
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cardiovasc. Med. , 19 November 2021
Sec. Cardiovascular Therapeutics
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.770421
This article is part of the Research Topic New Strategies to Inhibit Cell Death in Myocardial Ischemia-Reperfusion Injury: How to succeed? View all 17 articles
 Magda C. Díaz-Vesga1,2,3†
Magda C. Díaz-Vesga1,2,3† Úrsula Zúñiga-Cuevas3†
Úrsula Zúñiga-Cuevas3† Andrés Ramírez-Reyes3
Andrés Ramírez-Reyes3 Nicolas Herrera-Zelada3
Nicolas Herrera-Zelada3 Iván Palomo4,5
Iván Palomo4,5 Roberto Bravo-Sagua3,5,6
Roberto Bravo-Sagua3,5,6 Jaime A. Riquelme3,5*
Jaime A. Riquelme3,5*Despite important advances in the treatment of myocardial infarction that have significantly reduced mortality, there is still an unmet need to limit the infarct size after reperfusion injury in order to prevent the onset and severity of heart failure. Multiple cardioprotective maneuvers, therapeutic targets, peptides and drugs have been developed to effectively protect the myocardium from reperfusion-induced cell death in preclinical studies. Nonetheless, the translation of these therapies from laboratory to clinical contexts has been quite challenging. Comorbidities, comedications or inadequate ischemia/reperfusion experimental models are clearly identified variables that need to be accounted for in order to achieve effective cardioprotection studies. The aging heart is characterized by altered proteostasis, DNA instability, epigenetic changes, among others. A vast number of studies has shown that multiple therapeutic strategies, such as ischemic conditioning phenomena and protective drugs are unable to protect the aged heart from myocardial infarction. In this Mini-Review, we will provide an updated state of the art concerning potential new cardioprotective strategies targeting the aging heart.
Although the available treatments of acute myocardial infarction (MI) have improved in the past decades, the consequent heart failure is consistently rising and thus, effective protection of the heart to preserve ventricular function and reduce myocardial remodeling is still an unmet need (1). Several years of research have established that cardiomyocytes have signaling pathways that, upon activation, can reduce the damage elicited by ischemia/reperfusion (I/R) injury, such as the Reperfusion Injury Salvage Kinases (RISK) and the Survivor Activating Enhancement Factor (SAFE) (2–4). There are numerous cardioprotective strategies that can be induced by different ischemic conditioning procedures and drugs with successful results in preclinical studies (5). Nonetheless, translation of cardioprotection from bench to bedside has been highly challenging (6). Remote ischemic conditioning (RIC) is an important example of this. This cardioprotective maneuver has been thoroughly established as a potent therapeutic strategy in multiple preclinical studies, as well as small clinical trials (5). However, the CONDI-2/ERIC-PPCI trial showed solid evidence that RIC does not provide protection after 1 year in patients with ST-elevation myocardial infarction treated with primary percutaneous coronary intervention (7). It is currently proposed that this discrepancy between preclinical and clinical settings may be attributed to factors such as comorbidities and comedications (8), as well as sex and age (9, 10).
In the context of myocardial infarction and cardioprotection, aging is increasingly attracting attention, due to its rising prevalence. Over the past decades, life expectancy has been steadily increasing (11). It has been estimated that by 2050, a quarter of the global population will be over 65 years old, with the exception of Africa (12). Importantly, although life expectancy has increased, disease-free lifespan has not been markedly increased as compared with lifespan, thereby increasing the burden of age-related diseases, such as cancer, neurodegenerative and cardiovascular diseases (11). Indeed, aging is considered a significant risk factor for cardiovascular diseases (13, 14) and particularly, for MI (15). Accordingly, chronic low-grade inflammation associated with aging is known to promote endothelial and vascular smooth muscle dysfunction, thus exacerbating atherosclerosis and increasing the risk of plaque rupture and thrombus formation (16).
The hallmarks of myocardial aging may account for the reduced tolerance against myocardial I/R injury in preclinical studies (17) and thus, its understanding may enable the development of new and effective therapies to reduce cardiac damage after MI in the context of aging. This mini review aims to discuss potential cardioprotective approaches in the aging heart such as the use of senolytics, as well as therapeutic strategies that aim to decrease mitochondrial damage and reduce inflammation.
One of the key features of the aging heart is mitochondrial dysfunction, characterized by increased mitochondrial fragmentation, ROS production and mitochondrial permeability transition pore opening, as well as reduced mitochondrial biogenesis and electron transport chain activity (17–19). Furthermore, senescent cardiomyocytes also undergo DNA instability, mostly related with telomere shortening and mutations of nuclear and mitochondrial DNA (20). Altered proteostasis is another important feature of aging cardiomyocytes, whereby a reduction in protein synthesis/turnover is associated with a decreased efficiency of the ubiquitin-proteasome system and autophagy (21). The main features of the aged cardiomyocyte are summarized in Figure 1A. In addition, Ruiz-Meana et al. provide an in depth analysis and discussion of the aged cardiomyocyte in the context of cardioprotection (17).
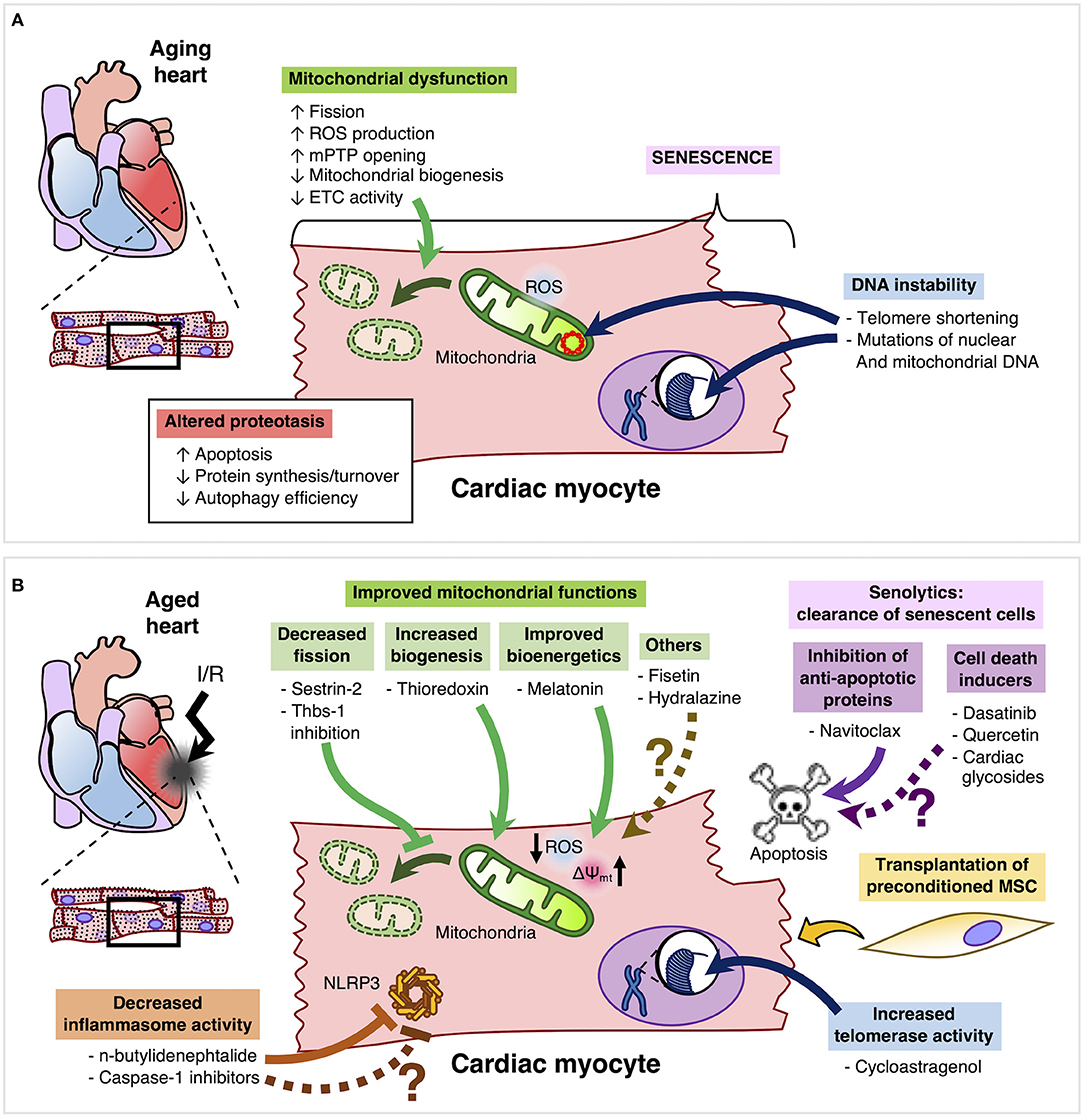
Figure 1. (A) Aging cardiomyocytes are mainly characterized by mitochondrial dysfunction, senescence, altered proteostasis and DNA instability. (B) Improvement of mitochondrial dysfunction in aged cardiomyocytes may be achieved by inhibition of Thbs-1 and Sestrin-2, which can decrease Drp-1-mediated mitochondrial fission. Moreover, thioredoxin can promote mitochondrial biogenesis and melatonin can improve bioenergetics by reducing ROS production and improving mitochondrial membrane potential. Other compounds such as Fisetin and hydralazine can confer cardioprotection by reducing mitochondrial dysfunction, but whether these compounds can effectively limit the infarct size and improve left ventricular function of the aging heart remains to be elucidated. Senolytics can clear senescent cells by inhibiting anti-apoptotic proteins. However, only Navitoclax has been tested in the aged heart. However, other senolytics such as Dasatinib, Quercetin, and cardiac glycosides are yet to be evaluated in the settings of cardioprotection and aging. Furthermore, activation of telomerase in order to preserve telomere length and integrity is a promising candidate for cardioprotection in the aging heart. Another emergent target is to inhibit the inflammasome by compounds such as n-butylidenephtalide. Finally, the transplantation of preconditioned mesenchymal cells may have an important therapeutic role in the protection of the aged myocardium.
Senescent cells increase in aged tissues, which has been associated with the progression of age-related diseases (22). In this context, senescence markers are augmented in aged cardiomyocytes (23), which has been linked to higher risk of cardiovascular diseases (24). Senolytics are agents that can selectively target pro-survival proteins of senescent cells, inducing cell death. Regarding the heart, there are three major senolytics that have been widely studied in vivo and in vitro; Dasatinib, Quercetin, and Navitoclax (24). These senolytics have been shown to improve vascular function (25, 26). Importantly, a study performed by Walaszczyk et al. showed that oral administration of navitoclax to aged mice before in vivo myocardial infarction reduced morality, as well as age-related myocardial remodeling and improved left ventricular function (27).
Despite the beneficial effects of senolytics on a preclinical level, there are only two clinical studies showing that these drugs can decrease senescent cells in humans (28, 29). Justice et al., showed that a combination of Dasatinib + Quercetin in patients with idiopathic pulmonary fibrosis can improve physical function (29), while Hickson et al. observed that administration of Dasatinib + Quercetin in patients with diabetic kidney disease elicited a decrease in senescent cells of adipose tissue (28). Despite showing efficacy in treatment of myocardial infarction in the aged heart, Navitoclax has yet to be tested in humans with cardiovascular diseases. Overall, senolytics are a promising pharmacological approach, which appears to effectively reduce senescent cells in humans, but additional preclinical evidence is warranted before they can be translated into the clinical setting of cardioprotection in the elder population.
Currently, there are ongoing efforts to identify new or old compounds that can decrease the number of senescent cells (30). For instance, cardiac glycosides (CGs), have been recently identified as potential senolytic compounds that can successfully decrease the number of senescent cells in the context of cancer in old mice (31) and senescence-induced lung fibrosis (32) in mice. Both of these studies showed that the senolytic effect exerted by CGs depends on the inhibition of Na+/K+ ATPase pump (31, 32). Nonetheless, despite the novelty of using these old drugs to clear senescent cells, there are currently no studies showing whether CGs can protect the adult or aged myocardium from I/R injury and thus, this research field is still in preliminary stages in the context of cardioprotection. Moreover, Ouabain is a cardiac glycoside that has been shown to impair growth of proliferative cells in human epithelial cells in a context of oncogene-induced senescence (33) and thus, the specificity of these drugs for senescent cells must be thoroughly evaluated before considering their use in MI.
Recently, an interesting non-pharmacological approach has been investigated. Amor et al. showed that the urokinase-type plasminogen activator receptor (uPAR)-a cell-surface marker that is overexpressed in senescent cells- is a target for chimeric antigen receptor (CAR) T cells, which can clear senescent cells in mice both in vitro and in vivo in the context of lung adenocarcinoma and liver fibrosis (34), thus providing a promising proof of concept of therapeutic agents for the treatment of senescence-associated diseases, such as MI of the aging heart. However, concerns have been raised over the adverse effects of this therapy. The most frequently reported side effects are cytokine release syndrome and neurotoxicity, but cardiovascular toxic effects, such as systolic dysfunction, arrhythmias, or hypotension have also been reported and thereby, a thorough assessment of cardiac imaging and evaluation of reliable biomarkers is needed to identify at-risk population and establish the safety of this therapy (35).
The mitochondria have been identified as an important target to reduce myocardial ischemia/reperfusion injury in the aged heart (36). The mitochondria in aging cardiomyocytes shows elevated ROS production, higher fragmentation and reduced biogenesis, thus producing mitochondrial dysfunction, which can contribute to the increased susceptibility of the aged heart to ischemic injury (17). Therefore, therapies targeting the mitochondria are an attractive area of research in cardioprotection.
It has been reported that Thrombospondin-1 (Thbs-1), which is known to induce ROS production after I/R, regulates Drp-1 dependent mitochondrial fission after MI in both young and old mice (37). Thus, inhibition of Thbs-1 levels after I/R may reduce mitochondrial fission and ROS production, thereby protecting the aging heart against I/R injury. Furthermore, it has been shown that overexpression of thioredoxin -a cytosolic redox protein with reducing properties- in aged mice subjected to in vivo I/R, improved mitochondrial function and biogenesis, as well as reducing the infarct size and left ventricular dysfunction (38). Another potential target for cardioprotection is Sestrin2. This protein is induced by stress and forms a complex with AMP-activated protein kinase (AMPK), thus promoting autophagy. Importantly, its cellular levels drop with age, and it's been shown that a Sestrin2-KO promotes mitochondrial fission and damage after myocardial I/R in mice (39). Moreover, overexpression of Sestrin2 in aged mice hearts improved the heart's response to MI (39). Finally, melatonin is a molecule secreted by the pineal gland in humans. It exerts antioxidant and anti-inflammatory properties that may play a cardioprotective role (40). A study in aged rats showed that administration of melatonin and nicotinamide mononucleotide reduces the infarct size and improves left ventricular function after I/R injury by reducing mitochondrial ROS and improving mitochondrial membrane potential (41). Moreover, it has been suggested that melatonin protects against I/R injury via the RISK and SAFE pathways (42). Interestingly, a small clinical trial has provided proof of concept that the timing of administration of melatonin in infarcted patients undergoing percutaneous coronary intervention is key for its effectiveness (43). However, this observation needs to be confirmed in large randomized controlled clinical trials.
There are multiple strategies and treatments that target the mitochondria to achieve cardioprotection that have not been yet studied in the context of aging. We will discuss two of them: fisetin and hydralazine. Fisetin is a flavonoid found in many fruits with senolytic properties (44). Recently it's been shown that it can also confer protection in I/R settings. A preliminary study showed that fisetin can inhibit H9c2 cardiomyoblast apoptosis after hypoxia/reoxygenation, as well as to reduce mitochondrial ROS production and inhibit the activation of caspases 3 and 9 (45). In addition, adult rats pre-treated with fisetin and subjected to ex vivo I/R showed a reduced infarct size, reduced mitochondrial oxidative stress and improved mitochondrial structure and function by a mechanism that appears to involve inhibition of GSK3β activity (46). Hydralazin, is an FDA approved drug currently used as an anti-hypertensive agent and in the treatment of chronic heart failure. Recently, it has been described that it can confer cardioprotection in mice using in vivo, ex vivo and in vitro experimental models of I/R injury via inhibition of Drp-1-mediated mitochondrial fission (47). These two molecules have in common their capacity to protect the heart from I/R injury by improving mitochondrial function and thus, these therapeutic agents are promising candidates to protect the aging heart from MI. Therefore, future studies should explore this possibility.
During MI, a pathogen/antigen-independent inflammatory response, known as sterile inflammation, takes place (48). Due to the rupture in the cellular structure that occurs during MI, damage-associated molecular patterns (DAMPS) mediators are released and are recognized by pattern recognition receptors (PRRs), which in turn mediate the initiation of the inflammatory response (49). NLPR3 inflammasome is a multiprotein complex formed by the activation of PRRs, thereby increasing the production and release of proinflammatory cytokines via activation of caspase-1 (50). NLPR3 has attracted attention as a mediator of damage produced by I/R injury (51). Mastracola et al. tested the role of NLPR3 in C57B1 male mice fed with a high fat high fructose diet (HFHF) using an ex vivo I/R model. The authors found that the HFHF diet upregulated NLPR3 protein content, and this elevation downregulated the RISK/HIF-2alpha pathways (51–53). Furthermore, NLPR3 has also been proposed as a target in the aging myocardium. Lee et al. has explored this hypothesis in a recent study where they use n-butylidenephthalide as a preconditioning agent and tested the capacity of human adipose–derived stem cell (hADSC) engraftment in the recovery of infarcted myocardium of young and old Wistar rats (54). The study showed NLPR3 activity and ROS production was significantly increased in old rats after MI, as compared with young rats. Transplantation of hADSC reduced NLPR3 activation and ROS levels in both young and old rats, but this response was significantly more effective in young rats. The authors discovered that the NLPR3 inflammasome mediates the difference of the response to the engraftments in old and young rats. They also reported that n-butylidenephthalide reversed the complex microenvironment that impedes engraftment success (54).
Interestingly, pharmacological inhibition of caspase-1 reduced the infarct size in isolated rat hearts (55). Also, caspase-1 inhibition was also shown to provide additional protection when combined with remote ischemic preconditioning in rats subjected to in vivo myocardial infarction (56). This study is particularly interesting, since these experiments were performed using a co-medication model, consisting of co-administration with an opioid agonist, heparin and a platelet-inhibitor to mimic a clinical setting, thereby highlighting the translational value of this cardioprotective therapy (56). This promising evidence merits further research to evaluate whether this approach may have a significant impact in the cardioprotection of the aged heart.
Telomeres are repeated hexanucleotide sequences at the end of eukaryotic chromosomes. Their presence is associated with DNA protection during cell division. Division of the cell as well as oxidative stress shortens these structures, leading the cell to a senescent state or apoptosis (57). Telomere length has been associated with coronary artery disease and therefore, it has been proposed as a biomarker for cardiovascular diseases (58). Telomerase is a key regulator of telomere length and integrity (20) and as such, has gained attention for its potential benefits in age-related cardiovascular diseases. For instance, absence of telomerase has been associated with increased susceptibility to ischemic injury (59). By the same token, overexpression of telomerase can confer cardioprotection in mice hearts (60). Furthermore, overexpression of growth differentiation factor 11 (GDF11) in mice subjected to in vivo I/R injury showed a reduced infarct size, activation of telomerase, longer telomeres and increased mitochondrial biogenesis (61). In a clinical context, Gupta et al. have published a pilot study that proposes telomere length as a screening tool for patients with acute myocardial infarction (62). Their study revealed that telomere length was reduced in young patients without risk factors who underwent MI, as compared with young patients without MI (62). Moreover, Maier et al. have published the design of a pilot trial that uses the telomerase activator TA-65MD, which is a purified and encapsulated form of cycloastragenol, in elderly patients with acute coronary syndrome (63). The aim of this study is to assess whether this therapy can reverse inmmunosenescence, which is associated to the pathophysiological progression of coronary artery disease. The results to this trial are scheduled to be published in 2021 (63). Although more preclinical data is required to accurately assess whether this therapeutic strategy can reduce cardiac damage in aging, telomerase appears to be an essential player in cardioprotection.
In addition to the aforementioned strategies, there are other approaches that may wield cardioprotective effects in aging conditions. An interesting study has suggested that necroptosis may contribute to ischemic susceptibility in the aging heart and that metformin reduces necroptosis elicited by alterations of autophagy during myocardial aging (64). Another potential therapeutic target may be the JAK/STAT pathway. It has been reported that inhibition of JAK in old mice has been linked with reduced senescence-associated secretory phenotype and frailty (65). Moreover, deletion of Kcne4 in old mice has been found to sex-specifically induce arrhythmias by a mechanism that involves defective signaling of the RISK/SAFE pathway mediated by testosterone (66). Furthermore, it has been observed that the JAK/STAT pathway is activated in aging humans, which is associated with chronic inflammation and increased cardiovascular risk (67). However, whether targeting the JAK/STAT pathway may restore the effectiveness of protective strategies in the context of MI remains to be elucidated.
Aging has also been described to alter mesenchymal stem cell function, which impairs their cardioprotective effects (68). It has been recently identified that miR-155-5p can regulate mesenchymal stem cell senescence (69). This study demonstrated that inhibition of miR-155-5p reduced senescence in aged mesenchymal stem cell, thus improving their ability to protect the aged heart from MI in mice (69).
Finally, a study by Crewe et al. revealed that adipocytes can secrete small extracellular vesicles that can transport damaged mitochondria, which can precondition the heart by inducing a short period of mitochondrial oxidative stress, which eventually leads to an antioxidant effect that can effectively protect the heart from MI (70). While this effect has not been evaluated in the aged heart, protection elicited by transport of mitochondria by small extracellular vesicles via hormesis may be an effective therapy, given the importance of this organelle in the aging myocardium.
The aged heart is more susceptible to I/R injury. Moreover, multiple cardioprotective maneuvers have been shown to be ineffective under these conditions, highlighting the need to develop therapies that can provide clinical efficacy in the reduction of infarct size. Clearance of senescent cells by the senolytic drug navitoclax has shown clear protection of aged hearts from MI, but the current evidence is still preliminary and more senolytics need to be tested to establish the real potential of these agents. Moreover, the clinical evidence regarding the use of these drugs is still in early stages, especially since there are no studies testing their effects in MI patients. Cardioprotective approaches targeting the mitochondria in the aged heart are promising, although the current evidence is mainly centered in preclinical studies, suggesting that the translational value of these therapies remains to be tested in clinical settings. Similarly, targeting of the inflammasome or telomerase activity are research areas of interest, but further studies are certainly required to evaluate whether these therapeutic strategies can be translated from bench to bedside. Currently, cardioprotection in aging can be tested using primary cells, ex vivo and in vivo animal models to evaluate the effect of protective drugs or conditioning maneuvers (17). The use of cell lines may be useful to assess stress-induced premature senescence and thus generate a preliminary proof-of-concept regarding a potential senolytic therapy, but while this allows to avoid the use of animals, this approach only elicits a senescence-like phenotype (71). Therefore, this model may have a low translational value. Regarding cardioprotection research, preclinical models are solid in the early stages, but clinical studies are strong in the follow-up process (72). In this context, Heusch has proposed to use integrative large animal models and study the whole process: from acute myocardial infarction to a 12 months follow-up (72) and this idea is certainly valid for the study of cardioprotection in aging as well. Furthermore, it's crucial to consider the importance of generating reliable evidence to drive cardioprotection research forward by adhering to proposed guidelines to ensure rigor and reproducibility, such as the ones presented by Bøtker et al. (73). Finally, while the potential therapies we have reviewed are promising (Figure 1B), it is important to acknowledge a lack of studies in this field. For instance, while aging can impair the cardioprotective effect of conditioning strategies in preclinical studies (8) a retrospective study showed that age did not disrupt cardioprotection in coronary artery bypass graft patients with or without RIC (74). Thus, while the evidence showing aging as a confounding factor in cardioprotection is thorough, clinical evidence is mainly obtained from retrospective secondary analyses (8), highlighting the distance between bench and bedside, which is barrier that needs to be overcome to protect the elderly from myocardial infarction.
MD-V, ÚZ-C, AR-R, NH-Z, IP, RB-S, and JR participated in the conception and design of the manuscript and made significant contributions to the analysis of the evidence discussed. All authors wrote the manuscript and gave the final approval of the submitted manuscript.
This work was supported by CONICYT FONDAP grant 15130011 (to JR and RB-S), FONDECYT Iniciación 11181000 (to JR) and 11201267 (to RB-S), Subvención a la Instalación PAI 77170004 (to RB-S), Universidad de Chile grants ABCvital 02-2018 and U-Inicia UI-006/19 (to RB-S), and ANID fellowship 21191341 (to MD-V).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Hausenloy DJ, Garcia-Dorado D, Botker HE, Davidson SM, Downey J, Engel FB, et al. Novel targets and future strategies for acute cardioprotection: position paper of the European Society of Cardiology Working Group on cellular biology of the heart. Cardiovasc Res. (2017) 113:564–85. doi: 10.1093/cvr/cvx049
2. Jovanovic A. Cardioprotective signalling: past, present and future. Eur J Pharmacol. (2018) 833:314–9. doi: 10.1016/j.ejphar.2018.06.029
3. Rossello X, Yellon DM. The RISK pathway and beyond. Basic Res Cardiol. (2018) 113:2. doi: 10.1007/s00395-017-0662-x
4. Hadebe N, Cour M, Lecour S. The SAFE pathway for cardioprotection: is this a promising target? Basic Res Cardiol. (2018) 113:9. doi: 10.1007/s00395-018-0670-5
5. Heusch G. Myocardial ischaemia-reperfusion injury and cardioprotection in perspective. Nat Rev Cardiol. (2020) 17:773–89. doi: 10.1038/s41569-020-0403-y
6. Rossello X, Yellon DM. Cardioprotection: the disconnect between bench and bedside. Circulation. (2016) 134:574–5. doi: 10.1161/CIRCULATIONAHA.116.022829
7. Hausenloy DJ, Kharbanda RK, Moller UK, Ramlall M, Aaroe J, Butler R, et al. Effect of remote ischaemic conditioning on clinical outcomes in patients with acute myocardial infarction (CONDI-2/ERIC-PPCI): a single-blind randomised controlled trial. Lancet. (2019) 394:1415–24. doi: 10.1016/S0140-6736(19)32039-2
8. Kleinbongard P, Botker HE, Ovize M, Hausenloy DJ, Heusch G. Co-morbidities and co-medications as confounders of cardioprotection-Does it matter in the clinical setting? Br J Pharmacol. (2020) 177:5252–69. doi: 10.1111/bph.14839
9. Heinen A, Behmenburg F, Aytulun A, Dierkes M, Zerbin L, Kaisers W, et al. The release of cardioprotective humoral factors after remote ischemic preconditioning in humans is age- and sex-dependent. J Transl Med. (2018) 16:112. doi: 10.1186/s12967-018-1480-0
10. Ferdinandy P, Hausenloy DJ, Heusch G, Baxter GF, Schulz R. Interaction of risk factors, comorbidities, and comedications with ischemia/reperfusion injury and cardioprotection by preconditioning, postconditioning, and remote conditioning. Pharmacol Rev. (2014) 66:1142–74. doi: 10.1124/pr.113.008300
11. Partridge L, Deelen J, Slagboom PE. Facing up to the global challenges of ageing. Nature. (2018) 561:45–56. doi: 10.1038/s41586-018-0457-8
12. Khan HTA. Population ageing in a globalized world: risks and dilemmas? J Eval Clin Pract. (2019) 25:754–60. doi: 10.1111/jep.13071
13. Nanayakkara S, Marwick TH, Kaye DM. The ageing heart: the systemic and coronary circulation. Heart. (2018) 104:370–6. doi: 10.1136/heartjnl-2017-312114
14. Ward SA, Parikh S, Workman B. Health perspectives: international epidemiology of ageing. Best Pract Res Clin Anaesthesiol. (2011) 25:305–17. doi: 10.1016/j.bpa.2011.05.002
15. Shih H, Lee B, Lee RJ, Boyle AJ. The aging heart and post-infarction left ventricular remodeling. J Am Coll Cardiol. (2011) 57:9–17. doi: 10.1016/j.jacc.2010.08.623
16. Liberale L, Montecucco F, Tardif JC, Libby P, Camici GG. Inflamm-ageing: the role of inflammation in age-dependent cardiovascular disease. Eur Heart J. (2020) 41:2974–82. doi: 10.1093/eurheartj/ehz961
17. Ruiz-Meana M, Bou-Teen D, Ferdinandy P, Gyongyosi M, Pesce M, Perrino C, et al. Cardiomyocyte ageing and cardioprotection: consensus document from the ESC working groups cell biology of the heart and myocardial function. Cardiovasc Res. (2020) 116:1835–49. doi: 10.1093/cvr/cvaa132
18. Boengler K, Kosiol M, Mayr M, Schulz R, Rohrbach S. Mitochondria and ageing: role in heart, skeletal muscle and adipose tissue. J Cachexia Sarcopenia Muscle. (2017) 8:349–69. doi: 10.1002/jcsm.12178
19. Fernandez-Sanz C, Ruiz-Meana M, Castellano J, Miro-Casas E, Nunez E, Inserte J, et al. Altered FoF1 ATP synthase and susceptibility to mitochondrial permeability transition pore during ischaemia and reperfusion in aging cardiomyocytes. Thromb Haemost. (2015) 113:441–51. doi: 10.1160/TH14-10-0901
20. Moslehi J, DePinho RA, Sahin E. Telomeres and mitochondria in the aging heart. Circ Res. (2012) 110:1226–37. doi: 10.1161/CIRCRESAHA.111.246868
21. Henning RH, Brundel B. Proteostasis in cardiac health and disease. Nat Rev Cardiol. (2017) 14:637–53. doi: 10.1038/nrcardio.2017.89
22. de Magalhaes JP, Passos JF. Stress, cell senescence and organismal ageing. Mech Ageing Dev. (2018) 170:2–9. doi: 10.1016/j.mad.2017.07.001
23. Anderson R, Lagnado A, Maggiorani D, Walaszczyk A, Dookun E, Chapman J, et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 2019 38. doi: 10.15252/embj.2018100492
24. Dookun E, Passos JF, Arthur HM, Richardson GD. Therapeutic potential of senolytics in cardiovascular disease. Cardiovasc Drugs Ther. (2020). doi: 10.1007/s10557-020-07075-w. [Epub ahead of print].
25. Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, Thalji NM, et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell. (2016) 15:973–7. doi: 10.1111/acel.12458
26. Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science. (2016) 354:472–7. doi: 10.1126/science.aaf6659
27. Walaszczyk A, Dookun E, Redgrave R, Tual-Chalot S, Victorelli S, Spyridopoulos I, et al. Pharmacological clearance of senescent cells improves survival and recovery in aged mice following acute myocardial infarction. Aging Cell. (2019) 18:e12945. doi: 10.1111/acel.12945
28. Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, Hashmi SK, et al. Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine. (2019) 47:446–56. doi: 10.1016/j.ebiom.2019.08.069
29. Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, Hashmi SK, et al. Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. EBioMedicine. (2019) 40:554–63. doi: 10.1016/j.ebiom.2018.12.052
30. Robbins PD, Jurk D, Khosla S, Kirkland JL, LeBrasseur NK, Miller JD, et al. Senolytic drugs: reducing senescent cell viability to extend health span. Annu Rev Pharmacol Toxicol. (2021) 61:779–803. doi: 10.1146/annurev-pharmtox-050120-105018
31. Guerrero A, Herranz N, Sun B, Wagner V, Gallage S, Guiho R, et al. Cardiac glycosides are broad-spectrum senolytics. Nat Metab. (2019) 1:1074–88. doi: 10.1038/s42255-019-0122-z
32. Triana-Martinez F, Picallos-Rabina P, Da Silva-Alvarez S, Pietrocola F, Llanos S, Rodilla V, et al. Identification and characterization of cardiac glycosides as senolytic compounds. Nat Commun. (2019) 10:4731. doi: 10.1038/s41467-019-12888-x
33. Lallet-Daher H, Wiel C, Gitenay D, Navaratnam N, Augert A, Le Calve B, et al. Potassium channel KCNA1 modulates oncogene-induced senescence and transformation. Cancer Res. (2013) 73:5253–65. doi: 10.1158/0008-5472.CAN-12-3690
34. Amor C, Feucht J, Leibold J, Ho YJ, Zhu C, Alonso-Curbelo D, et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature. (2020) 583:127–32. doi: 10.1038/s41586-020-2403-9
35. Ghosh AK, Chen DH, Guha A, Mackenzie S, Walker JM, Roddie C, et al. Cell therapy-related cardiovascular outcomes and management: systemic disease or direct cardiotoxicity? JACC CardioOncol. (2020) 2:97–109. doi: 10.1016/j.jaccao.2020.02.011
36. Lesnefsky EJ, He D, Moghaddas S, Hoppel CL. Reversal of mitochondrial defects before ischemia protects the aged heart. FASEB J. (2006) 20:1543–5. doi: 10.1096/fj.05-4535fje
37. Kelm NQ, Beare JE, Weber GJ, LeBlanc AJ. Thrombospondin-1 mediates Drp-1 signaling following ischemia reperfusion in the aging heart. FASEB Bioadv. (2020) 2:304–14. doi: 10.1096/fba.2019-00090
38. Subramani J, Kundumani-Sridharan V, Das KC. Thioredoxin protects mitochondrial structure, function and biogenesis in myocardial ischemia-reperfusion via redox-dependent activation of AKT-CREB- PGC1alpha pathway in aged mice. Aging (Albany NY). (2020) 12:19809–27. doi: 10.18632/aging.104071
39. Quan N, Sun W, Wang L, Chen X, Bogan JS, Zhou X, et al. Sestrin2 prevents age-related intolerance to ischemia and reperfusion injury by modulating substrate metabolism. FASEB J. (2017) 31:4153–67. doi: 10.1096/fj.201700063R
40. Fu Z, Jiao Y, Wang J, Zhang Y, Shen M, Reiter RJ, et al. Cardioprotective role of melatonin in acute myocardial infarction. Front Physiol. (2020) 11:366. doi: 10.3389/fphys.2020.00366
41. Hosseini L, Vafaee MS, Badalzadeh R. Melatonin and nicotinamide mononucleotide attenuate myocardial ischemia/reperfusion injury via modulation of mitochondrial function and hemodynamic parameters in aged rats. J Cardiovasc Pharmacol Ther. (2020) 25:240–50. doi: 10.1177/1074248419882002
42. Lochner A, Marais E, Huisamen B. Melatonin and cardioprotection against ischaemia/reperfusion injury: what's new? A review J Pineal Res. (2018) 65:e12490. doi: 10.1111/jpi.12490
43. Dominguez-Rodriguez A, Abreu-Gonzalez P, de la Torre-Hernandez JM, Consuegra-Sanchez L, Piccolo R, Gonzalez-Gonzalez J, et al. Usefulness of early treatment with melatonin to reduce infarct size in patients with ST-segment elevation myocardial infarction receiving percutaneous coronary intervention (From the melatonin adjunct in the acute myocardial infarction treated with angioplasty trial). Am J Cardiol. (2017) 120:522–6. doi: 10.1016/j.amjcard.2017.05.018
44. Yousefzadeh MJ, Zhu Y, McGowan SJ, Angelini L, Fuhrmann-Stroissnigg H, Xu M, et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine. (2018) 36:18–28. doi: 10.1016/j.ebiom.2018.09.015
45. Rodius S, de Klein N, Jeanty C, Sanchez-Iranzo H, Crespo I, Ibberson M, et al. Fisetin protects against cardiac cell death through reduction of ROS production and caspases activity. Sci Rep. (2020) 10:2896. doi: 10.1038/s41598-020-59894-4
46. Shanmugam K, Ravindran S, Kurian GA, Rajesh M. Fisetin confers cardioprotection against myocardial ischemia reperfusion injury by suppressing mitochondrial oxidative stress and mitochondrial dysfunction and inhibiting glycogen synthase kinase 3beta activity. Oxid Med Cell Longev. (2018) 2018:9173436. doi: 10.1155/2018/9173436
47. Kalkhoran SB, Kriston-Vizi J, Hernandez-Resendiz S, Crespo-Avilan GE, Rosdah AA, Lees JG, et al. Hydralazine protects the heart against acute ischemia/reperfusion injury by inhibiting Drp1-mediated mitochondrial fission. Cardiovasc Res. (2021). doi: 10.1093/cvr/cvaa343. [Epub ahead of print].
48. Toldo S, Mezzaroma E, Mauro AG, Salloum F, Van Tassell BW, Abbate A. The inflammasome in myocardial injury and cardiac remodeling. Antioxid Redox Signal. (2015) 22:1146–61. doi: 10.1089/ars.2014.5989
49. Chen GY, Nunez G. Sterile inflammation: sensing and reacting to damage. Nat Rev Immunol. (2010) 10:826–37. doi: 10.1038/nri2873
50. Toldo S, Abbate A. The NLRP3 inflammasome in acute myocardial infarction. Nat Rev Cardiol. (2018) 15:203–14. doi: 10.1038/nrcardio.2017.161
51. Mastrocola R, Collino M, Penna C, Nigro D, Chiazza F, Fracasso V, et al. Maladaptive modulations of NLRP3 inflammasome and cardioprotective pathways are involved in diet-induced exacerbation of myocardial ischemia/reperfusion injury in mice. Oxid Med Cell Longev. (2016) 2016:3480637. doi: 10.1155/2016/3480637
52. Gao R, Zhao H, Wang X, Tang B, Cai Y, Zhang X, et al. Mild hypothermia therapy lowers the inflammatory level and apoptosis rate of myocardial cells of rats with myocardial ischemia-reperfusion injury via the NLRP3 inflammasome pathway. Comput Math Methods Med. (2021) 2021:6415275. doi: 10.1155/2021/6415275
53. Penna C, Aragno M, Cento AS, Femmino S, Russo I, Bello FD, et al. Ticagrelor conditioning effects are not additive to cardioprotection induced by direct NLRP3 inflammasome inhibition: role of RISK, NLRP3, and redox cascades. Oxid Med Cell Longev. (2020) 2020:9219825. doi: 10.1155/2020/9219825
54. Lee TM, Harn HJ, Chiou TW, Chuang MH, Chen CH, Chuang CH, et al. Host pre-conditioning improves human adipose-derived stem cell transplantation in ageing rats after myocardial infarction: role of NLRP3 inflammasome. J Cell Mol Med. (2020) 24:12272–84. doi: 10.1111/jcmm.15403
55. Do Carmo H, Arjun S, Petrucci O, Yellon DM, Davidson SM. The caspase 1 inhibitor VX-765 Protects the Isolated Rat Heart via the RISK Pathway. Cardiovasc Drugs Ther. (2018) 32:165–8. doi: 10.1007/s10557-018-6781-2
56. He Z, Davidson SM, Yellon DM. The importance of clinically relevant background therapy in cardioprotective studies. Basic Res Cardiol. (2020) 115:69. doi: 10.1007/s00395-020-00830-y
57. von Zglinicki T. Role of oxidative stress in telomere length regulation and replicative senescence. Ann N Y Acad Sci. (2000) 908:99–110. doi: 10.1111/j.1749-6632.2000.tb06639.x
58. Vecoli C, Borghini A, Andreassi MG. The molecular biomarkers of vascular aging and atherosclerosis: telomere length and mitochondrial DNA(4977) common deletion. Mutat Res Rev Mutat Res. (2020) 784:108309. doi: 10.1016/j.mrrev.2020.108309
59. Ait-Aissa K, Heisner JS, Norwood Toro LE, Bruemmer D, Doyon G, Harmann L, et al. Telomerase deficiency predisposes to heart failure and ischemia-reperfusion injury. Front Cardiovasc Med. (2019) 6:31. doi: 10.3389/fcvm.2019.00031
60. Bar C, Bernardes de. Jesus B, Serrano R, Tejera A, Ayuso E, Jimenez V, et al. Telomerase expression confers cardioprotection in the adult mouse heart after acute myocardial infarction. Nat Commun. (2014) 5:5863. doi: 10.1038/ncomms6863
61. Chen L, Luo G, Liu Y, Lin H, Zheng C, Xie D, et al. Growth differentiation factor 11 attenuates cardiac ischemia reperfusion injury via enhancing mitochondrial biogenesis and telomerase activity. Cell Death Dis. (2021) 12:665. doi: 10.1038/s41419-021-03954-8
62. Gupta MD, Miglani M, Bansal A, Jain V, Arora S, Kumar S, et al. Telomere length in young patients with acute myocardial infarction without conventional risk factors: a pilot study from a South Asian population. Indian Heart J. (2020) 72:619–22. doi: 10.1016/j.ihj.2020.11.006
63. Maier R, Bawamia B, Bennaceur K, Dunn S, Marsay L, Amoah R, et al. Telomerase activation to reverse immunosenescence in elderly patients with acute coronary syndrome: protocol for a randomized pilot trial. JMIR Res Protoc. (2020) 9:e19456. doi: 10.2196/19456
64. Li C, Mu N, Gu C, Liu M, Yang Z, Yin Y, et al. Metformin mediates cardioprotection against aging-induced ischemic necroptosis. Aging Cell. (2020) 19:e13096. doi: 10.1111/acel.13096
65. Xu M, Tchkonia T, Ding H, Ogrodnik M, Lubbers ER, Pirtskhalava T, et al. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. Proc Natl Acad Sci U S A. (2015) 112:E6301–10. doi: 10.1073/pnas.1515386112
66. Hu Z, Wei W, Zhou L, Chen M, Abbott GW. Kcne4 deletion sex-specifically predisposes to cardiac arrhythmia via testosterone-dependent impairment of RISK/SAFE pathway induction in aged mice. Sci Rep. (2018) 8:8258. doi: 10.1038/s41598-018-26599-8
67. Shen-Orr SS, Furman D, Kidd BA, Hadad F, Lovelace P, Huang YW, et al. Defective signaling in the JAK-STAT pathway tracks with chronic inflammation and cardiovascular risk in aging humans. Cell Syst. (2016) 3:374–84 e4. doi: 10.1016/j.cels.2016.09.009
68. Liu Y, Liu T, Han J, Yang Z, Xue X, Jiang H, et al. Advanced age impairs cardioprotective function of mesenchymal stem cell transplantation from patients to myocardially infarcted rats. Cardiology. (2014) 128:209–19. doi: 10.1159/000360393
69. Hong Y, He H, Jiang G, Zhang H, Tao W, Ding Y, et al. miR-155-5p inhibition rejuvenates aged mesenchymal stem cells and enhances cardioprotection following infarction. Aging Cell. (2020) 19:e13128. doi: 10.1111/acel.13128
70. Crewe C, Funcke JB, Li S, Joffin N, Gliniak CM, Ghaben AL, et al. Extracellular vesicle-based interorgan transport of mitochondria from energetically stressed adipocytes. Cell Metab. (2021) 33:1853–68.e11. doi: 10.1016/j.cmet.2021.08.002
71. Debacq-Chainiaux F, Ben Ameur R, Bauwens E, Dumortier E, Toutfaire M, Toussaint O. Stress-induced (Premature) senescence. In: Rattan SIS, Hayflick L, editors. Cellular Ageing and Replicative Senescence. Cham: Springer International Publishing (2016). p. 243–62. doi: 10.1007/978-3-319-26239-0_13
72. Heusch G. Cardioprotection research must leave its comfort zone. Eur Heart J. (2018) 39:3393–5. doi: 10.1093/eurheartj/ehy253
73. Bøtker HE, Hausenloy D, Andreadou I, Antonucci S, Boengler K, Davidson SM, et al. Practical guidelines for rigor and reproducibility in preclinical and clinical studies on cardioprotection. Basic Res Cardiol. (2018) 113:39. doi: 10.1007/s00395-018-0696-8
Keywords: cardioprotection, aging, ischemia/reperfusion injury, mitochondria, senolytics
Citation: Díaz-Vesga MC, Zúñiga-Cuevas Ú, Ramírez-Reyes A, Herrera-Zelada N, Palomo I, Bravo-Sagua R and Riquelme JA (2021) Potential Therapies to Protect the Aging Heart Against Ischemia/Reperfusion Injury. Front. Cardiovasc. Med. 8:770421. doi: 10.3389/fcvm.2021.770421
Received: 03 September 2021; Accepted: 29 October 2021;
Published: 19 November 2021.
Edited by:
Stéphanie Barrere-Lemaire, INSERM U1191 Institut de Génomique Fonctionnelle (IGF), FranceReviewed by:
Pasquale Pagliaro, University of Turin, ItalyCopyright © 2021 Díaz-Vesga, Zúñiga-Cuevas, Ramírez-Reyes, Herrera-Zelada, Palomo, Bravo-Sagua and Riquelme. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jaime A. Riquelme, cmlxdWVsbWVAY2lxLnVjaGlsZS5jbA==
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.