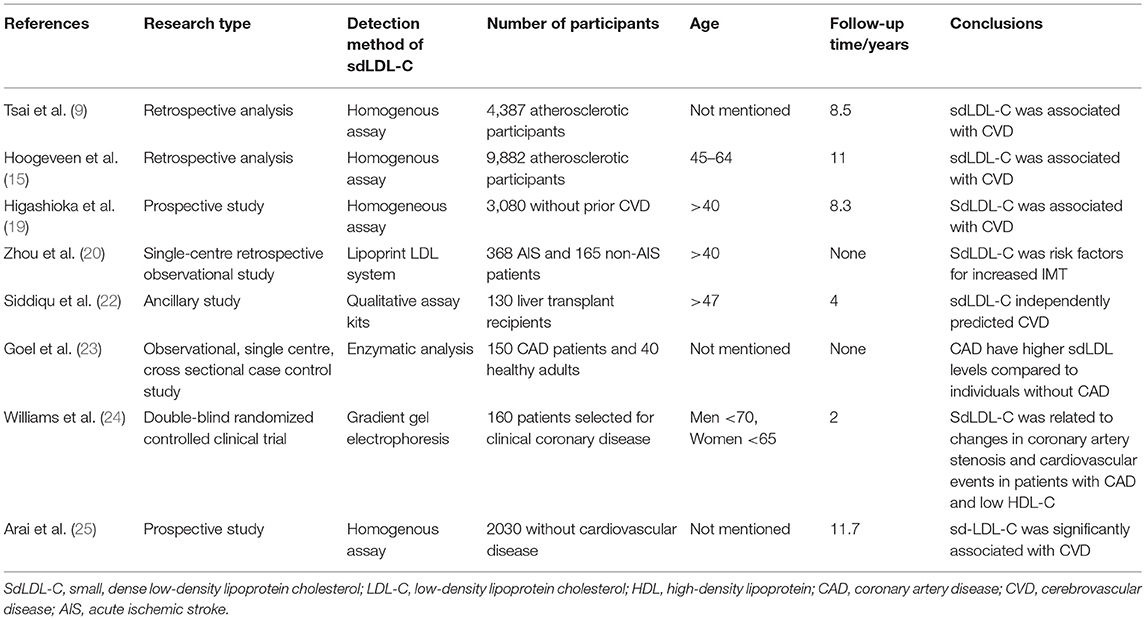
Table 1. The relationship between sdLDL-C and AS.
The term “induration of the arteries” refers to a condition where lipids and alternative substances deposit in and on the artery walls (referred to as “plaques”) that limit traditional blood flow (26). AS and the pathology of related ischemic organs are the primary causes of mortality worldwide (27). Currently, there are many theories regarding the mechanisms of AS. It is generally believed that AS is a chronic inflammatory process involving multiple cell types and cytokines (3, 28). AS begins with the deposition of LDL-C, endothelial dysfunction, the accumulation of foam cells under the endothelium, and the formation of fat streaks through the activation of pro-inflammatory f actors (1).
The metabolism of cholesterol esters (CEs) is regulated by the macrophage gene network. Studies have shown that genes such as ATF3 (activating transcription factor 3) and EGR2 affect AS by regulating lipid metabolism (29–32). ATF3 is a member of the mammalian activation transcription factor/cAMP response element binding (CREB) family (33, 34). SdLDL-C can reduce the ability of ATF3 to induce type B scavenger receptor (SR-BI) by down-regulating the expression of ATF3, and promote hepatic cholesterol 12α hydroxylase (CYP8B1) by interacting with p53 and hepatocyte nuclear factor 4α, thus reducing the uptake of high-density lipoprotein, promoting visceral fat and cholesterol absorption, and inhibiting the reverse cholesterol transport of phagocytes (32). EGR2 is another gene related to cholesterol metabolism, which is involved in the synthesis of free cholesterol (FC) and lipid droplets (LD) (35).
There were some differences in the content of components in different subgroups of LDL-C. SdLD-C showed a significant decrease in free cholesterol (FC), cholesterol ester (CE) and phospholipid (PL) than large and light LDL-C. The study of biofilm and lipid bilayer shows that the incorporation of cholesterol affects the permeability of metabolites. With the increase of FC content, the accessibility of oxidants to lipid core decreased, which may be a reasonable explanation for the protective effect of LDL particles on oxidation susceptibility. The increase of FC content in LDL particles may directly regulate the susceptibility to oxidative stress and help to prevent LDL particles from undergoing subsequent oxidative modification (36). Studies have shown that the introduction of sdLDL-C into traditional M2 macrophages can inhibit the expression of EGR2, resulting in the increase of CE production and FC efflux (29), therefore, the sensitivity of sdLDL-C to lipid peroxidation increased with the decrease of FC content in each particle. Ohmura evaluated the sensitivity to lipid peroxidation modification of low density lipoprotein by the formation of conjugated diene induced by copper, which also confirmed that sdLDL-C was more sensitive to lipid peroxidation modification (36). On the other hand, due to the efflux of FC, when macrophages accumulate a large amount of FC, it is a powerful apoptosis inducer, which will lead to the release of intracellular contents and thrombosis (37). In addition, studies have shown that the plasma retention time of apolipoproteinB-100 (apoB-100) on the surface of sdLDL-C is significantly longer than that of apoB-100 on the surface of IbLDL, which increased the possibility of oxidation (38). LDL is cleared after binding to LDL-C receptors, but apoB-100 on the surface of sdLDL-C molecules and LDL-C receptors has a low affinity, which makes it difficult for the receptors to recognize sdLDL-C, and is more likely to be absorbed by phagocytes, which develop into foam cells and promote the occurrence and development of AS (39).
Current research shows that the increase of sdLDL-C level will reduce the generation of very-long-chain fatty acid (VLCFA), the ability to regulate peroxisome function and interact with peroxisome proliferator activated receptor (PPAR) was weakened (40), thus affecting lipid metabolism, fatty acid β oxidation, plasminogen (PL) biosynthesis and so on (41). This may also be one of the reasons why sdLDL-C has a stronger ability to cause AS. In addition, quantitative reverse transcriptase polymerase chain reaction (qRT-PCR) was used to detect serum miR-126 and 122 levels in 78 patients with CAD and 60 patients without CAD in one study. There, it was suggested that miR-126 may play a role in the cholesterol metabolism of sdLDL-C. However, the mechanism by which the decrease in circulating miR-126 levels in patients with CAD is proportional to the increase in sdLDL-C is not fully understood (42). In 2012, miRNAs were reported to be important regulators of HDL metabolism and reverse cholesterol transport, including direct targeting of cellular cholesterol efflux, HDL biogenesis, liver high-density lipoprotein uptake, and the synthesis of bile acid and secretion-related genes (43). It has also been reported that miR-126 attenuates oxidized LDL (ox-LDL)-induced endothelial cell injury by inhibiting signal transduction to delay AS (44, 45). Therefore, a larger sample cohort is needed to further explain the role of miR-126 in sdLDL-C cholesterol metabolism and to understand the development of the disease (42) (Figure 2).
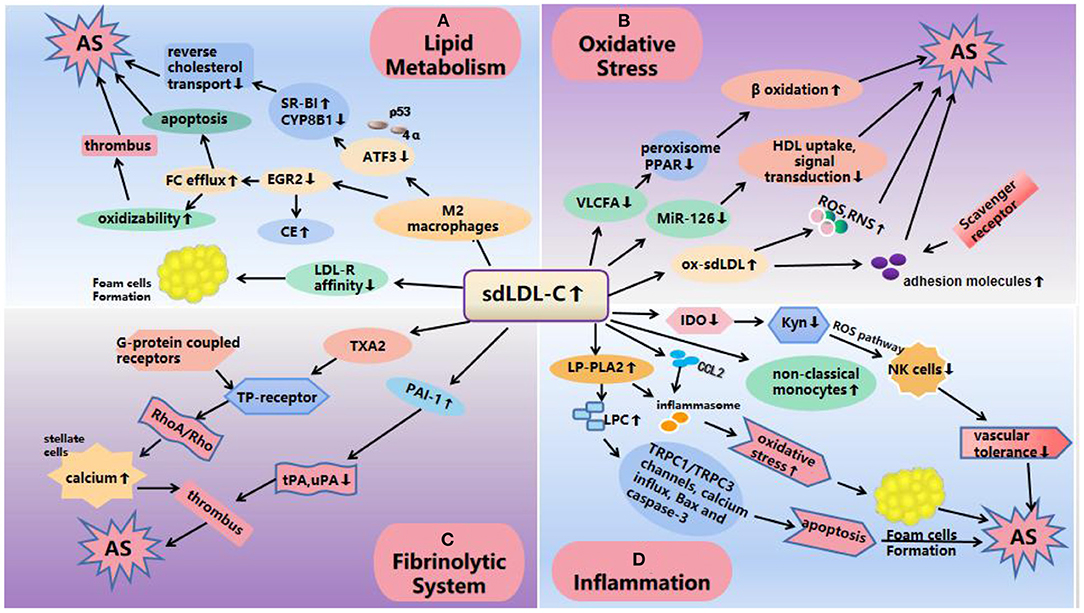
Figure 2. Mechanisms of atherosclerosis induced by sdLDL-C. (A) Lipid metabolism: SdLDL-C reduces the expression of ATF3 and EGR2, ATF3 decreases the ability of SR-BI by interacting with p53 and 4α and promotes CYP8B1 to inhibit cholesterol reverse transport; EGR2 leads to an increase in CE production and FC outflow, thus increasing the oxidation sensitivity of sdLDL-C. On the other hand, the low affinity of apoB-100 on the surface of LDL-C receptors and sdLDL-C makes it difficult for the receptors to recognize sdLDL-C and is more easily absorbed by phagocytes to form foam cells and promote the occurrence and development of AS. (B) Oxidative stress: The increase of sdLDL-C level reduces the production of VLCFA and miR-126, which affects lipid metabolism and fatty acid β oxidation; miR-126 affects HDL uptake, and enhances signal transduction resulting in AS. In addition, ox-sdLDL can also increase the expression of adhesion molecules and induce excessive production of ROS and RNS, resulting in the enhancement of oxidative stress to cause AS. (C) Fibrinolytic system: SdLDL-C increases the levels of PAI-1 and TXA2. PAI-1 inhibits the function of u-PA and t-PA, which easily leads to thrombosis. TXA2 activates TP receptor and activates RhoA/Rho21 kinase pathway through its G protein coupled receptor, and increases calcium levels in hepatic stellate cells, resulting in vasoconstriction, platelet aggregation, thrombosis and AS. (D) Inflammation: SdLDL-C levels reduces IDO, causing a decrease in vascular tolerance by affecting the Kyn pathway; LP-PLA2 increased that activated TRPC1/TRPC3 channels, calcium influx, Bax and caspase-3 pathways to cause apoptosis; increased expression of inflammatory cytokines and the formation of foam cell, suggesting an inflammatory response. SdLDL-C, small, dense low-density lipoprotein cholesterol; AS, atherosclerosis; CE, cholesterol ester; FC, free cholesterol; VLCFA, very-long-chain fatty acid; ATF3, activating transcription factor 3; LDs, lipid droplets; IDO, indoleamine 2,3-dioxygenase; LP-PLA2, lipoprotein-associated phospholipase A2; LPCs, lysophosphatidylcholine; ROS, reactive oxygen species; RNS, reactive nitrogen species; PAI-1, plasminogen activator inhibitor 1; TXA2, thromboxane A2; t-PA, tissue type plasminogen activator; u-PA, urokinase type plasminogen activator.
Monocytes play an important role in the early formation and maturation of plaques. They are drawn to the arteries by chemokines, such as CCL2, which are secreted by activated epithelial tissue cells (46–49) and take up lipids among the subendothelial tissue to differentiate into foam cells (50). In addition, they can also engulf precipitated cholesterol crystals (51) and oxidized lipid species (52–54) that activate the inflammasome, resulting to cell death in a highly inflammatory form called prolapse, as well as the induction of innate immune responses (51). Human monocytes are mainly divided into three types: classical (CD14+CD16−), non-classical (CD14−CD16+), and intermediate (CD14+CD16+) (55). Supported by proof from murine studies (56, 57), as well as current human observations (58), classical monocytes are believed to have the ability to differentiate into monocyte-derived macrophages and monocyte-derived dendritic cells (59) and play an indispensable role in the formation and regression of tissue inflammation. Related studies have shown that the production of sdLDL-C is related to an increase the number of non-classical monocytes and a decrease in the number of classic monocytes (60). The specific mechanism has not been detailed, but we speculate that the effect of sdLDL-C on AS may be related to the inflammatory response of monocytes. In 2017, monocytes from healthy people (stenosis degree <5%) and patients (stenosis degree >70%; single-vessel disease, two-vessel disease, three-vessel disease) were separated using a Rosette Sepkit, and macrophage colony stimulating factor (M-CSF) was used to induce them to differentiate into M2 macrophages. qRT-PCR and ELISA were used to detect MRC1 gene expression and histamine levels, respectively. After sdLDL-C treatment, the expression level of MRC1 in normal human M2 macrophages was significantly increased (P = 0.05), while the expression level of MRC1 gene was decreased in patients with single-vessel disease (P = 0.05), two-vessel disease (P = 0.01), and three-vessel disease (P = 0.9). The histamine levels secreted by M2 macrophages (after treatment for 7 day) in the case group were higher than that in the healthy control group (>3-fold, P = 0.02). The results illustrated that sdLDL-C granules decreased the expression of MRC1 in differentiated M2 macrophages from patients with CHD. In addition, they have a strong ability to secrete histamine (61).
Hassanpour et al. detected the effect of sdLDL-C on the changes in IDO in differentiated macrophages using RT-qPCR, colorimetric, and ELISA methods. Their results show that sdLDL-C reduces the expression and activity of IDO in macrophages (62), IDO is the first and rate-limiting enzyme in the tryptophan (Trp)-degraded kynurenine (Kyn) pathway, and its downstream metabolite is collectively called kynurenines (63). The expression of IDO was inhibited, the metabolic pathway of Trp was blocked, the formation of Kyn was decreased, the ability of cell death mediated by kyn through reactive oxygen species (ROS) pathway in natural killer (NK) cells decreased (64), vascular tolerance decreased, and promoted the occurrence and development of inflammation and AS (65, 66). In addition, ATF3 (67, 68) and EGR2 (69, 70) play crucial roles in signal transduction in the process of anti-apoptosis, anti-migration, and anti-inflammation. sdLDL-C also promotes inflammation and accelerates AS by inhibiting the expression of AFT3 and EGR2 (32).
Lipoprotein-associated phospholipase A2 (Lp-PLA2) hydrolyzes phospholipids and releases pro-inflammatory products; therefore, it is considered to be a new biomarker of vascular risk (71–74). Among the phenotypes of LDL, Lp-PLA2 preferentially binds to small, dense LDL particles (72) to produce lysophosphatidylcholine (LPC). LPC can promote the expression of inflammatory factors (75), damage arterial relaxation, increase oxidative stress, induce endothelial activation and atherosclerosis (76). SdLDL-C granules contain more Lp-PLA2. Studies have confirmed that LPC can induce apoptosis of human coronary artery smooth muscle cells by activating TRPC1/TRPC3 channels, calcium influx, Bax and caspase-3, and lead to atherosclerosis and coronary artery disease (77), which may also be a mechanism of AS induced by sdLDL-C. In addition, higher concentrations of LPC can destroy the integrity of mitochondria and enhance the release of cytochrome C in hepatocytes (78). We speculate that sdLDL-C may lead to AS through mitochondrial damage, which may be a potential mechanism (Figure 2).
Vascular epithelial tissue cell pathology plays an important role in the initiation and development of AS (79, 80). Endothelial cell injury increases intimal permeability and leukocyte adhesion, promoting thrombus formation and rapid malady progression (81). Both ox-LDL and cholesterol cause functional damage to the arterial intima, change the surface characteristics of endothelial cells and leukocytes (monocytes and lymphocytes), and increase the expression of adhesion molecules. The number of monocytes adhering to endothelial cells increases and gets transferred from endothelial cells to subintimal macrophages, which are then transformed into foam cells via scavenger receptor phagocytosis of ox-LDL, forming the earliest lipid streaks of atherosclerotic lesions (82–88).
Ox-LDL induces an excessive production of reactive oxygen species (ROS) and reactive nitrogen species (RNS) in vivo, which induces antioxidant defense; however, the degree of oxidation exceeds the scavenging ability of oxides, resulting in tissue damage (89–92). SdLDL-C particles have the characteristics of a small size, low density, large surface area, long retention time in vivo, and induces great damage to endothelial cells, which leads to their increased permeability, chemotaxis of monocytes in blood vessels to form macrophages, and phagocytosis of oxidized LDL-C to form foam cells, thus further supporting the formation of AS (93). In addition, there are a few polar molecules on the surface of sdLDL-C particles, and their affinity with proteoglycans on arterial intima is enhanced, so they can easily adhere to the vascular wall and enter vascular endothelial cells, resulting in vascular endothelial damage and promoting the occurrence and development of AS (38, 94). This was verified by an experiment on the modification of LDL by methylglyoxal (MG). The modification of LDL by MG resulted in a significant decrease in the particle size of LDL similar to that of sdLDL. Vortex-stimulation showed that sdLDL-C showed higher PG aggregation rate and degree than unmodified LDL (95).
SdLDL-C not only injures the vascular endothelium but also activates the fibrinolytic system and produces plasminogen activator inhibitor 1 (PAI-1) (96–99) and vasoconstrictor thromboxane A2 (TXA2) (100, 101), thus promoting AS (93). A previous study found that plasma PAI-1 levels were positively correlated with the concentration of sdLDL-C (102). PAI-1 inhibits the function of t-PA and u-PA by binding to tissue-type plasminogen activator (t-PA) and urokinase-type plasminogen activator (u-PA) in a ratio of 1:1, The increase of PAI-1 expression in vivo will inhibit the normal fibrinolytic system, which is easy to lead to thrombosis (103). TXA2 activates TP receptors, activates RhoA/Rho kinase pathway through its G-protein coupled receptors, and increases calcium levels in hepatic stellate cells (HSC), resulting in vasoconstriction, platelet aggregation, thrombosis and atherosclerosis (104). Therefore, compared with LDL-C, sdLDL-C has a stronger effect on AS, and the damage it induces to the blood vessel wall lasts longer (105) (Figure 2).
SdLDL-C levels are closely associated with meal compositions and dietary habits. Almonds have been shown to reduce LDL-C levels; however, there is limited data regarding their effects on dyslipidemia characterized by accrued levels of VLDL and sdLDL-C particles that are related to abdominal fat and high carbohydrate intake (106). A current meta-analysis of randomized controlled clinical trials found that consumption of almonds can reduce plasma TC concentration by 0.15 mmol/L, TG concentration by 0.07 mmol/L, and LDL-C concentration by 0.12 mmol/L (107). Studies have shown that almonds and almonds with dark chocolate and cocoa ingested for four weeks have a good effect on the levels of lipids, lipoproteins, and apolipoproteins, and the combined consumption of dark chocolate, cocoa, and almonds significantly reduces levels of sdLDL-C, apoB, and the ratio of apoB/apoAI, which in turn are expected to reduce the risk of CHD (108). Avocados are a nutritious source of monounsaturated fatty acids (MUFAs), which are rich in antioxidants. Avocados have an extra effect of reducing LDL-C levels, especially sdLDL-C particles, which are prone to oxidation in the body and are associated with an increased risk of CVD (109). In another randomized, crossover, controlled feeding study of patients with elevated LDL-C levels, compared with a daily intake of pistachios (32–63 g), a twice-daily intake of pistachios (63–126 g) significantly decreased sdLDL-C levels within four weeks. This experiment adopted a double-blind crossover design in which 30 postmenopausal women with moderate hypercholesterolemia were randomly assigned to two 35-day diets supplemented with corn oil or partially hydrogenated soybean oil to diets providing energy intake for weight maintenance. The results illustrated that the decrease in sdLDL-C concentration was positively correlated with a decrease in TG (110). In a randomized, double-blind, crossover study, subjects ate 10 grams of flaxseed oil or corn oil at dinner once a day, containing 5.49 and 0.09 g α-linolenic acid, respectively. Blood samples were collected at 0.4 and 12 weeks for the analysis of serum lipids, lipid-related proteins, serum fatty acids and serum sdLDL-C. Flaxseed oil supplementation significantly decreased the concentration of sdLDL at 4 and 12 weeks (111).
A study by Mendoza et al. strengthened the relationship between weight loss and reduced sdLDL-C levels. After weight loss, the concentration of apoC-III decreased, and the average BMI decreased from 27 to 25 kg m2, which was related to an increase in the peak particle diameter of LDL and a decrease in serum concentration of sdLDL-type (112). In addition to dietary intervention, exercise can effectively reduce the risk of cardiovascular disease (113). One hundred participants from the RESOLE trial (ages 50–70) were followed up for a year, starting with a three-week accommodation program that combined high exercise (15–20 h per week), diet restriction (500 kcal/day), and education. Forty age-matched healthy controls were recruited as a baseline reference. Lipoprint® electrophoresis was used to evaluate the distribution of lipoprotein subfractions in these subjects, allowing separation, and the results showed that sdLDL-C concentration decreased significantly after a 3-week residence plan (114). Another study conducted a six-month intervention on 30 hyperlipidemic subjects (12 males, 18 females; mean age, 64 years), focusing on moderate increases in physical activity. Clinical data before and after the intervention were observed. In addition to determining the average particle size of LDL and diacron reactive oxygen metabolites (d-ROMs) via gel electrophoresis, the risk factors for AS were also determined. The average LDL particle size after intervention was significantly larger than that before intervention (26.9 ± 0.3 vs. 27.1 ± 0.4 nm, mean ± SD, P < 0.01), whereas the level of sdLDL-C decreased significantly (115) (Table 2).
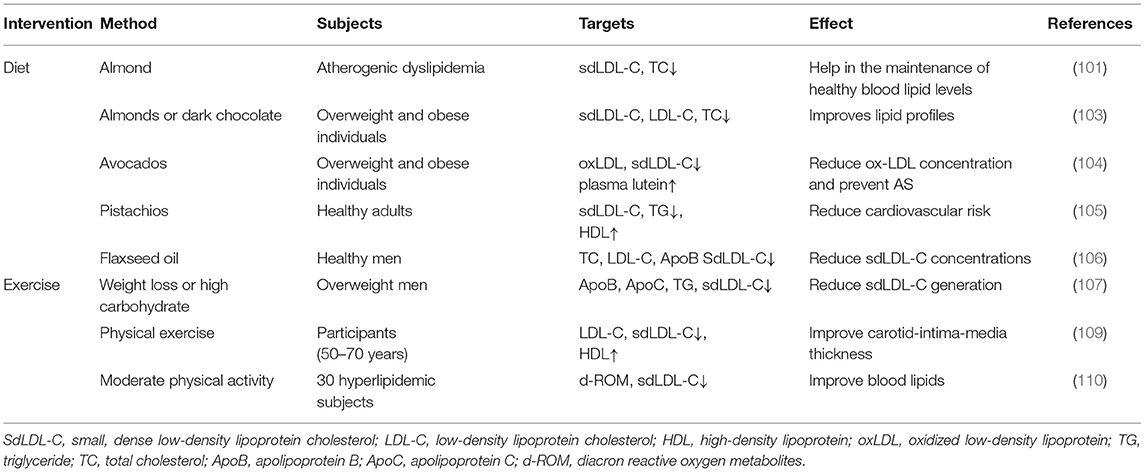
Table 2. Studies of non-medicinal intervention of sdLDL-C against AS.
It is well-known that statins can effectively regulate blood lipid levels and delay the process of atherosclerosis. A study recruited 12 white men with metabolic syndrome. All subjects were treated with pitavastatin (4 mg/day) and their blood lipid levels were measured after 180 days. The results found that pitavastatin not only lowered LDL-C (−38%), sdLDL-C (sd-LDL4) and (sd-LDL5) are also effectively reduced (−27 and−33%, respectively). However, the sample size of this experiment is too small, and the sample size needs to be expanded to prove this point of view (116). A prospective, randomized, open-label, multicenter, parallel grouping comparative trial was conducted in Japan from October 2011 to November 2012. Eligible subjects (people at high cardiovascular risk over the age of 20) were treated with high-dose statins and conventional statins, respectively. The high-dose treatment group took 5 mg of rosuvastatin a day for the first four weeks, and then 10 mg a day for 8 consecutive weeks, and the low-dose statin group took 2.5 mg of rosuvastatin a day for 12 consecutive weeks. Lipid measurements were taken before, 4 and 12 weeks. The results show that both groups can reduce oxidized low-density lipoprotein cholesterol and sdLDL-C, and the effect of the high-dose group is more obvious (117). Although the high-dose group of statin therapy is effective in improving blood lipid levels, some cardiovascular events continue to occur. Therefore, high-dose statin therapy is recommended for the initial treatment of patients with high risk of atherosclerotic vascular disease (118). Another in vitro experiment evaluated the effect of combination therapy of Eicosapentaenoic Acid (EPA) and atorvastatin on endothelial cell function under oxidative stress conditions by measuring the release of NO and peroxynitrite (ONOO–) from human umbilical vein endothelial cells (HUVECS) to examine the comparative and time-dependent effects of these agents on endothelial dysfunction. Data shows that the combined treatment of EPA and atorvastatin can effectively reduce the level of sdLDL-C, thereby improving endothelial dysfunction, which may be because EPA contains substances that inhibit the oxidation of ApoB particles, which has a stronger antioxidant effect (119). However, some studies have shown that compared with patients who received atorvastatin for <6 days, patients who received atorvastatin for more than 90 days had significantly lower total cholesterol and LDL-C levels, but slightly lower sdLDL-C levels increased, but not significant (p = 0.06) (120).
Fibrates are agonists of peroxisome proliferator-activated receptor-α (PPAR-α), which regulate lipoprotein metabolism through transcription factors. Fibrates have shown effects in reducing fasting and postprandial TG and TG-rich lipoprotein residual particles (121). A meta-analysis of 13 studies illustrated that fibrates can reduce triglyceride levels, increase HDL-C levels, reduce the proportion of sdLDL-C, fibrates could be effective in secondary prevention considering a compound objective of non-fatal stroke, non-fatal myocardial infarction, and death of cardiovascular origin, and have fewer side effects; the most widely used drug of this class is fenofibrate (122). Other studies have also proved this point (123, 124). A retrospective study included 72 patients with type 2 diabetes. All patients received pemafibrate 0.2 mg (0.1 mg twice daily) for 24 weeks. During the entire study period, all patients did not change their exercise or diet regimens. The results show that Pemafibrate significantly reduces the levels of TG and sd-LDL-C, improves the composition of LDL and may reduce the risk of cardiovascular disease (125). It also has a better benefit-risk balance than conventional fibrates and can be applied to patients who find it difficult to use existing fibrates, such as those taking statins or those who have renal insufficiency (126).
Ezetimibe is a novel drug used for the treatment of dyslipidemia, which resists cholesterol absorption by inhibiting Niemann pick C1 like protein (NPC1L1) (127). Ezetimibe alone or in combination with statins can reduce the level of sdLDL-C (128). From October 2014 to November 2015, the author recruited patients with type 2 diabetes who had normal LDL-C levels and received statin therapy at the outpatient clinic of their institution. A total of 50 patients (31 men and 19 women) were enrolled in this study, and all subjects were randomly assigned to receive statins (statin group) or fenofibrate (160 mg/day) and ezetimibe treatment (10 mg/day), the results showed that the combination of fenofibrate and ezetimibe can effectively control the levels of sdLDL-C and TG, increase the level of HDL-C, and improve the vascular function of patients with type 2 diabetes. The effect of this combination is even better than treatment with statins alone (129). Therefore, to reduce the level of blood lipids using ezetimibe, it may be more beneficial if it is combined with other drugs. Current studies have illustrated that fenofibrate combined with ezetimibe can improve sdLDL-C levels and vascular function compared with statins (129).
Niacin inhibits AS by activating the anti-inflammatory G protein-coupled receptor Gpr109a, also known as hydroxycarboxylic acid receptor 2 (HCA2), expressed on immune cells, inactivating the immune response and adventitious inflammatory cell infiltration (130). Niacin treatment was shown to decrease total cholesterol, triglyceride (20–50% decrease), and LDL-C levels. Additionally, niacin decreased sdLDL-C levels, leading to a shift to massive buoyant LDL particles, delaying the progression of AS (7). However, the negative effects of nicotinic acid were in accordance with the results of the HPS2THRIVE and AIM-HIGH trials, which suggests that its clinical application requires further study (131, 132).
Omega-3 fatty acids are essential fatty acids found in certain fish and vegetables. These are necessary for growth and development. Numerous studies have reported that omega-3 fatty acids scale back plasma triglycerides and increase HDL levels. They have been reported to inhibit blood platelet aggregation, improve endothelial function, decrease oxidative stress, and act as a potent medication agent (133). Changes in blood lipid and lipoprotein profiles were also observed after omega-3 fatty acid treatment for 8 weeks. The results of one study also showed that sdLDL-C levels decreased significantly after intake of omega-3 fatty acids (134). The omega-3 fatty acid EPA has substiantial antioxidant activity and can protect the membrane structure, which may promote scavenging of free radicals in sdLDL-C and membrane bilayers (135).
Proprotein convertase subtilisin/kexin type 9 (PCSK9) belongs to the proprotein convertase family of enzymes that degrades LDL-R, which directly mediates the degradation of LDL-R in lysosome, which in turn increases plasma LDL level (136, 137). PCSK9 is positively correlated with sdLDL-C levels (138). At present, PCSK9 inhibitors (PCSK-9i) have been clinically used to reduce cholesterol levels and cardiovascular events in patients (139). In addition, resin and orlistat can also reduce sdLDL-C levels (140). Baricitinib treatment can also increase LDL levels and reduce sdLDL-C particles. One of the mechanisms by which baricitinib and related interventions increase the particle size of LDL-C may be the increased activities of phospholipase A2, liver lipase, lipoprotein lipase, and endothelial lipase. It has been reported that these enzymes are increased in a state of chronic inflammation (141).
Lipid metabolism disorder is an important factor leading to AS. A large amount of evidence shows the pathogenic role of increased LDL-C in AS, and SdLDL-C, as a subgroup of LDL-C, has been proved to be a specific index for the detection of AS. Compared with traditional lipid monitoring, sdLDL-C monitoring has better sensitivity and specificity, and has better clinical value in predicting AS (7, 9). At present, there are many methods for the detection of sdLDL-C, but most of them have some limitations which cannot be widely used in the laboratory and clinic because they require expensive equipment, are time consuming, labor-intensive, and other reasons. The Lipoprint LDL system is currently the mainstream method for detection of sdLDL-C because it utilizes linear polyacrylamide gel electrophoresis to separate low-density lipoprotein according to particle size and charge, which has advantages of high efficiency, high speed, and low materials consumption (6).
Because of its small size and higher density compared to larger LDL-C particles, sdLDL-C has a greater ability to penetrate the artery wall, in addition to having a longer half-life and greater susceptibility to oxidative modification. Cardiovascular diseases caused by abnormal sdLDL-C are reflected in many clinical cases. SdLDL-C plays a variety of roles in the process of AS, such as affecting lipid metabolism, promoting the release of inflammatory factors leading to inflammatory reaction, releasing excessive ROS and RNS to produce oxidative stress, activating fibrinolytic system to produce thrombus. At present, there are non-drug interventions, such as regulating diet (low carbohydrates, soybeans, corn oil, etc.). Proper exercise can effectively improve the level of sdLDL-C in patients. Medical interventions, such as statins, fibrates, ezetimibe, niacin and omega-3 fatty acids, can reduce sdLDL-C levels in the body. The main mechanism is to improve the level of blood lipids and vascular endothelial function. In recent years, Pemabet and PCSK9 inhibitors have become the focus of current research as new interventions to prevent AS.
The specific mechanism of atherosclerosis caused by sdLDL-C has not been fully explained, and it is still being explored. Further exploration of the specific mechanism and intervention measures of sdLDL-C may provide a new direction for clinical prevention, evaluation and treatment of AS. Some studies have shown that the concentration of sdLDL-C is related to the changes of gene loci, and we speculate that gene detection may provide a new reference for the study of sdLDL-C. We know that intestinal microflora is significantly associated with lipid metabolism. A randomized controlled trial shows that changing intestinal microflora in patients with hyperlipidemia can effectively reduce sdLDL-C levels in patients with hyperlipidemia (142). However, the related research on the effect of intestinal flora on sdLDL-C is insufficient, whether it can reduce the level of sdLDL-C by improving intestinal flora, so as to reduce AS, is worthy of further study. In conclusion, these studies on the role of sdLDL-C in AS may provide information regarding new targets for the prevention and treatment of AS.
XJ and MW designed the article and wrote the manuscript. SY and JL searched the literature and aided in the design of the illustrations. All authors contributed to the article and approved the submitted version.
The work was supported by the National Natural Science Foundation of China (Grant Nos. 81202805 and 82074254), the Beijing Natural Science Foundation (No. 7172185).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, Bittencourt MS, et al. Atherosclerosis. Nat Rev Dis Primers. (2019) 5:56. doi: 10.1038/s41572-019-0106-z
2. Chen J, Zhang S, Wu J, Wu S, Xuand G, Wei D. Essential role of nonessential amino acid glutamine in atherosclerotic cardiovascular disease. DNA Cell Biol. (2020) 39:8–15. doi: 10.1089/dna.2019.5034
3. Geovanini G.R., Libby P.. Atherosclerosis and inflammation: overview and updates. Clin Sci. (2018) 132:1243–52. doi: 10.1042/CS20180306
4. Wang HH, Garruti G, Liu M, Portincasaand P, Wang DQ. Cholesterol and lipoprotein metabolism and atherosclerosis: recent advances in reverse cholesterol transport. Ann Hepatol. (2017) 16:s27–42. doi: 10.5604/01.3001.0010.5495
5. Khosravi M, Poursaleh A, Ghasempour G, Farhadand S, Najafi M. The effects of oxidative stress on the development of atherosclerosis. Biol Chem. (2019) 400:711–32. doi: 10.1515/hsz-2018-0397
6. Balling M, Nordestgaard BG, Langsted A, Varbo A, Kamstrupand PR, Afzal S. Small dense low-density lipoprotein cholesterol predicts atherosclerotic cardiovascular disease in the copenhagen general population study. J Am Coll Cardiol. (2020) 75:2873–5. doi: 10.1016/j.jacc.2020.03.072
7. Ivanova EA, Myasoedova VA, Melnichenko AA, Grechkoand AV, Orekhov AN. Small dense low-density lipoprotein as biomarker for atherosclerotic diseases. Oxid Med Cell Longev. (2017) 2017:1273042. doi: 10.1155/2017/1273042
8. Kanonidou C. Small dense low-density lipoprotein: analytical review. Clin Chim Acta. (2021) 520:172–78. doi: 10.1016/j.cca.2021.06.012
9. Tsai MY, Steffen BT, Guan W, McClelland RL, Warnick R, McConnell J, et al. New automated assay of small dense low-density lipoprotein cholesterol identifies risk of coronary heart disease: the multi-ethnic study of atherosclerosis. Arterioscler Thromb Vasc Biol. (2014) 34:196–201. doi: 10.1161/ATVBAHA.113.302401
10. Marin MT, Dasari PS, Tryggestad JB, Aston CE, Teagueand AM, Short KR. Oxidized HDL and LDL in adolescents with type 2 diabetes compared to normal weight and obese peers. J Diabetes Compl. (2015) 29:679–85. doi: 10.1016/j.jdiacomp.2015.03.015
11. Ito Y, Fujimura M, Ohtaand M, Hirano T. Development of a homogeneous assay for measurement of small dense LDL cholesterol. Clin Chem. (2011) 57:57–65. doi: 10.1373/clinchem.2010.149559
12. Sampson M, Wolska A, Warnick R, Luceroand D, Remaley AT. A new equation based on the standard lipid panel for calculating small dense low-density lipoprotein-cholesterol and its use as a risk-enhancer test. Clin Chem. (2021) 67:987–97. doi: 10.1093/clinchem/hvab048
13. Sumino H, Nakajimaand K, Murakami M. [Possibility of new circulating atherosclerosis-related lipid markers measurement in medical and complete medical checkups: small dense low-density lipoprotein cholesterol and lipoprotein lipase]. Rinsho Byori. (2016) 64:298–307.
14. Musunuru K, Orho-Melander M, Caulfield MP, Li S, Salameh WA, Reitz RE, et al. Ion mobility analysis of lipoprotein subfractions identifies three independent axes of cardiovascular risk. Arterioscler Thromb Vasc Biol. (2009) 29:1975–80. doi: 10.1161/ATVBAHA.109.190405
15. Hoogeveen RC, Gaubatz JW, Sun W, Dodge RC, Crosby JR, Jiang J, et al. Small dense low-density lipoprotein-cholesterol concentrations predict risk for coronary heart disease: the atherosclerosis risk in communities (ARIC) study. Arterioscler Thromb Vasc Biol. (2014) 34:1069–77. doi: 10.1161/ATVBAHA.114.303284
16. Liu F, Wang Z, Cao X, Pan Y, Zhang E, Zhou J, et al. Relationship between small dense low-density lipoprotein cholesterol with carotid plaque in Chinese individuals with abnormal carotid artery intima-media thickness. BMC Cardiovasc Disord. (2021) 21:216. doi: 10.1186/s12872-021-02023-4
17. Duran EK, Aday AW, Cook NR, Buring JE, Ridkerand PM, Pradhan AD. Triglyceride-rich lipoprotein cholesterol, small dense LDL cholesterol, and incident cardiovascular disease. J Am Coll Cardiol. (2020) 75:2122–35. doi: 10.1016/j.jacc.2020.02.059
18. Ikezaki H, Furusyo N, Yokota Y, Ai M, Asztalos BF, Murata M, et al. Small dense low-density lipoprotein cholesterol and carotid intimal medial thickness progression. J Atheroscler Thromb. (2020) 27:1108–22. doi: 10.5551/jat.54130
19. Higashioka M, Sakata S, Honda T, Hata J, Shibata M, Yoshida D, et al. The association of small dense low-density lipoprotein cholesterol and coronary heart disease in subjects at high cardiovascular risk. J Atheroscler Thromb. (2021) 28:79–89. doi: 10.5551/jat.55350
20. Zhou P, Shen Y, Wang L, Cao Z, Feng W, Liu J, et al. Association between carotid intima media thickness and small dense low-density lipoprotein cholesterol in acute ischaemic stroke. Lipids Health Dis. (2020) 19:177. doi: 10.1186/s12944-020-01353-0
21. Liou L, Kaptoge S. Association of small, dense LDL-cholesterol concentration and lipoprotein particle characteristics with coronary heart disease: a systematic review and meta-analysis. PLoS ONE. (2020) 15:e0241993. doi: 10.1371/journal.pone.0241993
22. Siddiqui MB, Arshad T, Patel S, Lee E, Albhaisi S, Sanyal AJ, et al. Small dense low-density lipoprotein cholesterol predicts cardiovascular events in liver transplant recipients. Hepatology. (2019) 70:98–107. doi: 10.1002/hep.30518
23. Goel PK, Ashfaq F, Khanna R, Rameshand V, Pandey CM. The association between small dense low density lipoprotein and coronary artery disease in north indian patients. Indian J Clin Biochem. (2017) 32:186–92. doi: 10.1007/s12291-016-0592-7
24. Williams PT, Zhao XQ, Marcovina SM, Brownand BG, Krauss RM. Levels of cholesterol in small LDL particles predict atherosclerosis progression and incident CHD in the HDL-atherosclerosis treatment study (HATS). PLoS ONE. (2013) 8:e56782. doi: 10.1371/journal.pone.0056782
25. Arai H, Kokubo Y, Watanabe M, Sawamura T, Ito Y, Minagawa A, et al. Small dense low-density lipoproteins cholesterol can predict incident cardiovascular disease in an urban Japanese cohort: the Suita study. J Atheroscler Thromb. (2013) 20:195–203. doi: 10.5551/jat.14936
26. Munjal A, Khandia R. Atherosclerosis: orchestrating cells and biomolecules involved in its activation and inhibition. Adv Protein Chem Struct Biol. (2020) 120:85–122. doi: 10.1016/bs.apcsb.2019.11.002
27. Ziegler T, Abdel Rahman F, Jurisch V, Kupatt C. Atherosclerosis and the capillary network; pathophysiology and potential therapeutic strategies. Cells. (2019) 9:50. doi: 10.3390/cells9010050
28. Saigusa R, Winkels H, Ley K. T cell subsets and functions in atherosclerosis. Nat Rev Cardiol. (2020) 17:387–401. doi: 10.1038/s41569-020-0352-5
29. Hosseini-Fard SR, Khosravi M, Yarnazari A, Hassanpour P, Amirfarhangi A, Tajik N, et al. ATF3 and EGR2 gene expression levels in sdLDL-treated macrophages of patients with coronary artery stenosis. Laboratoriums Medizin. (2018) 42:23–9. doi: 10.1515/labmed-2017-0138
30. Kim S, Song NJ, Bahn G, Chang SH, Yun UJ, Ku JM, et al. Atf3 induction is a therapeutic target for obesity and metabolic diseases. Biochem Biophys Res Commun. (2018) 504:903–8. doi: 10.1016/j.bbrc.2018.09.048
31. Chou CL, Li CH, Lin H, Liao MH, Wu CC, Chen JS, et al. Role of activating transcription factor 3 in fructose-induced metabolic syndrome in mice. Hypertens Res. (2018) 41:589–97. doi: 10.1038/s41440-018-0058-9
32. Xu Y, Li Y, Jadhav K, Pan X, Zhu Y, Hu S, et al. Hepatocyte ATF3 protects against atherosclerosis by regulating HDL and bile acid metabolism. Nat Metab. (2021) 3:59–74. doi: 10.1038/s42255-020-00331-1
33. Zhou H, Li N, Yuan Y, Jin YG, Guo H, Deng W, et al. Activating transcription factor 3 in cardiovascular diseases: a potential therapeutic target. Basic Res Cardiol. (2018) 113:37. doi: 10.1007/s00395-018-0698-6
34. Qin W, Yang H, Liu G, Bai R, Bian Y, Yang Z, et al. Activating transcription factor 3 is a potential target and a new biomarker for the prognosis of atherosclerosis. Hum Cell. (2021) 34:49–59. doi: 10.1007/s13577-020-00432-9
35. Korber M, Klein I, Daum G. Steryl ester synthesis, storage and hydrolysis: a contribution to sterol homeostasis. Biochim Biophys Acta Mol Cell Biol Lipids. (2017) 1862:1534–45. doi: 10.1016/j.bbalip.2017.09.002
36. Ohmura H, Mokuno H, Sawano M, Hatsumi C, Mitsugi Y, Watanabe Y, et al. Lipid compositional differences of small, dense low-density lipoprotein particle influence its oxidative susceptibility: possible implication of increased risk of coronary artery disease in subjects with phenotype B. Metabolism. (2002) 51:1081–7. doi: 10.1053/meta.2002.34695
37. Morita SY. Metabolism and modification of apolipoprotein B-containing lipoproteins involved in dyslipidemia and atherosclerosis. Biol Pharm Bull. (2016) 39:1–24. doi: 10.1248/bpb.b15-00716
38. Thongtang N, Diffenderfer MR, Ooi EMM, Barrett PHR, Turner SM, Le NA, et al. Metabolism and proteomics of large and small dense LDL in combined hyperlipidemia: effects of rosuvastatin. J Lipid Res. (2017) 58:1315–24. doi: 10.1194/jlr.M073882
39. Chapman MJ, Orsoni A, Tan R, Mellett NA, Nguyen A, Robillard P, et al. LDL subclass lipidomics in atherogenic dyslipidemia: effect of statin therapy on bioactive lipids and dense LDL. J Lipid Res. (2020) 61:911–32. doi: 10.1194/jlr.P119000543
40. Yamazaki Y, Kondo K, Maeba R, Nishimukai M, Nezuand T, Hara H. Proportion of nervonic acid in serum lipids is associated with serum plasmalogen levels and metabolic syndrome. J Oleo Sci. (2014) 63:527–37. doi: 10.5650/jos.ess13226
41. Lodhi IJ, Semenkovich CF. Peroxisomes: a nexus for lipid metabolism and cellular signaling. Cell Metab. (2014) 19:380–92. doi: 10.1016/j.cmet.2014.01.002
42. Mishra S, Rizvi A, Pradhan A, Perrone MA, Ali W. Circulating microRNA-126 &122 in patients with coronary artery disease: Correlation with small dense LDL. Prostaglandins Other Lipid Mediat. (2021) 153:106536. doi: 10.1016/j.prostaglandins.2021.106536
43. Rottiers V, Näär AM. MicroRNAs in metabolism and metabolic disorders. Nat Rev Mol Cell Biol. (2012) 13:239–50. doi: 10.1038/nrm3313
44. Tang F, Yang TL. MicroRNA-126 alleviates endothelial cells injury in atherosclerosis by restoring autophagic flux via inhibiting of PI3K/Akt/mTOR pathway. Biochem Biophys Res Commun. (2018) 495:1482–89. doi: 10.1016/j.bbrc.2017.12.001
45. Chen Z, Pan X, Sheng Z, Yan G, Chen L, Ma G. Baicalin suppresses the proliferation and migration of Ox-LDL-VSMCs in atherosclerosis through upregulating miR-126-5p. Biol Pharm Bull. (2019) 42:1517–23. doi: 10.1248/bpb.b19-00196
46. Jaipersad AS, Shantsila A, Lip GY, Shantsila E. Expression of monocyte subsets and angiogenic markers in relation to carotid plaque neovascularization in patients with pre-existing coronary artery disease and carotid stenosis. Ann Med. (2014) 46:530–8. doi: 10.3109/07853890.2014.931101
47. Schiopu A, Bengtsson E, Gonçalves I, Nilsson J, Fredrikson GN, Björkbacka H. Associations between macrophage colony-stimulating factor and monocyte chemotactic protein 1 in plasma and fifirst-time coronary events: a nested case-control study. J Am Heart Assoc. (2016) 5:E002851. doi: 10.1161/JAHA.115.002851
48. Bäck M, Yurdagul Jr A, Tabas I, Öörni K, Kovanen PT. Inflammation and its resolution in atherosclerosis: mediators and therapeutic opportunities. Nat Rev Cardiol. (2019) 16:389–406. doi: 10.1038/s41569-019-0169-2
49. Weber C, Noels H. Atherosclerosis: current pathogenesis and therapeutic options. Nat Med. (2011) 17:1410–22. doi: 10.1038/nm.2538
50. Mehta S, Dhawan V. Exposure of cigarette smoke condensate activates NLRP3 inflammasome in THP-1 cells in a stage-specific manner: an underlying role of innate immunity in atherosclerosis. Cell Signal. (2020) 72:109645. doi: 10.1016/j.cellsig.2020.109645
51. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. (2010) 464:1357–61. doi: 10.1038/nature08938
52. Bekkering S, Quintin J, Joosten LA, van der Meer JW, Netea MG, Riksen NP. Oxidized low-density lipoprotein induces long-term proinflammatory cytokine production and foam cell formation via epigenetic reprogramming of monocytes. Arterioscler Thromb Vasc Biol. (2014) 34:1731–8. doi: 10.1161/ATVBAHA.114.303887
53. van der Valk FM, Bekkering S, Kroon J, Yeang C, Van den Bossche J, van Buul JD, et al. Oxidized phospholipids on lipoprotein(a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation. (2016) 134:611–24. doi: 10.1161/CIRCULATIONAHA.116.020838
54. Que X, Hung MY, Yeang C, Gonen A, Prohaska TA, Sun X, et al. Oxidized phospholipids are proinflammatory and proatherogenic in hypercholesterolaemic mice. Nature. (2018) 558:301–6. doi: 10.1038/s41586-018-0198-8
55. Kapellos TS, Bonaguro L, Gemünd I, Reusch N, Saglam A, Hinkley ER, et al. Human monocyte subsets and phenotypes in major chronic inflammatory diseases. Front Immunol. (2019) 10:2035. doi: 10.3389/fimmu.2019.02035
56. Guilliams M, Mildner A, Yona S. Developmental and functional heterogeneity of monocytes. Immunity. (2018) 49:595–613. doi: 10.1016/j.immuni.2018.10.005
57. Jakubzick CV, Randolph GJ, Henson PM. Monocyte differentiation and antigen-presenting functions. Nat Rev Immunol. (2017) 17:349–62. doi: 10.1038/nri.2017.28
58. Sander J, Schmidt SV, Cirovic B, McGovern N, Papantonopoulou O, Hardt AL, et al. Cellular differentiation of human monocytes is regulated by time-dependent interleukin-4 signaling and the transcriptional regulator NCOR2. Immunity. (2017) 47:1051–66.e12. doi: 10.1016/j.immuni.2017.11.024
59. Menezes S, Melandri D, Anselmi G, Perchet T, Loschko J, Dubrot J, et al. The heterogeneity of Ly6C(hi) monocytes controls their differentiation into iNOS(+) macrophages or monocyte-derived dendritic cells. Immunity. (2016) 45:1205–18. doi: 10.1016/j.immuni.2016.12.001
60. Krychtiuk KA, Kastl SP, Pfaffenberger S, Lenz M, Hofbauer SL, Wonnerth A, et al. Association of small dense LDL serum levels and circulating monocyte subsets in stable coronary artery disease. PLoS ONE. (2015) 10:e0123367. doi: 10.1371/journal.pone.0123367
61. Yarnazari A, Hassanpour P, Hosseini-Fard SR, Amirfarhangi A, Najafi M. The sdLDL reduces MRC1 expression level and secretion of histamin e in differentiated M2-macrophages from patients with coronary artery stenosis. Cardiovasc Hematol Disord Drug Targets. (2017) 17:28–32. doi: 10.2174/1871529X17666170106095554
62. Hassanpour P, Amirfarhangi A, Hosseini-Fard SR, Yarnazari A, Najafi M. Interleukin 6 may be related to indoleamine 2,3-dioxygense function in M2 macrophages treated with small dense LDL particles. Gene. (2017) 626:442–6. doi: 10.1016/j.gene.2017.05.063
63. Polyzos KA, Ovchinnikova O, Berg M, Baumgartner R, Agardh H, Pirault J, et al. Inhibition of indoleamine 2,3-dioxygenase promotes vascular inflammation and increases atherosclerosis in Apoe-/- mice. Cardiovasc Res. (2015) 106:295–302. doi: 10.1093/cvr/cvv100
64. Song H, Park H, Kim YS, Kim KD, Lee HK, Cho DH, et al. L-kynurenine-induced apoptosis in human NK cells is mediated by reactive oxygen species. Int Immunopharmacol. (2011) 11:932–8. doi: 10.1016/j.intimp.2011.02.005
65. Yun TJ, Lee JS, Machmach K, Shim D, Choi J, Wi YJ, et al. Indoleamine 2,3-dioxygenase-expressing aortic plasmacytoid dendritic cells protect against atherosclerosis by induction of regulatory T cells. Cell Metab. (2016) 23:852–66. doi: 10.1016/j.cmet.2016.11.008
66. Forteza MJ, Polyzos KA, Baumgartner R, Suur BE, Mussbacher M, Johansson DK, et al. Activation of the regulatory t-cell/indoleamine 2,3-dioxygenase axis reduces vascular inflammation and atherosclerosis in hyperlipidemic mice. Front Immunol. (2018) 9:950. doi: 10.3389/fimmu.2018.00950
67. Lin H, Cheng CF. Activating transcription factor 3, an early cellular adaptive responder in ischemia/reperfusion-induced injury. Ci Ji Yi Xue Za Zhi. (2018) 30:61–65. doi: 10.4103/tcmj.tcmj_37_18
68. Teasdale JE, Hazell GG, Peachey AM, Sala-Newby GB, Hindmarch CC, McKay TR, et al. Cigarette smoke extract profoundly suppresses TNFα-mediated proinflammatory gene expression through upregulation of ATF3 in human coronary artery endothelial cells. Sci Rep. (2017) 7:39945. doi: 10.1038/srep39945
69. Omodho B, Miao T, Symonds ALJ, Singh R, Li S, Wang P. Transcription factors early growth response gene (Egr) 2 and 3 control inflammatory responses of tolerant T cells. Immun Inflamm Dis. (2018) 6:221–33. doi: 10.1002/iid3.210
70. Symonds AL, Zheng W, Miao T, Wang H, Wang T, Kiome R, et al. Egr2 and 3 control inflflammation, but maintain homeostasis, of PD- 1(high) memory phenotype CD4 T cells. Life Sci Alliance. (2020) 3:E202000766. doi: 10.26508/lsa.202000766
71. Keleşoglu M, Kizilay F, Barutçuoglu B, Başol G, Saraç F, Mutaf I, et al. The relationship between lipoprotein-associated phospholipase A2 with cardiovascular risk factors in testosterone deficiency. Turk J Urol. (2018) 44:103–8. doi: 10.5152/tud.2017.30633
72. Sofogianni A, Alkagiet S, Tziomalos K. Lipoprotein-associated phospholipase A2 and coronary heart disease. Curr Pharm Des. (2018) 24:291–6. doi: 10.2174/1381612824666180111110550
73. De Stefano A, Mannucci L, Tamburi F, Cardillo C, Schinzari F, Rovella V, et al. Lp-PLA(2), a new biomarker of vascular disorders in metabolic diseases. Int J Immunopathol Pharmacol. (2019) 33:2058738419827154. doi: 10.1177/2058738419827154
74. Santoso A, Heriansyah T, Rohman MS. Phospholipase A2 is an inflammatory predictor in cardiovascular diseases: is there any spacious room to prove the causation? Curr Cardiol Rev. (2020) 16:3–10. doi: 10.2174/1573403X15666190531111932
75. Chang MC, Lee JJ, Chen YJ, Lin SI, Lin LD, Jein-Wen Liou E, et al. Lysophosphatidylcholine induces cytotoxicity/apoptosis and IL-8 production of human endothelial cells: related mechanisms. Oncotarget. (2017) 8:106177–6189. doi: 10.18632/oncotarget.22425
76. Li X, Wang L, Fang P, Sun Y, Jiang X, Wang H, et al. Lysophospholipids induce innate immune transdifferentiation of endothelial cells, resulting in prolonged endothelial activation. J Biol Chem. (2018) 293:11033–45. doi: 10.1074/jbc.RA118.002752
77. Wang Y, Wang Y, Li GR. TRPC1/TRPC3 channels mediate lysophosphatidylcholine-induced apoptosis in cultured human coronary artery smooth muscles cells. Oncotarget. (2016) 7:50937–51. doi: 10.18632/oncotarget.10853
78. Law SH, Chan ML, Marathe GK, Parveen F, Chen CH, Ke LY. An updated review of lysophosphatidylcholine metabolism in human diseases. Int J Mol Sci. (2019) 20:1149. doi: 10.3390/ijms20051149
79. Wang J, Wang WN, Xu SB, Wu H, Dai B, Jian DD, et al. MicroRNA-214-3p: a link between autophagy and endothelial cell dysfunction in atherosclerosis. Acta Physiol. (2018) 222:e12973. doi: 10.1111/apha.12973
80. Yang X, Yin G, Sun H, Zhao G. Physcion 8-O-β-Glucopyranoside alleviates oxidized low-density lipoprotein-induced human umbilical vein endothelial cell injury by inducing autophagy through AMPK/SIRT1 signaling. J Cardiovasc Pharmacol. (2019) 74:53–61. doi: 10.1097/FJC.0000000000000680
81. Wang R, Wang M, Ye J, Sun G, Sun X. Mechanism overview and target mining of atherosclerosis: endothelial cell injury in atherosclerosis is regulated by glycolysis (Review). Int J Mol Med. (2021) 47:65–76. doi: 10.3892/ijmm.2020.4798
82. Bar A, Targosz-Korecka M, Suraj J, Proniewski B, Jasztal A, Marczyk B, et al. Degradation of glycocalyx and multiple manifestations of endothelial dysfunction coincide in the early phase of endothelial dysfunction before atherosclerotic plaque development in apolipoprotein e/low-density lipoprotein receptor-deficient mice. J Am Heart Assoc. (2019) 8:e011171. doi: 10.1161/JAHA.118.011171
83. Geng J, Xu H, Yu X, Xu G, Cao H, Lin G, et al. Rosuvastatin protects against oxidized low-density lipoprotein-induced endothelial cell injury of atherosclerosis in vitro. Mol Med Rep. (2019) 19:432–440. doi: 10.3892/mmr.2018.9666
84. Xu X, Ma C, Duan Z, Du Y, Liu C. lncRNA ZEB1-AS1 mediates oxidative low-density lipoprotein-mediated endothelial cells injury by post-transcriptional stabilization of NOD2. Front Pharmacol. (2019) 10:397. doi: 10.3389/fphar.2019.00397
85. Zhao D, Sun X, Lv S, Sun M, Guo H, Zhai Y, et al. Salidroside attenuates oxidized low-density lipoprotein-induced endothelial cell injury via promotion of the AMPK/SIRT1 pathway. Int J Mol Med. (2019) 43:2279–90. doi: 10.3892/ijmm.2019.4153
86. Bian W, Jing X, Yang Z, Shi Z, Chen R, Xu A, et al. Downregulation of LncRNA NORAD promotes Ox-LDL-induced vascular endothelial cell injury and atherosclerosis. Aging. (2020) 12:6385–400. doi: 10.18632/aging.103034
87. Hang L, Peng Y, Xiang R, Li X, Li Z. Ox-LDL causes endothelial cell injury through ASK1/NLRP3-mediated inflammasome activation via endoplasmic reticulum stress. Drug Des Devel Ther. (2020) 14:731–44. doi: 10.2147/DDDT.S231916
88. Peng K, Jiang P, Du Y, Zeng D, Zhao J, Li M, et al. Oxidized low-density lipoprotein accelerates the injury of endothelial cells via circ-USP36/miR-98-5p/VCAM1 axis. IUBMB Life. (2021) 73:177–87. doi: 10.1002/iub.2419
89. Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O. Oxidative stress and antioxidant defense. World Allergy Organ J. (2012) 5:9–19. doi: 10.1097/WOX.0b013e3182439613
90. Suciu CF, Prete M, Ruscitti P, Favoino E, Giacomelli R, Perosa F. Oxidized low density lipoproteins: the bridge between atherosclerosis and autoimmunity. Possible implications in accelerated atherosclerosis and for immune intervention in autoimmune rheumatic disorders. Autoimmun Rev. (2018) 17:366–75. doi: 10.1016/j.autrev.2017.11.028
91. Yao J, Zou Z, Wang X, Ji X, Yang J. Pinoresinol diglucoside alleviates oxLDL-induced dysfunction in human umbilical vein endothelial cells. Evid Based Complement Alternat Med. (2016) 2016:3124519. doi: 10.1155/2016/3124519
92. Kattoor AJ, Kanuri SH, Mehta JL. Role of Ox-LDL and LOX-1 in atherogenesis. Curr Med Chem. (2019) 26:1693–700. doi: 10.2174/0929867325666180508100950
93. Li G, Wu HK, Wu XW, Cao Z, Tu YC, Ma Y, et al. Small dense low density lipoprotein-cholesterol and cholesterol ratios to predict arterial stiffness progression in normotensive subjects over a 5-year period. Lipids Health Dis. (2018) 17:27. doi: 10.1186/s12944-018-0671-2
94. Klisic A, Kavaric N, Vujcic S, Mihajlovic M, Zeljkovic A, Ivanisevic J, et al. Inverse association between serum endocan levels and small LDL and HDL particles in patients with type 2 diabetes mellitus. Eur Rev Med Pharmacol Sci. (2020) 24:8127–35. doi: 10.26355/eurrev_202008_22499
95. Rabbani N, Godfrey L, Xue M, Shaheen F, Geoffrion M, Milne R, et al. Glycation of LDL by methylglyoxal increases arterial atherogenicity: a possible contributor to increased risk of cardiovascular disease in diabetes. Diabetes. (2011) 60:1973–80. doi: 10.2337/db11-0085
96. Iida K, Tani S, Atsumi W, Yagi T, Kawauchi K, Matsumoto N, et al. Association of plasminogen activator inhibitor-1 and low-density lipoprotein heterogeneity as a risk factor of atherosclerotic cardiovascular disease with triglyceride metabolic disorder: a pilot cross-sectional study. Coron Artery Dis. (2017) 28:577–87. doi: 10.1097/MCA.0000000000000521
97. Song C, Burgess S, Eicher JD, O'Donnell CJ, Johnson AD. Causal effffect of plasminogen activator inhibitor type 1 on coronary heart disease. J Am Heart Assoc. (2017) 6:E004918. doi: 10.1161/JAHA.116.004918
98. Corban MT, Prasad A, Nesbitt L, Loeffler D, Herrmann J, Lerman LO, et al. Local production of soluble urokinase plasminogen activator receptor and plasminogen activator inhibitor-1 in the coronary circulation is associated with coronary endothelial dysfunction in humans. J Am Heart Assoc. (2018) 7:e009881. doi: 10.1161/JAHA.118.009881
99. Maloberti A, Farina F, Carbonaro M, Piccinelli E, Bassi I, Pansera F, et al. In healthy normotensive subjects age and blood pressure better predict subclinical vascular and cardiac organ damage than atherosclerosis biomarkers. Blood Press. (2018) 27:262–70. doi: 10.1080/08037051.2018.1461010
100. Huang SW, Lien JC, Kuo SC, Huang TF. Inhibitory effects of an orally active thromboxane A2 receptor antagonist, nstpbp5185, on atherosclerosis in apoe-deficient mice. Thromb Haemost. (2018) 118:401–14. doi: 10.1160/TH17-07-0519
101. Zhao Z, Hu J, Gao X, Liang H, Yu H, Liu S, et al. Hyperglycemia via activation of thromboxane A2 receptor impairs the integrity and function of blood-brain barrier in microvascular endothelial cells. Oncotarget. (2017) 8:30030–38. doi: 10.18632/oncotarget.16273
102. Somodi S, Seres I, Lorincz H, Harangi M, Fülöp P, Paragh G. Plasminogen activator inhibitor-1 level correlates with lipoprotein subfractions in obese nondiabetic subjects. Int J Endocrinol. (2018) 2018:9596054. doi: 10.1155/2018/9596054
103. Jung RG, Simard T, Labinaz A, Ramirez FD, Di Santo P, Motazedian P, et al. Role of plasminogen activator inhibitor-1 in coronary pathophysiology. Thromb Res. (2018) 164:54–62. doi: 10.1016/j.thromres.2018.02.135
104. Rosado E, Rodríguez-Vilarrupla A, Gracia-Sancho J, Tripathi D, García-Calderó H, Bosch J, et al. Terutroban, a TP-receptor antagonist, reduces portal pressure in cirrhotic rats. Hepatology. (2013) 58:1424–35. doi: 10.1002/hep.26520
105. Bansal SK, Yadav R. A study of the extended lipid profile including oxidized LDL, small dense LDL, lipoprotein (a) and apolipoproteins in the assessment of cardiovascular risk in hypothyroid patients. J Clin Diagn Res. (2016) 10:Bc04–8. doi: 10.7860/JCDR/2016/19775.8067
106. Williams PT, Bergeron N, Chiu S, Krauss RM. A randomized, controlled trial on the effects of almonds on lipoprotein response to a higher carbohydrate, lower fat diet in men and women with abdominal adiposity. Lipids Health Dis. (2019) 18:83. doi: 10.1186/s12944-019-1025-4
107. Musa-Veloso K, Paulionis L, Poon T, Lee HY. The effects of almond consumption on fasting blood lipid levels: a systematic review and meta-analysis of randomised controlled trials. J Nutr Sci. (2016) 5:e34. doi: 10.1017/jns.2016.19
108. Lee Y, Berryman CE, West SG, Chen CO, Blumberg JB, Lapsley KG, et al. Effffects of dark chocolate and almonds on cardiovascular risk factors in overweight and obese individuals: a randomized controlled-feeding trial. J Am Heart Assoc. (2017) 6:E005162. doi: 10.1161/JAHA.116.005162
109. Wang L, Tao L, Hao L, Stanley TH, Huang KH, Lambert JD, et al. A moderate-fat diet with one avocado per day increases plasma antioxidants and decreases the oxidation of small, dense LDL in adults with overweight and obesity: a randomized controlled trial. J Nutr. (2020) 150:276–84. doi: 10.1093/jn/nxz231
110. Holligan SD, West SG, Gebauer SK, Kay CD, Kris-Etherton PM. A moderate-fat diet containing pistachios improves emerging markers of cardiometabolic syndrome in healthy adults with elevated LDL levels. Br J Nutr. (2014) 112:744–52. doi: 10.1017/S0007114514001561
111. Kawakami Y, Yamanaka-Okumura H, Naniwa-Kuroki Y, Sakuma M, Taketani Y, Takeda E. Flaxseed oil intake reduces serum small dense low-density lipoprotein concentrations in Japanese men: a randomized, double blind, crossover study. Nutr J. (2015) 14:39. doi: 10.1186/s12937-015-0023-2
112. Mendoza S, Trenchevska O, King SM, Nelson RW, Nedelkov D, Krauss RM, et al. Changes in low-density lipoprotein size phenotypes associate with changes in apolipoprotein C-III glycoforms after dietary interventions. J Clin Lipidol. (2017) 11:224–33.e2. doi: 10.1016/j.jacl.2016.12.009
113. Fiuza-Luces C, Santos-Lozano A, Joyner M, Carrera-Bastos P, Picazo O, Zugaza JL, et al. Exercise benefits in cardiovascular disease: beyond attenuation of traditional risk factors. Nat Rev Cardiol. (2018) 15:731–43. doi: 10.1038/s41569-018-0065-1
114. Dutheil F, Walther G, Chapier R, Mnatzaganian G, Lesourd B, Naughton G, et al. Atherogenic subfractions of lipoproteins in the treatment of metabolic syndrome by physical activity and diet - the RESOLVE trial. Lipids Health Dis. (2014) 13:112. doi: 10.1186/1476-511X-13-112
115. Kotani K, Tsuzaki K, Sakane N, Taniguchi N. The correlation between small dense LDL and reactive oxygen metabolites in a physical activity intervention in hyperlipidemic subjects. J Clin Med Res. (2012) 4:161–6. doi: 10.4021/jocmr870w
116. Chapman MJ, Orsoni A, Robillard P, Therond P, Giral P. Duality of statin action on lipoprotein subpopulations in the mixed dyslipidemia of metabolic syndrome: quantity vs. quality over time and implication of CETP. J Clin Lipidol. (2018) 12:784–800.e4. doi: 10.1016/j.jacl.2018.02.001
117. Nishikido T, Oyama J, Keida T, Ohira H, Node K. High-dose statin therapy with rosuvastatin reduces small dense LDL and MDA-LDL: the standard versus high-dose therApy with rosuvastatin for lipiD lowering (SARD) trial. J Cardiol. (2016) 67:340–6. doi: 10.1016/j.jjcc.2015.05.017
118. Stone NJ, Robinson JG, Lichtenstein AH, Merz CNB, Blum CB, Eckel RH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults. J Am Coll Cardiol. (2014) 63:2889–934. doi: 10.1016/j.jacc.2013.11.002
119. Mason RP, Dawoud H, Jacob RF, Sherratt SCR, Malinski T. Eicosapentaenoic acid improves endothelial function and nitric oxide bioavailability in a manner that is enhanced in combination with a statin. Biomed Pharmacother. (2018) 103:1231–7. doi: 10.1016/j.biopha.2018.04.118
120. Shahsavari G, Raoufi A, Toolabi A, Hosseninejadmir N, Ahmadv H, Safariebrahimsarabie M. The effect of atorvastatin treatment duration on oxidative stress markers and lipid profile in patients with coronary artery diseases: a case series study. ARYA Atheroscler. (2017) 13:282–7.
121. Mach F, Baigent C, Catapano AL, Koskinas KC, Casula M, Badimon L, et al. 2019 ESC/EAS guidelines for the management of dyslipidaemias: lipid modification to reduce cardiovascular risk. Eur Heart J. (2020) 41:111–88. doi: 10.1093/eurheartj/ehz455
122. Millan J, Pintó X, Brea A, Blasco M, Hernández-Mijares A, Ascaso J, et al. Fibrates in the secondary prevention of cardiovascular disease (infarction and stroke). Results of a systematic review and meta-analysis of the Cochrane collaboration. Clin Investig Arterioscler. (2018) 30:30–5. doi: 10.1016/j.artere.2018.02.001
123. Wang D, Liu B, Tao W, Hao Z, Liu M. Fibrates for secondary prevention of cardiovascular disease and stroke. Cochrane Database Syst Rev. (2015) 2015:Cd009580. doi: 10.1002/14651858.CD009580.pub2
124. Brea A, Millán J, Ascaso JF, Blasco M, Díaz A, Hernández-Mijares A, et al. Fibrates in primary prevention of cardiovascular disease. Comments on the results of a systematic review of the cochrane collaboration. Clin Investig Arterioscler. (2018) 30:188–92. doi: 10.1016/j.artere.2018.06.002
125. Komiya I, Yamamoto A, Sunakawa S, Wakugami T. Pemafibrate decreases triglycerides and small, dense LDL, but increases LDL-C depending on baseline triglycerides and LDL-C in type 2 diabetes patients with hypertriglyceridemia: an observational study. Lipids Health Dis. (2021) 20:17. doi: 10.1186/s12944-021-01434-8
126. Yamashita S, Masuda D, Matsuzawa Y. Pemafibrate, a new selective PPARα modulator: drug concept and its clinical applications for dyslipidemia and metabolic diseases. Curr Atheroscler Rep. (2020) 22:5. doi: 10.1007/s11883-020-0823-5
127. Yunoki K, Nakamura K, Miyoshi T, Enko K, Kohno K, Morita H, et al. Ezetimibe improves postprandial hyperlipemia and its induced endothelial dysfunction. Atherosclerosis. (2011) 217:486–91. doi: 10.1016/j.atherosclerosis.2011.04.019
128. Farnier M. Ezetimibe/statin combination therapy to treat patients with type 2 diabetes. Atheroscler Suppl. (2015) 17:2–8. doi: 10.1016/S1567-5688(15)50002-0
129. Shinnakasu A, Yamamoto K, Kurano M, Arimura H, Arimura A, Kikuti A, et al. The combination therapy of fenofibrate and ezetimibe improved lipid profile and vascular function compared with statins in patients with type 2 diabetes. J Atheroscler Thromb. (2017) 24:735–48. doi: 10.5551/jat.39446
130. Horimatsu T, Blomkalns AL, Ogbi M, Moses M, Kim D, Patel S, et al. Niacin protects against abdominal aortic aneurysm formation via GPR109A independent mechanisms: role of NAD+/nicotinamide. Cardiovasc Res. (2020) 116:2226–38. doi: 10.1093/cvr/cvz303
131. Haynes R, Jiang L, Hopewell JC, Li J, Chen F, Parish S, et al. HPS2-THRIVE randomized placebo-controlled trial in 25 673 high-risk patients of ER niacin/laropiprant: trial design, pre-specifified muscle and liver outcomes, and reasons for stopping study treatment. Eur Heart J. (2013) 34:1279–91. doi: 10.1093/eurheartj/eht055
132. Teo KK, Goldstein LB, Chaitman BR, Grant S, Weintraub WS, Anderson DC, et al. Extended-release niacin therapy and risk of ischemic stroke in patients with cardiovascular disease: the atherothrombosis intervention in metabolic syndrome with low HDL/High triglycerides: impact on global health outcome (AIM-HIGH) trial. Stroke. (2013) 44:2688–93. doi: 10.1161/STROKEAHA.113.001529
133. Shibabaw T. Omega-3 polyunsaturated fatty acids: anti-inflammatory and anti-hypertriglyceridemia mechanisms in cardiovascular disease. Mol Cell Biochem. (2021) 476:993–1003. doi: 10.1007/s11010-020-03965-7
134. Masuda D, Miyata Y, Matsui S, Yamashita S. Omega-3 fatty acid ethyl esters improve low-density lipoprotein subclasses without increasing low-density lipoprotein-cholesterol levels: a phase 4, randomized study. Atherosclerosis. (2020) 292:163–70. doi: 10.1016/j.atherosclerosis.2019.11.014
135. Sherratt SCR, Juliano RA, Mason RP. Eicosapentaenoic acid (EPA) has optimal chain length and degree of unsaturation to inhibit oxidation of small dense LDL and membrane cholesterol domains as compared to related fatty acids in vitro. Biochim Biophys Acta Biomembr. (2020) 1862:183254. doi: 10.1016/j.bbamem.2020.183254
136. Liu Y, Wang X, Han J, Liu L, Jin Y, Jin L, et al. PCSK9 positively correlates with plasma sdLDL in community-dwelling population but not in diabetic participants after confounder adjustment. Medicine. (2019) 98:e15062. doi: 10.1097/MD.0000000000015062
137. Garside B, Ho JH, Kwok S, Liu Y, Dhage S, Donn R, et al. Changes in PCSK 9 and apolipoprotein B100 in Niemann-Pick disease after enzyme replacement therapy with olipudase alfa. Orphanet J Rare Dis. (2021) 16:107. doi: 10.1186/s13023-021-01739-y
138. Li S, Zhao X, Zhang Y, Zhu CG, Guo YL, Wu NQ, et al. Novel circulating lipid measurements for current dyslipidemias in non-treated patients undergoing coronary angiography: PCSK9, apoC3 and sdLDL-C. Oncotarget. (2017) 8:12333–41. doi: 10.18632/oncotarget.12471
139. Di Minno A, Gentile M, Iannuzzo G, Calcaterra I, Tripaldella M, Porro B, et al. Endothelial function improvement in patients with familial hypercholesterolemia receiving PCSK-9 inhibitors on top of maximally tolerated lipid lowering therapy. Thromb Res. (2020) 194:229–36. doi: 10.1016/j.thromres.2020.07.049
140. Santos HO, Earnest CP, Tinsley GM, Izidoro LFM, Macedo RCO. Small dense low-density lipoprotein-cholesterol (sdLDL-C): analysis, effects on cardiovascular endpoints and dietary strategies. Prog Cardiovasc Dis. (2020) 63:503–9. doi: 10.1016/j.pcad.2020.04.009
141. Taylor PC, Kremer JM, Emery P, Zuckerman SH, Ruotolo G, Zhong J, et al. Lipid profile and effect of statin treatment in pooled phase II and phase III baricitinib studies. Ann Rheum Dis. (2018) 77:988–95. doi: 10.1136/annrheumdis-2017-212461
142. Michael DR, Jack AA, Masetti G, Davies TS, Loxley KE, Kerry-Smith J, et al. A randomised controlled study shows supplementation of overweight and obese adults with lactobacilli and bifidobacteria reduces bodyweight and improves well-being. Sci Rep. (2020) 10:4183. doi: 10.1038/s41598-020-60991-7
Keywords: small dense low-density lipoprotein-cholesterol, atherosclerosis, lipid metabolism, inflammation, endothelial injury, review
Citation: Jin X, Yang S, Lu J and Wu M (2022) Small, Dense Low-Density Lipoprotein-Cholesterol and Atherosclerosis: Relationship and Therapeutic Strategies. Front. Cardiovasc. Med. 8:804214. doi: 10.3389/fcvm.2021.804214
Received: 29 October 2021; Accepted: 02 December 2021;
Published: 10 February 2022.
Edited by:
Nathalie Pamir, Oregon Health and Science University, United StatesReviewed by:
Konstantinos Tziomalos, Aristotle University of Thessaloniki, GreeceCopyright © 2022 Jin, Yang, Lu and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Min Wu, d3VtaW4xOTc2MjAwMEAxMjYuY29t
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.