
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med., 06 January 2022
Sec. Cardiovascular Metabolism
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.788852
This article is part of the Research TopicMetabolism Linking Immunity and Inflammatory Phenotypes in Cardiovascular DiseaseView all 13 articles
 Preethi Basavaraju1Rubadevi Balasubramani1Divya Sri Kathiresan1Ilakkiyapavai Devaraj1Kavipriya Babu1Vasanthakumar Alagarsamy1
Preethi Basavaraju1Rubadevi Balasubramani1Divya Sri Kathiresan1Ilakkiyapavai Devaraj1Kavipriya Babu1Vasanthakumar Alagarsamy1 Vinayaga Moorthi Puthamohan2*
Vinayaga Moorthi Puthamohan2*Apolipoproteins (APO proteins) are the lipoprotein family proteins that play key roles in transporting lipoproteins all over the body. There are nearly more than twenty members reported in the APO protein family, among which the A, B, C, E, and L play major roles in contributing genetic risks to several disorders. Among these genetic risks, the single nucleotide polymorphisms (SNPs), involving the variation of single nucleotide base pairs, and their contributing polymorphisms play crucial roles in the apolipoprotein family and its concordant disease heterogeneity that have predominantly recurred through the years. In this review, we have contributed a handful of information on such genetic polymorphisms that include APOE, ApoA1/B ratio, and A1/C3/A4/A5 gene cluster-based population genetic studies carried throughout the world, to elaborately discuss the effects of various genetic polymorphisms in imparting various medical conditions, such as obesity, cardiovascular, stroke, Alzheimer's disease, diabetes, vascular complications, and other associated risks.
Apolipoproteins (APO proteins) are the major class of proteins that play vital roles in the binding and transport of lipid molecules to form lipoproteins (1). There are six major classes of APO proteins: A, B, C, D, E, and H and F and G (2). The human genome encodes for nearly more than 21 APO genes, out of which APOA (APOA1, APOA2, APOA4, and APOA5), APOC (APOC1, APOC2, and APOC3), APOB, and APOE (APOE2, APOE3, and APOE4) contribute to coding proteins that regulate triglycerides (TGs) and cholesterol metabolism, transport, and management at cellular levels (3–5). The genetic variations studied in these coding genes have a wide range of contributions toward the medical conditions that provoke a phenomenon known as ‘multiple heterogeneities (6). The genetic studies allow us to unravel the aetiological heterogeneity of specific gene polymorphisms at a molecular level to understand its genotype variations and concordant risks associated with several disease conditions throughout the genome (7). In the present review, we have shed light on the major polymorphisms and mutations that are widely studied as genotypic expressions associated with the different APO genes, linked to a wide range of disease severities and risks (8).
The major genetic risks associated with the different APO proteins are listed in Table 1. Analysis of genetic variations predominantly involves the screening of single nucleotide polymorphisms (SNPs) residing along the upstream regions of the gene (9). The APO gene family polymorphisms are linked to several diseases, such as APOE genotypes with circulating lipid levels and coronary heart risks, cardiovascular disease (CVD), and with the ε4 homozygous genotypes showing increased risk of Alzheimer's disease (AD) (OMIM: 104310), APOA5 variants and the hypertriglyceridemia (OMIM: 145750), APOA polymorphisms with familial hypercholesterolemia (OMIM: 144010), APOB gene alterations, and ischemic heart disease (OMIM: 601367) increased risks (10–13).

Table 1. The different lipoproteins expressed and their major genotypic risks associated with concordant disease conditions.
Several studies on the genetic polymorphisms of the APO genes have been reported to date, but we do largely shortfall on the cumulative data that on the evident support of future understanding of molecular backgrounds and their contributions toward the aetiological heterogeneity of various diseases linked with APO gene family metabolism and expression. In the current review, we have gathered cumulative information on the various APO gene family-based genotype and polymorphism studies scattered into pieces on population-based criteria to review the backgrounds of the APO gene family polymorphisms and their risks for various clinical conditions all over the world.
The plasma lipoprotein segregates into seven classes, namely, the chylomicrons, chylomicron remnants, very low-density lipoprotein (VLDL), intermediate-density lipoprotein (IDL), low-density lipoprotein (LDL), high-density lipoprotein (HDL), and lipoprotein (a) [Lp (a)], among which the HDL levels are designated to be ~1.063–1.21 g/ml as the control parameter, and it mainly comprises of diverse groups of tiny spherical disc shaped particles classified based on their size, charge, density, electrophoretic mobility, and Apolipoprotein compositions (14). The dense composition of the APO proteins comprises the APOA1 that is primarily synthesized in the liver and small intestine (15), playing key roles in the signaling of reverse cholesterol transport mechanisms. Apart from the APOA1, the HDL family proteins comprise the ApoA-II, ApoA-IV, ApoA-V, ApoC-I, ApoC-II, ApoC-III, ApoD, ApoE, ApoJ, ApoL, and ApoM (16).
Apolipoprotein A4 (APOA4) is one of the first identified protein components of chylomicrons and HDL family (17, 18) whose physiological role remains unclear, but some of the recent studies have insisted their protective effects against the formation of diet-induced aortic lesions in mice models (19). APOA5 is the newly identified member of the apolipoprotein gene family by the comparison of human and mouse DNA sequences (20). It is expressed predominantly in the liver, playing crucial roles in maintaining the TG levels. The APOC3 is another protein component that belongs to the TG-rich lipoproteins and HDL family, which is mainly synthesized in the liver and intestine (21), playing major physiological roles in inhibiting lipoprotein lipases (LPLs).
These APO proteins ApoA-I, ApoA-IV, ApoA-V, and ApoC-III are coded by the APOA1 (OMIM: 107680), APOA4 (OMIM: 107690), APOA5 (OMIM: 606368), and APOC3 (OMIM: 107720) genes that collectively contribute to the A1/C3/A4/A5 complex which is an incomplete gene cluster with numerous interactions transcribing different factors functioning as immediate upstream regions of each adjacent genes located on the chromosome 11. This gene cluster contributes to ~38% of major genetic mutations that lead to plasma lipoprotein alternating levels (22). The cluster extends up to 17 Kb lengths, where the APOA1 and APOA4 genes are transcribed along the same direction, and the APOC3 gene is transcribed in the opposite direction (20) as shown in Figure 1. Almost at a distance of 30 kb upstream along this cluster is the APOA5 gene, transcribed through the same direction of APOA1 and APOA4 genes. The entire gene cluster covers ~60 kb, comprising of the strong Linkage disequilibrium and polymorphism sites at different loci (3). Three of the four genes belonging to this cluster contain polymorphisms within the upstream region of the gene and are described as the promoter region specific SNPs exhibiting strong associations with several lipid-based alternative variants (20).

Figure 1. The APOA1/C3/A4/A5 gene cluster residing on chromosome 11 spanning various polymorphisms.
The Apolipoprotein family encoding gene cluster and associated enzyme based phenotypic expressions are believed to be one of the underlying mechanisms for maintaining meticulous actions, such as antioxidant, anti-inflammatory, and anti-thrombotic effects (23). The plasma levels of Apo-B and Apo-C (III) are the key predictors of risk factors associated with coronary heart diseases (24), and whereas the ApoB levels alone are interlinked to the intermediate interactions involved in LDL and arterial wall pressure mechanisms, thereby infuriating the development of atherosclerosis conditions at increased levels (25, 26). Hence, the genetic mutations screened within the APOA1/C3/A4/A5 gene cluster are extensively examined in concordance to the biochemical alterations of serum lipid levels and cardiovascular risks (27–29). The major polymorphisms of the gene cluster span for seven major SNPs give rise to the haplotype alterations, as shown in Table 2 and Figure 2.
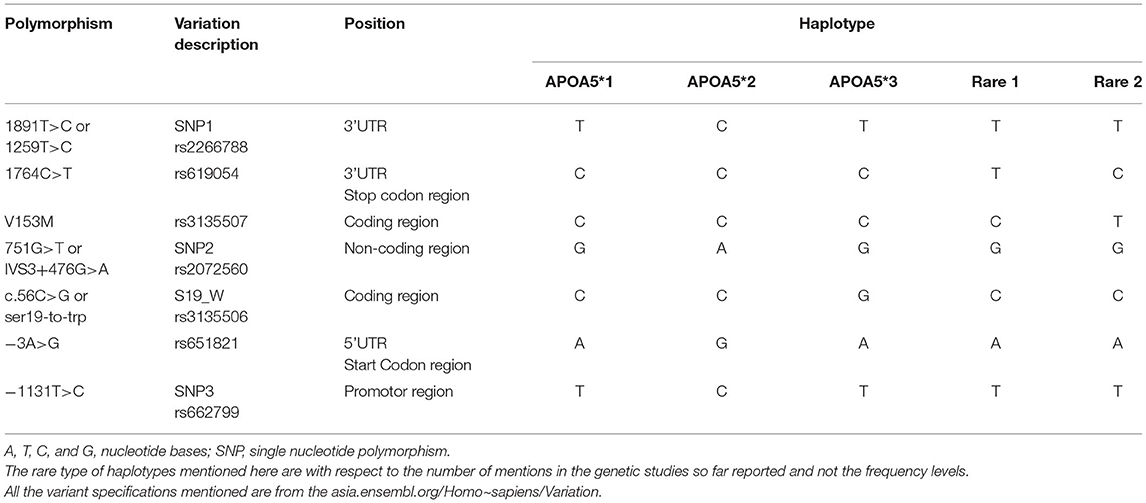
Table 2. The APOA5 gene polymorphisms that contribute to the nucleotide variations in the five major haplotypes.
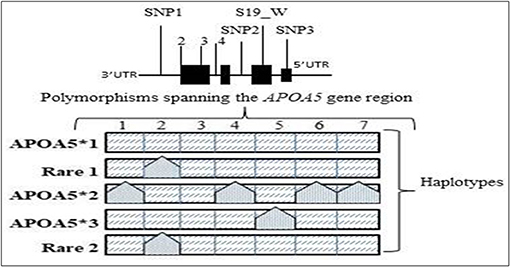
Figure 2. The APOA5 variants and common haplotypes: specific APOA5 polymorphisms found on the genetic interval contributing to the five major haplotypes seen in various populations worldwide.
The gene that codes for the APOA1 protein has APOA1 276 → G/A point mutation widely seen in the promoter region and is associated with altered postprandial lipid metabolism defects (30, 31). Apart from this major mutation, the SNPs APOA1−2630G/A, APOA1−2803G/A, and −3012A/G are seen in the promoter region and are associated with fasting lipid levels alterations (32).
Some mutations seen in the APOA5 gene, such as T-1131C and Ser19Trp, are linked to the altered fasting, and postprandial TG levels (33, 34), and this gene also contributes to three main haplotypes based on five polymorphisms by APOA5, APOA5, and APOA5: 1131T>C, c.3A>G, c.56C>G, IVS3+476G>A, and c.1259T>C, respectively. Several SNPs within APOC3 promoter regions have been studied for the association with altered TGs levels (35–38), out of which the three SNPs APOC3 rs5128 (SNP 41) Sst1, associated with altered fasting triglycerides, APOC3 rs2854117 (SNP38)−482C/T, associated with longevity and HDL levels (39). The APOC3 rs2854116 (SNP 39)−455T/C, is classified as type II and widely studied polymorphs strongly associated with the increased TG levels.
The APOA1/C3/A4/A5 gene cluster has been assumed to be the roots of various diseases that include TG and TC levels, among them a study reported on English families consisted of 84 known occurrences of familial combined hyperlipidaemia (FCH) subjects and their relative spouses totalling a sample size of ~200 and more were analyzed a span of eight SNPs (APOA5 58892C > T, APOA5 56C > G, APOA5 12238T > C, APOC3 386C >G, and APOA1 3031C > T) encountered within the gene cluster to show that a single haplotype of the APOA5 gene caused by a missense mutation was seen 3-folds higher in the FCH probands when compared with the relative spouses. Similarly, the secondary haplotypes of the APOC3 and APOA5 genes had relatively higher occurrences in the spouse subjects compared with that of the actual probands (40). Later a similar linkage and association study on 128 Dutch FCH families showed that the two independent alleles of APOA5 or APOC3 genes (APOA556C > G and APOC3 386C >G) belonging to the APOA1/C3/A4/A5 gene cluster were overexpressed in all subjects irrespective of the common haplotype of the APOA1/C3/A4/A5 gene cluster being expressed at a very low transmission level (41). These results have spotlighted the effects of APOA1/C3/A4/A5 gene cluster haplotypes that have stronger evidence over the plasma TG and LDL levels, thereby contributing to the known recurrences of the FCH families in an orderly pattern.
An ancestral study on Sri Lankan population comprising of 95 men, screened for ApoA-I/C-III/A-IV-SstI associated SNPs showed higher risks for CHD (42), and the reports of African-American type 2 diabetic population consisting of 168 subjects had confirmed APOC III levels alone serve as the major risk factors of metabolic syndromes with decreased TG and HDL levels contributing to the high risks of CHD and dyslipidemia in the Tri-ethnic groups chosen (white Non–Hispanics, African-American, and Hispanics) (43). Similarly, various other studies have reported the plasma and serum levels of the TG molecules and their concordant associations for the risks of CVD, growth hormone deficiency (GHD), and CHD increasingly in APOC3 and APOA5 rs2854116 and rs662799 polymorphisms in the European population but contrast show lower risks for the Triethnic groups (Non–Hispanics, African-American, and Hispanics) in C allele with the higher HDL levels (44–47). Whereas, a study conducted on the American population predominantly comprising of white subjects totalling several ~1,200 men and women with known CVD risks were analyzed for several SNPs harboring within the APOA1/C3/A4/A5 gene cluster and showed that the APOA5 gene associated 56C > G and the1,131T > C SNPs were having higher transmission rates and expressed high TG, remnant-like particles, large and intermediate VLDL levels in both men and women CVD subjects (48). In addition, the study concluded with several new haplotype expressions and showed women with the 1131C allele were associated with higher risk assessments of CVD complications.
The gender and ethnicity-based study on Asian women (n = 973) having dyslipidaemia complications compared with that of the European women by the same team also showed stronger influences for CVD and CHD risks via the APOA5T-1131C promoter polymorphism, depicting the minor−1131C allele for both homozygotes and heterozygotes conditions that were associated with elevated plasma TG, VLDL-TG, LDL-TG, HDL-TG, and VLDL-cholesterol levels. Concomitantly, a study on Singapore population comprising of the Chinese, Indian, and Malay groups totalling a sum of ~3,971 subjects screened for the interactions among plasma lipids levels and the APOA5 polymorphisms showed increased levels of plasma TG and TC in association with the C allele of the APOA5 polymorphism, where the levels of HDL and HDL-C remained same across the polymorphisms for all the ethnic groups. The study also suggested that the APOA5 genotypes play major roles in plasma TG, TC, and HDL cholesterol exhibiting ethnic differences (49).
Further, the study on the genetic modulation of serum fasting triacylglycerol levels and the APOA5 gene alterations had shown the elevation of postprandial triacylglycerol concentrations in ~158 non-obese subjects habituating to the Korean population in association with the APOA5T-1131C promoter polymorphism serving to be one of the standard markers of oxidation and inflammation risks leading to major CVD risks. As an insight to add more information on the APOA1/C3/A4/A5 gene cluster interlinked cardiovascular disorders, APOC3 genotype based report on Spanish (Costa Dica) population including 336 individuals screened for the APOC3 [promoter region 455-T > C, T-625del and C3238G 3' untranslated region (UTR)] polymorphisms associated risks for CVD showed no evident interactions among plasma lipoproteins, and saturated fat intake in APOC3-455C-625del allele carriers neither did the 3'UTR C3238G have any observed association for the CVD risks. In contrast, a habitat based low saturated fat diet was found beneficial for the plasma lipoprotein levels among the APOC3 homozygotes of the chosen polymorphisms (50). Another study on the Ely (European) cohort including ~500 individuals screened for two variants of the insulin responsive element (IRE) of the APOC3 promoter region (−455T > C and −482C > T) were in Hardy Weinberg equilibrium, and the differences between the polymorphisms were observed to be less with reduced insulin secretion in male −482T/T carriers followed by the increase in insulin levels later.
The study concluded with milder links between the altered non-esterified fatty acid (NEFA) in men for both the T and C allele of the polymorphism and higher risks of diabetic onsets along with CHD for the APOC3−482C/T polymorphism (51). Similarly, the serum LDL cholesterol in men has a strong risk associated with the A allele of APOA1-75G/A polymorphism as per the study conducted on ~800 Calabria (southern Italy) population for the risk of CVD whereas, showing a decreased risk of the G allele among the three genotypes screened in the middle aged and elderly population (52). These cumulative results suggest that the risk associated with the increase and decrease of the biochemical markers of the CHD, CVD, and coronary artery disease (CAD) based diseases are not only significantly linked to the corresponding genetic polymorphisms seen in the APOA1/C3/A4/A5 gene cluster but also have strongly unpinned associations with the aetiological heterogeneity of their habituations.
The APO proteins are strong biomarkers of several cardiovascular events, and we have been listing all the major literature that has lined up in the row of finding their roles. Apart from the gene polymorphism studies, the Apolipoprotein A1/B ratio levels in the serum and plasma samples play a vital biochemical role in diagnosing an atherogenic risk of an individual (53). The Apolipoprotein A1/B ratio signifies the equilibrium seen among atherogenic and anti-atherogenic lipoproteins mainly observed in the serum and is estimated to be Apolipoprotein A (mg/dl)/Apolipoprotein B (mg/dl): <0.8 for individuals aged <18 years and 0.7–0.9 for individuals aged >18 years in men and <0.8 for individuals aged <18 years and 0.6–0.8 for individuals aged >18 years in women (54).
The APOB gene localized on human chromosome 2 encodes for the protein (Apo) B-100 (UniProt:P04114) mainly constituting of chylomicron apo B-48, LDL apo B-100, and VLDL apo B-100, which are some of the principal plasma protein components used to estimate the LDL and LDL receptor cellular binding and internalization of LDL molecules from the liver and peripheral cells, thereby, playing a crucial role in cholesterol homeostasis mechanisms (55–57). Whereas the APOA1 gene on the human chromosome 11 encodes for a serum and/or plasma protein ApoA-I (UniProt: P02647) that acts in the reverse transport events of lipid and cholesterol molecules from tissues to the liver, promoting excretion of cholesterol through efflux mechanism by acting as a cofactor for lecithin cholesterol acyltransferase (LCAT), a key enzyme that regulates the lipoprotein metabolism (58). The alterations in Apolipoprotein A1/B ratio levels are directly linked to the plasma cholesterol levels and are substantially increased via the decreased clearance of the LDL molecules by the un-functional expression of APOB/E receptors (59).
A study comprising of Jing and Han ethnic groups of the Chinese population screened for testing the link between genetic polymorphisms in the dedicator of cytokinesis 7 (DOCK7), one of the trans-membrane pro-protein convertase subtilisin/kexin type 9 (PCSK9) enzyme and serum lipid levels. The study consisted of ~1,869 CVD and control subjects of the middle age groups tested for genotype analysis showing Apo-A1: rs1997947 polymorphism predominantly only in the Jing ethnic group and ApoA1: rs11206517 and rs4846913 only in the Han nationals. The study had Apo-B: rs7552841 polymorphism in both the ethnic groups but Apo-B: rs10889332 only in the Han nationals, and the ApoA1/ApoB ratio polymorphisms showed rs7552841 for Jing and rs4846913 in the Han ethnic groups, respectively. Similarly, APOA1/B gene based polymorphisms had significant correlations with the plasma TC, TG, and LDL cholesterol levels thereby, imparting a specific new rare haplotype (G-C-G-C-T-G-C-C-A) that had diminishing effects on TC (4.90 ± 0.86 mmol/L), TG (1.30 (1.08) mmol/L), LDL (2.80 ± 0.40 mmol/L) cholesterol, and APOB (1.02 ± 0.22 g/L) levels alone when compared with that of the other 27 haplotypes seen (60).
Later, a subsequent study by the same author concluded that there is strong evidence to confirm, Han ethnic groups of the Chinese population, including 1,640 individuals, show lower risks for the increased levels of TG [1.37 (1.09) mmol/L), LDL (2.85 ± 0.43 mmol/L), APOB (1.05 ± 0.24 g/L], and serum lipid levels thereby, providing a masking effect for the CVD risks and moreover, this effect is specific for certain haplotype (CAPN3- FRMD5 T-A, T-C, C-A, and C-C) carriers only (61). In line with these results, the haplotype-based studies on the ApoA1/B ratio polymorphisms have shown supporting evidence on CVD/CHD, stroke, and/or other cardiovascular events, diabetes, dyslipidaemia at varying levels of risk issues in human samples of different populations including (Han, Mulao, and Maonan) Chinese population.
A study by Berkinbayev et al. (62) showed that the decreased levels of Apo A1 (136.3 ± 5.5 mg/d) and increased levels of the Apo B (88.98 ± 2.63 mg/d) in CHD individuals (n = 448) belonging to the Kazakhs have Apo E levels significantly (< 0.05) lower than that of the healthy Uighurs population (62). The risk of CHD/CVD along with stroke and diabetes showed evident occurrences along with the rare haplotypes: SRGAP2 rs2483058C-rs2580520G of ApoA1 and ApoA1/ApoB ratio, thereby contributing to the development of breast cancer in the Han population having greater risks over the subjects presenting increased levels of circulating oestrogens. In addition, the study also showed that the rs2483058C-rs2580520G haplotype is associated with an increased risk of dyslipidaemia in the Han and Maonan groups (n = 2,444), showing consistent risks of increased serum TC (5.32 ± 1.06 mmol/L), HDL-C (1.71 ± 0.58 mmol/L), ApoA1 (1.22 ± 0.14 g/L), and the ApoA1 to ApoB ratio (1.47 ± 0.47 g/L) (63).
The APOB gene polymorphism 8046C > T (SNP: rs1367117) is one of the major genetic-based studies that has been carried out worldwide, and it encounters strong influence over the plasma of LDL cholesterol concentrations. A population-based study on the Shanghai (Chinese) groups consisting of ~2,000 and more cumulative subjects having gallbladder, extra hepatic bile duct, ampulla of Vater cancer, and biliary stone defects was compared for their plasma lipoprotein levels with the control groups. The study also concluded that the homozygous C allele of the polymorphism is strongly associated with ~1.5-fold higher risks for bile duct related cancers and was seen predominant for 8046C > T among the two major haplotypes (64).
Similarly, two studies insisting over the multiple genetic determinants screening study associated with that of plasma lipid levels in the Caribbean Hispanic population consisting of ~477 subjects and Chinese Shanghai population comprising of 799 randomly selected healthy subjects showed that the 114 and 21 SNPs screened, respectively, related to lipid metabolism events and revealed three genes including APOB rs1367117 had strong influence over the HDL-C/TG, TC, and LDL-C level variations (65, 66). Later, a study on the Young adult subjects (n ~1,250) belonging to the Jerusalem population also showed that the APOB (rs1367117) polymorphism have a significant maternally-derived effect on body mass index (BMI) and waist circumference (WC) parameters providing evidence that the APOB gene polymorphism 8046C > T (SNP: rs1367117) plays a strong and effective role on regulatory mechanisms (67).
Along with this information, the recent APOB gene polymorphism studies report that they are strongly associated with CHD and CVD risks. In addition, a study carried out on the Pakistani Asian population including ~1,960 control and CHD subjects showed that the APOB gene 4181 E>K polymorphism (SNP: rs1042031) has an evident influence on the risks for developing CHD. Simultaneously, a study on the same population consisting of ~625 subjects screened for the rs1042031 genotype showed atherogenic blood lipid associations with the gene polymorphism when analyzed for the significant positive link with the genetic risk score (GRS). Similarly, an insertion/deletion (ins/del) polymorphism study carried out on signal peptide region of rs1042031 4154 G>A on exon 29 of the gene alters the binding efficiency of LDL to LDL receptor, causing an increase in LDL-c levels, thereby having higher risks of cardiovascular defective events (37, 68). The cumulative report on the Apolipoprotein A1/C3/A4/A5 gene cluster-based studies and the associated gene polymorphism studies having various effects on the wide range of medical complications are listed in Table 3.
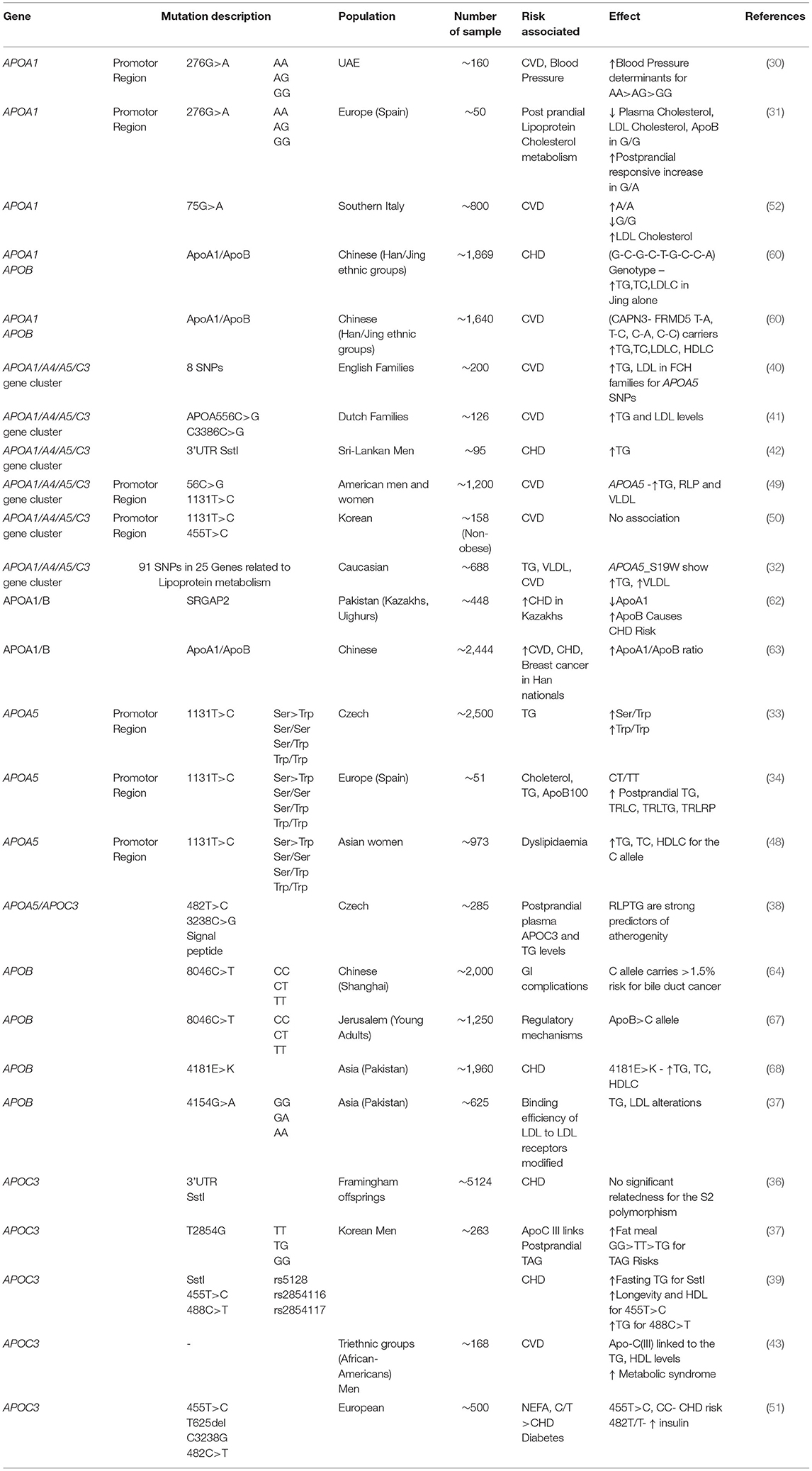
Table 3. List of genetic studies related to the APOA1, APOA5, APOC3, and APOA1/A4/A5/C3 gene cluster, presented with their mutation, population, sample size, and contributing medical risks.
Apolipoprotein E or APOE is a 34 kDa (kilo Dalton) protein comprising 299 amino acid residues that transport lipid molecules (69). The protein is encoded by the APOE gene (OMIM: 107741) located in chromosome 19, having 4 exons (70). The APOE gene variants generate from the exon 3 region contributing to the rs7412 C/T and rs429358 C/T, SNPs generating: ε2 (APOE2), ε3 (APOE3), and ε4 (APOE4) (71, 72) major haplotypes as shown in Figure 3. These isoforms ε3 have cysteine and arginine residues in 112th and 158th positions, respectively, and ε4 has two cysteine and two arginine residues, respectively (74). It contributes to six major genotypes, such as ε2/ε2, ε3/ε3, and ε4/ε4 that are homozygous forms and ε2/ε3, ε3/ε4, and ε2/ε4 heterozygous forms. Over the years, it has been strongly believed that the ε4 allelic expression in a population is the most prevalent modulator associated with AD risk. A cumulative correlation of APOE polymorphisms and associated risks are tabulated in Table 4. The clinical characteristics and biochemical associations witnessed for various APOE polymorphisms are depicted in Figure 4 respectively.
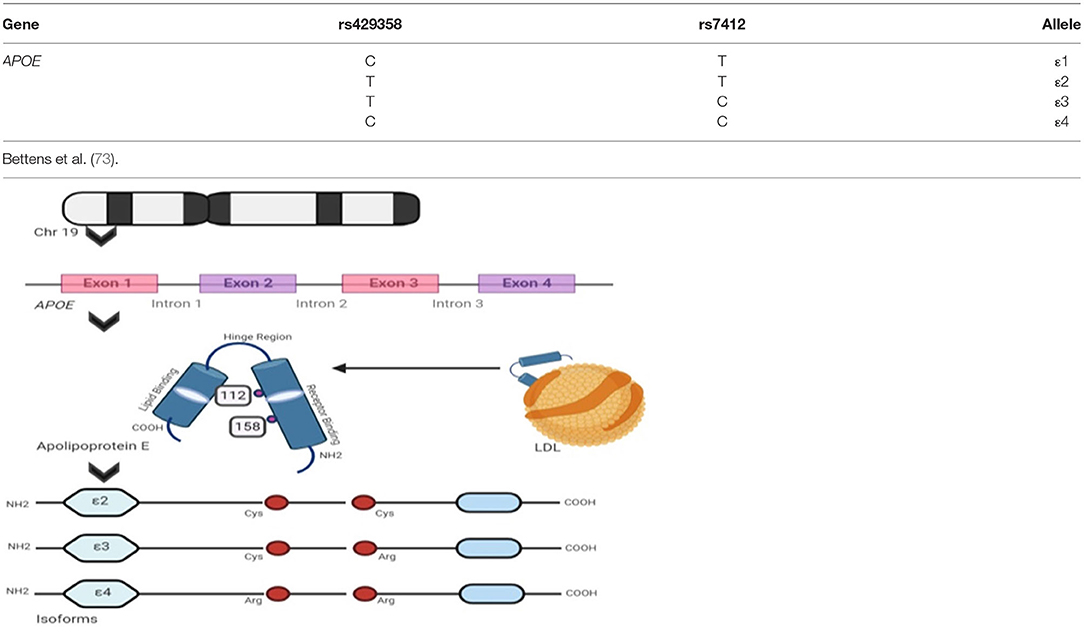
Figure 3. Apolipoprotein E isoforms and its genetic integrity: the figure shows APOE allelic variants encoded by the APOE gene that transcribes Apolipoprotein E isoforms and the table lists all the major genotype specific single nucleotide polymorphism (SNP) variants and their nucleotide transformations seen, respectively.
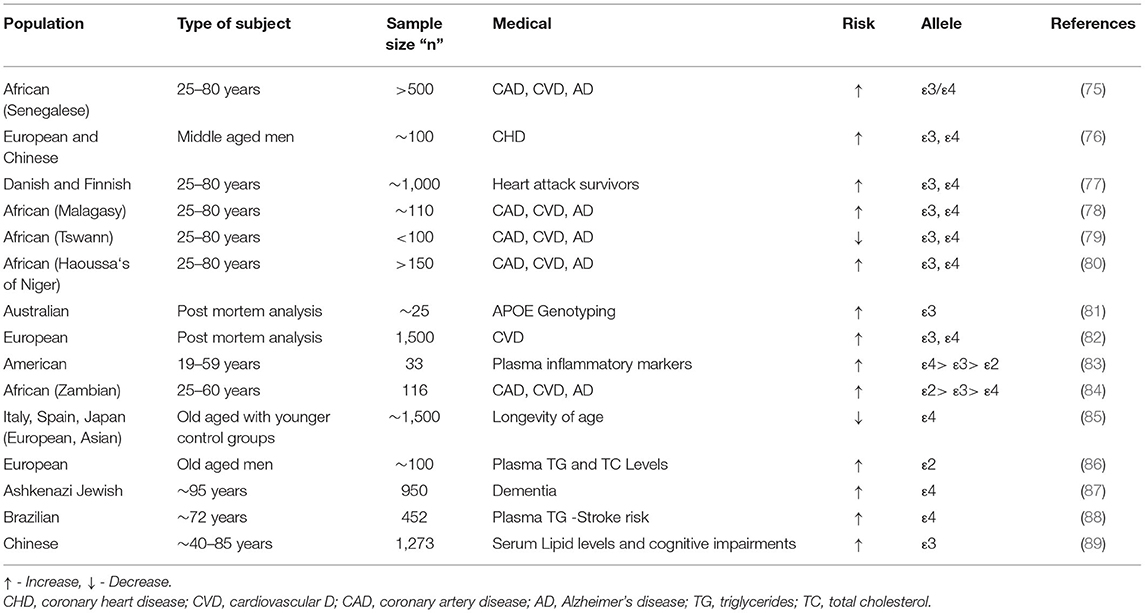
Table 4. The APOε isoforms related genetic studies depicting the risks of various medical conditions.
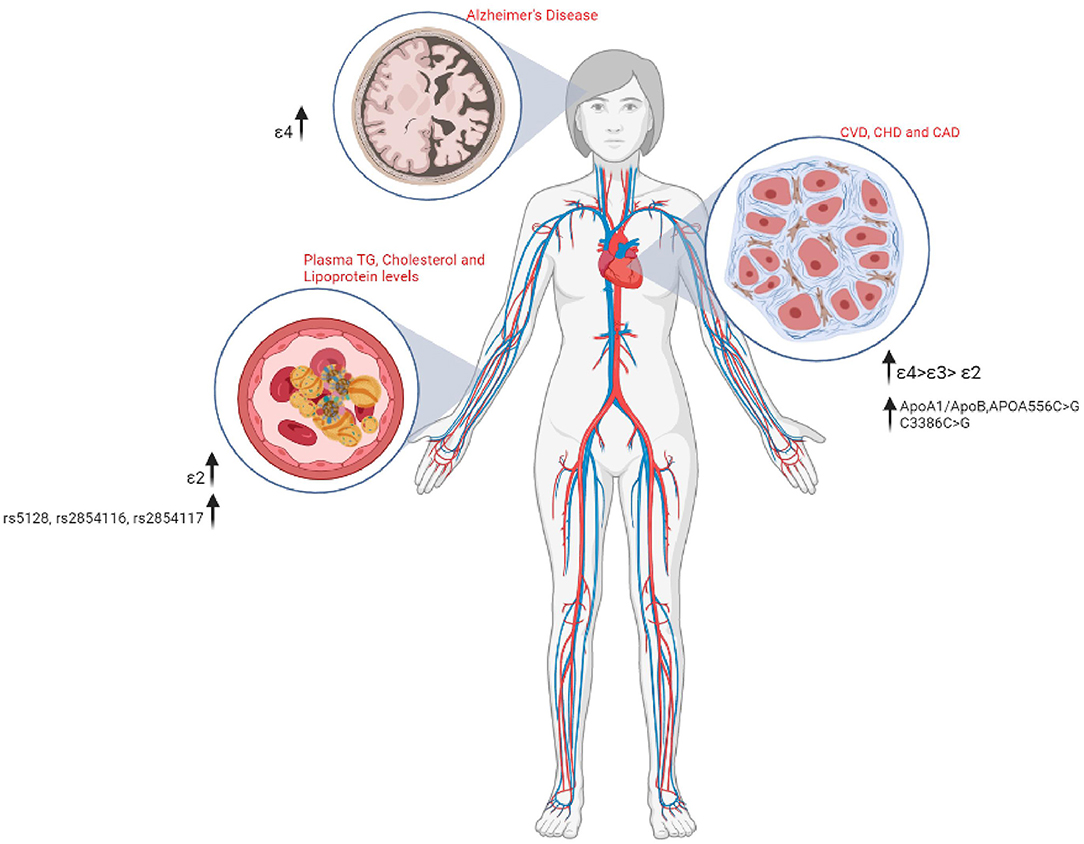
Figure 4. A schematic representation of the various Apolipoprotein polymorphisms and their associated medical risks.
Dementia is a generic term for memory, language, problem-solving, and other thinking abilities that are bad enough to cause daily difficulties (90, 91). Heterozygous recessive inheritance of the ε4 allele is associated with increased genetic susceptibility to the risk of dementia that ranges from an ~3 to ~15 increase in heterozygous and homozygous individuals, respectively (73, 92, 93). The ε4 allele is also associated with AD, Aortic valve disease (AVD), CAD, LDL-C levels, and longevity with the APOE isoforms are portrayed on a stepwise increase in risk from ε2>ε3>ε4. The strongest evidence for the role of APOE-ε4 isoforms in non-AD neurodegenerative diseases can be mapped to dementia with Lewy bodies (DLB) as in the case of Parkinson's disease (PD) (94).
The common APOE alleles: ε2, ε3, and ε4 are predominantly located in the CpG islands and these genotype expressions analyzed by means of the three alleles have an impact on the magnitude of the CpG di-nucleotides, contributing to the expression of the DNA methylation of respective gene (95). A study on DNA methylation profiles of these genomic regions assists to play a major role in differentiating the AD brain defects from control brains (96), and several other studies conducted on the African-American population suggested that DNA methylation screened in blood cells is a clear indicator of risks for developing dementia which may, later on, lead to increased risks for AD pathologies. The results showed that the methylation seen in eight CpGs demonstrated 8.7% of gene variance associated with memory defects within the APOE ε4 carriers (97). Whereas, the genotyping analysis study on post-mortem brain hippocampus and cerebellum reports of pathologically confirmed AD cases consisting of ~25 AD and control groups showed the presence of soluble protein fragments of APOE in APO ε3 expressing individuals, measuring 2- to 3-folds higher than that of the APO ε4 subjects (81). More significantly, a study that included 116 subjects between the age groups of 25–60 years belonging to 72 ethnic groups found in the native African population analyzed for the associations of CAD, CVD, and AD complications showed that the APOE isoform (ε4 carriers) recurrences inferred for the Zambian tribe's ancestry varied significantly between Zambian and the North African populations, such as Moroccans and Tunisians. This study suggested that the variation may be due to the extensive intermixing of the North African populations with Arab, Jewish, and other Western Eurasian populations, as reported by Wozniak et al. (78). Comparatively analysis carried out on the Zambian and other African populations (Tswana, Botswana) for the APOE allele frequencies showed 7 and 11.1% in Haoussa's of Niger and 4% (Malagasy) to 34.4% (Senegalese), respectively, as of ε2 and ε3 alleles.
Some of the diseases affecting the circulatory system, such as CVD, are major causes of morbidity and mortality worldwide. An accurate prediction and screening of the genetic predisposition of an individual will incur the risks of an individual being affected by such genetic alterations. More importantly, the risk for CVD is determined by a combination of modifiable and non-modifiable genetic risks. It would be embarrassing if these reasons were not rectified and met in advance. The genetic factors involved in lipid metabolism playing key roles in CVD risks presenting increased susceptibility for the disease are accurately predicted beforehand. The genetic susceptibility for E4 allele occurrence and its strong associations with higher LDL-C concentration and total cholesterol were some of the evident initial research outcomes (98). These correlations were then studied with reference to CVD risks later, but as days scored, the association of the E4 allele with the risk of dementia was more evident in various populations around the world, making it less observed for LDL-concentrations and associated vascular complications (99). The APOE-based genetic polymorphisms are also strongly associated with plasma lipid profiles that correlate to increased risks of vascular complications in later stages of life, but the APOE polymorphisms are directly correlated with CVDs, aging, and longevity in humans (100).
A report on nine ethnic groups of the Asian population (eight European and one Chinese ancestry), such as middle-aged men, showed that variations in relative frequencies of ε4 allele occurrence could predict 40–75% risks for CHD (76). Similarly, a follow-up study on the Scandinavian population belonging to the Northern European regions that included 1,000 Danish and Finnish heart attack survivors report denoted the risk for carrying ε4 variant is one of the prognostic elements for CVD risks, as these subjects show an 80% increase risk for recurrences and death (77). Similarly, the inference presented by a pilot study that included the post-mortem analysis of 1,500 individuals who died of natural causes, performed at the Oslo University Hospital (Norway) showed that the group of patients presenting the risks of CVD (35% of the total), were significantly more susceptible for ε4 homozygous cohorts (34% against 29%) than the rest of the group having lesser chances of ε2 homozygous or heterozygous carriers (12% against 14%) for chosen p-values (p < 0.05) (82). The ε4 allele frequency between the Zambian and other African populations was as low as 1.8% in Zambian, and whereas for the Tswana population, it was as high as 23.7%. The difference between the Zambian and Senegalese populations showed 13.8, 59.5, and 26.7% for the ε2, ε3, and ε4 allelic frequencies, respectively. The ε4 allele frequency was depicted to be lower in patients with CVD, CHD, and CAD comparatively from various studies (75, 78–80, 84).
The transport and clearance mechanism of chylomicron remnants, VLDL, and HDL from the bloodstream is brought about by the action of the APOC3 gene (57). The −482 C/T (rs2854117) and −455 T/C (rs2854116) polymorphisms of APOC3 are actively associated with non-alcoholic fatty liver disease (NAFLD) and insulin resistance (IR) (101). Sometimes, it is also considered that the ApoCIII promotes hypertriglyceridemia (HTG), inhibits the activity of LPL, disturbing lipids lipolysis, and interferes with apoE or apoB binding to hepatic receptors causing delayed catabolism of TRL remnants, regulates the VLDL assembly, and secretion in the liver through different mechanisms (102, 103). In such cases, the ApoCIII and lipoproteins that carry apoCIII are studied as the key regulators in the TG metabolism and independent predictors of CVD risk. Since the APOC3 brings in a phenotypic change in the effect on lipid metabolism, it is also believed to contribute to an increased risk of CAD (104). Similarly, the APOA5 plays a key role in modulating TG metabolism, thereby acting as a potential regulator of plasma TG causing CVD risks (105).
A similar pattern of study provoked from the highlighted presumed associations of APOE and extreme lifespan longevity focused on three independent cohorts of older aged groups from Italy, Spain, and Japan, compared for healthy and younger controls. This study confirmed that the occurrence of the ε4 allele has a negative association with that of the extreme longevity in all three cases after adjustment for gender based correlations. This served to be one of the major genes based positive association studies that concluded the presence of the same traits of the ε2 allele in Japanese and Italian cohorts. This strongly highlighted that the ε4 variants validate to decrease the probability of the increasing risks of ages across ethnicity and geographic origins (85). Later, in a study on Ashkenazi Jewish ancestry subjects, ~1,000 individuals consisting of older groups having occurrences of familial longevity history (~95 years of age) and younger aged control groups were analyzed for the full-length expressions of APOE gene regions. The analysis showed the occurrence of two common regulatory variants (rs405509 and rs769449), playing regulatory functions via promoter regions of the gene. The results showed significantly depleted expressions of ε2 allele (p = 0.003) and the ε2/ε3 genotype (p = 0.005) in the elderly groups for the designated p values (p <0.036) and evident amelioration of the ε3/ε4 genotype (p = 0.005) when compared with the control groups seen with increased longevity characteristics (87).
The APOε genotypes were not only believed to have recurrences with AD and heart diseases but have also been associated with the fluctuating levels of unsaturated and saturated circulating fatty acids (106). People who are obese possess increased blood or plasma cholesterol, glucose, and insulin conditions that lead to several other diseases, such as diabetes and other metabolic syndromes, independently associated with unique cognitive deficits (107–109). The first study reported on the Brazilian population that included 220 subjects above the age of 85 years, 232 controls averaging to the age of 72 years comprising of the oldest-olds, investigated the association among FOXO3, SOD2, SIRT1, and APOE known gene variants. The investigation showed that the only association seen for the gender-based TG levels was confirmed with that of the APOE isoforms ε2 and ε3 but for the rest was normal. Moreover, the report also suggested that expansion of samples size with gender- and age-based criteria for the chosen population would yield better confirmative results (88).
In the European population, a study conducted for analyzing the small portion of ε2 allele variant, the rarest amongst the six genotypes, showed that the known variant recurrences have risks for developing lipid metabolisms related disorders, mainly contributing to clinical pathogenesis known as the Type III hyperlipoproteinemia that is characterized by extreme elevations of TGs and TC levels.
Several studies suggest that the occurrence of the ε2 homozygous genotype has higher risks for CAD, palmar xanthomata (orange creases in the palms), and tuberous xanthomas (nodular lipid deposits) on skin, knees, and elbows (86). In addition, the ε2 allele does not appear to be protective against ischemic stroke (IS) as reported by the study that was carried on European subjects aged above 75 years with recurrent cardiovascular events (110). Whereas in terms of the APO ε4 allele expressions, a study reported with the Chinese population consisting of 1,273 subjects (aged between 40 and 85 years) analyzed for APOE genotype and concordant Serum lipid levels showed no significant correlations between the serum LDL—TG/TC levels and the APO ε4 allele expressions having the odds ratio (OR) of OR = 20.094 for OR@95% with the estimated p-value of 0.039 (p ≤ 0.005). The study reported that the homozygous ε4 carriers have lower serum HDL with positively concordant cognitive impairments, showing that the APOE isoforms play major roles in serum lipid levels and the assessments of cognitive impairments and memory (89).
Moreover, the APOE isoforms are believed to play a key role in modulating the immune responses of inflammatory mediators secreted by the microglia cells through HDL and LDL levels of the brain in an APOε4>APOε3>APOε2 pattern (111). Where obese patients present increased activated macrophages and pro-inflammatory cytokines, tumor necrosis factor-α (TNF)-α, and interleukin (IL)-6 secretion from adipose tissue. Moreover, APOE isoforms are directly linked with the expression of the plasma inflammatory markers where the exogenous expression of APOε3 acts as a critical suppressor of inflammatory processes, and the APOε4 expressions are linked with overexpression of the pro-inflammatory TNF-α, IL-1β, IL-6, and IL-12 immune cells being major phenotype markers of JNK, MAPK p38, and casein kinase 2 (CK2) pathways (112). APOE genotype study carried out on 33 middle aged cohorts (19–59 years of age) of the American population related to the plasma signaling protein levels showed a concomitant expression of the inflammatory markers I-309, IL-1, IL-3, IL-7, IL-12p40, IL-13, and IL-15, to be the lowest in ε2 allele followed by the APOε3>APOε4 patterns (83).
The effects of APOE genotype variants in obese populations are based on several phenotypical features, such as sex, age, and co-inherited gene variants. The E4 midlife obese carriers showed increased susceptibility for AD risks in later stages of life, whereas the increased waist to hip ratios were associated with impaired cognitive deficits in middle-aged cohorts expressing increased white matter hyper-intensities (113, 114). Similarly, weight loss in older Prospective Population Study of Women (PPSW) in Sweden showed an increased risk of cognitive decline concerning declined BMI and E4 allele recurrences (115). The mechanism of cellular obesity occurring during the middle-ages is strongly associated with decreased blood-brain barrier (BBB) integrity, tau phosphorylation, and decreased amyloid precursor protein (APP) levels. The gender-based susceptibility of AD risk is higher in the case of obese women presenting deficits in BBB integrity, and high BMI associated inflammation at an older age (116, 117). More interestingly, the APOE4 carriers are comparatively more sensitive to metabolic alterations associated with obesity (118). Apart from which the recycling mechanism of APOE appears to depend on the surface expression of LDL receptors and ATP binding cassette 1 (ABCA1) activities. The E4 carriers are strongly associated with lower BMI but increased susceptibility to higher insulin associated metabolic syndromes (119).
The accumulation of pathological proteins in AD brains, being a prominent feature, induces the activation of glial cells, leading to prolonged neuro-inflammation that might induce neuronal injury or death in multiple ways (120). This cascade reaction involves the initial activation of glia producing toxic compounds that fall under two major categories, i.e., the reactive oxygen species (ROS) or nitric oxide (NO) that damage neurons. Sometimes, this reaction is also replaced by the sublethal pathological inflammatory stress that can damage viable neurons to express apoptotic signals that cause phagocytosis of the damaged neurons through a process called phagoptosis (121). The neuro-inflammation process is the major hallmark that contributes to the onset and progression of AD pathogenesis. The activated microglia responded to the pro-inflammatory mechanisms through the release of cytokines, such as TNFα and interleukin 1β (IL-1β), additionally inducing damage to other surrounding glial and anchoring cells (122).
The expression of APOE suppresses the proliferation of T cells and activation of a neutrophil by regulating and facilitating macrophage functions and lipid antigen presentation by the CD1 molecules. Naturally, the cellular ApoE regulates the lipopolysaccharides (LPS) induced lethality by attenuating inflammatory responses, where deficiency of ApoE causes impaired immune responses leading to the production of significantly higher pro-inflammatory cytokines, such as IL-1, IL-6, and TNF- (123). On the other hand, the ApoE suppresses the production of pro-inflammatory cytokines, through a descending order as E2>E3>E4 isoforms. The crucial initiation process of the innate and adaptive immunities relies on macrophage activation, and this is achieved by the classical activation of the inflammatory stimuli via the pro-inflammatory signals that are accompanied by the downregulation of ApoE expression. One of the conventional identities of activated macrophages is observed by the production of NO. Several animal studies on Human TG mice show that the increased ApoE treatment induces the production of macrophages and microglia with higher NO secretion. Recent studies also show that the B cells utilize ApoE-mediated pathways for lipid antigen presentation rather than the dendritic cells and these ApoE based CD1-mediated self-lipid presentation to NK-cells are widely involved in autoimmune diseases, such as atherosclerosis, multiple sclerosis, and others (124, 125).
A stroke is a form of disability, characterized by the blockage of blood vessels causing detrimental effects that lead to the second largest form of death occurring in the older population worldwide (90, 126–129). The age-based risk for stroke increases with age and is doubled over the age of 55 years in both men and women (130, 131). Some of the subtypes of stroke, such as the lobar intracerebral hemorrhage, are associated with an increased risk on the occurrences of ε2 or ε4 homozygous alleles, where the ApoEε4 allele carriers are reported with higher volumes of subcortical white matter lesions presenting increased risks for IS and associated medical complications (132).
The ApoE ε4 allele carriers were said to exhibit significant risk for IS, and an increased risk of ICH was associated with the presence of ε2 allele by a meta-analysis report in 2006 (133). The meta-analysis reports from Zhang et al. (134) showed that the ε4 allele carriers present increased risks for ICH but not the ε2 allele carriers. But the actual association of the ApoE polymorphism and IS or ICH in the Chinese ethnic population is still an outstanding question. Similarly, a meta-analysis study carried out later on 18 different reports that have worked in various Chinese provinces suggested that there is a significant association between the ε4 allele carriers and increased ICH risk when compared with that of the ε3 and ε2 alleles. Similarly, the study showed a significant association for ε4 carriers and ε3ε3 genotype carriers in IS risk with an OR of 2.41 and 2.00–2.89 for (95% CI at p < 0.001) (135). A study reported on South Indian population also showed that the frequency of HLA-DRB1/DQB1alleles were higher in IS patients but not in control, predicting DRB1*10-DQB1*05 haplotype existence to be protective over IS (136).
The current review has put forth the progressive population genetic data that have been great insights in understanding the role of genetic polymorphisms that are correlated to several APO protein-based medical risks. From the cumulative knowledge perceived, the contribution of genetic polymorphisms that influence the lipoprotein levels appear to be associated with selective risks, such as cardiovascular associated disorders, neurodegenerative disorders, metabolic syndromes, and other lipidaemias, respectively, across several population groups. From future perspectives, the comprehensive understanding of such molecular events correlated with several environmental and dietary factors can unravel novel perceptions of various therapeutic approaches interlinked to the abovementioned clinical manifestations. In addition, the review strongly suggests that a broad range of molecular analysis studies considering factors, such as patient's history of ethnicity and habitual traits with a huge sample size across various populations will be a challenging field to entangle in the near future, and this can further develop the understandings of unspotted haplotype-based studies and narrowing down the complications in several metabolic disorders associated risk assessments.
RB: raw data and basic information collection. PB: conceptualization and writing—original draft preparation. DK: data curation and formal analysis. VP: guidance, validation, and supervision. All authors contributed to the article and approved the submitted version.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We are very thankful for:
• BioRender for offering a wonderful platform to create our conceptual ideas into figures illustrated; and
• The Biomaterials and Nanomedicine Laboratory team for supporting us in this contribution.
APO proteins, Apolipoproteins; SNP, single nucleotide polymorphism; CVD, cardiovascular disease; AD, Alzheimer's disease; HDL, high-density lipoprotein; IDL, intermediate-density lipoprotein; LDL, low-density lipoprotein; VLDL, very low-density lipoprotein; CHD, coronary heart disease; PVD, peripheral vascular disease; APOE, Apolipoprotein E; APOA, Apolipoprotein A; APOB, Apolipoprotein B; APOC, Apolipoprotein C; CAD, coronary artery disease; PD, Parkinson's disease; DLB, dementia with Lewy bodies; LDL-C, low-density lipoprotein-cholesterol; AVD, aortic valve disease; CK2, casein kinase 2; Lp (a), lipoprotein (a); TG, triglycerides; FCH, familial combined hyperlipidemia; HDLC, high-density lipoprotein cholesterol; GHD, growth hormone deficiency; TC, total cholesterol; IRE, insulin responsive element; UTR, untranslated region; NEFA, non-esterified fatty acid; LCAT, lecithin cholesterol acyltransferase; PCSK9, proprotein convertase subtilisin/kexin type 9; DOCK7, dedicator of cytokinesis 7; GRS, genetic risk score; UAE, United Arab Emirates; RLP, remnants of lipoprotein elements; ROS, reactive oxygen species or nitric oxide (NO).
1. Atkinson RA, Gaysina D, Jennifer MR. Responses to executive demand in young adulthood differ by APOE genotype. Behav Brain Res. (2018) 33:360. doi: 10.1016/j.bbr.2018.11.033
2. Genest J. Genetics and prevention: a new look at high-density lipoprotein cholesterol. Cardiol Rev. (2002) 10:61–71. doi: 10.1097/00045415-200201000-00011
3. Haddad IA, Ordovas JM, Fitzpatrick T. Structural analysis of human apolipoprotein A-I variants. J Biol Chem. (1986) 261:13268–77. doi: 10.1016/S0021-9258(18)69300-7
4. Zhou Y, Mägi R, Milani L, Lauschke VM. Global genetic diversity of human apolipoproteins and effects on cardiovascular disease risk. J Lipid Res. (2018) 59:1987–2000. doi: 10.1194/jlr.P086710
5. Kanter JE, Shao B, Kramer F. Increased apolipoprotein C3 drives cardiovascular risk in type 1 diabetes. J Clin Investig. (2019) 129:27308. doi: 10.1172/JCI127308
6. Sluis S, Verhage M, Posthuma D. Phenotypic complexity, measurement bias, and poor phenotypic resolution contribute to the missing heritability problem in genetic association studies. PLoS ONE. (2010) 5:e13929. doi: 10.1371/journal.pone.0013929
7. Manchia M, Cullis J, Turecki G. The impact of phenotypic and genetic heterogeneity on results of genome wide association studies of complex diseases. PLoS ONE. (2013) 8:e076295. doi: 10.1371/journal.pone.0076295
8. IBC 50 K CAD Consortiums. Large-scale gene-centric analysis identifies novel variants for coronary artery disease. PLoS Genet. (2011) 7:e1002260. doi: 10.1371/journal.pgen.1002260
9. Hernandez LM. Institute of Medicine (US) Committee on Assessing Interactions Among Social, Behavioral, and Genetic Factors in Health; Genes, Behavior, and the Social Environment: Moving Beyond the Nature/Nurture Debate. Washington, DC: National Academies Press (US) (2006).
10. Tybjaerg-Hansen A, Steffensen R, Meinertz H. Association of mutations in the apolipoprotein B gene with hypercholesterolemia and the risk of ischemic heart disease. N Engl J Med. (1998) 338:1577–84. doi: 10.1056/NEJM199805283382203
11. Pennacchio LA, Olivier M, Hubacek JA. Two independent apolipoprotein A5 haplotypes influence human plasma triglyceride levels. Hum Mol Genet. (2002) 11:3031–8. doi: 10.1093/hmg/11.24.3031
12. Bennet AM, Di Angelantonio E, Ye Z, Wensley F, Dahlin A, Ahlbom A, et al. Association of apolipoprotein E genotypes with lipid levels and coronary risk. JAMA. (2007) 298:1300–11. doi: 10.1001/jama.298.11.1300
13. Yu JT, Tan L, Hardy J. Apolipoprotein E in Alzheimer's disease: an update. Annu Rev Neurosci. (2014) 37:79–100. doi: 10.1146/annurev-neuro-071013-014300
14. Matsunaga A. Apolipoprotein A-I mutations and clinical evaluation. In: Tsugikazu K, editor. The HDL Handjournal. Elsevier: Academic Press (2014). doi: 10.1016/B978-0-12-407867-3.00002-0
15. Sorci-Thomas M, Prack MM, Dashti N. Differential effects of dietary fat on the tissue-specific expression of the apolipoprotein AI gene: relationship to plasma concentration of high density lipoproteins. J Lipid Res. (1989) 30:1397–403. doi: 10.1016/S0022-2275(20)38254-7
16. Gauthamadasa K, Rosales C, Pownall HJ, Macha S, Jerome WG, Huang R, et al. Speciated human high-density lipoprotein protein proximity profiles. Biochem. (2010) 49:10656–65. doi: 10.1021/bi1015452
17. Swaney JB, Braithwaite F, Eder HA. Characterization of the apolipoproteins of rat plasma lipoproteins. Biochem. (1977) 16:271–8. doi: 10.1021/bi00621a018
18. Beisiegel U, Utermann G. An apolipoprotein homolog of rat apolipoprotein A-IV in human plasma: isolation and partial characterisation. Eur J Biochem. (1979) 93:601–8. doi: 10.1111/j.1432-1033.1979.tb12860.x
19. Siri-Tarino PW, Krauss RM. The early years of lipoprotein research: from discovery to clinical application. J Lipid Res. (2016) 57:1771–7. doi: 10.1194/jlr.R069575
20. Pennacchio LA, Olivier M, Hubacek JA. An apolipoprotein influencing triglycerides in humans and mice revealed by comparative sequencing. Science. (2001) 294:169–73. doi: 10.1126/science.1064852
21. Karathanasis SK. Apolipoproteinmultigene family: tandem organization of human apolipoprotein AI, CIII, AIV genes. Proc Natl Acad Sci USA. (1985) 82:6374–8. doi: 10.1073/pnas.82.19.6374
22. Talmud PJ, Drenos F, Shah S. Gene-centric association signals for lipids and apolipoproteins identified via the HumanCVDBeadChip. Am J Hum Genet. (2009) 85:628–42. doi: 10.1016/j.ajhg.2009.10.014
23. Wu Y, Yu Y, Zhao T. Interactions of environmental factors and APOA1-APOC3-APOA4-APOA5 gene cluster gene polymorphisms with metabolic syndrome. PLoS ONE. (2016) 11:e0147946. doi: 10.1371/journal.pone.0147946
24. Wang Y, Yin X, Li L. Association of apolipoprotein C3 genetic polymorphisms with the risk of ischemic stroke in the Northern Chinese Han Population. PLoS ONE. (2016) 11:e0163910. doi: 10.1371/journal.pone.0163910
25. Ou HJ, Huang G, Liu W. Relationship of the APOA5/A4/C3/A1 gene cluster and APOB gene polymorphisms with dyslipidemia. Genet Mol Res. (2015) 14:9277–90. doi: 10.4238/2015.August.10.8
26. Martin S, Nicaud V, Humphries SE. Contribution of APOA5 gene variants to plasma triglyceride determination and to the response to both fat and glucose tolerance challenges. Biochim Biophys Acta. (2003) 1637:217–25. doi: 10.1016/S0925-4439(03)00033-4
27. Liu Y, Ordovas JM, Gao G. Pharmacogenetic association of the APOA1/C3/A4/A5 gene cluster and lipid responses to fenofibrate: the genetics of lipid-lowering drugs and diet network study. Pharmacogenet Genomics. (2009) 19:161–9. doi: 10.1097/FPC.0b013e32831e030e
28. Pollin TI, Damcott CM, Shen H. A null mutation in human APOC3 confers a favorable plasmid lipid profile and apparent cardioprotection. Science. (2008) 322:1702–5. doi: 10.1126/science.1161524
29. Schaefer EJ, Santos RD, Asztalos BF. Marked HDL deficiency and premature coronary heart disease. Curr Opin Lipid. (2010) 21:289–97. doi: 10.1097/MOL.0b013e32833c1ef6
30. Sadaf A, Siddiqui S, Lestringant GG. Apolipoprotein AI promoter variant in blood pressure determination. Clin Genet. (2002) 61:314–6. doi: 10.1034/j.1399-0004.2002.610414.x
31. Marin C, López-Miranda J, Gómez P. Effects of the human apolipoprotein AI promoter GA mutation on postprandial lipoprotein metabolism. Am J Clin Nutr. (2002) 76:319–25. doi: 10.1093/ajcn/76.2.319
32. Smith JA, Arnett DK, Kelly RJ. The genetic architecture of fasting plasma triglyceride response to fenofibrate treatment. Eur J Hum Genet. (2008) 16:603–13. doi: 10.1038/sj.ejhg.5202003
33. Vrablik M, Horínek A, Ceska R, Adámková V, Poledne R, Hubacek JA, et al. Ser19 → Trp polymorphism within the apolipoprotein AV gene in hypertriglyceridaemic people. J Med Genet. (2003) 40:e105. doi: 10.1136/jmg.40.8.e105
34. Moreno R, Perez-Jimenez F, Marin C. A single nucleotide polymorphism of the apolipoprotein A-V gene– 1131T> C modulates postprandial lipoprotein metabolism. Atherosclerosis. (2006) 189:163–8. doi: 10.1016/j.atherosclerosis.2005.11.029
35. Lopez-Miranda J, Williams C, Lairon D. Dietary, physiological, genetic and pathological influences on postprandial lipid metabolism. Br J Nutr. (2007) 98:458–73. doi: 10.1017/S000711450774268X
36. Russo GT, Meigs JB, Cupples LA. Association of the Sst-I polymorphism at the APOC3 gene locus with variations in lipid levels, lipoprotein subclass profiles and coronary heart disease risk: the Framingham offspring study. Atherosclerosis. (2001) 158:173–81. doi: 10.1016/S0021-9150(01)00409-9
37. Woo SK, Kang HS. The apolipoprotein CIII T2854G variants are associated with postprandial triacylglycerol concentrations in normolipidemic Korean men. J Hum Genet. (2003) 48:551–5. doi: 10.1007/s10038-003-0074-7
38. Waterworth DM, Hubacek JA, Pitha J. Plasma levels of remnant particles are determined in part by variation in the APOC3 gene insulin response element and the APOCI-APOE cluster. J Lipid Res. (2000) 41:1103–9. doi: 10.1016/S0022-2275(20)32016-2
39. Atzmon G, Rincon M, Schechter CB. Lipoprotein genotype and conserved pathway for exceptional longevity in humans. PLoS Biol. (2006) 4:40113. doi: 10.1371/journal.pbio.0040113
40. Mar R, Pajukanta P, Allayee H. Association of the APOLIPOPROTEIN A1/C3/A4/A5 gene cluster with triglyceride levels and LDL particle size in familial combined hyperlipidemia. Circ Res. (2004) 94:993–9. doi: 10.1161/01.RES.0000124922.61830.F0
41. Eichenbaum-Voline S, Olivier M, Jones EL, Naoumova RP, Jones B, Gau B, et al. Linkage and association between distinct variants of the APOA1/C3/A4/A5 gene cluster and familial combined hyperlipidemia. Arteriosclerosis. (2004) 24:167–74. doi: 10.1161/01.ATV.0000099881.83261.D4
42. Mendis S, Shepherd J, Packard CJ. Genetic variation in the cholesteryl ester transfer protein and apolipoprotein AI genes and its relation to coronary heart disease in a Sri Lankan population. Atherosclerosis. (1990) 83:21–7. doi: 10.1016/0021-9150(90)90126-4
43. Florez H, Mendez A, Casanova-Romero P, Larreal-Urdaneta C, Castillo-Florez S, Lee DP, et al. Increased apolipoprotein C-III levels associated with insulin resistance contribute to dyslipidemia in normoglycemic and diabetic subjects from a triethnic population. Atherosclerosis. (2006) 188:134–41. doi: 10.1016/j.atherosclerosis.2005.10.013
44. Olivieri O, Bassi A, Stranieri C. Apolipoprotein C-III, metabolic syndrome, and risk of coronary artery disease. J Lipid Res. (2003) 44:2374–81. doi: 10.1194/jlr.M300253-JLR200
45. Austin MA, Talmud PJ, Farin FM. Association of apolipoprotein A5 variants with LDL particle size and triglyceride in Japanese Americans. Mol Basis Dis. (2004) 1688:1–9. doi: 10.1016/j.bbadis.2003.10.003
46. Masana L, Ribalta J, Salazar J. The apolipoprotein AV gene and diurnal triglyceridaemia in normolipidaemic subjects. Clin Chem Lab Med. (2003) 41:517–21. doi: 10.1515/CCLM.2003.078
47. Lee KW, Ayyobi AF, Frohlich JJ, Hill JS. APOA5 gene polymorphism modulates levels of triglyceride, HDL cholesterol and FERHDL but is not a risk factor for coronary artery disease. Atherosclerosis. (2004) 176:165–72. doi: 10.1016/j.atherosclerosis.2004.04.024
48. Lai CQ, Demissie S, Cupples LA. Influence of the APOA5 locus on plasma triglyceride, lipoprotein subclasses, and CVD risk in the Framingham Heart Study. J Lipid Res. (2004) 45:2096–105. doi: 10.1194/jlr.M400192-JLR200
49. Lai CQ, Tai ES, Tan CE. The APOA5 locus is a strong determinant of plasma triglyceride concentrations across ethnic groups in Singapore. J Lipid Res. (2003) 44:2365–73. doi: 10.1194/jlr.M300251-JLR200
50. Brown S, Ordovás JM, Campos H. Interaction between the APOC3 gene promoter polymorphisms, saturated fat intake and plasma lipoproteins. Atherosclerosis. (2003) 170:307–13. doi: 10.1016/S0021-9150(03)00293-4
51. Waterworth DM, Talmud PJ, Luan J. Variants in the APOC3 promoter insulin responsive element modulate insulin secretion and lipids in middle-aged men. Biochim Biophys Acta-Mol Basis Dis. (2003) 1637:200–6. doi: 10.1016/S0925-4439(03)00021-8
52. Garasto S, Rose G, Derango F, Berardelli M, Corsonello A, Feraco E, et al. The study of APOA1, APOC3 and APOA4 variability in healthy ageing people reveals another paradox in the oldest old subjects. Ann Hum Genet. (2003) 67:54–62. doi: 10.1046/j.1469-1809.2003.00008.x
53. Tian M, Li R, Shan Z. Comparison of Apolipoprotein B/A1 ratio, Framingham risk score and TC/HDL-c for predicting clinical outcomes in patients undergoing percutaneous coronary intervention. Lipids Health Dis. (2019) 18:202. doi: 10.1186/s12944-019-1144-y
54. Jacobson TA, Ito MK, Maki KC, Orringer CE, Bays HE, Jones PH, et al. National Lipid Association recommendations for patient-centered management of dyslipidemia: Part 1-executive summary. J Clin Lipidol. (2014) 8:473–88. doi: 10.1016/j.jacl.2014.07.007
55. Benn M, Nordestgaard BG, Jensen JS, Nilausen K, Meinertz H, Tybjaerg-Hansen A, et al. Mutation in apolipoprotein B associated with hypobetalipoproteinemia despite decreased binding to the low density lipoprotein receptor. J Biol Chem. (2005) 280:21052–60. doi: 10.1074/jbc.M413877200
56. Soria LF, Ludwig EH, Clarke HRG. Association between a specific apolipoprotein B mutation and familial defective apolipoprotein B-100. Proc Natl Acad Sci USA. (1989) 86:587–91. doi: 10.1073/pnas.86.2.587
57. Ooi EM, Barrett PHR, Chan DC. Dose-dependent effect of rosuvastatin on apolipoprotein B-100 kinetics in the metabolic syndrome. Atherosclerosis. (2008) 197:139–46. doi: 10.1016/j.atherosclerosis.2007.03.004
58. Aakerloef E, Joernvall H, Slotte H. Identification of apolipoprotein A1 and immunoglobulin as components of a serum complex that mediates activation of human sperm motility. Biochemistry. (1991) 30:8986–90. doi: 10.1021/bi00101a011
59. Pullinger CR, Hennessy LK, Chatterton JE. Familial ligand-defective apolipoprotein B. Identification of a new mutation that decreases LDL receptor binding affinity. J Clin Invest. (1995) 95:1225–34. doi: 10.1172/JCI117772
60. Guo T, Yin RX, Huang F, Yao LM, Lin WX, Pan SL, et al. Association between the DOCK7, PCSK9 and GALNT2 gene polymorphisms and serum lipid levels. Sci Rep. (2016) 6:19079. doi: 10.1038/srep19079
61. Guo T, Yin RX, Pan L, Yang S, Miao L, Huang F, et al. Integrative variants, haplotypes and diplotypes of the CAPN3 and FRMD5 genes and several environmental exposures associate with serum lipid variables. Sci Rep. (2017) 7:45119. doi: 10.1038/srep45119
62. Berkinbayev S, Rysuly M, Mussayev A, Blum K, Baitasova N, Mussagaliyeva A, et al. Apolipoprotein gene polymorphisms (APOB, APOC111, APOE) in the development of coronary heart disease in ethnic groups of Kazakhstan. J Gene Syndr Gene Therapy. (2014) 5:216. doi: 10.4172/2157-7412.1000216
63. Miao L, Yin RX, Wu JZ. The SRGAP2 SNPs, their haplotypes and G × E interactions on serum lipid traits. Sci Rep. (2017) 7:1–15. doi: 10.1038/s41598-017-10950-6
64. Andreotti G, Chen J, Gao YT. Polymorphisms of genes in the lipid metabolism pathway and risk of biliary tract cancers and stones: a population-based case-control study in Shanghai, China. Cancer Epidemiol Biomarkers Prev. (2008) 17:525–34. doi: 10.1158/1055-9965.EPI-07-2704
65. Liao YC, Lin HF, Rundek T. Multiple genetic determinants of plasma lipid levels in Caribbean Hispanics. Clin Biochem. (2008) 41:306–12. doi: 10.1016/j.clinbiochem.2007.11.011
66. Andreotti G, Menashe I, Chen J. Genetic determinants of serum lipid levels in Chinese subjects: a population-based study in Shanghai, China. Eur J Epidemiol. (2009) 24:763–74. doi: 10.1007/s10654-009-9402-3
67. Hochner H, Allard C, Granot-Hershkovitz E, Chen J, Sitlani CM, Sazdovska S, et al. Parent-of-origin effects of the APOB gene on adiposity in young adults. PLoS Genet. (2015) 11:e1005573. doi: 10.1371/journal.pgen.1005573
68. Shahid SU, Shabana, Cooper JA, Beaney KE, Li K, Rehman A, et al. Effect of SORT1, APOB and APOE polymorphisms on LDL-C and coronary heart disease in Pakistani subjects and their comparison with Northwick Park Heart Study II. Lipids Health Dis. (2016) 15:83. doi: 10.1186/s12944-016-0253-0
69. Mahley RW, Huang Y, Rall SC. Pathogenesis of type III hyperlipoproteinemia (dysbetalipoproteinemia). Questions, quandaries, and paradoxes. J Lipid Res. (1999) 40:1933–49. doi: 10.1016/S0022-2275(20)32417-2
70. Das HK, McPherson J, Bruns GA, Karathanasis SK, Breslow JL. Isolation, characterization, and mapping to chromosome 19 of the human apolipoprotein E gene. J Biol Chem. (1985) 260:6240–7. doi: 10.1016/S0021-9258(18)88963-3
71. Weisgraber KH, Rall SC, Mahley RW. Human E apoprotein heterogeneity. Cysteine-arginine interchanges in the amino acid sequence of the apo-E isoforms. J Biol Chem. (1981) 256:9077–83. doi: 10.1016/S0021-9258(19)52510-8
72. Weisgraber KH. Apolipoprotein E distribution among human plasma lipoproteins: role of the cysteine-arginine interchange at residue 112. J Lipid Res. (1990) 31:1503–11. doi: 10.1016/S0022-2275(20)42621-5
73. Bettens K, Sleegers K, Van Broeckhoven C. Genetic insights in Alzheimer's disease. Lancet Neurol. (2013) 12:92–104. doi: 10.1016/S1474-4422(12)70259-4
74. Chetty PS, Mayne L, Lund-Katz S, Englander SW, Phillips MC, et al. Helical structure, stability, and dynamics in human apolipoprotein E3 and E4 by hydrogen exchange and mass spectrometry. Proc Natl Acad Sci USA. (2017) 114:968–73. doi: 10.1073/pnas.1617523114
75. Zekraoui L, Lagarde JP, Raisonnier A. High frequency of the apolipoprotein E4 allele in African pygmies and most of the African populations in sub-Saharan Africa. Human Biol. (1997) 1997:575–81.
76. Stengard JH, Weiss KM, Sing CF. An ecological study of association between coronary heart disease mortality rates in men and the relative frequencies of common allelic variations in the gene coding for apolipoprotein E. Hum Genet. (1998) 103:234–41. doi: 10.1007/s004390050811
77. Gerdes LU, Gerdes C, Kervinen K, Savolainen M, Klausen IC, Hansen PS, et al. The apolipoprotein ε4 allele determines prognosis and the effect on prognosis of simvastatin in survivors of myocardial infarction: a substudy of the Scandinavian simvastatin survival study. Circulation. (2000) 101:1366–71. doi: 10.1161/01.CIR.101.12.1366
78. Wozniak MA, Faragher EB, Todd JA, Koram KA, Riley EM, Itzhaki RF, et al. Does apolipoprotein E polymorphism influence susceptibility to malaria? J Med Genet. (2003) 40:348–51. doi: 10.1136/jmg.40.5.348
79. Bahri R, Esteban E, Moral P, Hassine M, Ben Hamda K, et al. Apolipoprotein gene polymorphisms and plasma levels in healthy Tunisians and patients with coronary artery disease. Lipids Health Dis. (2008) 7:46. doi: 10.1186/1476-511X-7-46
80. They-They TP, Hamzi K, Moutawafik MT. Prevalence of angiotensin-converting enzyme, methylenetetrahydrofolatereductase, Factor V Leiden, prothrombin and apolipoprotein E gene polymorphisms in Morocco. Ann Hum Biol. (2010) 37:767–77. doi: 10.3109/03014461003738850
81. Elliott DA, Tsoi K, Holinkova S, Chan SL, Kim WS, Halliday GM, et al. Isoform-specific proteolysis of apolipoprotein-E in the brain. Neurobiol Aging. (2011) 32:257–71. doi: 10.1016/j.neurobiolaging.2009.02.006
82. Kumar NT, Liestol K, Loberg EM, Reims HM, Brorson SH, Maehlen J, et al. The apolipoprotein E polymorphism and cardiovascular diseases - an autopsy study. Cardiovasc Pathol. (2012) 21:461–9. doi: 10.1016/j.carpath.2012.02.005
83. Ringman JM, Elashoff D, Geschwind DH. Plasma signaling proteins in persons at genetic risk for Alzheimer disease: influence of APOE genotype-plasma biomarkers and AD genetic risk. Arch Neurol. (2012) 69:757–64. doi: 10.1001/archneurol.2012.277
84. Atadzhanov M, Mwaba MH, Mukomena PN, Lakhi S, Mwaba P. Frequency of APOE. MTHFR and ACE polymorphisms in the Zambian population. BMC Res Notes. (2014) 7:194. doi: 10.1186/1756-0500-7-194
85. Garatachea N, Emanuele E, Calero M, Fuku N, Arai Y, Abe Y, et al. ApoE gene and exceptional longevity: insights from three independent cohorts. Experi Gerontol. (2014) 53:16–23. doi: 10.1016/j.exger.2014.02.004
86. Marais AD, Solomon GAE, Blom DJ. Dysbetalipoproteinaemia: a mixed hyperlipidaemia of remnant lipoproteins due to mutations in apolipoprotein E. Crit Rev Clin Lab Sci. (2014) 51:46–62. doi: 10.3109/10408363.2013.870526
87. Ryu S, Atzmon G, Barzilai N, Raghavachari N, Suh Y. Genetic landscape of APOE in human longevity revealed by high-throughput sequencing. Mech Ageing Dev. (2016) 155:7–9. doi: 10.1016/j.mad.2016.02.010
88. Silva-Sena GG, Camporez D, Santos LRD. An association study of FOXO3 variant and longevity. Genet Mol Biol. (2018) 41:386–96. doi: 10.1590/1678-4685-gmb-2017-0169
89. Wei S, Gao L, Jiang Y, Shang S, Chen C, Dang L, et al. The apolipoprotein E ε4 allele-dependent relationship between serum lipid levels and cognitive function: a population-based cross-sectional study. Front Aging Neurosci. (2020) 12:44. doi: 10.3389/fnagi.2020.00044
90. Vijayan M, Reddy PH. Stroke, vascular dementia, and Alzheimer's disease: molecular links. J Alzheimers Dis. (2016) 54:427–43. doi: 10.3233/JAD-160527
91. Vijayan M, Kumar S, Bhatti JS, Reddy PH. Molecular links and biomarkers of stroke, vascular dementia, Alzheimer's disease. Prog Mol Biol Transl Sci. (2017) 146:95–126. doi: 10.1016/bs.pmbts.2016.12.014
92. Corder EH, Saunders AM, Risch NJ, Strittmatter WJ, Schmechel DE, Gaskell PC, et al. Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat Genet. (1994) 7:180–4. doi: 10.1038/ng0694-180
93. Strittmatter WJ, Weisgraber KH, Huang DY. Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc Natl Acad Sci USA. (1993) 90:8098–102. doi: 10.1073/pnas.90.17.8098
94. Simon-Sanchez J, Schulte C, Bras JM. Genome-wide association study reveals genetic risk underlying Parkinson's disease. Nat Genet. (2009) 41:1308.
95. Lee EG, Tulloch J, Chen S. Redefining transcriptional regulation of the APOE gene and its association with Alzheimer's disease. PLoS ONE. (2020) 15:e0227667. doi: 10.1371/journal.pone.0227667
96. Foraker J, Millard SP, Leong L, Thomson Z, Chen S, Keene CD, et al. The APOE gene is differentially methylated in Alzheimer's disease. JAD. (2015) 48:745–55. doi: 10.3233/JAD-143060
97. Liu J, Zhao W, Ware EB, Turner ST, Mosley TH, Smith JA, et al. DNA methylation in the APOE genomic region is associated with cognitive function in African Americans. BMC Med Genom. (2018) 11:43. doi: 10.1186/s12920-018-0363-9
98. Wroolie T, Roat-Shumway S, Watson K, Reiman E, Rasgon N. Effects of LDL cholesterol and statin use on verbal learning and memory in older adults at genetic risk for Alzheimer's disease. J Alzheimer's Dis. (2020) 75:903–10. doi: 10.3233/JAD-191090
99. Hu P, Qin YH, Lei FY, Pei J, Hu B, Lu L. Variable frequencies of apolipoprotein E genotypes and its effect on serum lipids in the Guangxi Zhuang and Han children. Int J Mol Sci. (2011) 12:5604–15. doi: 10.3390/ijms12095604
100. Corella D, Ordovás JM. Aging and cardiovascular diseases: The role of gene-diet interactions. Ageing Res Rev. (2014) 18:53–73. doi: 10.1016/j.arr.2014.08.002
101. Li XL, Sui JQ, Lu LL. Gene polymorphisms associated with non-alcoholic fatty liver disease and coronary artery disease: a concise review. Lipids Health Dis. (2016) 15:53. doi: 10.1186/s12944-016-0221-8
102. Sundaram M, Zhong S, Bou Khalil M, Links PH, Zhao Y, Iqbal J, et al. Expression of apolipoprotein C-III in McA-RH7777 cells enhances VLDL assembly and secretion under lipid-rich conditions. J Res. (2010) 51:150–61. doi: 10.1194/jlr.M900346-JLR200
103. Yao Z. Human apolipoprotein C-III - a new intrahepatic protein factor promoting assembly and secretion of very low density lipoproteins. Cardiovasc Hematol Disord Drug Targets. (2012) 12:133–40. doi: 10.2174/1871529X11202020133
104. Luo M, Peng D. The emerging role of apolipoprotein C-III: beyond effects on triglyceride metabolism. Lipids Health Dis. (2016) 15:184. doi: 10.1186/s12944-016-0352-y
105. Su X, Kong Y, Peng D. New insights into apolipoprotein A5 in controlling lipoprotein metabolism in obesity and the metabolic syndrome patients. Lipids Health Dis. (2018) 17:174. doi: 10.1186/s12944-018-0833-2
106. Satizabal CL, Samieri C, Davis-Plourde KL. APOE and the association of fatty acids with the risk of stroke, CHD and mortality. Stroke. (2018) 49:2822. doi: 10.1161/STROKEAHA.118.022132
107. Mathieu P, Poirier P, Pibarot P, Lemieux I, Despres JP. Visceral obesity: The link among inflammation, hypertension, cardiovascular disease. Hypertension. (2009) 53:577–84. doi: 10.1161/HYPERTENSIONAHA.108.110320
108. Neth BJ, Craft S. Insulin resistance and Alzheimer's disease: bioenergetic linkages. Front Aging Neurosci. (2017) 9:345. doi: 10.3389/fnagi.2017.00345
109. Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Investig. (2003) 112:1821–30. doi: 10.1172/JCI200319451
110. Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, et al. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. (2012) 485:512–6. doi: 10.1038/nature11087
111. Keene CD, Cudaback E, Li X. Apolipoprotein E isoforms and regulation of the innate immune response in brain of patients with Alzheimer's disease. Curr Opin Neurobiol. (2011) 21:920–8. doi: 10.1016/j.conb.2011.08.002
112. Zhang H, Wu LM, Wu J. Cross-talk between apolipoprotein E and cytokines. Mediators Inflamm. (2011) 2011:949072. doi: 10.1155/2011/949072
113. Ghebranious N, Mukesh B, Giampietro PF, Glurich I, Mickel SF, Waring SC, et al. A pilot study of gene/gene and gene/environment interactions in Alzheimer disease. Clin Med Res. (2011) 9:17–25. doi: 10.3121/cmr.2010.894
114. Zade D, Beiser A, McGlinchey R, Au R, Seshadri S, Palumbo C, et al. Apolipoprotein epsilon 4 allele modifies waist-to-hip ratio effects on cognition and brain structure. J Stroke Cerebrovasc. Dis. (2013) 22:119–25. doi: 10.1016/j.jstrokecerebrovasdis.2011.06.020
115. Backman K, Joas E, Waern M, Östling S, Guo X, Blennow K, et al. 37 years of body mass index and dementia: effect modification by the APOE genotype: observations from the prospective population study of Women in Gothenburg, Sweden. J Alzheimers Dis. (2015) 48:1119–27. doi: 10.3233/JAD-150326
116. Cherbuin N, Sargent-Cox K, Fraser M, Sachdev P, Anstey KJ. Being overweight is associated with hippocampal atrophy: The PATH Through Life Study. Int J Obes. (2015) 39:1509–14. doi: 10.1038/ijo.2015.106
117. Gustafson DR, Karlsson C, Skoog I, Rosengren L, Lissner L, Blennow K. Mid-life adiposity factors relate to blood-brain barrier integrity in late life. J Int Med. (2007) 262:643–50. doi: 10.1111/j.1365-2796.2007.01869.x
118. Moser VA, Pike CJ. Obesity accelerates Alzheimer-related pathology in APOE4 but not APOE3 Mice. Eneuro. (2017) 4:17. doi: 10.1523/ENEURO.0077-17.2017
119. Yassine HN, Finch CE. APOE alleles and diet in brain aging and Alzheimer's Disease. Front Aging Neurosci. (2020) 12:150. doi: 10.3389/fnagi.2020.00150
120. Brown GC, Neher JJ. Microglial phagocytosis of live neurons. Nat Rev Neurosci. (2014) 15:209–16. doi: 10.1038/nrn3710
121. Shi Y, Holtzman DM. Interplay between innate immunity and Alzheimer disease: APOE and TREM2 in the spotlight. Nat Rev Immunol. (2018) 18:759–72. doi: 10.1038/s41577-018-0051-1
122. Houtman J, Freitag K, Gimber N, Schmoranzer J, Heppner FL, Jendrach M. Beclin1-driven autophagy modulates the inflammatory response of microglia via NLRP3. EMBO J. (2019) 38:e99430. doi: 10.15252/embj.201899430
123. Zhang HL, Wu J, Zhu J. The immune-modulatory role of apolipoprotein E with emphasis on multiple sclerosis and experimental autoimmune encephalomyelitis. Clin Dev Immunol. (2010) 2010:1–10. doi: 10.1155/2010/186813
124. Centa M, Prokopec KE, Garimella MG, Habir K, Hofste L, Stark JM, et al. Acute loss of apolipoprotein E triggers an autoimmune response that accelerates atherosclerosis. Arteriosclerosis. (2018) 38:e145–58. doi: 10.1161/ATVBAHA.118.310802
125. Fazekas F, Strasser-Fuchs S, Kollegger H, Berger T, Kristoferitsch W, Schmidt H, et al. Apolipoprotein E ε4 is associated with rapid progression of multiple sclerosis. Neurol. (2001) 57:853–7. doi: 10.1212/WNL.57.5.853
126. Vijayan M, Chinniah R, Ravi PM, Mosses Joseph AK, Vellaiappan NA, Krishnan JI, et al. ACE-II genotype and I allele predicts ischemic stroke among males in south India. Meta Gene. (2014) 2:661–9. doi: 10.1016/j.mgene.2014.09.003
127. Vijayan M, Chinniah R, Ravi PM, Sivanadham R, Mosses Joseph AK, Vellaiappan NA, et al. MTHFR (C677T) CT genotype and CT-apoE3/3 genotypic combination predisposes the risk of ischemic stroke. Gene. (2016) 591:465–70. doi: 10.1016/j.gene.2016.06.062
128. Vijayan M, Reddy PH. Peripheral biomarkers of stroke: Focus on circulatory microRNAs. Biochim Biophys Acta. (2016) 1862:1984–93. doi: 10.1016/j.bbadis.2016.08.003
129. Vijayan M, Kumar S, Yin X, Zafer D, Chanana V, Cengiz P, et al. Identification of novel circulatory microRNA signatures linked to patients with ischemic stroke. Hum Mol Genet. (2018) 27:2318–29. doi: 10.1093/hmg/ddy136
130. GBD 2016 Stroke Collaborators. Global, regional, and national burden of stroke, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. (2019) 18:439–58. doi: 10.1016/S1474-4422(19)30034-1
131. Kuriakose D, Xiao Z. Pathophysiology and treatment of stroke: present status and future perspectives. Int J Mol Sci. (2020) 21:7609. doi: 10.3390/ijms21207609
132. Nyberg L, Salami A. The APOE ε4 allele in relation to brain white-matter microstructure in adulthood and aging. Scand J Psychol. (2014) 55:263–7. doi: 10.1111/sjop.12099
133. Lv P, Zheng Y, Huang J, Ke J, Zhang H. Association of apolipoprotein E gene polymorphism with ischemic stroke in coronary heart disease patients treated with medium-intensity statins. Neuropsychiatr Dis Treat. (2020) 16:2459–66. doi: 10.2147/NDT.S265194
134. Zhang R, Wang X, Tang Z, Liu J, Yang S, Zhang Y, et al. Apolipoprotein E gene polymorphism and the risk of intracerebral hemorrhage: a meta-analysis of epidemiologic studies. Lipids Health Dis. (2014) 13:47. doi: 10.1186/1476-511X-13-47
135. Chen C, Hu Z. ApoE polymorphisms and the risk of different subtypes of stroke in the Chinese population: a comprehensive meta-analysis. Cerebrovasc Dis. (2016) 41:119–38. doi: 10.1159/000442678
Keywords: apolipoproteins, polymorphisms, haplotypes, medical complications, global population
Citation: Basavaraju P, Balasubramani R, Kathiresan DS, Devaraj I, Babu K, Alagarsamy V and Puthamohan VM (2022) Genetic Regulatory Networks of Apolipoproteins and Associated Medical Risks. Front. Cardiovasc. Med. 8:788852. doi: 10.3389/fcvm.2021.788852
Received: 03 October 2021; Accepted: 22 November 2021;
Published: 06 January 2022.
Edited by:
Murali Vijayan, Texas Tech University Health Sciences Center, United StatesReviewed by:
Aravind Ramanathan, Eunice Kennedy Shriver National Institute of Child Health and Human Development (NICHD), United StatesCopyright © 2022 Basavaraju, Balasubramani, Kathiresan, Devaraj, Babu, Alagarsamy and Puthamohan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vinayaga Moorthi Puthamohan, dmluYXlwdXRodUBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.