
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 15 October 2021
Sec. Cardiovascular Genetics and Systems Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.771349
This article is part of the Research Topic Women in Cardiovascular Genetics and Systems Medicine View all 12 articles
 Sara D'Imperio1†
Sara D'Imperio1† Michelle M. Monasky1†
Michelle M. Monasky1† Emanuele Micaglio1
Emanuele Micaglio1 Giuseppe Ciconte1
Giuseppe Ciconte1 Luigi Anastasia2
Luigi Anastasia2 Carlo Pappone1,2*
Carlo Pappone1,2*Brugada syndrome (BrS) is a hereditary disorder, characterized by a specific electrocardiogram pattern and highly related to an increased risk of sudden cardiac death. BrS has been associated with other cardiac and non-cardiac pathologies, probably because of protein expression shared by the heart and other tissue types. In fact, the most commonly found mutated gene in BrS, SCN5A, is expressed throughout nearly the entire body. Consistent with this, large meals and alcohol consumption can trigger arrhythmic events in patients with BrS, suggesting a role for organs involved in the digestive and metabolic pathways. Ajmaline, a drug used to diagnose BrS, can have side effects on non-cardiac tissues, such as the liver, further supporting the idea of a role for organs involved in the digestive and metabolic pathways in BrS. The BrS electrocardiogram (ECG) sign has been associated with neural, digestive, and metabolic pathways, and potential biomarkers for BrS have been found in the serum or plasma. Here, we review the known associations between BrS and various organ systems, and demonstrate support for the hypothesis that BrS is not only a cardiac disorder, but rather a systemic one that affects virtually the whole body. Any time that the BrS ECG sign is found, it should be considered not a single disease, but rather the final step in any number of pathways that ultimately threaten the patient's life. A multi-omics approach would be appropriate to study this syndrome, including genetics, epigenomics, transcriptomics, proteomics, metabolomics, lipidomics, and glycomics, resulting eventually in a biomarker for BrS and the ability to diagnose this syndrome using a minimally invasive blood test, avoiding the risk associated with ajmaline testing.
Brugada syndrome (BrS) is a hereditary disorder, highly related to an increased risk of sudden cardiac death (1), characterized by a type 1 (coved type) ST-segment elevation ≥2 mm followed by a negative T-wave in ≥1 of the right precordial leads V1 to V2 on the electrocardiogram (ECG) (2), which can occur spontaneously or after pharmacologically induced. The typical symptoms of BrS are syncope and cardiac arrest due to ventricular fibrillation (VF) or ventricular tachycardia (VT), in the absence of overt cardiac structural changes, typically between 30 and 50 years of age (3, 4). BrS has been associated with several other cardiac disorders, such as early repolarization syndrome (5), arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D) (6–9), progressive cardiac conduction defect (Lenègre syndrome) (10, 11), LQTS (12–14), Wolff-Parkinson-White (15, 16), hypertrophic cardiomyopathy (17, 18), atrial flutter (19), and atrial fibrillation (20–23). In addition, BrS has also been associated with non-cardiac pathologies, such as epilepsy, thyroid disorders, cancer, skeletal muscle sodium channelopathies, laminopathies, and diabetes. One reason for this may be due to similar ion channel expression shared by the heart and other tissue types (24–27). In fact, the most commonly found mutated gene in BrS, SCN5A (28), is expressed throughout the entire body, with the largest protein expression levels found in the plasma, heart, and pancreatic juice (29). Consistent with this, large meals, alcohol, specific drugs, and fever can trigger arrhythmic events in patients with BrS, suggesting a role for organs involved in the digestive and metabolic (including mitochondrial) pathways (28, 30).
Drugs used to induced the type 1 BrS ECG pattern include Ajmaline, Flecainide, and Procainamide, which are often thought of as sodium channel blockers (4, 31, 32). Many centers prefer the use of Ajmaline because of its lower false-negative rate (33). However, several studies have highlighted the complex mechanism of Ajmaline, which does not act solely as a sodium channel blocker, but rather acts additionally on potassium and calcium channels (34, 35). Ajmaline can have side effects on non-cardiac tissues, such as the liver and mitochondria (36), further supporting the idea of a role for organs involved in the digestive and metabolic pathways in BrS.
In addition to cardiac, neural, digestive, and metabolic involvement, BrS may also affect other areas of the body. Potential biomarkers for BrS have been reportedly found in the serum or plasma of BrS patients (37, 38), although the conclusions are disputed (39). Indeed, a multi-omics approach would be appropriate to study this syndrome, including not only genetics, but also epigenomics, transcriptomics, proteomics, metabolomics, lipidomics, and glycomics (28). Here, we review the known associations between BrS and various systems and demonstrate support for the hypothesis that BrS is not only a cardiac disorder, but rather a systemic one that affects virtually the whole body.
BrS was once viewed as a pure monogenic Mendelian disorder, caused by a single variant in a single gene. In some families, this may in fact still be the case. However, the view of BrS genetics has more recently evolved to recognize, in most cases, that BrS is an oligogenic syndrome, which can result from the combined effect of multiple variants, even in multiple genes, that occur in the same person (28, 40, 41). Much research is underway to determine what genes may be involved. Candidate genes (2, 28) commonly involve those encoding for sodium, potassium, and calcium channels (42–46), as well as, less commonly, sarcomeric and structural proteins (17, 18, 45, 47, 48) and mitochondrial genes (49) (Table 1).
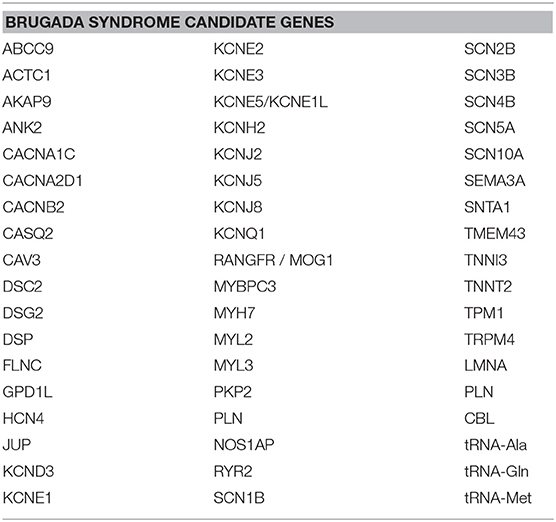
Table 1. BrS candidate genes.
There are several candidate genes currently under investigation, reviewed elsewhere (2, 28, 40). Here, we do not seek to exhaustively list every single candidate gene that exists, but rather, we focus on a selected number of them to highlight the concept that these genes are expressed not only in the heart, but throughout the body, and thus mutations in any of these genes could have pathogenic effects not only on the heart, but also on other organ systems. The most widely screened BrS candidate genes encoding for sodium channels include SCN5A, SCN10A, SCN1B, SCN2B, and SCN3B, while genes encoding for potassium channels include HCN4, KCND2, KCND3, KCNE3, KCNE5, KCNH2, and KCNJ8, and genes encoding for calcium channels include CACNA1C, CACNA2D1, CACNB2, PLN, and TRPM4 (2, 28, 40). These genes are expressed not only in the heart, but also throughout the rest of the body. Figure 1 shows the organs in which the protein level encoded by certain sodium, potassium, and calcium genes described in BrS is highly expressed. However, many of these genes encode for proteins that are expressed to a lesser extent in several other organs. Despite being expressed to a lesser extent, the low expression of these proteins in other cell types can influence disease expression and contribute to overlap pathologies. For more details on the organs and cell types in which these genes are expressed to a lower extent, see GeneCards (29).
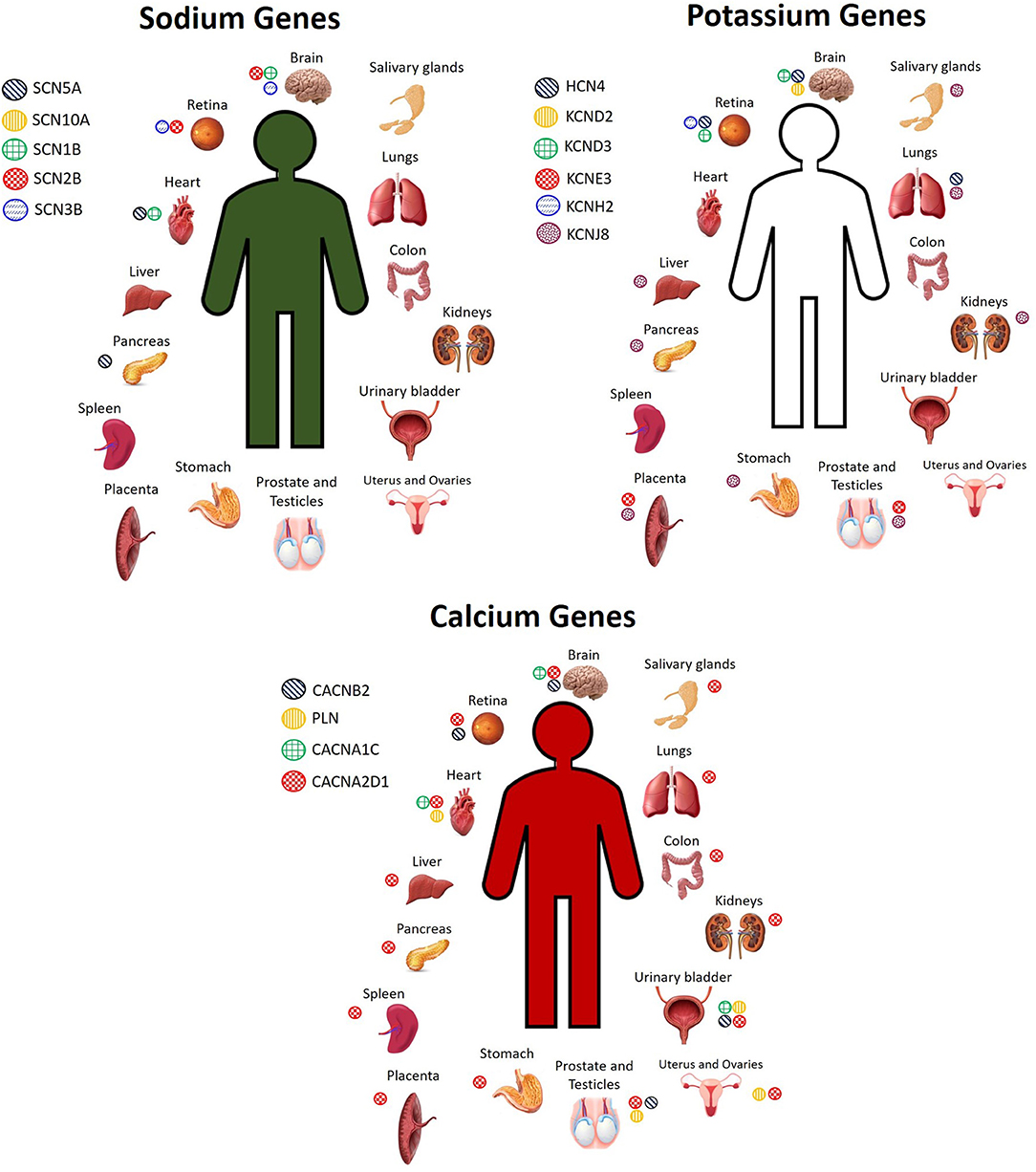
Figure 1. Cell and tissue types in which sodium, potassium, and calcium channel BrS candidate genes encode for high levels of protein expression. Not pictured: Sodium genes: SCN5A: plasma, pancreas juice. SCN10A: monocytes, neutrophil, B cells, T cells, skin. SCN2B: spinal cord, fetal heart, fetal brain. SCN3B: fetal brain. Potassium genes: HCN4: plasma, PBMCs, fetal heart. KCND2: fetal brain. KCND3: fetal brain, bones. KCNJ8: lymph node, bone marrow, stromal cells, nasopharynx, rectum, thyroid, adrenal, breast, gallbladder, skin, cervix, seminal vesicles Calcium genes: CACNA2D1: serum, plasma, PBMCs, lymph nodes, tonsil, bones, skin, bone marrow, esophagus, adipocytes, fetal brain, frontal cortex, cerebral cortex, cerebral spinal fluid, spinal cord, fetal heart, oral epithelial, cardia, fetal gut, rectum, thyroid, adrenal, gallbladder, urine, fetal ovary, fetal testis, seminal vesicles. PLN: tonsil, fetal heart, esophagus, urinary bladder, seminal vesicles. CACNB2: fetal brain. CACNA1C: frontal cortex, fetal heart, urinary bladder. TRPM4: Spinal cord, nasal respiratory epithelium, rectum, breast (29).
Calcium is the connection between excitation and contraction, and in fact, arrhythmias and sudden death can occur as a result of calcium mishandling, including altered calcium signaling resulting from sarcomeric mutations (50). Recent studies have suggested a possible involvement of sarcomeric mutations in BrS (17, 45, 51) that alter calcium signaling (18, 42). BrS sarcomeric gene candidates include TPM1, MYBPC3, DSG2, PKP2, LMNA, MYH7, TTN, and GATA4 (17, 45, 47, 48, 52). In BrS, it has been proposed that loss of the action potential, and consequent reduced calcium channel current and cardiomyocyte calcium depletion, could result in wall motion abnormalities, dilation of the right ventricular outflow tract (RVOT) region, and reduced ejection fraction (EF) (31, 42, 53, 54), which have, in fact, been recently observed clinically in patients affected by BrS (55). BrS candidate genes encoding for proteins important in cell signaling include AKAP9, ABCC9, and CBL (40, 45, 56), and these, like the other BrS gene candidates, are expressed not only in the heart, but additionally throughout the rest of the body. The cell or tissue types in which these genes are highly expressed are shown in Figure 2. For a more comprehensive assessment of all the tissue types in which they are expressed even to a lesser extent, please see GeneCards (29).
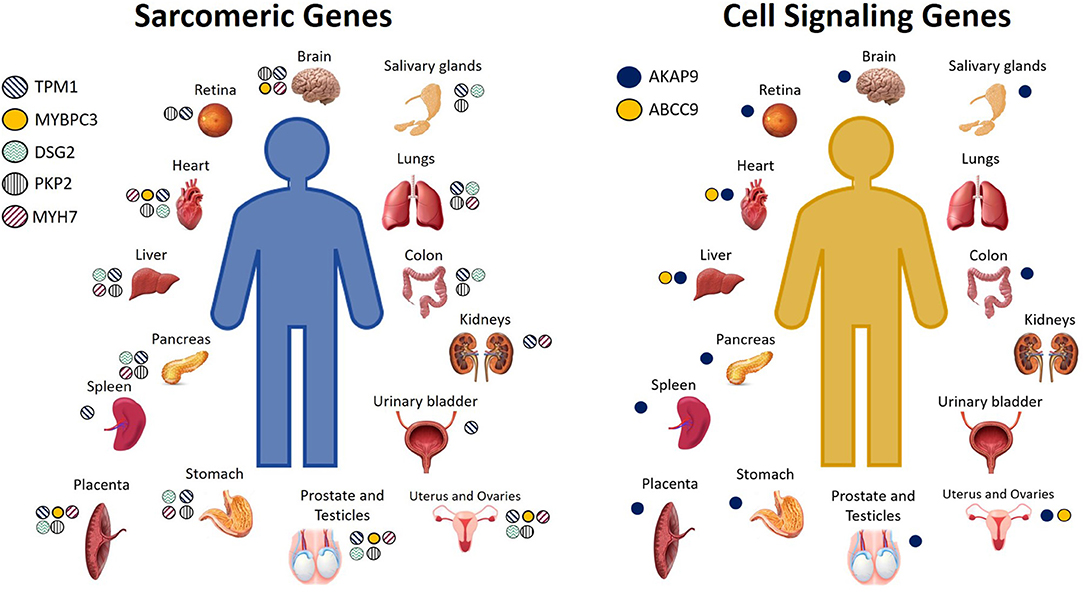
Figure 2. Cell and tissue types in which sarcomeric and cell signaling BrS candidate genes encode for high levels of protein expression. Not pictured: Sarcomeric genes: TPM1: Serum, plasma, monocytes, neutrophil, B lymphocytes, CD8 T cells, NK cells, PBMCs, platelets, lymph nodes, tonsils, bone marrow, fetal brain, spinal cord, fetal heart, bone, oral epithelial, nasopharynx, esophagus, cardia, gut fetal, rectum, fetal liver, adipocytes, amniocytes, thyroid, adrenal, breast, milk, gallbladder, urine, skin, fetal ovary, fetal testis, seminal vesicles. MYBPC3: fetal brain, fetal heart, oral epithelium, fetal liver, adrenal, fetal testis. DSG2: serum, plasma, platelets, tonsils, fetal heart, oral epithelia, esophagus, cardia, fetal gut, rectum, fetal liver, adipocytes, synovial fluid, amniocytes, thyroid, breast, gallbladder, urine, urinary bladder, skin, fetal ovary. PKP2: platelets, spinal cord, fetal heart, fetal gut, rectum, amniocytes, adrenal, breast, gallbladder, skin, fetal ovary, fetal testis. MYH7: platelets, tonsil, fetal brain, spinal cord, fetal heart, esophagus, rectum, fetal liver, adipocytes, fetal ovaries, fetal testis. Cell signaling genes: AKAP9: monocytes, PBMCs, platlets, lymph node, brain fetal, esophagus, cardia, rectum, fetal liver, adipocytes, amniocytes, adrenal, pancreatic juice, islet of Langherans, gallbladder, ovary fetal, testis fetal. ABCC9: plasma, bone marrow, heart fetal, fetal liver (29).
Genes identified in BrS genetics research as candidate genes related to the mechanism of BrS are expressed throughout the human body (Figures 1, 2), and variants identified in any one of them could modify not only heart function, but the function of any tissue type in which they are expressed. To what extent is uncertain, and we summarize below studies which support the idea that we are only seeing the tip of the iceberg in what can be learned from BrS research, namely, the molecular connections between multiple pathologies throughout the body.
An overlap between cardiac arrhythmia and epilepsy has been described, possibly because of common mutations in genes encoding for ion channels. Several studies point toward the possibility of co-expression of ion channels in both the heart and brain, leading to the manifestation of both cardiac arrhythmia and epilepsy (57–59). Many of these studies have identified gene mutations known to cause cardiac arrhythmia syndromes such as LQTS and BrS. For example, idiopathic epilepsy and LQTS may be linked by mutations in the potassium channel gene, KCNQ1 (58). Also the sodium channel gene, SCN5A, usually associated with SCD and BrS (60–62), has been of particular interest to understand the effects of specific mutations (63–68) or even polymorphisms (69–73) in this gene, and their relation with BrS, and this gene has been linked to sudden unexplained death in epilepsy (SUDEP) (74). In fact, this gene has already been implicated in an overlap syndrome involving both epilepsy and BrS (24). Indeed, up to 18% of patients affected by epilepsy die of SUDEP (59). Unfortunately, autopsy performed on SUDEP patients does not reveal the cause of death nor any evidence related to pulmonary or cardiac pathology (75); however, SUDEP and sudden cardiac death due to cardiac arrhythmia share a few risk factors: age and sex (76). Furthermore, it has also been shown through EEG/ECG combined studies that patients with true epileptic seizures have a high prevalence (33–44%) of cardiac arrhythmias (58, 77–81), including, at least, LQTS, BrS, QT dispersion, sinus tachycardia, T-wave alternans, bradyarrhythmia, or asystole, sometimes through common genetic mutations (25–27, 82–88). Potential gene candidates for SUDEP include FBN1, HCN1, SCN4A, EFHC1, CACNA1A, SCN11A, and SCN10A (89). SCN10A is also a gene candidate for BrS (70, 90, 91), and patients with this syndrome and variants in this gene have been shown to have similar clinical presentations to patients with variants in the gene SCN5A (43).
Episodes of cardiac arrhythmia in patients affected by epilepsy may also be induced by antiepileptic drugs, such as carbamazepine and lamotrigine, which are known to target sodium voltage-gated channels (92–94). Carbamazepine, an anti-epileptic drug used also to treat schizophrenia, induced the BrS ECG pattern in a patient with schizophrenia (95). In a patient with epilepsy and intellectual disability, the drug Lamotrigine induced the BrS ECG pattern, possibly in conjunction with a genetic predisposition for cardiac arrhythmia due to a variant in the SCN9A gene, also possibly in association with a genetic variant in the gene AKAP9 (96). The overlap between epilepsy and cardiac arrhythmias demonstrated in several studies suggest that patients with either of these conditions should be checked for the other. Many of the variants thought to be responsible for cardiac arrhythmias occur in genes that have a low expression in the brain. However, their expression is nevertheless present, and, as can be seen from the literature, very relevant. Thus, future studies certainly should investigate these associations.
An elevated ST segment, which looks identical to the ECG pattern used to diagnose BrS, has been described as a result of potassium or calcium imbalances in the blood. Often, this type of ECG pattern discovered as a result of electrolyte imbalances is described as a BrS “phenocopy.” However, so much is not known about the mechanism of BrS, and given the fact that BrS has historically been defined more for what it is not than for what it is, it may actually be premature to discount ST segment elevations occurring in the presence of electrolyte imbalances in the quest to unveil the mechanism of BrS (41). Several studies have described hypokalemia (97–99), hyperkalemia (100–104), or hypercalcemia (105–107) in association with the BrS ECG pattern, or in association with an elevated ST segment that appears identical to the BrS pattern. Diseases related to these conditions, such as Gitelman syndrome, which results in potassium depletion, usually attributed to the SLC12A3 gene, can lend new candidate genes to the field of BrS (108). In patients with severe hyperkalemia, the ST segment elevation, even if described at the time as a “phenocopy,” was described in association with malignant arrhythmias secondary to resting potential depolarization, reduced sodium current availability, and fibrosis at the right ventricular outflow tract (102), signs which are now typically described also in association with “true BrS,” whatever that is. Hypercalcaemia can be responsible for the ST segment elevation, resembling the BrS ECG pattern, and ventricular fibrillation, secondary to calcium-dependent loss of sodium channel function (106). This mechanism too is consistent with current theories about the possible mechanisms of “true BrS.” Furthermore, hypercalcaemia is associated with hyperthyroidism (109), which is additionally implicated in the development of an ST segment elevation resembling the BrS ECG pattern.
In addition to electrolyte imbalances, abnormalities of the blood occurring even from blood transfusion can result in the BrS ECG pattern. The type 1 BrS ECG pattern appeared after cardiac iron overload after blood transfusion (110). Another patient developed the BrS ECG pattern during febrile neutropenia after undergoing high dose chemotherapy followed by autologous peripheral blood cell transplantation (ABSCT) for acute myeloid leukemia (111). These cases of apparent transmission of the BrS could provide further insight into the mechanisms of the development of the BrS ECG pattern.
Plasmic proteomic changes have been observed in patients with BrS, including increased levels of apolipoprotein E, prothrombin, vitronectin, complement-factor H, vitamin-D-binding protein, voltage-dependent anion-selective channel protein 3, and clusterin (37). Similarly, decreased protein levels were observed for alpha-1-antitrypsin, fibrinogen, and angiotensinogen, and post-translational modifications of antithrombin-III were observed (37).
An elevated ST segment identical to the BrS ECG pattern can also be elicited by environmental factors, which ultimately act on the heart. It has been established that metals could induce this pattern. Several studies reported the manifestation of the BrS ECG pattern after the consumption of metal phosphides, which are mainly found in rodenticide, insecticide, and fumigant, and they are used as suicidal agents. These substances, such as aluminum phosphide (ALP), zinc phosphide (ZnP), calcium phosphide, and magnesium phosphide, are very toxic because they release phosphine gas as soon as they interact with hydrochloric acid in the stomach. The phosphine gas is a non-competitive inhibitor of cytochrome c oxidase in the mitochondria, and the mechanism of toxicity includes the formation of highly reactive free hydroxyl radicals (112). It can result in myocardial toxicity and cardia arrhythmia, which eventually can lead to death (113). Moreover, the intoxication from metal phosphides can result in several ECG abnormalities, including ST segment elevation in leads V1-V3, like the BrS ECG pattern. For example, three different cases of patients who ingested rat poison containing aluminum phosphide manifested significant abnormalities, including the ST segment elevation in leads V1-V3 (114–116). Moreover, another case reported a patient who intoxicated himself with rodenticide, which contained ZnP. The ECG of the patient showed a type 1 BrS pattern, which then normalized after 24 h (117). Finally, a male patient, after the ingestion of 7.5 g of Ratol paste, which is another rodenticide, showed prolonged PR interval, prolonged QRS duration, and the BrS ECG pattern (118). The manifestation of the BrS ECG pattern was related to the yellow phosphorous (YP), which is a protoplasmic poison contained in the paste, and it can cause damage to the liver, kidney, pancreas, brain, and heart (119). These reports about metal poisoning eliciting a BrS ECG pattern are also consistent with the fact that metals, such as zinc and copper, can act as endogenous regulators of sodium, potassium, and calcium channels, including NaV1.5, the sodium channel most implicated in BrS, through mechanisms that could be important not only for the heart, but also for diseases such as Alzheimer's disease and epilepsy (120). Thus, metals could play an important clue in understanding the various ways in which the BrS ECG pattern can be developed.
Whether through genetics, electrolyte imbalance, blood transfusion, poisoning, or other factors, the ECG pattern exhibiting an elevated ST segment can be developed in a number of different ways. However, regardless of the many initiating factors, all these pathways ultimately converge into one: the BrS ECG pattern, and we must not lose sight of the fact that, whatever we want to call it—“true BrS” or “phenocopy”—all of these initiating factors have converged into the same life-threatening arrhythmogenesis pathway, manifesting as the BrS ECG pattern, and the patient is at increased risk of ventricular fibrillation and SCD.
Several studies have identified and described the relationship between thyroid hormone imbalances and cardiovascular diseases (121). It is known that thyroid hormone receptors are expressed in many different cell types, including heart (122), and, therefore, systemic vascular resistance, cardiac contractility, blood pressure, myocardial oxygen consumption, and heart rhythm can be disrupted by both hypothyroidism and hyperthyroidism (123, 124). Indeed, both of these thyroid dysfunctions can lead to cardiovascular disorders, including atrial fibrillation and ventricular arrhythmia (121). These correlations were also confirmed by cardiac electrophysiological studies focused on the activity of multiple ion channel subunits, transporters, and exchangers (121, 125). Moreover, a possible overlap of hypothyroidism and BrS has been reported in three case reports. All cases showed three men affected by hypothyroidism with a BrS ECG pattern in leads V1-V3. The BrS ECG pattern disappeared in all patients when the thyroid function was normalized (126–128). Therefore, the normalization of the ECG was considered directly related with the restoration of thyroid hormone balance. Moreover, one of the three patients with hypothyroidism was also affected by liver dysfunction, which returned to normal after thyroid function went back to normal (128). Only one of the patients underwent genetic testing. This patient resulted positive for three common variants in the SCN5A gene (127). Furthermore, a possible overlap between hyperthyroidism and BrS has been reported. A male patient suffered a cardiac arrest, and after his resuscitation, his ECG showed a type 1 BrS pattern. The laboratory tests showed also low K+ levels, low TSH levels, and high FT4 levels. However, the BrS ECG pattern was only transient, it normalized after carbimazole administration, and the patient resulted negative to ajmaline and flecainide tests. Genetic testing was not performed (129). Moreover, another patient with Graves' hyperthyroidism developed ventricular fibrillation, and she was implanted with an ICD. An ECG that was performed after she was re-admitted for fever and pleural effusion exhibited the type 2 BrS ECG pattern, although the report is unclear whether she performed ECG at the time of the actual fever. Genetic testing revealed a likely pathogenic mutation in the SCN5A gene (130). Therefore, genetic studies could be helpful to better understand the link between the manifestation of the BrS ECG pattern and thyroid dysfunction.
The overexpression of voltage-gated sodium channels has been shown to be associated with metastatic behavior in a variety of human cancers, including breast cancer, prostate cancer, lung cancer, colorectal cancer, cervical cancer, lymphoma, and neuroblastoma (131, 132). Overexpression of the neonatal isoform of the voltage-gated sodium channel, Nav1.5 (nNav1.5), is associated with aggressive human breast cancer metastasis and patient death (131, 133, 134). Nav1.5 overexpression increases metastasis and invasiveness of breast cancer cells by altering H+ efflux and promoting epithelial-to-mesenchymal transition and the expression of cysteine cathepsin (132), possibly due to reduced expression of salt-inducible kinase 1 (SIK1) (135). Both neonatal and adult (aNav1.5) forms of Nav1.5 are expressed at the messenger RNA level in colorectal cancer (136) and the SCN5A gene encoding for the Nav1.5 channel protein is an important regulator of the colon cancer invasion network, involving genes that encompass Wnt signaling, cell migration, ectoderm development, response to biotic stimulus, steroid metabolic process, and cell cycle control (137). Therefore, in the field of cancer, drug treatments that aim to block Nav1.5-dependent inward currents are of interest. However, sodium channel blockers used to treat patients with cancer may exacerbate underlying predispositions for cardiac arrhythmias, as sodium channel blockers, especially those targeting the Nav1.5 channel, may provoke arrhythmias such as the BrS ECG pattern. In fact, drugs studied for possible use in cancer therapy, such as Ranolazine (138) or Tetrodotoxin (136), are known to act not only on cancer cells, but on other cells in the body. Ranolazine acts on cardiac sodium channels and sarcomeric proteins (139). Recent studies have implicated cardiomyocyte sarcomeric alterations as possible mechanisms of BrS (28). Tetrodotoxin, believed to be one of the most selective inhibitors of voltage-gated fast sodium channels, actually acts additionally on other channels, including cardiac L-type calcium channels (140). Calcium channels and their related proteins have been implicated in a number of cardiac pathologies, including catecholaminergic polymorphic ventricular tachycardia, congenital long QT syndrome, idiopathic ventricular fibrillation, hypertrophic cardiomyopathy, arrhythmogenic cardiomyopathy, dilated cardiomyopathy, heart failure, atrial fibrillation (141), and BrS (28, 42). Thus, the possible development of cardiac arrhythmias should be considered for any drugs used for cancer treatment.
Although equally transmitted to both genders (142), BrS seems to be more predominant in males (143), possibly due to hormonal action, among other factors (42). Specifically, males with BrS have been described to have higher testosterone, serum sodium, potassium, and chloride levels compared to controls, after adjusting for age, exercise, stress, smoking, and medication for hypertension, diabetes, and hyperlipidemia (144). In the same study, males with BrS had significantly lower body-mass index and body fat percentage than controls (144). Thus, a link between high levels of plasma testosterone, BrS, and prostate cancer was suggested (145). Men with Brugada-like electrocardiogram patterns have a higher risk of prostate cancer independent of age, smoking habit, and radiation exposure, and thus, men with either a Brugada-like ECG or prostate cancer should be checked for the other (145). The BrS ECG pattern can disappear after surgical castration for prostate cancer (146). The hypothesis for hormonal involvement in the development of BrS is further supported by cases in which BrS signs and symptoms manifest only in adulthood in patients who tested ajmaline negative in childhood (147). Moreover, healthy men are known to have differences in the ECG, compared to healthy women, for example, shorter and faster repolarization and a shorter duration of QTc and JTc intervals (148), supporting the idea that ventricular repolarization can be disrupted by androgens. Furthermore, no sex-differences in repolarization are observed in neonates or in children before puberty, probably due to low levels of testosterone (149). Cases in which BrS is found in pre-adolescent children, including also infants or when it is expected to be the cause of spontaneous abortions, may be due to certain genetic factors found in those families. For example, certain SCN5A mutations are known to be so detrimental that they can lead to sudden infant death (150), or they have been suspected as the cause of spontaneous abortions (151). In these cases, testosterone may play less (if any) of a role. However, testosterone is certainly one of the many factors that may contribute to the development of the BrS ECG pattern in many patients. Thus, future studies should investigate further the relationship between testosterone levels and the development of both BrS and prostate cancer.
Mitochondria influence cellular physiology in a variety of ways (41), and are involved in many pathologies, including epilepsy (152, 153), thyroid function (154), cancer (155–158), diabetes (159, 160), and BrS (41), among others. Mitochondria produce adenosine triphosphate (ATP), which is required for protein phosphorylation, a process that is required for normal protein function (161). Protein phosphorylation is a normal regulatory process used by the cell to control the force-frequency relationship (the heart beats harder and relaxes faster to accommodate the need for higher heart rates) (162), the length-dependent relationship (Frank-Starling relationship, the heart beats harder when the muscle is stretched) (163), and other necessary intracellular communications (164–168). Proteins that are normally phosphorylated under certain conditions include several of those known to be important for arrhythmia development, including the sodium channel Nav1.5 (encoded by the SCN5A gene; important for BrS) (41), L-type calcium channels (encoded by the CACNA1C gene; important for BrS) (28), ryanodine receptors [encoded by the RYR2 gene; important for catecholaminergic polymorphic ventricular tachycardia (169)], phospholamban [encoded by the PLN gene; important for arrhythmogenic cardiomyopathy and dilated cardiomyopathy (170)], cardiac myosin-binding protein-C (encoded by the MYBPC3 gene; BrS candidate gene) (48), tropomyosin (encoded by the TPM1 gene; BrS candidate gene) (28), and other sarcomeric proteins that regulate cardiac function (50, 163–166, 171), such as troponin I (encoded by the TNNI3 gene), myosin light chain-2 (encoded by the MLC2 gene), and troponin T (encoded by the TNNT2 gene). Therefore, mitochondrial dysfunction that results in ATP depletion would alter the function of these proteins, possibly resulting in cardiac arrhythmias (50, 172–174). Furthermore, mitochondria are additionally responsible for the production of reactive oxygen species (ROS), and a mutation in the GPD1-L protein, encoded by an important BrS candidate gene (175), has been shown to promote ROS production, which could be detrimental to the sodium current (176). BrS has been associated with mutations in mitochondrial transfer RNA (tRNA) genes (49) and a specific mitochondrial DNA (mtDNA) allelic combination and a high number of mtDNA single nucleotide polymorphisms (SNPs) (177, 178). Thus, mitochondrial genes and function are important for BrS. Mitochondrial products that can lead to arrhythmias are shown in Figure 3.
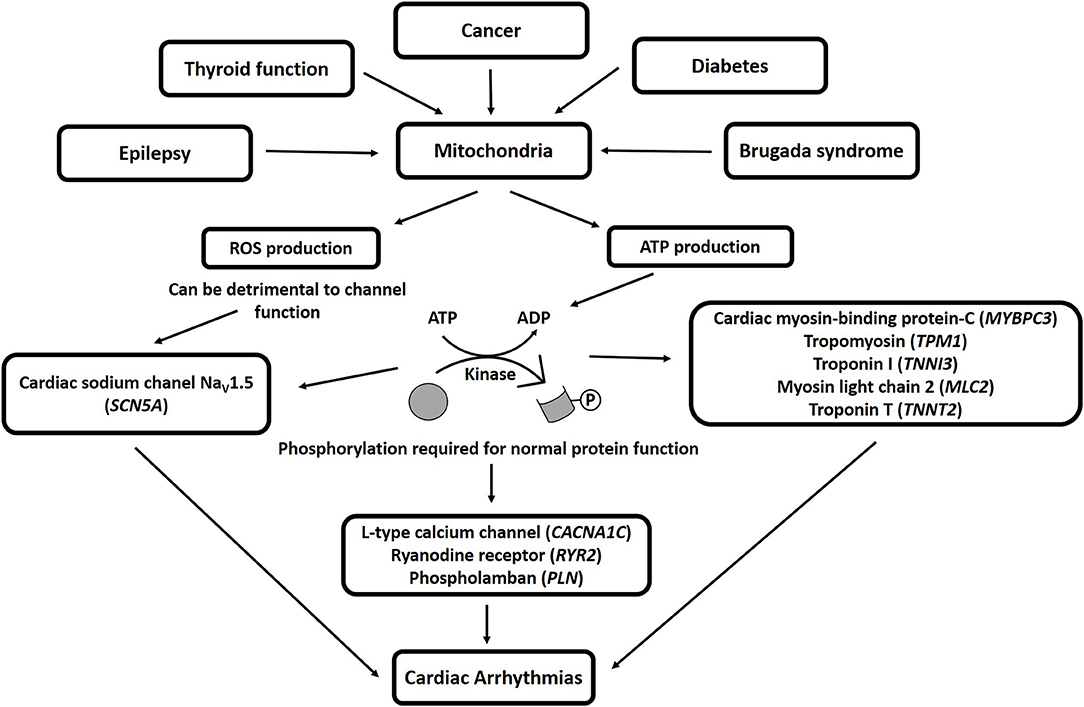
Figure 3. Mitochondrial products that can lead to arrhythmias. ROS, reactive oxygen species; ATP, adenosine triphosphate; ADP, adenosine diphosphate.
Ion channel dysfunction has been associated with diabetes mellitus (DM), leading to the development of a heart disorder called diabetic cardiomyopathy (179, 180), characterized by contractile dysfunction, abnormal cardiac electrical activity, mitochondrial dysfunction, arrhythmia, and sudden cardiac death (159, 160). Several patients affected by DM have been reported to exhibit the BrS ECG pattern, whether it was considered “true BrS” or a “BrS phenocopy” (104, 181–184), although a difference between “true BrS” and “BrS phenocopy” may not actually exist (41). A case report described a 16-year-old boy affected by DM and a mutation in the GPD1L gene who died suddenly during the night (185). The autopsy examination excluded hypoglycemia as the cause of death due to a full stomach and normal glucose levels in the vitreous humor. Indeed, the GPD1L gene is important for BrS, and may have been the cause of death (186). Variants in this gene have also been linked to sudden infant death syndrome (SIDS) (187). Therefore, a genetic link may exist between diabetes and BrS that could result in a phenotypic overlap. However, diabetes can also be caused by non-genetic factors, such as life style, food, persistent organic pollutants, and the gut ecosystem (188), and diabetes can cause fibrosis leading to diastolic heart failure with preserved ejection fraction, which may deteriorate into the development of arrhythmias and sudden death (189). Therefore, we cannot rule out that disease overlap is always caused by genetic factors, but instead the situation may be the evolution of one disease eventually resulting in the other, or there may be no mechanistic overlap at all in some patients (two different mutations, or a mutation and an environmental influence). In any case, an overlap exists between diabetes, cardiac arrhythmias, and sudden death that will be the subject of future investigations.
The SCN4A gene encodes for the voltage-gated sodium channel NaV1.4, and its mutations are usually related to non-dystrophic myotonia, periodic paralysis, and myasthenic syndrome (190). This gene has also been implicated in overlap between BrS and skeletal muscle sodium channelopathies (122). Indeed, several patients with pathogenic mutations in the SCN4A gene and cardiac electrophysiological abnormalities have been described (191, 192). For example, a patient affected by non-dystrophic myotonia with a mutation in the SCN4A gene resulted positive to BrS with both flecainide and ajmaline challenges (192). Additionally, in a different study, the authors investigated whether BrS can be part of the clinical phenotype associated with SCN4A variants, and whether patients with BrS present with non-dystrophic myotonia or periodic paralysis and related gene mutations. Three patients from families with an SCN4A-associated non-dystrophic myotonia had also BrS. Also, the authors found a high prevalence of myotonic features in the families with BrS, involving other genes (191). NaV1.4 and NaV1.5 are both expressed in the skeletal muscle during embryogenesis (193), and they have 65% sequence identity (194). However, during the development, the relative NaV1.5 expression decreases, making NaV1.4 the most abundantly expressed sodium channel in skeletal muscle (193, 195). Taken together, patients with BrS or skeletal muscles channelopathies should be evaluated for the other.
Lamin A and C are intermediate filament proteins of the nuclear protein, and they are encoded by the LMNA gene. Diseases associated with the LMNA gene include Hutchinson-Gilford Progeria Syndrome, Emery-Dreifuss Muscular Dystrophy 2, Autosomal Dominant (29), and inherited cardiomyopathies, such as arrhythmogenic cardiomyopathy and dilated cardiomyopathy (125). A patient who experienced a cardiac arrest and manifested a BrS ECG pattern resulted positive for a variant in the LMNA gene, suggesting an overlap between BrS and laminopathies (125). Thus, this gene could be investigated in patients with BrS in future studies.
There have been several excellent studies regarding the overlap between BrS and other cardiac pathologies, including early repolarization syndrome (5), arrhythmogenic right ventricular cardiomyopathy/dysplasia (ARVC/D) (6–9), progressive cardiac conduction defect (Lenègre syndrome) (10, 11), LQTS (12–14), Wolff-Parkinson-White (15, 16), hypertrophic cardiomyopathy (17, 18), atrial flutter (19), and atrial fibrillation (20–23). Therefore, whatever mechanisms that ultimately lead to the development of the BrS ECG pattern may also be involved in other pathways that result in other cardiac pathologies. These mechanisms may include genetic variants, fibrosis, altered calcium signaling, and anatomical substrate overlap.
BrS has been hypothesized to be like focal epicardial arrhythmogenic cardiomyopathy, and the final common pathway “reduced RVOT conduction reserve,” regardless of the genetics or non-genetic factors that may have been responsible. In that study, the patient's intrinsic RVOT conduction reserve was hypothesized to be age- and sex-dependent, with marginal reserves able to be exposed by the use of certain drugs or altered vagal tone (196).
The genetics of BrS remain elusive, as the most commonly found mutated gene, SCN5A, is only found to be mutated in a minority of patients. However, this gene is widely studied, and is the only one considered by some groups to have definitive evidence to be involved in BrS, while the involvement of the other genes is disputed (197). Since then, several other studies have been published supporting the role of other genes, although research is still underway to understand fully the genetic background of patients with BrS (44). The discovery of genes involved in BrS may be complicated by the possibility of BrS to be transmitted as a polygenic or oligogenic disease, making the data more complicated to interpret, and thus impeding the ability to reclassify these variants (40).
There is already a lot of evidence for genetic overlap between BrS and other cardiac pathologies. Overlap between BrS and LQTS type 3 has been described (198), even sharing the same mutation in the SCN5A gene as the likely molecular cause (13, 199). In fact, variants in the SCN5A gene have been described as causative for a number of other cardiac pathologies, such as ARVC/D, atrial standstill, atrial fibrillation, left ventricular non-compaction, dilated cardiomyopathy, LQTS, sick sinus syndrome, idiopathic ventricular fibrillation, and heart block (200, 201). In addition, several other genes associated with BrS are also associated with other cardiac pathologies, as previously reviewed elsewhere (202), such as PKP2, DSC2, JUP, DSG2, and RYR2 in ARVC (108, 202), SCN1B and CACNA1C in LQTS, CACNA1C, CACNA2D1, and CACNB2 in short QT syndrome, and CACNA1C, CACNA2D1, CACNB2, HCN4, and KCNJ8 in early repolarization syndrome (202). Atrial fibrillation is associated with a number of genes that have been suspected in BrS, including SCN5A, SCN1B, SCN2B, SCN3B, SCN4B, SCN10A, ABCC9, HCN4, KCNQ1, KCNJ2, KCNJ5, KCNJ8, KCNE1, KCNE2, KCNE3, KCNE5, KCNH2, KCND3, RYR2, and CACNB2 (203). Therefore, genetics may play more of a role in the overlap of these pathologies than currently understood.
Disturbances of the connexome, the complex of structures of the intercalated disc that connect cardiomyocytes, could be involved in the mechanism of overlap between BrS and both arrhythmogenic cardiomyopathy and ARVC (9, 41). In addition to the overlap of several causative genes, the extracellular matrix and fibrosis may play a role in the overlap of several of these pathologies, as it is found to be present in BrS (204, 205), early repolarization syndrome (5), ARVC/D (206), Lenègre syndrome (207), LQTS (208–210), Wolff-Parkinson-White (211, 212), hypertrophic cardiomyopathy (213), atrial flutter and atrial fibrillation (214, 215). Thus, disturbances of the connexome and fibrosis can be important elements in the overlap of BrS with other cardiac pathologies.
There may be an effect of anatomical overlap between BrS and other cardiac pathologies. For example, sarcomeric alterations, which are known to cause hypertrophic or dilated cardiomyopathy, are able to cause calcium mishandling that can lead to sudden death (216, 217), and so, over time, overlap may occur as the pathology progresses and affects other areas of the cell.
An arrhythmogenic substrate (AS) is consistently identified in all patients with BrS. This AS is commonly found in the RVOT of the right ventricle (RV) epicardium. Ajmaline administration reveals the full extent of the AS, which is useful for radiofrequency ablation, which results in ECG normalization and the patients are no longer inducible for VT/VF during electrophysiological study (EPS) (4, 218). The size of the AS has been independently associated with arrhythmia inducibility, with a substrate area of greater than 4 cm2 predicting patient inducibility to VT/VF during the EPS (219). The size of the AS can be altered not only by ajmaline, but by also a number of other factors, including genetics, temperature, anesthetics (220), and other factors known to trigger syncopic episodes or the BrS ECG sign. A larger AS is associated with reduced contractility and mechanical dysfunction (51). Patients harboring SCN5A variants exhibit more pronounced epicardial electrical abnormalities (60, 62), and the AS is comparable in patients with SCN10A variants (43). The procedure of epicardial mapping together with ajmaline challenge can be used to identify and ablate the AS in patients with other cardiomyopathies, thus preventing the recurrence of VT/VF in those patients as well (48).
While BrS was described for decades as a simple monogenic disease originating only as a channelopathy, more recent studies have begun to recognize that actually BrS is an oligogenic disease with altered metabolics (28, 40). BrS has traditionally been described more for what it is not than for what it is, being described as a particular ECG pattern in the absence of certain criteria, but we believe that whenever the BrS ECG pattern is present, the sign should be taken for what it really is: a warning of increased risk of sudden death of the patient (41). It does not matter how the sign arose—from a channelopathy, a cardiomyopathy, an electrolyte imbalance, heavy meals, alcohol intake, or even poisoning—the BrS mechanism is a particular ECG warning of the increased risk of sudden death. Rather than being thought of as a single disease, it should be thought of as the final step in many pathways in many diseases or after exposure to certain environmental situations, that ultimately manifests as the BrS ECG pattern—a mechanism that can cause sudden death.
The BrS ECG sign does not always indicate solely cardiac dysfunction, but, in fact, this ECG sign could be the result of a larger systemic problem. Genetic mutations that are expressed in multiple cell types may contribute to overlap pathologies. We hypothesize that, with time, more overlap phenotypes will emerge in a host of other cell types, especially since many of the genes suspected to be involved in BrS are expressed throughout the body. Thus, future studies may focus on the effects of these genetic mutations in various cell and tissue types, identifying other problems for which the patients must be treated, as well as possible mechanisms for doing so. This could connect a whole magnitude of other pathologies to BrS, lending further evidence to the mechanism of BrS, arriving finally at better therapeutic options.
Ideally, where available, whole genome sequencing should be performed for the identification of new candidate genes. The effects of these specific genetic mutations in specific patients should then be assessed on a case-by-case basis to try to understand the effects of these mutations on other tissue types, in a move toward personalized medicine. Additionally, a multi-omics approach would be appropriate to study this syndrome, including not only genetics, but also epigenomics, transcriptomics, proteomics, metabolomics, lipidomics, and glycomics, resulting eventually in a biomarker for BrS and the ability to diagnose this syndrome using a minimally invasive blood test, avoiding the risk associated with ajmaline testing.
SD'I and MM: conceptualization, literature search, and writing—original draft preparation. CP: funding acquisition. SD'I, MM, and CP: writing—draft revision. SD'I, MM, EM, GC, LA, and CP: reviewed and provided comments. All authors have read and agreed to the published version of the manuscript.
This study was partially supported by Ricerca Corrente funding from Italian Ministry of Health to IRCCS Policlinico San Donato.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Fernandez-Falgueras A, Sarquella-Brugada G, Brugada J, Brugada R, Campuzano O. Cardiac channelopathies and sudden death: recent clinical and genetic advances. Biology. (2017) 6:7. doi: 10.3390/biology6010007
2. Brugada J, Campuzano O, Arbelo E, Sarquella-Brugada G, Brugada R. Present status of brugada syndrome: JACC state-of-the-art review. J Am Coll Cardiol. (2018) 72:1046–59. doi: 10.1016/j.jacc.2018.06.037
3. Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Brugada R, Corrado D, et al. Brugada syndrome: report of the second consensus conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. (2005) 111:659–70. doi: 10.1161/01.CIR.0000152479.54298.51
4. Pappone C, Santinelli V. Brugada syndrome: progress in diagnosis and management. Arrhythm Electrophysiol Rev. (2019) 8:13–8. doi: 10.15420/aer.2018.73.2
5. Boukens BJ, Potse M, Coronel R. Fibrosis and conduction abnormalities as basis for overlap of brugada syndrome and early repolarization syndrome. Int J Mol Sci. (2021) 22:1570. doi: 10.3390/ijms22041570
6. Perez Riera AR, Antzelevitch C, Schapacknik E, Dubner S, Ferreira C. Is there an overlap between Brugada syndrome and arrhythmogenic right ventricular cardiomyopathy/dysplasia? J Electrocardiol. (2005) 38:260–3. doi: 10.1016/j.jelectrocard.2005.03.009
7. Corrado D, Zorzi A, Cerrone M, Rigato I, Mongillo M, Bauce B, et al. Relationship between arrhythmogenic right ventricular cardiomyopathy and brugada syndrome: new insights from molecular biology and clinical implications. Circ Arrhythm Electrophysiol. (2016) 9:e003631. doi: 10.1161/CIRCEP.115.003631
8. Kataoka S, Serizawa N, Kitamura K, Suzuki A, Suzuki T, Shiga T, et al. An overlap of Brugada syndrome and arrhythmogenic right ventricular cardiomyopathy/dysplasia. J Arrhythm. (2016) 32:70–3. doi: 10.1016/j.joa.2015.10.007
9. Moncayo-Arlandi J, Brugada R. Unmasking the molecular link between arrhythmogenic cardiomyopathy and Brugada syndrome. Nat Rev Cardiol. (2017) 14:744–56. doi: 10.1038/nrcardio.2017.103
10. Probst V, Allouis M, Sacher F, Pattier S, Babuty D, Mabo P, et al. Progressive cardiac conduction defect is the prevailing phenotype in carriers of a Brugada syndrome SCN5A mutation. J Cardiovasc Electrophysiol. (2006) 17:270–5. doi: 10.1111/j.1540-8167.2006.00349.x
11. Shimizu W. Does an overlap syndrome really exist between Brugada syndrome and progressive cardiac conduction defect (Lenegre syndrome)? J Cardiovasc Electrophysiol. (2006) 17:276–8. doi: 10.1111/j.1540-8167.2006.00406.x
12. Kotta CM, Anastasakis A, Stefanadis C. Effects of mutations and genetic overlap in inherited long-QT and Brugada arrhythmia syndromes. Hellenic J Cardiol. (2012) 53:439–46.
13. Kanters JK, Yuan L, Hedley PL, Stoevring B, Jons C, Bloch Thomsen PE, et al. Flecainide provocation reveals concealed brugada syndrome in a long QT syndrome family with a novel L1786Q mutation in SCN5A. Circ J. (2014) 78:1136–43. doi: 10.1253/circj.CJ-13-1167
14. Nakaya H. SCN5A mutations associated with overlap phenotype of long QT syndrome type 3 and Brugada syndrome. Circ J. (2014) 78:1061–2. doi: 10.1253/circj.CJ-14-0319
15. Jaiswal A, Heretis K, Goldbarg S. Coexistent Brugada Syndrome and Wolff-Parkinson-White syndrome: what is the optimal management? Indian Pacing Electrophysiol J. (2013) 13:173–7. doi: 10.1016/S0972-6292(16)30669-6
16. Erdogan O. Coexistence of Wolff-Parkinson-White and Brugada ECG. Turk Kardiyol Dern Ars. (2018) 46:433–4. doi: 10.5543/tkda.2018.75271
17. Mango R, Luchetti A, Sangiuolo R, Ferradini V, Briglia N, Giardina E, et al. Next generation sequencing and linkage analysis for the molecular diagnosis of a novel overlapping syndrome characterized by hypertrophic cardiomyopathy and typical electrical instability of Brugada syndrome. Circ J. (2016) 80:938–49. doi: 10.1253/circj.CJ-15-0685
18. Monasky MM, Ciconte G, Anastasia L, Pappone C. Commentary: next generation sequencing and linkage analysis for the molecular diagnosis of a novel overlapping syndrome characterized by hypertrophic cardiomyopathy and typical electrical instability of Brugada syndrome. Front Physiol. (2017) 8:1056. doi: 10.3389/fphys.2017.01056
19. Hothi SS, Ara F, Timperley J. p.Y1449C SCN5A mutation associated with overlap disorder comprising conduction disease. Brugada syndrome, and atrial flutter. J Cardiovasc Electrophysiol. (2015) 26:93–7. doi: 10.1111/jce.12470
20. Francis J, Antzelevitch C. Atrial fibrillation and Brugada syndrome. J Am Coll Cardiol. (2008) 51:1149–53. doi: 10.1016/j.jacc.2007.10.062
21. Kewcharoen J, Rattanawong P, Kanitsoraphan C, Mekritthikrai R, Prasitlumkum N, Putthapiban P, et al. Atrial fibrillation and risk of major arrhythmic events in Brugada syndrome: a meta-analysis. Ann Noninvasive Electrocardiol. (2019) 24:e12676. doi: 10.1111/anec.12676
22. Tse G, Lee S, Mok NS, Liu T, Chang D. Incidence and predictors of atrial fibrillation in a Chinese cohort of Brugada syndrome. Int J Cardiol. (2020) 314:54–7. doi: 10.1016/j.ijcard.2020.05.007
23. Vlachos K, Mascia G, Martin CA, Bazoukis G, Frontera A, Cheniti G, et al. Atrial fibrillation in Brugada syndrome: current perspectives. J Cardiovasc Electrophysiol. (2020) 31:975–84. doi: 10.1111/jce.14361
24. Parisi P, Oliva A, Coll Vidal M, Partemi S, Campuzano O, Iglesias A, et al. Coexistence of epilepsy and Brugada syndrome in a family with SCN5A mutation. Epilepsy Res. (2013) 105:415–8. doi: 10.1016/j.eplepsyres.2013.02.024
25. Sandorfi G, Clemens B, Csanadi Z. Electrical storm in the brain and in the heart: epilepsy and Brugada syndrome. Mayo Clin Proc. (2013) 88:1167–73. doi: 10.1016/j.mayocp.2013.06.019
26. Camacho Velasquez JL, Rivero Sanz E, Velazquez Benito A, Mauri Llerda JA. Epilepsy and Brugada syndrome. Neurologia. (2017) 32:58–60. doi: 10.1016/j.nrl.2015.03.010
27. Abdelghani MS, Chapra A, Asaad N, Hayat SA. Epilepsy and Brugada Syndrome: association or uncommon presentation? Heart Views. (2020) 21:114–7. doi: 10.4103/HEARTVIEWS.HEARTVIEWS_34_20
28. Monasky MM, Micaglio E, Ciconte G, Pappone C. Brugada Syndrome: OLIGOGENIC OR MENDELIAN DISEASe? Int J Mol Sci. (2020) 21:1687. doi: 10.3390/ijms21051687
29. GeneCards: The Human Gene Database [Online] (2021). Available online at: https://www.genecards.org/ (accessed July 22, 2021).
30. D'Imperio S, Monasky MM, Micaglio E, Negro G, Pappone C. Impact of dietary factors on Brugada syndrome and long QT syndrome. Nutrients. (2021) 13:2482. doi: 10.3390/nu13082482
31. Antzelevitch C. Brugada syndrome. Pacing Clin Electrophysiol. (2006) 29:1130–59. doi: 10.1111/j.1540-8159.2006.00507.x
32. Obeyesekere MN, Klein GJ, Modi S, Leong-Sit P, Gula LJ, Yee R, et al. How to perform and interpret provocative testing for the diagnosis of Brugada syndrome, long-QT syndrome, and catecholaminergic polymorphic ventricular tachycardia. Circ Arrhythm Electrophysiol. (2011) 4:958–64. doi: 10.1161/CIRCEP.111.965947
33. McMillan MR, Day TG, Bartsota M, Mead-Regan S, Bryant R, Mangat J, et al. Feasibility and outcomes of ajmaline provocation testing for Brugada syndrome in children in a specialist paediatric inherited cardiovascular diseases centre. Open Heart. (2014) 1:e000023. doi: 10.1136/openhrt-2013-000023
34. Fish JM, Antzelevitch C. Role of sodium and calcium channel block in unmasking the Brugada syndrome. Heart Rhythm. (2004) 1:210–7. doi: 10.1016/j.hrthm.2004.03.061
35. Kiesecker C, Zitron E, Lück S, Bloehs R, Scholz EP, Kathöfer S, et al. Class Ia anti-arrhythmic drug ajmaline blocks HERG potassium channels: mode of action. Naunyn Schmiedebergs Arch Pharmacol. (2004) 370:423–35. doi: 10.1007/s00210-004-0976-8
36. Chukhlova EA, Sadykov I, Kholmukhamedov EL, Grenader AK. [Effect of trimecaine, ajmaline, stenopril and chloracizine on fluctuations in K+ currents in rat liver mitochondria]. Ukr Biokhim Zh. (1984) 56:207–10.
37. Di Domenico M, Scumaci D, Grasso S, Gaspari M, Curcio A, Oliva A, et al. Biomarker discovery by plasma proteomics in familial Brugada Syndrome. Front Biosci. (2013) 18:564–71. doi: 10.2741/4120
38. Chatterjee D, Pieroni M, Fatah M, Charpentier F, Cunningham KS, Spears DA, et al. An autoantibody profile detects Brugada syndrome and identifies abnormally expressed myocardial proteins. Eur Heart J. (2020) 41:2878–90. doi: 10.1093/eurheartj/ehaa383
39. Wilde AM, Lodder EM. A highly specific biomarker for Brugada syndrome. Also too good to be true? Eur Heart J. (2020) 41:2891–3. doi: 10.1093/eurheartj/ehaa468
40. Campuzano O, Sarquella-Brugada G, Cesar S, Arbelo E, Brugada J, Brugada R. Update on genetic basis of Brugada syndrome: monogenic, polygenic or oligogenic? Int J Mol Sci. (2020) 21:7155. doi: 10.3390/ijms21197155
41. Monasky MM, Micaglio E, Locati ET, Pappone C. Evaluating the use of genetics in Brugada syndrome risk stratification. Front Cardiovasc Med. (2021) 8:652027. doi: 10.3389/fcvm.2021.652027
42. Monasky MM, Pappone C, Piccoli M, Ghiroldi A, Micaglio E, Anastasia L. Calcium in Brugada syndrome: questions for future research. Front Physiol. (2018) 9:1088. doi: 10.3389/fphys.2018.01088
43. Monasky MM, Micaglio E, Vicedomini G, Locati ET, Ciconte G, Giannelli L, et al. Comparable clinical characteristics in Brugada syndrome patients harboring SCN5A or novel SCN10A variants. Europace. (2019) 21:1550–8. doi: 10.1093/europace/euz186
44. Di Mauro V, Ceriotti P, Lodola F, Salvarani N, Modica J, Bang ML, et al. Peptide-based targeting of the L-type calcium channel corrects the loss-of-function phenotype of two novel mutations of the CACNA1 gene associated with Brugada syndrome. Front Physiol. (2020) 11:616819. doi: 10.3389/fphys.2020.616819
45. Pappone C, Micaglio E, Locati ET, Monasky MM. The omics of channelopathies and cardiomyopathies: what we know and how they are useful. Eur Heart J Suppl. (2020) 22:L105–9. doi: 10.1093/eurheartj/suaa146
46. Monasky MM, Rutigliani C, Micaglio E, Pappone C. Commentary: Peptide-based targeting of the L-type calcium channel corrects the loss-of-function phenotype of two novel mutations of the CACNA1 gene associated with Brugada syndrome. Front Physiol. (2021) 12:682567. doi: 10.3389/fphys.2021.682567
47. Di Resta C, Pietrelli A, Sala S, Della Bella P, De Bellis G, Ferrari M, et al. High-throughput genetic characterization of a cohort of Brugada syndrome patients. Hum Mol Genet. (2015) 24:5828–35. doi: 10.1093/hmg/ddv302
48. Pappone C, Monasky M, Ciconte G. Epicardial ablation in genetic cardiomyopathies: a new frontier. Eur Heart J Suppl. (2019) 21:B61–6. doi: 10.1093/eurheartj/suz028
49. Tafti MF, Khatami M, Rezaei S, Heidari MM, Hadadzadeh M. Novel and heteroplasmic mutations in mitochondrial tRNA genes in Brugada syndrome. Cardiol J. (2018) 25:113–9. doi: 10.5603/CJ.a2017.0104
50. Yar S, Monasky MM, Solaro RJ. Maladaptive modifications in myofilament proteins and triggers in the progression to heart failure and sudden death. Pflugers Arch. (2014) 466:1189–97. doi: 10.1007/s00424-014-1457-7
51. Pappone C, Monasky MM, Micaglio E, Ciconte G. Right ventricular electromechanical abnormalities in Brugada syndrome: is this a cardiomyopathy? Eur Heart J Suppl. (2020) 22:E101–4. doi: 10.1093/eurheartj/suaa071
52. Han H, Wang Y, Zhou F, Chen X. [Analysis of DSG2, TTN and GATA4 gene variants in patients with Brugada syndrome from Henan]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. (2021) 38:488–91. doi: 10.3760/cma.j.cn511374-20200416-00275
53. Antzelevitch C. Brugada syndrome: historical perspectives and observations. Eur Heart J. (2002) 23:676–8. doi: 10.1053/euhj.2001.3145
54. Antzelevitch C, Yan GX, Ackerman MJ, Borggrefe M, Corrado D, Guo J, et al. J-Wave syndromes expert consensus conference report: emerging concepts and gaps in knowledge. J Arrhythm. (2016) 32:315–39. doi: 10.1016/j.joa.2016.07.002
55. Pappone C, Mecarocci V, Manguso F, Ciconte G, Vicedomini G, Sturla F, et al. New electromechanical substrate abnormalities in high-risk patients with Brugada syndrome. Heart Rhythm. (2020) 17:637–45. doi: 10.1016/j.hrthm.2019.11.019
56. Allegue C, Coll M, Mates J, Campuzano O, Iglesias A, Sobrino B, et al. Genetic analysis of arrhythmogenic diseases in the era of NGS: the complexity of clinical decision-making in Brugada syndrome. PLoS ONE. (2015) 10:e0133037. doi: 10.1371/journal.pone.0133037
57. Aurlien D, Leren TP, Tauboll E, Gjerstad L. New SCN5A mutation in a SUDEP victim with idiopathic epilepsy. Seizure. (2009) 18:158–60. doi: 10.1016/j.seizure.2008.07.008
58. Goldman AM, Glasscock E, Yoo J, Chen TT, Klassen TL, Noebels JL. Arrhythmia in heart and brain: KCNQ1 mutations link epilepsy and sudden unexplained death. Sci Transl Med. (2009) 1:2ra6. doi: 10.1126/scitranslmed.3000289
59. Chahal CA, Salloum MN, Alahdab F, Gottwald JA, Tester DJ, et al. Systematic review of the genetics of sudden unexpected death in epilepsy: potential overlap with sudden cardiac death and arrhythmia-related genes. J Am Heart Assoc. (2020) 9:e012264. doi: 10.1161/JAHA.119.012264
60. Ciconte G, Monasky MM, Santinelli V, Micaglio E, Vicedomini G, Anastasia L, et al. Brugada syndrome genetics is associated with phenotype severity. Eur Heart J. (2021) 42:1082–90. doi: 10.1093/eurheartj/ehaa942
61. Monasky MM, Micaglio E, Ciconte G, Rivolta I, Borrelli V, Ghiroldi A, et al. Novel SCN5A p.Val1667Asp missense variant segregation and characterization in a family with severe Brugada syndrome and multiple sudden deaths. Int J Mol Sci. (2021) 22:4700. doi: 10.3390/ijms22094700
62. Pappone C, Ciconte G, Micaglio E, Monasky MM. Common modulators of Brugada syndrome phenotype do not affect SCN5A prognostic value. Eur Heart J. (2021) 42:1273–4. doi: 10.1093/eurheartj/ehab071
63. Micaglio E, Monasky MM, Ciconte G, Vicedomini G, Conti M, Mecarocci V, et al. SCN5A Nonsense mutation and NF1 frameshift mutation in a family with Brugada Syndrome and neurofibromatosis. Front Genet. (2019) 10:50. doi: 10.3389/fgene.2019.00050
64. Micaglio E, Monasky MM, Ciconte G, Vicedomini G, Conti M, Mecarocci V, et al. Novel SCN5A frameshift mutation in brugada syndrome associated with complex arrhythmic phenotype. Front Genet. (2019) 10:547. doi: 10.3389/fgene.2019.00547
65. Micaglio E, Monasky MM, Resta N, Bagnulo R, Ciconte G, Gianelli L, et al. Novel SCN5A p.W697X Nonsense mutation segregation in a family with Brugada syndrome. Int J Mol Sci. (2019) 20:4920. doi: 10.3390/ijms20194920
66. Monasky MM, Micaglio E, Ciconte G, Benedetti S, Di Resta C, Vicedomini G, et al. Genotype/phenotype relationship in a consanguineal family with Brugada syndrome harboring the R1632C missense variant in the SCN5A gene. Front Physiol. (2019) 10:666. doi: 10.3389/fphys.2019.00666
67. Monasky MM, Micaglio E, Giachino D, Ciconte G, Giannelli L, Locati ET, et al. Genotype-phenotype correlation in a family with brugada syndrome harboring the novel p.Gln371* nonsense variant in the SCN5A gene. Int J Mol Sci. (2019) 20:5522. doi: 10.3390/ijms20225522
68. Monasky MM, Micaglio E, Ciconte G, Borrelli V, Giannelli L, Vicedomini G, et al. Novel SCN5A p.V1429M variant segregation in a family with Brugada syndrome. Int J Mol Sci. (2020) 21:5902. doi: 10.3390/ijms21165902
69. Lizotte E, Junttila MJ, Dube MP, Hong K, Benito B, Zutter MDE, et al. Genetic modulation of Brugada syndrome by a common polymorphism. J Cardiovasc Electrophysiol. (2009) 20:1137–41. doi: 10.1111/j.1540-8167.2009.01508.x
70. Bezzina CR, Barc J, Mizusawa Y, Remme CA, Gourraud JB, Simonet F, et al. Common variants at SCN5A-SCN10A and HEY2 are associated with Brugada syndrome, a rare disease with high risk of sudden cardiac death. Nat Genet. (2013) 45:1044–9. doi: 10.1038/ng.2712
71. Makarawate P, Chaosuwannakit N, Vannaprasaht S, Sahasthas D, Koo SH, Lee EJD, et al. SCN5A genetic polymorphisms associated with increased defibrillator shocks in Brugada syndrome. J Am Heart Assoc. (2017) 6:e005009. doi: 10.1161/JAHA.116.005009
72. Matsumura H, Nakano Y, Ochi H, Onohara Y, Sairaku A, Tokuyama T, et al. H558R, a common SCN5A polymorphism, modifies the clinical phenotype of Brugada syndrome by modulating DNA methylation of SCN5A promoters. J Biomed Sci. (2017) 24:91. doi: 10.1186/s12929-017-0397-x
73. Wijeyeratne YD, Tanck MW, Mizusawa Y, Batchvarov V, Barc J, Crotti L, et al. SCN5A Mutation type and a genetic risk score associate variably with Brugada Syndrome phenotype in SCN5A families. Circ Genom Precis Med. (2020) 13:e002911. doi: 10.1161/CIRCGEN.120.002911
74. Ravindran K, Powell KL, Todaro M, O'Brien TJ. The pathophysiology of cardiac dysfunction in epilepsy. Epilepsy Res. (2016) 127:19–29. doi: 10.1016/j.eplepsyres.2016.08.007
75. Cihan E, Devinsky O, Hesdorffer DC, Brandsoy M, Li L, Fowler DR, et al. Temporal trends and autopsy findings of SUDEP based on medico-legal investigations in the United States. Neurology. (2020) 95:e867–77. doi: 10.1212/WNL.0000000000009996
76. Sveinsson O, Andersson T, Carlsson S, Tomson T. The incidence of SUDEP: A nationwide population-based cohort study. Neurology. (2017) 89:170–7. doi: 10.1212/WNL.0000000000004094
77. Nei M, Ho RT, Sperling MR. EKG abnormalities during partial seizures in refractory epilepsy. Epilepsia. (2000) 41:542–8. doi: 10.1111/j.1528-1157.2000.tb00207.x
78. Opherk C, Coromilas J, Hirsch LJ. Heart rate and EKG changes in 102 seizures: analysis of influencing factors. Epilepsy Res. (2002) 52:117–27. doi: 10.1016/S0920-1211(02)00215-2
79. Zijlmans M, Flanagan D, Gotman J. Heart rate changes and ECG abnormalities during epileptic seizures: prevalence and definition of an objective clinical sign. Epilepsia. (2002) 43:847–54. doi: 10.1046/j.1528-1157.2002.37801.x
80. Britton JW, Ghearing GR, Benarroch EE, Cascino GD. The ictal bradycardia syndrome: localization and lateralization. Epilepsia. (2006) 47:737–44. doi: 10.1111/j.1528-1167.2006.00509.x
81. McKeon A, Vaughan C, Delanty N. Seizure versus syncope. Lancet Neurol. (2006) 5:171–80. doi: 10.1016/S1474-4422(06)70350-7
82. Fauchier L, Babuty D, Cosnay P. Epilepsy, Brugada syndrome and the risk of sudden unexpected death. J Neurol. (2000) 247:643–4. doi: 10.1007/s004150070135
83. Girard S, Cadena A, Acharya Y. Woman with Brugada syndrome and epilepsy: a unifying diagnosis? South Med J. (2008) 101:1150–3. doi: 10.1097/SMJ.0b013e318182095b
84. Surges R, Sander JW. Sudden unexpected death in epilepsy: mechanisms, prevalence, and prevention. Curr Opin Neurol. (2012) 25:201–7. doi: 10.1097/WCO.0b013e3283506714
85. Zimmer T, Haufe V, Blechschmidt S. Voltage-gated sodium channels in the mammalian heart. Glob Cardiol Sci Pract. (2014) 2014:449–63. doi: 10.5339/gcsp.2014.58
86. Tiron C, Campuzano O, Perez-Serra A, Mademont I, Coll M, Allegue C, et al. Further evidence of the association between LQT syndrome and epilepsy in a family with KCNQ1 pathogenic variant. Seizure. (2015) 25:65–7. doi: 10.1016/j.seizure.2015.01.003
87. Gigli L, Bertero G, Vidal MC, Iglesias A, Campuzano O, Striano P, et al. Juvenile myoclonic epilepsy and Brugada type 1 ECG pattern associated with (a novel) plakophillin 2 mutation. J Neurol. (2017) 264:792–5. doi: 10.1007/s00415-017-8414-2
88. García Gozalo M, Bermejo Arnedo I, de Vera McMullan P. KCNQ1 gene mutation and epilepsy in patient with long QT syndrome. Med Clin. (2020). doi: 10.1016/j.medcli.2020.09.008. [Epub ahead of print].
89. Coll M, Allegue C, Partemi S, Mates J, Del Olmo B, Campuzano O, et al. Genetic investigation of sudden unexpected death in epilepsy cohort by panel target resequencing. Int J Legal Med. (2016) 130:331–9. doi: 10.1007/s00414-015-1269-0
90. Hu D, Barajas-Martinez H, Pfeiffer R, Dezi F, Pfeiffer J, Buch T, et al. Mutations in SCN10A are responsible for a large fraction of cases of Brugada syndrome. J Am Coll Cardiol. (2014) 64:66–79. doi: 10.1016/j.jacc.2014.04.032
91. Behr ER, Savio-Galimberti E, Barc J, Holst AG, Petropoulou E, Prins BP, et al. Role of common and rare variants in SCN10A: results from the Brugada syndrome QRS locus gene discovery collaborative study. Cardiovasc Res. (2015) 106:520–9. doi: 10.1093/cvr/cvv042
92. Farber NB, Jiang XP, Heinkel C, Nemmers B. Antiepileptic drugs and agents that inhibit voltage-gated sodium channels prevent NMDA antagonist neurotoxicity. Mol Psychiatry. (2002) 7:726–33. doi: 10.1038/sj.mp.4001087
93. Cronin NB, O'Reilly A, Duclohier H, Wallace BA. Binding of the anticonvulsant drug lamotrigine and the neurotoxin batrachotoxin to voltage-gated sodium channels induces conformational changes associated with block and steady-state activation. J Biol Chem. (2003) 278:10675–82. doi: 10.1074/jbc.M208356200
94. Nakatani Y, Masuko H, Amano T. Effect of lamotrigine on Na(v)1.4 voltage-gated sodium channels. J Pharmacol Sci. (2013) 123:203–6. doi: 10.1254/jphs.13116SC
95. Ota H, Kawamura Y, Sato N, Hasebe N. A Carbamazepine-induced Brugada-type electrocardiographic pattern in a patient with schizophrenia. Intern Med. (2017) 56:3047–50. doi: 10.2169/internalmedicine.8875-17
96. Banfi P, Coll M, Oliva A, Alcalde M, Striano P, Mauri M, et al. Lamotrigine induced Brugada-pattern in a patient with genetic epilepsy associated with a novel variant in SCN9A. Gene. (2020) 754:144847. doi: 10.1016/j.gene.2020.144847
97. Araki T, Konno T, Itoh H, Ino H, Shimizu M. Brugada syndrome with ventricular tachycardia and fibrillation related to hypokalemia. Circ J. (2003) 67:93–5. doi: 10.1253/circj.67.93
98. Swe T, Dogar MH. Type 1 Brugada pattern electrocardiogram induced by hypokalemia. J Family Med Prim Care. (2016) 5:709–11. doi: 10.4103/2249-4863.197295
99. Baranchuk A, Antiperovitch P, Gottschalk BH. Brugada phenocopy due to hypokalemia. J Family Med Prim Care. (2018) 7:1141–2. doi: 10.4103/jfmpc.jfmpc_64_17
100. Maheshwari A, Von Wald L, Krishnan B, Benditt DG. Hyperkalemia-induced Brugada phenocopy. JACC Clin Electrophysiol. (2017) 3:1058–9. doi: 10.1016/j.jacep.2016.12.012
101. Tomcsanyi K, Tomcsanyi J. Brugada sign in a patient with hyperkalemia due to rhabdomyolysis in hypothermia. J Electrocardiol. (2017) 50:375–7. doi: 10.1016/j.jelectrocard.2016.12.006
102. Rivera-Juarez A, Hernandez-Romero I, Puertas C, Zhang-Wang S, Sanchez-Alamo B, Martins R, et al. Clinical characteristics and electrophysiological mechanisms underlying Brugada ECG in patients with severe hyperkalemia. J Am Heart Assoc. (2019) 8:e010115. doi: 10.1161/JAHA.118.010115
103. Zhuo M, Li J, Lecker SH. Brugada phenocopy induced by hyperkalemia. Kidney Int. (2019) 95:471. doi: 10.1016/j.kint.2018.07.014
104. Pfirman KS, Donley CJ, Fryman EB, Champaneria SU, Gatewood WT. Brugada pattern manifesting during hyperkalemia, diabetic ketoacidosis, and acute alcohol intoxication. Am J Case Rep. (2021) 22:e932048. doi: 10.12659/AJCR.932048
105. Mehta S, Parameswaran AC, Greenspan A, Figueredo VM. Hypercalcemia due to rhabdomyolysis mimicking Brugada syndrome. Pacing Clin Electrophysiol. (2009) 32:e14–5. doi: 10.1111/j.1540-8159.2009.02520.x
106. Zeb M, McKenzie DB, Naheed B, Gazis T, Morgan JM, Staniforth AD. Hypercalcaemia and a Brugada-like ECG: an independent risk factor for fatal arrhythmias. Resuscitation. (2010) 81:1048–50. doi: 10.1016/j.resuscitation.2010.04.025
107. Sonoda K, Watanabe H, Hisamatsu T, Ashihara T, Ohno S, Hayashi H, et al. High frequency of early repolarization and Brugada-type electrocardiograms in hypercalcemia. Ann Noninvasive Electrocardiol. (2016) 21:30–40. doi: 10.1111/anec.12303
108. Online Mendelian Inheritance in Man [Online] (2021). Available online at: https://www.omim.org/ (accessed August 5, 2021).
109. Chen K, Xie Y, Zhao L, Mo Z. Hyperthyroidism-associated hypercalcemic crisis: a case report and review of the literature. Medicine. (2017) 96:e6017. doi: 10.1097/MD.0000000000006017
110. Sun XR, Liu T, Liu XP. Type 1 Brugada phenotype induced by cardiac iron overload after blood transfusion. HeartRhythm Case Rep. (2020) 6:187–90. doi: 10.1016/j.hrcr.2019.12.011
111. Matsubara E, Fujisaki T, Minamoto Y, Aoki K, Yokota E. [Brugada syndrome occurring after autologous peripheral blood stem cell transplantation for acute myeloid leukemia]. Rinsho Ketsueki. (2004) 45:481–3.
112. Nath NS, Bhattacharya I, Tuck AG, Schlipalius DI, Ebert PR. Mechanisms of phosphine toxicity. J Toxicol. (2011) 2011:494168. doi: 10.1155/2011/494168
113. Bumbrah GS, Krishan K, Kanchan T, Sharma M, Sodhi GS. Phosphide poisoning: a review of literature. Forensic Sci Int. (2012) 214:1–6. doi: 10.1016/j.forsciint.2011.06.018
114. Nayyar S, Nair M. Brugada pattern in toxic myocarditis due to severe aluminum phosphide poisoning. Pacing Clin Electrophysiol. (2009) 32:e16–7. doi: 10.1111/j.1540-8159.2009.02522.x
115. Udriste AS, Dumitriu S, Ceausu M, Radu D, Capatina CO. Aluminium phosphide fatal poisoning associated with a Brugada-like ECG pattern. Rom J Leg Med. (2013) 21:249–52. doi: 10.4323/rjlm.2013.249
116. Guru S, Kumar R, Behera A, Patra S, Kumar P Jr. Aluminium phosphide-induced expression of covertly present Brugada pattern in electrocardiogram: a rare case report. Cureus. (2020) 12:e10552. doi: 10.7759/cureus.10552
117. Prabhu MA, Agustinus R, Shenthar J. Suicidal Zinc Phosphide poisoning unmasking Brugada Syndrome and triggering near fatal ventricular arrhythmia. Pacing Clin Electrophysiol. (2016) 39:198–201. doi: 10.1111/pace.12749
118. Dharanipradab M, Viswanathan S, Kumar GR, Krishnamurthy V, Stanley DD. Yellow phosphorus-induced Brugada phenocopy. J Electrocardiol. (2018) 51:129–31. doi: 10.1016/j.jelectrocard.2017.08.033
119. Mohideen SK, Kumar KS. Should ratol paste be banned? Indian J Crit Care Med. (2015) 19:128–9. doi: 10.4103/0972-5229.151026
120. Mathie A, Sutton GL, Clarke CE, Veale EL. Zinc and copper: pharmacological probes and endogenous modulators of neuronal excitability. Pharmacol Ther. (2006) 111:567–83. doi: 10.1016/j.pharmthera.2005.11.004
121. Cappola AR, Desai AS, Medici M, Cooper LS, Egan D, Sopko G, et al. Thyroid and cardiovascular disease research agenda for enhancing knowledge, prevention, and treatment. Circulation. (2019) 139:2892–909. doi: 10.1161/CIRCULATIONAHA.118.036859
122. Flamant F, Gauthier K. Thyroid hormone receptors: the challenge of elucidating isotype-specific functions and cell-specific response. Biochim Biophys Acta. (2013) 1830:3900–7. doi: 10.1016/j.bbagen.2012.06.003
123. Udovcic M, Pena RH, Patham B, Tabatabai L, Kansara A. Hypothyroidism and the heart. Methodist Debakey. Cardiovasc J. (2017) 13:55–9. doi: 10.14797/mdcj-13-2-55
124. Razvi S, Jabbar A, Pingitore A, Danzi S, Biondi B, Klein I, et al. Thyroid hormones and cardiovascular function and diseases. J Am Coll Cardiol. (2018) 71:1781–96. doi: 10.1016/j.jacc.2018.02.045
125. Tribulova N, Knezl V, Shainberg A, Seki S, Soukup T. Thyroid hormones and cardiac arrhythmias. Vascul Pharmacol. (2010) 52:102–12. doi: 10.1016/j.vph.2009.10.001
126. Kitahara A, Hirai R, Matsui Y, Ikeda Y, Nakamura H. A case of hypothyroidism with brugada electrocardiographic waveforms. Endocr J. (2008) 55:589–94. doi: 10.1507/endocrj.K07E-024
127. Taira K, Fujino A, Watanabe T, Ogyu A, Ashikawa K, Shimizu W. Brugada-type electrocardiogram in a patient with hypothyroidism. J Cardiol Cases. (2010) 2:e147–50. doi: 10.1016/j.jccase.2010.05.011
128. Zhao YH, Zhang JY, Xu Y, Zhang XY. Hypothyroid patient with Brugada electrocardiographic waveforms: case report. Pacing Clin Electrophysiol. (2012) 35:e222–5. doi: 10.1111/j.1540-8159.2010.02960.x
129. Elgara M, Khalil MO, Raza T. Hyperthyroidism precipitating cardiac arrest in a patient with Brugada pattern. BMJ Case Rep. (2021) 14:e240038. doi: 10.1136/bcr-2020-240038
130. Stawiarski K, Clarke JD, Pollack A, Winslow R, Majumdar S. Ventricular fibrillation in Graves disease reveals a rare SCN5A mutation with W1191X variant associated with Brugada syndrome. HeartRhythm Case Rep. (2021) 7:95–9. doi: 10.1016/j.hrcr.2020.11.010
131. Dutta S, Lopez Charcas O, Tanner S, Gradek F, Driffort V, Roger S, et al. Discovery and evaluation of nNav1.5 sodium channel blockers with potent cell invasion inhibitory activity in breast cancer cells. Bioorg Med Chem. (2018) 26:2428–36. doi: 10.1016/j.bmc.2018.04.003
132. Luo Q, Wu T, Wu W, Chen G, Luo X, Jiang L, et al. The functional role of voltage-gated sodium channel Nav15 in metastatic breast cancer. Front Pharmacol. (2020) 11:1111. doi: 10.3389/fphar.2020.01111
133. Yamaci RF, Fraser SP, Battaloglu E, Kaya H, Erguler K, Foster CS, et al. Neonatal Nav1.5 protein expression in normal adult human tissues and breast cancer. Pathol Res Pract. (2017) 213:900–7. doi: 10.1016/j.prp.2017.06.003
134. Murtadha AH, Azahar MII, Sharudin NA, Che Has AT, Mokhtar NF. Influence of nNav1.5 on MHC class I expression in breast cancer. J Biosci. (2021) 46:70. doi: 10.1007/s12038-021-00196-w
135. Gradek F, Lopez-Charcas O, Chadet S, Poisson L, Ouldamer L, Goupille C, et al. Sodium channel Nav15 controls epithelial-to-mesenchymal transition and invasiveness in breast cancer cells through its regulation by the salt-inducible kinase-1. Sci Rep. (2019) 9:18652. doi: 10.1038/s41598-019-55197-5
136. Guzel RM, Ogmen K, Ilieva KM, Fraser SP, Djamgoz MBA. Colorectal cancer invasiveness in vitro: predominant contribution of neonatal Nav1.5 under normoxia and hypoxia. J Cell Physiol. (2019) 234:6582–93. doi: 10.1002/jcp.27399
137. House CD, Vaske CJ, Schwartz AM, Obias V, Frank B, Luu T, et al. Voltage-gated Na+ channel SCN5A is a key regulator of a gene transcriptional network that controls colon cancer invasion. Cancer Res. (2010) 70:6957–67. doi: 10.1158/0008-5472.CAN-10-1169
138. Driffort V, Gillet L, Bon E, Marionneau-Lambot S, Oullier T, Joulin V, et al. Ranolazine inhibits NaV1.5-mediated breast cancer cell invasiveness and lung colonization. Mol Cancer. (2014) 13:264. doi: 10.1186/1476-4598-13-264
139. Lovelock JD, Monasky MM, Jeong EM, Lardin HA, Liu H, Patel BG, et al. Ranolazine improves cardiac diastolic dysfunction through modulation of myofilament calcium sensitivity. Circ Res. (2012) 110:841–50. doi: 10.1161/CIRCRESAHA.111.258251
140. Hegyi B, Komaromi I, Kistamas K, Ruzsnavszky F, Vaczi K, Horvath B, et al. Tetrodotoxin blockade on canine cardiac L-type Ca(2)(+) channels depends on pH and redox potential. Mar Drugs. (2013) 11:2140–53. doi: 10.3390/md11062140
141. Landstrom AP, Dobrev D, Wehrens XHT. Calcium Signaling and Cardiac Arrhythmias. Circ Res. (2017) 120:1969–93. doi: 10.1161/CIRCRESAHA.117.310083
142. Meregalli PG, Wilde AA, Tan HL. Pathophysiological mechanisms of Brugada syndrome: depolarization disorder, repolarization disorder, or more? Cardiovasc Res. (2005) 67:367–78. doi: 10.1016/j.cardiores.2005.03.005
143. Nademanee K, Veerakul G, Nimmannit S, Chaowakul V, Bhuripanyo K, Likittanasombat K, et al. Arrhythmogenic marker for the sudden unexplained death syndrome in Thai men. Circulation. (1997) 96:2595–600. doi: 10.1161/01.CIR.96.8.2595
144. Shimizu W, Matsuo K, Kokubo Y, Satomi K, Kurita T, Noda T, et al. Sex hormone and gender difference–role of testosterone on male predominance in Brugada syndrome. J Cardiovasc Electrophysiol. (2007) 18:415–21. doi: 10.1111/j.1540-8167.2006.00743.x
145. Haruta D, Matsuo K, Ichimaru S, Soda M, Hida A, Sera N, et al. Men with Brugada-like electrocardiogram have higher risk of prostate cancer. Circ J. (2009) 73:63–8. doi: 10.1253/circj.CJ-08-0680
146. Matsuo K, Akahoshi M, Seto S, Yano K. Disappearance of the Brugada-type electrocardiogram after surgical castration: a role for testosterone and an explanation for the male preponderance. Pacing Clin Electrophysiol. (2003) 26:1551–3. doi: 10.1046/j.1460-9592.2003.t01-1-00227.x
147. Conte G, De Asmundis C, Ciconte G, Julia J, Sieira J, Chierchia GB, et al. Follow-up from childhood to adulthood of individuals with family history of Brugada syndrome and normal electrocardiograms. JAMA. (2014) 312:2039–41. doi: 10.1001/jama.2014.13752
148. Bidoggia H, Maciel JP, Capalozza N, Mosca S, Blaksley EJ, Valverde E, et al. Sex differences on the electrocardiographic pattern of cardiac repolarization: possible role of testosterone. Am Heart J. (2000) 140:678–83. doi: 10.1067/mhj.2000.109918
149. Stramba-Badiale M, Spagnolo D, Bosi G, Schwartz PJ. Are gender differences in QTc present at birth? MISNES Investigators. Multicenter Italian Study on Neonatal Electrocardiography and Sudden Infant Death Syndrome. Am J Cardiol. (1995) 75:1277–8. doi: 10.1016/S0002-9149(99)80781-4
150. Wedekind H, Smits JPP, Schulze-Bahr E, Arnold R, Veldkamp MW, Bajanowski T, et al. De novo mutation in the SCN5A gene associated with early onset of sudden infant death. Circulation. (2001) 104:1158–64. doi: 10.1161/hc3501.095361
151. Najafi K, Mehrjoo Z, Ardalani F, Ghaderi-Sohi S, Kariminejad A, Kariminejad R, et al. Identifying the causes of recurrent pregnancy loss in consanguineous couples using whole exome sequencing on the products of miscarriage with no chromosomal abnormalities. Sci Rep. (2021) 11:6952. doi: 10.1038/s41598-021-86309-9
152. Waldbaum S, Patel M. Mitochondria, oxidative stress, and temporal lobe epilepsy. Epilepsy Res. (2010) 88:23–45. doi: 10.1016/j.eplepsyres.2009.09.020
153. Kovac S, Dinkova Kostova AT, Herrmann AM, Melzer N, Meuth SG, Gorji A. Metabolic and homeostatic changes in seizures and acquired epilepsy-mitochondria, calcium dynamics and reactive oxygen species. Int J Mol Sci. (2017) 18:1935. doi: 10.3390/ijms18091935
154. Cioffi F, Senese R, Lanni A, Goglia F. Thyroid hormones and mitochondria: with a brief look at derivatives and analogues. Mol Cell Endocrinol. (2013) 379:51–61. doi: 10.1016/j.mce.2013.06.006
156. Yang Y, Karakhanova S, Hartwig W, D'Haese JG, Philippov PP, Werner J, et al. Mitochondria and mitochondrial ROS in cancer: novel targets for anticancer therapy. J Cell Physiol. (2016) 231:2570–81. doi: 10.1002/jcp.25349
157. Zong WX, Rabinowitz JD, White E. Mitochondria and cancer. Mol Cell. (2016) 61:667–76. doi: 10.1016/j.molcel.2016.02.011
158. Burke PJ. Mitochondria, bioenergetics and apoptosis in cancer. Trends Cancer. (2017) 3:857–70. doi: 10.1016/j.trecan.2017.10.006
159. Shen GX. Oxidative stress and diabetic cardiovascular disorders: roles of mitochondria and NADPH oxidase. Can J Physiol Pharmacol. (2010) 88:241–8. doi: 10.1139/Y10-018
160. Song J, Yang R, Yang J, Zhou L. Mitochondrial dysfunction-associated arrhythmogenic substrates in diabetes mellitus. Front Physiol. (2018) 9:1670. doi: 10.3389/fphys.2018.01670
161. Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res. (2013) 113:709–24. doi: 10.1161/CIRCRESAHA.113.300376
162. Varian KD, Janssen PM. Frequency-dependent acceleration of relaxation involves decreased myofilament calcium sensitivity. Am J Physiol Heart Circ Physiol. (2007) 292:H2212–9. doi: 10.1152/ajpheart.00778.2006
163. Monasky MM, Biesiadecki BJ, Janssen PM. Increased phosphorylation of tropomyosin, troponin I, and myosin light chain-2 after stretch in rabbit ventricular myocardium under physiological conditions. J Mol Cell Cardiol. (2010) 48:1023–8. doi: 10.1016/j.yjmcc.2010.03.004
164. Bers DM, Despa S. Cardiac myocytes Ca2+ and Na+ regulation in normal and failing hearts. J Pharmacol Sci. (2006) 100:315–22. doi: 10.1254/jphs.CPJ06001X
165. Solaro RJ, Kobayashi T. Protein phosphorylation and signal transduction in cardiac thin filaments. J Biol Chem. (2011) 286:9935–40. doi: 10.1074/jbc.R110.197731
166. Monasky MM, Taglieri DM, Patel BG, Chernoff J, Wolska BM, Ke Y, et al. p21-activated kinase improves cardiac contractility during ischemia-reperfusion concomitant with changes in troponin-T and myosin light chain 2 phosphorylation. Am J Physiol Heart Circ Physiol. (2012) 302:H224–30. doi: 10.1152/ajpheart.00612.2011
167. Herren AW, Bers DM, Grandi E. Post-translational modifications of the cardiac Na channel: contribution of CaMKII-dependent phosphorylation to acquired arrhythmias. Am J Physiol Heart Circ Physiol. (2013) 305:H431–445. doi: 10.1152/ajpheart.00306.2013
168. Monasky MM, Taglieri DM, Jacobson AK, Haizlip KM, Solaro RJ, Janssen PM. Post-translational modifications of myofilament proteins involved in length-dependent prolongation of relaxation in rabbit right ventricular myocardium. Arch Biochem Biophys. (2013) 535:22–9. doi: 10.1016/j.abb.2012.10.005
169. Venetucci L, Denegri M, Napolitano C, Priori SG. Inherited calcium channelopathies in the pathophysiology of arrhythmias. Nat Rev Cardiol. (2012) 9:561–75. doi: 10.1038/nrcardio.2012.93
170. Hof IE, Van Der Heijden JF, Kranias EG, Sanoudou D, De Boer RA, Van Tintelen JP, et al. Prevalence and cardiac phenotype of patients with a phospholamban mutation. Neth Heart J. (2019) 27:64–9. doi: 10.1007/s12471-018-1211-4
171. Heijman J, Dewenter M, El-Armouche A, Dobrev D. Function and regulation of serine/threonine phosphatases in the healthy and diseased heart. J Mol Cell Cardiol. (2013) 64:90–8. doi: 10.1016/j.yjmcc.2013.09.006
172. Anderson ME. Ca2+-dependent regulation of cardiac L-type Ca2+ channels: is a unifying mechanism at hand? J Mol Cell Cardiol. (2001) 33:639–50. doi: 10.1006/jmcc.2000.1354
173. Eisner DA, Kashimura T, Venetucci LA, Trafford AW. From the ryanodine receptor to cardiac arrhythmias. Circ J. (2009) 73:1561–7. doi: 10.1253/circj.CJ-09-0478
174. Kumar M, Haghighi K, Kranias EG, Sadayappan S. Phosphorylation of cardiac myosin-binding protein-C contributes to calcium homeostasis. J Biol Chem. (2020) 295:11275–91. doi: 10.1074/jbc.RA120.013296
175. Brugada R, Campuzano O, Sarquella-Brugada G, Brugada P, Brugada J, Hong K. Brugada Syndrome In: GeneReviews(R). Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Ledbetter N, Mefford HC, Smith RJH, Stephens K, editors. Seattle, WA (1993).
176. Liu M, Sanyal S, Gao G, Gurung IS, Zhu X, Gaconnet G, et al. Cardiac Na+ current regulation by pyridine nucleotides. Circ Res. (2009) 105:737–45. doi: 10.1161/CIRCRESAHA.109.197277
177. Stocchi L, Polidori E, Potenza L, Rocchi MB, Calcabrini C, Busacca P, et al. Mutational analysis of mitochondrial DNA in Brugada syndrome. Cardiovasc Pathol. (2016) 25:47–54. doi: 10.1016/j.carpath.2015.10.001
178. Polidori E, Stocchi L, Potenza D, Cucchiarini L, Stocchi V, Potenza L. A high number of ‘natural’ mitochondrial DNA polymorphisms in a symptomatic Brugada syndrome type 1 patient. J Genet. (2020) 99:66. doi: 10.1007/s12041-020-01228-4
179. Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res. (2018) 122:624–38. doi: 10.1161/CIRCRESAHA.117.311586
180. Hegyi B, Ko CY, Bossuyt J, Bers DM. Two-hit mechanism of cardiac arrhythmias in diabetic hyperglycemia: reduced repolarization reserve, neurohormonal stimulation and heart failure exacerbate susceptibility. Cardiovasc Res. (2021) cvab006. doi: 10.1093/cvr/cvab006
181. Postema PG, Vlaar AP, Devries JH, Tan HL. Familial Brugada syndrome uncovered by hyperkalaemic diabetic ketoacidosis. Europace. (2011) 13:1509–10. doi: 10.1093/europace/eur151
182. Alanzalon RE, Burris JR, Vinocur JM. Brugada phenocopy associated with diabetic ketoacidosis in two pediatric patients. J Electrocardiol. (2018) 51:323–6. doi: 10.1016/j.jelectrocard.2017.10.017
183. Haseeb S, Kariyanna PT, Jayarangaiah A, Thirunavukkarasu G, Hegde S, Marmur JD, et al. Brugada pattern in diabetic ketoacidosis: a case report and scoping study. Am J Med Case Rep. (2018) 6:173–9. doi: 10.12691/ajmcr-6-9-2
184. Kalkan S, Guner A, Kalcik M, Gecmen C, Ozkan M. Type I Brugada pattern associated with diabetic ketoacidosis in a patient with type II diabetes mellitus. Anatol J Cardiol. (2019) 21:E1. doi: 10.14744/AnatolJCardiol.2018.43789
185. Skinner JR, Marquis-Nicholson R, Luangpraseuth A, Cutfield R, Crawford J, Love DR. Diabetic dead-in-bed syndrome: a possible link to a cardiac ion channelopathy. Case Rep Med. (2014) 2014:647252. doi: 10.1155/2014/647252
186. London B, Michalec M, Mehdi H, Zhu X, Kerchner L, Sanyal S, et al. Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias. Circulation. (2007) 116:2260–8. doi: 10.1161/CIRCULATIONAHA.107.703330
187. Van Norstrand DW, Valdivia CR, Tester DJ, Ueda K, London B, Makielski JC, et al. Molecular and functional characterization of novel glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) mutations in sudden infant death syndrome. Circulation. (2007) 116:2253–9. doi: 10.1161/CIRCULATIONAHA.107.704627
188. Mambiya M, Shang M, Wang Y, Li Q, Liu S, Yang L, et al. The play of genes and non-genetic factors on type 2 diabetes. Front Public Health. (2019) 7:349. doi: 10.3389/fpubh.2019.00349
189. Russo I, Frangogiannis NG. Diabetes-associated cardiac fibrosis: cellular effectors, molecular mechanisms and therapeutic opportunities. J Mol Cell Cardiol. (2016) 90:84–93. doi: 10.1016/j.yjmcc.2015.12.011
190. Nicole S, Fontaine B. Skeletal muscle sodium channelopathies. Curr Opin Neurol. (2015) 28:508–14. doi: 10.1097/WCO.0000000000000238
191. Bissay V, Van Malderen SC, Keymolen K, Lissens W, Peeters U, Daneels D, et al. SCN4A variants and Brugada syndrome: phenotypic and genotypic overlap between cardiac and skeletal muscle sodium channelopathies. Eur J Hum Genet. (2016) 24:400–7. doi: 10.1038/ejhg.2015.125
192. Cavalli M, Fossati B, Vitale R, Brigonzi E, Ricigliano VAG, Saraceno L, et al. Flecainide-induced Brugada syndrome in a patient with skeletal muscle sodium channelopathy: a case report with critical therapeutical implications and review of the literature. Front Neurol. (2018) 9:385. doi: 10.3389/fneur.2018.00385
193. Mannikko R, Wong L, Tester DJ, Thor MG, Sud R, Kullmann DM, et al. Dysfunction of NaV1.4. A skeletal muscle voltage-gated sodium channel, in sudden infant death syndrome: a case-control study. Lancet. (2018) 391:1483–92. doi: 10.1016/S0140-6736(18)30021-7
194. Loussouarn G, Sternberg D, Nicole S, Marionneau C, Le Bouffant F, Toumaniantz G, et al. Physiological and pathophysiological insights of Nav1.4 and Nav1.5 comparison. Front Pharmacol. (2015) 6:314. doi: 10.3389/fphar.2015.00314
195. Cannon SC. Sodium channelopathies of skeletal muscle. Handb Exp Pharmacol. (2018) 246:309–30. doi: 10.1007/164_2017_52
196. Behr ER, Ben-Haim Y, Ackerman MJ, Krahn AD, Wilde AAM. Brugada syndrome and reduced right ventricular outflow tract conduction reserve: a final common pathway?. Eur Heart J. (2001) 42:1073–81. doi: 10.1093/eurheartj/ehaa1051
197. Hosseini SM, Kim R, Udupa S, Costain G, Jobling R, Liston E, et al. Reappraisal of reported genes for sudden arrhythmic death: evidence-based evaluation of gene validity for Brugada syndrome. Circulation. (2018) 138:1195–205. doi: 10.1161/CIRCULATIONAHA.118.035070
198. D'Imperio S, Monasky MM, Micaglio E, Negro G, Pappone C. Early morning QT prolongation during hypoglycemia: only a matter of glucose? Front Cardiovasc Med. (2021) 8:688875. doi: 10.3389/fcvm.2021.688875
199. Bezzina C, Veldkamp MW, Van Den Berg MP, Postma AV, Rook MB, Viersma JW, et al. A single Na(+) channel mutation causing both long-QT and Brugada syndromes. Circ Res. (1999) 85:1206–13. doi: 10.1161/01.RES.85.12.1206
200. Gosselin-Badaroudine P, Moreau A, Chahine M. Nav 1.5 mutations linked to dilated cardiomyopathy phenotypes: is the gating pore current the missing link? Channels. (2014) 8:90–4. doi: 10.4161/chan.27179
201. Zaklyazminskaya E, Dzemeshkevich S. The role of mutations in the SCN5A gene in cardiomyopathies. Biochim Biophys Acta. (2016) 1863:1799–805. doi: 10.1016/j.bbamcr.2016.02.014
202. Sarquella-Brugada G, Campuzano O, Arbelo E, Brugada J, Brugada R. Brugada syndrome: clinical and genetic findings. Genet Med. (2016) 18:3–12. doi: 10.1038/gim.2015.35
203. Campuzano O, Perez-Serra A, Iglesias A, Brugada R. Genetic basis of atrial fibrillation. Genes Dis. (2016) 3:257–62. doi: 10.1016/j.gendis.2016.09.003
204. Campuzano O, Brugada R. Age, genetics, and fibrosis in the Brugada syndrome. J Am Coll Cardiol. (2015) 66:1987–9. doi: 10.1016/j.jacc.2015.09.006
205. Nademanee K, Raju H, De Noronha SV, Papadakis M, Robinson L, Rothery S, et al. Fibrosis, connexin-43, and conduction abnormalities in the Brugada syndrome. J Am Coll Cardiol. (2015) 66:1976–86. doi: 10.1016/j.jacc.2015.08.862
206. Cipriani A, Bauce B, De Lazzari M, Rigato I, Bariani R, Meneghin S, et al. Arrhythmogenic right ventricular cardiomyopathy: characterization of left ventricular phenotype and differential diagnosis with dilated cardiomyopathy. J Am Heart Assoc. (2020) 9:e014628. doi: 10.1161/JAHA.119.014628
207. King JH, Huang CL, Fraser JA. Determinants of myocardial conduction velocity: implications for arrhythmogenesis. Front Physiol. (2013) 4:154. doi: 10.3389/fphys.2013.00154
208. Pietrasik G, Zareba W. QRS fragmentation: diagnostic and prognostic significance. Cardiol J. (2012) 19:114–21. doi: 10.5603/CJ.2012.0022
209. Foo B, Williamson B, Young JC, Lukacs G, Shrier A. hERG quality control and the long QT syndrome. J Physiol. (2016) 594:2469–81. doi: 10.1113/JP270531
210. Chadda KR, Ahmad S, Valli H, Den Uijl I, Al-Hadithi AB, Salvage SC, et al. The effects of ageing and adrenergic challenge on electrocardiographic phenotypes in a murine model of long QT syndrome type 3. Sci Rep. (2017) 7:11070. doi: 10.1038/s41598-017-11210-3
211. Bartlett TG, Friedman PL. Current management of the Wolff-Parkinson-White syndrome. J Card Surg. (1993) 8:503–15. doi: 10.1111/j.1540-8191.1993.tb00401.x
212. Lee HJ, Uhm JS, Hong YJ, Hur J, Choi BW, Joung B, et al. Altered myocardial characteristics of the preexcited segment in Wolff-Parkinson-White syndrome: a pilot study with cardiac magnetic resonance imaging. PLoS ONE. (2018) 13:e0198218. doi: 10.1371/journal.pone.0198218
213. Raman B, Ariga R, Spartera M, Sivalokanathan S, Chan K, Dass S, et al. Progression of myocardial fibrosis in hypertrophic cardiomyopathy: mechanisms and clinical implications. Eur Heart J Cardiovasc Imaging. (2019) 20:157–67. doi: 10.1093/ehjci/jey135
214. Everett THT, Olgin JE. Atrial fibrosis and the mechanisms of atrial fibrillation. Heart Rhythm. (2007) 4:S24–7. doi: 10.1016/j.hrthm.2006.12.040
215. Sohns C, Marrouche NF. Atrial fibrillation and cardiac fibrosis. Eur Heart J. (2020) 41:1123–31. doi: 10.1093/eurheartj/ehz786
216. Marian AJ, Braunwald E. Hypertrophic cardiomyopathy: genetics, pathogenesis, clinical manifestations, diagnosis, and therapy. Circ Res. (2017) 121:749–70. doi: 10.1161/CIRCRESAHA.117.311059
217. Geske JB, Ommen SR, Gersh BJ. Hypertrophic cardiomyopathy: clinical update. JACC Heart Fail. (2018) 6:364–75. doi: 10.1016/j.jchf.2018.02.010
218. Pappone C, Brugada J, Vicedomini G, Ciconte G, Manguso F, Saviano M, et al. Electrical substrate elimination in 135 consecutive patients with Brugada syndrome. Circ Arrhythm Electrophysiol. (2017) 10:e005053. doi: 10.1161/CIRCEP.117.005053
219. Pappone C, Ciconte G, Manguso F, Vicedomini G, Mecarocci V, Conti M, et al. Assessing the malignant ventricular arrhythmic substrate in patients with Brugada syndrome. J Am Coll Cardiol. (2018) 71:1631–46. doi: 10.1016/j.jacc.2018.02.022
Keywords: Brugada syndrome (BrS), sudden cardiac death (SCD), arrhythmia, genetics, epilepsy, ajmaline, thyroid, cancer
Citation: D'Imperio S, Monasky MM, Micaglio E, Ciconte G, Anastasia L and Pappone C (2021) Brugada Syndrome: Warning of a Systemic Condition? Front. Cardiovasc. Med. 8:771349. doi: 10.3389/fcvm.2021.771349
Received: 06 September 2021; Accepted: 23 September 2021;
Published: 15 October 2021.
Edited by:
George W. Booz, University of Mississippi Medical Center School of Dentistry, United StatesReviewed by:
Hariharan Raju, Macquarie University, AustraliaCopyright © 2021 D'Imperio, Monasky, Micaglio, Ciconte, Anastasia and Pappone. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carlo Pappone, Y2FybG8ucGFwcG9uZUBhZi1hYmxhdGlvbi5vcmc=
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.