 Weikang Bian
Weikang Bian Zhicheng Wang
Zhicheng Wang Chongxiu Sun2*
Chongxiu Sun2* Dai-Min Zhang
Dai-Min Zhang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 23 December 2021
Sec. Cardiovascular Biologics and Regenerative Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.765419
Calcified aortic valve disease (CAVD) was previously regarded as a passive process associated with valve degeneration and calcium deposition. However, recent studies have shown that the occurrence of CAVD is an active process involving complex changes such as endothelial injury, chronic inflammation, matrix remodeling, and neovascularization. CAVD is the ectopic accumulation of calcium nodules on the surface of the aortic valve, which leads to aortic valve thickening, functional stenosis, and ultimately hemodynamic disorders. CAVD has become an important cause of death from cardiovascular disease. The discovery of therapeutic targets to delay or block the progression of CAVD and the clinical application of transcatheter aortic valve implantation (TAVI) provide new ideas for the prevention and treatment of CAVD. This article summarizes the pathogenesis of CAVD and provides insight into the future directions of CAVD diagnosis and treatment.
Valvular heart disease decreases activity tolerance of physical function and longevity. The death rate of rheumatic heart disease has remained fairly static since 2000, while deaths from calcific aortic stenosis continued to rise over the past 20 years (1). Calcified aortic valve disease (CAVD) is a common valvular disease that progresses from early valvular sclerosis without hemodynamic influence to severe calcified aortic valve stenosis requiring valve replacement (2). The main characteristics are inflammation, fibrosis, and calcification (3). In developed countries, calcified aortic valve disease is the most common cause of adult aortic valve stenosis, and the prevalence increases non-linearly with age. Currently, CAVD is considered to be an active and adjustable process accompanied by initiation factors that can promote disease occurrence, including clinical and genetic susceptibility and imbalances in molecules and cellular pathways (4).
Cardiovascular disease-related studies have shown that the incidence of all-cause death from cardiovascular disease caused by aortic valve sclerosis has increased by 35% (5). The early occurrence of aortic valve calcification affects patient prognosis and increases the risk of valve replacement surgery (6). As the most effective treatment for heart valve disease is valve replacement surgery, thus far, there has been no drug to treat or slow disease progression. At present, there is a lack of effective drugs for CAVD, so surgical replacement of calcified or stenotic valves is the only effective treatment (7). It is very important to determine the severity of the disease, analyze the calcification of the aortic valve, and assess the risk of cardiovascular disease to understand the opportunities and methods to improve patient prognosis (2). Based on these issues, this paper reviews the immune molecular process of CAVD and the effects of mechanical pressure and flow rate on the development of CAVD.
According to the statistical committee of the American Heart Association (AHA) in the United States, the prevalence of CAVD in people over 65 years old is 20–30 and 48–57% in people over 85 years old; thus, CAVD has become the third most common cardiovascular disease after coronary heart disease and hypertension, and calcified aortic valve stenosis (CAS) has become the primary indication for aortic valve replacement (8). According to the results of a European Society of Cardiology (ESC) analysis, 81.9% of aortic stenosis in Europe is caused by calcification (9).
CAVD can be divided into primary and secondary CAVD. Primary CAVD is also known as senile aortic valve calcification or degenerative aortic valve calcification. Secondary CAVD calcification occurs on the basis of the original disease and is more common in rheumatic heart disease, infective endocarditis, dissecting aneurysm, congenital bicuspid valve, or single leaf aortic valve. Depending on the degree of aortic valve calcification, the condition can be divided into aortic valve calcification (AVC) and CAS. Under ultrasound evaluation, AVC shows valve thickening, strong echo shadows, valve orifice areas >3 cm2, cross valve blood flow rates <2.5 m/s, and unrestricted leaflet activity. With disease progression, in the context of AVC, the valve orifice area becomes narrower, the rate of cross valve blood flow becomes faster, and leaflet activity is limited; then, CAS occurs (10). Therefore, AVC is the early stage of CAS.
Macroscopically, a normal aortic valve can be divided into two margins, two sides, and three layers: the free margin and basal margin, the aortic surface and ventricular surface, and the ventricular layer, fibrous layer, and spongy layer (Figure 1). Microscopically, the valve is mainly composed of valve endothelial cells, valve interstitial cells, and extracellular matrix components, such as collagen, dextran, and elastin.
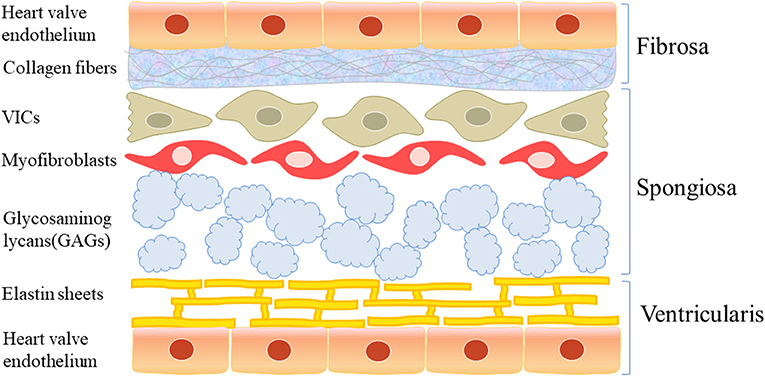
Figure 1. The structure of aortic valve. Macroscopically, the normal aortic valve is roughly divided into two edges, two sides, and three layers: free edge and basal edge, aortic surface and ventricular surface, ventricular layer, fibrous layer and sponge layer. Microscopically, it is mainly composed of valve endothelial cells and valve stromal cells and extracellular matrix such as collagen, glucan, and elastin.
Accumulating evidence has shown that calcified aortic valve disease is an active inflammatory disease caused by various factors. The pathological mechanism is complex and includes endothelial injury, inflammatory reactions, and oxidative stress that cause changes in cell composition in the valve, which is characterized by local thickening of the valve, the deposition of calcium salt, and the formation of calcium nodules, resulting in dysfunctional valve activity and hemodynamic changes.
The progression of calcified aortic valve disease is divided into two stages. The first stage is aortic valve sclerosis, in which extracellular matrix secretion is increased and some inflammatory cells infiltrate; the second stage is called aortic valve calcification, in which a large amount of calcium salts are deposited, forming calcium nodules, extracellular matrix components are abnormally increased, the valve leaflet is stiff and deformed, the number of interstitial cells in the valve is reduced, and neovascularization occurs (10–13).
CAVD is often associated with aging, degenerative changes, and calcium deposition and is regarded as an irreversible passive process. However, in recent years, it has been gradually recognized that CAVD is an active process involving endothelial injury, lipid infiltration, chronic inflammation, matrix remodeling, fibrosis, cell differentiation, progressive calcification, and neovascularization (Figure 2). Qiao et al. (14) identified severe hug genes and multiple potential miRNAs. These promising biomarkers and pathways for CAVD may provide novel molecular markers for diagnosis and targeted therapy. The pathological changes are similar to those of atherosclerosis in the early stage but may be similar to those of bone formation in the late stage.
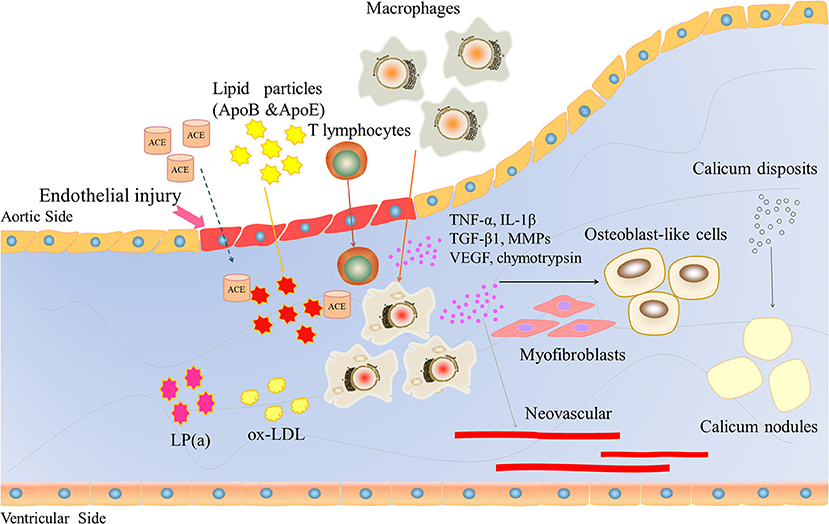
Figure 2. The role of inflammatory factors in regulating CAVD development and progression. Endothelial injury induces lipid particles (ApoB and ApoE), T lymphocytes and macrophages into the valve and releases large inflammatory factors, such as TNF-a, IL-1β, TGFβ1, and VEGF. VEGF induces neovascularization. These inflammatory factors lead to osteoblast-like cells, ultimately inducing calcium deposition.
The normal aortic valve is composed of three thin, smooth cusp valves. The specific physical and mechanical structure allows the valve to effectively adapt to the mechanical stress of blood flow. Therefore, under normal circumstances, most of the pressure on endothelial cells becomes laminar shear force to avoid endothelial cell damage. However, when the body exhibits abnormal conditions, such as a long-term increase in blood pressure or an increase in cardiac load caused by various reasons, a series of hemodynamic changes occur, such as turbulence, eddy currents, and other blood flow disorders. At this time, the stress on endothelial cells changes, and it is difficult for mechanical stress to become laminar shear force. Under the continuous impact of disturbed blood flow and the direct effect of mechanical stress, endothelial cells are damaged. The basement membrane is broken, damaging the protective barrier's function and resulting in dysfunction.
Structurally, the valves of patients with congenital bicuspid aortic valve malformation are composed of two cusp valves. Compared with the normal trilobal active valve, the bicuspid aortic valve will bear higher and unbalanced mechanical stress. Interestingly, in valve replacement and valve repair, most of the bicuspid aortic valves have calcifications. Studies have shown that on average, bicuspid aortic valve calcification occurs nearly 20 years earlier than tricuspid active valve calcification, and it very easily progresses to CAS (15). The most common site of aortic valve calcification is the fibrous layer of the aortic valve. The possible reason for this occurrence is that there is no blood flow in the mitral valve during diastole, while the fibrous layer of the aortic valve is adjacent to the aortic root, which is prone to hemodynamic changes such as turbulence. Common changes in the two valves are that the effect of laminar shear is reduced or even disappears, and the mechanical stress on endothelial cells increases (16). These findings provide the best evidence supporting the theory of endothelial injury.
Due to mechanical stress, after endothelial cell injury and basement membrane fracture, lipid components in the plasma will deposit at the fracture site and form scattered lipid points, and the main lipid components are apolipoproteins B and E. Renin-angiotensin converting enzyme (ACE) can be detected in the lesions, and ACE is usually associated with apolipoprotein B, which confirms that lipid particles bring ACE into the plasma (17). Endothelial cell injury and the formation of lipid points can trigger a chronic inflammatory response, attracting a large number of macrophages and a small number of T lymphocytes to gather near the lipid point through adhesion molecules. Immuno-histochemical staining and frozen section observation of diseased valves collected during surgery showed the presence of macrophages, which phagocytose mast cells formed by lipid transformation, scattered T lymphocytes, and a small number of smooth muscle cells (18, 19). A large amount of oxidized low-density lipoprotein (ox-LDL) and activated inflammatory cells could also be detected. The oxidized products are highly cytotoxic and stimulate inflammation and ossification in the later stage. Then, infiltrating inflammatory cells begin to act, releasing inflammatory factors, chemokines, growth factors, and cathepsin, as well as tumor necrosis factor α (TNF-α), transforming growth factor β1 (TGF-β1), vascular endothelial growth factor (VEGF), interleukin-1β (IL-1β), matrix metalloproteinase (MMPs), and chymotrypsin, which have been confirmed to be closely associated with calcification and stenosis of the aortic valve (20).
Cyclooxygenase (COX), also known as prostaglandin internal oxidase reductase, is a bifunctional enzyme with cyclooxygenase and catalase activities. It is the key enzyme that catalyzes the conversion of arachidonic acid to prostaglandins. Cyclooxygenase has two COX-1 and COX-2 isozymes. COX-1 mainly exists in blood vessels, the stomach, and kidney. It is involved in the regulation of vasomotor contraction, platelet aggregation, gastric mucosal blood flow, gastric mucus secretion, and renal function. COX1 is related to the protection of the gastrointestinal mucus membrane, the regulation of platelet aggregation, the regulation of peripheral vascular resistance and the regulation of renal blood flow distribution. COX2 is inducible. Various damaging chemical, physical and biological factors activate phospholipase A2 to hydrolyze cell membrane phospholipids to produce arachidonic acid. The latter generates prostaglandins through COX-2 catalytic oxygenation. In normal tissue/cells, the activity of COX-2 is very low. When cells are stimulated by inflammation, their expression level in inflammatory cells can increase to 10–80 times the normal level, resulting in an increase in the contents of PEG2, PGI2, and PGE1 in inflammatory sites and resulting in an inflammatory response and tissue damage. Vieceli Dalla Sega et al. (21) reported that COX-2 expression in human aortic valve interstitial cells (AVICs) is decreased in patients with calcific valves. Celecoxib, a COX-2 inhibitor, induced AVICs toward a myofibroblast phenotype and increased the expression of TGF-β to promote the formation of calcific nodules. These data support that celecoxib may facilitate CAVD progression. Treatment with celecoxib increased dystrophic calcification of aortic valve interstitial cells in vitro, while dimethyl celecoxib prevented TGF-β1-mediated calcification of AVICs (22).
Under the stimulation of inflammatory mediators, chemokines, growth factors, and cathepsins released by inflammatory cells, normal collagen fibers and elastic fibers in the aortic valve are destroyed. Furthermore, fibroblasts in the aortic valve are induced to differentiate into myofibroblasts and are activated for a long time, producing a large amount of collagen. The interstitial layer of the aortic valve was originally composed of type I and type III collagen. The regular arrangement of fibers and elastic fibers is destroyed. Furthermore, the ratio of collagen and elastin is disrupted, extracellular collagen levels are increased, and the matrix is reconstructed, which results in continuous thickening and weakened elasticity in the valve. Finally, the thin, elastic valve becomes fibrous, thickened, and hardened, and loses elasticity (17). MMPs and tissue inhibitors of matrix metalloproteinases (TIMPs) play important roles in this process. In normal tissues, there is a dynamic balance between MMPs and TIMPs to maintain extracellular matrix homeostasis. In the calcified valve interstitium, this balance is completely broken. The expression of MMP-1, MMP-2, and MMP-9 was significantly higher than that in the normal valve. In vitro cell experiments showed that the activity of MMP-2 was regulated by inflammatory factors (TNF-α, IL-1β) (20), which further verified that matrix remodeling was a pathological change in response to chronic inflammation.
Myofibroblasts in the aortic valve are phenotypically between smooth muscle cells and fibroblasts and have dual properties. Under the stimulation of TGF-β1, bone matrix protein and other cytokines, myofibroblasts differentiate into osteoblast-like cells, and then heterotopic calcification occurs in the valve. Active osteogenic changes and bone resorption can be observed. With disease development, even messy lamellar bone tissue can appear. Recently, a large-scale clinical study analyzed valves removed from CAS patients and found that more than 10% of patients had bone or cartilage formation in valves, and the ossification in cartilage was similar to the repair of normal fractures (21). In addition, the theory of myofibroblast differentiation into osteoblast-like cells has been confirmed at the cell culture and molecular levels. During the process of bone formation, a variety of signaling molecules and signal transduction pathways are involved. Bone matrix proteins, including osteopontin (OPN), bone sialoprotein (BSP), bone morphogenetic protein (BMP), osteocalcin (OCN), and alkaline phosphatase (ALP), are markers of osteoblast differentiation and maturation and are closely related to bone formation. These factors are significantly increased in the valve tissues of patients with CAVD (22), while some miRNAs that can inhibit the expression of OPN and BMP, such as miRNA-141, are significantly reduced in the diseased valve (23).
There are no microvessels in normal heart valves. The nutrients and oxygen needed by valve cells are mainly supplied by the diffusion of blood. However, when the valve exhibits inflammatory infiltration and ossification, the delicate balance between angiogenic factors and vascular inhibitors is broken, resulting in the formation of new microvessels. Studies have shown that when valve lesions occur, the expression of VEGF and its receptor, secreted protein acidic and rich in cysteine (SPARC) and bone connexin, is greatly increased, and the expression of the vascular growth inhibitor chondromodulin-1 is decreased, which eventually leads to the formation of new blood vessels (24–26). Neovascularization not only creates conditions for the infiltration of inflammatory cells and plasma lipids but also promotes endochondral osteogenesis in valves (17). However, we all know that there are no blood vessels in normal valves, so it is worth discussing where the endothelial cells in new microvessels come from. Human valve interstitial cells (VICs) are involved in aortic valve angiogenesis. These effects were inhibited by blocking VEGF-A with a blocking antibody or siRNA in a VEGF-A-dependent mechanism (27).
The aortic valve is mechanically stimulated by shear stress, strain, and tension/pressure in lobular tissue. The aortic valve mainly faces two types of pressure: oscillatory flow from the fibrous membrane surface of the valve and laminar flow from the ventricular muscle surface (28). The fibrous membrane of the valve is mainly subjected to oscillatory shear and is more prone to calcification than the ventricular muscle, which is sensitive to laminar shear. Simmons et al. (28) showed that endothelial cells passing through the fibrous membrane could express osteogenic factors, while cells in ventricular muscle could express calcification inhibitors. However, it is not clear whether the diversity of the valve endothelial cell (VEC) population and location and origin of VEC development affect mRNA expression under different biomechanical pressures. Using oscillatory flow and laminar flow models, Moore et al. (29) verified the specificity of aortic anatomy in different patients, and these differences could produce different flow patterns, which were related to the formation of local calcium nodules. These findings may explain the dramatic changes in flow rate in some populations, including patients with bicuspid aortic valve (BAV) who have a bicuspid rather than a normal tricuspid valve. Based on a computational growth and remodeling (G&R) framework, Sadrabadi et al. (30) developed an algorithm to evaluate the effects of aging and calcification on aortic valve dynamics. The patterns in geometric orifice area reduction and an increase valve stress during local and global growth and remodeling of the aortic valve.
In the pathological progress of calcific aortic valves, glycosaminoglycans (GAGs) shift from the valve spongiosa to the collagen I-rich fibrosa layer near calcified nodules. In a complete model of aortic valve disease, including endothelial cells, interstitial cells, and disease-like ECM, the glycosaminoglycan chondroitin sulfate (CS) increases the expression of pro-calcific genes and calcified nodule formation (31).
Fisher et al. (32) used the Flexcell tension system to place VICS in a tension environment, used TGF-β1 to treat calcified nodules, and compared the outcomes to the effect of no pressure (with TGF-β1 treatment). The finite element method also showed that mechanical tension has a positive regulatory effect on calcification (33). However, it is not clear whether mechanical pressure can initiate changes in valve shape and stiffness before osteogenesis or whether other small changes lead to changes in the flow pattern.
Clonal hematopoiesis of indeterminate potential (CHIP) is a blood disorder with some gene variants from a hematopoietic stem cell. CHIP itself does not denote a malignancy, but the individual with CHIP associates with higher risks of malignant blood disorders and atherosclerotic cardiovascular diseases. The threshold for CHIP was set a variant allele fraction of 2%. In vitro evaluation of Tet2, a gene known to have a role in inflammation regulation, germline sites may help to identify mutations that interfere with the function of the Tet2 distal enhancer. Disrupting the Tet2 distal enhancer will increase the self-renewal level of hematopoietic stem cells. Mutations in germline genetics shape hematopoietic stem cell function and induce CHIP formation (34).
Mas-Peiro et al. (35) reported the incidence of CHIP increased in an age-dependent manner. Patients with DNMT3A or Tet2-CHIP driver mutations had significantly increased medium-term all-cause mortality following TAVI. Mutation of CHIP driver genes is associated with increased pro-inflammatory leucocyte subsets and increased mortality following successful TAVI. Recent evidence has shown that CHIP has an increased risk of developing cardiovascular disease (36). A study observed eight patients with severe degenerative aortic value stenosis, six with chronic post-infarction heart failure, and three normal participants (37). Patients who carry CHIP driver sequence variations and have cardiovascular disease are susceptible to a greater inflammatory response.
The role of inflammation in cardiovascular events has always been a research hotspot. Recently, a large prospective randomized clinical trial (Cantos) conducted anti-inflammatory therapy targeting interleukin-1β in patients with existing myocardial infarction and continuous increase of inflammatory marker C-reactive protein. The comparative study of monoclonal antibody canakinumab and placebo showed that patients in the intervention group had fewer recurrent events (38). The enrolled samples of patients in the study are being tested to determine whether those with CHIP benefit the most from the intervention. If so, it means finding a potential tool to reduce the vascular risk of CHIP. Considering the prevalence of CHIP in the elderly and the high incidence of stroke and myocardial infarction, CHIP vascular risk is a greater public health risk than hematological malignancies.
In CAVD, the main treatment strategy is to control the related pathogenic factors, such as antihypertension, smoking cessation, and body weight control. Although a small retrospective analysis showed that the use of bisphosphonates for anti-osteoporosis treatment and ACEI drugs to inhibit the effect of angiotensin could delay the progression of CAVD (39, 40), there is a lack of large-scale prospective clinical trials, and the mechanism and target effect of bisphosphonates need to be strengthened. Surgical valve replacement remains the main treatment for CAVD patients with obvious symptoms or very severe asymptomatic CAS patients. Recent evidence has shown that transcatheter aortic valve replacement (TAVR) is expected to become a treatment for CAVD patients who cannot tolerate surgery (41, 42). TAVR is rapidly expanding the treatment of CAVD and bicuspid aortic valves (43). Evogliptin inhibits CAVD by reducing inflammation, fibrosis, and calcification, suggesting its potential as a targeted therapeutic agent for inhibiting CAVD progression (44).
Although aortic valve replacement has been shown to be effective in the late stage of CAVD, it is costly and not optimal for elderly patients. These issues drive increasing interest in non-invasive therapies for patients with CAVD. Macrophages, T lymphocytes, and B lymphocytes are increased in the aortic valve with the progression of valve calcification, as well as cytokine signaling. This evidence indicates that modulation of adaptive immune cell signaling may be a strategy for treating CAVD (45). With further understanding of the molecular immune mechanism of CAVD, well-designed drugs for treating CAVD will bring more hope to patients.
An increasing number of studies have begun to find new immune molecules associated with the pathogenesis of CAVD and explore the role of mechanical pressure and flow on disease occurrence. Although elucidating the pathogenesis of CAVD is helpful for treatment, its biological complexity remains the main challenge for individualized therapy.
The pathophysiological changes in CAVD are similar to those in atherosclerosis in the early stage but may be similar to those in bone formation in the late stage and involve abnormal signal transduction pathways and gene variation. However, the promoting factors, the relationship between each pathway, and the specific molecular mechanism need to be further studied. CAVD pathogenesis can be summarized as endothelial injury, lipid infiltration, and chronic inflammation. Of course, the development and continuous improvements in TAVI technology, as well as the discovery of new and effective therapeutic targets, have brought hope to patients. However, more clinical trials and long-term follow-up are needed to further verify this hypothesis. It is believed that with the consistent efforts of researchers, breakthroughs will be made in both basic research and control methods of CAVD in the future.
WB and ZW contributed to literature search. DMZ and CS drafted the manuscript and contributed to review design. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (81870355 and 81970342) and Jiangsu Provincial Key Research and Development Program (BE2018611).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank all lab members for helpful discussion.
1. Coffey S, Roberts-Thomson R, Brown A, Carapetis J, Chen M, Enriquez-Sarano M, et al. Global epidemiology of valvular heart disease. Nat Rev Cardiol. (2021) 18:853–64. doi: 10.1038/s41569-021-00570-z
2. Otto CM, Prendergast B. Aortic-valve stenosis–from patients at risk to severe valve obstruction. N Engl J Med. (2014) 371:744–56. doi: 10.1056/NEJMra1313875
3. Otto CM, Kuusisto J, Reichenbach DD, Gown AM, O'Brien KD. Characterization of the early lesion of 'degenerative' valvular aortic stenosis. Histological and immunohistochemical studies. Circulation. (1994) 90:844–53. doi: 10.1161/01.CIR.90.2.844
4. Ginghină C, Florian A, Beladan C, Iancu M, Călin A, Popescu BA, et al. Calcific aortic valve disease and aortic atherosclerosis-two faces of the same disease? Rom J Intern Med. (2009) 47:319–29.
5. Eveborn GW, Schirmer H, Heggelund G, Lunde P, Rasmussen K. The evolving epidemiology of valvular aortic stenosis. the Troms study. Heart. (2013) 99:396–400. doi: 10.1136/heartjnl-2012-302265
6. Rosenhek R, Binder T, Porenta G, Lang I, Christ G, Schemper M, et al. Predictors of outcome in severe, asymptomatic aortic stenosis. N Engl J Med. (2000) 343:611–7. doi: 10.1056/NEJM200008313430903
7. Bonow RO, Leon MB, Doshi D, Moat N. Management strategies and future challenges for aortic valve disease. Lancet. (2016) 387:1312–23. doi: 10.1016/S0140-6736(16)00586-9
8. Furukawa K. Recent advances in research on human aortic valve calcification. J Pharmacol Sci. (2014) 124:129–37. doi: 10.1254/jphs.13R05CR
9. Thanassoulis G, Campbell CY, Owens DS, Smith JG, Smith AV, Peloso GM, et al. Genetic associations with valvular calcification and aortic stenosis. N Engl J Med. (2013) 368:503–12. doi: 10.1056/NEJMoa1109034
10. Yutzey KE, Demer LL, Body SC, Huggins GS, Towler DA, Giachelli CM, et al. Calcific aortic valve disease: a consensus summary from the Alliance of Investigators on Calcific Aortic Valve Disease. Arterioscler Thromb Vasc Biol. (2014) 34:2387–93. doi: 10.1161/ATVBAHA.114.302523
11. Izquierdo-Gómez MM, Hernández-Betancor I, García-Niebla J, Marí-López B, Laynez-Cerdeña I, Lacalzada-Almeida J. Valve calcification in aortic stenosis: etiology and diagnostic imaging techniques. Biomed Res Int. (2017) 2017:5178631. doi: 10.1155/2017/5178631
12. Pasipoularides A. Calcific aortic valve disease: part 1-molecular pathogenetic aspects, hemodynamics, and adaptive feedbacks. J Cardiovasc Transl Res. (2016) 9:102–18. doi: 10.1007/s12265-016-9679-z
13. Zeng YI, Sun R, Li X, Liu M, Chen S, Zhang P. Pathophysiology of valvular heart disease. Exp Ther Med. (2016) 11:1184–8. doi: 10.3892/etm.2016.3048
14. Qiao E, Huang Z, Wang W. Exploring potential genes and pathways related to calcific aortic valve disease. Gene. (2022). 808:145987. doi: 10.1016/j.gene.2021.145987
15. Dweck MR, Boon NA, Newby DE. Calcific aortic stenosis: a disease of the valve and the myocardium. J Am Coll Cardiol. (2012) 60:1854–63. doi: 10.1016/j.jacc.2012.02.093
16. Sage AP, Tintut Y, Demer LL. Regulatory mechanisms in vascular calcification. Nat Rev Cardiol. (2010) 7:528–36. doi: 10.1038/nrcardio.2010.115
17. Towler DA. Molecular and cellular aspects of calcific aortic valve disease. Circ Res. (2013) 113:198–208. doi: 10.1161/CIRCRESAHA.113.300155
18. Heistad DD, Shanahan C, Demer LL. Introduction to the Compendium on calcific aortic valve disease. Circ Res. (2013) 113:176–8. doi: 10.1161/CIRCRESAHA.113.301833
19. Park JY, Ryu SK, Choi JW, Ho KM, Jun JH, Rha SW, et al. Association of inflammation, myocardial fibrosis and cardiac remodelling in patients with mild aortic stenosis as assessed by biomarkers and echocardiography. Clin Exp Pharmacol Physiol. (2014) 41:185–91. doi: 10.1111/1440-1681.12206
20. Fondard O, Detaint D, Iung B, Choqueux C, Adle-Biassette H, Jarraya M, et al. Extracellular matrix remodelling in human aortic valve disease: the role of matrix metalloproteinases and their tissue inhibitors. Eur Heart J. (2005) 26:1333–41. doi: 10.1093/eurheartj/ehi248
21. Vieceli Dalla Sega F, Fortini F, Cimaglia P, Marracino L, Tonet E, Antonucci A, et al. COX-2 is downregulated in human stenotic aortic valves and its inhibition promotes dystrophic calcification. Int J Mol Sci. (2020) 21:8917. doi: 10.3390/ijms21238917
22. Bowler MA, Raddatz MA, Johnson CL, Lindman BR, Merryman WD. Celecoxib is associated with dystrophic calcification and aortic valve stenosis. JACC Basic Transl Sci. (2019) 4:135–43. doi: 10.1016/j.jacbts.2018.12.003
23. Yanagawa B, Lovren F, Pan Y, Garg V, Quan A, Tang G, et al. miRNA-141 is a novel regulator of BMP-2-mediated calcification in aortic stenosis. J Thorac Cardiovasc Surg. (2012) 144:256–62. doi: 10.1016/j.jtcvs.2011.10.097
24. Xu XH, Xu J, Xue L, Cao HL, Liu X, Chen YJ. VEGF attenuates development from cardiac hypertrophy to heart failure after aortic stenosis through mitochondrial mediated apoptosis and cardiomyocyte proliferation. J Cardiothorac Surg. (2011) 6:54. doi: 10.1186/1749-8090-6-54
25. Moreno PR, Astudillo L, Elmariah S, Purushothaman KR, Purushothaman M, Lento PA, et al. Increased macrophage infiltration and neovascularization in congenital bicuspid aortic valve stenosis. J Thorac Cardiovasc Surg. (2011) 142:895–901. doi: 10.1016/j.jtcvs.2011.03.002
26. Toutouzas K, Drakopoulou M, Synetos A, Tsiamis E, Agrogiannis G, Kavantzas N, et al. In vivo aortic valve thermal heterogeneity in patients with nonrheumatic aortic valve stenosis the: first in vivo experience in humans. J Am Coll Cardiol. (2008) 52:758–63. doi: 10.1016/j.jacc.2008.04.057
27. Gendron N, Rosa M, Blandinieres A, Sottejeau Y, Rossi E, Van Belle E, et al. Human aortic valve interstitial cells display proangiogenic properties during calcific aortic valve disease. Arterioscler Thromb Vasc Biol. (2021) 41:415–29. doi: 10.1161/ATVBAHA.120.314287
28. Arjunon S, Rathan S, Jo H, Yoganathan AP. Aortic valve: mechanical environment and mechanobiology. Ann Biomed Eng. (2013) 41:1331–46. doi: 10.1007/s10439-013-0785-7
29. Moore BL, Dasi LP. Coronary flow impacts aortic leaflet mechanics and aortic sinus hemodynamics. Ann Biomed Eng. (2015) 43:2231–41. doi: 10.1007/s10439-015-1260-4
30. Sadrabadi MS, Eskandari M, Feigenbaum HP, Arzani A. Local and global growth and remodeling in calcific aortic valve disease and aging. J Biomech. (2021) 128:110773. doi: 10.1016/j.jbiomech.2021.110773
31. Dahal S, Bramsen JA, Alber BR, Murray BT, Huang P, Chen MH, et al. Chondroitin sulfate promotes interstitial cell activation and calcification in an in vitro model of the aortic valve. Cardiovasc Eng Technol. (2021). doi: 10.1007/s13239-021-00586-z. [Epub ahead of print].
32. Fisher CI, Chen J, Merryman WD. Calcific nodule morphogenesis by heart valve interstitial cells is strain dependent. Biomech Model Mechanobiol. (2013) 12:5–17. doi: 10.1007/s10237-012-0377-8
33. Arzani A, Mofrad MRK. A strain-based finite element model for calcification progression in aortic valves. J Biomech. (2017) 65:216–20. doi: 10.1016/j.jbiomech.2017.10.014
34. Bick AG, Weinstock JS, Nandakumar SK, Fulco CP, Bao EL, Zekavat SM, et al. Inherited causes of clonal haematopoiesis in 97,691 whole genomes. Nature. (2020) 586:763–8. doi: 10.1038/s41586-020-2819-2
35. Mas-Peiro S, Hoffmann J, Fichtlscherer S, Dorsheimer L, Rieger MA, Dimmeler S, et al. Clonal haematopoiesis in patients with degenerative aortic valve stenosis undergoing transcatheter aortic valve implantation. Eur Heart J. (2020) 41:933–9. doi: 10.1093/eurheartj/ehz591
36. Papa V, Marracino L, Fortini F, Rizzo P, Campo G, Vaccarezza M, et al. Translating evidence from clonal hematopoiesis to cardiovascular disease: a systematic review. J Clin Med. (2020) 9:2480. doi: 10.3390/jcm9082480
37. Abplanalp WT, Mas-Peiro S, Cremer S, John D, Dimmeler S, Zeiher AM. Association of clonal hematopoiesis of indeterminate potential with inflammatory gene expression in patients with severe degenerative aortic valve stenosis or chronic postischemic heart failure. JAMA Cardiol. (2020) 5:1170–5. doi: 10.1001/jamacardio.2020.2468
38. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, et al. Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. (2017) 377:1119–31. doi: 10.1056/NEJMoa1707914
39. Ngo DT, Sverdlov AL, Horowitz JD. Prevention of aortic valve stenosis: a realistic therapeutic target? Pharmacol Ther. (2012) 135:78–93. doi: 10.1016/j.pharmthera.2012.04.001
40. Dalsgaard M, Iversen K, Kjaergaard J, Grande P, Goetze JP, Clemmensen P, et al. Short-term hemodynamic effect of angiotensin-converting enzyme inhibition in patients with severe aortic stenosis: a placebo-controlled, randomized study. Am Heart J. (2014) 167:226–34. doi: 10.1016/j.ahj.2013.11.002
41. Horne A Jr, Reineck EA, Hasan RK, Resar JR, Chacko M. Transcatheter aortic valve replacement: historical perspectives, current evidence, and future directions. Am Heart J. (2014) 168:414–23. doi: 10.1016/j.ahj.2014.07.017
42. Adams DH, Popma JJ, Reardon MJ, Yakubov SJ, Coselli JS, Deeb GM, et al. Transcatheter aortic-valve replacement with a self-expanding prosthesis. N Engl J Med. (2014) 370:1790–8. doi: 10.1056/NEJMoa1400590
43. Vincent F, Ternacle J, Denimal T, Shen M, Redfors B, Delhaye C, et al. Transcatheter aortic valve replacement in bicuspid aortic valve stenosis. Circulation. (2021) 143:1043–61. doi: 10.1161/CIRCULATIONAHA.120.048048
44. Choi B, Kim EY, Kim JE, Oh S, Park SO, Kim SM, et al. Evogliptin suppresses calcific aortic valve disease by attenuating inflammation, fibrosis, and calcification. Cells. (2021) 10:57. doi: 10.3390/cells10010057
Keywords: calcified aortic valve disease, aortic stenosis, inflammation, matrix remodeling, osteogenesis
Citation: Bian W, Wang Z, Sun C and Zhang D-M (2021) Pathogenesis and Molecular Immune Mechanism of Calcified Aortic Valve Disease. Front. Cardiovasc. Med. 8:765419. doi: 10.3389/fcvm.2021.765419
Received: 31 August 2021; Accepted: 30 November 2021;
Published: 23 December 2021.
Edited by:
Maurizio Pesce, Monzino Cardiology Center (IRCCS), ItalyReviewed by:
Atze Van Der Pol, Eindhoven University of Technology, NetherlandsCopyright © 2021 Bian, Wang, Sun and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chongxiu Sun, Y3hzdW5AbmptdS5lZHUuY24=; Dai-Min Zhang, ZGFpbWluemhAMTI2LmNvbQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.