 Michelle M. Monasky
Michelle M. Monasky Emanuele Micaglio
Emanuele Micaglio Sara D'Imperio
Sara D'Imperio Carlo Pappone
Carlo Pappone- 1Arrhythmology Department, IRCCS Policlinico San Donato, San Donato Milanese, Milan, Italy
- 2Vita-Salute San Raffaele University, Milan, Italy
Ajmaline is an anti-arrhythmic drug that is used to unmask the type-1 Brugada syndrome (BrS) electrocardiogram pattern to diagnose the syndrome. Thus, the disease is defined at its core as a particular response to this or other drugs. Ajmaline is usually described as a sodium-channel blocker, and most research into the mechanism of BrS has centered around this idea that the sodium channel is somehow impaired in BrS, and thus the genetics research has placed much emphasis on sodium channel gene mutations, especially the gene SCN5A, to the point that it has even been suggested that only the SCN5A gene should be screened in BrS patients. However, pathogenic rare variants in SCN5A are identified in only 20–30% of cases, and recent data indicates that SCN5A variants are actually, in many cases, prognostic rather than diagnostic, resulting in a more severe phenotype. Furthermore, the misconception by some that ajmaline only influences the sodium current is flawed, in that ajmaline actually acts additionally on potassium and calcium currents, as well as mitochondria and metabolic pathways. Clinical studies have implicated several candidate genes in BrS, encoding not only for sodium, potassium, and calcium channel proteins, but also for signaling-related, scaffolding-related, sarcomeric, and mitochondrial proteins. Thus, these proteins, as well as any proteins that act upon them, could prove absolutely relevant in the mechanism of BrS.
Introduction
Ajmaline is used as a pharmacologic test to diagnose Brugada syndrome (BrS) and identify people who are at higher risk of developing life-threatening arrhythmias and sudden cardiac death. Many patients are ultimately implanted with an implantable cardioverter-defibrillator that can save their lives. The BrS is an inherited disease characterized by a coved-type ST-segment elevation in the right precordial leads (V1-V3) on the electrocardiogram (ECG). The true prevalence of BrS is unknown, since many people are asymptomatic. In fact, the syndrome may not even be suspected until an incidence of cardiac arrest. Certain “trigger situations,” such as fever, drug use, or consumption of alcohol or large meals can elicit the BrS ECG pattern (1). Since the systematic introduction of sodium-channel blockers to screen for the syndrome, the diagnosis, and thus the perceived incidence, of BrS has increased (2).
Sodium channel blockers, such as ajmaline, flecainide, or procainamide can be used to provoke the type-1 BrS ECG pattern, which is said to affirmatively diagnose the syndrome (3, 4). Thus, the disease is defined at its core as a particular response to these drugs. Some clinicians prefer the use of ajmaline, which appears to have a lower false negative rate, due to its higher sensitivity (5, 6). This higher sensitivity of ajmaline, compared to flecainide, may be due to flecainide's greater inhibition of Ito, which then renders it less effective (5). Whole-cell patch clamp experiments demonstrated a reduced Ito total charge with an IC50 of 216 and 15.2 μM for ajmaline and flecainide, respectively, while sodium channel current was affected similarly by both drugs, as suggested by equivalent changes in QRS and PQ intervals (5). However, reports have cautioned about ajmaline's false positive rate, stating that a positive ajmaline test does not always mean that a patient has BrS (7–10). In fact, ajmaline metabolism is very complex (11) for several reasons relating to the liver metabolism, kidney metabolism, plasma proteins binding, and variability in the expression of ajmaline-metabolizing enzymes (12). Ajmaline undergoes some major metabolic pathways: mono- and di-hydroxylation of the benzene ring with subsequent O-methylation, reduction of the C-21, oxidation of both C-17 and C-21-hydroxyl function and N-oxidation (13). Consequently, one of the major genes controlling ajmaline metabolism is CYP2D6, encoding for a cytochrome C component. Thus, it is not surprising that patients harboring variants or even polymorphisms in the CYP2D6 gene might display a different capability to metabolize ajmaline (14). To date, more than 70 allelic variants of the CYP2D6 gene have been reported, and altered CYP2D6 function has been associated with both adverse drug reactions and reduced drug efficacy (15). This is the main reason why poor metabolizer alleles can be important as a possible cause of false positivity during ajmaline challenge test.
Ajmaline challenges must be conducted in specialized centers due to the potential development of life-threatening ventricular arrhythmias, such as polymorphic ventricular tachycardia (VT) or ventricular fibrillation (VF) (16–18). Ajmaline infusion should be done carefully, stopping as soon as the result is positive or when QRS broadens to ≥130% of baseline or frequent pre-mature ventricular complexes occur (17, 19, 20).
Ajmaline is usually described as a sodium-channel blocker (3), and most research into the mechanism of BrS has centered around this idea that the sodium channel is somehow impaired in BrS (21, 22), and thus the genetics research has placed much emphasis on sodium channel gene mutations, especially the gene SCN5A, whereas systematic studies on other genes are lacking (23). The research up until this point has focused so much on the SCN5A gene that it has even been suggested that only the SCN5A gene should be screened in BrS patients (23), something that has been hotly debated (24–26), as many argue that research is needed to understand the possible role of several other genes in this disease (27–32). However, pathogenic rare variants in SCN5A are identified in only 20–30% of ajmaline-positive cases (33–36), and recent data indicates that mutations in SCN5A are actually, in many cases, prognostic rather than diagnostic, resulting in a more severe phenotype (26, 35, 37–39). Furthermore, the misconception by some that ajmaline only influences the sodium current, and thus sodium channels should be the only channels of interest in BrS, is flawed, in that ajmaline actually acts additionally on potassium and calcium currents, as well as mitochondria and metabolic pathways. Thus, potassium channels, calcium channels, mitochondrial proteins, and metabolic pathway proteins, or factors that act upon these proteins, could prove absolutely relevant, as their function is directly influenced by the very drug that is used to diagnose the disease in the first place.
Multiple Binding Sites of Ajmaline On Na+, K+, and Ca2+ Channels
Ajmaline has multiple sites of action, including sodium, potassium, and calcium channels. Plant alkaloids, including ajmaline, act on at least six receptor sites on voltage-gated Na+ channels (40). In single intact amphibian skeletal muscle fibers, it appeared that ajmaline has multiple sites of action, including the positively charged S4 voltage-sensing segment of Na+ and K+ channels (40). However, ajmaline also blocks channels that do not have a voltage sensor (e.g., KATP) (40).
In human embryonic kidney (HEK) cells, ajmaline has an inhibitory effect on human ether a-go-go related gene (HERG) potassium channels in the open, but not in the closed states, and probably binds at aromatic residues Tyr-652 and Phe-656 in the channel pore cavity (41). The inhibitory effect was stronger at higher frequencies (41). Ajmaline is an open channel inhibitor at therapeutic concentrations of cardiac potassium KV1.5 and KV4.3 channels, responsible for cardiac IKur and Ito current, respectively (42). Ajmaline potently blocks glibenclamide-sensitive K+ channels in Xenopus oocytes in a concentration-dependent manner (43). There is an effect of ajmaline on the inhibition of K+ outflow from rat liver mitochondria (44). In rat right ventricular myocytes, the decreased amplitude and time integral of Ito by ajmaline is dependent on concentration, but not frequency or use (45). In rat right ventricular myocytes, ajmaline blocks the transient outward potassium current (Ito) when the channel is in the open state and there is fast recovery from the block at resting voltage (45).
Whole cell patch clamp technique used to determine the effect of ajmaline on action potential (AP) and ionic current components in rat right ventricular myocytes demonstrated an inhibitory effect on sodium current (INa), L-type calcium current (ICa−L), transient outward potassium current (Ito), the current measured at the end of 300 ms depolarizing pulse (IK,end), and ATP-sensitive potassium current [IK(ATP)] (46). The inhibition of INa causes both the decreased rate of rise of depolarizing phase and the lowered amplitude of AP (46). Additionally, Ito inhibition was responsible for AP prolongation after ajmaline administration (46). In isolated guinea pig ventricular cardiomyocytes, ajmaline suppressed calcium currents (ICa) in a dose-dependent manner without affecting the steady-state inactivation kinetics and the voltage dependency of the current-voltage relationship, inhibited inwardly rectifying potassium current (IK1), and decreased the delayed rectifier potassium current (IK) without altering the activation or deactivation time courses (47). A study recording intracellular action potentials and transmural ECG in canine RV wedge preparations suggested that combined sodium and calcium channel block may be more effective than sodium channel block alone in unmasking the BrS pattern (48). The study used terfenadine to block both sodium and calcium current, which resulted in the loss of the epicardial AP dome, ST segment elevation, phase 2 reentry, and spontaneous polymorphic VT/VF (48). This effect of terfenadine was normalized with 4-aminopyridine, which inhibits Ito (48). The drugs flecainide, ajmaline, and procainamide alone did not generate polymorphic VT, but they did so together with the calcium channel blocker verapamil (48).
N-propyl ajmaline (NPA) is the quaternary derivative of ajmaline. The permanently charged NPA and protonated ajmaline both act mainly with open channels, while unprotonated ajmaline acts mainly on inactivated Na+ channels in frog myelinated fibers (49). In frog myelinated fibers, sodium and potassium currents are inhibited by ajmaline and NPA, for sodium in both directions, but for potassium, only the outward potassium current, not the inward potassium current (49). The location of the binding sites have been suggested to be in the inner mouths of Na+ and K+ channels (49). In voltage clamp experiments using frog nodes of Ranvier, the binding site for NPA has been described to be located in the inner mouth of the Na+ channels, and it becomes available to the charged blocker (NPA) only after opening of the activation gate (50). NPA in enzymatically isolated cells of adult rats inhibits INa due to a voltage-dependent interaction with open Na+ channels, and NPA has similar blocking effects on Na+ channels in myocardial cells and nerve fibers (51).
Genetics of Channels Implicated by Functional Studies
Functional studies have identified several molecular targets of ajmaline. Many of these molecular targets are encoded for by genes that have been associated with BrS in clinical studies. Table 1 lists the known molecular targets of ajmaline and their related genes. Figure 1 shows a schematic of ajmaline targets in the cell, as demonstrated by functional studies.
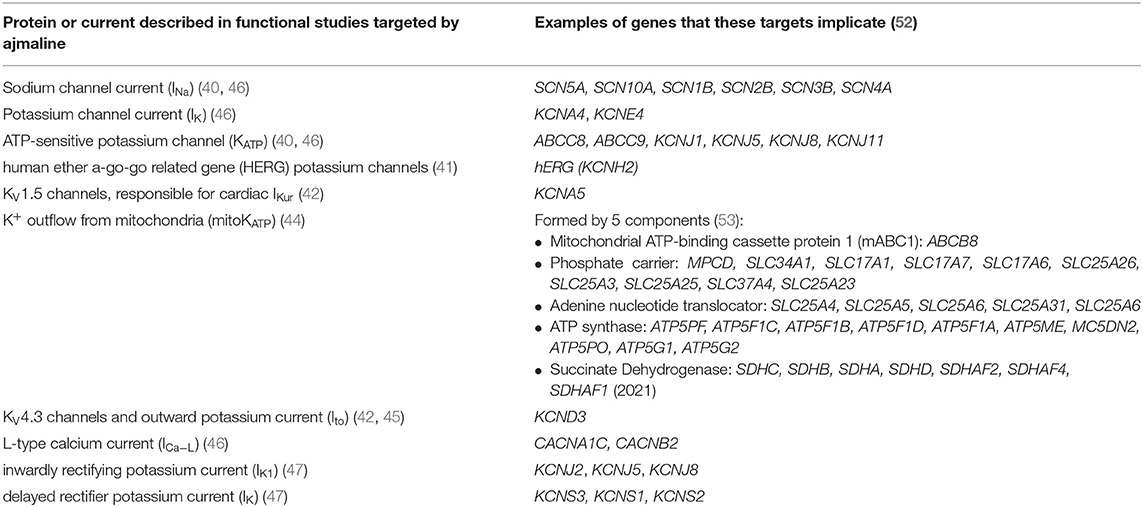
Table 1. Known molecular targets of ajmaline and potential genes that they implicate.
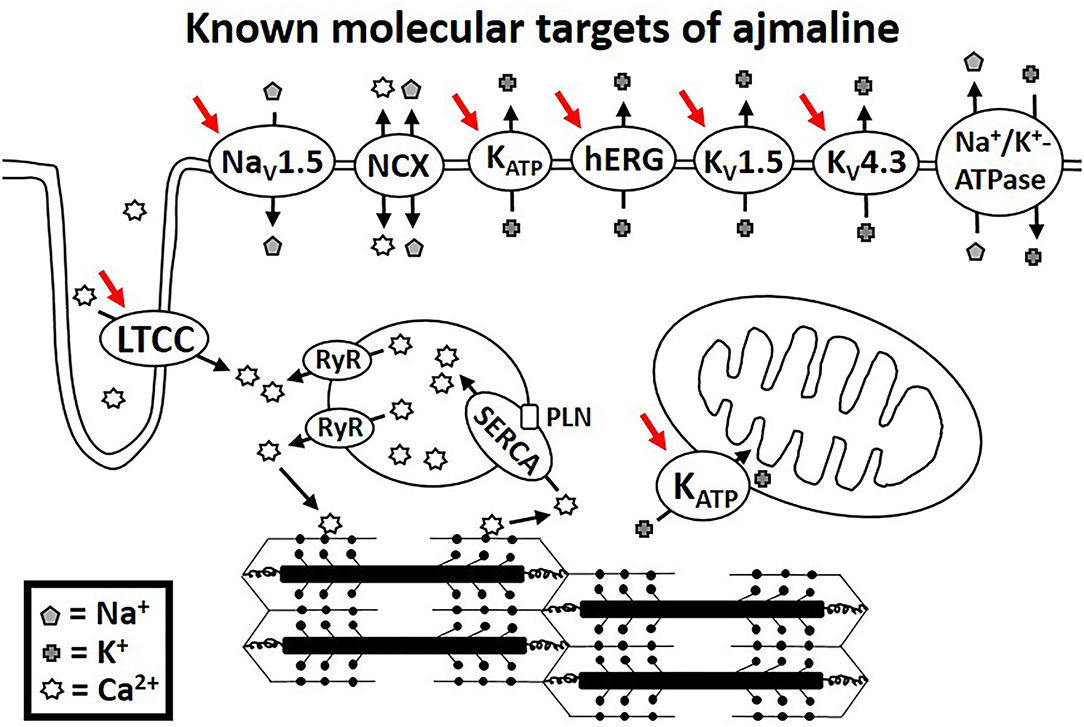
Figure 1. Molecular targets of ajmaline as suggested by functional studies indicated by red arrows.
A Unique Role for Calcium: Excitation-Contraction Coupling in Brugada Syndrome
Calcium signaling is responsible for connecting the electrical signaling of the cell to the mechanical force of contraction and relaxation of the sarcomeric proteins. Thus, calcium imbalances in the cell could result in alterations to the force production. In porcine epicardial shavings, excitation failure by current-to-load mismatch was shown to cause ST segment elevation modulated by Ito and ICaL (54). A study by Biamino et al. demonstrated a relaxing effect of ajmaline on vascular smooth muscle using aortic helical strips, attributing the effect possibly to a reduction in Ca2+ and probably Na+ conductance (55). In BrS patients, ajmaline administration results in a decrease of right ventricular ejection fraction and minimum principal strain in the right ventricular outflow tract and right ventricular anterior wall (56, 57). In fact, it has been previously suggested that the electromechanical coupling in BrS, including calcium handling and sarcomeric alterations, should be investigated (28, 57). Reduced intracellular calcium, which may result in a reduction of force production, has been proposed as a possible mechanism in BrS (8, 28, 58, 59). Additionally, administration of pharmaceuticals that act on outer cell membrane receptors can result in signaling changes within the cell (60, 61). It would be interesting to see in future studies whether ajmaline affects intracellular processes, such as signaling pathways that lead to post-translational modifications, affecting various proteins, such as those located in the sarcoplasmic reticulum or the myofilaments.
Genetics of Channels Implicated by Clinical Studies
The genetics of BrS remains a hotly debated subject. More than 20 genes are currently included in diagnostic genetic testing panels, previously reviewed in detail elsewhere (32), although the significance of variants in all but the SCN5A gene are disputed, since most studies to-date have focused on understanding better variants in the SCN5A gene, while studies on the other genes are generally lacking (23). However, pathogenic rare variants in SCN5A are identified in only 20–30% of ajmaline-positive cases (33–36), and recent data indicates that mutations in SCN5A are actually, in many cases, prognostic rather than diagnostic, resulting in a more severe phenotype (26, 35, 37–39). Several important studies of other genes are now available, and more are needed to better understand the mechanism of ajmaline in provoking the type-1 BrS ECG pattern.
Sodium channel-related genes other than SCN5A that have been previously implicated in BrS, and they include SCN10A, SCN1B, SCN2B, SCN3B, SCN4B, RANGRF (MOG1), and GPD1L. Potassium-related genes previously associated in BrS include KCND2, KCND3, KCNE1, KCNE2, KCNE3, KCNE5, KCNH2, KCNJ2, KCNJ5, KCNJ8, KCNQ1, ABCC9, and HCN4, while calcium-related genes previously described in BrS include CACNA1C, CACNA2D1, CACNB2, RYR2, and TRPM4 (32, 62). In addition, the gene PKP2 has been associated with BrS, and studies have shown a relationship between PKP2 and both sodium and potassium channels. For example, in a study by Cerrone et al., loss of PKP2 caused decreased INa and NaV1.5 (63). Hong et al. demonstrated an interaction between PKP2 and KATP channels in rat heart (64).
Sarcomeric properties have been directly linked to arrythmogenic sudden death (61, 65), and variants in myofilament genes, including TPM1 and MYBPC3, have been found in BrS patients (27, 66, 67). Several other genes, encoding signaling and scaffolding proteins, including AKAP9, ANK2, CASQ2, CAV3, CBL, DSC2, DSG2, DSP, FGF12, HEY2, JUP, LMNA, LRRC10, NOS1AP, SEMA3A, SLMAP, SNTA1, and TMEM43, have been suggested as candidate genes (32, 62, 68). The function of proteins that are affected by protein kinase A or reactive oxygen species (ROS), such as the protein products of many of the genes listed above, may be altered by changes in mitochondrial function, which is responsible for ATP and ROS production (26). In fact, studies have implicated a direct role for mitochondria in BrS, specifically, severe cases have been associated with a particular mitochondrial DNA (mtDNA) allelic combination and a high number of mtDNA single nucleotide polymorphisms (69, 70), and a role for mitochondrial transfer RNA genes has been suggested (71). Thus, in addition to SCN5A, various other genes have been suggested to have a role in BrS, including other sodium channel-related genes, as well as several potassium-related, calcium-related, signaling-related, scaffolding-related, sarcomeric, and mitochondrial genes, consistent with the identified molecular targets of the ajmaline drug used to unmask and diagnosis the syndrome.
Although it is generally agreed that variants in the SCN5A gene are involved in BrS, it is important to think of variants even within this gene as individual variants with specific effects, rather than thinking of all SCN5A variants collectively, as some may be benign, while others pathogenic (26). Along these lines, several studies have sought to understand the effect of specific SCN5A variants (37, 72–80). It has been recently suggested that variants in the SCN5A gene may actually be prognostic, rather than diagnostic (35, 38, 39).
Studies to better understand the role of variants in each of the above-mentioned genes will be an important area of future research. A recent study by Di Mauro et al. demonstrated an important role for CACNA1C (31), highlighting the importance of functional studies of genes that may be involved in BrS, but for which we currently lack the proof (81). Recent studies have also focused on the roles of the genes SCN10A and HEY2 in BrS (29, 82). However, much work remains to be done before we can understand the role of each of the protein products of these genes, as well as the role of the proteins that signal to them and alter their function. Currently, the understanding of genetics in BrS is in its infancy, and genetic testing alone should not be used for diagnostic purposes, but rather, diagnosis of BrS should be based upon an arrhythmological examination by a specialized cardiologist (26). The presence of a variant in the SCN5A gene, however, may be relevant for prognostic purposes (35, 38).
Limitations and Future Studies
Most of the studies to better understand the mechanism of ajmaline have been performed in cellular models using non-cardiomyocyte cell types or in animal models that are sometimes not even mammalian. While these models give us some insight, each model comes with its own set of advantages and limitations (83). The functional studies performed to-date indicate that ajmaline does not act solely on sodium channels and suggests that clinical genetic testing could be expanded for research purposes to include, for example, genes that encode for potassium and calcium channels. Thus, the mechanism of BrS could be researched from also this clinical direction. Regarding future functional studies, it would be interesting to quantify ajmaline signaling to, and effects on, particular sodium, potassium, and calcium channels and the resulting effect of sodium, potassium, and calcium handling, to ultimately understand the mechanism behind the altered ECG.
Conclusion
The misconception by some that ajmaline only influences the sodium current, and thus sodium channels should be the only channels of interest in BrS, is flawed, in that ajmaline actually acts additionally on potassium and calcium currents, as well as mitochondria and metabolic pathways. Clinical studies have implicated several candidate genes in BrS, encoding not only for sodium, potassium, and calcium channel proteins, but also for signaling-related, scaffolding-related, sarcomeric, and mitochondrial proteins. Thus, these proteins, as well as any proteins that act upon them, could prove absolutely relevant in the mechanism of BrS.
Author Contributions
MM: conceptualization and writing—original draft preparation. MM, EM, and SD'I: literature search and writing—draft revision. CP: funding acquisition. MM, EM, SD'I, and CP: reviewed and provided comments. All authors have read and agreed to the published version of the manuscript.
Funding
This study was partially supported by Ricerca Corrente funding from Italian Ministry of Health to IRCCS Policlinico San Donato. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript, or in the decision to publish the results.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
AP, action potential; BrS, Brugada syndrome; Ca2+, calcium; ECG, electrocardiogram; HEK, human embryonic kidney; HERG, human ether a-go-go related gene; ICa−L, L-type calcium current; IK, delayed rectifier potassium current; IK1, inwardly rectifying potassium current; IK(ATP), ATP-sensitive potassium current; IK,end, the current measured at the end of 300 ms depolarizing pulse; IKur, ultrarapid outward potassium current; INa, sodium current; Ito, transient outward potassium current; K+, potassium; KATP, ATP-sensitive potassium channel; Na+, sodium; NCX, sodium-calcium exchanger; NPA, N-propyl ajmaline, a quaternary derivative of ajmaline; PVCs, pre-mature ventricular complexes; RV, right ventricle; Vas, ventricular arrhythmias; VF, ventricular fibrillation; VT, ventricular tachycardia.
References
1. D'imperio S, Monasky MM, Micaglio E, Negro G, Pappone C. Impact of dietary factors on Brugada syndrome and long QT syndrome. Nutrients. (2021) 13:2482. doi: 10.3390/nu13082482
2. Priori SG, Blomstrom-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: the task force for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. (2015) 36:2793–867. doi: 10.1093/eurheartj/ehv316
3. Brugada R, Brugada J, Antzelevitch C, Kirsch GE, Potenza D, Towbin JA, et al. Sodium channel blockers identify risk for sudden death in patients with ST-segment elevation and right bundle branch block but structurally normal hearts. Circulation. (2000) 101:510–5. doi: 10.1161/01.CIR.101.5.510
4. Therasse D, Sacher F, Petit B, Babuty D, Mabo P, Martins R, et al. Sodium-channel blocker challenge in the familial screening of Brugada syndrome: safety and predictors of positivity. Heart Rhythm. (2017) 14:1442–8. doi: 10.1016/j.hrthm.2017.06.031
5. Wolpert C, Echternach C, Veltmann C, Antzelevitch C, Thomas GP, Spehl S, et al. Intravenous drug challenge using flecainide and ajmaline in patients with Brugada syndrome. Heart Rhythm. (2005) 2:254–60. doi: 10.1016/j.hrthm.2004.11.025
6. Cheung CC, Mellor G, Deyell MW, Ensam B, Batchvarov V, Papadakis M, et al. Comparison of ajmaline and procainamide provocation tests in the diagnosis of brugada syndrome. JACC Clin Electrophysiol. (2019) 5:504–12. doi: 10.1016/j.jacep.2019.01.026
7. Viskin S, Rosso R, Friedensohn L, Havakuk O, Wilde AA. Everybody has Brugada syndrome until proven otherwise? Heart Rhythm. (2015) 12:1595–8. doi: 10.1016/j.hrthm.2015.04.017
8. Antzelevitch C, Yan GX, Ackerman MJ, Borggrefe M, Corrado D, Guo J, et al. J-Wave syndromes expert consensus conference report: emerging concepts and gaps in knowledge. Heart Rhythm. (2016) 13:e295–324. doi: 10.1016/j.hrthm.2016.05.024
9. Tadros R, Nannenberg EA, Lieve KV, Skoric-Milosavljevic D, Lahrouchi N, Lekanne Deprez RH, et al. Yield and pitfalls of ajmaline testing in the evaluation of unexplained cardiac arrest and sudden unexplained death: single-center experience with 482 families. JACC Clin Electrophysiol. (2017) 3:1400–8. doi: 10.1016/j.jacep.2017.04.005
10. Viskin S, Rosso R. Read my lips: a positive ajmaline test does not always mean you have brugada syndrome. JACC Clin Electrophysiol. (2017) 3:1409–11. doi: 10.1016/j.jacep.2017.05.016
11. Yasuhara M, Hashimoto Y, Okumura K, Hori R, Sakurai T, Kawai C. Kinetics of ajmaline disposition and pharmacologic response in beagle dogs. J Pharmacokinet Biopharm. (1987) 15:39–55. doi: 10.1007/BF01062938
12. Padrini R, Piovan D, Javarnaro A, Cucchini F, Ferrari M. Pharmacokinetics and electrophysiological effects of intravenous ajmaline. Clin Pharmacokinet. (1993) 25:408–14. doi: 10.2165/00003088-199325050-00006
13. Koppel C, Tenczer J, Arndt I. Metabolic disposition of ajmaline. Eur J Drug Metab Pharmacokinet. (1989) 14:309–16. doi: 10.1007/BF03190117
14. Zhou SF. Polymorphism of human cytochrome P450 2D6 and its clinical significance: part I. Clin Pharmacokinet. (2009) 48:689–723. doi: 10.2165/11318030-000000000-00000
15. Taylor C, Crosby I, Yip V, Maguire P, Pirmohamed M, Turner RM. A Review of the important role of CYP2D6 in Pharmacogenomics. Genes (Basel). (2020) 11:1295. doi: 10.3390/genes11111295
16. Conte G, Sieira J, Sarkozy A, De Asmundis C, Di Giovanni G, Chierchia GB, et al. Life-threatening ventricular arrhythmias during ajmaline challenge in patients with Brugada syndrome: incidence, clinical features, and prognosis. Heart Rhythm. (2013) 10:1869–74. doi: 10.1016/j.hrthm.2013.09.060
17. Poli S, Toniolo M, Maiani M, Zanuttini D, Rebellato L, Vendramin I, et al. Management of untreatable ventricular arrhythmias during pharmacologic challenges with sodium channel blockers for suspected Brugada syndrome. Europace. (2018) 20:234–42. doi: 10.1093/europace/eux092
18. Ciconte G, Monasky MM, Vicedomini G, Borrelli V, Giannelli L, Pappone C. Unusual response to ajmaline test in Brugada syndrome patient leads to extracorporeal membrane oxygenator support. Europace. (2019) 21:1574. doi: 10.1093/europace/euz139
19. Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Brugada R, Corrado D, et al. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. (2005) 111:659–70. doi: 10.1161/01.CIR.0000152479.54298.51
20. Nault I, Champagne J. How safe is ajmaline challenge in patients with suspected Brugada syndrome? Heart Rhythm. (2013) 10:1875–6. doi: 10.1016/j.hrthm.2013.10.047
21. Bebarova M, O'hara T, Geelen JL, Jongbloed RJ, Timmermans C, Arens YH, et al. Subepicardial phase 0 block and discontinuous transmural conduction underlie right precordial ST-segment elevation by a SCN5A loss-of-function mutation. Am J Physiol Heart Circ Physiol. (2008) 295:H48–58. doi: 10.1152/ajpheart.91495.2007
22. Petitprez S, Jespersen T, Pruvot E, Keller DI, Corbaz C, Schlapfer J, et al. Analyses of a novel SCN5A mutation (C1850S): conduction vs. repolarization disorder hypotheses in the Brugada syndrome. Cardiovasc Res. (2008) 78:494–504. doi: 10.1093/cvr/cvn023
23. Hosseini SM, Kim R, Udupa S, Costain G, Jobling R, Liston E, et al. Reappraisal of reported genes for sudden arrhythmic death. Circulation. (2018) 138:1195–205. doi: 10.1161/CIRCULATIONAHA.118.035070
24. London B. Reappraisal of reported genes for sudden arrhythmic death: evidence-based evaluation of gene validity for brugada syndromeCirculation. (2019) 139:1758–9. doi: 10.1161/CIRCULATIONAHA.118.036889
25. Wilde AM, Gollob MH. Reappraisal of Reported Genes for Sudden Arrhythmic Death: Evidence-Based Evaluation of Gene Validity for Brugada Syndrome. Circulation. (2019) 139:1760–1. doi: 10.1161/CIRCULATIONAHA.119.039065
26. Monasky MM, Micaglio E, Locati ET, Pappone C. Evaluating the use of genetics in brugada syndrome risk stratification. Front Cardiovasc Med. (2021) 8:652027. doi: 10.3389/fcvm.2021.652027
27. Monasky MM, Ciconte G, Anastasia L, Pappone C. Commentary: next generation sequencing and linkage analysis for the molecular diagnosis of a novel overlapping syndrome characterized by hypertrophic cardiomyopathy and typical electrical instability of Brugada syndrome. Front Physiol. (2017) 8:1056. doi: 10.3389/fphys.2017.01056
28. Monasky MM, Pappone C, Piccoli M, Ghiroldi A, Micaglio E, Anastasia L. Calcium in Brugada syndrome: questions for future research. Front Physiol. (2018) 9:1088. doi: 10.3389/fphys.2018.01088
29. Monasky MM, Micaglio E, Vicedomini G, Locati ET, Ciconte G, Giannelli L, et al. Comparable clinical characteristics in Brugada syndrome patients harboring SCN5A or novel SCN10A variants. Europace. (2019) 21:1550–8. doi: 10.1093/europace/euz186
30. Campuzano O, Sarquella-Brugada G, Cesar S, Arbelo E, Brugada J, Brugada R. Update on genetic basis of brugada syndrome: monogenic, polygenic or oligogenic? Int J Mol Sci. (2020) 21:7155. doi: 10.3390/ijms21197155
31. Di Mauro V, Ceriotti P, Lodola F, Salvarani N, Modica J, Bang ML, et al. Peptide-based targeting of the L-type calcium channel corrects the loss-of-function phenotype of two novel mutations of the CACNA1 gene associated with brugada syndrome. Front Physiol. (2020) 11:616819. doi: 10.3389/fphys.2020.616819
32. Monasky MM, Micaglio E, Ciconte G, Pappone C. Brugada syndrome: oligogenic or mendelian disease? Int J Mol Sci. (2020) 21:1687. doi: 10.3390/ijms21051687
33. Probst V, Wilde AA, Barc J, Sacher F, Babuty D, Mabo P, et al. SCN5A mutations and the role of genetic background in the pathophysiology of Brugada syndrome. Circ Cardiovasc Genet. (2009) 2:552–7. doi: 10.1161/CIRCGENETICS.109.853374
34. Kapplinger JD, Tester DJ, Alders M, Benito B, Berthet M, Brugada J, et al. An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart Rhythm. (2010) 7:33–46. doi: 10.1016/j.hrthm.2009.09.069
35. Ciconte G, Monasky MM, Santinelli V, Micaglio E, Vicedomini G, Anastasia L, et al. Brugada syndrome genetics is associated with phenotype severity. Eur Heart J. (2021) 42:1082–92. doi: 10.1093/eurheartj/ehaa942
36. Wijeyeratne YD, Tanck MW, Mizusawa Y, Batchvarov V, Barc J, Crotti L, et al. SCN5A mutation type and a genetic risk score associate variably with brugada syndrome phenotype in SCN5A families. Circ Genom Precis Med. (2020) 13:e002911. doi: 10.1161/CIRCGEN.120.002911
37. Denham NC, Pearman CM, Ding WY, Waktare J, Gupta D, Snowdon R, et al. Systematic re-evaluation of SCN5A variants associated with Brugada syndrome. J Cardiovasc Electrophysiol. (2019) 30:118–27. doi: 10.1111/jce.13740
38. Pappone C, Ciconte G, Micaglio E, Monasky MM. Common modulators of Brugada syndrome phenotype do not affect SCN5A prognostic value. Eur Heart J. (2021) 42:1273–4. doi: 10.1093/eurheartj/ehab071
39. Postema PG, Walsh R, Bezzina CR. Illuminating the path from genetics to clinical outcome in Brugada syndrome. Eur Heart J. (2021) 42:1091–3. doi: 10.1093/eurheartj/ehaa994
40. Friedrich OFVW, Wink M, Fink RH. NA+- and K+-channels as molecular targets of the alkaloid ajmaline in skeletal muscle fibres. Br J Pharmacol. (2007) 151:82–93. doi: 10.1038/sj.bjp.0707194
41. Kiesecker C, Zitron E, Luck S, Bloehs R, Scholz EP, Kathofer S, et al. Class Ia anti-arrhythmic drug ajmaline blocks HERG potassium channels: mode of action. Naunyn Schmiedebergs Arch Pharmacol. (2004) 370:423–35. doi: 10.1007/s00210-004-0976-8
42. Fischer F, Vonderlin N, Zitron E, Seyler C, Scherer D, Becker R, et al. Inhibition of cardiac Kv1.5 and Kv4.3 potassium channels by the class Ia anti-arrhythmic ajmaline: mode of action. Naunyn Schmiedebergs Arch Pharmacol. (2013) 386:991–9. doi: 10.1007/s00210-013-0901-0
43. Sakuta H, Okamoto K, Watanabe Y. Blockade by antiarrhythmic drugs of glibenclamide-sensitive K+ channels in Xenopus oocytes. Br J Pharmacol. (1992) 107:1061–7. doi: 10.1111/j.1476-5381.1992.tb13407.x
44. Chukhlova EA, Sadykov I, Kholmukhamedov EL, Grenader AK. Effect of trimecaine, ajmaline, stenopril and chloracizine on fluctuations in K+ currents in rat liver mitochondria. Ukr Biokhim Zh (1978). (1984) 56:207–10.
45. Bebarova M, Matejovic P, Pasek M, Simurdova M, Simurda J. Effect of ajmaline on transient outward current in rat ventricular myocytes. Gen Physiol Biophys. (2005) 24:27–45.
46. Bebarova M, Matejovic P, Pasek M, Simurdova M, Simurda J. Effect of ajmaline on action potential and ionic currents in rat ventricular myocytes. Gen Physiol Biophys. (2005) 24:311–25.
47. Enomoto K, Imoto M, Nagashima R, Kaneko T, Maruyama T, Kaji Y, et al. Effects of ajmaline on non-sodium ionic currents in guinea pig ventricular myocytes. Jpn Heart J. (1995) 36:465–76. doi: 10.1536/ihj.36.465
48. Fish JM, Antzelevitch C. Role of sodium and calcium channel block in unmasking the Brugada syndrome. Heart Rhythm. (2004) 1:210–7. doi: 10.1016/j.hrthm.2004.03.061
49. Khodorov BI, Zaborovskaya LD. Blockade of sodium and potassium channels in the node of Ranvier by ajmaline and N-propyl ajmaline. Gen Physiol Biophys. (1983) 2:233–68.
50. Zaborovskaia LD, Khodorov BI. Localization of the binding site for quaternary derivatives of ajmaline in the Na channel of an excitable membrane. Biull Eksp Biol Med. (1985) 100:297–9. doi: 10.1007/BF00839409
51. Zil'berter Iu I, Khodorov BI, Shubert B. Stimulus-dependent blockade of the Na channels of isolated myocardial cells in the rat by the anti-arrhythmia agent N-propyl ajmaline (neogiluritmal). Biull Eksp Biol Med. (1983) 95:55–8. doi: 10.1007/BF00838861
52. Online Mendelian Inheritance in Man (2021). Available online at: https://www.omim.org/ (accessed August 5, 2021).
53. Ardehali H, Chen Z, Ko Y, Mejia-Alvarez R, Marban E. Multiprotein complex containing succinate dehydrogenase confers mitochondrial ATP-sensitive K+ channel activity. Proc Natl Acad Sci USA. (2004) 101:11880–5. doi: 10.1073/pnas.0401703101
54. Hoogendijk MG, Potse M, Vinet A, De Bakker JM, Coronel R. ST segment elevation by current-to-load mismatch: an experimental and computational study. Heart Rhythm. (2011) 8:111–8. doi: 10.1016/j.hrthm.2010.09.066
55. Biamino G, Wessel HJ, Noring J. Ajamline-induced changes in mechanical and electrical activity of vascular smooth muscle. Blood Vessels. (1975) 12:68–80. doi: 10.1159/000158039
56. Pappone C, Mecarocci V, Manguso F, Ciconte G, Vicedomini G, Sturla F, et al. New electromechanical substrate abnormalities in high-risk patients with Brugada syndrome. Heart Rhythm. (2020) 17:637–45. doi: 10.1016/j.hrthm.2019.11.019
57. Pappone C, Monasky MM, Micaglio E, Ciconte G. Right ventricular electromechanical abnormalities in Brugada syndrome: is this a cardiomyopathy? Eur Heart J Suppl. (2020) 22:E101–4. doi: 10.1093/eurheartj/suaa071
58. Antzelevitch C. Brugada syndrome: historical perspectives and observations. Eur Heart J. (2002) 23:676–8. doi: 10.1053/euhj.2001.3145
59. Antzelevitch C. Brugada syndrome. Pacing Clin Electrophysiol. (2006) 29:1130–59. doi: 10.1111/j.1540-8159.2006.00507.x
60. Monasky MM, Taglieri DM, Henze M, Warren CM, Utter MS, Soergel DG, et al. The beta-arrestin-biased ligand TRV120023 inhibits angiotensin II-induced cardiac hypertrophy while preserving enhanced myofilament response to calcium. Am J Physiol Heart Circ Physiol. (2013) 305:H856–866. doi: 10.1152/ajpheart.00327.2013
61. Yar S, Monasky MM, Solaro RJ. Maladaptive modifications in myofilament proteins and triggers in the progression to heart failure and sudden death. Pflugers Arch. (2014) 466:1189–97. doi: 10.1007/s00424-014-1457-7
62. Allegue C, Coll M, Mates J, Campuzano O, Iglesias A, Sobrino B, et al. Genetic analysis of arrhythmogenic diseases in the era of NGS: the complexity of clinical decision-making in Brugada syndrome. PLoS ONE. (2015) 10:e0133037. doi: 10.1371/journal.pone.0133037
63. Cerrone M, Lin X, Zhang M, Agullo-Pascual E, Pfenniger A, Chkourko Gusky H, et al. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype. Circulation. (2014) 129:1092–103. doi: 10.1161/CIRCULATIONAHA.113.003077
64. Hong M, Bao L, Kefaloyianni E, Agullo-Pascual E, Chkourko H, Foster M, et al. Heterogeneity of ATP-sensitive K+ channels in cardiac myocytes: enrichment at the intercalated disk. J Biol Chem. (2012) 287:41258–67. doi: 10.1074/jbc.M112.412122
65. Baudenbacher F, Schober T, Pinto JR, Sidorov VY, Hilliard F, Solaro RJ, et al. Myofilament Ca2+ sensitization causes susceptibility to cardiac arrhythmia in mice. J Clin Invest. (2008) 118:3893–903. doi: 10.1172/JCI36642
66. Mango R, Luchetti A, Sangiuolo R, Ferradini V, Briglia N, Giardina E, et al. Next generation sequencing and linkage analysis for the molecular diagnosis of a novel overlapping syndrome characterized by hypertrophic cardiomyopathy and typical electrical instability of brugada syndrome. Circ J. (2016) 80:938–49. doi: 10.1253/circj.CJ-15-0685
67. Pappone C, Monasky M, Ciconte G. Epicardial ablation in genetic cardiomyopathies: a new frontier. Eur Heart J Suppl. (2019) 21:B61–6. doi: 10.1093/eurheartj/suz028
68. Pappone C, Micaglio E, Locati ET, Monasky MM. The omics of channelopathies and cardiomyopathies: what we know and how they are useful. Eur Heart J Suppl. (2020) 22:L105–9. doi: 10.1093/eurheartj/suaa146
69. Stocchi L, Polidori E, Potenza L, Rocchi MB, Calcabrini C, Busacca P, et al. Mutational analysis of mitochondrial DNA in Brugada syndrome. Cardiovasc Pathol. (2016) 25:47–54. doi: 10.1016/j.carpath.2015.10.001
70. Polidori E, Stocchi L, Potenza D, Cucchiarini L, Stocchi V, Potenza L. A high number of ‘natural' mitochondrial DNA polymorphisms in a symptomatic Brugada syndrome type 1 patient. J Genet. (2020) 99:66. doi: 10.1007/s12041-020-01228-4
71. Tafti MF, Khatami M, Rezaei S, Heidari MM, Hadadzadeh M. Novel and heteroplasmic mutations in mitochondrial tRNA genes in Brugada syndrome. Cardiol J. (2018) 25:113–9. doi: 10.5603/CJ.a2017.0104
72. Samani K, Ai T, Towbin JA, Brugada R, Shuraih M, Xi Y, et al. A nonsense SCN5A mutation associated with Brugada-type electrocardiogram and intraventricular conduction defects. Pacing Clin Electrophysiol. (2009) 32:1231–6. doi: 10.1111/j.1540-8159.2009.02470.x
73. Kapplinger JD, Giudicessi JR, Ye D, Tester DJ, Callis TE, Valdivia CR, et al. Enhanced classification of Brugada syndrome-associated and long-QT syndrome-associated genetic variants in the SCN5A-encoded Na(v)1.5 cardiac sodium channel. Circ Cardiovasc Genet. (2015) 8:582–95. doi: 10.1161/CIRCGENETICS.114.000831
74. Micaglio E, Monasky MM, Ciconte G, Vicedomini G, Conti M, Mecarocci V, et al. SCN5A Nonsense mutation and NF1 frameshift mutation in a family with Brugada syndrome and neurofibromatosis. Front Genet. (2019) 10:50. doi: 10.3389/fgene.2019.00050
75. Micaglio E, Monasky MM, Ciconte G, Vicedomini G, Conti M, Mecarocci V, et al. Novel SCN5A frameshift mutation in Brugada syndrome associated with complex arrhythmic phenotype. Front Genet. (2019) 10:547. doi: 10.3389/fgene.2019.00547
76. Micaglio E, Monasky MM, Resta N, Bagnulo R, Ciconte G, Gianelli L, et al. Novel SCN5A p.W697X nonsense mutation segregation in a family with Brugada syndrome. Int J Mol Sci. (2019) 20:4920. doi: 10.3390/ijms20194920
77. Monasky MM, Micaglio E, Ciconte G, Benedetti S, Di Resta C, Vicedomini G, et al. Genotype/phenotype relationship in a consanguineal family with Brugada syndrome harboring the R1632C missense variant in the SCN5A gene. Front Physiol. (2019) 10:666. doi: 10.3389/fphys.2019.00666
78. Monasky MM, Micaglio E, Giachino D, Ciconte G, Giannelli L, Locati ET, et al. Genotype-phenotype correlation in a family with Brugada syndrome harboring the novel p.Gln371* nonsense variant in the SCN5A gene. Int J Mol Sci. (2019) 20:5522. doi: 10.3390/ijms20225522
79. Monasky MM, Micaglio E, Ciconte G, Borrelli V, Giannelli L, Vicedomini G, et al. Novel SCN5A p.V1429M variant segregation in a family with brugada syndrome. Int J Mol Sci. (2020) 21:5902. doi: 10.3390/ijms21165902
80. Monasky MM, Micaglio E, Ciconte G, Rivolta I, Borrelli V, Ghiroldi A, et al. Novel SCN5A p.Val1667Asp missense variant segregation and characterization in a family with severe Brugada syndrome and multiple sudden deaths. Int J Mol Sci. (2021) 22:4700. doi: 10.3390/ijms22094700
81. Monasky MM, Rutigliani C, Micaglio E, Pappone C. Commentary: peptide-based targeting of the L-type calcium channel corrects the loss-of-function phenotype of two novel mutations of the CACNA1 gene associated with Brugada syndrome. Front Physiol. (2021) 12:682567. doi: 10.3389/fphys.2021.682567
82. Bezzina CR, Barc J, Mizusawa Y, Remme CA, Gourraud JB, Simonet F, et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet. (2013) 45:1044–9. doi: 10.1038/ng.2712
Keywords: Brugada syndrome (BrS), ajmaline, arrhythmias, sudden cardiac death (SCD), sodium channel, potassium channel, calcium channel, mitochondria
Citation: Monasky MM, Micaglio E, D'Imperio S and Pappone C (2021) The Mechanism of Ajmaline and Thus Brugada Syndrome: Not Only the Sodium Channel!. Front. Cardiovasc. Med. 8:782596. doi: 10.3389/fcvm.2021.782596
Received: 24 September 2021; Accepted: 29 November 2021;
Published: 23 December 2021.
Edited by:
Alexander Pott, Ulm University Medical Center, GermanyReviewed by:
Ranjan K. Dash, Medical College of Wisconsin, United StatesGerrit Frommeyer, University Hospital Münster, Germany
Copyright © 2021 Monasky, Micaglio, D'Imperio and Pappone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carlo Pappone, Y2FybG8ucGFwcG9uZUBhZi1hYmxhdGlvbi5vcmc=