 Michelle Lin1
Michelle Lin1 Robyn A. Roth
Robyn A. Roth Beth A. Kozel
Beth A. Kozel Carmen M. Halabi
Carmen M. Halabi
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
BRIEF RESEARCH REPORT article
Front. Cardiovasc. Med. , 01 November 2021
Sec. Atherosclerosis and Vascular Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.782138
This article is part of the Research Topic Insights in Atherosclerosis and Vascular Medicine: 2021 View all 22 articles
There is ample evidence supporting a role for angiotensin II type 2 receptor (AT2R) in counterbalancing the effects of angiotensin II (ang II) through the angiotensin II type 1 receptor by promoting vasodilation and having anti-inflammatory effects. Elastin insufficiency in both humans and mice results in large artery stiffness and systolic hypertension. Unexpectedly, mesenteric arteries from elastin insufficient (Eln+/−) mice were shown to have significant vasoconstriction to AT2R agonism in vitro suggesting that AT2R may have vasoconstrictor effects in elastin insufficiency. Given the potential promise for the use of AT2R agonists clinically, the goal of this study was to determine whether AT2R has vasoconstrictive effects in elastin insufficiency in vivo. To avoid off-target effects of agonists and antagonists, mice lacking AT2R (Agtr2−/Y) were bred to Eln+/− mice and cardiovascular parameters were assessed in wild-type (WT), Agtr2−/Y, Eln+/−, and Agtr2−/Y;Eln+/− littermates. As previously published, Agtr2−/Y mice were normotensive at baseline and had no large artery stiffness, while Eln+/− mice exhibited systolic hypertension and large artery stiffness. Loss of AT2R in Eln+/− mice did not affect large artery stiffness or arterial structure but resulted in significant reduction of both systolic and diastolic blood pressure. These data support a potential vasocontractile role for AT2R in elastin insufficiency. Careful consideration and investigation are necessary to determine the patient population that might benefit from the use of AT2R agonists.
Elastin (ELN), the main component of elastic fibers, is responsible for conduit arteries' elastic recoil. This recoil is necessary to dampen the pulsatile flow of ventricular ejection at the level of the ascending aorta and transform it into continuous flow at the level of arterioles or small resistance arteries. Elastic fibers are organized into fenestrated concentric sheets or lamellae in blood vessels. Decreased elasticity of large arteries with aging is attributed to fragmentation and thinning of these lamellae and results in increased pulse wave velocity leading to a greater augmentation of the central aortic systolic and pulse pressures (1, 2). Similarly, genetic reduction of elastin through deletion of a single copy of the gene ELN (supravalvular aortic stenosis—SVAS, OMIM #185500) or deletion of ELN as part of a 25–27 coding gene microdeletion of chromosome 7 (Williams syndrome, OMIM #194050) leads to increased pulse wave velocity and hypertension (3–5).
Similar to humans with SVAS and Williams syndrome, mice hemizygous for the elastin gene (Eln+/−) develop large artery stiffness and systolic hypertension (6, 7). Interestingly, the increased large artery stiffness in Eln+/− mice precedes the appearance of hypertension (8) and is not affected by commonly used anti-hypertensives (9). Increased large artery stiffness and central systolic and pulse pressures often lead to structural and functional changes in small resistance arteries that further exacerbate hypertension and a vicious cycle ensues (10). This appears to be the case in elastin insufficiency as recent studies showed altered resistance vessel reactivity that is vascular bed-specific (11–13). Mesenteric arteries (MAs) and middle cerebral arteries (MCAs), but not gastrocnemius feed arteries (GFAs), were found to have impaired endothelial-dependent dilation to acetylcholine due to decreased nitric oxide availability resulting from increased oxidative stress (11, 12, 14). Furthermore, MAs and MCAs, but not GFAs, had an increased contractile response to angiotensin II (ang II) (11, 13). Interestingly, the hypercontractile response of mesenteric arteries to ang II was mediated, at least in part, by the angiotensin II type 2 receptor (AT2R) as blockade of AT2R with the antagonist PD123319 decreased the contractile response of MAs to ang II while its activation with novokinin resulted in vasoconstriction (11).
Given the multitude of evidence suggesting a vasodilatory role for AT2R particularly in disease states and the consideration for the use of AT2R agonists for patients with COVID-19 and idiopathic pulmonary fibrosis among others (clinicaltrials.gov), we sought to determine the cardiovascular role of AT2R in elastin insufficiency in vivo. We bred elastin insufficient (Eln+/−) mice to AT2R knock-out (Agtr2−/Y) mice and examined cardiovascular endpoints. While loss of AT2R did not affect large artery structure or function, it lowered blood pressure in elastin insufficient mice, suggesting that AT2R plays a vasocontractile role in elastin insufficiency. This observation has significant therapeutic implications since AT2R agonists, which may be beneficial in some conditions such as stroke (15, 16), aneurysm formation (17, 18) and myocardial fibrosis (19), would not be appropriate in patients with elastin insufficiency.
Eln+/− mice backcrossed into the 129X1/SvJ background (14) over 10 times and the genetic background confirmed by single nucleotide polymorphism genotyping were bred to Agtr2−/− mice maintained on the FVB/n background (20). The Agtr2−/− mice were obtained from Dr. Curt Sigmund, with permission from Dr. Victor Dzau. Tail DNA was used to genotype the mice. Genotyping for Eln was done as previously described (21). The following primers were used in one PCR reaction to genotype for Agtr2: AT2-F GTGGTCTCACTGTTTTGTTGTC, AT2-R-WT GTATTCAATGGTTCTGACATCC, and AT2-R-KO TGCAATCCATCTTGTTCAATGGC, resulting in a 374 bp product in the WT case and a 570 bp product in the knock-out case. Since Agtr2 is on the X chromosome and littermates were used for the studies, male mice were used for the physiologic studies to reduce the number of animals needed. Mice were housed under standard conditions with free access to food and water. All surgical procedures were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of Washington University School of Medicine.
While sedation is known to lower blood pressure and heart rate, invasive blood pressure measurement provides a more accurate assessment of central arterial pressure compared to tail cuff measurement. Unfortunately, the small caliber and tortuosity of Eln+/− carotid arteries makes blood pressure measurement via telemetry technically challenging, therefore we measured central arterial pressure invasively under sedation. The anesthetic used, isoflurane, has the least effects on the cardiovascular system among commonly used anesthetics (22). Briefly, 3–4 month-old mice were anesthetized with 2% isoflurane and maintained at 37°C using a heating pad and a rectal thermometer for monitoring. The right common carotid artery was exposed and a Millar pressure transducer (model SPR-671) was introduced and advanced to the ascending aorta. After instrumentation was complete, isoflurane anesthesia was reduced to 1.5% and systolic blood pressure, diastolic blood pressure, and heart rate were recorded using the PowerLab data acquisition system (ADInstruments). The average of a 3-min period of stable recording was reported. The data were analyzed using LabChart 8 for Mac software (ADInstruments).
Ascending aorta and left common carotid artery of 3–4 month-old mice were excised and placed in physiologic saline solution (PSS) composed of 130 mM NaCl, 4.7 mM KCl, 1.6 mM CaCl2, 1.18 mM MgSO4-7H2O, 1.17 mM KH2PO4, 14.8 mM NaHCO3, 5.5 mM dextrose, and 0.026 mM EDTA (pH 7.4). Vessels were cleaned of surrounding fat, mounted on a pressure arteriograph (Danish Myo Technology) and maintained in PSS at 37°C. Vessels were visualized with an inverted microscope connected to a CCD camera and a computerized system, which allows continuous recording of vessel diameter. Intravascular pressure was increased from 0 to 175 mmHg by 25-mmHg increments, the vessel outer diameter was recorded at each step (12 s per step). The average of three measurements at each pressure was reported.
Ascending aorta were dissected and frozen in optimal cutting temperature (OCT) compound (Sakura Finetek) at −80°C. Using a cryostat, 3-μm sections were obtained and fixed in 4% paraformaldehyde for 10 min at 4°C. Sections were washed twice with 1 × PBS for 5 min each and then incubated in 1:1,000 of a 2 mM Alexa Fluor 633 hydrazide (Life Technologies) stock in 1% bovine serum albumin (BSA)/1% fish gelatin/0.05% Triton-X in 1 × PBS for 5 min at room temperature. Sections were then washed twice with 1 × PBS for 5 min each. Slides were mounted with DAPI Fluoromount-G (SouthernBiotech) and coverslipped. Images were obtained using a Zeiss Axioskop 50 microscope and QCapture Pro software (Media Cybernetics Inc.).
After isolation, mesenteric arteries from 3 to 4 month-old mice were fixed in 2.5% glutaraldehyde and 0.1 M sodium cacodylate at 4°C overnight. Vessels were then sent to Washington University's Center for Cellular Imaging for processing and thin sectioning. Images were taken using a JEOL JEM-1400 Plus transmission electron microscope that is equipped with an Advanced Microscopy Techniques XR111 high-speed, 4,000 × 2,000–pixel, phosphor-scintillated, 12-bit charge-coupled device (CCD) camera.
One-way or two-way analysis of variance with Tukey's multiple comparisons test was used to determine differences between genotypes, as indicated in each figure legend. Statistical analyses were run using Prism 9 for Mac OS X (GraphPad Software Inc.). Data are presented as means ± SD. Differences were considered statistically significant when P was equal to or less than 0.05.
To determine the role, if any, of AT2R in blood pressure regulation in elastin insufficiency, we bred Agtr2−/Y to Eln+/− mice. As previously reported, loss of AT2R did not affect blood pressure at baseline (20) and Eln+/− mice exhibited systolic hypertension compared to wild-type (WT) littermates (6) (Figures 1A–C). Interestingly, loss of AT2R in elastin insufficient mice (Agtr2−/Y;Eln+/−) resulted in reduction of not only systolic, but also diastolic blood pressure (Figures 1A–C). Heart rate, body weight and heart weight were not different among the genotypes (Figures 1D–F).
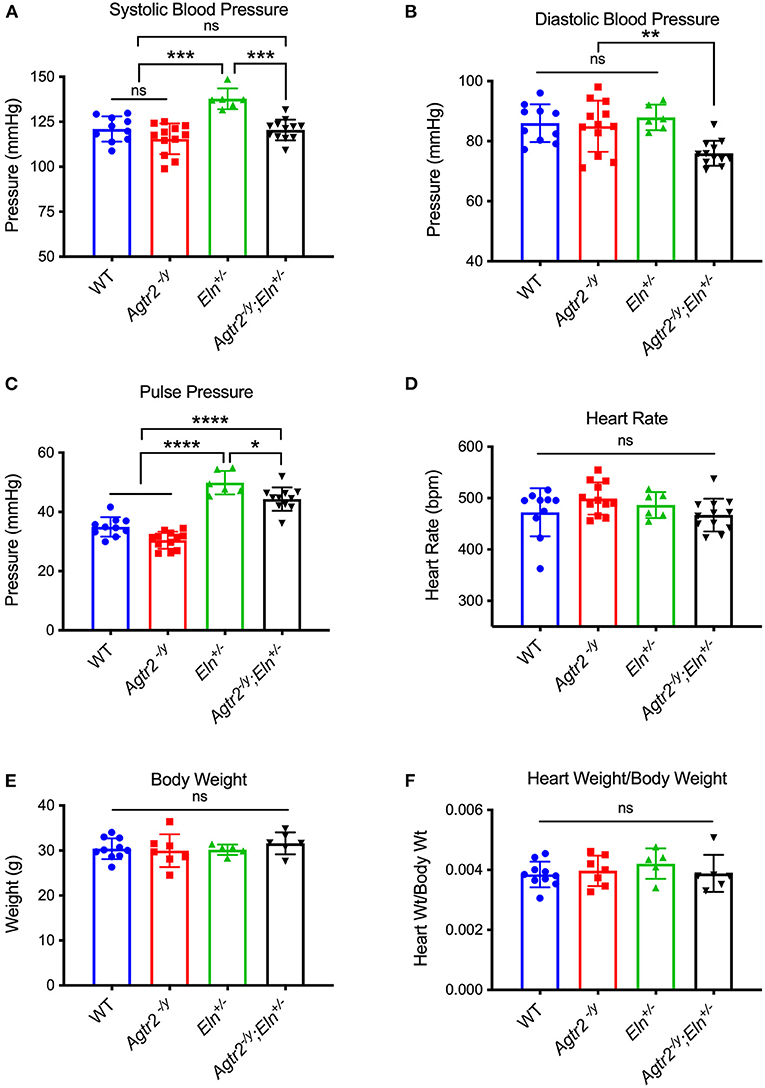
Figure 1. Loss of AT2R leads to a reduction in systolic and diastolic blood pressure in elastin insufficient mice. Systolic (A), diastolic (B), and pulse pressure (C) [calculated as systolic–diastolic blood pressure], heart rate (D), body weight (E) and heart weight/body weight (F) of WT, Agtr2−/Y, Eln+/− and Agtr2−/Y;Eln+/− mice. Data are presented as mean ± standard deviation. One-way analysis of variance with Tukey's multiple comparison test was performed to compare all groups. Significant difference: *P < 0.05, **P < 0.005, ***P < 0.001, and ****P < 0.0001, between indicated groups.
One of the characteristic features of elastin insufficiency is large artery stiffness assessed by pressure-diameter curves experimentally in Eln+/− mice and by pulse wave velocity in humans with Williams syndrome (4, 6). To determine whether the improvement in blood pressure in Agtr2−/Y;Eln+/− mice was related to an improvement in large artery stiffness, we assessed ascending aorta and carotid artery mechanics in mutant and littermate control mice. As seen in Figure 2, loss of AT2R alone had no effect on large artery stiffness or compliance and it did not ameliorate the large artery stiffness seen in elastin insufficiency.
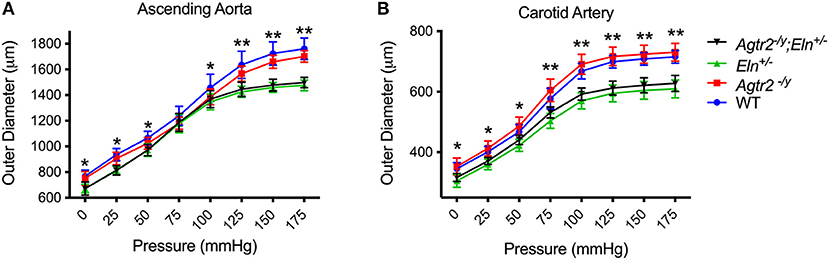
Figure 2. Loss of AT2R does not affect large artery stiffness. Pressure-diameter relationships of ascending aorta (A) and carotid arteries (B) from WT (n = 9–10), Agtr2−/Y (n = 10–11), Eln+/− (n = 6) and Agtr2−/Y;Eln+/− (n = 15) mice. Data are presented as mean ± standard deviation. Two-way analysis of variance with Tukey's multiple comparison test was performed to compare all groups. Significant difference: *P < 0.05 and **P < 0.005 between WT or Agtr2−/Y vs. Eln+/− or Agtr2−/Y;Eln+/−.
Ascending aorta of elastin insufficient mice have, on average, two additional lamellar units (7). We examined whether loss of AT2R has any consequences on large and small artery structure. Fluorescence microscopy using Alexa-633 hydrazide staining of ascending aorta showed that, like WT ascending aorta, Agtr2−/Y ascending aorta have 8–9 lamellar units while Eln+/− ascending aorta have 10–11. Loss of AT2R did not affect lamellar unit number in elastin insufficiency as Agtr2−/Y;Eln+/− ascending aortae had 10–11 lamellar units. Representative images are shown in Figure 3A. Ultrastructural examination of mesenteric arteries by transmission electron microscopy did not identify an effect of AT2R on arteriolar wall structure. As previously described, the internal elastic lamina of Eln+/− mesenteric arteries was thinner compared to WT mesenteric arteries, a finding that was not affected by loss of AT2R (Figure 3B).
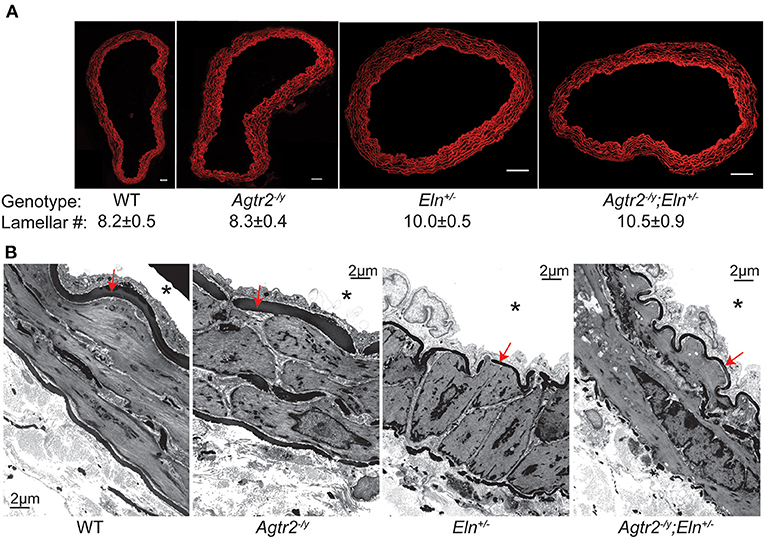
Figure 3. Loss of AT2R does not affect arterial structure. Representative cross sections of Alexa-633 hydrazide-stained ascending aorta from WT, Agtr2−/Y, Eln+/−, and Agtr2−/Y;Eln+/− mice along with the respective average lamellar number ± standard error of the mean, n = 3–4 per group, scale bar = 50 μm (A). Transmission electron micrographs of mesenteric arteries from all genotypes, *indicates vessel lumen and red arrow indicated internal elastic lamina (B).
Ang II, the principal effector of the renin-angiotensin system, exerts its functions in physiological and pathological states mainly through two receptors, AT1R and AT2R. In hypertension, the pathologic remodeling that occurs, including vasoconstriction, fibrosis, proliferation, and inflammation, has been attributed to ang II's actions through AT1R. Over the past two to three decades, a great deal of effort has focused on understanding the role of the more elusive AT2R. Evidence suggests that while its levels are low in the adult cardiovascular system at baseline, AT2R expression increases significantly in pathological conditions and it is thought to counter-balance the effects of ang II by promoting a vasodilatory, anti-fibrotic, apoptotic, and anti-inflammatory phenotype (23, 24). Often the vasodilatory effect of AT2R is only evident when the vasoconstrictor action of AT1R is blocked. At baseline, AT2R knock-out mice were normotensive but showed an increased pressor response to ang II infusion (20). With the availability of several non-peptide AT2R agonists, their use is being investigated as a potential therapeutic option in several disease conditions. In this report, based on in vitro data suggesting a vasocontractile role for AT2R in elastin insufficiency, we sought to determine whether AT2R contributes to elastin insufficiency-mediated hypertension in vivo. Using mouse models with genetic loss or insufficiency of AT2R and ELN, we show that, unlike its protective role in heart failure, myocardial infarction and aneurysms, in the context of elastin insufficiency loss of AT2R improves blood pressure making its activation a potentially detrimental therapeutic strategy in this disease state.
While initially surprising, the observation that AT2R may play a vasocontractile role has been made in other models of hypertension. For instance, Touyz et al. (25) showed an enhanced contractile response to ang II in mesenteric arteries from spontaneously hypertensive rats (SHR) compared to Wystar-Kyoto rats (WKY). This response was reduced by AT2R blockade in young but not old SHR. Similarly, coronary arteries from SHR were found to have enhanced constriction to ang II, that was attributed to the absence of counter-regulatory AT2R-mediated relaxation and/or a change in the AT2R phenotype from dilatory to contractile (26).
An interesting observation from our study is that loss of AT2R decreases both systolic and diastolic blood pressure, while pulse pressure, an indicator of conduit artery stiffness, remains significantly elevated in Agtr2−/y;Eln+/− mice compared to WT and Agtr2−/y mice. These data support the large artery pressure-diameter measurements showing that loss of AT2R does not affect large artery mechanics in elastin insufficiency. Rather, loss of AT2R likely affects peripheral vascular resistance leading to a reduction in both systolic and diastolic blood pressure.
Activation of AT2R by ang II has been shown to increase nitric oxide (NO) production, which activates guanylate cyclase to generate cyclic guanosine monophosphate (cGMP) leading to vasodilation (27, 28). The mechanism by which AT2R leads to vasoconstriction in elastin insufficiency is unclear at this time. Similar to WT, Agtr1 is expressed at higher levels than Agtr2 in Eln+/− vessels, and both Agtr1 and Agtr2 expression levels were unchanged in aortae and reduced in mesenteric arteries of Eln+/− mice (11), making relative changes in receptor levels an unlikely explanation for the observed blood pressure response. It is interesting to note however that, while usually thought of as monomers, G protein-coupled receptors like AT1R, AT2R and bradykinin receptor (B2R) have been shown to heterodimerize and adopt either an enhanced or an altered function. For instance, heterodimerization of AT1R and B2R led to increased activation of Gαq and Gαi, the two major signaling proteins activated by AT1R (29). This AT1R-B2R heterodimerization was shown to contribute to ang II hypersensitivity in pre-eclampsia (30). AT2R has been shown to dimerize with B2R leading to enhanced NO and cGMP (27). Since AT2R expression was reduced in elastin insufficient mesenteric arteries (11), it is interesting to speculate that AT2R-B2R dimer formation may be affected, or alternatively, that AT2R heterodimerizes with AT1R in elastin insufficiency, resulting in vasoconstriction rather than vasodilation; hypotheses that will be the focus of future investigation.
In summary, using a mouse model of elastin insufficiency-mediated hypertension, here we show that loss of AT2R improves blood pressure in this model. While the process of elastin insufficiency is distinct, with normal aging older adults develop vascular elastic fiber thinning, systolic hypertension with widened pulse pressure and large artery stiffness, all characteristics of elastin insufficient mice. Therefore, if AT2R agonists are to be considered for clinical use, carefully designed randomized clinical trials with special attention to patient population and endpoints will be necessary to ensure that they are not contributing to disease, particularly hypertension. AT2R agonists will likely be useful in a context-specific manner.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by Institutional Animal Care and Use Committee of Washington University School of Medicine.
RM, BK, and CH conceived the study design. ML, RR, and CH performed experiments, generated and analyzed data. CH drafted the manuscript. All authors have read and approved the final manuscript.
This work was supported by National Institutes of Health grants K08 HL135400 to CH and R01-HL53325 to RM. Funding for BK came from the Division of Intramural Research of the NIH, ZIA HL006210. Funds were also provided by the Ines Mandl Research Foundation to RM.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Dr. Curt Sigmund for providing the Agtr2−/Y mice with permission from Dr. Victor Dzau. We thank James Fitzpatrick and Greg Strout from Washington University's Center for Cellular Imaging that is supported by the Children's Discovery Institute of Washington University and St. Louis Children's Hospital (CDI-CORE-2015-505) for their expertise and assistance with electron microscopy.
1. Jadidi M, Razian SA, Habibnezhad M, Anttila E, Kamenskiy A. Mechanical, structural, and physiologic differences in human elastic and muscular arteries of different ages: comparison of the descending thoracic aorta to the superficial femoral artery. Acta Biomater. (2021) 119:268–83. doi: 10.1016/j.actbio.2020.10.035
2. Laurent S, Boutouyrie P, Asmar R, Gautier I, Laloux B, Guize L, et al. Aortic stiffness is an independent predictor of all-cause and cardiovascular mortality in hypertensive patients. Hypertension. (2001) 37:1236–41. doi: 10.1161/01.HYP.37.5.1236
3. Kozel BA, Barak B, Kim CA, Mervis CB, Osborne LR, Porter M, et al. Williams syndrome. Nat Rev Dis Primers. (2021) 7:42. doi: 10.1038/s41572-021-00276-z
4. Kozel BA, Danback JR, Waxler JL, Knutsen RH, de Las Fuentes L, Reusz GS, et al. Williams syndrome predisposes to vascular stiffness modified by antihypertensive use and copy number changes in NCF1. Hypertension. (2014) 63:74–9. doi: 10.1161/HYPERTENSIONAHA.113.02087
5. Pober BR, Johnson M, Urban Z. Mechanisms and treatment of cardiovascular disease in Williams-Beuren syndrome. J Clin Investig. (2008) 118:1606–15. doi: 10.1172/JCI35309
6. Faury G, Pezet M, Knutsen RH, Boyle WA, Heximer SP, McLean SE, et al. Developmental adaptation of the mouse cardiovascular system to elastin haploinsufficiency. J Clin Investig. (2003) 112:1419–28. doi: 10.1172/JCI19028
7. Li DY, Faury G, Taylor DG, Davis EC, Boyle WA, Mecham RP, et al. Novel arterial pathology in mice and humans hemizygous for elastin. J Clin Investig. (1998) 102:1783–7. doi: 10.1172/JCI4487
8. Le VP, Knutsen RH, Mecham RP, Wagenseil JE. Decreased aortic diameter and compliance precedes blood pressure increases in postnatal development of elastin-insufficient mice. Am J Physiol Heart Circ. (2011) 301:H221–9. doi: 10.1152/ajpheart.00119.2011
9. Halabi CM, Broekelmann TJ, Knutsen RH, Ye L, Mecham RP, Kozel BA. Chronic antihypertensive treatment improves pulse pressure but not large artery mechanics in a mouse model of congenital vascular stiffness. Am J Physiol Heart Circ. (2015) 309:H1008–16. doi: 10.1152/ajpheart.00288.2015
10. Laurent S, Boutouyrie P. The structural factor of hypertension: large and small artery alterations. Circ Res. (2015) 116:1007–21. doi: 10.1161/CIRCRESAHA.116.303596
11. Osei-Owusu P, Knutsen RH, Kozel BA, Dietrich HH, Blumer KJ, Mecham RP. Altered reactivity of resistance vasculature contributes to hypertension in elastin insufficiency. Am J Physiol Heart Circ. (2014) 306:H654–66. doi: 10.1152/ajpheart.00601.2013
12. Walker AE, Henson GD, Reihl KD, Morgan RG, Dobson PS, Nielson EI, et al. Greater impairments in cerebral artery compared with skeletal muscle feed artery endothelial function in a mouse model of increased large artery stiffness. J Physiol. (2015) 593:1931–43. doi: 10.1113/jphysiol.2014.285338
13. Walker AE, Kronquist EK, Chinen KT, Reihl KD, Li DY, Lesniewski LA, et al. Cerebral and skeletal muscle feed artery vasoconstrictor responses in a mouse model with greater large elastic artery stiffness. Exp Physiol. (2019) 104:434–42. doi: 10.1113/EP087453
14. Kozel BA, Knutsen RH, Ye L, Ciliberto CH, Broekelmann TJ, Mecham RP. Genetic modifiers of cardiovascular phenotype caused by elastin haploinsufficiency act by extrinsic noncomplementation. J Biol Chem. (2011) 286:44926–36. doi: 10.1074/jbc.M111.274779
15. Jackson-Cowan L, Eldahshan W, Dumanli S, Dong G, Jamil S, Abdul Y, et al. Delayed administration of angiotensin receptor (AT2R) agonist C21 improves survival and preserves sensorimotor outcomes in female diabetic rats post-stroke through modulation of microglial activation. Int J Mol Sci. (2021) 22:1356. doi: 10.3390/ijms22031356
16. Shan BS, Mogi M, Iwanami J, Bai HY, Kan-No H, Higaki A, et al. Attenuation of stroke damage by angiotensin II type 2 receptor stimulation via peroxisome proliferator-activated receptor-gamma activation. Hypertens Res. (2018) 41:839–48. doi: 10.1038/s41440-018-0082-9
17. Habashi JP, Doyle JJ, Holm TM, Aziz H, Schoenhoff F, Bedja D, et al. Angiotensin II type 2 receptor signaling attenuates aortic aneurysm in mice through ERK antagonism. Science. (2011) 332:361–5. doi: 10.1126/science.1192152
18. Lange C, Sommerfeld M, Namsolleck P, Kintscher U, Unger T, Kaschina E. AT2R (Angiotensin AT2 Receptor) agonist, compound 21, prevents abdominal aortic aneurysm progression in the rat. Hypertension. (2018) 72:e20–9. doi: 10.1161/HYPERTENSIONAHA.118.11168
19. Dopona EPB, Rocha VF, Furukawa LNS, Oliveira IB, Heimann JC. Myocardial hypertrophy induced by high salt consumption is prevented by angiotensin II AT2 receptor agonist. Nutr Metab Cardiovasc Dis. (2019) 29:301–5. doi: 10.1016/j.numecd.2018.11.001
20. Hein L, Barsh GS, Pratt RE, Dzau VJ, Kobilka BK. Behavioural and cardiovascular effects of disrupting the angiotensin II type-2 receptor in mice. Nature. (1995) 377:744–7. doi: 10.1038/377744a0
21. Hirano E, Knutsen RH, Sugitani H, Ciliberto CH, Mecham RP. Functional rescue of elastin insufficiency in mice by the human elastin gene: implications for mouse models of human disease. Circ Res. (2007) 101:523–31. doi: 10.1161/CIRCRESAHA.107.153510
22. Zhao X, Ho D, Gao S, Hong C, Vatner DE, Vatner SF. Arterial pressure monitoring in mice. Curr Protoc Mouse Biol. (2011) 1:105–22. doi: 10.1002/9780470942390.mo100149
23. Lax CJ, Domenighetti AA, Pavia JM, Di Nicolantonio R, Curl CL, Morris MJ, et al. Transitory reduction in angiotensin AT2 receptor expression levels in postinfarct remodelling in rat myocardium. Clin Exp Pharmacol Physiol. (2004) 31:512–7. doi: 10.1111/j.1440-1681.2004.04034.x
24. Nio Y, Matsubara H, Murasawa S, Kanasaki M, Inada M. Regulation of gene transcription of angiotensin II receptor subtypes in myocardial infarction. J Clin Investig. (1995) 95:46–54. doi: 10.1172/JCI117675
25. Touyz RM, Endemann D, He G, Li JS, Schiffrin EL. Role of AT2 receptors in angiotensin II-stimulated contraction of small mesenteric arteries in young SHR. Hypertension. (1999) 33(1 Pt 2):366–72. doi: 10.1161/01.HYP.33.1.366
26. Moltzer E, Verkuil AV, van Veghel R, Danser AH, van Esch JH. Effects of angiotensin metabolites in the coronary vascular bed of the spontaneously hypertensive rat: loss of angiotensin II type 2 receptor-mediated vasodilation. Hypertension. (2010) 55:516–22. doi: 10.1161/HYPERTENSIONAHA.109.145037
27. Abadir PM, Periasamy A, Carey RM, Siragy HM. Angiotensin II type 2 receptor-bradykinin B2 receptor functional heterodimerization. Hypertension. (2006) 48:316–22. doi: 10.1161/01.HYP.0000228997.88162.a8
28. Li J, Zhao X, Li X, Lerea KM, Olson SC. Angiotensin II type 2 receptor-dependent increases in nitric oxide synthase expression in the pulmonary endothelium is mediated via a G alpha i3/Ras/Raf/MAPK pathway. Am J Physiol Cell Physiol. (2007) 292:C2185–96. doi: 10.1152/ajpcell.00204.2006
29. AbdAlla S, Lother H, Quitterer U. AT1-receptor heterodimers show enhanced G-protein activation and altered receptor sequestration. Nature. (2000) 407:94–8. doi: 10.1038/35024095
Keywords: angiotensin II type 2 receptor, elastin insufficiency, hypertension, vascular stiffness, vascular biology
Citation: Lin M, Roth RA, Kozel BA, Mecham RP and Halabi CM (2021) Loss of Angiotensin II Type 2 Receptor Improves Blood Pressure in Elastin Insufficiency. Front. Cardiovasc. Med. 8:782138. doi: 10.3389/fcvm.2021.782138
Received: 23 September 2021; Accepted: 11 October 2021;
Published: 01 November 2021.
Edited by:
Anthony Wayne Orr, Louisiana State University Health Shreveport, United StatesReviewed by:
Mark C. Chappell, Wake Forest School of Medicine, United StatesCopyright © 2021 Lin, Roth, Kozel, Mecham and Halabi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carmen M. Halabi, Y2hhbGFiaUB3dXN0bC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.