 Rohan Kulkarni
Rohan Kulkarni Elizabeth Andraska1
Elizabeth Andraska1 Ryan McEnaney
Ryan McEnaney
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 05 November 2021
Sec. Atherosclerosis and Vascular Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.761007
This article is part of the Research Topic Arteriogenesis and Collateral Remodelling in Ischaemic Disease View all 7 articles
Lower extremity arterial occlusive disease (AOD) results in significant morbidity and mortality for the population, with up to 10% of patients ultimately requiring amputation. An alternative method for non-surgical revascularization which is yet to be fully understood is the optimization of the body's own natural collateral arterial network in a process known as arteriogenesis. Under conditions of conductance vessel stenosis or occlusion resulting in increased flow, shear forces, and pressure gradients within collaterals, positive remodeling occurs to increase the diameter and capacity of these vessels. The creation of a distal arteriovenous fistula (AVF) will drive increased arteriogenesis as compared to collateral formation with the occlusion of a conductance vessel alone by further increasing flow through these arterioles, demonstrating the capacity for arteriogenesis to form larger, more efficient collaterals beyond what is spontaneously achieved after arterial occlusion. Arteries rely on an extracellular matrix (ECM) composed of elastic fibers and collagens that provide stability under hemodynamic stress, and ECM remodeling is necessary to allow for increased diameter and flow conductance in mature arterial structures. When positive remodeling occurs, digestion of lamella and the internal elastic lamina (IEL) by matrix metalloproteinases (MMPs) and other elastases results in the rearrangement and thinning of elastic structures and may be replaced with disordered elastin synthesis without recovery of elastic function. This results in transmission of wall strain to collagen and potential for aneurysmal degeneration along collateral networks, as is seen in the pancreaticoduodenal artery (PDA) after celiac occlusion and inferior mesenteric artery (IMA) with concurrent celiac and superior mesenteric artery (SMA) occlusions. Further understanding into the development of collaterals is required to both better understand aneurysmal degeneration and optimize collateral formation in AOD.
The incidence of lower extremity arterial occlusive disease (AOD) has continued to increase over the past several decades resulting in significant morbidity and mortality for the population. Symptoms progress slowly after onset, however between 5 and 10 years after diagnosis 20–30% of patients will experience progressive symptoms requiring intervention with up to 10% requiring amputation (1, 2). Non-surgical therapies for symptomatic patients include behavioral and pharmacological risk factor modification and exercise therapy. Revascularization, however, depends on invasive interventions like endoluminal angioplasty and stenting or surgical bypass, as examples. Despite improving methods and technologies, revascularization procedures pose some risk to the individual and have anatomical requirements. As a result, some patients are not suitable candidates for revascularization.
Fortunately, individuals with AOD often benefit from some level of natural adaptation which manifests as development of collateral arterial networks. When large conductance arteries become obstructed, flow patterns immediately change and distal perfusion becomes increasingly dependent on collateral development (3). This process is known as arteriogenesis and involves the outward remodeling and growth of pre-existing arterioles to create an effective collateral network (4). Compared with angiogenesis, which results in the local growth and development of de novo capillaries in ischemic beds, arteriogenesis is the primary means by which blood flow is recovered to distal tissue (5). Effective, functional collateral arteries may minimize clinical symptoms of AOD and allow for conservative management of symptoms (6, 7). Functional coronary collateral networks associate with reduced mortality as well (3, 8–10). Collateral artery networks can readily be identified on arteriograms obtained from patients with peripheral AOD as demonstrated in Figure 1.
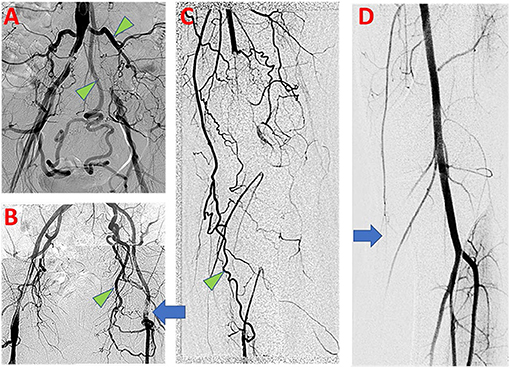
Figure 1. Clinical findings of PAD in symptomatic patients. (A) Aortoiliac arteriogram demonstrates bilateral common iliac artery occlusive disease, with evidence of compensation by large lumbar and inferior mesenteric arterial collaterals (Green arrowhead). (B) Left common femoral artery occlusive disease (blue arrow) with prominent developed left obturator artery collateral (Green arrowhead). (C) Left lower limb popliteal artery occlusion with numerous collateral arteries, including developed branches of the descending genicular artery (Green arrowhead). (D) Normal left lower limb arteriogram shows small size and limited opacification of branches of the descending genicular artery at baseline (Blue arrow).
Unfortunately, collateral vessels formed do not spontaneously restore maximal conductance to levels equal to that of the occluded artery they seek to replace. In an early experimental model of arterial occlusion, spontaneously developing collaterals only restore 35–40% of maximum conductance of the occluded artery (11) and does not exceed 50% (12). We have previously shown, however, that collateral capacity has a significantly higher ceiling than what may be achieved spontaneously after large artery occlusion, suggesting potential utility for pharmacotherapies that may augment collateral network development (13). In the following review, we describe current knowledge of mechanisms of arteriogenesis.
Collateral arteries naturally develop from pre-existing arterial connections across arterial territories that span an occluded conductance vessel (14). Although not readily identifiable by conventional arteriogram in the absence of pathology (Figure 1D), collateral anastomoses are widely present among healthy individuals and persist from development (15). Such arterial anastomoses have been shown to exist in all arterial territories, but may vary by tissue and species, leading to differences in perfusion protection in the case of arterial occlusion (16). Among mice strains, the differences in baseline collateral connections have been shown to be genetically determined (17–19). In human coronary disease patients, Hollander et al. showed that improved human coronary collateral flow indices was associated with higher palmar collateral flow indices, indicating coherence of collateral connections between different circulatory beds within an individual (20).
By Longland's classification, collateral arterial pathways are comprised of three components; the proximal, communicating, and the distal branches, more conveniently referred to as the stem, the midzone, and the re-entrant arteries (21). The midzone is the area of greatest focus because this is where the greatest degree of outward structural remodeling occurs, effectively transforming resistance vessels such as arterioles into collateral arteries which have a role in blood flow conductance.
The arterial extracellular matrix (ECM) is primarily composed of elastic fibers and various collagens and is necessary to provide structural stability to the vessel under conditions of hemodynamic stress. Elastin stores energy and distributes stress along the vessel wall while collagen, which is primarily located in the adventitia, prevents over distension of the vessel and provides a resilient framework, though it is unable to store energy (22). In the setting of excessive wall strain, such as what occurs in mid-zone collaterals during arterial occlusive disease, collagen in the adventitia limits increases in arterial diameter and over-stretching of elastic tissues. This is evidenced in the straightening of collagen fibers and is known as the “two-phase” material property which supports arterial function and stability (23–27). Unfortunately, this sets limits on potential outward diameter expansion and must be overcome for collateral enlargement to occur.
Elastin fibers consist of a heavily crosslinked, dense elastin protein core surrounded by peripherally oriented proteins and proteoglycans. These fibers have an estimated half-life approaching the human lifespan and are created predominantly in neonatal and early postnatal life with very little new elastin made during adulthood (28, 29). Their construction consists of tropopelastin monomers produced by vascular smooth muscle cells (VSMCs) and endothelial cells which then self-assemble in the extracellular space, assisted by a microfibril scaffold. Lysine residues within tropoelastin are modified by the enzyme lysyl oxidase (LOX) which leads to covalent cross-link formation, greatly contributing to the elastic polymer's resilience and durability (28–30). When damaged physiologically or pathologically, elastic fibers can be salvaged if integrity is preserved. Lost elastic fiber integrity may be replaced with disordered elastin synthesis without recovery of elastic function (31–35).
Elastic fibers are present throughout the arterial wall but are most prominently featured in the dense sheets separating rows of resident VSMCs known as lamellae. These lamellae are present within the tunica media. Between the tunica intima and media, a prominent lamina underpins the endothelium and is better known as the internal elastic lamina (IEL). These elastic lamellae contain fenestrations which vary in size and frequency depending on the arterial branch order, and allow for cellular communication, diffusion, and molecular transport (36–39). More importantly, these lamellae become active sites of arterial remodeling during postnatal growth of arterial structures and arteriogenesis (40, 41). Large elastic arteries (such as the common femoral artery) consist of thick, wrinkled elastin with small and rounded fenestrations, while secondary and more distal vessels have a fine meshwork of fibers in place of an established IEL (39). Under low pressure the IEL appears wrinkled and wavy due to redundancy and is observed to flatten at higher pressures as the artery distends.
In small arteries Type IV collagen forms the basement membrane while type I collagen bundles are abundant in the adventitia. Collagen fibers have a similar wavy appearance in all vessels. This baseline variability in ECM structure is significant in that it may have clinical consequences regarding remodeling capacity of these vessels. Alternatively, collagen fibers have half-lives as low as 2 weeks under experimental conditions of hypertension, and likely must be continually synthesized and replaced (42, 43).
As arterial occlusive disease progresses, flow patterns automatically adapt as blood flow follows the path of least resistance. This may result in large flow rate increases among inter-territorial connections. The endothelial cells are influenced by the resulting shear stress elevations imparted by this increased flow and may become activated (5, 44). Arteriogenesis is initiated in response to sustained elevations in shear force gradients (45). Through a mechanism that is not well-elucidated, vascular endothelial cells transduce the increased shear forces and initially respond with endothelial nitric oxide (NO) gene expression as well as cytokine and adhesion molecule release (46).
Assuming laminar conditions, the primary forces acting on the vascular wall are fluid shear stress (FSS) and circumferential wall stress (CWS). FSS is experienced as the frictional force of blood exerted against the vascular wall, specifically the endothelium, which is believed to act as the primary modulator of this input (47). The force of increased flow as interpreted by the endothelium is widely regarded as the initial event leading to vasodilation and downstream chronic vascular remodeling (48). The initial abrupt elevation in shear force followed by a gradual normalization is subsequently associated with remodeling of all three layers of the vascular wall, extracellular matrix, and ultimately yields vessel diameter expansion (49, 50).
Increased volume flow through a vessel has been shown to induce outward remodeling and diameter growth (51–53). As luminal diameter increases, fluid shear stress necessarily drops precipitously, and may provide a “set point” for growth. This self-regulating mechanism, which has been described as the shear stress “set point theory,” states that fluid shear stress at the endothelial level essentially will return to normal, signaling resolution of remodeling. Increases in the vessel radius leads to decreases in wall shear stress and provides the system with a negative feedback autoregulatory loop (48). The mechanism for this process is incompletely understood, however there are theories related to epigenetics and DNA hypermethylation which could influence mechanosensitivity, as well as influences from VEGFR3 in setting a vessels innate setpoint (54, 55).
Loss of a conductance artery necessarily contributes to altered pathways for blood flow due to shifting in pressure gradients. Pre-existing collateral connections between territories separated by a conductance artery occlusion will be subjected to increased flow and shear stress. The endothelium within these collateral pathways detects the shear alterations through mechanotransduction cascades that are complex and incompletely understood. The endothelial glycocalyx has been shown to be important in endothelial mechanotransduction and its absence leads to diminished arteriogenesis (56, 57). Nucleotides are released extracellularly in response to shear stress and subsequent purinergic receptor activation leads to endothelial cell mediated vasodilation (58–60). Caveolae are sites of signaling activity in response to FSS alterations in cultured endothelial cells and are necessary for flow-mediated remodeling responses (61, 62). Various integrins are implicated in shear force transduction and endothelial-mediated vasodilatory response (63, 64), and the functional remodeling required for arteriogenesis including production of elastase (64–67).
Nitric oxide has been implicated in arteriogenesis, but its role is complex. Endothelia respond to increased shear stress with increased endothelial nitric oxide synthase (eNOS) expression and subsequent production of NO, a potent vasodilator (68, 69). Purinergic receptors activated in response to elevated FSS and are necessary for flow-mediated NO production and subsequent vasodilation (59, 60, 70). Some described arteriogenesis experiments have demonstrated that early perfusion recovery depends on vasodilatory mechanisms, and NO production (71). Additionally, while loss of eNOS alone does not alter arteriogenesis, loss of inducible nitric oxide synthase (iNOS) does inhibit collateral development (72). NO has a role in remodeling via MMP activation, andinhibition of NOS results in a significant decrease in MMP activity (73). Chronic inhibition of NOS reduces diameter enlargement relative to controls and impairs vessel autoregulation to its shear stress set point (74). Notably, loss of endothelium decreases vessel response to chronic flow alterations (75).
Although short-lived vasoactive signals may produce immediate vasoconstriction or vasodilation mediated by VSMC shortening or lengthening, cessation of the signal results in return to baseline vessel diameter. Sustained signal, however, produces additional adaptation of resident VSMCs through “length autoregulation,” reorienting cell-cell and cell-ECM adhesion and thus increasing or decreasing VSMC overlap (76). Such adaptations result in maximal allowable diameter increase of the arterial segment without breakdown of the ECM, referred to as mechanoadaptation (76, 77).
Endothelial cells activated by prolonged elevations in shear stress begin to express increased levels of vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule 1 (ICAM-1). Adhesion molecule expression recruits circulating leukocytes to the developing collateral artery by promoting adherence and transmigration into the developing vessel wall (78–81). Inhibition of ICAM-1 via monoclonal antibodies directly reduces leukocyte migration, and ICAM-1 deficiency reduces collateral perfusion in response to arterial occlusion (82, 83). Endothelial VCAM-1 and ICAM-1 expression in response to flow alterations is regulated in part by thy P2Y2 purinergic receptor (84–86), and its absence results in reduced inflammatory cell recruitment and diminished collateral development (87).
Shear stress induced endothelial activation also promotes the release of several cytokines including monocyte chemoattractant protein-1 (MCP-1), TNF-α, and granulocyte macrophage colony stimulating factor (GM-CSF) which importantly attract monocytes (46, 88, 89). MCP-1 attracts monocytes to areas of active remodeling as well as upregulation of cellular adhesion molecule ICAM-1 (79). Local infusions of MCP-1 to developing collateral arteries have been shown to improve arteriogenesis. However, in an experimental model of arteriogenesis it was found that local tissue macrophage proliferation in response to MCP-1 was more important than blood-borne monocyte recruitment for mediating collateral development (90).
Adhesion molecule expression by activated endothelium and local cytokine production recruits a broad population of inflammatory cells to participate in arteriogenesis. Neutrophils appear to be recruited first, and produce cytokines such as midkine, which has been suggested to mediate VEGF release (91, 92). We have also shown that within the first 48 h, there is a significant upregulation of neutrophil elastase transcription in whole vessel analysis, suggesting that neutrophil presence may be important in initiation of ECM remodeling. Neutrophils have a short time presence outside of the circulation, however, and are generally absent later in developing collaterals, suggesting their role may be limited. Natural killer cells and CD4+ T cells have been implicated in arteriogenesis as well (93, 94).
Additionally, administration of lipopolysaccharide (LPS) stimulates TNF-a and is also capable of increasing perivascular concentrations of monocytes/macrophages leading to collateral growth and development (95). Once activated, monocytes, as well as T lymphocytes, release MMPs along with TNF-a and growth factors such as b-FGF and PDGF which serve to induce smooth muscle cell phenotype switching from a contractile to a proliferative phenotype, permitting migration (95–98). It has been shown that intra-arterial injection of MCP-1 and GM-CSF are capable of increasing collateral diameter due to recruitment of circulating monocytes, and is inhibited by monocyte depletion (82, 99, 100). Macrophages have been demonstrated as being important mediators of arteriogenesis (101). Of course, macrophages have complex functions depending on their phenotypes, and as a result may play pro-inflammatory or reparative roles. Even in the absence of flow induced changes, macrophages have been shown to promote degradation of the IEL, a key occurrence to allow for outward remodeling (78, 101, 102).
Collateral arterial growth occurs with the proliferation of resident tissue cells, which can expand the vessel mass nearly 25 times the original (103). Activated endothelium release mitogenic factors as well as promote local proliferation and ECM remodeling. Endothelial proliferation has been shown to precede that of VSMCs and may be directly induced by shear stress activation (95). VSMCs are not exposed to shear forces, and direct control of the phenotypic modulation and proliferation necessary are less well-defined. Typically, VSMCs are maintained in what is known as a contractile phenotype (oriented circumferentially around a vessel) which is typically the differentiated, quiescent form for VSMCs. These cells are separated from the intima by the IEL and contained within their local microenvironment by a basal lamina which envelopes these cells (104). VSMCs will dedifferentiate, reverting to a synthetic phenotype capable of proliferation and migration during the expansion of arteriogenesis (105). FGF and PDGF have been found to be important growth factors involved in upregulating vascular smooth muscle cell (VSMC) growth and differentiation leading to phenotype switching, actin polymerization, and maturation (106–108). Increased PDGF resulting from increased shear is suggested to be an important early factor involved in the cellular adaptation of vessels to flow mediated via the endothelium (109).
As VSMC populations migrate and expand, a neointima forms in the developing collateral artery, appearing as early as 3 days following occlusion (79). Formation of the neointima depends on ECM modifications to remove barriers to migration. VSMCs may release MMPs and plasmin activators (which convert the pro-enzyme plasminogen to active plasmin) and degrade several ECM components (110). Experimental evidence has suggested that VSMC migration and proliferation depend on MMP activity and IEL degradation (111). Unlike instances of neointima formation in intimal hyperplasia or atherosclerosis, in arteriogenesis the increased wall thickness is balanced by overall increase in luminal diameter. Eventually luminal diameter increases enough to reduce local FSS back to within an acceptable range, the mitogenic stimuli dissipate, and VSMCs return to a contractile phenotype.
Diameter increase along with ECM remodeling requires the rearrangement of elastic tissue in the internal elastic lamina. Ultimately, elastolysis allows for vessel diameter enlargement, and ECM remodeling is necessary to allow for increased diameter and flow conductance in mature arterial structures (112). In the developing collateral artery, this appears as fenestration enlargement of the IEL, such that outward remodeling during collateral development is achieved while simultaneously maintaining IEL continuity. Others have demonstrated that the fenestrations are the active sites of both outward and inward remodeling of the IEL (40, 41, 113, 114). We have found that after femoral artery ligation with distal arteriovenous fistula creation (FAL + AVF), an initially dense elastin network transforms into a loose meshwork with the general pattern of IEL reorganization, demonstrating increases in fenestration size bordered by cords of branching elastic fibers (Figure 2). Increases in circulating desmosine (an elastin breakdown product) within 1 week after FAL + AVF were found which disappear after 2 weeks, (Andraska et al. submitted to the current issue) supporting that elastin degradation is important early in arteriogenesis. It has been shown that collateral artery development requires activated MMP-2 and MMP-9, and no fragmentation of the IEL noted when administered MMP inhibitors (115). Cathespins, also involved in vascular remodeling, have elastolytic properties as well and are found to be upregulated in developing collateral arteries (116–119).
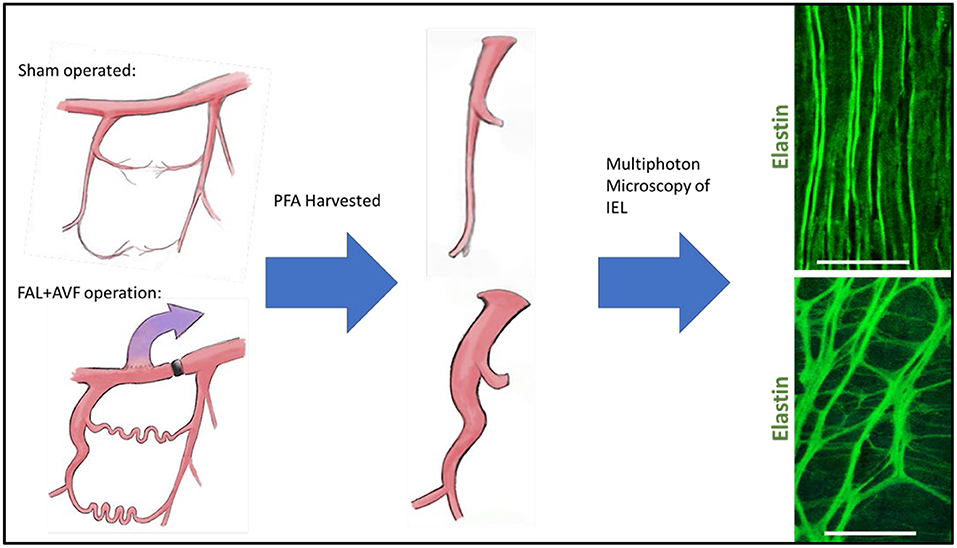
Figure 2. Internal elastic lamina (IEL) remodeling in a rodent model of arteriogenesis. Rat hindlimbs were rendered ischemic by placement of femoral artery ligation with distal arteriovenous fistula or treated as controls with sham operation (incision and exposure without further arterial manipulation) as we have previously described (13). Profunda femoral arteries were harvested at 12 weeks and imaged with multiphoton microscopy. Note the wrinkled topology of the IEL with small regular fenestrations in the control vessel, which is lost in the remodeled vessel as the fenestrations are greatly enlarged. Images acquired with Olympus FV1000MPE utilizing 830 nm laser. Bar = 50 um. Multiphoton microscopy methods described previously (39).
The damage to the elastic fibers during arteriogenesis must be limited in order to maintain fiber integrity and prevent loss of IEL continuity. Tissue inhibitor of metalloproteinases (TIMP1) and plasminogen activator inhibitor 1 (PAI-1) also play vital roles in collateral development by inhibiting MMP function and preventing excessive breakdown of ECM (111). It would seem that elastic fiber preservation and repair is necessary during arteriogenesis, given that construction of new elastic fibers construction is unlikely to occur.
Cross linking of elastin and collagen fibers is mediated via the enzyme lysyl oxidase (LOX) and is essential to maintaining the integrity of the ECM. LOX expression is increased in remodeling of FAL + AVF arterioles and we found that inhibiting LOX [using β-aminopropionitrile (BAPN)] resulted in rapid fragmentation and loss of continuity of the IEL from collateral arteries. This suggests the role of LOX in the repair and stabilization of proteolytically digested elastic fibers during arteriole remodeling. This suggests the proteolytic balance between breakdown and repair during remodeling, possibly related to cross linking of newly synthesized tropoelastin monomers in the later stages of arteriogenesis (31–34). Notably, increased tropoelastin expression has been seen in models of vascular remodeling previously (120). This may be necessary for ECM stabilization in a setting where whole new fibers cannot be constructed. As such, repaired elastin polymers may not achieve full strength as some original peptide bonds cannot be recovered, and total elastin content cannot keep pace with increasing vessel size resulting in thinning of the IEL (32).
Loss of elastic laminar definition is also a consistent histologic feature of arterial aneurysm development. In some cases, the development of specific types of arterial aneurysms have been linked to flow-mediated remodeling of the IEL, sometimes occurring within collateral arterial networks. For instance, aneurysm degeneration has been observed in mesenteric arterial collaterals in response to isolated celiac occlusion [resulting in pancreaticoduodenal arterial aneurysms (121–123)] or concurrent celiac and superior mesenteric arterial occlusion [resulting in inferior mesenteric arterial aneurysms (124, 125)]. Disruption in the IEL and changes in the media are essential features in the development and propagation of human cerebral aneurysms (126–128). Morphologic assessments of the aneurysm wall have demonstrated malalignment in medial smooth muscle cells and accumulation of macrophages, MMP-9, and myeloperoxidase, essential components of elastin and IEL degradation (129, 130). Degeneration of the IEL and longitudinal elongation (conversion from a contractile to a “synthetic” phenotype) is similarly found at sites of intracerebral aneurysms as well (131).
In the case of cerebral aneurysm pathogenesis, focally increased fluid shear stress provides the local impetus for IEL degradation and enlargement of fenestrations (132–134). It is possible that this would mechanically weaken the vessel wall and predispose to aneurysmal degeneration (132). Notably, cerebral aneurysms are frequently associated with elevated shear stress, and occur more frequently in association with a carotid artery occlusion (130, 135–137). Given the irreplaceable nature of elastic fibers, aggressive diameter expansion risks exhausting local baseline elastin content which can create weakened and aneurysm prone collaterals. The underlying pathology of what may otherwise appear to be disparate manifestations of aneurysmal disease may relate to mechanisms of remodeling like those of arteriogenesis.
The use of arterial ligation in animal models of arteriogenesis typically creates an acute ischemia and is an important limitation regarding extrapolating animal models to human disease which tends to develop chronically. Numerous studies in larger animals, however, have employed surgically placed ameroid constrictors as a method to simulate more gradual arterial occlusion. Human arterial occlusive disease is variable, with (perhaps most commonly) a slowly worsening stenosis in the case of chronic atherosclerosis, but also via sudden arterial occlusion with thromboembolism or in situ thrombosis, or even arterial transection in trauma. Evidence of collateral artery formation may be found in all instances.
Interventions are currently being directed toward improving arteriogenesis as well as angiogenesis and are under clinical investigation with the hope that these will lead to more effective and non-surgical therapies for AOD. Unfortunately, however, current knowledge of arteriogenesis is limited, and methods to enhance the inflammation and positive remodeling of collateral arteries through growth factor or cytokine supplementation are known to have opposing effects by exacerbating atherosclerosis (138). Developing effective therapies to augment arteriogenesis yet not promote atherogenesis requires more detailed understanding of the molecular mechanisms involved.
Arteriogenesis is a complex mechanism for collateral arterial pathways to develop quickly into larger and higher capacity vessels capable of effectively perfusing tissues distal to a conductance vessel occlusion (Figure 3). Increased fluid shear stress, typically caused by flow adaptations in AOD due to large vessel occlusion, initiates endothelial activation ultimately resulting in a cascade of inflammation, cellular proliferation, migration, and tissue remodeling. While details of the molecular signaling processes underpinning arteriogenesis are continually emerging, effective methods to improve collateral development as a clinically useful therapy remain elusive. Continued efforts aimed at manipulating and enhancing functional collaterals promises to reveal possible therapies to medically revascularize patients suffering from practically any manifestation of AOD. However, caution will likely be necessary once adapting to clinical use, as there are shared pathways between arteriogenesis and atherogenesis, and outward remodeling of arteriogenesis may produce weakened ECM structures that theoretically would be at risk for aneurysmal degeneration. Further investigations are required into this field to fully appreciate the molecular cascades involved in linking these processes and constitute potential avenues for continued investigation.
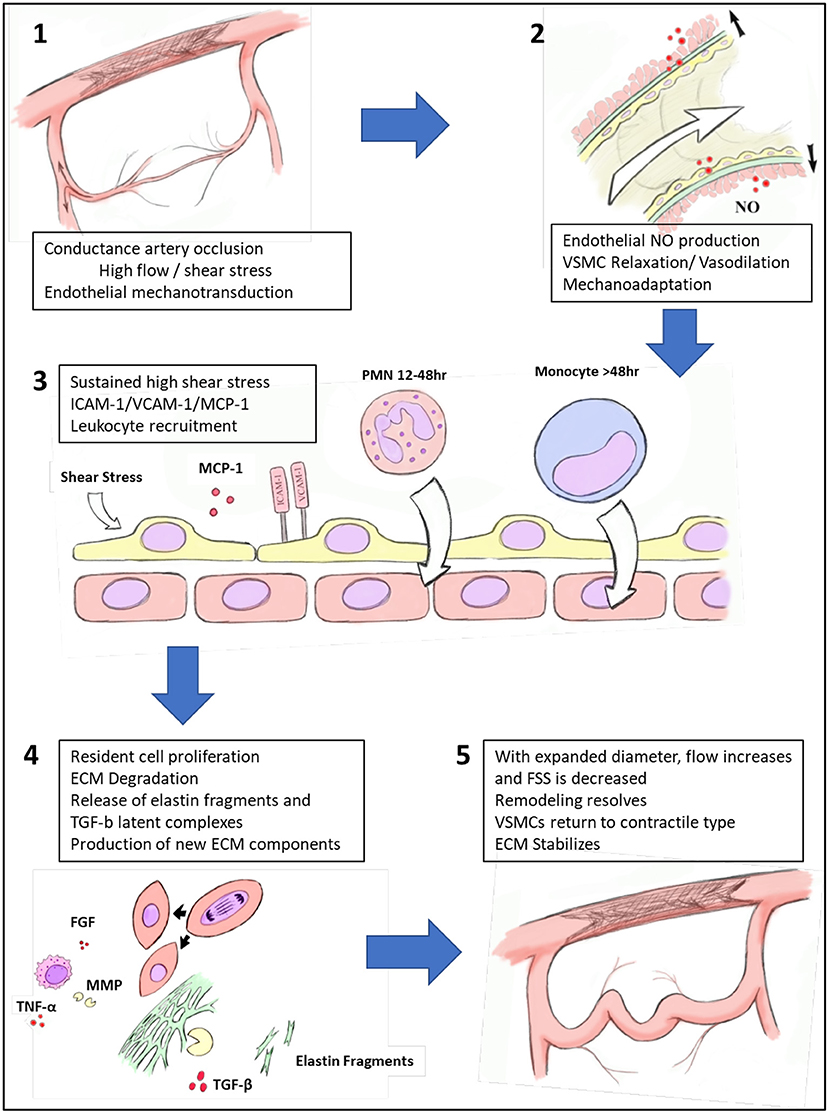
Figure 3. Process of collateral artery recruitment and remodeling. Following occlusion of a conductance vessel, there is immediate increase in flow across pre-existing collaterals. The resulting elevated fluid shear stress (FSS) is recognized by endothelial cells via mechanoreceptors, which leads to nitric oxide (NO) production and subsequent relaxation of vascular smooth muscle cells (VSMCs) and then vasodilation. Prolonged vasodilation produces mechanoadaptation of collaterals. With sustained elevation of FSS, endothelial cells are activated to express intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1) and monocyte chemoattractant protein-1 (MCP1) which recruit inflammatory cells to the developing collateral artery. With the aid of a population of perivascular macrophages, growth factors and cytokines such as tumor necrosis factor-alpha (TNF-α), fibroblast growth factor (FGF) are produced, increasing phenotypic modification of VSMCs and proliferation. Elastolytic enzymes such as matrix metalloproteinases (MMPs) are produced, which partially degrade the elastic framework, releasing latent transforming growth factor-beta (TGF-β) complexes. As diameter expands, FSS decreases and the pressure for outward remodeling dissipates. VSMCs return to differentiated phenotype and collateral artery extracellular matrix (ECM) stabilizes.
Conceptualization and design of the study were performed by RM and RK. Original draft written by RK. RM and EA wrote sections of the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by a grant from the Department of Veterans Affairs (IK-2BX003509) and in part by the Vascular Cures Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Cronenwett JL, Warner KG, Zelenock GB, Whitehouse WM, Graham LM, Lindenauer SM, et al. Intermittent claudication: current results of nonoperative management. Arch Surg. (1984) 119:430–6. doi: 10.1001/archsurg.1984.01390160060012
2. Imparato AM, Kim G-E, Davidson T, Crowley JG. Intermittent claudication: its natural course. Surgery. (1975) 78:795–9.
3. Berry C, Balachandran KP, L'Allier PL, Lespérance J, Bonan R, Oldroyd KG. Importance of collateral circulation in coronary heart disease. Eur Heart J. (2007) 28:278–91. doi: 10.1093/eurheartj/ehl446
4. He L, Liu Q, Hu T, Huang X, Zhang H, Tian X, et al. Genetic lineage tracing discloses arteriogenesis as the main mechanism for collateral growth in the mouse heart. Cardiovasc Res. (2016) 109:419–30. doi: 10.1093/cvr/cvw005
5. Scholz D, Ziegelhoeffer T, Helisch A, Wagner S, Friedrich C, Podzuweit T, et al. Contribution of arteriogenesis and angiogenesis to postocclusive hindlimb perfusion in mice. J Mol Cell Cardiol. (2002) 34:775–87. doi: 10.1006/jmcc.2002.2013
6. Komshian SR, Lu K, Pike SL, Siracuse JJ. Infrainguinal open reconstruction: a review of surgical considerations and expected outcomes. Vasc Health Risk Manag. (2017) 13:161–8. doi: 10.2147/VHRM.S106898
8. Schaper W. Collateral vessels reduce mortality. Eur Heart J. (2012) 33:564–6. doi: 10.1093/eurheartj/ehr385
9. Meier P, Hemingway H, Lansky AJ, Knapp G, Pitt B, Seiler C. The impact of the coronary collateral circulation on mortality: a meta-analysis. Eur Heart J. (2012) 33:614–21. doi: 10.1093/eurheartj/ehr308
10. Meier P, Gloekler S, Zbinden R, Beckh S, de Marchi SF, Zbinden S, et al. Beneficial effect of recruitable collaterals: a 10-year follow-up study in patients with stable coronary artery disease undergoing quantitative collateral measurements. Circulation. (2007) 116:975–83. doi: 10.1161/CIRCULATIONAHA.107.703959
11. Schaper W, Flameng W, Winkler B, Wüsten B, Türschmann W, Neugebauer G, et al. Quantification of collateral resistance in acute and chronic experimental coronary occlusion in the dog. Circ Res. (1976) 39:371–7. doi: 10.1161/01.RES.39.3.371
12. Schaper W. Quo vadis collateral blood flow? A commentary on a highly cited paper. Cardiovasc Res. (2000) 45:220–3. doi: 10.1016/S0008-6363(99)00332-6
13. McEnaney RM, McCreary D, Tzeng E. A modified rat model of hindlimb ischemia for augmentation and functional measurement of arteriogenesis. J Biol Methods. (2018) 5:e89. doi: 10.14440/jbm.2018.234
15. Faber JE, Chilian WM, Deindl E, van Royen N, Simons M. A brief etymology of the collateral circulation. Arterioscler Thromb Vasc Biol. (2014) 34:1854–9. doi: 10.1161/ATVBAHA.114.303929
16. Helisch A, Schaper W. Arteriogenesis: the development and growth of collateral arteries. Microcirculation. (2003) 10:83–97. doi: 10.1080/mic.10.1.83.97
17. Sealock R, Zhang H, Lucitti JL, Moore SM, Faber JE. Congenic fine-mapping identifies a major causal locus for variation in the native collateral circulation and ischemic injury in brain and lower extremity. Circ Res. (2014) 114:660–71. doi: 10.1161/CIRCRESAHA.114.302931
18. Wang S, Zhang H, Dai X, Sealock R, Faber JE. Genetic architecture underlying variation in extent and remodeling of the collateral circulation. Circ Res. (2010) 107:558–68. doi: 10.1161/CIRCRESAHA.110.224634
19. Dokun A, Keum S, Hazarika S, Li Y, Lamonte G, Wheeler F, et al. A quantitative trait locus (LSq-1) on mouse chromosome 7 is linked to the absence of tissue loss after surgical hindlimb ischemia. Circulation. (2008) 117:1207–15. doi: 10.1161/CIRCULATIONAHA.107.736447
20. Hollander MR, Jansen MF, Schumacher SP, Stuijfzand WJ, van Leeuwen MAH, van de Ven PM, et al. Coronary collateral flow index is correlated with the palmar collateral flow index: indicating systemic collateral coherence in individual patients-brief report. Arterioscler Thromb Vasc Biol. (2021) 41:1830–6. doi: 10.1161/ATVBAHA.121.316092
21. Longland C. The collateral circulation of the limb: arris and gale lecture delivered at the royal college of surgeons of england on 4th February, 1953. Ann Royal Coll Surg Engl. (1953) 13:161.
22. Wolinsky H, Glagov S. Structural basis for the static mechanical properties of the aortic media. Circ Res. (1964) 14:400–13. doi: 10.1161/01.RES.14.5.400
23. Wagenseil JE, Mecham RP. Vascular extracellular matrix and arterial mechanics. Physiol Rev. (2009) 89:957–89. doi: 10.1152/physrev.00041.2008
24. Shadwick RE. Mechanical design in arteries. J Exp Biol. (1999) 202(Pt 23):3305–13. doi: 10.1242/jeb.202.23.3305
25. Roach MR, Burton AC. The reason for the shape of the distensibility curves of arteries. Can J Biochem Physiol. (1957) 35:681–90. doi: 10.1139/o57-080
26. Krasny W, Morin C, Magoariec H, Avril S. A comprehensive study of layer-specific morphological changes in the microstructure of carotid arteries under uniaxial load. Acta Biomater. (2017) 57:342–51. doi: 10.1016/j.actbio.2017.04.033
27. Chow MJ, Turcotte R, Lin CP, Zhang Y. Arterial extracellular matrix: a mechanobiological study of the contributions and interactions of elastin and collagen. Biophys J. (2014) 106:2684–92. doi: 10.1016/j.bpj.2014.05.014
28. Shapiro SD, Endicott SK, Province MA, Pierce JA, Campbell EJ. Marked longevity of human lung parenchymal elastic fibers deduced from prevalence of D-aspartate and nuclear weapons-related radiocarbon. J Clin Invest. (1991) 87:1828–34. doi: 10.1172/JCI115204
29. Wagenseil JE, Mecham RP. New insights into elastic fiber assembly. Birth Defects Res C Embryo Today. (2007) 81:229–40. doi: 10.1002/bdrc.20111
30. Schräder CU, Heinz A, Majovsky P, Karaman Mayack B, Brinckmann J, Sippl W, et al. Elastin is heterogeneously cross-linked. J Biol Chem. (2018) 293:15107–19. doi: 10.1074/jbc.RA118.004322
31. Morris SM, Thomas KM, Rich CB, Stone PJ. Degradation and repair of elastic fibers in rat lung interstitial fibroblast cultures. Anat Rec. (1998) 250:397–407. doi: 10.1002/(SICI)1097-0185(199804)250:4<397::AID-AR2>3.0.CO;2-V
32. Stone PJ, Morris SM, Martin BM, McMahon MP, Faris B, Franzblau C. Repair of protease-damaged elastin in neonatal rat aortic smooth muscle cell cultures. J Clin Invest. (1988) 82:1644–54. doi: 10.1172/JCI113776
33. Stone PJ, Morris SM, Thomas KM, Schuhwerk K, Mitchelson A. Repair of elastase-digested elastic fibers in acellular matrices by replating with neonatal rat-lung lipid interstitial fibroblasts or other elastogenic cell types. Am J Respir Cell Mol Biol. (1997) 17:289–301. doi: 10.1165/ajrcmb.17.3.2597
34. Alves C, Araujo AD, Oliveira CL, Imsirovic J, Bartolak-Suki E, Andrade JS, et al. Homeostatic maintenance via degradation and repair of elastic fibers under tension. Sci Rep. (2016) 6:27474. doi: 10.1038/srep27474
35. Lavin B, Lacerda S, Andia ME, Lorrio S, Bakewell R, Smith A, et al. Tropoelastin: an in vivo imaging marker of dysfunctional matrix turnover during abdominal aortic dilation. Cardiovasc Res. (2020) 116:995–1005. doi: 10.1093/cvr/cvz178
36. Tada S, Tarbell JM. Internal elastic lamina affects the distribution of macromolecules in the arterial wall: a computational study. Am J Physiol Heart Circ Physiol. (2004) 287:H905–13. doi: 10.1152/ajpheart.00647.2003
37. Sandow SL, Gzik DJ, Lee RM. Arterial internal elastic lamina holes: relationship to function? J Anat. (2009) 214:258–66. doi: 10.1111/j.1469-7580.2008.01020.x
38. Kirby BS, Bruhl A, Sullivan MN, Francis M, Dinenno FA, Earley S. Robust internal elastic lamina fenestration in skeletal muscle arteries. PLoS ONE. (2013) 8:e54849. doi: 10.1371/journal.pone.0054849
39. McCreary DD, Skirtich NO, Andraska EA, Tzeng E, McEnaney RM. Survey of the extracellular matrix architecture across the rat arterial tree. JVS Vasc Sci. (2021). doi: 10.1016/j.jvssci.2021.08.001
40. Roach MR. The pattern of elastin in the aorta and large arteries of mammals. Ciba Found Symp. (1983) 100:37–55. doi: 10.1002/9780470720813.ch4
41. Wong LC, Langille BL. Developmental remodeling of the internal elastic lamina of rabbit arteries: effect of blood flow. Circ Res. (1996) 78:799–805. doi: 10.1161/01.RES.78.5.799
42. Nissen R, Cardinale GJ, Udenfriend S. Increased turnover of arterial collagen in hypertensive rats. Proc Natl Acad Sci USA. (1978) 75:451–3. doi: 10.1073/pnas.75.1.451
43. Ooshima A, Fuller GC, Cardinale GJ, Spector S, Udenfriend S. Increased collagen synthesis in blood vessels of hypertensive rats and its reversal by antihypertensive agents. Proc Natl Acad Sci USA. (1974) 71:3019–23. doi: 10.1073/pnas.71.8.3019
44. Gray C, Packham IM, Wurmser F, Eastley NC, Hellewell PG, Ingham PW, et al. Ischemia is not required for arteriogenesis in zebrafish embryos. Arterioscl Thromb Vasc Biol. (2007) 27:2135–41. doi: 10.1161/ATVBAHA.107.143990
45. Buschmann I, Schaper W. The pathophysiology of the collateral circulation (arteriogenesis). J Pathol. (2000) 190:338–42. doi: 10.1002/(SICI)1096-9896(200002)190:3<338::AID-PATH594>3.0.CO;2-7
46. Hoefer IE, den Adel B, Daemen MJ. Biomechanical factors as triggers of vascular growth. Cardiovasc Res. (2013) 99:276–83. doi: 10.1093/cvr/cvt089
47. Girard PR, Nerem RM. Shear stress modulates endothelial cell morphology and F-actin organization through the regulation of focal adhesion-associated proteins. J Cell Physiol. (1995) 163:179–93. doi: 10.1002/jcp.1041630121
48. Kamiya A, Togawa T. Adaptive regulation of wall shear stress to flow change in the canine carotid artery. Am J Physiol Heart Circ Physiol. (1980) 239:H14–21. doi: 10.1152/ajpheart.1980.239.1.H14
49. Tulis DA, Unthank JL, Prewitt RL. Flow-induced arterial remodeling in rat mesenteric vasculature. Am J Physiol Heart Circ Physiol. (1998) 274:H874–82. doi: 10.1152/ajpheart.1998.274.3.H874
50. Tuttle JL, Nachreiner RD, Bhuller AS, Condict KW, Connors BA, Herring BP, et al. Shear level influences resistance artery remodeling: wall dimensions, cell density, and eNOS expression. Am J Physiol Heart Circ Physiol. (2001) 281:H1380–9. doi: 10.1152/ajpheart.2001.281.3.H1380
51. Holman E, Taylor G. Problems in the dynamics of blood flow: II. Pressure relations at site of an arteriovenous fistula. Angiology. (1952) 3:415–30. doi: 10.1177/000331975200300601
52. Liebow A. Situations which lead to changes in vascular patterns. Am Physiol Soc Handbook Physiol Circ. (1963) 1251–76.
53. Schenk W, Martin J, Leslie M, Portin B. The regional hemodynamics of chronic experimental arteriovenous fistulas. Plastic Reconst Surg. (1960) 25:387. doi: 10.1097/00006534-196004000-00029
54. Baeyens N, Nicoli S, Coon BG, Ross TD, Van den Dries K, Han J, et al. Vascular remodeling is governed by a VEGFR3-dependent fluid shear stress set point. Elife. (2015) 4:e04645. doi: 10.7554/eLife.04645
55. Heuslein JL, Gorick CM, Song J, Price RJ. DNA methyltransferase 1–dependent DNA hypermethylation constrains arteriogenesis by augmenting shear stress set point. J Am Heart Assoc. (2017) 6:e007673. doi: 10.1161/JAHA.117.007673
56. Grundmann S, Schirmer SH, Hekking LH, Post JA, Ionita MG, de Groot D, et al. Endothelial glycocalyx dimensions are reduced in growing collateral arteries and modulate leucocyte adhesion in arteriogenesis. J Cell Mol Med. (2009) 13:3463–74. doi: 10.1111/j.1582-4934.2009.00735.x
57. Potter DR, van Teeffelen J, Vink H, van den Berg BM. Perturbed mechanotransduction by endothelial surface glycocalyx modification greatly impairs the arteriogenic process. Am J Physiol Heart Circ Physiol. (2015) 309:H711–7. doi: 10.1152/ajpheart.00257.2015
58. Wang S, Iring A, Strilic B, Juarez J, Kaur H, Troidl K, et al. P2Y2 and Gq/G11 control blood pressure by mediating endothelial mechanotransduction. J Clin Invest. (2015) 125:3077–86. doi: 10.1172/JCI81067
59. Raqeeb A, Sheng J, Ao N, Braun AP. Purinergic P2Y2 receptors mediate rapid Ca(2+) mobilization, membrane hyperpolarization and nitric oxide production in human vascular endothelial cells. Cell Calcium. (2011) 49:240–8. doi: 10.1016/j.ceca.2011.02.008
60. Yamamoto K, Sokabe T, Matsumoto T, Yoshimura K, Shibata M, Ohura N, et al. Impaired flow-dependent control of vascular tone and remodeling in P2X4-deficient mice. Nat Med. (2006) 12:133–7. doi: 10.1038/nm1338
61. Boyd NLPH, Yi H, Boo YC, Sorescu GP, Sykes M, Jo H. Chonic shear induces caveolae formation and alters ERK and Akt responses in endothelial cells. Am J Physiol Heart Circ Physiol. (2003) 285:H1113–22. doi: 10.1152/ajpheart.00302.2003
62. Yu J, Bergaya S, Murata T, Alp IF, Bauer MP, Lin MI, et al. Direct evidence for the role of caveolin-1 and caveolae in mechanotransduction and remodeling of blood vessels. J Clin Invest. (2006) 116:1284–91. doi: 10.1172/JCI27100
63. Lucitti JL, Mackey JK, Morrison JC, Haigh JJ, Adams RH, Faber JE. Formation of the collateral circulation is regulated by vascular endothelial growth factor-A and a disintegrin and metalloprotease family members 10 and 17. Circ Res. (2012) 111:1539–50. doi: 10.1161/CIRCRESAHA.112.279109
64. Henning C, Branopolski A, Schuler D, Dimitroulis D, Huelsemann P, Nicolaus C, et al. Requirement of beta1 integrin for endothelium-dependent vasodilation and collateral formation in hindlimb ischemia. Sci Rep. (2019) 9:16931. doi: 10.1038/s41598-019-53137-x
65. Hennig T, Mogensen C, Kirsch J, Pohl U, Gloe T. Shear stress induces the release of an endothelial elastase: role in integrin α(v)β(3)-mediated FGF-2 release. J Vasc Res. (2011) 48:453–64. doi: 10.1159/000327009
66. Cai WJ, Li MB, Wu X, Wu S, Zhu W, Chen D, et al. Activation of the integrins a5b1 and avb3 and focal adhesion kinase (FAK) during arteriogenesis. Mol Cell Biochem. (2009) 322:161–9. doi: 10.1007/s11010-008-9953-8
67. Gloe T, Sohn HY, Meininger GA, Pohl U. Shear stress-induced release of basic fibroblast growth factor from endothelial cells is mediated by matrix interaction via integrin alpha(v)beta3. J Biol Chem. (2002) 277:23453–8. doi: 10.1074/jbc.M203889200
68. Ignarro LJ, Napoli C, Loscalzo J. Nitric oxide donors and cardiovascular agents modulating the bioactivity of nitric oxide: an overview. Circ Res. (2002) 90:21–8. doi: 10.1161/hh0102.102330
69. Topper JN, Cai J, Falb D, Gimbrone MA Jr. Identification of vascular endothelial genes differentially responsive to fluid mechanical stimuli: cyclooxygenase-2, manganese superoxide dismutase, and endothelial cell nitric oxide synthase are selectively up-regulated by steady laminar shear stress. Proc Natl Acad Sci USA. (1996) 93:10417–22. doi: 10.1073/pnas.93.19.10417
70. Rosenmeier JB, Yegutkin GG, González-Alonso J. Activation of ATP/UTP-selective receptors increases blood flow and blunts sympathetic vasoconstriction in human skeletal muscle. J Physiol. (2008) 586:4993–5002. doi: 10.1113/jphysiol.2008.155432
71. Mees B, Wagner S, Ninci E, Tribulova S, Martin S, van Haperen R, et al. Endothelial nitric oxide synthase activity is essential for vasodilation during blood flow recovery but not for arteriogenesis. Arterioscler Thromb Vasc Biol. (2007) 27:1926–33. doi: 10.1161/ATVBAHA.107.145375
72. Troidl K, Tribulova S, Cai WJ, Rüding I, Apfelbeck H, Schierling W, et al. Effects of endogenous nitric oxide and of DETA NONOate in arteriogenesis. J Cardiovasc Pharmacol. (2010) 55:153–60. doi: 10.1097/FJC.0b013e3181c9556f
73. Tronc F, Mallat Z, Lehoux S, Wassef M, Esposito B, Tedgui A. Role of matrix metalloproteinases in blood flow-induced arterial enlargement: interaction with NO. Arterioscler Thromb Vasc Biol. (2000) 20:E120–6. doi: 10.1161/01.ATV.20.12.e120
74. Tronc F, Wassef M, Esposito B, Henrion D, Glagov S, Tedgui A. Role of NO in flow-induced remodeling of the rabbit common carotid artery. Arterioscler Thromb Vasc Biol. (1996) 16:1256–62. doi: 10.1161/01.ATV.16.10.1256
75. Lasch M, Kleinert EC, Meister S, Kumaraswami K, Buchheim JI, Grantzow T, et al. Extracellular RNA released due to shear stress controls natural bypass growth by mediating mechanotransduction in mice. Blood. (2019) 134:1469–79. doi: 10.1182/blood.2019001392
76. Martinez-Lemus LA, Hill MA, Bolz SS, Pohl U, Meininger GA. Acute mechanoadaptation of vascular smooth muscle cells in response to continuous arteriolar vasoconstriction: implications for functional remodeling. Faseb J. (2004) 18:708–10. doi: 10.1096/fj.03-0634fje
77. Martinez-Lemus LA, Hill MA, Meininger GA. The plastic nature of the vascular wall: a continuum of remodeling events contributing to control of arteriolar diameter and structure. Physiology. (2009) 24:45–57. doi: 10.1152/physiol.00029.2008
78. Kadam AA, Gersch RP, Rosengart TK, Frame MD. Inflammatory monocyte response due to altered wall shear stress in an isolated femoral artery model. J Biol Meth. (2019) 6:e109. doi: 10.14440/jbm.2019.274
79. Scholz D, Ito W, Fleming I, Deindl E, Sauer A, Wiesnet M, et al. Ultrastructure and molecular histology of rabbit hind-limb collateral artery growth (arteriogenesis). Virchows Arch. (2000) 436:257–70. doi: 10.1007/s004280050039
80. O'Keeffe LM, Muir G, Piterina AV, McGloughlin T. Vascular cell adhesion molecule-1 expression in endothelial cells exposed to physiological coronary wall shear stresses. J Biomech Eng. (2009) 131:081003. doi: 10.1115/1.3148191
81. Pipp F, Boehm S, Cai W, Adili F, Ziegler B, Karanovic G, et al. Elevated fluid shear stress enhances postocclusive collateral artery growth and gene expressioinin the pig hind limb. Arterioscler Thromb Vasc Biol. (2004) 24:1664–8. doi: 10.1161/01.ATV.0000138028.14390.e4
82. Hoefer IE, van Royen N, Rectenwald JE, Deindl E, Hua J, Jost M, et al. Arteriogenesis proceeds via ICAM-1/Mac-1- mediated mechanisms. Circ Res. (2004) 94:1179–85. doi: 10.1161/01.RES.0000126922.18222.F0
83. Nie Q, Fan J, Haraoka S, Shimokama T, Watanabe T. Inhibition of mononuclear cell recruitment in aortic intima by treatment with anti-ICAM-1 and anti-LFA-1 monoclonal antibodies in hypercholesterolemic rats: implications of the ICAM-1 and LFA-1 pathway in atherogenesis. Lab Invest. (1997) 77:469–82.
84. Seye CI, Yu N, Jain R, Kong Q, Minor T. The P2Y2 nucleotide receptor mediates UTP-induced vascular cell adhesion molecule-1 expression in coronary artery endothelial cells. J Biol Chem. (2003) 278:24960–5. doi: 10.1074/jbc.M301439200
85. Seye C, Yu N, Gonzalez F, Erb L, Weisman G. The P2Y2 nucleotide receptor mediates vascular cell adhesion molecule-1 expression through interaction with VEGF receptor-2 (KDR/Flk-1). J Biol Chem. (2004) 279:35679–86. doi: 10.1074/jbc.M401799200
86. Seye CI, Agca Y, Agca C, Derbigny W. P2Y2 receptor-mediated lymphotoxin-alpha (LTA) secretioin regulates intercellular cell adhesion molecule (ICAM)-1 expression in vascular smooth muscle cells. J Biol Chem. (2012) 287:10535–43. doi: 10.1074/jbc.M111.313189
87. McEnaney R, Shukla A, Madigan M, Sachdev U, Tzeng E. P2Y2 nucleotide receptor mediates arteriogenesis in a murine model of hind limb ischemia. J Vasc Surg. (2014) 63:216–25. doi: 10.1016/j.jvs.2014.06.112
88. Shyy Y-J, Wickham LL, Hagan JP, Hsieh H-J, Hu Y-L, Telian SH, et al. Human monocyte colony-stimulating factor stimulates the gene expression of monocyte chemotactic protein-1 and increases the adhesion of monocytes to endothelial monolayers. J Clin Invest. (1993) 92:1745–51. doi: 10.1172/JCI116762
89. Schaper J, König R, Franz D, Schaper W. The endothelial surface of growing coronary collateral arteries. Intimal margination and diapedesis of monocytes. Virchows Archiv A. (1976) 370:193–205. doi: 10.1007/BF00427580
90. Khmelewski E, Becker A, Meinertz T, Ito WD. Tissue resident cells play a dominant role in arteriogenesis and concomitant macrophage accumulation. Circ Res. (2004) 95:E56–64. doi: 10.1161/01.RES.0000143013.04985.E7
91. Lautz T, Lasch M, Borgolte J, Troidl K, Pagel JI, Caballero-Martinez A, et al. Midkine controls arteriogenesis by regulating the bioavailability of vascular endothelial growth factor a and the expression of nitric oxide synthase 1 and 3. EBio Med. (2018) 27:237–46. doi: 10.1016/j.ebiom.2017.11.020
92. Ohki Y, Heissig B, Sato Y, Akiyama H, Zhu Z, Hicklin DJ, et al. Granulocyte colony-stimulating factor promotes neovascularization by releasing vascular endothelial growth factor from neutrophils. Faseb J. (2005) 19:2005–7. doi: 10.1096/fj.04-3496fje
93. Stabile E, Burnett MS, Watkins C, Kinnaird T, Bachis A, la Sala A, et al. Impaired arteriogenic response to acute hindlimb ischemia in CD4-knockout mice. Circulation. (2003) 108:205–10. doi: 10.1161/01.CIR.0000079225.50817.71
94. van Weel V, Toes RE, Seghers L, Deckers MM, de Vries MR, Eilers PH. Natural killer cells and CD4+ T-cells modulate collateral artery development. Arterioscler Thromb Vasc Biol. (2007) 27:2310–8. doi: 10.1161/ATVBAHA.107.151407
95. Arras M, Ito WD, Scholz D, Winkler B, Schaper J, Schaper W. Monocyte activation in angiogenesis and collateral growth in the rabbit hindlimb. J Clin Invest. (1998) 101:40–50. doi: 10.1172/JCI119877
96. Heil M, Ziegelhoeffer T, Wagner S, Fernández B, Helisch A, Martin S, et al. Collateral artery growth (arteriogenesis) after experimental arterial occlusion is impaired in mice lacking CC-chemokine receptor-2. Circ Res. (2004) 94:671–7. doi: 10.1161/01.RES.0000122041.73808.B5
97. Kusch A, Tkachuk S, Lutter S, Haller H, Dietz R, Lipp M, et al. Monocyte-expressed urokinase regulates human vascular smooth muscle cell migration in a coculture model. Biol Chem. (2002) 383:217–21. doi: 10.1515/BC.2002.022
98. Rensen S, Doevendans P, Van Eys G. Regulation and characteristics of vascular smooth muscle cell phenotypic diversity. Netherlands Heart J. (2007) 15:100–8. doi: 10.1007/BF03085963
99. Ito WD, Arras M, Winkler B, Scholz D, Schaper J, Schaper W. Monocyte chemotactic protein-1 increases collateral and peripheral conductance after femoral artery occlusion. Circ Res. (1997) 80:829–37. doi: 10.1161/01.RES.80.6.829
100. Hoefer IE, van Royen N, Buschmann IR, Piek JJ, Schaper W. Time course of arteriogenesis following femoral artery occlusion in the rabbit. Cardiovasc Res. (2001) 49:609–17. doi: 10.1016/S0008-6363(00)00243-1
101. Bergmann CE, Hoefer IE, Meder B, Roth H, van Royen N, Breit SM, et al. Arteriogenesis depends on circulating monocytes and macrophage accumulation and is severely depressed in op/op mice. J Leukoc Biol. (2006) 80:59–65. doi: 10.1189/jlb.0206087
102. Chillo O, Kleinert EC, Lautz T, Lasch M, Pagel JI, Heun Y, et al. Perivascular mast cells govern shear stress-induced arteriogenesis by orchestrating leukocyte function. Cell Rep. (2016) 16:2197–207. doi: 10.1016/j.celrep.2016.07.040
103. Scholz D, Cai WJ, Schaper W. Arteriogenesis, a new concept of vascular adaptation in occlusive disease. Angiogenesis. (2001) 4:247–57. doi: 10.1023/A:1016094004084
104. Pauly RR, Passaniti A, Bilato C, Monticone R, Cheng L, Papadopoulos N, et al. Migration of cultured vascular smooth muscle cells through a basement membrane barrier requires type IV collagenase activity and is inhibited by cellular differentiation. Circ Res. (1994) 75:41–54. doi: 10.1161/01.RES.75.1.41
105. Cai WJ, Kocsis E, Scholz D, Luo X, Schaper W, Schaper J. Presence of Cx37 and lack of desmin in smooth muscle cells are early markers for arteriogenesis. Mol Cell Biochem. (2004) 262:17–23. doi: 10.1023/B:MCBI.0000038201.43148.20
106. Faries PL, Marin ML, Veith FJ, Ramirez JA, Suggs WD, Parsons RE, et al. Immunolocalization and temporal distribution of cytokine expression during the development of vein graft intimal hyperplasia in an experimental model. J Vasc Surg. (1996) 24:463–71. doi: 10.1016/S0741-5214(96)70203-3
107. Miyake H, Yoshimura K, Hara I, Eto H, Arakawa S, Kamidono S. Basic fibroblast growth factor regulates matrix metalloproteinases production and in vitro invasiveness in human bladder cancer cell lines. J Urol. (1997) 157:2351–5. doi: 10.1097/00005392-199706000-00112
108. Van Leeuwen R. Extracellular proteolysis and the migrating vascular smooth muscle cell. Fibrinolysis. (1996) 10:59–74. doi: 10.1016/S0268-9499(96)80081-6
109. Langille BL, Bendeck MP, Keeley FW. Adaptations of carotid arteries of young and mature rabbits to reduced carotid blood flow. Am J Physiol. (1989) 256(4 Pt 2):H931–9. doi: 10.1152/ajpheart.1989.256.4.H931
110. Mignatti P, Rifkin DB. Biology and biochemistry of proteinases in tumor invasion. Physiol Rev. (1993) 73:161–95. doi: 10.1152/physrev.1993.73.1.161
111. Cai W, Vosschulte R, Afsah-Hedjri A, Koltai S, Kocsis E, Scholz D, et al. Altered balance between extracellular proteolysis and antiproteolysis is associated with adaptive coronary arteriogenesis. J Mol Cell Cardiol. (2000) 32:997–1011. doi: 10.1006/jmcc.2000.1137
112. Tyagi SC, Kumar S, Cassatt S, Parker JL. Temporal expression of extracellular matrix metalloproteinases and tissue plasminogen activator in the development of collateral vessels in the canine model of coronary occlusion. Can J Physiol Pharmacol. (1996) 74:983–95. doi: 10.1139/y96-095
113. Masuda H, Zhuang YJ, Singh TM, Kawamura K, Murakami M, Zarins CK, et al. Adaptive remodeling of internal elastic lamina and endothelial lining during flow-induced arterial enlargement. Arterioscler Thromb Vasc Biol. (1999) 19:2298–307. doi: 10.1161/01.ATV.19.10.2298
114. Jackson ZS, Gotlieb AI, Langille BL. Wall tissue remodeling regulates longitudinal tension in arteries. Circ Res. (2002) 90:918–25. doi: 10.1161/01.RES.0000016481.87703.CC
115. Dodd T, Jadhav R, Wiggins L, Stewart J, Smith E, Russell JC, et al. MMPs 2 and 9 are essential for coronary collateral growth and are prominently regulated by p38 MAPK. J Mol Cell Cardiol. (2011) 51:1015–25. doi: 10.1016/j.yjmcc.2011.08.012
116. Wu H, Cheng XW, Hu L, Takeshita K, Hu C, Du Q, et al. Cathepsin S activity controls injury-related vascular repair in mice via the TLR2-mediated p38MAPK and PI3K-Akt/p-HDAC6 signaling pathway. Arterioscler Thromb Vasc Biol. (2016) 36:1549–57. doi: 10.1161/ATVBAHA.115.307110
117. Wang Y, Tang C, Qin Y. Cathepsins: a new culprit behind abdominal aortic aneurysm. Regen Med Res. (2013) 1:5. doi: 10.1186/2050-490X-1-5
118. Chang CJ, Chen CC, Hsu LA, Chang GJ, Ko YH, Chen CF, et al. Degradation of the internal elastic laminae in vein grafts of rats with aortocaval fistulae: potential impact on graft vasculopathy. Am J Pathol. (2009) 174:1837–46. doi: 10.2353/ajpath.2009.080795
119. Lohoefer F, Reeps C, Lipp C, Rudelius M, Zimmermann A, Ockert S, et al. Histopathological analysis of cellular localization of cathepsins in abdominal aortic aneurysm wall. Int J Exp Pathol. (2012) 93:252–8. doi: 10.1111/j.1365-2613.2012.00819.x
120. Protack CD, Foster TR, Hashimoto T, Yamamoto K, Lee MY, Kraehling JR, et al. Eph-B4 regulates adaptive venous remodeling to improve arteriovenous fistula patency. Sci Rep. (2017) 7:15386. doi: 10.1038/s41598-017-13071-2
121. Corey MR, Ergul EA, Cambria RP, Patel VI, Lancaster RT, Kwolek CJ, et al. The presentation and management of aneurysms of the pancreaticoduodenal arcade. J Vasc Surg. (2016) 64:1734–40. doi: 10.1016/j.jvs.2016.05.067
122. Kalva SP, Athanasoulis CA, Greenfield AJ, Fan CM, Curvelo M, Waltman AC, et al. Inferior pancreaticoduodenal artery aneurysms in association with celiac axis stenosis or occlusion. Eur J Vasc Endovasc Surg. (2007) 33:670–5. doi: 10.1016/j.ejvs.2006.12.021
123. Brocker JA, Maher JL, Smith RW. True pancreaticoduodenal aneurysms with celiac stenosis or occlusion. Am J Surg. (2012) 204:762–8. doi: 10.1016/j.amjsurg.2012.03.001
124. Tsukioka K, Nobara H, Nishimura K. A case of inferior mesenteric artery aneurysm with an occlusive disease in superior mesenteric artery and the celiac artery. Ann Vasc Dis. (2010) 3:160–3. doi: 10.3400/avd.cr01006
125. Tan C, Reul R. Inferior mesenteric artery aneurysm in the setting of celiac and superior mesenteric artery occlusion. J Vasc Surg Cases Innov Tech. (2019) 5:197–9. doi: 10.1016/j.jvscit.2018.07.001
126. Hashimoto N, Handa H, Hazama F. Experimentally induced cerebral aneurysms in rats. Surg Neurol. (1978) 10:3–8.
127. Frösen J, Piippo A, Paetau A, Kangasniemi M, Niemelä M, Hernesniemi J, et al. Remodeling of saccular cerebral artery aneurysm wall is associated with rupture: histological analysis of 24 unruptured and 42 ruptured cases. Stroke. (2004) 35:2287–93. doi: 10.1161/01.STR.0000140636.30204.da
128. Kataoka K, Taneda M, Asai T, Kinoshita A, Ito M, Kuroda R. Structural fragility and inflammatory response of ruptured cerebral aneurysms. A comparative study between ruptured and unruptured cerebral aneurysms. Stroke. (1999) 30:1396–401. doi: 10.1161/01.STR.30.7.1396
129. Chu Y, Wilson K, Gu H, Wegman-Points L, Dooley SA, Pierce GL, et al. Myeloperoxidase is increased in human cerebral aneurysms and increases formation and rupture of cerebral aneurysms in mice. Stroke. (2015) 46:1651–6. doi: 10.1161/STROKEAHA.114.008589
130. Shimizu K, Kataoka H, Imai H, Yamamoto Y, Yamada T, Miyata H, et al. Hemodynamic force as a potential regulator of inflammation-mediated focal growth of saccular aneurysms in a rat model. J Neuropathol Exp Neurol. (2021) 80:79–88. doi: 10.1093/jnen/nlaa131
131. Kataoka H, Yagi T, Ikedo T, Imai H, Kawamura K, Yoshida K, et al. Hemodynamic and histopathological changes in the early phase of the development of an intracranial aneurysm. Neurol Med Chir. (2020) 60:319–28. doi: 10.2176/nmc.st.2020-0072
132. Campbell GJ, Roach MR. A physical model for the formation of evaginations: a prospective precursor to the creation of saccular aneurysms. Stroke. (1984) 15:642–52. doi: 10.1161/01.STR.15.4.642
133. Wang S, Dai D, Kolumam Parameswaran P, Kadirvel R, Ding Y-H, Robertson AM, et al. Rabbit aneurysm models mimic histologic wall types identified in human intracranial aneurysms. J Neuro Int Surg. (2018) 10:411–5. doi: 10.1136/neurintsurg-2017-013264
134. Cebral JR, Duan X, Gade PS, Chung BJ, Mut F, Aziz K, et al. Regional mapping of flow and wall characteristics of intracranial aneurysms. Ann Biomed Eng. (2016) 44:3553–67. doi: 10.1007/s10439-016-1682-7
135. Meng H, Wang Z, Hoi Y, Gao L, Metaxa E, Swartz DD, et al. Complex hemodynamics at the apex of an arterial bifurcation induces vascular remodeling resembling cerebral aneurysm initiation. Stroke. (2007) 38:1924–31. doi: 10.1161/STROKEAHA.106.481234
136. Shakur SF, Alaraj A, Mendoza-Elias N, Osama M, Charbel FT. Hemodynamic characteristics associated with cerebral aneurysm formation in patients with carotid occlusion. J Neurosurg. (2018) 130:917–22. doi: 10.3171/2017.11.JNS171794
137. Werner C, Mathkour M, Scullen T, McCormack E, Dumont AS, Amenta PS. Multiple flow-related intracranial aneurysms in the setting of contralateral carotid occlusion: coincidence or association? Brain Circ. (2020) 6:87–95. doi: 10.4103/bc.bc_1_20
Keywords: arteriogenesis, extracellular matrix, elastic fiber, outward remodeling, collateral arteries, arterial occlusive disease
Citation: Kulkarni R, Andraska E and McEnaney R (2021) Structural Remodeling of the Extracellular Matrix in Arteriogenesis: A Review. Front. Cardiovasc. Med. 8:761007. doi: 10.3389/fcvm.2021.761007
Received: 19 August 2021; Accepted: 11 October 2021;
Published: 05 November 2021.
Edited by:
Junxi Wu, University of Strathclyde, United KingdomReviewed by:
Mingyi Wang, National Institutes of Health (NIH), United StatesCopyright © 2021 Kulkarni, Andraska and McEnaney. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ryan McEnaney, bWNlbmFuZXlybUB1cG1jLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.