 Xiao Lin1
Xiao Lin1 Muhammad Hasnain Ehsan Ullah2
Muhammad Hasnain Ehsan Ullah2 Xiong Wu1
Xiong Wu1 Feng Xu2Su-Kang Shan2
Feng Xu2Su-Kang Shan2 Li-Min Lei2
Li-Min Lei2 Ling-Qing Yuan2*
Ling-Qing Yuan2* Jun Liu1,3,4*
Jun Liu1,3,4*- 1Department of Radiology, The Second Xiangya Hospital, Central South University, Changsha, China
- 2Department of Endocrinology and Metabolism, National Clinical Research Center for Metabolic Diseases, The Second Xiangya Hospital, Central South University, Changsha, China
- 3Clinical Research Center for Medical Imaging in Hunan Province, Changsha, China
- 4Department of Radiology Quality Control Center in Hunan Province, Changsha, China
Primary aldosteronism (PA) is the most common type of endocrine hypertension, and numerous experimental and clinical evidence have verified that prolonged exposure to excess aldosterone is responsible for an increased risk of cerebro-cardiovascular events and target organ damage (TOD) in patients with PA. Therefore, focusing on restoring the toxic effects of excess aldosterone on the target organs is very important to reduce cerebro-cardiovascular events. Current evidence convincingly demonstrates that both surgical and medical treatment strategies would benefit cerebro-cardiovascular outcomes and mortality in the long term. Understanding cerebro-cardiovascular risk in PA would help clinical doctors to achieve both early diagnosis and treatment. Therefore, in this review, we will summarize the cerebro-cardiovascular risk in PA, focusing on the TOD of aldosterone, including brain, heart, vascular system, renal, adipose tissues, diabetes, and obstructive sleep apnea (OSA). Furthermore, the various treatment outcomes of adrenalectomy and medical treatment for patients with PA will also be discussed. We hope this knowledge will help improve cerebro-cardiovascular prognosis and reduce the incidence and mortality of cerebro-cardiovascular events in patients with PA.
Introduction
Primary aldosteronism (PA) is a clinical syndrome mainly characterized by hypertension, suppressed levels of plasma renin, and autonomous plasma aldosterone overproduction. Recognition of the prevalence of PA has increased from <1% to over 10% of patients with hypertension, with the in-depth knowledge and application of the screening-confirmation-typing system (1). Moreover, the prevalence of PA in patients with refractory hypertension ranges from 8.9 to 33% (2). Therefore, PA is the most common type of endocrine hypertension. However, PA is often underestimated, and patients with untreated (or inappropriately treated) PA have an increased risk of cardiovascular events and target organ damage (TOD). A substantial body of clinical studies have demonstrated that undiagnosed PA is associated with stroke, heart failure (HF), diabetes mellitus (DM), obstructive sleep apnea (OSA), renal failure, and other consequential cardiovascular events, along with poorer health-related quality of life (QoL), even premature death (3–8). Prolonged exposure to excess aldosterone has a toxic effect on target organs, including the brain, heart, vascular system, kidney, adipose tissues, and OSA, which could increase the incidence and mortality of cerebro-cardiovascular events in patients with PA (4, 5). Most importantly, the rate of TOD, to a large extent, could be reversed via removing the toxic effects of excess aldosterone with either adrenalectomy (ADX) or treatment with a mineral ocorticoid receptor antagonist (MRA) (9). Therefore, this review aims to discuss the new evidence linking aldosterone to the TOD and treatment outcome of PA, thereby hoping to help decrease the risk of developing cerebro-cardiovascular and renal complications in patients with PA.
Mechanisms of Aldosterone on Target Organs
Aldosterone, a mineral corticoid hormone, is synthesized in the zona glomerulosa of the adrenal glands. Aldosterone is one of the effector molecules of the renin-angiotensin-aldosterone system (RAAS), whose synthesis and secretion are stimulated by angiotensin II (Ang-II) through the angiotensin I receptor (ATI-R) in the adrenal cortex (10). The genomic effects of aldosterone occur through binding to the mineral ocorticoid receptor (MR), translocating to the nucleus, interacting with DNA, and thus promoting the transcription of genes that regulate the transport of sodium and potassium and fluid balance (11). Aldosterone induces rapid cellular responses by modulating intracellular calcium () and cyclic adenosine monophosphate (cAMP) levels; sodium/hydrogen (Na+/H+) exchanger activity; and phosphorylation of signaling molecules, including protein kinase C (PKC), epidermal growth factor receptor (EGFR), mitogen-activated protein kinases (MAPKs), (including c-Jun NH2-terminal kinase), and extracellular signal-regulated kinases (ERKs) 1/2 (12–14). Recent insights into sodium and potassium have demonstrated that excess aldosterone promotes epithelial sodium channel (ENaC) activity and facilitates renal outer medullary K+ channel (ROMK) activity to increase the sodium chloride cotransporter (NCC) and pendrinapical abundance in the late but not in the early distal convoluted tubule, which allows sodium/potassium exchange through the ENaC, ROMK, and big potassium channel (15). Wu et al. demonstrated that oral co-administration of fludrocortisone acetate (a potent mineral ocorticoid) and KCl in patients with PA is associated with reduced pendrin and enhanced ROMK in urinary extracellular vesicles (16). Based on this, Stavropoulos et al. assumed that pendrin inhibition might confer an efficacious therapeutic option for patients with PA (17).
Besides, the aldosterone non-genomic pathways are through the AT1-R, G-protein-coupled receptor, and EGFR (18). These receptors include the MAPK/ERK1/2/p38 signaling pathways, mediating vascular remodeling, inflammation, hemodynamic alterations, nephrosclerosis, and fibrosis (19, 20), as well as being involved in cardiovascular, renal, and metabolic diseases (21, 22). Therefore, the outcome of aldosterone excess would induce structural and functional alterations in the heart, kidney, and vascular system, which leads to the development of cardiac hypertrophy, stroke, coronary heart disease, nephrosclerosis, vascular inflammation, sclerosis, myocardial infarction, fibrosis, and tissue remodeling (5, 23–25). The major pathophysiological non-genomic mechanisms involved in the impact of aldosterone on target organs are summarized in Figure 1, and part of the detailed mechanisms are discussed as follows.
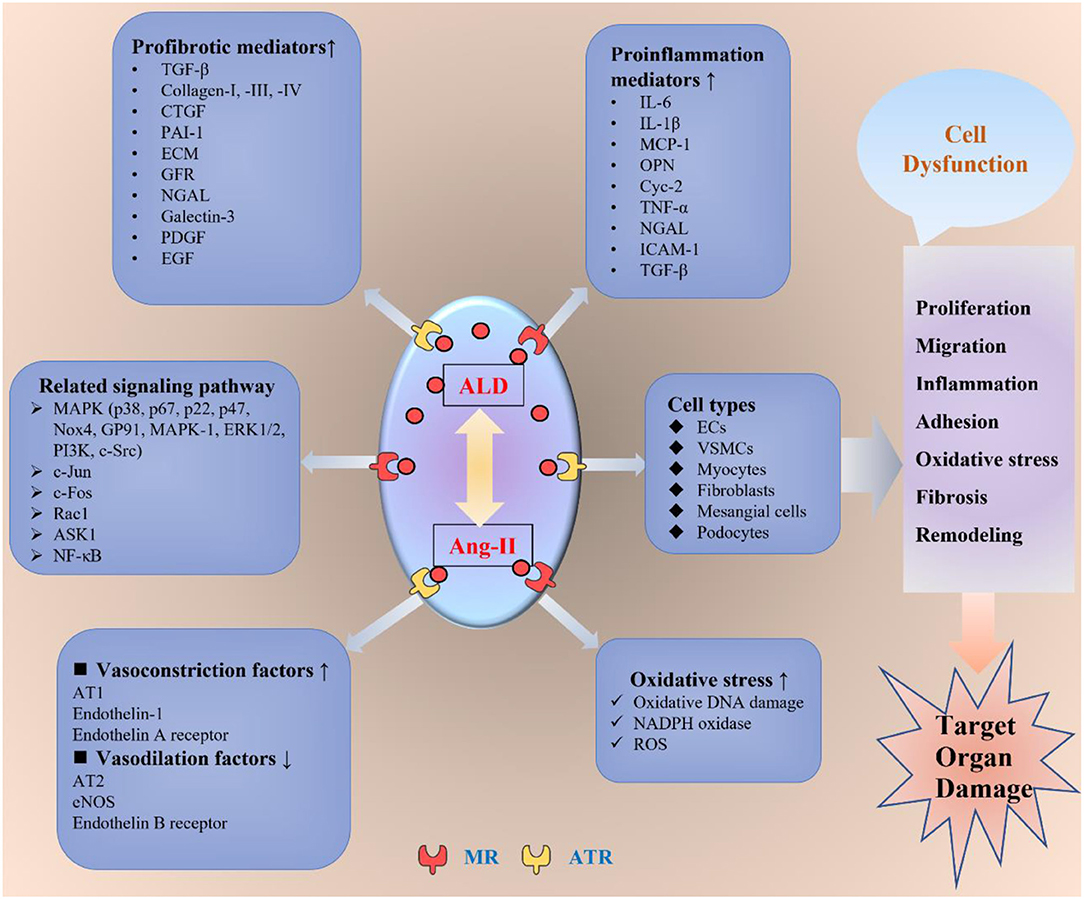
Figure 1. The non-genomic mechanisms of aldosterone-modulating TOD. ALD interacts with Ang-II by binding to the MR or ATR to exert its toxic effect on the target organs. ALD promotes inflammation, oxidative stress, fibrosis, migration, proliferation, adhesion, endothelial dysfunction, and vascular remodeling through different kinds of mediators and signaling pathways in various cells, including ECs, VSMCs, myocytes, fibroblasts, mesangial cells, and podocytes. ALD, aldosterone; Ang-II, angiotensin II; ATR, angiotensin receptor; MR, mineral ocorticoid receptor; AT1, angiotensin receptor 1; AT2, angiotensin receptor 2; ECs, endothelial cells; VSMCs, vascular smooth muscle cells; eNOS, endothelial nitric oxide synthase; TGF-β, transforming growth factor β; CTGF, connective tissue growth factor; PAI-1, plasminogen activator inhibitor 1; ECM, extracellular matrix; GFR, growth factor receptor; NGAL, neutrophil gelatinase-associated lipocalin; PDGF, platelet-derived growth factor; EGF, epidermal growth factor; IL-1β, interleukin 1β; IL-6, interleukin 6; MCP-1, monocyte chemoattractant protein-1; OPN, osteopontin; TNF-α, tumor necrosis factor-α; ICAM-1, intercellular adhesion molecule-1; NADPH, nicotinamide adenine dinucleotide phosphate; ROS, reactive oxygen species; ASK1, apoptosis signal-regulated kinase 1; ERK1/2, extracellular signal-regulated kinase 1/2; PI3K, phosphoinositide 3-kinase; NF-κB, NF-kappaB; TOD, target organ damage.
Aldosterone and Oxidative Stress
Multiple animal (rats and mice) studies have demonstrated that aldosterone and Ang-II potentiate each other's action in inducing oxidative stress via elevating levels of oxidative stress markers (malondialdehyde, procollagen type 1 amino-terminal propeptide et al.), leading to detrimental consequences in the target organs of patients with PA (26–28). Human red blood cells (RBCs) are anucleated cells and are particularly sensitive to oxidative assault. Bordin et al. verified that RBCs from patients with PA displayed membrane alterations and increased senescence in vitro, which were accompanied by increased high molecular weight aggregates and diamide-induced band 3 Tyr-P levels (29). Moreover, in an in vivo study, they demonstrated that, due to the MR-mediated response involving ligand specificity, aldosterone would induce MR activation and lead to RBC membrane alterations and IgG binding in patients with PA (30).
Aldosterone has been reported to activate nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and increase reactive oxygen species (ROS) levels in several kinds of cells, including myocardial cells, endothelial cells (ECs), vascular smooth muscle cells (VSMCs), and mesangial cells in rats (31, 32). Petramala et al. firstly demonstrated increased oxidative stress characterized by increased serum levels of NADPH oxidase (Nox-2-derived peptide) and urinary excretion of isoprostanes in patients with PA (33). Besides, aldosterone would interact with Ang-II to activate NADPH oxidase through the phosphorylation and activation of p47phox and Rac1 (27, 28, 32), as well as c-Src-dependent mechanisms, in mice (26). Aldosterone increased rat myocyte ROS production by the non-genomic activation of NADPH oxidase, which in turn triggers apoptosis of myocyte associated with the activation of apoptosis signal-regulating kinase 1 (ASK1) (34). Besides, aldosterone activating the MR in ECs and VSMCs, derived from mice, rats, or humans, is associated with ROS production through increasing the expression and activity of NADPH oxidases in the heart (26, 35–37). Aldosterone decreases the expression of glucose-6-phosphate dehydrogenase (G6PD), shifting the balance toward increased oxidative stress in bovine and human ECs (38). In human pulmonary artery ECs, aldosterone increases ROS, which in turn modifies the cysteinyl thiols in the endothelial nitric oxide synthase (eNOS) activating region of the endothelin B receptor, thus decreasing endothelin-1-stimulated eNOS activity (37). In addition, MRs in SMCs contribute to Ang-II-induced vascular oxidative stress both in mice and humans (39). Moreover, oxidative stress, in turn, promotes rat SMC senescence (40). Another important signaling molecule in VSMCs, c-Src, is a critical proximal regulator of NAD(P)H oxidase (41), and aldosterone rapidly increases activation of MAPKs (p38MAPK, c-Jun NH2-terminal kinase, and ERK1/2) through c-Src-dependent pathways in mice (26). In addition, chronic treatment with aldosterone will induce MAPK activation via a ROS-dependent pathway and then, increase the expression of p67phox, p22phox, Nox4, Gp91phox, p47phox, and Rac1 in the rat kidney (42–44), and it also induces apoptosis in rat mesangial cells (45).
Aldosterone and Inflammation
Early study has demonstrated that rats treated with aldosterone have perivascular leucocyte infiltration and increased expression of interleukin (IL)6, IL-1β, osteopontin (OPN), monocyte chemoattractant protein 1 (MCP-1), and cyc-2 in the kidney (46). Besides, aldosterone participated in the process of inflammation in mice by increasing the expression of adhesion molecule-1 and the activation of c-Jun and c-Fos in response to pro-inflammatory stimuli (47). Eissler et al. also demonstrated that aldosterone causes the overexpression of toll-like receptor4 (TLR4) and the higher expression of inflammatory cytokines (TNF-α, IL-1, and MCP-1) in rat cardiac tissue (48). Moreover, aldosterone had pro-inflammatory effects by increasing neutrophil gelatinase-associated lipocalin (NGAL) expression in mice dendritic cells, macrophages, and peripheral blood mononuclear cells (49). In mice dendritic cells, aldosterone also induces the secretion of the pro-inflammatory cytokines IL-6 and transforming growth factor β (TGF-β) via MR activation (50).
Aldosterone and Fibrosis
In experiments with rats and mice, aldosterone administration has been associated with an increase in TGF-β, connective tissue growth factor (CTGF), and collagen gene expression that is accompanied by kidney fibrosis (51–54). Aldosterone has been reported to increase the production of plasminogen activator inhibitor-1 (PAI-1) and subsequent extracellular matrix (ECM) accumulation in the development of glomerulo sclerosis and SMC stiffness (55–58). Besides, aldosterone has been reported to stimulate collagen gene expression and synthesis in cultured fibroblasts via activation of cellular ERK1/2 phosphorylation (59) and increase the mRNA levels of collagens I, III, and IV in rat glomerular mesangial cells (60). Moreover, aldosterone stimulates fibronectin synthesis through MR-dependent activation and phosphorylation of the c-Jun N-terminal kinase in rats (61). Aldosterone also induces expression of OPN in rat fibroblasts (62) and stimulates rat fibroblasts, resulting in rapid activation of growth-factor receptors (GFRs) and the induction of phosphoinositide 3-kinase/mitogen-activated protein kinase (PI3K/MAPK) signaling, which stimulates proliferation of fibroblasts (63). In addition, in human studies, NGAL has been shown to increase the production of galectin-3 and collagen I through the NF-κB pathway (64, 65) and acted through the TGF-β signaling, epidermal growth factor (EGF), platelet-derived growth factor (PDGF), and their receptors, thus contributing to the development of fibrosis (66, 67).
Aldosterone and TOD
With this new evidence, excessive aldosterone is regarded as an important determinant of the cerebro-cardiovascular risk profile in patients with PA, which has a toxic effect on the cerebro-cardiovascular system. Moreover, aldosterone is associated with severe TOD (including the brain, heart, kidney, vascular system, adipose tissues, and OSA) as shown in Figure 2, independently from blood pressure (BP) levels (7, 25).
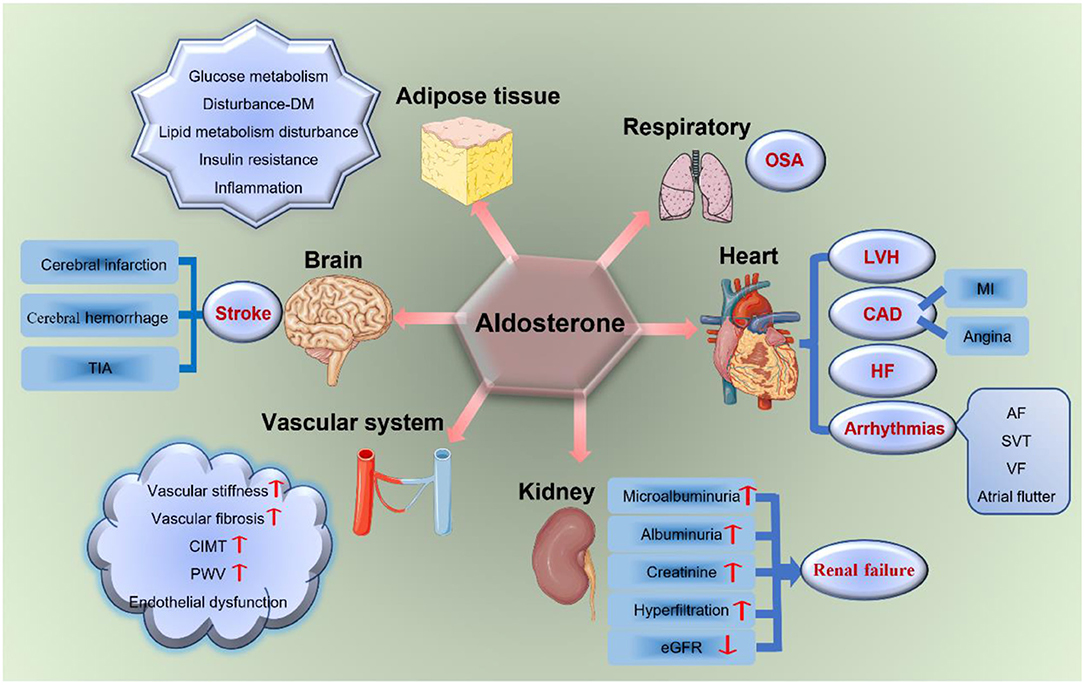
Figure 2. Aldosterone and TOD. Aldosterone has a toxic effect on target organs, including the brain, heart, vascular system, kidney, respiratory system, and adipose tissues. It increases the incidence of stroke, LVH, CAD, HF, arrhythmias, renal failure, vascular fibrosis and stiffness, endothelial dysfunction, glucose and lipid metabolism disturbances, insulin resistance, OSA, and so on. TIA, transient ischemic attack; DM, diabetes mellitus; LVH, left ventricular hypertrophy; CAD, coronary artery disease; MI, myocardial infarction; HF, heart failure; AF, atrial fibrillation; SVT, sustained ventricular tachycardia; VF, ventricular fibrillation; eGFR, estimated glomerular filtration rate; CIMT, carotid intima-media thickness; PWV, pulse wave velocity; OSA, obstructive sleep apnea; TOD, target organ damage.
Toxic Effects of Aldosterone on Stroke
Stroke is defined as ischemic or hemorrhagic cerebrovascular disease, including cerebral infarction, cerebral hemorrhage, or transient ischemic attack (TIA) (9, 68). Experimental and human studies have verified that excess aldosterone promotes cerebral vascular oxidative stress, inflammation, and endothelial dysfunction, which increases the risk of stroke, independent of BP and other risk factors (9, 43, 68, 69). Cerebrovascular accidents or TIAs are more frequent in patients with PA than essential hypertension (EH), and cerebral infarction is the most frequent of the cerebrovascular events (70).
In animal studies, as early as 1992, Kim et al. found that plasma aldosterone was increased in a 25-week-old stroke-prone spontaneously hypertensive rat (SHRSP) (71). Accordingly, Enea et al. reported that plasma aldosterone levels were significantly increased and enhanced stroke development in SHRSP rats (from 442 ± 56.5 pg/ml to 739 ± 125.7 pg/ml) vs. Wistar-Kyoto rats (72, 73). Other studies have reported that rats fed aldosterone chronically experienced strokes, and treatment with a MRA ameliorated the effects (43, 74). A mechanism study demonstrated that Nox2-containing NADPH oxidase-mediated aldosterone induced increases in ROS production and endothelial dysfunction in cerebral arteries from mice, independently of BP changes (75).
In clinical studies, as early as 1998, Litchfield et al. found that patients with glucocorticoid-remediable aldosteronism tended to have an early onset of stroke (cerebral hemorrhage), which was related to increased aldosterone levels (68). Accordingly, Cristiana et al. verified that prolonged exposure to elevated aldosterone levels could increase the incidence of stroke or TIA in patients with PA when compared to patients with EH (9). Moreover, Takeda et al. investigated the incidence of cerebrovascular complications in 224 cases of PA in Japan and found that there were 14 cases of cerebral hemorrhage and 10 cases of cerebral infarction (76). Another retrospective study showed that stroke and TIA were increased significantly in patients with PA compared to those with EH (10.4 vs. 4.9%) (77), and the latest multicenter study also demonstrated that the prevalence of stroke was about 7.4% in Japan (78). In addition, Milliez et al. reported 12.9% of patients with PA had a history of stroke (4). However, Miyaji et al. discovered the incidence of PA in patients with acute stroke was just 4.% (79). The reasons to underestimate this incidence of stroke in patients with PA might be due to the demographic bias and non-standard confirmatory test strategies.
Toxic Effects of Aldosterone on the Heart
Structural and functional abnormalities of the heart are common consequences of hypertensive states (80), and about 14 to 35% of patients with PA have cardiovascular complications, including myocardial hypertrophy, myocardial fibrosis, coronary artery disease (CAD), heart failure (HF), and atrial fibrillation (AF) (7, 81–83). Moreover, several large studies from different parts of the world have demonstrated that among patients with hypertension of equal severity and duration, those with PA have significantly more cardiovascular pathology than those with essential hypertension (5, 84, 85). Experimental and clinical studies have also demonstrated that both cardiomyocytes and cardiac fibroblasts express MR with a high affinity for aldosterone, and they could be activated by high aldosterone levels (86, 87). Based on that, aldosterone has been proved to affect both the structure and function of the heart and is regarded as a potential cardiovascular risk factor in patients with PA (86–88). The role of aldosterone in left ventricular hypertrophy (LVH), CAD, HF, and arrhythmias will be discussed as follows.
Aldosterone and LVH
Left ventricular (LV) remodeling is a process of changes in LV size, shape, texture, and function regulated by various stimuli including aldosterone, and it includes two parts in PA: LVH and fibrosis (89). PA is associated with a higher degree of LVH and increased LV mass index (LVMI) when compared to matched EH, and LVH leads to changes in cardiac size, mass, geometry, and function (83). LVH is the most common cardiac structural abnormality induced by excess aldosterone, independently of its effects on BP (7, 74, 90, 91). Myocardial hypertrophy evaluated by electrocardiogram or echocardiography, especially LVH, was reported to be about two times as frequent in patients with PA when compared to that in otherwise similar patients with EH (4, 83, 92). The latest study demonstrated that autonomous aldosterone secretion levels, not the basal aldosterone concentration itself, were correlated significantly with LVMI, even after adjusting for age and BP (88). A recent meta-analysis by Monticone et al. (5) based on a pooled population from 20 studies, totaling 5,672 patients, documented that LV mass was higher in patients with PA compared to that in counterparts with EH (mean difference ranging from 20 to 69 g/m2 for LV mass/BSA and from 5 to 58 g/m2.7 for LV mass/h2.7).
In animal studies, the experimental study suggested that long-term aldosterone-salt treatment in rats displayed an increase in cardiac fibrosis and LVH (93). The endogenous aldosterone produced in the heart directly stimulates the hypertrophy of ventricular myocytes in neonatal rats via activation of ERK, JNK, and protein kinase C-α (PKC-α), affecting cardiac hypertrophy and function in hypertensive rats (94, 95). Besides, aldosterone has been shown to increase the size of rat cardiac myocytes, along with an increase in ROS and nitric oxide (NO) (96). Moreover, elevated aldosterone has been shown to promote myocardial hypertrophy and fibrosis in the rat model of aldosterone system overexpression (97).
In clinical studies, several cross-sectional studies have reported both LV wall thickness and LV dimension were increased in patients with PA (98–101). The mean interventricular septum and posterior wall thicknesses were increased, leading to higher rates of LVH in patients with PA relative to individuals with EH (101). The other two types of research have demonstrated that patients with PA had reduced diastolic function and increased LV wall thickness, even in the absence of arterial hypertension (102, 103). Exclusive analysis of a prospective study confirmed the association of PA with higher LV internal dimensions and higher LVMI compared with those in subjects with EH (104). Besides, the prevalence of LVH had no significant gender difference in patients with PA (105). What is more, Chen et al. demonstrated that patients with PA showed not only LV abnormalities but also impaired right ventricular function because of hyper aldosteronism (106).
Aldosterone and CAD
Coronary artery disease (CAD) often includes myocardial infarction (MI) and stenocardia, and MI is the most common manifestation of CAD. Mihailidou et al. demonstrated that aldosterone increased the incidence of MI, aggravating cardiac damage (107). Increasing production of cardiac aldosterone has been shown to increase the risk of MI, and in turn, MI could raise aldosterone synthase mRNA (the terminal enzyme of aldosterone synthesis) and the level of aldosterone in rats (108). Aldosterone has also been shown to induce a vascular inflammatory phenotype in rat heart and strongly increased cyclooxygenase 2 (COX-2) in ventricular cardiomyocytes after MI (109). Besides, increased levels of aldosterone have been shown to activate MR in the brain and then enhance apoptosis both in rat myocytes and nonmyocytes in the peri-infarct and infarct areas post-MI, contributing to the inflammatory response (110).
Patients with PA experienced MI or unstable angina, requiring angioplasty more frequently than those with EH, both at PA diagnosis, during follow-up, and in the overall period of the study (4, 9, 77, 111). Milliez et al. demonstrated non-fatal MI was diagnosed in 4.0% of 124 patients with PA and 0.6% of 456 patients with EH (4). Subsequently, Catena et al. demonstrated an increased risk of MI in patients with PA at diagnosis and during a 7-year follow-up study (9). In addition, a multicenter study in Japan involving 2,582 patients with PA found that the prevalence of CAD (including MI or angina) was 9.4% (78).
Aldosterone and HF
Heart failure (HF) is defined as, “a complex clinical syndrome that could result from any structural or functional cardiac disorder, which impairs the ability of the ventricle to fill or eject blood” (77, 111, 112). HF has been singled out as a clinical and public health problem and is associated with significant morbidity, mortality, and healthcare expenditures (112). The risks of developing HF requiring hospitalization and mortality were significantly higher in patients with PA than EH controls (111). Although Takeda et al. showed the incidence of congestive heart failure (CHF) in the PA group was a little lower than that in the EH group in 1995, this difference did not reach statistical significance (76). However, a large cohort study confirmed that the incidence of HF was much higher in patients with PA compared to that in patients with EH (111). Moreover, a multicenter study in Japan also found that the prevalence of HF (about 0.6%) was higher among patients with PA compared to that among EH controls (78).
Aldosterone and Arrhythmias
Atrial fibrillation (AF) is one of the most important and prevalent types of arrhythmias and imposes an increasing burden on the healthcare system, owing to the need for lifelong care and pharmacological treatment, which is associated with an increased risk of cardiovascular events (113). In patients with PA, aldosterone not only increased cardiac structural remodeling (such as atrial fibroblasts, LVH, and interstitial collagen) (114) but also promoted AF by altering repolarizing potassium currents, leading to action potential shortening in rats (115). Furthermore, Lu et al. reported that aldosterone enhanced the expression of Kv1.5, a promising target for the treatment of AF, by activating ROS-dependent phosphorylation of Smad 2/3 and ERK 1/2 in a rat AF model (116).
A growing body of evidence has demonstrated increased sustained arrhythmias, including AF, sustained ventricular tachycardia, atrial flutter, and ventricular fibrillation (VF), in patients with PA compared to those with EH (77, 111, 117, 118). Milliez et al. reported a history of AF in 7.3% of patients with PA (4), which was consistent with the latest study that showed a marked increase in the relative risk of AF (12.1-fold) in patients with PA compared to patients with EH (119). In addition, another study also reported that patients with PA had a 7.2-fold higher prevalence of history or current AF than patients with EH (101). Consistently, both a multicenter study and a large cohort study confirmed that the incidences of arrhythmias, especially AF, were all significantly higher among patients with PA compared to those among other hypertensive patients (78, 120). Besides, Catena et al. demonstrated an increased risk of sustained arrhythmias in patients with PA at diagnosis compared to patients with EH during a 7-year follow-up study (9). More seriously, PA with hypokalemia has been shown to increase the risk of life-threatening ventricular arrhythmias (121).
Toxic Effects of Aldosterone on the Vascular System
Aldosterone has been shown to regulate vascular contractility, cell growth, and apoptosis. Aldosterone-mediated EC growth and endothelium-mediated regulation of vasore activity via activation of G protein-coupled estrogen receptor (GPER) were observed in rats (122). Aldosterone has been shown to interact with Ang-II to increase apoptosis of rat microvascular ECs via upregulation of protein tyrosine phosphatase 1B (PTP1B) expression and inhibition of the PI3K/Akt pathway (123). In addition, in an in vitro study, aldosterone was shown to contribute to an increase of collagen synthesis and fibrosis in mice VSMCs (8, 26).
It is well documented that excess aldosterone is responsible for increased arterial wall stiffness due to the morphological and functional abnormalities of the blood vessel wall (3, 6, 8). The mechanisms leading to structural abnormalities include increases in both arterial wall stiffness (6) and carotid intima-media thickness (CIMT) (124), as well as abnormal endothelial function (8). The excess aldosterone generates oxidative stress to cause endothelial dysfunction and collagen remodeling or reduce the bioavailability of NO to directly affect EGFR, both of which lead to an increase in fibrosis and vascular stiffness (125, 126). Furthermore, increased CIMT was more frequent in patients with PA than in controls with EH (8, 127, 128). In addition, pulse wave velocity (PWV) is regarded to be another reliable marker of atherosclerosis and arterial stiffness. Several studies have demonstrated that PWV values, as well as brachial-ankle/heart-ankle PWV, increased in patients with PA compared to those in controls with EH, and this difference was independent of BP levels (125).
What is more, excess aldosterone has been shown to cause reduced endothelial progenitor cell (EPC) vascular elasticity, proliferation, differentiation, and migration in patients with PA (129), and the deficiency of EPCs may result in increased aortic stiffness and vascular damage (130). Moreover, endothelial inflammation contributed to the negative remodeling of the cerebral vasculature, which made the vessel wall less flexible and further impaired dilatation of the cerebral vessels during the stroke (131). Furthermore, pronounced fibrosis of small resistance arteries was also detected in patients with PA compared with BP-matched patients with EH (132).
Toxic Effects of Aldosterone on the Kidney
Clinical studies have indicated that plasma aldosterone concentration (PAC) was associated with renal dysfunction, and the incidence of renal complications was higher among patients with PA when compared to patients with EH at equivalent BPs (7, 133). A high PAC could affect more pronounced renal damage, including renal glomeruli and tubules (134–136). The latest histopathological analysis of patients with PA demonstrated that both mineral ocorticoid receptor and 11β-hydroxysteroid dehydrogenase type 2 were significantly higher in the renal tubules of patients with hyperaldosteronism, which resulted in interstitial fibrosis and segmental glomerulosclerosis. In the meantime, in those with hyperaldosteronism, glomerular size was significantly larger, luminal stenosis tended to be more marked, and arteriolar hyalinization was significantly more pronounced, but the intima-to-media ratio was significantly lower (134). In addition, the effective markers of kidney damage, including β2-microglobulin (β2-MG), urinary liver fatty acid-binding protein (L-FABP), and N-acetyl-β-D-glucosaminidase (NAG) for tubular damage, as well as urinary albumin-creatinine ratio (ACR) for glomerular damage, were significantly correlated with the PAC (136). In addition, severe vascular and glomerular sclerosis, fibrinoid necrosis and thrombosis, interstitial leukocyte infiltration, and tubular damage and regeneration were observed in aldosterone-treated rats, which leads to renal injury and fibrosis (46).
The structural renal damage induced by a high PAC in patients with PA may be associated with unfortunate outcomes, such as renal injury and renal failure (137, 138). Patients with PA presented more frequently with microalbuminuria and albuminuria, lower eGFR, and increased creatinine levels (111, 136, 139, 140). Studies have reported that albuminuria and renal failure were found in a range from 8 to 24% in subjects with PA (138, 141). Also, 24-h microalbuminuria was significantly greater in patients with PA compared to controls with EH in the PAPY study (142). Both Reincke et al. and Kawashima et al. found the prevalence of albuminuria was increased, and eGFR was lowered greatly in patients with PA, independent of other known risk factors (143, 144). Moreover, a recent meta-analysis study including 6,056 patients with PA also verified that patients with PA had an increased eGFR compared with other hypertensive patients [by 3.37 ml/min IQR (0.82–5.93)] and more severe albuminuria [standard mean difference.55 (0.19–0.91)], resulting in an association with microalbuminuria [odds ratio (OR): 2.09 (1.40; 3.12)] and proteinuria [OR: 2.68 (1.89; 3.79)] (145). The follow-up study further demonstrated that the greater risk of impairment of kidney function in patients with PA was independent of BP degree (146).
Another crucial influence of aldosterone excess in patients with PA is relative hyperfiltration (147). Most patients with long-standing PA have some degree of renal insufficiency, but aldosterone excess-induced glomerular hyperfiltration may mask mild to moderate underlying renal failure (148). The early sign of long-lasting hyperfiltration is microalbuminuria (149), and preventing the development of microalbuminuria by recognizing glomerular hyperfiltration becomes an important target in the management of PA (150). Therefore, the relative hyperfiltration beyond the effect of hypertension in PA could mislead clinicians as to the interpretation of normal and abnormal renal function by examining eGFR data only. Therefore, clinicians should be aware of possible underlying renal damage in patients with PA even when the eGFR is normal.
Toxic Effects of Aldosterone on Adipose Tissue
Adipose tissue is a complex, essential, and highly active metabolic and endocrine organ (151). It has been believed to have autocrine, paracrine, and endocrine functions and plays a key role in the pathogenesis of glucose and lipid metabolism disturbances and insulin resistance (IR) in patients with PA (152, 153). A high PAC may induce adipose tissue dysfunction and lead to inflammation, fibrosis, and a high incidence of metabolic syndrome in patients with PA (154, 155). Years ago, Rondinone et al. demonstrated that MRs were presented in adipocytes isolated from rats and aldosterone could induce differentiation of 3T3-L1 cells because of the presence of specific mineral ocorticoid-binding sites (156).
Both subcutaneous adipose tissue (SAT) and visceral adipose tissue (VAT) were observed to have a potential interplay with aldosterone in patients with PA (157–159). Studies have reported that the number and affinity of insulin receptors in SAT were reduced significantly, which resulted in glucose intolerance and reduced insulin sensitivity in patients with PA (160, 161). Besides, the expression of PCK1, ADIPOQ, PLIN, and PPARG in VAT were inversely correlated with excess aldosterone levels, which in turn contribute to the IR observed in patients with PA (159). Mechanism studies have demonstrated that aldosterone-reduced glucose uptake in human adipocytes by reducing GLUT4 cell-surface localization and phosphorylation of IRS1, PI3K, and AKT (162), as well as insulin signaling, was impaired in a rat model of PA (163). In addition, aldosterone excess was also associated with a reduction of both leptin and adiponectin expression in VAT in patients with aldosterone-producing adenoma (APA) (164, 165). Besides, elevated aldosterone levels are associated with elevated circulating resistin levels and cardiac morphological changes, independent of the presence of metabolic syndrome in patients with PA (154). However, other studies have shown that the correlation between the PAC and the percentage of VAT was not evident in patients with APA (158), and patients with APA had smaller visceral fat areas than their counterparts with EH (166).
Toxic Effects of Aldosterone on Diabetes Mellitus (DM)
Primary aldosteronism (PA) is frequently associated with impaired insulin sensitivity and an increased risk of developing DM. A multi-institutional, cross-sectional study demonstrated that DM was an independent risk factor in cardio-cerebrovascular events and renal complications in patients with PA (167). Elevated aldosterone levels were independently associated with IR, as observed by Kumagai et al. in a 10-year prospective study (168). Recent studies have further demonstrated that patients with APA presented with increased levels of not only aldosterone but also cortisol via the expression of both CYP11B1 and CYP11B2. CYP11B1 is required for the synthesis of cortisol and 11β-hydroxy and rostenedione, and CYP11B2 catalyzes aldosterone synthesis (169). Gerards et al. demonstrated that impaired glucose metabolism was associated with cortisol cosecretion, which increased the risk of type 2 DM in patients with PA (170). Besides, the coexistence of autonomous cortisol secretion was associated with poor clinical outcomes and psychological symptoms in patients with PA (171, 172). A recent prospective cohort study demonstrated that PA caused decreased insulin secretion and an increased rate of insulin clearance, which lead to glucose intolerance (173). Physically, insulin and glucose are delivered to different organs by the circulation, and they enter specific cells through NO-dependent transport (174). In patients with PA, excess aldosterone and MR activation increase ROS and promote endothelial remodeling, resulting in VSMC-related IR (175), skeletal muscle-related IR (176), and adipocyte-related IR (177). Furthermore, the relationship between PA and the increased prevalence of abnormal glucose metabolism was strengthened by two meta-analyses. Subjects with PA had a higher risk of developing DM (OR 1.33, 95% CI1.01–1.74) and a higher prevalence of DM compared to subjects with EH (OR 1.55, 95% CI 1.01–2.36, p = 0.04) (5). They also had an increased prevalence of impaired fasting glucose [31.2% (95% CI15.81–46.60%)], impaired glucose tolerance [26.19% (95% CI 15.17–37.21%)], and DM [15.22% (95% CI9.93–20.51%)] (178).
Aldosterone and OSA
Obstructive Sleep Apnea (OSA) is the most frequent secondary condition associated with resistant hypertension, and the association between OSA and PA has been a matter of debate (179). Recently, several efforts have been devoted to investigating the bidirectional relationship between aldosterone levels and OSA (180, 181). A small sample size (53 patients) study showed that 34% of hypertensive patients with OSA had PA (180). Besides, a prospective study that enrolled 207 patients with OSA showed the frequency of PA in patients with moderate-to-severe OSA was up to 21.3%, suggesting that moderate-to-severe OSA predicted the presence of PA (182). However, the Hypertensives with Primary Aldosteronism and OSA (HYPNOS) study demonstrated that the prevalence of PA in patients with OSA was 8.9% (11.8% of Caucasian ethnicity and 5.9% of Chinese ethnicity) (181), which is challenging the current recommendation of the 2016 Endocrine Society guidelines that recommend screening for PA in all hypertensive patients with OSA (183). But the latest study supported that rigorous screening for PA is cost-saving due to the aversion of cardiovascular risk even if screening was conservatively presumed to identify only 3% of new PA cases (184). What is more, aldosterone levels contributed to the severity of OSA in patients with PA (185). Accordingly, the HYPNOS study found the prevalence of OSA was 67.6% in the overall cohort of patients with PA (64.4% of Caucasian ethnicity, and 70.0% of Chinese ethnicity, respectively) (181). Another retrospective study in the Japanese population also showed that 55% of patients with PA were diagnosed with OSA (135). Interestingly, both of the studies by Wolley and Gaddam verified that treatment with a MRA in patients with PA significantly reduced the number of those with OSA (186, 187). Collectively, early screening of patients with moderate-to-severe OSA for PA could reduce TOD. Most importantly, improving the awareness of the impact of hyper aldosteronemia on OSA in patients with PA can help to reduce morbidity and mortality in patients with moderate-to-severe OSA.
Treatment Outcomes of PA
Targeted treatment with either adrenalectomy (ADX) and MRA are the two common, well-documented treatments to improve the outcomes of patients with PA. The current practice guidelines recommend ADX for lateralized aldosterone excess, whereas bilateral lesions are treated using a MRA (183, 188). When ADX was performed for lateralized aldosterone excess, the cure rate for hypertension (a composite of patients who are cured or experience a marked improvement) was up to 82% and that of the biochemical aspects of PA was close to 100%. Even when antihypertensive treatment cannot be withdrawn after ADX, the number or the doses of antihypertensive drugs can be markedly decreased, and/or resistant hypertension can be resolved (189). However, it should be noted that aldosterone levels, which are the occupation of aldosterone target-MR, are tightly regulated by the RAAS system, and treatment with a MRA cannot fully replicate physiological control. Interestingly, ADX was also associated with a considerable improvement in several indexes of QoL (190). For patients who are not candidates for surgery, treatment with anMRA is a reasonable alternative to ADX. However, it is difficult to assess response to therapy in PA due to a lack of standardized outcome measures. Although the majority of subjects benefited from both surgical and medical therapies in observational studies (120, 191, 192), the absence of unifying criteria for therapeutic response has limited comparative investigations. In 2017, the Primary Aldosteronism Surgical Outcome (PASO) study published explicit criteria for biochemical and clinical outcomes after ADX, but the complexity of the PASO criteria significantly limits their utility in routine clinical settings. Because the PASO criteria do not reflect the secondary outcomes associated with PA, including cardiovascular and renal dysfunction, which are arguable of significant clinical importance (193).
Recent investigations of therapeutic outcomes in PA have evaluated alternative endpoints, including target organ function, QoL, and overall survival (194). Although overall mortality after the diagnosis of PA and initiation of specific treatment is similar to that of matched controls with EH, available evidence, based on predominantly retrospective and a few prospective observational studies, shows that treatment of PA by ADX or treatment with anMRA reduced the all-cause morbidity of untreated PA, thereby reducing overall mortality (9, 195). Besides, the TOD induced by excess aldosterone could also be reversed, to a great degree, via ADX or treatment with anMRA (196, 197). Several recent studies (2011–2021) evaluating ADX vs. MRA therapy are discussed below and summarized in Table 1.
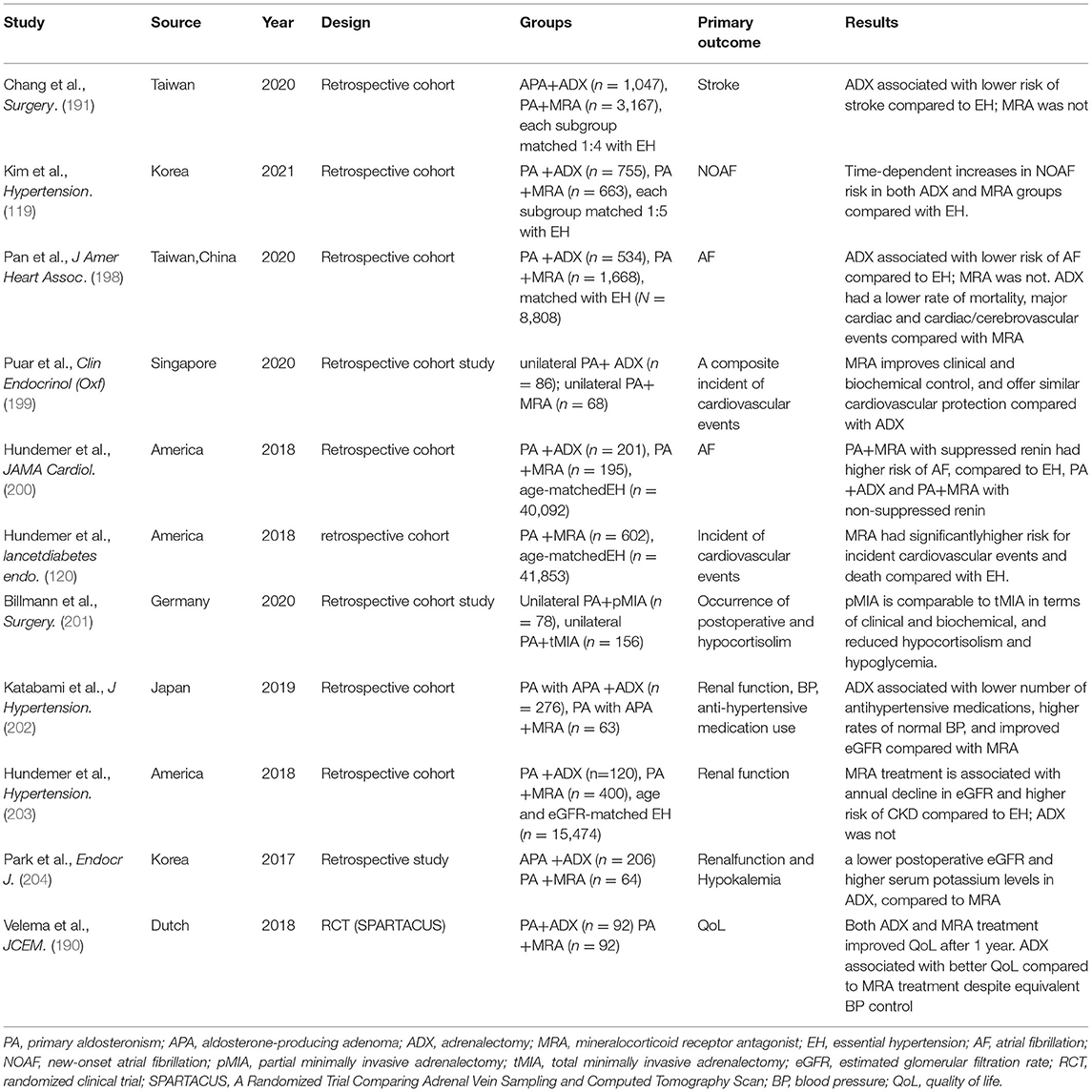
Table 1. A summary of selected recent studies (2011–2021) comparing medical and surgical treatments of PA.
Cerebro-Cardiovascular Outcomes
The cardiovascular system responds dynamically to aldosterone. PA is associated with vascular and cardiac remodeling beyond the degree of BP, which is the presumed mechanism for the higher rates of cardiovascular events (3, 4, 83). A retrospective cohort study showed that patients with PA treated with MRA had a higher risk of stroke (a competing hazard ratio = 1.83, P < 0.001), while ADX lowered the risk of incident stroke (competing for a hazard ratio = 0.75) compared to controls with EH (191). Catena et al. reported that both ADX and treatment with MRA (spironolactone) decreased LVM in patients with PA (196). Targeted surgical and medical treatment of PA-induced LVM via LV inward remodeling and ADX contributed to a prominent and persistent decrease in LVMI, while medical treatment resulted in a borderline significant fall (101). Besides, the worse LV diastolic function in patients with PA could be reversed after ADX (102), and ADX exerted a beneficial effect on LV geometry and structure by reducing LV concentric geometry and the burden of LVH in patients with PA (197). In addition, ADX lowers the risks of congestive heart failure (CHF) and all-cause mortality in a long-term follow-up (112).
However, studies have shown the different effects of medical or surgical treatment on AF in patients with PA. Pan et al. demonstrated that patients with PA who underwent ADX had a lower incidence of new-onset atrial fibrillation (NOAF), but this finding was not observed in patients with PA who received a lower dose of MRA therapy (198). The authors supposed the differences between the two strategies might be caused by the different doses of MRA therapy. A higher dosage of MRA may have higher clinical efficacy and reduce the differences between ADX and MRA therapy. However, this issue needs further large prospective randomized trials to figure this out. In addition, patients with PA treated with MRA (renin remained suppressed) had a significantly higher risk of AF, where treatment of PA with MRA substantially increases renin, or with surgical ADX was associated with no significant difference in reducing the risk for developing AF (120). Accordingly, Rossi et al. also demonstrated that treatment with MRA had a higher incidence of AF, compared with both ADX treatment for PA and the EH group (192), which was confirmed by a recent meta-analysis. This meta-analysis enrolled 2,705 patients with PA, and the results demonstrated that the incidence of NOAF among the patients with PA receiving treatment with MRA was much higher compared to those inpatients for PA who underwent ADX (pooled OR: 2.83, 95% CI: 1.76–4.57 in the random-effects model). The pooled OR was 1.91 (95% CI: 1.11–3.28) when compared to the patients with EH (205). These data suggested that persistent renin suppression may serve as a biomarker for inadequate MR blockade, leading to ongoing aldosterone exposure and consequent cardiometabolic effects. Taken together, surgical treatment seems to have a better protective effect on AF than medical treatment in patients with PA.
Renal Outcomes
The renal system is a direct target organ of hyper aldosteronism, and patients with PA often have an association with decreased eGFR and CKD (139, 142, 144). Effective treatment with either surgery or MRA therapy will unmask the underlying CKD in patients with PA (206). Short- and long-term follow-up after ADX consistently indicated that early involvement of the kidney in patients with PA was characterized by functional changes that would be largely reversible, with a significant decrease in urinary albumin excretion (207), and microalbuminuria was more likely to subside to normal levels (204). Besides, ADX had significantly lower utilization of antihypertensive medications, higher rates of normalization of BP, and improved eGFR compared with treatment with MRA (202). However, for elderly patients, MRA therapy may be more appropriate (204). Of course, a few major cardiovascular events and mortality events were also observed after surgical treatment (208). Studies found that kidney function further deteriorated after ADX in patients with PA (209, 210), and the incidence and risk of postoperative acute kidney injury were significantly higher in patients with PA after surgical ADX (211). MRAs must be dosed with care in the setting of CKD, as they may precipitate hyperkalemia [49]. Therefore, clinicians should pay more attention to postoperative renal function in patients with PA at elevated risk for a decline in kidney function, and it is important to evaluate the age, preoperative PAC, and preoperative potassium level before choosing the method of treatment.
Other Outcomes of the Vascular System
Arterial stiffness is one of the important factors in patients with PA and severe arterial stiffness before surgery was significantly associated with renal function decline and less LVM regression after ADX in patients with lateralized PA (212). Strauch et al. found that the PWV significantly decreased after ADX, whereas there are no changes in arterial stiffness (PWV, augmentation index) indices in patients treated with MRA (spironolactone) (213). Another study also showed both brachial-ankle PWV (baPWV) and heart-ankle PWV (haPWV) were significantly reduced after ADX in patients with APA, which suggested that ADX could reverse adverse vascular changes in patients with APA (127).
Major Limitations and Disadvantages of Treatments
The treatment objectives for patients with PA include resolution of hypokalemia and prevention of the morbidity and mortality associated with hypertension, progressive CKD, and further TOD (cerebro-cardiovascular, renal, and vascular systems). Although the surgery would improve the QoL of patients to normal population-adjusted values, it is more cost-effective in the long term, because the patients require fewer medications and less frequent clinic visits (190). Besides, surgery may also bring about superior cardiovascular, renal, and metabolic outcomes (214, 215). However, there exist some limitations and disadvantages to the surgical treatment (i.e., ADX) of PA. Firstly, not all patients with PA are suitable for ADX, and ADX is the best choice for common unilateral APA (aldosteronoma), primary unilateral adrenal hyperplasia, and multinodular unilateral adrenocortical hyperplasia. Secondly, there exists a risk of short-term hypoaldosteronism, leading to clinically important hyperkalemia after ADX surgery (216, 217). Thirdly, because glomerular hyperfiltration associated with aldosterone excess might mask mild-to-moderate underlying renal failure, most patients with long-standing PA have some degree of renal insufficiency. Some studies demonstrated that kidney function further deteriorated after ADX (209, 210) and the incidence and risk of postoperative acute kidney injury were significantly higher after ADX in patients with PA (211). For medical treatment, it is essential that the MRA dosage must be adequate to fully block the toxic effects of hyperaldosteronism. Nevertheless, for unselective MRA (spironolactone, canrenone, potassium canrenoate), painful gynaecomastia, erectile dysfunction, and decreased libido in men are induced in PA patients with spironolactone at dosages of more than 50 mg/day, because spironolactone and its metabolites are antagonists of the androgen receptor. They also are agonists of the progesterone receptor and thus interfere with the estrus cycle. These off-target effects occur at 25 mg/day after chronic use in some patients. Besides, agonist activity at the progesterone receptor may result in menstrual irregularity in women (218). Eplerenone, a competitive and selective MRA, is more expensive, weaker, and shorter-acting than the other MRAs. Most importantly, in patients with PA, lifetime use of MRA could increase the pill burden of antihypertensive medications significantly and reduce QoL greatly.
Conclusions and Perspectives
Primary aldosteronism (PA) is the most common cause of endocrine hypertension, and hyperaldosteronemia is one of the important characteristics for patients with PA (219). Increasing experimental and clinical data suggest prolonged exposure to elevated aldosterone concentrations in patients with PA is associated with increased TOD, including brain, heart, kidney, vessels, adipose tissues, and OSA, and therefore, leads to an increase in morbidity from cerebro-cardiovascular events. To attenuate the toxic effects of excess aldosterone, the first and key step is to screen for PA, especially in patients with hypertension that is severe and/or resistant to treatment. Early diagnosis is the fundamental and critical point to obtain benefits from the prevention of the development of TOD, because ADX for patients with unilateral could bring BP under control with the withdrawal or prominent reduction in the number and dosage of antihypertensive medications, as well as even reverse TOD (220). Besides, titration of MRA therapy to increase plasma renin levels may be an effective approach to avoid excessive cardiovascular risk in medically treated patients with PA. Furthermore, there are novel third-generation nonsteroidal MRAs (like eplerenone, the receptor selectivity of AZD-9977, esaxerenone, finerenone, and KBP-5074) currently in clinical trials and provide a cardiorenal benefit above that of current optimized standard-of-care treatment in a high-risk population with reduced renal function, and with a lower risk of hyperkalemia (221–224). These nonsteroidal MRAs generally exhibit both high affinity and good selectivity for the MR over other steroid receptors may have a better safety profile in patients with PA (222–226). For example, finerenone from Bayer is non-steroidal, and suggested to be cardiac preferring (over the kidney) in contrast with the steroidal MRAs (227). Esaxerenone from Daiichi-Sankyo showed over 10 times as potent as eplerenone in terms of lowering blood pressure: its particular clinical significance claims to be a safe and effective antihypertensive in patients with moderate renal dysfunction and/or T2DM and albuminuria (228). Therefore, these novel nonsteroidal MRAs have better therapeutic options for patients with PA in the future (222–224). In addition, more accurate methods to select patients with unilateral PA for ADX are needed in medically treated patients in the future (183). Taken together, there should be a low threshold for initiating screening for PA, since the early diagnosis of PA and early initiation of specific treatment strategies have a substantial impact on reducing cerebro-cardiovascular events and mortality in the long term.
Author Contributions
JL and L-QY conceived and designed the manuscript. XL collected the literature and wrote the manuscript. MU, XW, FX, S-KS, and L-ML were involved in data collection and made substantial contributions to data analysis. All the authors read and approved the manuscript and agreed to its publication.
Funding
This work was supported by grants from the National Natural Science Foundation of China (Nos. 82100944, 82100494, 82070910, and 81770881), Natural Science Foundation of Hunan Province (No. 2021JJ40842), and the Clinical Research Center for Medical Imaging in Hunan Province of China (No. 2020SK4001).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Parasiliti-Caprino M, Lopez C, Prencipe N, Lucatello B, Settanni F, Giraudo G, et al. Prevalence of primary aldosteronism and association with cardiovascular complications in patients with resistant and refractory hypertension. J Hypertens. (2020) 38:1841–8. doi: 10.1097/HJH.0000000000002441
2. Jaffe G, Gray Z, Krishnan G, Stedman M, Zheng Y, Han J, et al. Screening Rates for Primary Aldosteronism in Resistant Hypertension: A Cohort Study.Hypertension (Dallas, Tex.: 1979). (2020) 75:650–9. doi: 10.1161/HYPERTENSIONAHA.119.14359
3. Bernini G, Galetta F, Franzoni F, Bardini M, Taurino C, Bernardini M, et al. Arterial stiffness, intima-media thickness and carotid artery fibrosis in patients with primary aldosteronism. J Hypertens. (2008) 26:2399–405. doi: 10.1097/HJH.0b013e32831286fd
4. Milliez P, Girerd X, Plouin PF, Blacher J, Safar ME, Mourad JJ. Evidence for an increased rate of cardiovascular events in patients with primary aldosteronism. J Am Coll Cardiol. (2005) 45:1243–8. doi: 10.1016/j.jacc.2005.01.015
5. Monticone S, D'Ascenzo F, Moretti C, Williams TA, Veglio F, Gaita F, et al. Cardiovascular events and target organ damage in primary aldosteronism compared with essential hypertension: a systematic review and meta-analysis. Lancet. (2018) 6, 41–50. doi: 10.1016/S2213-8587(17)30319-4
6. Strauch B, Petrák O, Wichterle D, Zelinka T, Holaj R, Widimský JJr. Increased arterial wall stiffness in primary aldosteronism in comparison with essential hypertension. Am J Hypertens. (2006) 19:909–14. doi: 10.1016/j.amjhyper.2006.02.002
7. Vogt B, Burnier M. Aldosterone cardiovascular risk. Curr Hypertens Rep. (2009) 11:450–5. doi: 10.1007/s11906-009-0076-8
8. Widimsky JJr, Strauch B, Petrák O, Rosa J, Somloova Z, Zelinka T, et al. Vascular disturbances in primary aldosteronism: clinical evidence. Kidney Blood Press Res. (2012) 35:529–33. doi: 10.1159/000340031
9. Catena C, Colussi G, Nadalini E, Chiuch A, Baroselli S, Lapenna R, et al. Cardiovascular outcomes in patients with primary aldosteronism after treatment. Arch Intern Med. (2008) 168:80–5. doi: 10.1001/archinternmed.2007.33
10. Spät A, Hunyady L. Control of aldosterone secretion: a model for convergence in cellular signaling pathways. Physiol Rev. (2004) 84:489–539. doi: 10.1152/physrev.00030.2003
11. Muñoz-Durango N, Fuentes CA, Castillo AE, González-Gómez LM, Vecchiola A, Fardella CE, et al. Role of the renin-angiotensin-aldosterone system beyond blood pressure regulation: molecular and cellular mechanisms involved in end-organ damage during arterial hypertension. Int J Mol Sci. (2016) 17. doi: 10.3390/ijms17070797
12. Christ M, Günther A, Heck M, Schmidt BM, Falkenstein E, Wehling M. Aldosterone, not estradiol, is the physiological agonist for rapid increases in cAMP in vascular smooth muscle cells. Circulation. (1999) 99:1485–91. doi: 10.1161/01.CIR.99.11.1485
13. Stockand JD, Meszaros JG. Aldosterone stimulates proliferation of cardiac fibroblasts by activating Ki-RasA and MAPK1/2 signaling. Am J Physiol Heart Circ Physiol. (2003) 284:H176–84. doi: 10.1152/ajpheart.00421.2002
14. Wehling M, Ulsenheimer A, Schneider M, Neylon C, Christ M. Rapid effects of aldosterone on free intracellular calcium in vascular smooth muscle and endothelial cells: subcellular localization of calcium elevations by single cell imaging. Biochem Biophys Res Commun. (1994) 204:475–81. doi: 10.1006/bbrc.1994.2484
15. Rossi GM, Regolisti G, Peyronel F, Fiaccadori E. Recent insights into sodium and potassium handling by the aldosterone-sensitive distal nephron: implications on pathophysiology and drug discovery. J Nephrol. (2020) 33:447–66. doi: 10.1007/s40620-020-00700-9
16. Wu A, Wolley MJ, Wu Q, Gordon RD, Fenton RA, Stowasser M. The Cl(-)/HCO(3)(-) exchanger pendrin is downregulated during oral co-administration of exogenous mineralocorticoid and KCl in patients with primary aldosteronism. J Hum Hypertens. (2020) doi: 10.1038/s41371-020-00439-7
17. Stavropoulos K, Kassimatis E, Doumas M, Sarafidis P. Renal tubular transport protein regulation in primary aldosteronism: can large-scale proteomic analysis offer a new insight? J Hum Hypertens. (2021) doi: 10.1038/s41371-021-00537-0
18. Grossmann C, Benesic A, Krug AW, Freudinger R, Mildenberger S, Gassner B, et al. Human mineralocorticoid receptor expression renders cells responsive for nongenotropic aldosterone actions. Mol Endocrinol (Baltimore, Md). (2005) 19:1697–710. doi: 10.1210/me.2004-0469
19. Barrera-Chimal J, Girerd S, Jaisser F. Mineralocorticoid receptor antagonists and kidney diseases: pathophysiological basis. Kidney Int. (2019) 96:302–19. doi: 10.1016/j.kint.2019.02.030
20. Pacurari M, Kafoury R, Tchounwou PB, Ndebele K. The Renin-Angiotensin-aldosterone system in vascular inflammation and remodeling. Int J Inflam. (2014) 2014:689360. doi: 10.1155/2014/689360
21. Bauersachs J, Jaisser F, Toto R. Mineralocorticoid receptor activation and mineralocorticoid receptor antagonist treatment in cardiac and renal diseases. Hypertension (Dallas, Tex.: 1979). (2015) 65:257–63. doi: 10.1161/HYPERTENSIONAHA.114.04488
22. Buglioni A, Cannone V, Cataliotti A, Sangaralingham SJ, Heublein DM, Scott CG, et al. Circulating aldosterone and natriuretic peptides in the general community: relationship to cardiorenal and metabolic disease. Hypertension (Dallas, Tex.: 1979). (2015) 65:45–53. doi: 10.1161/HYPERTENSIONAHA.114.03936
23. Chen ZW, Hung CS, Wu VC, Lin YH. Primary aldosteronism and cerebrovascular diseases. Endocrinol Metab (Seoul, Korea). (2018) 33:429–34. doi: 10.3803/EnM.2018.33.4.429
24. Fardella CE, Mosso L, Gómez-Sánchez C, Cortés P, Soto J, Gómez L, et al. Primary hyperaldosteronism in essential hypertensives: prevalence, biochemical profile, molecular biology. J Clin Endocrinol Metab. (2000) 85:1863–7. doi: 10.1210/jc.85.5.1863
25. Prejbisz A, Warchoł-Celińska E, Lenders JW, Januszewicz A. Cardiovascular Risk in Primary Hyperaldosteronism. Horm Metab Res. (2015) 47:973–80. doi: 10.1055/s-0035-1565124
26. Callera GE, Touyz RM, Tostes RC, Yogi A, He Y, Malkinson S, et al. Aldosterone activates vascular p38MAP kinase and NADPH oxidase via c-Src. Hypertension (Dallas, Tex.: 1979). (2005) 45:773–9. doi: 10.1161/01.HYP.0000154365.30593.d3
27. Johar S, Cave AC, Narayanapanicker A, Grieve DJ, Shah AM. Aldosterone mediates angiotensin II-induced interstitial cardiac fibrosis via a Nox2-containing NADPH oxidase. FASEB J. (2006) 20:1546–8. doi: 10.1096/fj.05-4642fje
28. Keidar S, Kaplan M, Pavlotzky E, Coleman R, Hayek T, Hamoud S, et al. Aldosterone administration to mice stimulates macrophage NADPH oxidase and increases atherosclerosis development: a possible role for angiotensin-converting enzyme and the receptors for angiotensin II and aldosterone. Circulation. (2004) 109:2213–20. doi: 10.1161/01.CIR.0000127949.05756.9D
29. Bordin L, Don à G, Sabbadin C, Ragazzi E, Andrisani A, Ambrosini G, et al. Human red blood cells alterations in primary aldosteronism. J Clin Endocrinol Metab. (2013) 98:2494–501. doi: 10.1210/jc.2012-3571
30. Bordin L, Saccardi C, Don à G, Sabbadin C, Andrisani A, Ambrosini G, et al. Mineralocorticoid receptor is involved in the aldosterone pathway in human red blood cells. Am J Transl. (2016) 8:314–28.
31. Virdis A, Neves MF, Amiri F, Viel E, Touyz RM, Schiffrin EL. Spironolactone improves angiotensin-induced vascular changes and oxidative stress. Hypertension (Dallas, Tex.: 1979). (2002) 40:504–10. doi: 10.1161/01.HYP.0000034738.79310.06
32. Wei Y, Whaley-Connell AT, Habibi J, Rehmer J, Rehmer N, Patel K, et al. Mineralocorticoid receptor antagonism attenuates vascular apoptosis and injury via rescuing protein kinase B activation. Hypertension (Dallas, Tex.: 1979). (2009) 53:158–65. doi: 10.1161/HYPERTENSIONAHA.108.121954
33. Petramala L, Pignatelli P, Carnevale R, Zinnamosca L, Marinelli C, Settevendemmie A, et al. Oxidative stress in patients affected by primary aldosteronism. J Hypertens. (2014)32:2022–9. doi: 10.1097/HJH.0000000000000284
34. Hayashi H, Kobara M, Abe M, Tanaka N, Gouda E, Toba H, et al. Aldosterone nongenomically produces NADPH oxidase-dependent reactive oxygen species and induces myocyte apoptosis. Hypertens Res. (2008) 31:363–75. doi: 10.1291/hypres.31.363
35. Fiebeler A, Luft FC. The mineralocorticoid receptor and oxidative stress. Heart Fail Rev. (2005) 10:47–52. doi: 10.1007/s10741-005-2348-y
36. Iwashima F, Yoshimoto T, Minami I, Sakurada M, Hirono Y, Hirata Y. Aldosterone induces superoxide generation via Rac1 activation in endothelial cells. Endocrinology. (2008) 149:1009–14. doi: 10.1210/en.2007-0864
37. Taye A, Morawietz H. Spironolactone inhibits NADPH oxidase-induced oxidative stress and enhances eNOS in human endothelial cells. Iran J Pharm Res. (2011) 10:329–37.
38. Leopold JA, Dam A, Maron BA, Scribner AW, Liao R, Handy DE, et al. Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat Med. (2007) 13:189–97. doi: 10.1038/nm1545
39. McCurley A, Pires PW, Bender SB, Aronovitz M, Zhao MJ, Metzger D, et al. Direct regulation of blood pressure by smooth muscle cell mineralocorticoid receptors. Nat Med. (2012) 18:1429–33. doi: 10.1038/nm.2891
40. Min LJ, Mogi M, Iwanami J, Li JM, Sakata A, Fujita T, et al. Cross-talk between aldosterone and angiotensin II in vascular smooth muscle cell senescence. Cardiovasc Res. (2007) 76:506–16. doi: 10.1016/j.cardiores.2007.07.008
41. Touyz RM. Recent advances in intracellular signalling in hypertension. Curr Opin Nephrol Hypertens. (2003) 12:165–74. doi: 10.1097/00041552-200303000-00007
42. Miyata K, Rahman M, Shokoji T, Nagai Y, Zhang GX, Sun GP, et al. Aldosterone stimulates reactive oxygen species production through activation of NADPH oxidase in rat mesangial cells. J Am Soc Nephrol. (2005) 16:2906–12. doi: 10.1681/ASN.2005040390
43. Nishiyama A, Yao L, Nagai Y, Miyata K, Yoshizumi M, Kagami S, et al. Possible contributions of reactive oxygen species and mitogen-activated protein kinase to renal injury in aldosterone/salt-induced hypertensive rats. Hypertension (Dallas, Tex.: 1979). (2004) 43:841–8. doi: 10.1161/01.HYP.0000118519.66430.22
44. Shibata S, Nagase M, Yoshida S, Kawachi H, Fujita T. Podocyte as the target for aldosterone: roles of oxidative stress and Sgk1. Hypertension (Dallas, Tex.: 1979). (2007) 49:355–64. doi: 10.1161/01.HYP.0000255636.11931.a2
45. Mathew JT, Patni H, Chaudhary AN, Liang W, Gupta A, Chander PN, et al. Aldosterone induces mesangial cell apoptosis both in vivo and in vitro. Am J Physiol. (2008) 295:F73–81. doi: 10.1152/ajprenal.00435.2007
46. Blasi ER, Rocha R, Rudolph AE, Blomme EA, Polly ML, McMahon EG. (2003). Aldosterone/salt induces renal inflammation and fibrosis in hypertensive rats.Kidney international. 63, 1791–1800. doi: 10.1046/j.1523-1755.2003.00929.x
47. Sun JY, Li C, Shen ZX, Zhang WC, Ai TJ, Du LJ, et al. Mineralocorticoid Receptor Deficiency in Macrophages Inhibits Neointimal Hyperplasia and Suppresses Macrophage Inflammation Through SGK1-AP1/NF-κB Pathways. Arterioscler Thromb Vasc Biol. (2016) 36:874–85. doi: 10.1161/ATVBAHA.115.307031
48. Eissler R, Schmaderer C, Rusai K, Kühne L, Sollinger D, Lahmer T, et al. Hypertension augments cardiac Toll-like receptor 4 expression and activity. Hypertensi Res. (2011) 34:551–8. doi: 10.1038/hr.2010.270
49. Buonafine M, Martínez-Martínez E, Amador C, Gravez B, Ibarrola J, Fernández-Celis A, et al. Neutrophil Gelatinase-Associated Lipocalin from immune cells is mandatory for aldosterone-induced cardiac remodeling and inflammation. J Mol Cell Cardiol. (2018) 115:32–8. doi: 10.1016/j.yjmcc.2017.12.011
50. Herrada AA, Contreras FJ, Marini NP, Amador CA, González PA, Cortés CM, et al. Aldosterone promotes autoimmune damage by enhancing Th17-mediated immunity. J Immunol (Baltimore, Md.: 1950). (2010) 184:191–202. doi: 10.4049/jimmunol.0802886
51. Lai L, Chen J, Hao CM, Lin S, Gu Y. Aldosterone promotes fibronectin production through a Smad2-dependent TGF-beta1 pathway in mesangial cells. Biochem Biophys Res Commun. (2006) 348:70–5. doi: 10.1016/j.bbrc.2006.07.057
52. Ma LJ, Yang H, Gaspert A, Carlesso G, Barty MM, Davidson JM, et al. Transforming growth factor-beta-dependent and -independent pathways of induction of tubulointerstitial fibrosis in beta6(-/-) mice. Am J Pathol. (2003) 163:1261–73. doi: 10.1016/S0002-9440(10)63486-4
53. Martín-Fernández B, Rubio-Navarro A, Cortegano I, Ballesteros S, Alía M, Cannata-Ortiz P, et al. Aldosterone Induces Renal Fibrosis and Inflammatory M1-Macrophage Subtype via Mineralocorticoid Receptor in Rats. PLoS ONE. (2016) 11:e0145946. doi: 10.1371/journal.pone.0145946
54. Sun Y, Zhang J, Zhang JQ, Ramires FJ. Local angiotensin II and transforming growth factor-beta1 in renal fibrosis of rats. Hypertension (Dallas, Tex.: 1979). (2000) 35:1078–1084. doi: 10.1161/01.HYP.35.5.1078
55. Brown NJ, Nakamura S, Ma L, Nakamura I, Donnert E, Freeman M, et al. Aldosterone modulates plasminogen activator inhibitor-1 and glomerulosclerosis in vivo. Kidney Int. (2000) 58:1219–27. doi: 10.1046/j.1523-1755.2000.00277.x
56. Galmiche G, Pizard A, Gueret A, El Moghrabi S, Ouvrard-Pascaud A, Berger S, et al. Smooth muscle cell mineralocorticoid receptors are mandatory for aldosterone-salt to induce vascular stiffness. Hypertension (Dallas, Tex.: 1979). (2014) 63:520–6. doi: 10.1161/HYPERTENSIONAHA.113.01967
57. Huang W Xu C Kahng KW Noble NA Border WA Huang Aldosterone Y and TGF-beta1 synergistically increase PAI-1 and decrease matrix degradation in rat renal mesangial and fibroblast cells. Am J Physiol Renal Physiol. (2008) 294:F1287–95. doi: 10.1152/ajprenal.00017.2008
58. Pruthi D, McCurley A, Aronovitz M, Galayda C, Karumanchi SA, Jaffe IZ. Aldosterone promotes vascular remodeling by direct effects on smooth muscle cell mineralocorticoid receptors. Arterioscler Thromb Vasc Biol. (2014) 34:355–64. doi: 10.1161/ATVBAHA.113.302854
59. Zhou G, Kandala JC, Tyagi SC, Katwa LC, Weber KT. Effects of angiotensin II and aldosterone on collagen gene expression and protein turnover in cardiac fibroblasts. Mol Cell Biochem. (1996) 154:171–8. doi: 10.1007/BF00226785
60. Diah S, Zhang GX, Nagai Y, Zhang W, Gang L, Kimura S, et al. Aldosterone induces myofibroblastic transdifferentiation and collagen gene expression through the Rho-kinase dependent signaling pathway in rat mesangial cells. Exp Cell Res. (2008) 314:3654–62. doi: 10.1016/j.yexcr.2008.09.018
61. Chen D, Chen Z, Park C, Centrella M, McCarthy T, Chen L, et al. Aldosterone stimulates fibronectin synthesis in renal fibroblasts through mineralocorticoid receptor-dependent and independent mechanisms. Gene. (2013) 531:23–30. doi: 10.1016/j.gene.2013.08.047
62. Irita J, Okura T, Kurata M, Miyoshi K, Fukuoka T, Higaki J. Osteopontin in rat renal fibroblasts: functional properties and transcriptional regulation by aldosterone. Hypertension (Dallas, Tex.: 1979). (2008) 51:507–13. doi: 10.1161/HYPERTENSIONAHA.107.102640
63. Huang LL, Nikolic-Paterson DJ, Ma FY, Tesch GH. Aldosterone induces kidney fibroblast proliferation via activation of growth factor receptors and PI3K/MAPK signalling. Nephron Exp Nephrol. (2012) 120:e115–22. doi: 10.1159/000339500
64. Martínez-Martínez E, Buonafine M, Boukhalfa I, Ibarrola J, Fernández-Celis A, Kolkhof P, et al. Aldosterone Target NGAL (Neutrophil Gelatinase-Associated Lipocalin) Is Involved in Cardiac Remodeling After Myocardial Infarction Through NFκB Pathway. Hypertension (Dallas, Tex.: 1979). (2017) 70:1148–56. doi: 10.1161/HYPERTENSIONAHA.117.09791
65. Tarjus A, Martínez-Martínez E, Amador C, Latouche C, El Moghrabi S, Berger T, et al. Neutrophil gelatinase-associated lipocalin, a novel mineralocorticoid biotarget, mediates vascular profibrotic effects of mineralocorticoids. Hypertension (Dallas, Tex.: 1979). (2015) 66, 158–166. doi: 10.1161/HYPERTENSIONAHA.115.05431
66. Kok HM, Falke LL, Goldschmeding R, Nguyen TQ. Targeting CTGF, EGF and PDGF pathways to prevent progression of kidney disease. Nature reviews. Nephrology. (2014) 10:700–11. doi: 10.1038/nrneph.2014.184
67. Krug AW, Grossmann C, Schuster C, Freudinger R, Mildenberger S, Govindan MV, et al. Aldosterone stimulates epidermal growth factor receptor expression. J Biol Chem. (2003) 278:43060–6. doi: 10.1074/jbc.M308134200
68. Litchfield WR, Anderson BF, Weiss RJ, Lifton RP, Dluhy RG. Intracranial aneurysm and hemorrhagic stroke in glucocorticoid-remediable aldosteronism. Hypertension (Dallas, Tex.: 1979). (1998) 31:445–50. doi: 10.1161/01.HYP.31.1.445
69. Rocha R, Chander PN, Khanna K, Zuckerman A, Stier C. T. Jr. Mineralocorticoid blockade reduces vascular injury in stroke-prone hypertensive rats. Hypertension (Dallas, Tex.: 1979). (1998) 31:451–458. doi: 10.1161/01.HYP.31.1.451
70. Murata M, Kitamura T, Tamada D, Mukai K, Kurebayashi S, Yamamoto T, et al. Plasma aldosterone level within the normal range is less associated with cardiovascular and cerebrovascular risk in primary aldosteronism. J Hypertens. (2017) 35:1079–85. doi: 10.1097/HJH.0000000000001251
71. Kim S, Tokuyama M, Hosoi M, Yamamoto K. Adrenal and circulating renin-angiotensin system in stroke-prone hypertensive rats. Hypertension (Dallas, Tex.: 1979). (1992) 20:280–91. doi: 10.1161/01.HYP.20.3.280
72. Enea I, De Paolis P, Porcellini A, Piras O, Savoia C, Russo R, et al. Defective suppression of the aldosterone biosynthesis during stroke permissive diet in the stroke-prone phenotype of the spontaneously hypertensive rat. Basic Res Cardiol. (2000) 95:84–92. doi: 10.1007/s003950050168
73. MacLeod AB, Vasdev S, Smeda JS. The role of blood pressure and aldosterone in the production of hemorrhagic stroke in captopril-treated hypertensive rats. Stroke. (1997) 28, 1821–8. doi: 10.1161/01.STR.28.9.1821
74. Rocha R, Stier C. T. Jr. Pathophysiological effects of aldosterone in cardiovascular tissues. Trends Endocrinol Metab. (2001) 308–314. doi: 10.1016/S1043-2760(01)00432-5
75. Chrissobolis S, Drummond GR, Faraci FM, Sobey CG. Chronic aldosterone administration causes Nox2-mediated increases in reactive oxygen species production and endothelial dysfunction in the cerebral circulation. J Hypertens. (2014) 32:1815–21. doi: 10.1097/HJH.0000000000000259
76. Takeda R, Matsubara T, Miyamori I, Hatakeyama H, Morise T. Vascular complications in patients with aldosterone producing adenoma in Japan: comparative study with essential hypertension. The Research Committee of Disorders of Adrenal Hormones in Japan. J Endocrinol Invest. (1995) 18:370–3. doi: 10.1007/BF03347840
77. Mulatero P, Monticone S, Bertello C, Viola A, Tizzani D, Iannaccone A, et al. Long-term cardio- and cerebrovascular events in patients with primary aldosteronism. J Clin Endocrinol Metab. (2013) 98:4826–33. doi: 10.1210/jc.2013-2805
78. Ohno Y, Sone M, Inagaki N, Yamasaki T, Ogawa O, Takeda Y, et al. Prevalence of Cardiovascular Disease and Its Risk Factors in Primary Aldosteronism: A Multicenter Study in Japan. Hypertension (Dallas, Tex.: 1979). (2018) 71:530–7. doi: 10.1161/HYPERTENSIONAHA.117.10263
79. Kao CC, Wu CH, Lin YH, Chang CC, Chen HH, Wu MS, et al. Risk of ischemic stroke in primary aldosteronism patients. Clin Chim Acta. (2015) 438:86–9. doi: 10.1016/j.cca.2014.08.007
80. Petramala L, Concistr è A, Olmati F, Saracino V, Chimenti C, Frustaci A, et al. Cardiomyopathies and adrenal diseases. Int J Mol Sci. (2020) 21. doi: 10.3390/ijms21145047
81. Born-Frontsberg E, Reincke M, Rump LC, Hahner S, Diederich S, Lorenz R, et al. Cardiovascular and cerebrovascular comorbidities of hypokalemic and normokalemic primary aldosteronism: results of the German Conn's Registry. J Clin Endocrinol Metab. (2009) 94:1125–30. doi: 10.1210/jc.2008-2116
82. Marney AM, Brown NJ. Aldosterone and end-organ damage. Clinical science (London, England: 1979). (2007) 113:267–78. doi: 10.1042/CS20070123
83. Savard S, Amar L, Plouin PF, Steichen O. Cardiovascular complications associated with primary aldosteronism: a controlled cross-sectional study. Hypertension (Dallas, Tex.: 1979). (2013) 62:331–6. doi: 10.1161/HYPERTENSIONAHA.113.01060
84. Tanabe A, Naruse M, Naruse K, Hase M, Yoshimoto T, Tanaka M, et al. Left ventricular hypertrophy is more prominent in patients with primary aldosteronism than in patients with other types of secondary hypertension. Hypertens. Res. (1997) 20:85–90. doi: 10.1291/hypres.20.85
85. Turchi F, Ronconi V, di Tizio V, Ceccoli L, Boscaro M, Giacchetti G. Primary aldosteronism and essential hypertension: assessment of cardiovascular risk at diagnosis and after treatment. Nutr Metab Cardiovasc Dis. (2014) 24:476–82. doi: 10.1016/j.numecd.2013.09.009
86. Catena C, Colussi G, Marzano L, Sechi LA. Aldosterone and the heart: from basic research to clinical evidence. Horm Metab Res. (2012) 44:181–7. doi: 10.1055/s-0031-1291318
87. Rossi GP. Cardiac consequences of aldosterone excess in human hypertension. Am J Hypertens. (2006) 19:10–2. doi: 10.1016/j.amjhyper.2005.08.011
88. Ohno Y, Sone M, Inagaki N, Kawashima A, Takeda Y, Yoneda T, et al. Nadir aldosterone levels after confirmatory tests are correlated with left ventricular hypertrophy in primary aldosteronism. Hypertension (Dallas, Tex.: 1979). (2020) 75:1475–82. doi: 10.1161/HYPERTENSIONAHA.119.14601
89. Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling–concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. J Am Coll Cardiol. (2000) 35:569–82. doi: 10.1016/S0735-1097(99)00630-0
90. Connell JM, MacKenzie SM, Freel EM, Fraser R, Davies E. A lifetime of aldosterone excess: long-term consequences of altered regulation of aldosterone production for cardiovascular function. Endocr Rev. (2008) 29:133–54. doi: 10.1210/er.2007-0030
91. Weber KT, Brilla CG. Pathological hypertrophy and cardiac interstitium. Fibrosis and renin-angiotensin-aldosterone system. Circulation. (1991) 83:1849–65. doi: 10.1161/01.CIR.83.6.1849
92. Wu Q, Hong M, Xu J, Tang X, Zhu L, Gao P, et al. Diurnal blood pressure pattern and cardiac damage in hypertensive patients with primary aldosteronism. Endocrine. (2021) 72:835–43. doi: 10.1007/s12020-021-02606-3
93. Robert V, Silvestre JS, Charlemagne D, Sabri A, Trouv é P, Wassef M, et al. Biological determinants of aldosterone-induced cardiac fibrosis in rats. Hypertension (Dallas, Tex.: 1979). (1995) 26:971–8. doi: 10.1161/01.HYP.26.6.971
94. Okoshi MP, Yan X, Okoshi K, Nakayama M, Schuldt AJ, O'Connell TD, et al. Aldosterone directly stimulates cardiac myocyte hypertrophy. J Card Fail. (2004) 10:511–8. doi: 10.1016/j.cardfail.2004.03.002
95. Takeda Y, Yoneda T, Demura M, Miyamori I, Mabuchi H. Cardiac aldosterone production in genetically hypertensive rats. Hypertension (Dallas, Tex.: 1979). (2000) 36:495–500. doi: 10.1161/01.HYP.36.4.495
96. Montiel-Jaen MG, Monsalvo-Villegas A, Ávila G. Modulating ALDH2 reveals a differential dependence on ROS for hypertrophy and SR Ca(2+) release in aldosterone-treated cardiac myocytes. Biochem Biophys Res Commun. (2021) 536:7–13. doi: 10.1016/j.bbrc.2020.12.049
97. Habibi J, DeMarco VG, Ma L, Pulakat L, Rainey WE, Whaley-Connell AT, et al. Mineralocorticoid receptor blockade improves diastolic function independent of blood pressure reduction in a transgenic model of RAAS overexpression. Am J Physiol Heart Circ Physiol. (2011) 300:H1484–91. doi: 10.1152/ajpheart.01000.2010
98. Indra T, Holaj R, Zelinka T, Petrák O, Strauch B, Rosa J, et al. Left ventricle remodeling in men with moderate to severe volume-dependent hypertension. J Renin Angiotensin Aldosterone Syst. (2012) 13:426–34. doi: 10.1177/1470320312446240
99. Milan A, Magnino C, Fabbri A, Chiarlo M, Bruno G, Losano I, et al. Left heart morphology and function in primary aldosteronism. High Blood Press Cardiovasc Prev. (2012) 19:11–7. doi: 10.2165/11593690-000000000-00000
100. Muiesan ML, Salvetti M, Paini A, Agabiti-Rosei C, Monteduro C, Galbassini G, et al. Inappropriate left ventricular mass in patients with primary aldosteronism. Hypertension (Dallas, Tex.: 1979). (2008) 52:529–34. doi: 10.1161/HYPERTENSIONAHA.108.114140
101. Rossi GP, Cesari M, Cuspidi C, Maiolino G, Cicala MV, Bisogni V, et al. Long-term control of arterial hypertension and regression of left ventricular hypertrophy with treatment of primary aldosteronism. Hypertension (Dallas, Tex.: 1979). (2013) 62:62–9. doi: 10.1161/HYPERTENSIONAHA.113.01316
102. Chang YY, Liao CW, Tsai CH, Chen CW, Pan CT, Chen ZW, et al. Left Ventricular Dysfunction in Patients With Primary Aldosteronism: A Propensity Score-Matching Follow-Up Study With Tissue Doppler Imaging. J Am Heart Assoc. (2019) 8:e013263. doi: 10.1161/JAHA.119.013263
103. Stowasser M, Sharman J, Leano R, Gordon RD, Ward G, Cowley D, et al. Evidence for abnormal left ventricular structure and function in normotensive individuals with familial hyperaldosteronism type I. J Clin Endocrinol Metab. (2005) 90:5070–6. doi: 10.1210/jc.2005-0681
104. Yang Y, Zhu LM, Xu JZ, Tang XF, Gao PJ. Comparison of left ventricular structure and function in primary aldosteronism and essential hypertension by echocardiography. Hypertens Res. (2017) 40:243–50. doi: 10.1038/hr.2016.127
105. Aune A, Kokorina M, Grytaas MA, Midtbø H, Løvås K, Gerdts E. Preclinical cardiac disease in women and men with primary aldosteronism. Blood Press. (2021) 1–7. doi: 10.1080/08037051.2021.1904775
106. Chen YL, Xu TY, Xu JZ, Zhu LM, Li Y, Wang JG. A speckle tracking echocardiographic study on right ventricular function in primary aldosteronism. J Hypertens. (2020) 38:2261–9. doi: 10.1097/HJH.0000000000002527
107. Mihailidou AS, Loan Le, TY Mardini M, Funder JW. Glucocorticoids activate cardiac mineralocorticoid receptors during experimental myocardial infarction. Hypertension (Dallas, Tex.: 1979). (2009) 54:1306–12. doi: 10.1161/HYPERTENSIONAHA.109.136242
108. Silvestre JS, Heymes C, Oubénaïssa A, Robert V, Aupetit-Faisant B, Carayon A, et al. Activation of cardiac aldosterone production in rat myocardial infarction: effect of angiotensin II receptor blockade and role in cardiac fibrosis. Circulation. (1999) 99:2694–701. doi: 10.1161/01.CIR.99.20.2694
109. Rebsamen MC, Perrier E, Gerber-Wicht C, Benitah JP, Lang U. Direct and indirect effects of aldosterone on cyclooxygenase-2 and interleukin-6 expression in rat cardiac cells in culture and after myocardial infarction. Endocrinology. (2004) 145:3135–42. doi: 10.1210/en.2003-1544
110. Rafatian N, Westcott KV, White RA, Leenen FH. Cardiac macrophages and apoptosis after myocardial infarction: effects of central MR blockade. Am J Physiol Regul Integr Comp Physiol. (2014) 307:R879–87. doi: 10.1152/ajpregu.00075.2014
111. Monticone S, Burrello J, Tizzani D, Bertello C, Viola A, Buffolo F, et al. Prevalence and Clinical Manifestations of Primary Aldosteronism Encountered in Primary Care Practice. J Am Coll Cardiol. (2017) 69:1811–20. doi: 10.1016/j.jacc.2017.01.052
112. Huang WC, Chen YY, Lin YH, Chen L, Lin PC, Lin YF, et al. Incidental Congestive Heart Failure in Patients With Aldosterone-Producing Adenomas. J Am Heart Assoc. (2019) 8:e012410. doi: 10.1161/JAHA.119.012410
113. Seccia TM, Caroccia B, Maiolino G, Cesari M, Rossi GP. Arterial hypertension, aldosterone, atrial fibrillation. Curr Hypertens Rep. (2019) 21:94. doi: 10.1007/s11906-019-1001-4
114. Reil JC, Hohl M, Selejan S, Lipp P, Drautz F, Kazakow A, et al. Aldosterone promotes atrial fibrillation. Eur Heart J. (2012) 33:2098–108. doi: 10.1093/eurheartj/ehr266
115. Lammers C, Dartsch T, Brandt MC, Rottländer D, Halbach M, Peinkofer G, et al. Spironolactone prevents aldosterone induced increased duration of atrial fibrillation in rat. Cell Physiol Biochem. (2012) 29:833–40. doi: 10.1159/000178483
116. Lu G, Li J, Zhai Y, Li Q, Xie D, Zhang J, et al. Spironolactone suppresses aldosterone-induced Kv1.5 expression by attenuating mineralocorticoid receptor-Nox1/2/4-mediated ROS generation in neonatal rat atrial myocytes. Biochem Biophys Res Commun. (2019) 520:379–84. doi: 10.1016/j.bbrc.2019.10.039
117. Seccia TM, Letizia C, Muiesan ML, Lerco S, Cesari M, Bisogni V, et al. Atrial fibrillation as presenting sign of primary aldosteronism: results of the Prospective Appraisal on the Prevalence of Primary Aldosteronism in Hypertensive (PAPPHY) Study. J Hypertens. (2020) 38:332–9. doi: 10.1097/HJH.0000000000002250
118. Pan CT, Tsai CH, Chen ZW, Chang YY, Wu VC, Hung CS, et al. Atrial Fibrillation in Primary Aldosteronism. Horm Metab Res. (2020) 52:357–65. doi: 10.1055/a-1141-5989
119. Kim KJ, Hong N, Yu MH, Lee H, Lee S, Lim JS, et al. Time-Dependent Risk of Atrial Fibrillation in Patients With Primary Aldosteronism After Medical or Surgical Treatment Initiation. Hypertension (Dallas, Tex.: 1979). (2021) 77:1964–73. doi: 10.1161/HYPERTENSIONAHA.120.16909
120. Hundemer GL, Curhan GC, Yozamp N, Wang M, Vaidya A. Cardiometabolic outcomes and mortality in medically treated primary aldosteronism: a retrospective cohort study. Lancet Diabetes Endocrinol. (2018) 6:51–9. doi: 10.1016/S2213-8587(17)30367-4
121. Shao D, Wang S, Zhou S, Cai Q, Zhang R, Li H, et al. Aldosteronism with mild hypokalemia presenting as life-threatening ventricular arrhythmias: A case report. Medicine (Baltimore). (2018) 97:e13608. doi: 10.1097/MD.0000000000013608
122. Gros R, Ding Q, Liu B, Chorazyczewski J, Feldman RD. Aldosterone mediates its rapid effects in vascular endothelial cells through GPER activation. Am J Physiol Cell Physiol. (2013) 304:C532–40. doi: 10.1152/ajpcell.00203.2012
123. Wang Y, Fan Y, Song Y, Han X, Fu M, Wang J, et al. Angiotensin II induces apoptosis of cardiac microvascular endothelial cells via regulating PTP1B/PI3K/Akt pathway. In vitro cellular & developmental biology. Animal. (2019) 55:801–11. doi: 10.1007/s11626-019-00395-8
124. Holaj R, Zelinka T, Wichterle D, Petrák O, Strauch B, Widimský J. Jr. Increased intima-media thickness of the common carotid artery in primary aldosteronism in comparison with essential hypertension. J Hypertens. (2007) 25:1451–7. doi: 10.1097/HJH.0b013e3281268532
125. Briet M, Schiffrin EL. Vascular actions of aldosterone. J Vasc Res. (2013) 50:89–99. doi: 10.1159/000345243
126. Mulatero P, Monticone S, Veglio Diagnosis F, treatment of primary aldosteronism. Rev Endocr Metab Disord. (2011) 12:3–9. doi: 10.1007/s11154-011-9156-6
127. Lin YH, Lin LY, Chen A, Wu XM, Lee JK, Su TC, et al. Adrenalectomy improves increased carotid intima-media thickness and arterial stiffness in patients with aldosterone producing adenoma. Atherosclerosis. (2012) 221:154–9. doi: 10.1016/j.atherosclerosis.2011.12.003
128. Touboul PJ, Labreuche J, Vicaut E, Amarenco P. Carotid intima-media thickness, plaques, and Framingham risk score as independent determinants of stroke risk. Stroke. (2005) 36:1741–5. doi: 10.1161/01.STR.0000174490.23495.57
129. Chen ZW, Tsai CH, Pan CT, Chou CH, Liao CW, Hung CS, et al. Endothelial dysfunction in primary aldosteronism. Int J Mol Sci. (2019) 20. doi: 10.3390/ijms20205214
130. Wu VC, Lo SC, Chen YL, Huang PH, Tsai CT, Liang CJ, et al. Endothelial progenitor cells in primary aldosteronism: a biomarker of severity for aldosterone vasculopathy and prognosis. J Clin Endocrinol Metab. (2011) 96:3175–83. doi: 10.1210/jc.2011-1135
131. Feigin VL, Lawes CM, Bennett DA, Anderson CS. Stroke epidemiology: a review of population-based studies of incidence, prevalence, and case-fatality in the late 20th century. Lancet Neurol. (2003) 2:43–53. doi: 10.1016/S1474-4422(03)00266-7
132. Rizzoni D, Paiardi S, Rodella L, Porteri E, De Ciuceis C, Rezzani R, et al. Changes in extracellular matrix in subcutaneous small resistance arteries of patients with primary aldosteronism. J Clin Endocrinol Metab. (2006) 91:2638–42. doi: 10.1210/jc.2006-0101
133. Petramala L, Concistr è A, Circosta F, Gigante A, Sarlo F, Schina M, et al. Evaluation of Intra-Renal Stiffness in Patients with Primary Aldosteronism. High Blood Press Cardiovasc Prev. (2021) doi: 10.1007/s40292-021-00485-3
134. Ogata H, Yamazaki Y, Tezuka Y, Gao X, Omata K, Ono Y, et al. Renal injuries in primary aldosteronism: quantitative histopathological analysis of 19 patients with primary adosteronism. Hypertension (Dallas, Tex.: 1979). 78:411–21. (2021) doi: 10.1161/HYPERTENSIONAHA.121.17436
135. Nakamura Y, Kobayashi H, Tanaka S, Hatanaka Y, Fuke Y, Fukuda N, et al. Primary aldosteronism and obstructive sleep apnea: A single-center cross-sectional study of the Japanese population. Medicine. (2021) 100:e25049. doi: 10.1097/MD.0000000000025049
136. Nakamura Y, Kobayashi H, Tanaka S, Hatanaka Y, Fukuda N, Abe M. Association between plasma aldosterone and markers of tubular and glomerular damage in primary aldosteronism. Clin Endocrinol. (2021) 94:920–6. doi: 10.1111/cen.14434
137. Fox CS, Larson MG, Hwang SJ, Leip EP, Rifai N, Levy D, et al. Cross-sectional relations of serum aldosterone and urine sodium excretion to urinary albumin excretion in a community-based sample. Kidney Int. (2006) 69:2064–9. doi: 10.1038/sj.ki.5000378
138. Sechi LA, Colussi G, Di Fabio A, Catena C. Cardiovascular and renal damage in primary aldosteronism: outcomes after treatment. Am J Hypertens. (2010) 23:1253–60. doi: 10.1038/ajh.2010.169
139. Sechi LA, Novello M, Lapenna R, Baroselli S, Nadalini E, Colussi GL, et al. Long-term renal outcomes in patients with primary aldosteronism. JAMA. (2006) 295:2638–45. doi: 10.1001/jama.295.22.2638
140. Zhang SL, Gao JW, Guo Y, Feng QL, Tang JY, Yan L, et al. (2020). Associations between metabolic profiles and target-organ damage in chinese individuals with primary aldosteronism. Front Endocrinol. 11:547356. doi: 10.3389/fendo.2020.547356
141. Catena C, Colussi G, Sechi LA. Mineralocorticoid receptor antagonists and renal involvement in primary aldosteronism: opening of a new era. Eur J Endocrinol. (2013) 168:C1–5. doi: 10.1530/EJE-12-0899
142. Rossi GP, Bernini G, Desideri G, Fabris B, Ferri C, Giacchetti G, et al. Renal damage in primary aldosteronism: results of the PAPY study. Hypertension (Dallas, Tex.: 1979). (2006) 48:232–8. doi: 10.1161/01.HYP.0000230444.01215.6a
143. Kawashima A, Sone M, Inagaki N, Takeda Y, Itoh H, Kurihara I, et al. Renal impairment is closely associated with plasma aldosterone concentration in patients with primary aldosteronism. Eur J Endocrinol. (2019) 181:339–50. doi: 10.1530/EJE-19-0047
144. Reincke M, Rump LC, Quinkler M, Hahner S, Diederich S, Lorenz R, et al. Risk factors associated with a low glomerular filtration rate in primary aldosteronism. J Clin Endocrinol Metab. (2009) 94:869–75. doi: 10.1210/jc.2008-1851
145. Monticone S, Sconfienza E, D'Ascenzo F, Buffolo F, Satoh F, Sechi LA, et al. Renal damage in primary aldosteronism: a systematic review and meta-analysis. J Hypertens. (2020) 38:3–12. doi: 10.1097/HJH.0000000000002216
146. Fernández-Argüeso M, Pascual-Corrales E, Bengoa Rojano N, García Cano A, Jiménez Mendiguchía L, Araujo-Castro M. Higher risk of chronic kidney disease and progressive kidney function impairment in primary aldosteronism than in essential hypertension. Case-control study. Endocrine. 73:439–46 (2021) doi: 10.1007/s12020-021-02704-2
147. Kuo CC, Wu VC, Tsai CW, Wu KD. Relative kidney hyperfiltration in primary aldosteronism: a meta-analysis. J Renin Angiotensin Aldosterone Syst. (2011) 12:113–22. doi: 10.1177/1470320310391331
148. Sechi LA, Di Fabio A, Bazzocchi M, Uzzau A, Catena C. Intrarenal hemodynamics in primary aldosteronism before and after treatment. J Clin Endocrinol Metab. (2009) 94:1191–7. doi: 10.1210/jc.2008-2245
149. Palatini P, Mormino P, Dorigatti F, Santonastaso M, Mos L, De Toni R, et al. Glomerular hyperfiltration predicts the development of microalbuminuria in stage 1 hypertension: the HARVEST. Kidney Int. (2006) 70:578–84. doi: 10.1038/sj.ki.5001603
150. Pontremoli R, Leoncini G, Ravera M, Viazzi F, Vettoretti S, Ratto E, et al. Microalbuminuria, cardiovascular, and renal risk in primary hypertension. J Am Soc Nephrol. (2002) 13 Suppl 3:S169–72. doi: 10.1097/01.ASN.0000032601.86590.F7
151. Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab. (2004) 89:2548–56. doi: 10.1210/jc.2004-0395
152. Ronconi V, Turchi F, Appolloni G, di Tizio V, Boscaro M, Giacchetti G. Aldosterone, mineralocorticoid receptor and the metabolic syndrome: role of the mineralocorticoid receptor antagonists. Curr Vasc Pharmacol. (2012) 10:238–46. doi: 10.2174/157016112799304969
153. Vettor R, Milan G, Rossato M, Federspil G. Review article: adipocytokines and insulin resistance. Aliment Pharmacol Ther. (2005) 22 Suppl 2:3–10. doi: 10.1111/j.1365-2036.2005.02587.x
154. Iacobellis G, Petramala L, Cotesta D, Pergolini M, Zinnamosca L, Cianci R, et al. Adipokines and cardiometabolic profile in primary hyperaldosteronism. J Clin Endocrinol Metab. (2010) 95:2391–8. doi: 10.1210/jc.2009-2204
155. Wu C, Zhang H, Zhang J, Xie C, Fan C, Zhang H, et al. Inflammation and fibrosis in perirenal adipose tissue of patients with aldosterone-producing Adenoma. Endocrinology. (2018) 159:227–37. doi: 10.1210/en.2017-00651
156. Rondinone CM, Rodbard D, Baker ME. Aldosterone stimulated differentiation of mouse 3T3-L1 cells into adipocytes. Endocrinology. (1993) 132:2421–6. doi: 10.1210/endo.132.6.8504747
157. Jiang H, Ye XP, Yang ZY, Zhan M, Wang HN, Cao HM, et al. Aldosterone directly affects apelin expression and secretion in adipocytes. J Mol Endocrinol. (2013) 51:37–48. doi: 10.1530/JME-13-0025
158. Shibayama Y, Wada N, Baba S, Miyano Y, Obara S, Iwasaki R, et al. Relationship between visceral fat and plasma aldosterone concentration in patients with primary aldosteronism. J Endocr Soc. (2018) 2:1236–45. doi: 10.1210/js.2018-00187
159. Williams TA, Monticone S, Urbanet R, Bertello C, Giraudo G, Vettor R, et al. Genes implicated in insulin resistance are down-regulated in primary aldosteronism patients. Mol Cell Endocrinol. (2012) 355:162–8. doi: 10.1016/j.mce.2012.02.007
160. Corry DB, Tuck ML. The effect of aldosterone on glucose metabolism. Curr Hypertens Rep. (2003) 5:106–109. doi: 10.1007/s11906-003-0065-2
161. Giacchetti G, Sechi LA, Rilli S, Carey RM. The renin-angiotensin-aldosterone system, glucose metabolism and diabetes. Trends Endocrinol Metab. (2005) 16:120–6. doi: 10.1016/j.tem.2005.02.003
162. Urbanet R, Pilon C, Calcagno A, Peschechera A, Hubert EL, Giacchetti G, et al. Analysis of insulin sensitivity in adipose tissue of patients with primary aldosteronism. J Clin Endocrinol Metab. (2010) 95:4037–42. doi: 10.1210/jc.2010-0097
163. Jadhav A, Ndisang JF. Treatment with heme arginate alleviates adipose tissue inflammation and improves insulin sensitivity and glucose metabolism in a rat model of Human primary aldosteronism. Free Radic Biol Med. (2012) 53:2277–86. doi: 10.1016/j.freeradbiomed.2012.10.529
164. Fallo F, Della Mea P, Sonino N, Bertello C, Ermani M, Vettor R, et al. Adiponectin and insulin sensitivity in primary aldosteronism. Am J Hypertens. (2007) 20:855–61. doi: 10.1016/j.amjhyper.2007.03.012
165. Letizia C, Petramala L, Di Gioia CR, Chiappetta C, Zinnamosca L, Marinelli C, et al. Leptin and adiponectin mRNA expression from the adipose tissue surrounding the adrenal neoplasia. J Clin Endocrinol Metab. (2015) 100:E101–4. doi: 10.1210/jc.2014-2274
166. Er LK, Lin MC, Tsai YC, Hsiao JK, Yang CY, Chang CC, et al. Association of visceral adiposity and clinical outcome among patients with aldosterone producing adenoma. BMJ Open Diabetes Res Care. (2020) 8. doi: 10.1136/bmjdrc-2019-001153
167. Saiki A, Otsuki M, Tamada D, Kitamura T, Shimomura I, Kurihara I, et al. Diabetes mellitus itself increases cardio-cerebrovascular risk and renal complications in primary aldosteronism. J Clin Endocrinol Metab. (2020) 105. doi: 10.1210/clinem/dgaa177
168. Kumagai E, Adachi H, Jacobs D. R. Jr., Hirai Y, Enomoto M, Fukami A, et al. Plasma aldosterone levels and development of insulin resistance: prospective study in a general population. Hypertension (Dallas, Tex.: 1979). (2011) 58:1043–8. doi: 10.1161/HYPERTENSIONAHA.111.180521
169. Ahn CH, Na HY, Park SY, Yu HW, Kim SJ, Choi JY, et al. Expression of CYP11B1 and CYP11B2 in adrenal adenoma correlates with clinical characteristics of primary aldosteronism. Clini Endocrinol. (2021) 96:30–9. doi: 10.1111/cen.14628
170. Gerards J, Heinrich DA, Adolf C, Meisinger C, Rathmann W, Sturm L, et al. Impaired glucose metabolism in primary aldosteronism is associated with cortisol cosecretion. J Clin Endocrinol Metab. (2019) 104:3192–202. doi: 10.1210/jc.2019-00299
171. Gendreitzig P, Künzel HE, Adolf C, Handgriff L, Müller L, Holler F, et al. Autonomous cortisol secretion influences psychopathological symptoms in patients with primary aldosteronism. J Clin Endocrinol Metab. (2021) 106:e2423–33. doi: 10.1210/clinem/dgab099
172. Peng KY, Liao HW, Chan CK, Lin WC, Yang SY, Tsai YC, et al. Presence of subclinical hypercortisolism in clinical aldosterone-producing adenomas predicts lower clinical success. Hypertension (Dallas, Tex.: 1979). (2020) 76:1537–44. doi: 10.1161/HYPERTENSIONAHA.120.15328
173. Adler GK, Murray GR, Turcu AF, Nian H, Yu C, Solorzano CC, et al. Primary Aldosteronism Decreases Insulin Secretion and Increases Insulin Clearance in Humans. Hypertension (Dallas, Tex.: 1979). (2020) 75:1251–9. doi: 10.1161/HYPERTENSIONAHA.119.13922
175. Hitomi H, Kiyomoto H, Nishiyama A, Hara T, Moriwaki K, Kaifu K, et al. Aldosterone suppresses insulin signaling via the downregulation of insulin receptor substrate-1 in vascular smooth muscle cells. Hypertension (Dallas, Tex.: 1979). (2007) 50:750–5. doi: 10.1161/HYPERTENSIONAHA.107.093955
176. Selvaraj J, Sathish S, Mayilvanan C, Balasubramanian K. Excess aldosterone-induced changes in insulin signaling molecules and glucose oxidation in gastrocnemius muscle of adult male rat. Mol Cell Biochem. (2013) 372:113–26. doi: 10.1007/s11010-012-1452-2
177. Wada T, Ohshima S, Fujisawa E, Koya D, Tsuneki H, Sasaoka T. Aldosterone inhibits insulin-induced glucose uptake by degradation of insulin receptor substrate (IRS) 1 and IRS2 via a reactive oxygen species-mediated pathway in 3T3-L1 adipocytes. Endocrinology. (2009) 150:1662–9. doi: 10.1210/en.2008-1018
178. Chen W, Li F, He C, Zhu Y, Tan W. Elevated prevalence of abnormal glucose metabolism in patients with primary aldosteronism: a meta-analysis. Ir J Med Sci. (2014) 183:283–91. doi: 10.1007/s11845-013-1007-x
179. Pedrosa RP, Drager LF, Gonzaga CC, Sousa MG, de Paula LK, Amaro AC, et al. Obstructive sleep apnea: the most common secondary cause of hypertension associated with resistant hypertension. Hypertension (Dallas, Tex.: 1979). (2011) 58:811–7. doi: 10.1161/HYPERTENSIONAHA.111.179788
180. Di Murro A, Petramala L, Cotesta D, Zinnamosca L, Crescenzi E, Marinelli C, et al. Renin-angiotensin-aldosterone system in patients with sleep apnoea: prevalence of primary aldosteronism. J Renin Angiotensin Aldosterone Syst. (2010) 11:165–72. doi: 10.1177/1470320310366581
181. Wolf J, Narkiewicz K. Primary Aldosteronism and Obstructive Sleep Apnea: A Cross-Sectional Multi-Ethnic Study. Hypertension (Dallas, Tex.: 1979). (2019) 74:1305–1306. doi: 10.1161/HYPERTENSIONAHA.119.13935
182. Dobrowolski P, Kołodziejczyk-Kruk S, Warchoł-Celińska E, Kabat M, Ambroziak U, Wróbel A, et al. Primary aldosteronism is highly prevalent in patients with hypertension and moderate to severe obstructive sleep apnea. J Clin Sleep Med. (2021) 17:629–37. doi: 10.5664/jcsm.8960
183. Funder JW, Carey RM, Mantero F, Murad MH, Reincke M, Shibata H, et al. The Management of Primary Aldosteronism: Case Detection, Diagnosis, and Treatment: An Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. (2016) 101:1889–916. doi: 10.1210/jc.2015-4061
184. Chomsky-Higgins Menut K, Pearlstein SS, Conroy PC, Roman SA, Shen WT, Gosnell J, et al. Screening for primary aldosteronism in the hypertensive obstructive sleep apnea population is cost-saving. Surgery. (2021) 171:96–103. doi: 10.1016/j.surg.2021.05.052
185. Pratt-Ubunama MN, Nishizaka MK, Boedefeld RL, Cofield SS, Harding SM, Calhoun DA. Plasma aldosterone is related to severity of obstructive sleep apnea in subjects with resistant hypertension. Chest. (2007) 131:453–9. doi: 10.1378/chest.06-1442
186. Gaddam K, Pimenta E, Thomas SJ, Cofield SS, Oparil S, Harding SM, et al. Spironolactone reduces severity of obstructive sleep apnoea in patients with resistant hypertension: a preliminary report. J Hum Hypertens. (2010) 24:532–7. doi: 10.1038/jhh.2009.96
187. Wolley MJ, Pimenta E, Calhoun D, Gordon RD, Cowley D, Stowasser M. Treatment of primary aldosteronism is associated with a reduction in the severity of obstructive sleep apnoea. J Hum Hypertens. (2017) 31:561–7. doi: 10.1038/jhh.2017.28
188. Huang KH, Yu CC, Hu YH, Chang CC, Chan CK, Liao SC, et al. Targeted treatment of primary aldosteronism - The consensus of Taiwan Society of Aldosteronism. J Formos Med Assoc. (2019) 118:72–82. doi: 10.1016/j.jfma.2018.01.006
189. Rossi GP, Bolognesi M, Rizzoni D, Seccia TM, Piva A, Porteri E, et al. Vascular remodeling and duration of hypertension predict outcome of adrenalectomy in primary aldosteronism patients. Hypertension (Dallas, Tex.: 1979). (2008) 51:1366–71. doi: 10.1161/HYPERTENSIONAHA.108.111369
190. Velema M, Dekkers T, Hermus A, Timmers H, Lenders J, Groenewoud H, et al. Quality of Life in Primary Aldosteronism: A Comparative Effectiveness Study of Adrenalectomy and Medical Treatment. J Clin Endocrinol Metab. (2018) 103:16–24. doi: 10.1210/jc.2017-01442
191. Chang YH, Chung SD, Wu CH, Chueh JS, Chen L, Lin PC, et al. Surgery decreases the long-term incident stroke risk in patients with primary aldosteronism. Surgery. (2020) 167:367–77. doi: 10.1016/j.surg.2019.08.017
192. Rossi GP, Maiolino G, Flego A, Belfiore A, Bernini G, Fabris B, et al. Adrenalectomy lowers incident atrial fibrillation in primary aldosteronism patients at long term. Hypertension (Dallas, Tex.: 1979). (2018) 71:585–91. doi: 10.1161/HYPERTENSIONAHA.117.10596
193. Williams TA, Lenders JWM, Mulatero P, Burrello J, Rottenkolber M, Adolf C, et al. Outcomes after adrenalectomy for unilateral primary aldosteronism: an international consensus on outcome measures and analysis of remission rates in an international cohort. Lancet Diabetes Endocrinol. (2017) 5:689–99. doi: 10.1016/S2213-8587(17)30135-3
194. Wachtel H, Fraker DL. Therapeutic outcomes with surgical and medical management of primary aldosteronism. Curr Cardiol Rep. (2021) 23:89. doi: 10.1007/s11886-021-01516-0
195. Reincke M, Fischer E, Gerum S, Merkle K, Schulz S, Pallauf A, et al. Observational study mortality in treated primary aldosteronism: the German Conn's registry. Hypertension (Dallas, Tex.: 1979). (2012) 60:618–24. doi: 10.1161/HYPERTENSIONAHA.112.197111
196. Catena C, Colussi G, Lapenna R, Nadalini E, Chiuch A, Gianfagna P, et al. Long-term cardiac effects of adrenalectomy or mineralocorticoid antagonists in patients with primary aldosteronism. Hypertension (Dallas, Tex.: 1979). (2007) 50:911–8. doi: 10.1161/HYPERTENSIONAHA.107.095448
197. Cuspidi C, Tadic M, Sala C, Quarti-Trevano F, Gherbesi E, Mancia G, et al. Regression of left ventricular hypertrophy in primary aldosteronism after adrenalectomy: a meta-analysis of echocardiographic studies. J Hypertens. (2021) 39:775–83. doi: 10.1097/HJH.0000000000002679
198. Pan CT, Liao CW, Tsai CH, Chen ZW, Chen L, Hung CS, et al. Influence of Different Treatment Strategies on New-Onset Atrial Fibrillation Among Patients With Primary Aldosteronism: A Nationwide Longitudinal Cohort-Based Study. J Am Heart Assoc. (2020) 9:e013699. doi: 10.1161/JAHA.119.013699
199. Puar TH, Loh LM, Loh WJ, Lim DST, Zhang M, Tan PT, et al. Outcomes in unilateral primary aldosteronism after surgical or medical therapy. Clin Endocrinol. (2021) 94:158–67. doi: 10.1111/cen.14351
200. Hundemer GL, Curhan GC, Yozamp N, Wang M, Vaidya A. Incidence of Atrial Fibrillation and Mineralocorticoid Receptor Activity in Patients With Medically and Surgically Treated Primary Aldosteronism. JAMA Cardiol. (2018) 3:768–74. doi: 10.1001/jamacardio.2018.2003
201. Billmann F, Billeter A, Thomusch O, Keck T, El Shishtawi S, Langan EA, et al. Minimally invasive partial versus total adrenalectomy for unilateral primary hyperaldosteronism-a retrospective, multicenter matched-pair analysis using the new international consensus on outcome measures. Surgery. (2021) 169:1361–70. doi: 10.1016/j.surg.2020.09.005
202. Katabami T, Fukuda H, Tsukiyama H, Tanaka Y, Takeda Y, Kurihara I, et al. Clinical and biochemical outcomes after adrenalectomy and medical treatment in patients with unilateral primary aldosteronism. J Hypertens. (2019) 37:1513–20. doi: 10.1097/HJH.0000000000002070
203. Hundemer GL, Curhan GC, Yozamp N, Wang M, Vaidya A. Renal Outcomes in Medically and Surgically Treated Primary Aldosteronism. Hypertension (Dallas, Tex.: 1979). (2018) 72:658–66. doi: 10.1161/HYPERTENSIONAHA.118.11568
204. Park KS, Kim JH, Yang YS, Hong AR, Lee DH, Moon MK, et al. Outcomes analysis of surgical and medical treatments for patients with primary aldosteronism. Endocrine J. (2017) 64, 623–32. doi: 10.1507/endocrj.EJ16-0530
205. Tsai CH, Chen YL, Pan CT, Lin YT, Lee PC, Chiu YW, et al. New-onset atrial fibrillation in patients with primary aldosteronism receiving different treatment strategies: systematic review and pooled analysis of three studies. Front Endocrinol. (2021) 12:646933. doi: 10.3389/fendo.2021.646933
206. Young W. F. Jr. Diagnosis and treatment of primary aldosteronism: practical clinical perspectives. J Intern Med. (2019) 285:126–48. doi: 10.1111/joim.12831
207. Ribstein J, Du Cailar G, Fesler P, Mimran A. Relative glomerular hyperfiltration in primary aldosteronism. J Am Soc Nephrol. (2005) 16:1320–5. doi: 10.1681/ASN.2004100878
208. Huang WC, Chen YY, Lin YH, Chueh JS. Composite Cardiovascular Outcomes in Patients With Primary Aldosteronism Undergoing Medical Versus Surgical Treatment: A Meta-Analysis. Front Endocrinol (Lausanne). (2021) 12:644260. doi: 10.3389/fendo.2021.644260
209. Kim IY, Park IS, Kim MJ, Han M, Rhee H, Seong EY, et al. Change in kidney function after unilateral adrenalectomy in patients with primary aldosteronism: identification of risk factors for decreased kidney function. Int Urol Nephrol. (2018) 50:1887–95. doi: 10.1007/s11255-018-1887-9
210. Onohara T, Takagi T, Yoshida K, Iizuka J, Okumi M, Kondo T, et al. Assessment of postoperative renal function after adrenalectomy in patients with primary aldosteronism. Int J Urol. (2019) 26:229–33. doi: 10.1111/iju.13844
211. Lee JY, Kim H, Kim HW, Ryu GW, Nam Y, Lee S, et al. Incidence of acute kidney injury after adrenalectomy in patients with primary aldosteronism. Electrolyte Blood Press. (2019) 17:45–53. doi: 10.5049/EBP.2019.17.2.45
212. Chan CK, Yang WS, Lin YH, Huang KH, Lu CC, Hu YH, et al. Arterial stiffness is associated with clinical outcome and cardiorenal injury in lateralized primary aldosteronism. J Clin Endocrinol Metab. (2020) 105, doi: 10.1210/clinem/dgaa566
213. Strauch B, Petrák O, Zelinka T, Wichterle D, Holaj R, Kasalický M, et al. Adrenalectomy improves arterial stiffness in primary aldosteronism. Am J Hypertens. (2008) 21:1086–92. doi: 10.1038/ajh.2008.243
214. Sukor N, Kogovsek C, Gordon RD, Robson D, Stowasser M. Improved quality of life, blood pressure, and biochemical status following laparoscopic adrenalectomy for unilateral primary aldosteronism. J Clin Endocrinol Metab. (2010) 95:1360–4. doi: 10.1210/jc.2009-1763
215. Velema MS, de Nooijer AH, Burgers VWG, Hermus A, Timmers H, Lenders JWM, et al. Health-related quality of life and mental health in primary aldosteronism: a systematic review. Horm Metab Res. (2017) 49:943–50. doi: 10.1055/s-0043-121706
216. Chiang WF, Cheng CJ, Wu ST, Sun GH, Lin MY, Sung CC, et al. Incidence and factors of post-adrenalectomy hyperkalemia in patients with aldosterone producing adenoma. Clin Chim Acta. (2013) 424:114–8. doi: 10.1016/j.cca.2013.05.017
217. Tahir A, McLaughlin K, Kline G. Severe hyperkalemia following adrenalectomy for aldosteronoma: prediction, pathogenesis and approach to clinical management- a case series. BMC Endocr Disord. (2016) 16:43. doi: 10.1186/s12902-016-0121-y
218. Jeunemaitre X, Chatellier G, Kreft-Jais C, Charru A, DeVries C, Plouin PF, et al. Efficacy and tolerance of spironolactone in essential hypertension. Am J Cardiol. (1987) 60:820–5. doi: 10.1016/0002-9149(87)91030-7
219. Rossi GP. Primary Aldosteronism: JACC State-of-the-Art Review. J Am Coll Cardiol. (2019) 74:2799–811. doi: 10.1016/j.jacc.2019.09.057
220. Rossi GP. A comprehensive review of the clinical aspects of primary aldosteronism. Nature reviews. Endocrinology. (2011) 7:485–95. doi: 10.1038/nrendo.2011.76
221. Barrera-Chimal J, Kolkhof P, Lima-Posada I, Joachim A, Rossignol P, Jaisser F. Differentiation between emerging non-steroidal and established steroidal mineralocorticoid receptor antagonists: head-to-head comparisons of pharmacological and clinical characteristics. Expert Opin Investig Drugs. (2021) 1–17. doi: 10.1080/13543784.2021.2002844
222. Funder J. Primary aldosteronism: Treatment of the disease, and new therapeutic approaches. Best Pract Res Clin Endocrinol Metab. (2020) 34:101368. doi: 10.1016/j.beem.2019.101368
223. Funder JW. Primary aldosteronism: present and future. Vitam Horm. (2019) 109:285–302. doi: 10.1016/bs.vh.2018.10.006
224. Romero DG, Yanes Cardozo LL. Clinical practice guideline for management of primary aldosteronism: what is new in the 2016 update? Int J Endocrinol Metab Disord. (2016) 2. doi: 10.16966/2380-548X.129
225. Gomez-Sanchez EP. Third-generation Mineralocorticoid Receptor Antagonists: Why Do We Need a Fourth? J Cardiovasc Pharmacol. (2016) 67:26–38. doi: 10.1097/FJC.0000000000000329
226. Kolkhof P, Nowack C, Eitner F. Nonsteroidal antagonists of the mineralocorticoid receptor. Curr Opin Nephrol Hypertens. (2015) 24:417–24. doi: 10.1097/MNH.0000000000000147
227. Liu LC, Schutte E, Gansevoort RT, van der Meer P, Voors AA. Finerenone: third-generation mineralocorticoid receptor antagonist for the treatment of heart failure and diabetic kidney disease. Expert Opin Investig Drugs. (2015) 24:1123–35. doi: 10.1517/13543784.2015.1059819
Keywords: primary aldosteronism, aldosterone, cerebro-cardiovascular risk, target organ damage, mechanism, treatment
Citation: Lin X, Ullah MHE, Wu X, Xu F, Shan S-K, Lei L-M, Yuan L-Q and Liu J (2022) Cerebro-Cardiovascular Risk, Target Organ Damage, and Treatment Outcomes in Primary Aldosteronism. Front. Cardiovasc. Med. 8:798364. doi: 10.3389/fcvm.2021.798364
Received: 26 October 2021; Accepted: 20 December 2021;
Published: 02 February 2022.
Edited by:
Giuseppe Maiolino, University Hospital of Padua, ItalyReviewed by:
Elise Peery Gomez-Sanchez, University of Mississippi Medical Center, United StatesLuigi Petramala, Sapienza University of Rome, Italy
Copyright © 2022 Lin, Ullah, Wu, Xu, Shan, Lei, Yuan and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jun Liu, anVubGl1MTIzQGNzdS5lZHUuY24=; Ling-Qing Yuan, YWxsZW55bHFAY3N1LmVkdS5jbg==