
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med., 18 October 2021
Sec. Cardio-Oncology
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.761488
This article is part of the Research TopicCardio-Oncology and Reverse Cardio-Oncology: the Manifold Interconnections Between Heart Failure and CancerView all 8 articles
 Avirup Guha1,2*
Avirup Guha1,2* Xiaoling Wang3
Xiaoling Wang3 Ryan A. Harris3
Ryan A. Harris3 Anna-Gay Nelson4David Stepp5Zachary Klaassen6Priyanka Raval7Jorge Cortes7Steven S. Coughlin8Vladimir Y. Bogdanov9
Anna-Gay Nelson4David Stepp5Zachary Klaassen6Priyanka Raval7Jorge Cortes7Steven S. Coughlin8Vladimir Y. Bogdanov9 Justin X. Moore10Nihar Desai11,12D. Douglas Miller13Xin-Yun Lu14
Justin X. Moore10Nihar Desai11,12D. Douglas Miller13Xin-Yun Lu14 Ha Won Kim2,5
Ha Won Kim2,5 Neal L. Weintraub2,5*
Neal L. Weintraub2,5*Cardiovascular disease (CVD) and cancer often occur in the same individuals, in part due to the shared risk factors such as obesity. Obesity promotes adipose inflammation, which is pathogenically linked to both cardiovascular disease and cancer. Compared with Caucasians, the prevalence of obesity is significantly higher in African Americans (AA), who exhibit more pronounced inflammation and, in turn, suffer from a higher burden of CVD and cancer-related mortality. The mechanisms that underlie this association among obesity, inflammation, and the bidirectional risk of CVD and cancer, particularly in AA, remain to be determined. Socio-economic disparities such as lack of access to healthy and affordable food may promote obesity and exacerbate hypertension and other CVD risk factors in AA. In turn, the resulting pro-inflammatory milieu contributes to the higher burden of CVD and cancer in AA. Additionally, biological factors that regulate systemic inflammation may be contributory. Mutations in atypical chemokine receptor 1 (ACKR1), otherwise known as the Duffy antigen receptor for chemokines (DARC), confer protection against malaria. Many AAs carry a mutation in the gene encoding this receptor, resulting in loss of its expression. ACKR1 functions as a decoy chemokine receptor, thus dampening chemokine receptor activation and inflammation. Published and preliminary data in humans and mice genetically deficient in ACKR1 suggest that this common gene mutation may contribute to ethnic susceptibility to obesity-related disease, CVD, and cancer. In this narrative review, we present the evidence regarding obesity-related disparities in the bidirectional risk of CVD and cancer and also discuss the potential association of gene polymorphisms in AAs with emphasis on ACKR1.
Cardiovascular disease (CVD) and cancer often occur in the same individuals, in part because of shared risk factors (1). These risk factors can be classified as modifiable and non-modifiable (2–4). Non-modifiable risk factors include age, gender, genetic variants, and ancestry. Over the years, we have identified that modifiable risk factors- such as smoking, less physical activity, and obesity are independently and separately related to cardiovascular disease and cancer (5, 6). Although significant progress has been made in reducing these modifiable risk factors, rates of obesity continue to rise (7). Obesity contributes to the development of CVD, including atherosclerosis, abdominal aortic aneurysm, heart failure, and certain cancers, such as gastric cancer, through chronic low-grade inflammation (8–10). This inflammatory condition accelerates the onset or progress of carcinogenesis and atherosclerotic plaque formation, thus acting mechanistically as a shared risk factor.
The burden of certain types of CVD such as hypertension, heart failure, and ischemic stroke, and specific cancers such as prostate and breast cancer, as well as cancer-related mortality, is significantly higher in African Americans (AAs) compared to Caucasians (11, 12). At the same time, modifiable shared risk factors exhibit a racial skewing, with a higher proportion of AAs affected by obesity and obesity-associated diseases such as metabolic syndrome (7, 13, 14). Much of this risk can be explained by adverse social determinants of health (SDOH), often abbreviated as socio-economic disparities, that disproportionately afflict AAs, including poor quality diet, lack of safe places to exercise, insufficient access to health care, etc. (15–18).
On the other hand, genetic variability could potentially modify the bidirectional relationship between CVD and cancer. Many genetic variants have been individually linked to both CVD and cancer, but genes that modify the relationship between obesity and the bidirectional risk of CVD and cancer are poorly understood, particularly in AAs. A genetic basis for several diseases that are more common in AAs is well-established. The most noteworthy is sickle cell anemia, a condition resulting from a mutation in the β globin gene (HBB) (19). More recently, associations between variants in apolipoprotein L1 (APOL1) and chronic kidney disease in AAs have been established, and there is some evidence these variants play a role in increased CVD risk (20). Interestingly, these genetic variants in HBB and APOL1 confer resistance to life-threatening infections common in Sub-Saharan Africa, accounting for their prevalence in AAs (21, 22).
In this narrative review, we focus on the modifiable risk factor of obesity and present the data regarding differences in CVD and cancer risk in AAs compared to Caucasian Americans. We present the current literature on SDOH and its association with CVD and cancer. Additionally, we explore the role of ancestry, specifically a common variant in atypical chemokine receptor 1 [ACKR1, also known as the Duffy antigen receptor for chemokines (DARC)], in causation and outcomes in both conditions. Finally, we discuss the translational significance of these risk factors and associations and how this information might be used to identify potential biomarkers for CVD and cancer risk that can be used to help guide personalized therapeutic approaches.
Adiposity is traditionally classified based on body mass index (BMI), whereby the normal weight is defined as a BMI 18.5–24.9 kg/m2, overweight is defined as a BMI 25–29.9 kg/m2, and obesity is defined as a BMI ≥ 30 kg/m2 (23). Classes 2 and 3 obesity is defined as a BMI of 35–39.9 kg/m2 and ≥40 kg/m2, respectively. However, it is important to remember that although BMI is strongly correlated with body fat percentage in population studies, it is limited in its estimation of adiposity on an individual basis and significantly varies based on gender, age, race, ethnicity, and geography (24, 25). It is estimated that up to 50% of the world's population is either overweight or obese (5). In the United States, the prevalence of class 3 obesity is above 7% (7). This prevalence is highly variable across various races and ethnicities, with only a 5.5% prevalence in non-Hispanic white men compared to almost 17% in non-Hispanic Black women (7). It is essential to consider that some of these facts are based on obesity classification using BMI. In contrast, multiple studies have shown that waist circumference and body fat percentage measurements are better measurements of obesity in minority populations (26–28). In a study from the Cancer and Nutrition cohort, mortality increased by 17% in men with the same BMI with a 5 cm increase in waist circumference (29). The study by Kabakambira et al. evaluated African American men and women showed that waist circumference in non-Hispanic Black subjects tracked better with insulin resistance than BMI (27).
Obesity is estimated to have accounted for over 2.5 million additional CVD-related deaths in 2015 (30). Among individuals with obesity, 41% of deaths, and 34% of disability-adjusted life-years, are caused by excessive weight (10).
The understanding of the association between obesity and CVD has been evolving over the past four decades. It is certainly clear that obesity contributes to the development of atherosclerosis, heart failure, and arrhythmias in different ways (10). The obesity-associated CVD most relevant to this review is atherosclerosis. Obesity is frequently accompanied by other atherosclerosis risk factors, such as hypertension and dyslipidemia, which complicates precise attribution of risk related to obesity, as well as the underlying mechanisms (31). Evidence from research and clinical studies suggests that obesity promotes the development of atherosclerosis by augmenting insulin resistance and inflammation (32–34). Visceral adipose tissue depots are significantly more contributory to this inflammation than subcutaneous fat (35, 36). The liver, in turn, amplifies systemic inflammation by producing large quantities of inflammatory mediators such as various cytokines, complement factors, and C-reactive protein (37). Inflammation plays a key role in multiple aspects of atherosclerosis lesion formation and progression, including uptake of lipid into the blood vessel wall, vascular smooth muscle cell proliferation, vascular remodeling, and lesion destabilization (38). Additionally, endothelial dysfunction caused by an obesity-mediated decrease in endothelial nitric oxide bioavailability is a significant contributing factor (39). While adipose tissues are traditionally considered to contribute to atherosclerosis via indirect (remote) effects on the vascular wall, increasing evidence points toward a pathological role for perivascular adipose tissue, which expands in obesity and becomes more inflamed, thus accelerating vascular disease locally from the “outside-in” (40).
Obesity has been identified as a risk factor for several cancers, including a significant number of gastrointestinal cancers (8). The risk varies, with a relative risk of esophageal cancer as high as 4.8 when comparing class 2 obesity to individuals with normal BMI (8). In addition to esophageal cancer, obesity has been associated with increased risk of cancer occurrence for gastric, colorectal, liver, gallbladder, pancreas, breast (postmenopausal), uterine, ovarian, renal-cell, and thyroid cancers (8). By contrast, higher adiposity has been protective in multiple myeloma and meningioma in some studies (8). Nonetheless, 40% of cancer deaths in the US have been attributed to obesity (8, 41).
Changes in the adipose tissue microenvironment (ATME) may be mechanistically linked to cancer occurrence in obesity (9, 42, 43). As adipose tissues expand in obesity, adipocyte hypertrophy rather than hyperplasia predominates, and the ATME switches from type 2 [anti-inflammatory; e.g., interleukin (IL)-4, IL-10] to type 1 [pro-inflammatory; e.g., tumor necrosis factor (TNF), interferon (IFN)γ, IL-1β, and IL-6] cytokine generation (42, 44, 45). With further increases in adiposity, the adipocytes become mechanically stressed, and insufficient tissue vascularity leads to ischemia. Damage-associated molecular patterns (DAMPs) released from injured and dying adipocytes further promote infiltration of pro-inflammatory cells, such as dendritic cells and M1-macrophages, leading to the development of crown-like structure (CLS), a pathognomonic feature of adipocyte necrosis (9, 45). Numerous lymphoid cells, such as B-cells and CD8 T-cells, are also observed in the inflamed adipose tissue. This sustained inflammation is associated with a tumor environment that promotes cancer primarily through local rather than systemic stimulation of tumorigenesis, as cancers that are not directly surrounded by adipose tissues have a weaker association with obesity. The above crosstalk between adipocytes and cancer cells promotes tumorigenesis in obesity through complex mechanisms that have been reviewed elsewhere (9) and are summarized in Figure 1.
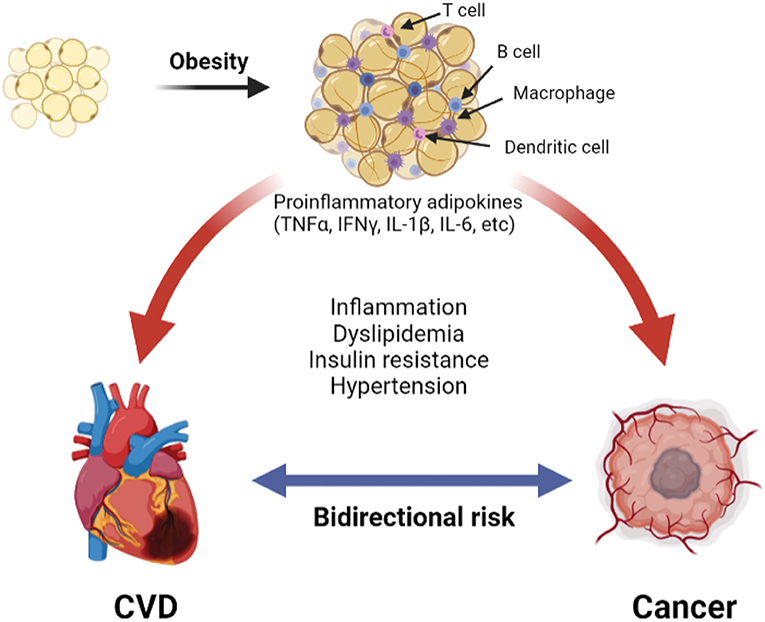
Figure 1. The pathologic adipose tissue microenvironment (ATME) has reduction in vascularity with hypertrophy. This releases damage-associated molecular patterns (DAMP) into the microenvironment, which trigger the infiltration with combination of pro-inflammatory macrophages, dendritic cells, B- and T-cells. Additionally, there is increase in pro-inflammatory cytokines (for example TNFα, IFNγ, etc.). A combination of these creates a chronic pro-inflammatory state that causes cardiovascular disease (31) and cancer (9).
Obesity-associated inflammation and insulin resistance promote both CVD and cancer. Additionally, CVD and cancer themselves can be developed from proinflammatory states (46, 47). Thus, we can speculate that either CVD or cancer is additive to obesity in promoting the other condition (e.g., those with CVD and obesity are likely at a higher risk of cancer and vice versa). Both epidemiological and mechanistic data demonstrate a potential increase in cancer development in those with CVD.
A Danish national registry study showed that patients with myocardial infarction were 14% more likely to develop cancer (48). Lau et al. using the Framingham 3rd offspring cohort, reported that patients with the highest CVD risk (>20% in 10-years, calculated using the pooled cohort equation) had a 3–4 times higher risk of cancer development than those at the lowest risk (<5% in 10-years) over a 15-year follow-up (49). To confirm these findings experimentally, Meijer et al. used a murine model of colon cancer and demonstrated accelerated cancer growth upon induction of myocardial infarction, and shotgun proteomics identified a number of pro-inflammatory cytokines that were associated with this phenomenon (50). Additionally, the authors confirmed that patients in a population cohort with congestive heart failure had elevated pro-inflammatory cytokine levels. Furthermore, the role of inflammation in cardiovascular disease in promoting carcinogenesis has also demonstrated this phenomenon in murine breast and lung cancer models (51, 52). There is also evidence to suggest that there is a role of the renin-angiotensin-aldosterone system (RAAS) in increasing the risk of cancer in patients with congestive heart failure (53). However, how obesity modifies this mechanistic pathway is to be determined.
A priori, we propose that increased adiposity can alter the rate of cardiovascular aging following the initial cancer diagnosis, which in turn can contribute to greater CVD risk over time. AAs exhibit greater adiposity compared to Caucasians and cancer mortality is also higher in the presence of obesity. Although the role of obesity in modifying the effects observed in these epidemiological or mechanistic studies has yet to be investigated, there is some evidence to support the role of obesity in carcinogenesis and cancer prognosis in AAs. Obesity has been shown to be independently associated with pancreatic cancer in AAs (54). Although obesity seems protective against multiple myeloma (8), obesity contributes to the increased incidence and mortality of multiple myeloma in AAs compared with other population groups (55). Although the role of adiposity in men with prostate cancer is somewhat controversial (56, 57); obesity, in general, can increase the risk of prostate cancer (58, 59). In fact, AA men exhibit more aggressive forms of prostate cancer (60), independent of BMI. In addition, AA women are almost twice as likely to be obese compared to Caucasian women (11) and greater adiposity has been shown to contribute to the increased prevalence of breast cancer (61). In addition, women with greater adiposity exhibit an increased risk of estrogen-receptor-positive (ER+) breast cancer and a decreased risk of triple-negative (TN) tumors (62) compared to average-weight women.
The relative contribution of obesity to CVD and cancer may depend on the population that is being investigated. Notably, in the United States, AAs are 1.5 times more likely to be obese compared with Caucasians (7). While several factors likely contribute, this trend primarily reflects socio-economic disparities (15, 18). Systemic racism has led to years of redlining in many parts of the country (63). Redlining is defined as the systematic denial of various services or goods by governmental agencies or the private sector, either directly or through the selective raising of prices which disadvantages the poor and minority communities (64, 65). This is a reason why many AAs live in food deserts with low availability of affordable, healthy foods (17, 66). In addition, certain areas have poor access to quality education and primary care physicians, leading to a lack of knowledge about weight management and healthy eating (67–69). Further, lack of access to quality education leads to lower-income and poverty, thereby reducing economic advancement opportunities (16, 70). The cycle of disparities related to these SDOH is self-perpetuating, leading to an exponential increase in obesity (16). It is predicted that one out of 4 AA adults will be severely obese by 2030 (7).
The direct effect of these disparities is reflected in poor cancer and CVD outcomes, particularly in the Southeastern United States, where the proportion of AAs in the rural regions is the highest (12, 71, 72). Although there has been a reduction in racial disparity in cancer mortality, the risk of mortality after cancer diagnosis remains 14% higher in AAs compared to Caucasians (12). Specifically, AA men are 111% more likely to die of prostate cancer when compared to Caucasian men, and AA women are 39% more likely to die of breast cancer compared to Caucasian women (12). AAs also have substantially higher rates of fatal atherosclerotic vascular disease compared with Caucasians [men: hazards ratio (HR), 2.18; women: HR, 1.63] (73).
In order to fully determine the role of adverse SDOH in mediating obesity-related disparities in the bidirectional relationship of cancer and CVD, it is important to objectively assess SDOH in a systematic epidemiological study. The World Health Organization (WHO) established the commission on SDOH (CSDH), which developed a framework with two major components (74). The first element of the framework is the socio-economic and political context. Table 1 summarizes the six aspects of the first element, which includes governance, macroeconomic policies, social policies, public policies, cultural, and societal values as well as epidemiological conditions. The second element of the framework is socio-economic position, which is operationalized by income, education, occupation, social classes, gender, and race/ethnicity. The CSDH framework posits that structural determinants generate or reinforce stratification in society, thus influencing the individual socio-economic position. The first and second elements lead to change in health through intermediary social factors, i.e., the SDOH. The SDOH are classified further into material-, psychosocial-, behavioral/biological-, and health system-related factors. While designing an epidemiological study, it is essential to realize that the SDOH are secondary features that need to be explained due to either the first or the second element. The standard measurements of the SDOH are also presented in Table 1, and the role of SDOH in relationship to obesity is illustrated in Figure 2. It is important to note that several of these indices are worse in AAs when compared to Caucasians.

Table 1. The World Health Organization framework for Social Determinants of Health.
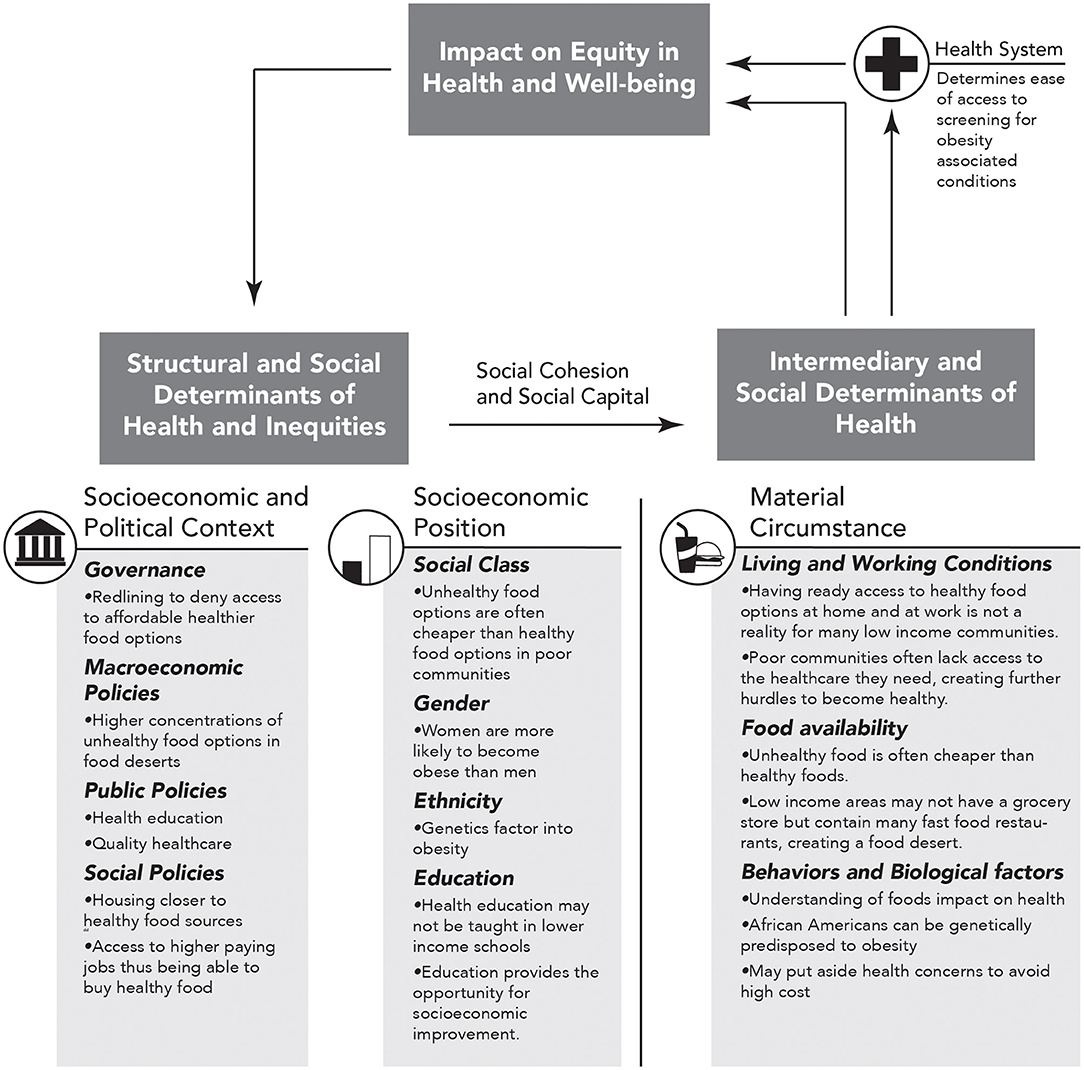
Figure 2. Obesity specific modification of figure A on page 6 of “A Conceptual Framework for Action on the Social Determinants of Health” document (74). Here we show how social determinants of health (SDOH) impact equity in health and well-being and how repetition within this cycle can increase in obesity and thus obesity associated CVD and cancer. The arrows represent directional association.
While SDOH undoubtedly accounts for much of the obesity-related disparities in the bidirectional risk of CVD and cancer in AAs, genetic factors can likely modify this relationship. Several genetic factors have been explored for their potential role (11, 93). Here, we will focus on two genes that may be of particular relevance: APOL1 and ACKR1 (or DARC).
The APOL1 gene regulates the process of autophagy and cell death, and APOL1 gene polymorphisms are relatively more common in those with West African ancestry (94). Two common polymorphisms, G1 and G2, have been associated with chronic kidney disease (95, 96). These APOL1 gene polymorphisms are located in the serum response associated (SRA) binding region and reduce the ability of SRA to bind APOL1 (94). The polymorphisms are associated with reduced risk for infection from the vector-borne ailment of African sleeping sickness, which is caused by Trypanosoma protozoans endemic to that part of the world, thus explaining their prevalence in the AA population (97). Indeed, as many as 21% of those from West Africa carry the G1 polymorphism, while 13% carry both G1 and G2, which are associated with increased risk of hypertension and renal disease (95, 97). With regard to CVD risk, population studies suggest that individuals with two APOL1 polymorphisms are at increased risk of cardiovascular mortality, surviving on average 3 years less than AAs harboring one or no APOL1 risk alleles (98). Further, the risk of myocardial infarction was estimated to be 80% higher in individuals carrying two of the risk alleles (98). Nonetheless, the mechanisms linking these alleles to CVD risk remain to be determined. Given the biological function of APOL1 in autophagy and cell death, its potential role in regulating cancer growth and outcomes seems more evident. In this regard, surprisingly little has been reported aside from a role in renal cell carcinoma, and a different polymorphism in the gene likely plays a role in this disparity (99). Further investigations into the role of APOL1 in both cancer and CVD are certainly warranted.
Mutations in ACKR-1, otherwise known as DARC, confer protection against malaria and are highly prevalent amongst AAs and other persons of color, including Hispanic Caucasians (100). Notably, ~70% of AAs harbor a T > C mutation at nucleotide−33 (GATA-1 binding site) in the promoter region that silences expression of ACKR-1 on red blood cells (RBC) (101). ACKR-1 is a promiscuous non-signaling chemokine receptor prominently expressed on RBC and to a lesser extent on other cell types such as endothelial cells and adipocytes. On RBC, ACKR-1 is thought to act primarily as a chemokine modulator by sequestering chemokines (i.e., “buffer-sink” function) or by regulating their local concentration at sites of inflammation (102). Among various chemokines, ACKR-1 has a strong binding affinity for CCL2/ monocyte chemoattractant protein (MCP-1), and ACKR-1 negative individuals are more sensitive to MCP-1-induced monocyte mobilization (102–104). Given the vital role that monocytes and inflammatory chemokines play in both CVD and cancer, common ACKR-1 polymorphisms would seem to be obvious candidates to modify the bidirectional risk between cardiovascular disease and cancer in AAs (105–107).
The association between ACKR-1 polymorphisms and inflammation is complex. Global ACKR-1 knockout mice exhibit enhanced susceptibility to diet induced-obesity/adipose tissue inflammation and increased severity of prostate cancer compared with wild-type mice, which is in line with clinical data in African Americans vs. Caucasians (106, 108, 109). Studies suggesting a role for pro-angiogenic ACKR-1 binding chemokines in the pathogenesis of prostate and breast cancer provide a mechanistic basis for how the loss of expression of ACKR-1 on RBC might promote tumor growth (110–112). On the contrary, the same ACKR-1 polymorphism common in AAs is also associated with a reduced number of neutrophils and neutrophil/lymphocyte ratio, a condition known as benign ethnic neutropenia (113). This could potentially function to offset the pro-inflammatory effects of the ACKR-1 gene mutation. Moreover, under some conditions, the absence of ACKR-1 may promote chemokine receptor desensitization, suggesting that loss of ACKR-1 expression may provoke either pro-inflammatory or anti-inflammatory responses on the type and chronicity of inflammation, local environmental factors, etc. There is evidence to suggest that ACKR-1 overexpression reduces the likelihood of metastatic disease in breast cancer (114). Similarly, global DARC knockout mice in the apolipoprotein E knockout background have been shown to exhibit reduced aortic atherosclerosis as compared to their wild-type counterparts (115). While the clinical relevance of this finding is unclear, it is important to point out that coronary artery calcifications, a pathognomonic feature of atherosclerosis, are consistently lower in AAs as compared with Caucasians (116).
Although there is mounting evidence to suggest that obesity is associated with the bidirectional risk of cancer and CVD, the precise level of risk, and the underlying mechanisms, are not fully understood. Moreover, it is apparent that the risk disproportionately affects certain ethnic groups, such as AAs. The SDOH plays a significant role in contributing to these health disparities, and addressing them is necessary to reduce the risk. Additionally, ancestry may contribute to the increased bidirectional risk, resulting in a maladaptive phenotype in AAs potentially due to the interaction of these two parameters. The role of obesity, SDOH, and ancestry should be investigated both independently and together in an effort to understand the relative importance of each of these elements in disease occurrence, disease outcomes, and therapeutic benefit. Examples of studies that would potentially address the disparities are sketched in Figure 3.
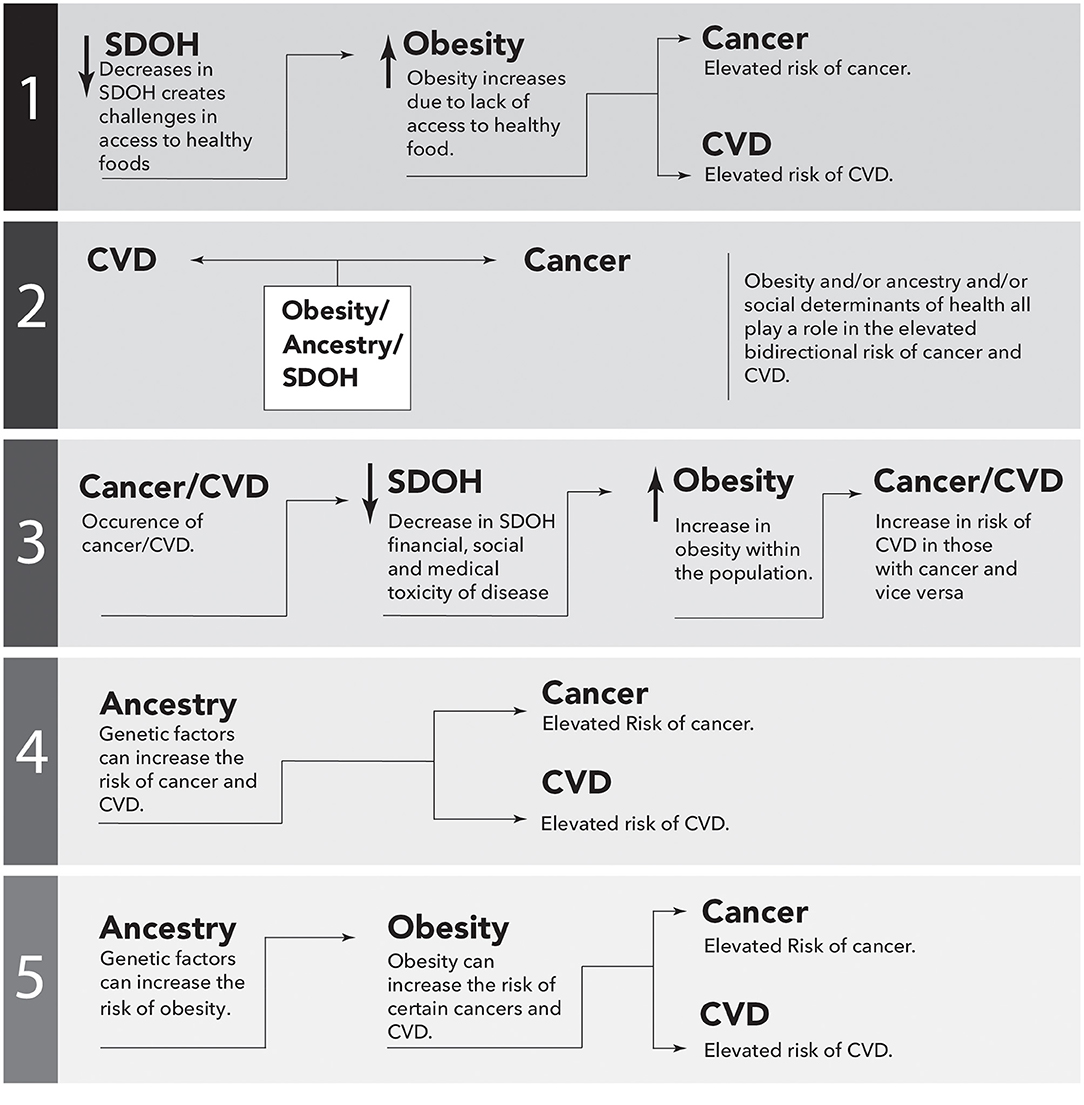
Figure 3. Simplistic causal directed acyclic graphs to illustrate possible epidemiological associations between cancer and cardiovascular disease (CVD). Performing these studies would potentially address the missing links between ancestry, Social Determinants of Health (SDOH), obesity and the bidirectional risk of CVD and cancer. The arrows represent directional association.
AG, NW, XW, and RH conceived the study concept. References were collected first by AG and verified by NW. AG drafted the first version of the manuscript. The study was conducted under the supervision of NW. All authors provided necessary revisions of the manuscript.
This work was supported in part by American Heart Association-Strategically Focused Research Network Grant 863622 in Disparities in Cardio-Oncology (AG, XW, RH, DS, VYB, and NW). NW was supported by NIH Grants HL124097, HL126949, HL134354, AR070029, and AG064895. RH was supported by Grants DK117365, DK125013, HL137087, CFFT Harris19A. JM was supported by Grant K01MD015304 from the National Institute on Minority Health and Health Disparities of the National Institutes of Health.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We would like to thank Allen Hagan and William Andrews of the Medical Illustration department in Augusta University for sketching (Figures 2, 3).
1. Koene RJ, Prizment AE, Blaes A, Konety SH. Shared risk factors in cardiovascular disease and cancer. Circulation. (2016) 133:1104–14. doi: 10.1161/CIRCULATIONAHA.115.020406
2. Stein C, Colditz G. Modifiable risk factors for cancer. Br J Cancer. (2004) 90:299–303. doi: 10.1038/sj.bjc.6601509
3. Fitzmaurice C, Abate D, Abbasi N, Abbastabar H, Abd-Allah F, Abdel-Rahman O, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 29 cancer groups, 1990 to 2017: a systematic analysis for the global burden of disease study. JAMA Oncol. (2019) 5:1749–68. doi: 10.1001/jamaoncol.2019.2996
4. Roth GA, Mensah GA, Johnson CO, Addolorato G, Ammirati E, Baddour LM, et al. Global burden of cardiovascular diseases and risk factors, 1990-2019: update from the GBD 2019 study. J Am Coll Cardiol. (2020) 76:2982–3021. doi: 10.1016/j.jacc.2020.11.010
5. Afshin A, Forouzanfar MH, Reitsma MB, Sur P, Estep K, Lee A, et al. Health effects of overweight and obesity in 195 countries over 25 years. N Engl J Med. (2017) 377:13–27. doi: 10.1056/NEJMoa1614362
6. GBD 2019 Tobacco Collaborators. Spatial, temporal, and demographic patterns in prevalence of smoking tobacco use and attributable disease burden in 204 countries and territories, 1990-2019: a systematic analysis from the Global Burden of Disease Study 2019. Lancet. (2021) 397:2337–60. doi: 10.1016/S0140-6736(21)01169-7
7. Ward ZJ, Bleich SN, Cradock AL, Barrett JL, Giles CM, Flax C, et al. Projected U.S. state-level prevalence of adult obesity and severe obesity. N Engl J Med. (2019) 381:2440–50. doi: 10.1056/NEJMsa1909301
8. Lauby-Secretan B, Scoccianti C, Loomis D, Grosse Y, Bianchini F, Straif K. Body fatness and cancer–viewpoint of the IARC working group. N Engl J Med. (2016) 375:794–8. doi: 10.1056/NEJMsr1606602
9. Quail DF, Dannenberg AJ. The obese adipose tissue microenvironment in cancer development and progression. Nat Rev Endocrinol. (2019) 15:139–54. doi: 10.1038/s41574-018-0126-x
10. Powell-Wiley TM, Poirier P, Burke LE, Després JP, Gordon-Larsen P, Lavie CJ, et al. Obesity and cardiovascular disease: a scientific statement from the American Heart Association. Circulation. (2021) 143:e984–1010. doi: 10.1161/CIR.0000000000000973
11. Carnethon MR, Pu J, Howard G, Albert MA, Anderson CAM, Bertoni AG, et al. Cardiovascular health in African Americans: a scientific statement from the American Heart Association. Circulation. (2017) 136:e393–423. doi: 10.1161/CIR.0000000000000534
12. DeSantis CE, Miller KD, Goding Sauer A, Jemal A, Siegel RL. Cancer statistics for African Americans, 2019. Cancer J Clinicians. (2019) 69:211–33. doi: 10.3322/caac.21555
13. Moore JX, Chaudhary N, Akinyemiju T. Metabolic syndrome prevalence by race/ethnicity and sex in the United States, national health and nutrition examination survey, 1988-2012. Prev Chronic Dis. (2017) 14:E24. doi: 10.5888/pcd14.160287
14. Akinyemiju T, Moore JX, Judd S, Lakoski S, Goodman M, Safford MM, et al. Metabolic dysregulation and cancer mortality in a national cohort of blacks and whites. BMC Cancer. (2017) 17:856. doi: 10.1186/s12885-017-3807-2
15. Hernandez DC, Reesor LM, Murillo R. Food insecurity and adult overweight/obesity: gender and race/ethnic disparities. Appetite. (2017) 117:373–8. doi: 10.1016/j.appet.2017.07.010
16. Assari S, Thomas A, Caldwell CH, Mincy RB. Blacks' diminished health return of family structure and socioeconomic status; 15 years of follow-up of a national urban sample of youth. J Urban Health. (2018) 95:21–35. doi: 10.1007/s11524-017-0217-3
17. Garcia MT, Sato PM, Trude AC, Eckmann T, Steeves ETA, Hurley KM, et al. Factors associated with home meal preparation and fast-food sources use among low-income urban African American adults. Ecol Food Nutr. (2018) 57:13–31. doi: 10.1080/03670244.2017.1406853
18. Wang Y, Beydoun MA, Min J, Xue H, Kaminsky LA, Cheskin LJ. Has the prevalence of overweight, obesity and central obesity levelled off in the United States? Trends, patterns, disparities, and future projections for the obesity epidemic. Int J Epidemiol. (2020) 49:810–23. doi: 10.1093/ije/dyz273
19. Solovieff N, Hartley SW, Baldwin CT, Klings ES, Gladwin MT, Taylor JG, et al. Ancestry of African Americans with sickle cell disease. Blood Cells Mol Dis. (2011) 47:41–5. doi: 10.1016/j.bcmd.2011.04.002
20. Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, et al. Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science. (2010) 329:841–5. doi: 10.1126/science.1193032
21. Allison AC. Protection afforded by sickle-cell trait against subtertian malarial infection. Br Med J. (1954) 1:290. doi: 10.1136/bmj.1.4857.290
22. Cooper A, Ilboudo H, Alibu VP, Ravel S, Enyaru J, Weir W, et al. APOL1 renal risk variants have contrasting resistance and susceptibility associations with African trypanosomiasis. elife. (2017) 6:e25461. doi: 10.7554/eLife.25461.022
23. WHO Consultation. Obesity: preventing and managing the global epidemic. World Health Organ Tech Rep Ser. (2000) 894:1–253.
24. Nevill AM, Stewart AD, Olds T, Holder R. Relationship between adiposity and body size reveals limitations of BMI. Am J Phys Anthropol. (2006) 129:151–6. doi: 10.1002/ajpa.20262
25. Romero-Corral A, Somers VK, Sierra-Johnson J, Thomas RJ, Collazo-Clavell ML, Korinek J, et al. Accuracy of body mass index in diagnosing obesity in the adult general population. Int J Obes. (2008) 32:959–66. doi: 10.1038/ijo.2008.11
26. Katzmarzyk PT, Bray GA, Greenway FL, Johnson WD, Newton RL Jr, Ravussin E, et al. Ethnic-specific BMI and waist circumference thresholds. Obesity. (2011) 19:1272–8. doi: 10.1038/oby.2010.319
27. Kabakambira JD, Baker RL Jr, Briker SM, Courville AB, Mabundo LS, DuBose CW, et al. Do current guidelines for waist circumference apply to black Africans? Prediction of insulin resistance by waist circumference among Africans living in America. BMJ Glob Health. (2018) 3:e001057. doi: 10.1136/bmjgh-2018-001057
28. Ross R, Neeland IJ, Yamashita S, Shai I, Seidell J, Magni P, et al. Waist circumference as a vital sign in clinical practice: a consensus statement from the IAS and ICCR working group on visceral obesity. Nat Rev Endocrinol. (2020) 16:177–89. doi: 10.1038/s41574-019-0310-7
29. Pischon T, Boeing H, Hoffmann K, Bergmann M, Schulze MB, Overvad K, et al. General and abdominal adiposity and risk of death in Europe. N Engl J Med. (2008) 359:2105–20. doi: 10.1056/NEJMoa0801891
30. Virani SS, Alonso A, Aparicio HJ, Benjamin EJ, Bittencourt MS, Callaway CW, et al. Heart disease and stroke statistics-2021 update: a report from the American Heart Association. Circulation. (2021) 143:e254–743. doi: 10.1161/CIR.0000000000000950
31. Piché ME, Tchernof A, Després JP. Obesity phenotypes, diabetes, cardiovascular diseases. Circ Res. (2020) 126:1477–500. doi: 10.1161/CIRCRESAHA.120.316101
32. Kahn BB, Flier JS. Obesity and insulin resistance. J Clin Investig. (2000) 106:473–81. doi: 10.1172/JCI10842
33. Rocha VZ, Libby P. Obesity, inflammation, and atherosclerosis. Nat Rev Cardiol. (2009) 6:399–409. doi: 10.1038/nrcardio.2009.55
34. de Heredia FP, Gómez-Martínez S, Marcos A. Obesity, inflammation and the immune system. Proc Nutr Soc. (2012) 71:332–8. doi: 10.1017/S0029665112000092
35. Abraham TM, Pedley A, Massaro JM, Hoffmann U, Fox CS. Association between visceral and subcutaneous adipose depots and incident cardiovascular disease risk factors. Circulation. (2015) 132:1639–47. doi: 10.1161/CIRCULATIONAHA.114.015000
36. Akinyemiju T, Moore JX, Pisu M, Goodman M, Howard VJ, Safford M, et al. Association of baseline inflammatory biomarkers with cancer mortality in the REGARDS cohort. Oncotarget. (2019) 10:4857–67. doi: 10.18632/oncotarget.27108
37. Lauridsen BK, Stender S, Kristensen TS, Kofoed KF, Køber L, Nordestgaard BG, et al. Liver fat content, non-alcoholic fatty liver disease, and ischaemic heart disease: Mendelian randomization and meta-analysis of 279 013 individuals. Eur Heart J. (2018) 39:385–93. doi: 10.1093/eurheartj/ehx662
38. Wolf D, Ley K. Immunity and inflammation in atherosclerosis. Circ Res. (2019) 124:315–27. doi: 10.1161/CIRCRESAHA.118.313591
39. Engin A. Endothelial dysfunction in obesity. Adv Exp Med Biol. (2017) 345–79. doi: 10.1007/978-3-319-48382-5_15
40. Kim HW, Shi H, Winkler MA, Lee R, Weintraub NL. Perivascular adipose tissue and vascular perturbation/atherosclerosis. Arterioscler Thromb Vasc Biol. (2020) 40:2569–76. doi: 10.1161/ATVBAHA.120.312470
41. Islami F, Goding Sauer A, Miller KD, Siegel RL, Fedewa SA, Jacobs EJ, et al. Proportion and number of cancer cases and deaths attributable to potentially modifiable risk factors in the United States. CA Cancer J Clin. (2018) 68:31–54. doi: 10.3322/caac.21440
42. Akinyemiju T, Moore JX, Judd SE, Pisu M, Goodman M, Howard VJ, et al. Pre-diagnostic biomarkers of metabolic dysregulation and cancer mortality. Oncotarget. (2018) 9:16099–109. doi: 10.18632/oncotarget.24559
43. Akinyemiju T, Moore JX, Pisu M, Judd SE, Goodman M, Shikany JM, et al. A prospective study of obesity, metabolic health, cancer mortality. Obesity. (2018) 26:193–201. doi: 10.1002/oby.22067
44. Braune J, Weyer U, Hobusch C, Mauer J, Bruning JC, Bechmann I, et al. IL-6 Regulates M2 polarization and local proliferation of adipose tissue macrophages in obesity. J Immunol. (2017) 198:2927–34. doi: 10.4049/jimmunol.1600476
45. Olson OC, Quail DF, Joyce JA. Obesity and the tumor microenvironment. Science. (2017) 358:1130–1. doi: 10.1126/science.aao5801
46. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. (2010) 140:883–99. doi: 10.1016/j.cell.2010.01.025
47. Alfaddagh A, Martin SS, Leucker TM, Michos ED, Blaha MJ, Lowenstein CJ, et al. Inflammation and cardiovascular disease: from mechanisms to therapeutics. Am J Prev Cardiol. (2020) 4:100130. doi: 10.1016/j.ajpc.2020.100130
48. Malmborg M, Christiansen CB, Schmiegelow MD, Torp-Pedersen C, Gislason G, Schou M. Incidence of new onset cancer in patients with a myocardial infarction - a nationwide cohort study. BMC Cardiovasc Disord. (2018) 18:198. doi: 10.1186/s12872-018-0932-z
49. Lau Emily S, Paniagua Samantha M, Liu E, Jovani M, Li Shawn X, Takvorian K, et al. Cardiovascular risk factors are associated with future cancer. JACC CardioOncol. (2021) 3:48–58. doi: 10.1016/j.jaccao.2020.12.003
50. Meijers WC, Maglione M, Bakker SJ, Oberhuber R, Kieneker LM, de Jong S, et al. Heart failure stimulates tumor growth by circulating factors. Circulation. (2018) 138:678–91. doi: 10.1161/CIRCULATIONAHA.117.030816
51. Avraham S, Abu-Sharki S, Shofti R, Haas T, Korin B, Kalfon R, et al. Early cardiac remodeling promotes tumor growth and metastasis. Circulation. (2020) 142:670–83. doi: 10.1161/CIRCULATIONAHA.120.046471
52. Koelwyn GJ, Newman AAC, Afonso MS, van Solingen C, Corr EM, Brown EJ, et al. Myocardial infarction accelerates breast cancer via innate immune reprogramming. Nat Med. (2020) 26:1452–8. doi: 10.1038/s41591-020-0964-7
53. Bertero E, Canepa M, Maack C, Ameri P. Linking heart failure to cancer. Circulation. (2018) 138:735–42. doi: 10.1161/CIRCULATIONAHA.118.033603
54. Bethea TN, Kitahara CM, Sonderman J, Patel AV, Harvey C, Knutsen SF, et al. A pooled analysis of body mass index and pancreatic cancer mortality in african americans. Cancer Epidemiol Biomarkers Prev. (2014) 23:2119–25. doi: 10.1158/1055-9965.EPI-14-0422
55. Sonderman JS, Bethea TN, Kitahara CM, Patel AV, Harvey C, Knutsen SF, et al. Multiple myeloma mortality in relation to obesity among African Americans. J Natl Cancer Inst. (2016) 108:djw120. doi: 10.1093/jnci/djw120
56. Porter MP, Stanford JL. Obesity and the risk of prostate cancer. Prostate. (2005) 62:316–21. doi: 10.1002/pros.20121
57. Pichardo MS, Smith CJ, Dorsey TH, Loffredo CA, Ambs S. Association of anthropometric measures with prostate cancer among African American men in the NCI-Maryland Prostate Cancer Case-Control Study. Cancer Epidemiol Biomarkers Prev. (2018) 27:936–44. doi: 10.1158/1055-9965.EPI-18-0242
58. Gong Z, Agalliu I, Lin DW, Stanford JL, Kristal AR. Obesity is associated with increased risks of prostate cancer metastasis and death after initial cancer diagnosis in middle-aged men. Cancer. (2007) 109:1192–202. doi: 10.1002/cncr.22534
59. Cao Y, Ma J. Body mass index, prostate cancer–specific mortality, and biochemical recurrence: a systematic review and meta-analysis. Cancer Prev Res. (2011) 4:486–501. doi: 10.1158/1940-6207.CAPR-10-0229
60. Su LJ, Arab L, Steck SE, Fontham ETH, Schroeder JC, Bensen JT, et al. Obesity and prostate cancer aggressiveness among African and Caucasian Americans in a population-based study. Cancer Epidemiol Biomarkers Prev. (2011) 20:844–53. doi: 10.1158/1055-9965.EPI-10-0684
61. Parekh N, Chandran U, Bandera EV. Obesity in cancer survival. Ann Rev Nutr. (2012) 32:311–42. doi: 10.1146/annurev-nutr-071811-150713
62. Bandera EV, Chandran U, Hong CC, Troester MA, Bethea TN, Adams-Campbell LL, et al. Obesity, body fat distribution, and risk of breast cancer subtypes in African American women participating in the AMBER Consortium. Breast Cancer Res Treat. (2015) 150:655–66. doi: 10.1007/s10549-015-3353-z
63. Schell CJ, Dyson K, Fuentes TL, Des Roches S, Harris NC, Miller DS, et al. The ecological and evolutionary consequences of systemic racism in urban environments. Science. (2020) 369:eaay4497. doi: 10.1126/science.aay4497
64. Harris R, Forrester D. The suburban origins of redlining: a Canadian case study, 1935-54. Urban Stud. (2003) 40:2661–86. doi: 10.1080/0042098032000146830
65. Rothstein R. The Color of Law: A Forgotten History of How Our Government Segregated America. Liveright Publishing (2017).
66. Hager ER, Cockerham A, O'Reilly N, Harrington D, Harding J, Hurley KM, et al. Food swamps and food deserts in Baltimore City, MD, USA: associations with dietary behaviours among urban adolescent girls. Public Health Nutr. (2017) 20:2598–607. doi: 10.1017/S1368980016002123
67. Cooksey-Stowers K, Schwartz MB, Brownell KD. Food swamps predict obesity rates better than food deserts in the United States. Int J Environ Res Public Health. (2017) 14:1366. doi: 10.3390/ijerph14111366
68. Gailey S, Bruckner TA. Obesity among black women in food deserts: an “omnibus” test of differential risk. SSM Popul Health. (2019) 7:100363. doi: 10.1016/j.ssmph.2019.100363
69. Testa A, Jackson DB. Food insecurity, food deserts, and waist-to-height ratio: variation by sex and race/ethnicity. J Community Health. (2019) 44:444–50. doi: 10.1007/s10900-018-00601-w
70. Arpey NC, Gaglioti AH, Rosenbaum ME. How socioeconomic status affects patient perceptions of health care: a qualitative study. J Prim Care Community Health. (2017) 8:169–75. doi: 10.1177/2150131917697439
71. Moss JL, Pinto CN, Srinivasan S, Cronin KA, Croyle RT. Persistent poverty and cancer mortality rates: an analysis of county-level poverty designations. Cancer Epidemiol Biomarkers Prev. (2020) 29:1949–54. doi: 10.1158/1055-9965.EPI-20-0007
72. Parcha V, Kalra R, Suri SS, Malla G, Wang TJ, Arora G, et al. Geographic variation in cardiovascular health among American adults. Mayo Clin Proc. (2021) 96:1770–81. doi: 10.1016/j.mayocp.2020.12.034
73. Safford MM, Brown TM, Muntner PM, Durant RW, Glasser S, Halanych JH, et al. Association of race and sex with risk of incident acute coronary heart disease events. JAMA. (2012) 308:1768–74. doi: 10.1001/jama.2012.14306
75. Corner L. Gender-sensitive and pro-poor indicators of good governance. In: Paper prepared as a background document for the UNDP-ICSSR technical workshop on Governance Indicators for Pro-poor and Gender-sensitive Policy Reform held in New Delhi. (2005). p. 20–2.
76. Chung H, Muntaner C. Welfare state matters: a typological multilevel analysis of wealthy countries. Health Policy. (2007) 80:328–39. doi: 10.1016/j.healthpol.2006.03.004
78. Kleczkowski BM, Roemer MI, Werff A. National Health Systems and Their Reorientation Towards Health for All: Guidelines for Policy-Making. World Health Organization (1984).
79. Solar O, Irwin A, Vega J. Equity in Health Sector Reform and Reproductive Health: Measurement Issues and the Health Systems Context. WHO Health Equity Team working paper (1984).
80. Murray SA, Manktelow K, Clifford C. The interplay between social and cultural context and perceptions of cardiovascular disease. J Adv Nurs. (2000) 32:1224–33. doi: 10.1046/j.1365-2648.2000.01593.x
81. Smith C, Levy I, Sabin C, Kaya E, Johnson M, Lipman M. Cardiovascular disease risk factors and antiretroviral therapy in an HIV-positive UK population. HIV Med. (2004) 5:88–92. doi: 10.1111/j.1468-1293.2004.00191.x
82. Seedat YK. Impact of poverty on hypertension and cardiovascular disease in sub-Saharan Africa. Cardiovasc J Afr. (2007) 18:316–20.
83. Gastwirth JL. The estimation of the Lorenz curve and Gini index. Rev Econ Stat.(1972) 54:306–16. doi: 10.2307/1937992
84. Albano JD, Ward E, Jemal A, Anderson R, Cokkinides VE, Murray T, et al. Cancer mortality in the United States by education level and race. J Natl Cancer Inst. (2007) 99:1384–94. doi: 10.1093/jnci/djm127
85. Winkleby MA, Jatulis DE, Frank E, Fortmann SP. Socioeconomic status and health: how education, income, and occupation contribute to risk factors for cardiovascular disease. Am J Public Health. (1992) 82:816–20. doi: 10.2105/AJPH.82.6.816
86. Muntaner C, Borrell C, Benach J, Pasarín MI, Fernandez E. The associations of social class and social stratification with patterns of general and mental health in a Spanish population. Int J Epidemiol. (2003) 32:950–8. doi: 10.1093/ije/dyg170
87. Hendifar A, Yang D, Lenz F, Lurje G, Pohl A, Lenz C, et al. Gender disparities in metastatic colorectal cancer survival. Clin Cancer Res. (2009) 15:6391–7. doi: 10.1158/1078-0432.CCR-09-0877
88. Chen L, Li CI. Racial disparities in breast cancer diagnosis and treatment by hormone receptor and HER2 status. Cancer Epidemiol Prev Biomarkers. (2015) 24:1666–72. doi: 10.1158/1055-9965.EPI-15-0293
89. Jacobs DE, Wilson J, Dixon SL, Smith J, Evens A. The relationship of housing and population health: a 30-year retrospective analysis. Environ Health Perspect. (2009) 117:597–604. doi: 10.1289/ehp.0800086
90. Suglia SF, Sapra KJ, Koenen KC. Violence and cardiovascular health: a systematic review. Am J Prev Med. (2015) 48:205–12. doi: 10.1016/j.amepre.2014.09.013
91. Newcomb PA, Carbone PP. The health consequences of smoking: cancer. Med Clin North Am. (1992) 76:305–31. doi: 10.1016/S0025-7125(16)30355-8
92. Bottle A, Gnani S, Saxena S, Aylin P, Mainous AG, Majeed A. Association between quality of primary care and hospitalization for coronary heart disease in England: national cross-sectional study. J Gen Intern Med. (2008) 23:135–41. doi: 10.1007/s11606-007-0390-2
93. Ragin C, Blackman E, Roberts R, Butler R, Gathere S, Halliday D, et al. Cancer in populations of African Ancestry: studies of the African Caribbean Cancer Consortium. Cancer Causes Control. (2017) 28:1173–6. doi: 10.1007/s10552-017-0974-z
94. Zhaorigetu S, Wan G, Kaini R, Wan G, Jiang Z, Hu CA. ApoL1, a BH3-only lipid-binding protein, induces autophagic cell death. Autophagy. (2008) 4:1079–82. doi: 10.4161/auto.7066
95. Freedman BI, Kopp JB, Langefeld CD, Genovese G, Friedman DJ, Nelson GW, et al. The apolipoprotein L1 (APOL1) gene and nondiabetic nephropathy in African Americans. J Am Soc Nephrol. (2010) 21:1422–6. doi: 10.1681/ASN.2010070730
96. Tzur S, Rosset S, Shemer R, Yudkovsky G, Selig S, Tarekegn A, et al. Missense mutations in the APOL1 gene are highly associated with end stage kidney disease risk previously attributed to the MYH9 gene. Hum Genet. (2010) 128:345–50. doi: 10.1007/s00439-010-0861-0
97. Thomson R, Genovese G, Canon C, Kovacsics D, Higgins MK, Carrington M, et al. Evolution of the primate trypanolytic factor APOL1. Proc Natl Acad Sci USA. (2014) 111:E2130–9. doi: 10.1073/pnas.1400699111
98. Mukamal KJ, Tremaglio J, Friedman DJ, Ix JH, Kuller LH, Tracy RP, et al. APOL1 genotype, kidney and cardiovascular disease, and death in older adults. Arterioscler Thromb Vasc Biol. (2016) 36:398–403. doi: 10.1161/ATVBAHA.115.305970
99. Hu CA, Klopfer EI, Ray PE. Human apolipoprotein L1 (ApoL1) in cancer and chronic kidney disease. FEBS Lett. (2012) 586:947–55. doi: 10.1016/j.febslet.2012.03.002
100. Langhi DM Jr, Bordin JO. Duffy blood group and malaria. Hematology. (2006) 11:389–98. doi: 10.1080/10245330500469841
101. Howes RE, Patil AP, Piel FB, Nyangiri OA, Kabaria CW, Gething PW, et al. The global distribution of the Duffy blood group. Nat Commun. (2011) 2:266. doi: 10.1038/ncomms1265
102. Mayr F, Spiel A, Leitner J, Firbas C, Schnee J, Hilbert J, et al. Influence of the Duffy antigen on pharmacokinetics and pharmacodynamics of recombinant monocyte chemoattractant protein (MCP-1, CCL-2) in vivo. Int J Immunopathol Pharmacol. (2009) 22:615–25. doi: 10.1177/039463200902200307
103. Hansell CA, Hurson CE, Nibbs RJ. DARC and D6: silent partners in chemokine regulation? Immunol Cell Biol. (2011) 89:197–206. doi: 10.1038/icb.2010.147
104. Wenzel P. Monocytes as immune targets in arterial hypertension. Br J Pharmacol. (2019) 176:1966–77. doi: 10.1111/bph.14389
105. Conti I, Rollins BJ. CCL2 (monocyte chemoattractant protein-1) and cancer. In: Seminars in cancer biology. Elsevier (2004). p. 149–154. doi: 10.1016/j.semcancer.2003.10.009
106. Benson TW, Weintraub DS, Crowe M, Yiew NK, Popoola O, Pillai A, et al. Deletion of the Duffy antigen receptor for chemokines (DARC) promotes insulin resistance and adipose tissue inflammation during high fat feeding. Mol Cell Endocrinol. (2018) 473:79–88. doi: 10.1016/j.mce.2018.01.006
107. Olingy CE, Dinh HQ, Hedrick CC. Monocyte heterogeneity and functions in cancer. J Leukoc Biol. (2019) 106:309–22. doi: 10.1002/JLB.4RI0818-311R
108. Shen H, Schuster R, Stringer KF, Waltz SE, Lentsch AB. The Duffy antigen/receptor for chemokines (DARC) regulates prostate tumor growth. FASEB J. (2006) 20:59–64. doi: 10.1096/fj.05-4764com
109. Kodama K, Tojjar D, Yamada S, Toda K, Patel CJ, Butte AJ. Ethnic differences in the relationship between insulin sensitivity and insulin response: a systematic review and meta-analysis. Diabetes Care. (2013) 36:1789–96. doi: 10.2337/dc12-1235
110. Soria G, Ben-Baruch A. The inflammatory chemokines CCL2 and CCL5 in breast cancer. Cancer Lett. (2008) 267:271–85. doi: 10.1016/j.canlet.2008.03.018
111. Izhak L, Wildbaum G, Uri W, Shaked Y, Alami J, Dumont D, et al. Predominant expression of CCL2 at the tumor site of prostate cancer patients directs a selective loss of immunological tolerance to CCL2 that could be amplified in a beneficial manner. J Immunol. (2010) 184:1092–101. doi: 10.4049/jimmunol.0902725
112. Walens A, DiMarco AV, Lupo R, Kroger BR, Damrauer JS, Alvarez JV. CCL5 promotes breast cancer recurrence through macrophage recruitment in residual tumors. Elife. (2019) 8:e43653. doi: 10.7554/eLife.43653.029
113. Haddy TB, Rana SR, Castro O. Benign ethnic neutropenia: what is a normal absolute neutrophil count? J Lab Clin Med. (1999) 133:15–22. doi: 10.1053/lc.1999.v133.a94931
114. Wang J, Ou Z, Hou Y, Luo J, Shen Z, Ding J, et al. Enhanced expression of Duffy antigen receptor for chemokines by breast cancer cells attenuates growth and metastasis potential. Oncogene. (2006) 25:7201–11. doi: 10.1038/sj.onc.1209703
115. Wan W, Liu Q, Lionakis MS, Marino AP, Anderson SA, Swamydas M, et al. Atypical chemokine receptor 1 deficiency reduces atherogenesis in ApoE-knockout mice. Cardiovasc Res. (2015) 106:478–87. doi: 10.1093/cvr/cvv124
Keywords: obesity, ancestry, cardiovascular disease, cancer, disparity, cardio-oncology
Citation: Guha A, Wang X, Harris RA, Nelson A-G, Stepp D, Klaassen Z, Raval P, Cortes J, Coughlin SS, Bogdanov VY, Moore JX, Desai N, Miller DD, Lu X-Y, Kim HW and Weintraub NL (2021) Obesity and the Bidirectional Risk of Cancer and Cardiovascular Diseases in African Americans: Disparity vs. Ancestry. Front. Cardiovasc. Med. 8:761488. doi: 10.3389/fcvm.2021.761488
Received: 19 August 2021; Accepted: 21 September 2021;
Published: 18 October 2021.
Edited by:
Alessandra Ghigo, University of Turin, ItalyReviewed by:
Aly Elezaby, Stanford Healthcare, United StatesCopyright © 2021 Guha, Wang, Harris, Nelson, Stepp, Klaassen, Raval, Cortes, Coughlin, Bogdanov, Moore, Desai, Miller, Lu, Kim and Weintraub. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Avirup Guha, YWd1aGFAYXVndXN0YS5lZHU=; Neal L. Weintraub, bndlaW50cmF1YkBhdWd1c3RhLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.