 Yi Luan
Yi Luan Kai-Di Ren2,3
Kai-Di Ren2,3 Ying Luan
Ying Luan Yang Yang
Yang Yang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 06 December 2021
Sec. Atherosclerosis and Vascular Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.770574
This article is part of the Research Topic Insights in Atherosclerosis and Vascular Medicine: 2021 View all 22 articles
Vascular diseases, particularly atherosclerosis, are associated with high morbidity and mortality. Endothelial cell (EC) or vascular smooth muscle cell (VSMC) dysfunction leads to blood vessel abnormalities, which cause a series of vascular diseases. The mitochondria are the core sites of cell energy metabolism and function in blood vessel development and vascular disease pathogenesis. Mitochondrial dynamics, including fusion and fission, affect a variety of physiological or pathological processes. Multiple studies have confirmed the influence of mitochondrial dynamics on vascular diseases. This review discusses the regulatory mechanisms of mitochondrial dynamics, the key proteins that mediate mitochondrial fusion and fission, and their potential effects on ECs and VSMCs. We demonstrated the possibility of mitochondrial dynamics as a potential target for the treatment of vascular diseases.
Cardiovascular disease (CVD) is the leading cause of death worldwide (1). Vascular diseases, particularly atherosclerosis, are initiated at an early stage in life and remain asymptomatic for a long period until they reach advanced stages (2). Among vascular diseases, atherosclerosis is a pathologic process of lipid accumulation, scarring, and inflammation in the vascular wall, particularly the subendothelial (intimal) space of arteries, which leads to vascular wall thickening, luminal stenosis, and calcification (3). Endothelial cell (EC) activation or dysfunction is an early symptom of vascular diseases that occur at the lesion-prone sites of arterial blood vessels, where ECs display pro-inflammatory and prothrombotic phenotypes and reduced barrier function. Notably, ECs are extremely sensitive to oxidative stress and respond rapidly to altered environments, such as changes in oxygen levels, pathogen stimulation, and damaging endogenous stimuli (4). In addition, the direct contact between ECs and circulating immune cells triggers immune reactions (5). Another substance that plays an important role in blood vessel function is nitric oxide (NO). NO is a signaling molecule in the vascular system, in which blood vessels control blood flow by sending signals to the vessels to vasodilate. NO could also slow the deposition of atherosclerotic plaque on the blood vessel wall (6).
Other mechanisms and stimuli also affect the function of blood vessels. Blood flow promotes the production of adhesive molecules, which recruit inflammatory cells (7). Besides, the migration of vascular smooth muscle cells (VSMCs) also facilitates atherosclerosis progression (5). Several vascular diseases ultimately lead to myocardial infarction, stroke, and peripheral artery disease (8). The etiology of vascular diseases is complex; thus, several risk factors may contribute to their progression, including dyslipidemia, diabetes, smoking, hypertension, oxidative stressors, angiotensin II, systemic infection, and inflammation (9). Nonetheless, an effective cure for vascular diseases still lacks partially because of the complex etiology of the diseases in spite of recent advances (4).
The occurrence of vascular diseases is related to the loss of energy metabolism; notably, the mitochondria are the core sites of cell energy metabolism (10). The mitochondria are important in endothelial and smooth muscle function (11, 12). Mitochondria are composed of a central mitochondrial matrix surrounded by two inner and outer mitochondrial membranes, and eukaryotic mitochondrial respiratory chain is composed of complex. Compounds I, II, III, IV and complex V (ATP synthase), ubiquinone, coenzyme Q and cytochrome C are located in the inner membrane of mitochondria. Mitochondrial respiratory chain oxidative phosphorylation is responsible for more than 90% of oxygen consumption and provides more than 95% of body energy. Supply, the mitochondrial matrix is the main site of the tricarboxylic acid cycle and fatty acid β oxidation. Apart from its capacity for ATP production, the mitochondria also modulate reactive oxygen species (ROS) generation, calcium regulation, cell death, and survival (13, 14). The function of the mitochondria is affected by mitochondrial dynamics, including fusion and fission, interaction with the endoplasmic reticulum (ER), and mitophagy (Figure 1). Mitochondrial dysfunction leads to cell senescence, inflammation, and apoptosis, which are characteristics of vascular diseases (15). In addition, mitochondrial dysfunction can be triggered by DNA damage, which is closely related to several risk factors of CVDs (16). In this review, we summarize the correlation between vascular diseases and mitochondrial dynamics with emphasis on the detailed function of mitochondrial dynamics in specific vascular disease forms and the potential therapeutic approach of mitochondrial dynamics in vascular diseases.
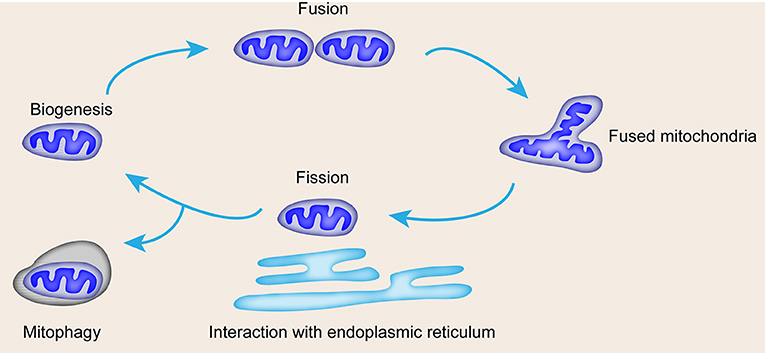
Figure 1. Mitochondrial life cycle and contribution of mitochondrial dynamics and mitophagy to quality control. Mitochondrial dynamics include biogenesis, fusion for mass increase, fission for number increase, interaction with ER, and mitophagy.
More and more evidences indicate the role of mitochondrial dynamics in vascular function and the pathogenesis of vascular diseases. The mitochondria are highly dynamic organelles whose structure and distribution affect metabolism despite being recognized as isolated organelles (17). The nature of the dynamic network depends on the proper balance between mitochondrial fusion and fission (18). Its balance can be destroyed by environmental stimuli, developmental status, and cellular metabolic demands. The recently identified molecular mediators of mitochondrial fusion and fission, as well as post-translational modification (PTM) by an extensive set of kinases, phosphatases, and ubiquitination mediators, bring a new slight on the mechanism of mitochondrial dynamics (Table 1) (19).
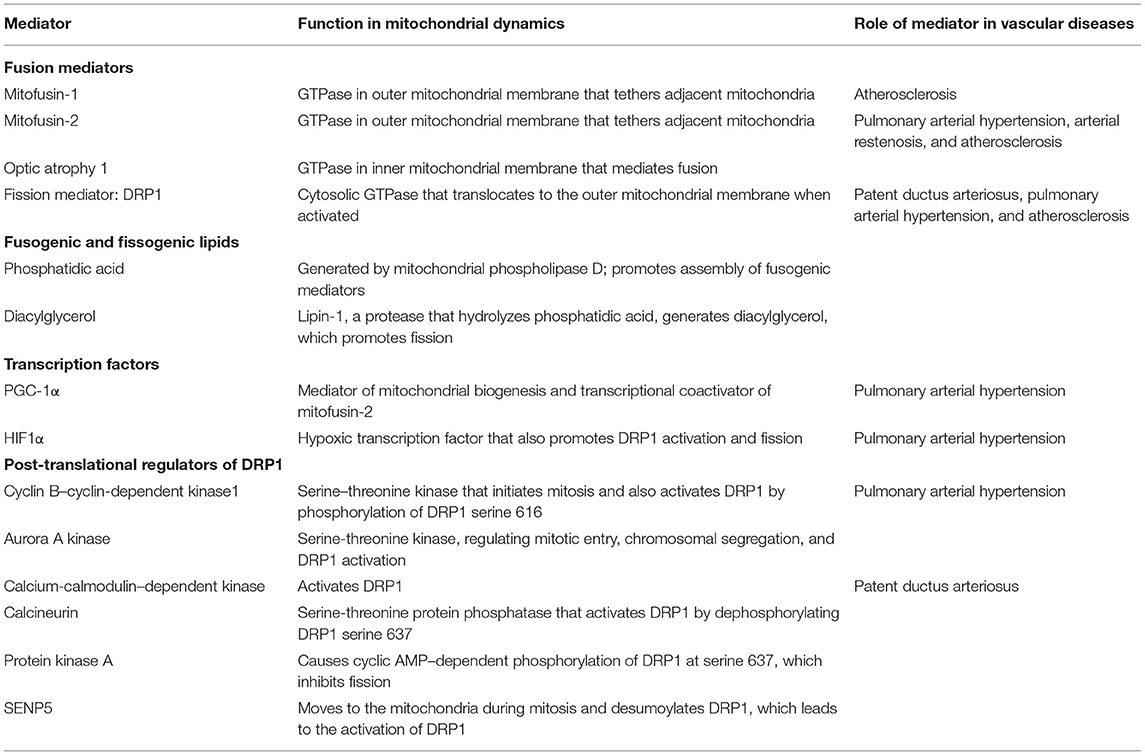
Table 1. Mediators involved in the regulation of mitochondria fission and fusion.
Mechanically, mitochondrial fusion at the outer mitochondrial membrane is controlled by the transmembrane GTPases, MFN 1 and MFN2, and fusion at the inner membrane is controlled by optic atrophy protein 1 (OPA1) (20, 21). Besides, fission is regulated by DRP1 and fission-1 (FIS1) (22, 23). Mitochondrial fusion is regulated by the coordinated action of conserved GTPase proteins, including MFN1 and MFN2, and these transmembrane GTPases located in the outer membrane of the mitochondria are responsible for the regulation of the mitochondrial fusion by forming homodimeric or heterodimeric, antiparallel, coiled-coil linkages between adjacent mitochondria and C-terminal domains (24) (Figure 2). MFN1 and MFN2 deficiencies lead to a remarkable decrease in mitochondrial fusion (25). Additionally, MFN2 mediates cell apoptosis and mitochondrial autophagy (26). OPA1, a dynamin-related GTPase embedded in the inner membrane or intermembrane of the mitochondria, is involved in mitochondrial intima fusion and mitochondrial cristae remodeling (20). OPA1 harbors two forms (i.e., long and short OPA1 proteins) with distinguished functions (27). The long form of OPA1 located in the inner membrane, which is responsible for intimal fusion, can be cleaved into short form under the digestion of the intestinal peptidase, OMA1, and the i-AAA proteolytic enzyme, YME1L, to induce mitochondrial fragmentation and fission in the membrane space (19).
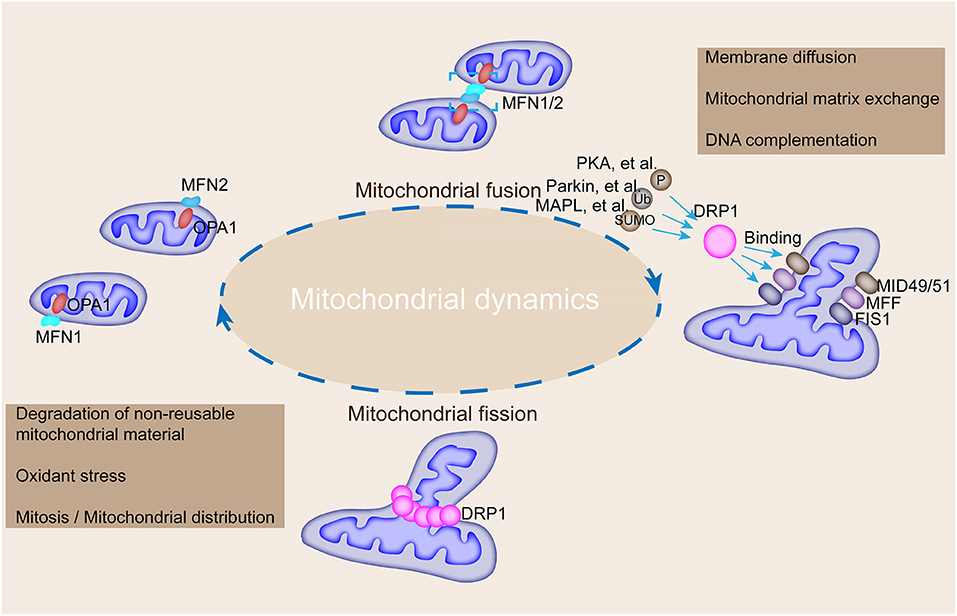
Figure 2. Mechanisms underlying the regulation of mitochondrial fusion and fission and their roles in modulating mitochondrial morphology. MFN1, MFN2, and OPA1 mediate mitochondrial fusion, whereas DRP1 interacts with FIS1, MFF, and MiD49/51 to participate in mitochondrial fission. DRP1 could be modified by phosphorylation, ubiquitination, and SUMOylation by corresponding enzymes to promote its binding with FIS1, MFF, and MiD49/51. Mitochondrial fusion is accompanied by membrane diffusion, matrix exchange, and DNA complementation, and fission is accompanied by the degradation of non-reusable mitochondrial material, oxidative stress, and mitosis.
Mitochondrial fission in mammalian cells is manipulated by DRP1, FIS1, mitochondrial fission factor (MFF), and mitochondrial dynamic proteins of 49 and 51 kDa (MiD49/51) as shown in Figure 2 (24). DRP1, a GTPase located in the cytoplasm, mediates mitochondrial fission at the outer membrane (28). DRP1 is encoded by the DNM1L gene and contains a GTPase region, an intermediate region, a polytropic region, and a GTPase effector region from the N-terminal to the C-terminal, which are involved in the physical constriction of the mitochondria (the early step of fission) (29). Notably, DRP1 needs to bind with other receptor proteins, such as FIS1, to embed on the outer membrane of the mitochondria because of its lack of lipid-interacting hydrophobic transmembrane domain (30). However, FIS1 depletion has minimal effect on the transfer of DRP1 to the mitochondria in mammalian cells (31). A multitude of receptors is involved in the recruitment of DRP1 to the mitochondria to trigger fission (32). Mid49/51 are involved in DRP1 translocation in this fission machinery (30). In addition, DRP1 activity is regulated by PTMs, such as acetylation and phosphorylation (32). DRP1 activity is modulated by two serine phosphorylation sites with opposing functions; that is, DRP1 activity can be activated by phosphorylation at serine 616 but inhibited by phosphorylation at serine 637 (33). Each serine phosphorylation is catalyzed by a different kinase and phosphatase; thus, mitochondrial fission is linked to key cellular processes (Table 1). For instance, the DRP1 phosphorylation at serine 616 mediated by the mitotic initiator, cyclin B1–cyclin-dependent kinase (CDK1), links mitochondrial fission to cell division (34). The phosphorylation mediated by calcium-calmodulin-dependent kinase (CamK) coordinates fission to intracellular calcium (35). The serine ratio between the 616 and 637 sites modulates DRP1 activity and reflects the polymerized influence of several kinases and phosphatases (36). DRP1 activity is also modulated by the ubiquitin ligases, membrane-associated RING-CH protein 5 and small ubiquitin-like modifier type 1 (37). DRP1 acetylation regulates the activity of itself and contributes to metabolic stress-associated cardiomyocyte death and dysfunction (38). Previous studies depicted that DRP1 forms a dimer or tetramer under basic conditions and further self-assembles into a larger polymer structure in the fission process.
Multiple PTMs of human mitochondrial proteins, such as phosphorylation (in MFN1, MFN2, and OPA1), acetylation (in MFN1, MFN2, and OPA1), methylation (in MFN1 and OPA1), and ubiquitination (in MFN1, MFN2, and OPA1) have been detected by mass spectrometry-based proteomics (39). However, the modulation of PTM in mitochondrial fusion proteins is largely uncharacterized compared with that in DRP1. For instance, the PTMs of MFN2 (phosphorylation and ubiquitination) are observed in hearts with cardiomyopathy. PINK1-dependent MFN2 phosphorylation induces Parkin translocation to the outer mitochondrial membrane upon membrane depolarization, which subsequently promotes Parkin-mediated MFN2 ubiquitination in adult cardiomyocytes (40). Also, Parkin-mediated MFN2 ubiquitination leads to MFN2 degradation, which results in the selective removal of damaged mitochondria by mitophagy in adult cardiomyocytes (41).
Apart from proteins, mitochondrial miRNAs (mitomiRs) modulate the translational activity of the mitochondrial genome and mitochondrial function (42). Mitochondrial fission/fusion can also be regulated by mitomiRs. Notably, miR-146a, miR-34a, and miR-181a may regulate mitochondrial dynamics by targeting Bcl-2 (42). Other mitomiRs can also directly target mitochondrial fusion/fission proteins. miR-484 suppresses FIS1-mediated fission and apoptosis in cardiomyocytes by decreasing FIS1 expression. Mitochondrial fission is also suppressed by the miR-30-mediated downregulation of DRP1 and p53 (43).
Mitochondrial fusion and fission can also be mediated by peroxisome proliferator-activated receptor γ co-activator 1α (PGC-1α), which is a modulator of mitochondrial fusion by acting as a transcriptional coactivator of MFN2 (44, 45). The assembly of fission apparatus also needs the assistance of the ER directly in contact with the mitochondria to form a microdomain that facilitates the assembly of DRP1, MEF, and proapoptotic proteins (46). The lipids produced by mitochondrial phospholipase D, especially phosphatidic acid, guide mitochondrial dynamics (47).
The cooperation of mitochondrial fusion and fission maintains the fundamental integrity and normal functioning of the mitochondria, including energy metabolism, ROS generation, and apoptosis regulation (48). Fusion favors mitochondrial interconnection, mitochondrial DNA mixing, signal transduction, and metabolite exchange (49). Mitochondrial fission facilitates the elimination of damaged mitochondria by dividing the mitochondria into daughter mitochondria to maintain the normal function of the mitochondria (50). However, the perturbation of mitochondrial fusion and fission breaks their balance and consequently leads to the accumulation of damaged and non-functional mitochondria (48).
Mitochondrial dynamics play an important role in the morphology, function, and distribution of mitochondria. Fusion and fission regulate mitochondrial shape, length, and number. The balance between mitochondrial fusion and fission controls mitochondrial morphology. Mitochondrial shape affects the ability of cells to distribute their mitochondria to specific subcellular locations. Fusion and fission allow the mitochondrial exchange of lipid membranes and intramitochondrial content, which is crucial for maintaining the health of a mitochondrial population (51). For instance, MFN1 and MFN2 ablation in fibroblasts induce reduced respiratory capacity and great heterogeneity in mitochondrial shape and membrane potential (52).
The proper function of the mitochondria in the arterial wall is critical in all atherogenesis-related key cell types, including ECs, VSMCs, and macrophages, which are responsible for massive lipid storage via phagocytosis, as well as pro-inflammatory status maintenance in a lesion (53). Normal endothelium is a dynamic organ that regulates vascular tone by balancing the production of vasodilators and vasoconstrictors in response to a variety of stimuli (54). The endothelial mitochondria act as critical signaling organelles that play a crucial role in endothelial function, including subcellular location, dynamics, biogenesis, mitophagy, autophagy, ROS; therefore, mitochondrial dysfunction facilitates atherosclerosis development (55, 56). Endothelial dysfunction is a pathological condition characterized by an imbalance between substances with vasodilating, antimitogenic, and antithrombogenic properties (endothelium-derived relaxing factors) and substances with vasoconstricting, prothrombotic, and proliferative characteristics (endothelium-derived contracting factors) (57). ECs play important roles in the maintenance of vascular homeostasis by modulating vasodilation, platelet activation, and leukocyte adhesion (58). Therefore, the dysfunction of EC leads to increased vascular tension and atherosclerosis, followed by systemic hypertension, and increased incidence of ischemia and stroke. Moreover, mitochondrial dysfunction is involved in the formation of oxidative stress conditions in atherosclerosis, which facilitate inflammatory response and lesion development (59).
In pulmonary ECs, DRP-1 activation, which induces mitochondrial fission, stimulates angiogenesis by promoting cell proliferation and migration and inhibiting apoptosis (60). Endothelial dysfunction contributes to the development of nearly all vascular diseases (10). Even though ECs have low mitochondrial content, mitochondrial dynamics act as a pivotal orchestrator of EC homeostasis under normal conditions; damage in mitochondrial dynamics participates in endothelial dysfunction and diverse vascular diseases. Endothelial dysfunction leads to altered mitochondrial morphology, reduced network extent, and increased FIS1 protein expression compared with ECs from healthy volunteers (61).
VSMCs are the main constitutive stromal cells of the vascular wall that engage in a variety of different structural and physiological functions (62). VSMCs are crucial components of blood vessels and the major determinants of vasotone (62). This critical and tightly regulated function is granted by the contractile phenotype of VSMCs. VSMCs can switch to a synthetic dedifferentiated phenotype characterized by increased proliferative and migratory capabilities in response to certain cues. The VSMC phenotypic switch is implicated in the pathogenesis of vascular diseases (63). During the progression of atherosclerosis, VSMCs are subjected to a phenotype switch that can internalize atherogenic LDL particles, such as oxidized LDL or desialylated LDL, for lipid accumulation to migrate to lesion sites (64, 65). Cells with lipid particle accumulation are recognized as “foam cells” and manifest as atherosclerotic plaques (66). The association between VSMCs and mitochondrial dysfunction in atherosclerosis has been discussed before.
Mitochondrial dysfunction characterized by decreased oxidative phosphorylation is a striking phenotype of VSMCs isolated from atherosclerosis (13). In addition, a multitude amount of energy and oxygen-free radicals are required for the impairment of nuclear and mitochondrial DNAs in VSMCs, which further promotes DNA damage, genomic instability, and mitochondrial damage (67). Mitochondrial fission and fusion also affect VSMC function (68). Mitochondrial fission is an integral process in cell migration, and controlling mitochondrial fission can limit VSMC migration and pathological intimal hyperplasia by altering mitochondrial energetics and ROS levels (69). For instance, mitofusin (MFN) 2 is an important suppressor of VSMC proliferation (70). In addition, the link between mitochondrial dynamics and VSMC senescence can be mediated by Krüppel-like factor 5 (Klf5), an essential transcriptional factor of cardiovascular remodeling. Klf5 downregulation induces VSMC senescence through eIF5a depletion and mitochondrial fission (71).
Macrophage mitochondrial fission is essential for the continued removal of apoptotic cells and plays a protective role in advanced atherosclerosis (72). In macrophage-enriched murine atherosclerosis lesion areas, the level of dynamin-related protein-1 (DRP1) is downregulated and MFN2 is upregulated as the lesion progresses. Inhibiting macrophage mitochondrial fission results in a dramatic increase in the necrotic core area and the accumulation of apoptotic cells in the advanced stage of atherosclerosis; thus, macrophage mitochondrial fusion/fission could be a potential therapeutic target to prevent lesion necrosis and stabilize advanced plaques (73). Human CD14+ monocytes exhibit reduced mitochondrial fission and increased mitochondrial fusion for metabolic adaptation upon lipopolysaccharide stimulation. Notably, mitochondrial dynamics affect the inflammatory responses of CD14+ monocytes.
Many studies have pointed out the beneficial effects of mitochondrial fusion in oxidative phosphorylation. Mitochondrial fusion maintains normal mitochondrial function by protecting from mitochondrial DNA loss and maintaining the synthesis of mitochondrial proteins (74). In addition, mitochondrial fusion events can attenuate the damage of DNA and protein contents and restore damaged mitochondria by “functional complementation” (75). Mitochondrial fusion damage can lead to increased mitochondrial fission and fragmentation, which induce oxidative phosphorylation and cell apoptosis attenuated by mitochondrial division (76). For example, DRP1 gene mutation in mice can damage mitochondrial function and induce mitophagy, which contribute to heart enlargement and failure (77). Mitochondrial division inhibitor 1 (Mdivi-1) is a selective cell-permeable inhibitor of mitochondrial division DRP1 and mitochondrial division dynamin I. Mdivi-1 attenuates mitophagy and enhances apoptosis. Also, DRP1 inhibition with Mdivi-1 protects the injured heart and brain from ischemia (78, 79). Mitochondrial fission seems harmful in this perspective; however, the deletion of myocardial DRP1 gene can lead to division disorders, which result in dysfunctional mitochondria and ultimately lead to heart failure and death (80). Mitochondrial dynamics proteins have been genetically alerted in vascular cells. For example, in VSMCs, the overexpression of the phospho-deficient mutation, MFN2-S442A, increases the inhibitory effects of MFN2 on cell proliferation, as well as neointimal hyperplasia and restenosis, in rat carotid artery balloon injury model (70).
Excessive mitochondrial fragmentation often occurs in most vascular diseases and thus could be a promising therapeutic target for these diseases (81). The promotion of mitochondrial fusion and the inhibition of mitochondrial fission guide the different fates of the heart (82). MFN2 upregulation, besides DRP1 downregulation, maintains mitochondrial function through the elimination of excessive mitochondrial fragmentation (83). Mitochondrial fusion promoter, M1 (2 mg/kg), as an intervention in rat ischemia–reperfusion (I/R), reduces infarct size and exerts a beneficial effect toward ischemia (84). This result demonstrated that increased mitochondrial fusion brings about a beneficial impact on myocardial I/R injury.
However, excessive mitochondrial fusion causes serious diseases (85). Point mutation in mitochondrial carrier protein, SLC25A46, promotes the protein's rapid degradation and the stable recruitment of MFN2 and MFN1 complexes to the mitochondria and ultimately leads to over-fusion and the phenotype of cerebellopontine hypoplasia (86). Additionally, excessive mitochondrial fusion results in elevated oxidative stress and abnormal Ca2+ homeostasis, which eventually cause arrhythmia, particularly atrial fibrillation (87). Therefore, the balance between mitochondrial fusion and fission plays a vital role in the normal function of the vascular system.
Structurally, the normal artery is composed of three layers (88). The inner layer lined by a monolayer of ECs is closely contacted with blood; the middle layer composed of VSMCs is located at the complex extracellular matrix; and the outer layer of arteries is composed of mast cells, nerve endings, and microvessels (89). Direct contact with blood makes ECs especially vulnerable to damages caused by molecules (90). ECs act as ideal protection because they sense alterations in external stimuli and directly respond or transmit signals; EC dysfunction leads to the pathogenesis of almost all types of vascular diseases (91). Despite the low mitochondrial content of ECs, mitochondrial dynamics is a key endothelial homeostasis coordinator under normal conditions (10).
Atherosclerosis is the leading cause of vascular diseases and responsible for almost 50% of all cardiovascular deaths, and the mechanism of atherosclerosis has been well studied. Its pathogenesis comprises respective mechanisms during different disease stages (8). It is initiated through atherosclerotic lesion formation with a phenotype of endothelial dysfunction (92). The endothelium provides the functional link between blood circulation and the vessel wall. Local disturbance to the arterial endothelium leads to cell activation, which promotes the recruitment of circulating immune cells and increases permeability for circulating lipoprotein particles (93). Low-density lipoprotein (LDL), especially in its modified atherogenic form, is the main source of lipids that accumulate in the arterial wall (94). Several studies have confirmed the close association between the mitochondria and the different stages of atherosclerosis (95).
Multiple factors are responsible for vascular diseases, including the infiltration, differentiation, and transformation of monocytes to active lipid foam cells, as well as VSMC migration to the intima (96). ROS production in the mitochondria is a key factor in vascular diseases (97).
Mitochondrial dynamics plays an important role in the progression of vascular diseases (Figure 3). Mitochondrial fragmentation and FIS1 expression are increased in patients with type 2 diabetes (98). DRP1 and FIS1 accumulate in human aortic ECs after high glucose treatment (99). Alterations in mitochondrial dynamics are correlated with the production of mitochondrial ROS, which affects the pathogenesis of vascular diseases (100). FIS1 and DRP1 inhibition can block the production of mitochondrial ROS and mitochondrial network; hence, mitochondrial fission has a vital role in vascular diseases (37).
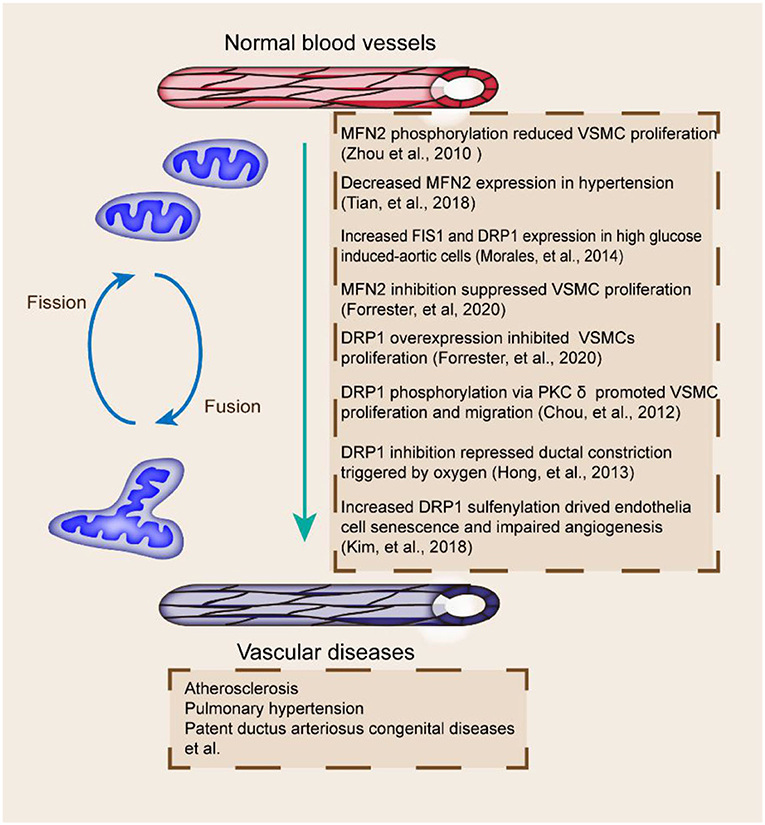
Figure 3. Mitochondrial fusion and fission proteins and vascular diseases. Fission and fusion imbalances are related to multiple vascular abnormalities, including MFN2 decrease and PTM, DRP1 inhibition and phosphorylation, and dysregulated FIS1 and DRP1 (70, 99, 104, 106, 114, 118, 122). The effects of proteins involved in the pathogenesis of vascular diseases are included.
The coordination of mitochondrial fusion and fission is essential for the maintenance of mitochondrial quantity and quality. Mitochondrial fragmentation occurs in vascular diseases (Table 2). Functional ductus arteriosus closure, initially induced by oxygen-dependent vasoconstriction shortly after birth, is dependent on mitochondrial fission (97). DRP1 perturbation is associated with endothelial dysfunction (101). DRP1-mediated mitochondrial fission exerts a critical function in the acute constriction of the ductus arteriosus to O2 and participates in the subsequent anatomic closure of the ductus arteriosus (102). Mitochondrial fission also seems indispensable for angiogenesis in ECs (103). The loss of protein disulfide isomerase active 1 in ECs induces mitochondrial fragmentation and mitochondrial ROS elevation by increasing Cys644 sulfenylation and DRP1 activity, which impair endothelium-dependent vasorelaxation and angiogenesis (104). DRP1 depletion in mice also leads to defective efferocytosis and has pathologic consequences in the thymus after dexamethasone treatment and in the advanced atherosclerotic lesions of fat-fed LDLR−/− mice (105). DRP1 overexpression or MFN2 inhibition also leads to endothelial dysfunction and the inhibition of VSMC proliferation (106). The decreased expression of MFN1 and MFN2 promotes atherosclerosis in animal models (107).
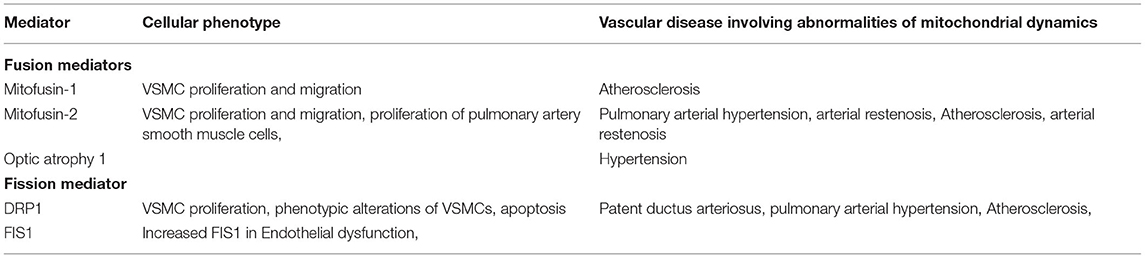
Table 2. Role of mitochondrial dynamics protein in vascular diseases.
The functions of MFN1 and MFN2 in ECs have also been addressed (108). Interestingly, the expression of MFNs could be stimulated in ECs when exposed to the angiogenic mitogen, vascular endothelial growth factor (VEGF) (109). The knockdown of MFN1 and MFN2 prevents the endothelial migration and differentiation induced by VEGF (110). Additionally, the diverse roles of MFNs in ECs were measured (111). MFN2 inhibition exclusively attenuates the production of basal and stress-induced ROS (96). MFN1 ablation particularly blocks VEGF signal transduction and suppresses NO production (23). Interestingly, the role of MFNs in vascular pathology is tightly related to metabolic stress (112).
Mitochondrial fission is indispensable for VSMC proliferation and migration, as well as pathophysiological processes, such as the premature closure of open arterial ducts and pulmonary hypertension (113). On the occasion of oxidative stress and angiotensin II stimulation, activated protein kinase C δ phosphorylates DRP1, which leads to mitochondrial fission and ROS-stimulated VSMC proliferation and migration (114). Therefore, the pharmacological inhibition of DRP1 could be used as a therapeutic target.
Emerging evidence implied that alteration in mitochondrial dynamics is accompanied by acute I/R. Several researches have observed that reduced OPA1 and MFN2 and increased DRP1 in cardiomyocytes simulate I/R. I/R stimulation in HL-1 cells induces mitochondrial fission through DRP1; the transfection of the fusion protein or DRP1 dominate-negative mutant protects from I/R injury (115). Moreover, OPA1 mild overexpression transgenic mice are resistant to muscular atrophy and I/R damage in the heart and brain. In spite of the well-known impact of mitochondrial fission and fusion balance on cardiac I/R injury, no study has shown a direct indication of their potential role in ischemic myopathy in peripheral artery disease.
Mitochondrial fusion and fission are also implicated in the abdominal aortic aneurysm (AAA). Angiotensin II stimulation (one of the main methods to induce AAA) in cultured rat aortic VSMCs induces mitochondrial fission. DRP1 expression was enhanced in human AAA samples compared with age-matched healthy controls (116). Furthermore, DRP1 inhibition protects from AAA development, as assessed by the diameters of the abdominal aorta as well as histological observation. Protection against AAA by DRP1 inhibition is accompanied by reduced stress response and senescence. Therefore, DRP1-mediated mitochondrial fission potentially stimulates the proinflammatory phenotypic alterations of VSMCs and contributes to the pathogenesis of AAA development.
As an impeditive vascular disease, pulmonary arterial hypertension is induced by several factors, including disordered oxygen sensing and dysregulated mitochondrial dynamics in pulmonary artery smooth muscle cells (117). Pulmonary arterial hypertension is believed to be contributed by excessive cell proliferation and impaired apoptosis accompanied by vasoconstriction, inflammation, and thrombosis (92). Pulmonary arterial hypertension is accompanied by reduced MFN2 and excessive DRP1 caused by increased hypoxia inducible factor 1 alpha (HIF-1α) activation and decreased PGC-1α activity (118). MFN2 mediates the proliferation of pulmonary artery smooth muscle cells in hypoxic pulmonary hypertension via the PI3K/Akt pathway (119). HIF-1α activation induces DRP1-dependent mitochondrial fission and an imbalance in fusion and fission in normal pulmonary artery smooth muscle cells (120). The decrease in MFN2 in pulmonary arterial hypertension leads to mitochondrial fragmentation and proliferation (121).
DRP1 inhibition represses the ductal constriction triggered by oxygen (122). Oxygen induces the PTM of DRP1 mediated by cyclin B1-CDK1 and CamK to trigger mitochondrial fission (123). Although the continuous inhibition of DRP1 impedes structural closure in an in vitro model of human open ductus arteriosus, the previous study still has not elucidated whether damaged mitochondria result in spontaneous patent ductus arteriosus (122).
MiRNA expression alteration contributes to ischemic heart disease by regulating the expression of various key mitochondrial elements involved in cell survival and death. MiR-762 and miR-210 are elevated whereas miR-1 is downregulated in myocardial infarction. miR-762 knockdown alleviates myocardial I/R injury in mice. The upregulation of miR-15/16 and miR-195 modulate cardiomyocyte survival and myocardial infarction by inhibiting ATP levels and inducing mitochondrial fusion. In addition, miR-15 inhibition protects against I/R injury in vivo by targeting pyruvate dehydrogenase kinase 4 and serum/glucocorticoid-regulated kinase 1, which are responsible for mitochondrial function and apoptosis, respectively (124). miRNAs also regulate foam cell formation and subsequent plaque formation. miR-302a suppresses foam cell formation, which would aggravate atheromatic plaque by increasing the activity of ABCA1, which induces the efflux of cholesterol out of macrophages.
Supporting materials relate mitochondrial fusion and fission to vascular diseases; emerging studies elucidated the protective effects of mitochondrial fusion and fission modulators on vascular diseases (Figure 3) (125). The pharmacological inhibition of DRP1 relieves plaque formation and lessens the accumulation of macrophages in the plaques of the ApoE−/− mouse model of carotid artery injury induced with wire (126). DRP1 seems to be a promising novel therapeutic target for atherosclerosis (106). As a selective cell-permeable inhibitor of mitochondrial division, Mdivi-1 treatment can dramatically reduce atherosclerotic lesion formation in streptozotocin-induced diabetic ApoE−/− mice (127). Mdivi-1 inhibits VSMC proliferation and migration through the attenuation of ROS production and DRP1 phosphorylation (128). Moreover, the anti-proliferation effect of Mdivi-1 is dependent on G2/M cell cycle arrest and independent on cyclin B1/CDK1-mediated DRP1 phosphorylation in arterial smooth muscle cells (118). Another example is ilexgenin A, a novel pentacyclic triterpenoid that exerts anti-atherosclerotic activity to reduce atherosclerosis in apolipoprotein E-deficient mice. Ilexgenin A hinders mitochondrial fission and induces DRP1 degradation dependent on Nrf2-induced proteasome subunit beta 5 in ECs, which contribute to the restraint of mitochondrial fission and thus relieve endothelial dysfunction (129). These findings provide the theoretical basis for the future development of ilexgenin A as a potential agent for atherosclerosis treatment. Mdivi-1 or congeners could also be used to maintain ductus arteriosus patency in infants awaiting congenital heart surgery (120). In addition, Mdivi-1 administration facilitates premature senescence and destroys the angiogenic function of human umbilical cord vein ECs by promoting the production of mitochondrial ROS and reducing autophagy flux (78). Therefore, DRP1 may be a promising therapeutic target for vascular repair (Figure 4).
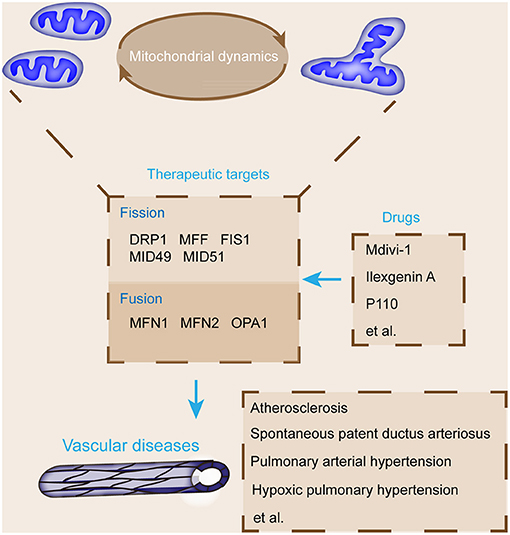
Figure 4. Summary of key mitochondrial dynamic regulatory proteins and major pharmacological agents that target these proteins for the treatment of vascular diseases. The targeting agents of mitochondrial fusion and fission proteins, such as DRP1 and MFN1, protect from vascular diseases, including pulmonary arterial hypertension, atherosclerosis, and ductus arteriosus closure.
Mitochondrial dynamics is associated with the pathogenesis of various vascular diseases and provides potential therapeutic targets (12, 56, 97, 98). Further identification prior to trial on potential therapeutic agents related to mitochondrial dynamics is indispensable to determine proper molecular targets and definitions and confirm the optimal and effective doses for mitochondrial fusion and fission modulators (130, 131). Extra modulators for mitochondrial fusion and fission are required (121, 132). For example, a recently designed inhibitor, P110, can inhibit DRP1 activation and fission by blocking the interaction between DRP1 and FIS1 (133).
In spite of the development of pharmacological agents that target fusion and fission for the prevention and treatment of vascular diseases, several obstacles remain to be solved to achieve this goal (87, 103, 104). First, the therapeutic agent needs to have specificity to target the organ and ascertain the duration time (134). Second, mitochondrial fusion and fission are vital for the proper functioning of the mitochondria and normal cells; hence, the manipulation of fusion and fission might have detrimental effects on normal cells. Besides, the application of such therapeutic agents is limited to temporary acute conditions rather than chronic conditions. Moreover, off-target effects should also be minimized. The off-target effects of these pharmacological agents are often caused by the recognition of the binding sites of the drug by other biomacromolecules, including receptors, enzymes, ion channels, transporters, and genes. This occurrence is still an important issue in the study of vascular related inhibitors.
In addition to energy metabolism, the mitochondria have multidimensional influence on cells and the vascular system. For example, the regulation of mitochondrial calcium homeostasis and mitophagy can affect vascular development and functional maintenance, but related molecular mechanisms still need further theoretical support (71, 105, 106). Whether mitochondrial homeostasis can cooperate with other mitochondrial functions to jointly affect vascular development and disease, as well as how key proteins play roles in this dynamic interaction process, needs further study. Mitochondrial dynamic regulatory proteins, such as FIS1, have a variety of functions. Their effects on vascular function still need to be studied in depth if these proteins will be used as therapeutic targets for vascular diseases.
YY and YL conceptualized, wrote the manuscript, and created Figures. YL, XC, and K-DR contributed to the writing of the manuscript. YY, XC, and YL reviewed and modified the manuscript. All authors approved the final version of the manuscript.
This work was supported by the National Natural Science Foundation of China (No. 31900502), the Henan Medical Science and Technology Joint Building Program (No. LHGJ20190236), and Key scientific Research project of Henan Universities (No. 21A310027).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Hammam N, Becher H, Andersen J, Manns PJ, Whittaker JL, Pritchard L. Early indicators of cardiovascular disease are evident in children and adolescents with cerebral palsy. Disabil Health J. (2021) 14:101112. doi: 10.1016/j.dhjo.2021.101112
2. Akawi N, Checa A, Antonopoulos AS, Akoumianakis I, Daskalaki E, Kotanidis CP, et al. Fat-secreted ceramides regulate vascular redox state and influence outcomes in patients with cardiovascular disease. J Am Coll Cardiol. (2021) 77:2494–513. doi: 10.1016/j.jacc.2021.03.314
3. Mitchell ME, Sidawy AN. The pathophysiology of atherosclerosis. Semin Vasc Surg. (1998) 11:134–41.
4. Deng HT, Liu HL, Zhai BB, Zhang K, Xu GC, Peng XM, et al. Vascular endothelial growth factor suppresses TNFSF15 production in endothelial cells by stimulating miR-31 and miR-20a expression via activation of Akt and Erk signals. FEBS Open Bio. (2017) 7:108–17. doi: 10.1002/2211-5463.12171
5. Kim J, Kang Y, Kojima Y, Lighthouse JK, Hu X, Aldred MA, et al. An endothelial apelin-FGF link mediated by miR-424 and miR-503 is disrupted in pulmonary arterial hypertension. Nat Med. (2013) 19:74–82. doi: 10.1038/nm.3040
6. Gimbrone MA Jr, Garcia-Cardena G. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ Res. (2016) 118:620–36. doi: 10.1161/CIRCRESAHA.115.306301
7. Fei Y, Hou J, Xuan W, Zhang C, Meng X. The relationship of plasma miR-503 and coronary collateral circulation in patients with coronary artery disease. Life Sci. (2018) 207:145–51. doi: 10.1016/j.lfs.2018.06.001
8. Bi R, Ding F, He Y, Jiang L, Jiang Z, Mei J, et al. miR-503 inhibits platelet-derived growth factor-induced human aortic vascular smooth muscle cell proliferation and migration through targeting the insulin receptor. Biomed Pharmacother. (2016) 84:1711–6. doi: 10.1016/j.biopha.2016.10.081
9. Ogita H, Kunimoto S, Kamioka Y, Sawa H, Masuda M, Mochizuki N. EphA4-mediated Rho activation via Vsm-RhoGEF expressed specifically in vascular smooth muscle cells. Circ Res. (2003) 93:23–31. doi: 10.1161/01.RES.0000079310.81429.C8
10. Tang X, Luo YX, Chen HZ, Liu DP. Mitochondria, endothelial cell function, and vascular diseases. Front Physiol. (2014) 5:175. doi: 10.3389/fphys.2014.00175
11. Kluge MA, Fetterman JL, Vita JA. Mitochondria and endothelial function. Circ Res. (2013) 112:1171–88. doi: 10.1161/CIRCRESAHA.111.300233
12. Park SY, Gifford JR, Andtbacka RH, Trinity JD, Hyngstrom JR, Garten RS, et al. Cardiac, skeletal, and smooth muscle mitochondrial respiration: are all mitochondria created equal? Am J Physiol Heart Circ Physiol. (2014) 307:H346–352. doi: 10.1152/ajpheart.00227.2014
13. Moulis M, Grousset E, Faccini J, Richetin K, Thomas G, Vindis C. The multifunctional sorting protein PACS-2 controls mitophagosome formation in human vascular smooth muscle cells through mitochondria-ER contact sites. Cells. (2019) 8:638. doi: 10.3390/cells8060638
14. Luan Y, Luan Y, Yuan RX, Feng Q, Chen X, Yang Y. Structure and Function of Mitochondria-Associated Endoplasmic Reticulum Membranes (MAMs) and Their Role in Cardiovascular Diseases. Oxid Med Cell Longev. (2021) 2021:4578809. doi: 10.1155/2021/4578809
15. Zhunina OA, Yabbarov NG, Grechko AV, Starodubova AV, Ivanova E, Nikiforov NG, et al. The role of mitochondrial dysfunction in vascular disease, tumorigenesis, and diabetes. Front Mol Biosci. (2021) 8:671908. doi: 10.3389/fmolb.2021.671908
16. Daiber A, Steven S, Vujacic-Mirski K, Kalinovic S, Oelze M, Di Lisa F, et al. Regulation of vascular function and inflammation via cross talk of reactive oxygen and nitrogen species from mitochondria or NADPH oxidase-implications for diabetes progression. Int J Mol Sci. (2020) 21:3405. doi: 10.3390/ijms21103405
17. Fukuda N, Yago N. Population dynamics of mitochondria. I A model for the role to ACTH in the degradation of adrenocortical mitochondria. J Theor Biol. (1974) 46:21–30. doi: 10.1016/0022-5193(74)90138-6
18. Bereiter-Hahn J, Voth M. Dynamics of mitochondria in living cells: shape changes, dislocations, fusion, and fission of mitochondria. Microsc Res Tech. (1994) 27:198–219. doi: 10.1002/jemt.1070270303
19. Liu X, Hajnoczky G. Altered fusion dynamics underlie unique morphological changes in mitochondria during hypoxia-reoxygenation stress. Cell Death Differ. (2011) 18:1561–72. doi: 10.1038/cdd.2011.13
20. Polyakov VY, Soukhomlinova MY, Fais D. Fusion, fragmentation, and fission of mitochondria. Biochemistry (Mosc). (2003) 68:838–49. doi: 10.1023/A:1025738712958
21. Vasquez-Trincado C, Garcia-Carvajal I, Pennanen C, Parra V, Hill JA, Rothermel BA, et al. Mitochondrial dynamics, mitophagy and cardiovascular disease. J Physiol. (2016) 594:509–25. doi: 10.1113/JP271301
22. Hunter AM, Kottachchi D, Lewis J, Duckett CS, Korneluk RG, Liston P, et al. novel ubiquitin fusion system bypasses the mitochondria and generates biologically active Smac/DIABLO. J Biol Chem. (2003) 278:7494–9. doi: 10.1074/jbc.C200695200
23. Lee JY, Kapur M, Li M, Choi MC, Choi S, Kim HJ, et al. MFN1 deacetylation activates adaptive mitochondrial fusion and protects metabolically challenged mitochondria. J Cell Sci. (2014) 127(Pt 22):4954–63. doi: 10.1242/jcs.157321
24. Bockler S, Chelius X, Hock N, Klecker T, Wolter M, Weiss M, et al. Fusion, fission, and transport control asymmetric inheritance of mitochondria and protein aggregates. J Cell Biol. (2017) 216:2481–98. doi: 10.1083/jcb.201611197
25. Arribat Y, Broskey NT, Greggio C, Boutant M, Conde Alonso S, Kulkarni SS, et al. Distinct patterns of skeletal muscle mitochondria fusion, fission and mitophagy upon duration of exercise training. Acta Physiol (Oxf). (2019) 225:e13179. doi: 10.1111/apha.13179
26. Ciarlo L, Vona R, Manganelli V, Gambardella L, Raggi C, Marconi M, et al. Recruitment of mitofusin 2 into “lipid rafts” drives mitochondria fusion induced by Mdivi-1. Oncotarget. (2018) 9:18869–84. doi: 10.18632/oncotarget.24792
27. Westermann B. Mitochondrial dynamics in model organisms: what yeasts, worms and flies have taught us about fusion and fission of mitochondria. Semin Cell Dev Biol. (2010) 21:542–9. doi: 10.1016/j.semcdb.2009.12.003
28. Varadi A, Johnson-Cadwell LI, Cirulli V, Yoon Y, Allan VJ, Rutter GA. Cytoplasmic dynein regulates the subcellular distribution of mitochondria by controlling the recruitment of the fission factor dynamin-related protein-1. J Cell Sci. (2004) 117(Pt 19):4389–400. doi: 10.1242/jcs.01299
29. Santel A, Frank S. Shaping mitochondria: The complex posttranslational regulation of the mitochondrial fission protein DRP1. IUBMB Life. (2008) 60:448–55. doi: 10.1002/iub.71
30. Zhou BH, Wei SS, Jia LS, Zhang Y, Miao CY, Wang HW. Drp1/Mff signaling pathway is involved in fluoride-induced abnormal fission of hepatocyte mitochondria in mice. Sci Total Environ. (2020) 725:138192. doi: 10.1016/j.scitotenv.2020.138192
31. Yoon YS, Yoon DS, Lim IK, Yoon SH, Chung HY, Rojo M, et al. Formation of elongated giant mitochondria in DFO-induced cellular senescence: involvement of enhanced fusion process through modulation of Fis1. J Cell Physiol. (2006) 209:468–80. doi: 10.1002/jcp.20753
32. Scarpelli PH, Tessarin-Almeida G, Vicoso KL, Lima WR, Borges-Pereira L, Meissner KA, et al. Melatonin activates FIS1, DYN1, and DYN2 Plasmodium falciparum related-genes for mitochondria fission: mitoemerald-GFP as a tool to visualize mitochondria structure. J Pineal Res. (2019) 66:e12484. doi: 10.1111/jpi.12484
33. Ko HJ, Tsai CY, Chiou SJ, Lai YL, Wang CH, Cheng JT, et al. The phosphorylation status of Drp1-Ser637 by PKA in mitochondrial fission modulates mitophagy via PINK1/Parkin to exert multipolar spindles assembly during mitosis. Biomolecules. (2021) 11:424. doi: 10.3390/biom11030424
34. Chen C, Huang J, Shen J, Bai Q. Quercetin improves endothelial insulin sensitivity in obese mice by inhibiting Drp1 phosphorylation at serine 616 and mitochondrial fragmentation. Acta Biochim Biophys Sin (Shanghai). (2019) 51:1250–7. doi: 10.1093/abbs/gmz127
35. Bo T, Yamamori T, Suzuki M, Sakai Y, Yamamoto K, Inanami O. Calmodulin-dependent protein kinase II (CaMKII) mediates radiation-induced mitochondrial fission by regulating the phosphorylation of dynamin-related protein 1 (Drp1) at serine 616. Biochem Biophys Res Commun. (2018) 495:1601–7. doi: 10.1016/j.bbrc.2017.12.012
36. Lee Y, Kwon I, Jang Y, Song W, Cosio-Lima LM, Roltsch MH. Potential signaling pathways of acute endurance exercise-induced cardiac autophagy and mitophagy and its possible role in cardioprotection. J Physiol Sci. (2017) 67:639–54. doi: 10.1007/s12576-017-0555-7
37. MacVicar TD, Lane JD. Impaired OMA1-dependent cleavage of OPA1 and reduced DRP1 fission activity combine to prevent mitophagy in cells that are dependent on oxidative phosphorylation. J Cell Sci. (2014) 127(Pt 10):2313–25. doi: 10.1242/jcs.144337
38. Hu Q, Zhang H, Gutierrez Cortes N, Wu D, Wang P, Zhang J, et al. Increased Drp1 acetylation by lipid overload induces cardiomyocyte death and heart dysfunction. Circ Res. (2020) 126:456–70. doi: 10.1161/CIRCRESAHA.119.315252
39. Adaniya SM. J OU, Cypress MW, Kusakari Y, Jhun BS. Posttranslational modifications of mitochondrial fission and fusion proteins in cardiac physiology and pathophysiology. Am J Physiol Cell Physiol. (2019) 316:C583–604. doi: 10.1152/ajpcell.00523.2018
40. Chen Y, Dorn GW 2nd. PINK1-phosphorylated mitofusin 2 is a parkin receptor for culling damaged mitochondria. Science. (2013) 340:471–5. doi: 10.1126/science.1231031
41. Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet. (2010) 19:4861–70. doi: 10.1093/hmg/ddq419
42. Song R, Hu XQ, Zhang L. Mitochondrial MiRNA in cardiovascular function and disease. Cells. (2019) 8:1475. doi: 10.3390/cells8121475
43. Li J, Donath S, Li Y, Qin D, Prabhakar BS Li P. miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet. (2010) 6:e1000795. doi: 10.1371/journal.pgen.1000795
44. Zhloba AA, Subbotina TF, Alekseevskaia ES, Moiseeva OM, Gavriluk ND, Irtuga OB. The metabolic and protein markers of dysfunction of mitochondria in patients with cardio-vascular diseases. Klin Lab Diagn. (2015) 60:35–41. doi: 10.18821/0860-2084-2015-60-7-35-41
45. Forte M, Schirone L, Ameri P, Basso C, Catalucci D, Modica J, et al. The role of mitochondrial dynamics in cardiovascular diseases. Br J Pharmacol. (2021) 178:2060–76. doi: 10.1111/bph.15068
46. Han XJ, Lu YF Li SA, Kaitsuka T, Sato Y, Tomizawa K, Nairn AC, et al. CaM kinase I alpha-induced phosphorylation of Drp1 regulates mitochondrial morphology. J Cell Biol. (2008) 182:573–85. doi: 10.1083/jcb.200802164
47. Luo J, Shen S. Lipoic acid alleviates schistosomiasis-induced liver fibrosis by upregulating Drp1 phosphorylation. Acta Trop. (2020) 206:105449. doi: 10.1016/j.actatropica.2020.105449
48. Arduino DM, Esteves AR, Cardoso SM. Mitochondrial fusion/fission, transport and autophagy in Parkinson's disease: when mitochondria get nasty. Parkinsons Dis. (2011) 2011:767230. doi: 10.4061/2011/767230
49. Karbowski M. Mitochondria on guard: role of mitochondrial fusion and fission in the regulation of apoptosis. Adv Exp Med Biol. (2010) 687:131–42. doi: 10.1007/978-1-4419-6706-0_8
50. Grohm J, Plesnila N, Culmsee C. Bid mediates fission, membrane permeabilization and peri-nuclear accumulation of mitochondria as a prerequisite for oxidative neuronal cell death. Brain Behav Immun. (2010) 24:831–8. doi: 10.1016/j.bbi.2009.11.015
51. Li D, Yang S, Xing Y, Pan L, Zhao R, Zhao Y, et al. Novel insights and current evidence for mechanisms of atherosclerosis: mitochondrial dynamics as a potential therapeutic target. Front Cell Dev Biol. (2021) 9:673839. doi: 10.3389/fcell.2021.673839
52. Baumann K. Organelle dynamics: fusing for stability. Nat Rev Mol Cell Biol. (2010) 11:391. doi: 10.1038/nrm2910
53. Masi S, Virdis A. Targeting mitochondria in age-related vascular changes: a new arrow to the bow of antioxidant treatment? Hypertension. (2018) 71:1023–5. doi: 10.1161/HYPERTENSIONAHA.118.10869
54. Pearson JD. Normal endothelial cell function. Lupus. (2000) 9:183–8. doi: 10.1191/096120300678828299
55. Wang X, Wang Y, Zhang L, Zhang D, Bai L, Kong W, et al. L-Cystathionine protects against homocysteine-induced mitochondria-dependent apoptosis of vascular endothelial cells. Oxid Med Cell Longev. (2019) 2019:1253289. doi: 10.1155/2019/1253289
56. Luan Y, Feng Q, Chen X, Ren KD, Yang Y. Emerging role of mitophagy in the heart: therapeutic potentials to modulate mitophagy in cardiac diseases. Oxid Med Cell Longev. (2021) 2021:13. doi: 10.1155/2021/3259963
57. Flammer AJ, Anderson T, Celermajer DS, Creager MA, Deanfield J, Ganz P, et al. The assessment of endothelial function: from research into clinical practice. Circulation. (2012) 126:753–67. doi: 10.1161/CIRCULATIONAHA.112.093245
58. Forte M, Stanzione R, Cotugno M, Bianchi F, Marchitti S, Rubattu S. Vascular ageing in hypertension: focus on mitochondria. Mech Ageing Dev. (2020) 189:111267. doi: 10.1016/j.mad.2020.111267
59. Markin AM, Khotina VA, Zabudskaya XG, Bogatyreva AI, Starodubova AV, Ivanova E, et al. Disturbance of mitochondrial dynamics and mitochondrial therapies in atherosclerosis. Life (Basel). (2021) 11:165. doi: 10.3390/life11020165
60. Caja S, Enriquez JA. Mitochondria in endothelial cells: sensors and integrators of environmental cues. Redox Biol. (2017) 12:821–7. doi: 10.1016/j.redox.2017.04.021
61. Shenouda SM, Widlansky ME, Chen K, Xu G, Holbrook M, Tabit CE, et al. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation. (2011) 124:444–53. doi: 10.1161/CIRCULATIONAHA.110.014506
62. Badran A, Nasser SA, Mesmar J, El-Yazbi AF, Bitto A, Fardoun MM, et al. Reactive oxygen species: modulators of phenotypic switch of vascular smooth muscle cells. Int J Mol Sci. (2020) 21:8764. doi: 10.3390/ijms21228764
63. Maddaluno M, Grassia G, Di Lauro MV, Parisi A, Maione F, Cicala C, et al. Bindarit inhibits human coronary artery smooth muscle cell proliferation, migration and phenotypic switching. PLoS ONE. (2012) 7:e47464. doi: 10.1371/journal.pone.0047464
64. Quintero M, Colombo SL, Godfrey A, Moncada S. Mitochondria as signaling organelles in the vascular endothelium. Proc Natl Acad Sci U S A. (2006) 103:5379–84. doi: 10.1073/pnas.0601026103
65. Lopes RA, Neves KB, Pestana CR, Queiroz AL, Zanotto CZ, Chignalia AZ, et al. Testosterone induces apoptosis in vascular smooth muscle cells via extrinsic apoptotic pathway with mitochondria-generated reactive oxygen species involvement. Am J Physiol Heart Circ Physiol. (2014) 306:H1485–1494. doi: 10.1152/ajpheart.00809.2013
66. Busija DW, Rutkai I, Dutta S, Katakam PV. Role of mitochondria in cerebral vascular function: energy production, cellular protection, and regulation of vascular tone. Compr Physiol. (2016) 6:1529–48. doi: 10.1002/cphy.c150051
67. Brock M, Samillan VJ, Trenkmann M, Schwarzwald C, Ulrich S, Gay RE, et al. AntagomiR directed against miR-20a restores functional BMPR2 signalling and prevents vascular remodelling in hypoxia-induced pulmonary hypertension. Eur Heart J. (2014) 35:3203–11. doi: 10.1093/eurheartj/ehs060
68. Zhang X, Chen W, Li J, Qi S, Hong S, Wang Y, et al. Involvement of mitochondrial fission in calcium sensing receptor-mediated vascular smooth muscle cells proliferation during hypertension. Biochem Biophys Res Commun. (2018) 495:454–60. doi: 10.1016/j.bbrc.2017.11.048
69. Wang L, Yu T, Lee H, O'Brien DK, Sesaki H, Yoon Y. Decreasing mitochondrial fission diminishes vascular smooth muscle cell migration and ameliorates intimal hyperplasia. Cardiovasc Res. (2015) 106:272–83. doi: 10.1093/cvr/cvv005
70. Zhou W, Chen KH, Cao W, Zeng J, Liao H, Zhao L, et al. Mutation of the protein kinase A phosphorylation site influences the anti-proliferative activity of mitofusin 2. Atherosclerosis. (2010) 211:216–23. doi: 10.1016/j.atherosclerosis.2010.02.012
71. Ma D, Zheng B, Liu HL, Zhao YB, Liu X, Zhang XH, et al. Klf5 down-regulation induces vascular senescence through eIF5a depletion and mitochondrial fission. PLoS Biol. (2020) 18:e3000808. doi: 10.1371/journal.pbio.3000808
72. Yarbro JR, Emmons RS, Pence BD. Macrophage immunometabolism and inflammaging: roles of mitochondrial dysfunction, cellular senescence, CD38, and NAD. Immunometabolism. (2020) 2:e200026. doi: 10.20900/immunometab20200026
73. Umezu R, Koga JI, Matoba T, Katsuki S, Wang L, Hasuzawa N, et al. Macrophage (Drp1) dynamin-related protein 1 accelerates intimal thickening after vascular injury. Arterioscler Thromb Vasc Biol. (2020) 40:e214–26. doi: 10.1161/ATVBAHA.120.314383
74. Li T, Zheng F, Cheung M, Wang F, Fu C. Fission yeast mitochondria are distributed by dynamic microtubules in a motor-independent manner. Sci Rep. (2015) 5:11023. doi: 10.1038/srep11023
75. Di Pietro V, Lazzarino G, Amorini AM, Signoretti S, Hill LJ, Porto E, et al. Fusion or fission: the destiny of mitochondria in traumatic brain injury of different severities. Sci Rep. (2017) 7:9189. doi: 10.1038/s41598-017-09587-2
76. Arimura SI. Fission and fusion of plant mitochondria, and genome maintenance. Plant Physiol. (2018) 176:152–61. doi: 10.1104/pp.17.01025
77. Chen WR, Zhou YJ, Sha Y, Wu XP, Yang JQ, Liu F. Melatonin attenuates vascular calcification by inhibiting mitochondria fission via an AMPK/Drp1 signalling pathway. J Cell Mol Med. (2020) 24:6043–54. doi: 10.1111/jcmm.15157
78. Sharp WW, Fang YH, Han M, Zhang HJ, Hong Z, Banathy A, et al. Dynamin-related protein 1 (Drp1)-mediated diastolic dysfunction in myocardial ischemia-reperfusion injury: therapeutic benefits of Drp1 inhibition to reduce mitochondrial fission. FASEB J. (2014) 28:316–26. doi: 10.1096/fj.12-226225
79. Zhang N, Wang S, Li Y, Che L, Zhao Q. A selective inhibitor of Drp1, mdivi-1, acts against cerebral ischemia/reperfusion injury via an anti-apoptotic pathway in rats. Neurosci Lett. (2013) 535:104–9. doi: 10.1016/j.neulet.2012.12.049
80. Wu J, Chen H, Qin J, Chen N, Lu S, Jin J, et al. Baicalin improves cardiac outcome and survival by suppressing Drp1-mediated mitochondrial fission after cardiac arrest-induced myocardial damage. Oxid Med Cell Longev. (2021) 2021:8865762. doi: 10.1155/2021/8865762
81. Tan DX, Chen XX, Bai TZ, Zhang J, Li ZF. Sevoflurane up-regulates microRNA-204 to ameliorate myocardial ischemia/reperfusion injury in mice by suppressing Cotl1. Life Sci. (2020) 259:118162. doi: 10.1016/j.lfs.2020.118162
82. Wang Z, Wang Z, Wang T, Yuan J, Wang X, Zhang Z. Inhibition of miR-34a-5p protected myocardial ischemia reperfusion injury-induced apoptosis and reactive oxygen species accumulation through regulation of Notch Receptor 1 signaling. Rev Cardiovasc Med. (2019) 20:187–97. doi: 10.31083/j.rcm.2019.03.545
83. Qin Y, Li A, Liu B, Jiang W, Gao M, Tian X, et al. Mitochondrial fusion mediated by fusion promotion and fission inhibition directs adult mouse heart function toward a different direction. FASEB J. (2020) 34:663–75. doi: 10.1096/fj.201901671R
84. Lees JG, Kong AM, Chen YC, Sivakumaran P, Hernandez D, Pebay A, et al. Mitochondrial fusion by M1 promotes embryoid body cardiac differentiation of human pluripotent stem cells. Stem Cells Int. (2019) 2019:6380135. doi: 10.1155/2019/6380135
85. Chen L, Chen H. Effect of Mahuang Gancao Ganjiang decoction on fusion and fission of mitochondria and apoptosis of lymphocytes in mice under cold stress. Evid Based Complement Alternat Med. (2017) 2017:5132963. doi: 10.1155/2017/5132963
86. Zhang ZQ, Zhang CZ, Shao B, Pang DH, Han GZ, Lin L. Effects of abnormal expression of fusion and fission genes on the morphology and function of lung macrophage mitochondria in SiO2-induced silicosis fibrosis in rats in vivo. Toxicol Lett. (2019) 312:181–7. doi: 10.1016/j.toxlet.2019.04.029
87. Wang X, Liu X, Chen Y, Wang H, Zhang R, Zhang Q, et al. Calreticulin regulated intrinsic apoptosis through mitochondria-dependent and independent pathways mediated by ER stress in arsenite exposed HT-22 cells. Chemosphere. (2020) 251:126466. doi: 10.1016/j.chemosphere.2020.126466
88. Yu E, Mercer J, Bennett M. Mitochondria in vascular disease. Cardiovasc Res. (2012) 95:173–82. doi: 10.1093/cvr/cvs111
89. Lo J, Plutzky J. The biology of atherosclerosis: general paradigms and distinct pathogenic mechanisms among HIV-infected patients. J Infect Dis. (2012) 205 Suppl 3:S368–374. doi: 10.1093/infdis/jis201
90. Lermant A, Murdoch CE. Cysteine glutathionylation acts as a redox switch in endothelial cells. Antioxidants (Basel). (2019) 8:315. doi: 10.3390/antiox8080315
91. Dedkova EN Ji X, Lipsius SL, Blatter LA. Mitochondrial calcium uptake stimulates nitric oxide production in mitochondria of bovine vascular endothelial cells. Am J Physiol Cell Physiol. (2004) 286:C406–415. doi: 10.1152/ajpcell.00155.2003
92. Lin CF, Chang YH Yu FC, Tsai CT, Chen CC, Liu HY, Chien LN. Risk of heart failure following drug-eluting stent implantation in patients with non-ST-elevation myocardial infarction. Atherosclerosis. (2021) 316:84–9. doi: 10.1016/j.atherosclerosis.2020.10.012
93. Gomibuchi H, Okazaki M, Iwai S, Kumai T, Kobayashi S, Oguchi K. Development of hyperfibrinogenemia in spontaneously hypertensive and hyperlipidemic rats: a potentially useful animal model as a complication of hypertension and hyperlipidemia. Exp Anim. (2007) 56:1–10. doi: 10.1538/expanim.56.1
94. Davidson SM, Duchen MR. Endothelial mitochondria: contributing to vascular function and disease. Circ Res. (2007) 100:1128–41. doi: 10.1161/01.RES.0000261970.18328.1d
95. Ballinger SW, Patterson C, Knight-Lozano CA, Burow DL, Conklin CA, Hu Z, et al. Mitochondrial integrity and function in atherogenesis. Circulation. (2002) 106:544–9. doi: 10.1161/01.CIR.0000023921.93743.89
96. Liu X, Zheng Z, Yu L, Hu J, Li X. Down-regulation of protein phosphatase 2A catalytic subunit involved in mitochondria fission/fusion dynamics imbalance and functional impairment induced by Human tau. Zhongguo Yi Xue Ke Xue Yuan Xue Bao. (2020) 42:297–306. doi: 10.3881/j.issn.1000-503X.12163
97. Khraiwesh H, Lopez-Dominguez JA, Lopez-Lluch G, Navas P, de Cabo R, Ramsey JJ, et al. Alterations of ultrastructural and fission/fusion markers in hepatocyte mitochondria from mice following calorie restriction with different dietary fats. J Gerontol A Biol Sci Med Sci. (2013) 68:1023–34. doi: 10.1093/gerona/glt006
98. Zheng J, Lu C. Oxidized LDL causes endothelial apoptosis by inhibiting mitochondrial fusion and mitochondria autophagy. Front Cell Dev Biol. (2020) 8:600950. doi: 10.3389/fcell.2020.600950
99. Morales PE, Torres G, Sotomayor-Flores C, Pena-Oyarzun D, Rivera-Mejias P, Paredes F, et al. GLP-1 promotes mitochondrial metabolism in vascular smooth muscle cells by enhancing endoplasmic reticulum-mitochondria coupling. Biochem Biophys Res Commun. (2014) 446:410–6. doi: 10.1016/j.bbrc.2014.03.004
100. Gbel J, Engelhardt E, Pelzer P, Sakthivelu V, Jahn HM, Jevtic M, et al. Mitochondria-endoplasmic reticulum contacts in reactive astrocytes promote vascular remodeling. Cell Metab. (2020) 31:791–808.e798. doi: 10.1016/j.cmet.2020.03.005
101. Li J, Wang Y, Wang Y, Wen X, Ma XN, Chen W, et al. Pharmacological activation of AMPK prevents Drp1-mediated mitochondrial fission and alleviates endoplasmic reticulum stress-associated endothelial dysfunction. J Mol Cell Cardiol. (2015) 86:62–74. doi: 10.1016/j.yjmcc.2015.07.010
102. Shang X, Li J, Yu R, Zhu P, Zhang Y, Xu J, et al. Sepsis-related myocardial injury is associated with Mst1 upregulation, mitochondrial dysfunction and the Drp1/F-actin signaling pathway. J Mol Histol. (2019) 50:91–103. doi: 10.1007/s10735-018-09809-5
103. She H, Zhu Y, Deng H, Kuang L, Fang H, Zhang Z, et al. Protective effects of dexmedetomidine on the vascular endothelial barrier function by inhibiting mitochondrial fission via ER/mitochondria contact. Front Cell Dev Biol. (2021) 9:636327. doi: 10.3389/fcell.2021.636327
104. Kim YM, Youn SW, Sudhahar V, Das A, Chandhri R, Cuervo Grajal H, et al. Redox regulation of mitochondrial fission protein Drp1 by protein disulfide isomerase limits endothelial senescence. Cell Rep. (2018) 23:3565–78. doi: 10.1016/j.celrep.2018.05.054
105. Wang Y, Subramanian M, Yurdagul A Jr, Barbosa-Lorenzi VC, Cai B, de Juan-Sanz J, et al. Mitochondrial fission promotes the continued clearance of apoptotic cells by macrophages. Cell. (2017) 171:331–345.e322. doi: 10.1016/j.cell.2017.08.041
106. Forrester SJ, Preston KJ, Cooper HA, Boyer MJ, Escoto KM, Poltronetti AJ, et al. Mitochondrial fission mediates endothelial inflammation. Hypertension. (2020) 76:267–76. doi: 10.1161/HYPERTENSIONAHA.120.14686
107. de Brito OM, Scorrano L. Mitofusin 2: a mitochondria-shaping protein with signaling roles beyond fusion. Antioxid Redox Signal. (2008) 10:621–33. doi: 10.1089/ars.2007.1934
108. Liu X, Liu Z, Hou W, Wang K, Ding W, Chen D, et al. Changes in mitochondria fusion protein-2 hepatic expression in conditions of liver cirrhosis and acute on chronic liver failure. Zhonghua Gan Zang Bing Za Zhi. (2014) 22:671–5. doi: 10.3760/cma.j.issn.1007-3418.2014.09.008
109. Garonna E, Botham KM, Birdsey GM, Randi AM, Gonzalez-Perez RR, Wheeler-Jones CP. Vascular endothelial growth factor receptor-2 couples cyclo-oxygenase-2 with pro-angiogenic actions of leptin on human endothelial cells. PLoS ONE. (2011) 6:e18823. doi: 10.1371/journal.pone.0018823
110. Lugus JJ, Ngoh GA, Bachschmid MM, Walsh K. Mitofusins are required for angiogenic function and modulate different signaling pathways in cultured endothelial cells. J Mol Cell Cardiol. (2011) 51:885–93. doi: 10.1016/j.yjmcc.2011.07.023
111. Trevisan T, Pendin D, Montagna A, Bova S, Ghelli AM, Daga A. Manipulation of mitochondria dynamics reveals separate roles for form and function in mitochondria distribution. Cell Rep. (2018) 23:1742–53. doi: 10.1016/j.celrep.2018.04.017
112. Chandhok G, Lazarou M, Neumann B. Structure, function, and regulation of mitofusin-2 in health and disease. Biol Rev Camb Philos Soc. (2018) 93:933–49. doi: 10.1111/brv.12378
113. Zeng JW, Chen BY, Lv XF, Sun L, Zeng XL, Zheng HQ, et al. Transmembrane member 16A participates in hydrogen peroxide-induced apoptosis by facilitating mitochondria-dependent pathway in vascular smooth muscle cells. Br J Pharmacol. (2018) 175:3669–84. doi: 10.1111/bph.14432
114. Chou CH, Lin CC, Yang MC, Wei CC, Liao HD, Lin RC, et al. GSK3beta-mediated Drp1 phosphorylation induced elongated mitochondrial morphology against oxidative stress. PLoS ONE. (2012) 7:e49112. doi: 10.1371/journal.pone.0049112
115. Jin JY, Wei XX, Zhi XL, Wang XH, Meng D. Drp1-dependent mitochondrial fission in cardiovascular disease. Acta Pharmacol Sin. (2021) 42:655–64. doi: 10.1038/s41401-020-00518-y
116. Wang X, Li S, Liu L, Jian Z, Cui T, Yang Y, et al. Role of the aryl hydrocarbon receptor signaling pathway in promoting mitochondrial biogenesis against oxidative damage in human melanocytes. J Dermatol Sci. (2019) 96:33–41. doi: 10.1016/j.jdermsci.2019.09.001
117. Chan DC. Mitochondrial dynamics and its involvement in disease. Annu Rev Pathol. (2020) 15:235–59. doi: 10.1146/annurev-pathmechdis-012419-032711
118. Tian L, Potus F, Wu D, Dasgupta A, Chen KH, Mewburn J, et al. Increased Drp1-mediated mitochondrial fission promotes proliferation and collagen production by right ventricular fibroblasts in experimental pulmonary arterial hypertension. Front Physiol. (2018) 9:828. doi: 10.3389/fphys.2018.00828
119. Li D, Li X, Guan Y, Guo X. Mitofusin-2-mediated tethering of mitochondria and endoplasmic reticulum promotes cell cycle arrest of vascular smooth muscle cells in G0/G1 phase. Acta Biochim Biophys Sin (Shanghai). (2015) 47:441–50. doi: 10.1093/abbs/gmv035
120. Feng W, Wang J, Yan X, Zhang Q, Chai L, Wang Q, et al. ERK/Drp1-dependent mitochondrial fission contributes to HMGB1-induced autophagy in pulmonary arterial hypertension. Cell Prolif. (2021) 54:e13048. doi: 10.1111/cpr.13048
121. Ryan J, Dasgupta A, Huston J, Chen KH, Archer SL. Mitochondrial dynamics in pulmonary arterial hypertension. J Mol Med (Berl). (2015) 93:229–42. doi: 10.1007/s00109-015-1263-5
122. Hong Z, Kutty S, Toth PT, Marsboom G, Hammel JM, Chamberlain C, et al. Role of dynamin-related protein 1 (Drp1)-mediated mitochondrial fission in oxygen sensing and constriction of the ductus arteriosus. Circ Res. (2013) 112:802–15. doi: 10.1161/CIRCRESAHA.111.300285
123. Xu S, Wang P, Zhang H, Gong G, Gutierrez Cortes N, Zhu W, et al. CaMKII induces permeability transition through Drp1 phosphorylation during chronic beta-AR stimulation. Nat Commun. (2016) 7:13189. doi: 10.1038/ncomms13189
124. Hullinger TG, Montgomery RL, Seto AG, Dickinson BA, Semus HM, Lynch JM, et al. Inhibition of miR-15 protects against cardiac ischemic injury. Circ Res. (2012) 110:71–81. doi: 10.1161/CIRCRESAHA.111.244442
125. Ong SB, Kalkhoran SB, Cabrera-Fuentes HA, Hausenloy DJ. Mitochondrial fusion and fission proteins as novel therapeutic targets for treating cardiovascular disease. Eur J Pharmacol. (2015) 763(Pt A):104–14. doi: 10.1016/j.ejphar.2015.04.056
126. Fernandez-Calle R, Vicente-Rodriguez M, Pastor M, Gramage E, Di Geronimo B, Zapico JM, et al. Pharmacological inhibition of Receptor Protein Tyrosine Phosphatase beta/zeta (PTPRZ1) modulates behavioral responses to ethanol. Neuropharmacology. (2018) 137:86–95. doi: 10.1016/j.neuropharm.2018.04.027
127. Cassidy-Stone A, Chipuk JE, Ingerman E, Song C, Yoo C, Kuwana T, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. (2008) 14:193–204. doi: 10.1016/j.devcel.2007.11.019
128. Lee DS, Kim JE. PDI-mediated S-nitrosylation of DRP1 facilitates DRP1-S616 phosphorylation and mitochondrial fission in CA1 neurons. Cell Death Dis. (2018) 9:869. doi: 10.1038/s41419-018-0910-5
129. Li Y, Yang J, Chen MH, Wang Q, Qin MJ, Zhang T, et al. Ilexgenin A inhibits endoplasmic reticulum stress and ameliorates endothelial dysfunction via suppression of TXNIP/NLRP3 inflammasome activation in an AMPK dependent manner. Pharmacol Res. (2015) 99:101–15. doi: 10.1016/j.phrs.2015.05.012
130. Cao J, Remaley AT, Guan W, Devaraj S, Tsai MY. Performance of novel low-density lipoprotein-cholesterol calculation methods in predicting clinical and subclinical atherosclerotic cardiovascular disease risk: the multi-ethnic study of atherosclerosis. Atherosclerosis. (2021) 327:1–4. doi: 10.1016/j.atherosclerosis.2021.04.018
131. Kuzmicic J, Del Campo A, Lopez-Crisosto C, Morales PE, Pennanen C, Bravo-Sagua R, et al. Mitochondrial dynamics: a potential new therapeutic target for heart failure. Rev Esp Cardiol. (2011) 64:916–23. doi: 10.1016/j.rec.2011.05.022
132. Gomez E, Vercauteren M, Kurtz B, Ouvrard-Pascaud A, Mulder P, Henry JP, et al. Reduction of heart failure by pharmacological inhibition or gene deletion of protein tyrosine phosphatase 1B. J Mol Cell Cardiol. (2012) 52:1257–64. doi: 10.1016/j.yjmcc.2012.03.003
133. Filichia E, Hoffer B, Qi X, Luo Y. Inhibition of Drp1 mitochondrial translocation provides neural protection in dopaminergic system in a Parkinson's disease model induced by MPTP. Sci Rep. (2016) 6:32656. doi: 10.1038/srep32656
134. Ishihara T, Ban-Ishihara R, Maeda M, Matsunaga Y, Ichimura A, Kyogoku S, et al. Dynamics of mitochondrial DNA nucleoids regulated by mitochondrial fission is essential for maintenance of homogeneously active mitochondria during neonatal heart development. Mol Cell Biol. (2015) 35:211–23. doi: 10.1128/MCB.01054-14
Keywords: cardiovascular disease (CVDs), vascular diseases, mitochondrial dynamics, fusion, fission
Citation: Luan Y, Ren K-D, Luan Y, Chen X and Yang Y (2021) Mitochondrial Dynamics: Pathogenesis and Therapeutic Targets of Vascular Diseases. Front. Cardiovasc. Med. 8:770574. doi: 10.3389/fcvm.2021.770574
Received: 04 September 2021; Accepted: 25 October 2021;
Published: 06 December 2021.
Edited by:
Masanori Aikawa, Brigham and Women's Hospital and Harvard Medical School, United StatesReviewed by:
Sudhahar Varadarajan, Augusta University, United StatesCopyright © 2021 Luan, Ren, Luan, Chen and Yang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yang Yang, eWFuZ3lhbmdiaW9AMTYzLmNvbQ==; Xing Chen, Y2hlbnhpbmcxMjEyQDE2My5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.