 Kang Fu
Kang Fu Huixia Lu
Huixia Lu Xiaoping Ji
Xiaoping Ji- The Key Laboratory of Cardiovascular Remodeling and Function Research, Chinese Ministry of Education, Chinese National Health Commission and Chinese Academy of Medical Sciences, The State and Shandong Province Joint Key Laboratory of Translational Cardiovascular Medicine, Qilu Hospital of Shandong University, Jinan, China
Type-1 cardiorenal syndrome refers to acute kidney injury induced by acute worsening cardiac function. Worsening renal function is a strong and independent predictive factor for poor prognosis. Currently, several problems of the type-1 cardiorenal syndrome have not been fully elucidated. The pathogenesis mechanism of renal dysfunction is unclear. Besides, the diagnostic efficiency, sensitivity, and specificity of the existing biomarkers are doubtful. Furthermore, the renal safety of the therapeutic strategies for acute heart failure (AHF) is still ambiguous. Based on these issues, we systematically summarized and depicted the research actualities and predicaments of the pathogenesis, diagnostic markers, and therapeutic strategies of worsening renal function in type-1 cardiorenal syndrome.
Introduction
Acute heart failure (AHF), characterized by acute or subacute worsening symptoms and signs of heart failure (HF), is an intractable clinical and public health problem with high morbidity, mortality, and economic burden. AHF commonly coexists with numerous complications and renal dysfunction may be the most frequent one with a prevalence of approximately 25–40% (1, 2). To better apprehend the disorders of the coexistence of the concomitant impairment of both the cardiac and renal function, whereby acute or chronic dysfunction in one organ may induce acute or chronic dysfunction of the other, the terminology “cardiorenal syndrome” (CRS) is nominated (3). Depending upon the chief culprit of the pernicious and bidirectional insufficiency of both organs, CRS can be categorized into five clinical subtypes and acute worsening renal function (WRF) caused by the acute deterioration of cardiac function is termed as type-1 CRS (CRS-1 or acute CRS) (4, 5).
Impaired kidney function is an important independent prognostic factor for adverse events including cardiovascular mortality, the longer length of in-hospital stay, and HF re-hospitalization for AHF patients (6–8). Related clinical studies have validated that even an insignificant increase (as low as 0.2 mg/dL) in serum creatinine correlates with a poor prognosis for AHF patients (9). CRS-1 is a tremendous obstacle to nephrologists and cardiologists. However, there are several problems of WRF in CRS-1 that need to be solved. First, there is still a scarcity of a precise and quantitative description of WRF while the criteria of a 0.3–0.5 mg/dl rise in serum creatinine concentration, a 25% increase in plasm creatinine levels, or a decline in glomerular filtration rate (GFR) of 9−15 ml/min during AHF admission is established and has been accepted by some clinical trials (10–13). Second, the etiologies of WRF in CRS-1 remain unclear. Numerous factors, including hemodynamic imbalance, neurohormonal activation, and oxidative stress, may be involved in the pathogenesis. The exhaustive mechanism has not been absolutely elucidated. Third, while an increasing number of biomarkers have been explored and utilized in the diagnosis of renal dysfunction, the diagnostic efficiency, sensitivity, and specificity are still skeptical in the clinical practice. Last, renal safety of therapeutic strategies for AHF, such as decongestant therapy, is still debatable and some agents may be kidney toxic. Taking into account these controversies, WRF in CRS-1 is a noteworthy topic.
In this review, we summarized the latest findings of WRF in CRS-1, including the pathogenesis, clinical parameters facilitating diagnosis, and treatment strategies. A comprehensive and deep understanding of WRF in CRS-1 may provide new insights for early identification and effective treatment.
Definition of WRF
Although the term WRF is commonly regarded as an acute and/or sub-acute change that occurs to kidney function, exhibiting an increase of serum creatinine concentration or a decline of the estimated glomerular filtration rate (eGFR) during AHF, a precise and unified definition of WRF in CRS-1 has not been given for that different renal injury biomarkers and different amplitude of variation which is considered significant were adopted in different studies. To tackle this issue, various definitions and criteria of WRF were proposed by three different consensus groups: Risk, Injury, Failure, Loss of kidney function, and End-stage kidney disease (RIFLE), Acute Kidney Injury Network (AKIN), and Kidney Disease: Improving Global Outcomes (KDIGO) (14–16). Different stages of acute kidney injuries (AKI) /WRF are classified in all sets of criteria, which are described in Table 1.
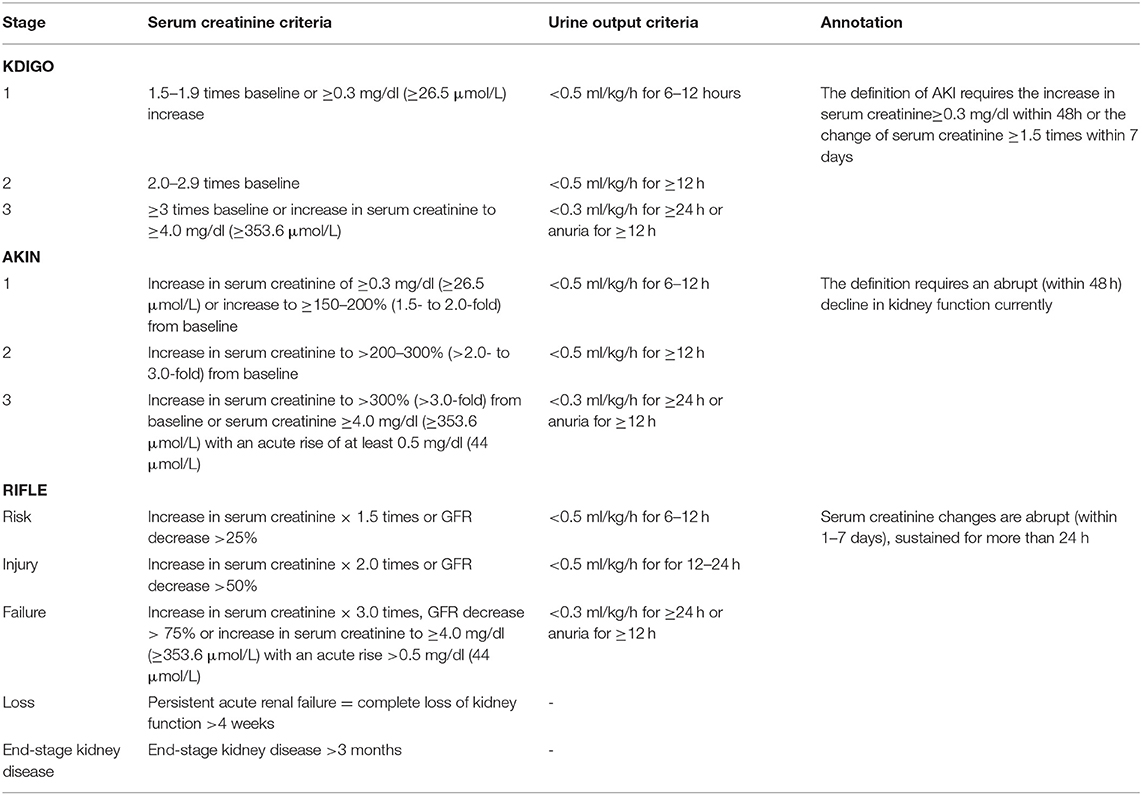
Table 1. Kidney Disease: Improving Global Outcomes (KDIGO), Acute Kidney Injury Network (AKIN), and Risk, Injury, Failure, Loss of kidney function, and End-stage kidney disease (RIFLE) criteria of AKI/WRF.
Pathogenesis
The etiologies and pathogenic mechanisms of WRF in CRS-1 patients are multifactorial and ambiguous. The hemodynamic imbalance, neurohormonal activation, sympathetic activity, pharmacological interventions, inflammation along with oxidative stress may be involved in the pathogenesis of WRF in CRS-1 (Figure 1) (17).
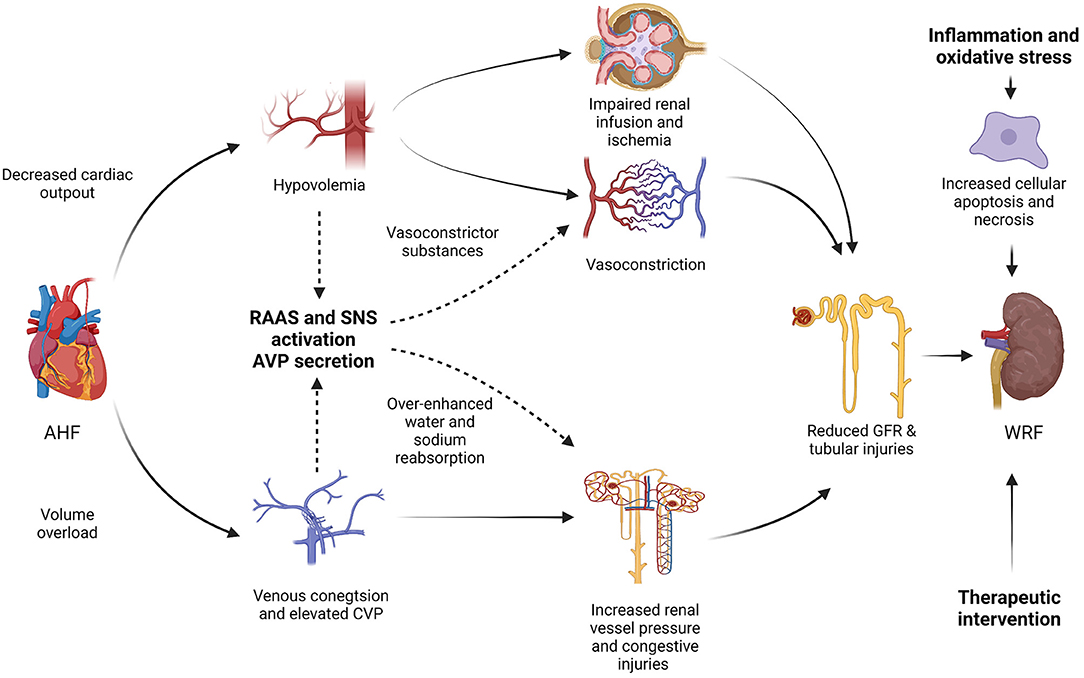
Figure 1. Pathogenesis mechanisms of worsening renal function (WRF) in type-1 cardiorenal syndrome (CRS-1). AHF, acute heart failure; RAAS, renin-angiotensin-aldosterone system; SNA, sympathetic nervous system; AVP, arginine vasopressin; CVP, central venous pressure; GFR, glomerular filtration rate; WRF, worsening renal function; CRS, cardiorenal syndrome.
Hemodynamic Imbalance
The hypovolemia caused by diminished cardiac output or over-quick decongestion therapy can lead to impaired renal infusion. The reduced renal perfusion, or so-called impaired renal “preload”, is regarded as the dominating etiology of WRF in CRS-1 according to the historical mainstream viewpoints (18). Kidney artery under-filling due to hypovolemic status can lead to renal cortical ischemia or infarction. Besides, inadequate kidney perfusion can induce acute ischemic tubular necrosis (19). Hypovolemia can also activate the neurohormonal activation as described hereinafter. The vasoconstriction and sodium-retaining neurohormones, including angiotensin II and renin, will be over-generated and over-secreted as an auto-compensatory mechanism (20). However, this hypothesis is oversimplified and can only partially explain WRF in CRS-1, especially for patients with AHF with obviously impaired left ventricular systolic function and decreased cardiac output. For patients with normal left ventricular systolic function, videlicet, persevered left ventricular ejection fraction (LVEF), or for those with isolated right ventricular failure and elevated pulmonary artery pressure, the theory is unpersuasive (21). In fact, the proportion of WRF in CRS-1 caused by diminished cardiac output and reduced renal vasculature perfusion may be relatively low. The clinical manifestation of the majority is a “warm and wet” pattern, rather than a “cold” pattern, which refers to the presentation of preserved left ventricular systolic function and/or normal blood volume (22). The Evaluation Study of Congestive Heart Failure and Pulmonary Artery Catheterization Effectiveness (ESCAPE) trial also revealed no relationship between hemodynamic parameters and WRF in congestive heart failure patients receiving pulmonary artery catheter–guided therapy (23). On this occasion, “forward failure” caused by low cardiac output should not be regarded as the main determinant of renal dysfunction in CRS-1 patients and renal congestion may play a more vital role.
Renal congestion induced by systemic congestion and volume overload may also participate in the pathogenesis (24). For patients with a “warm and wet” pattern, systemic congestion, increased pulmonary and/or renal congestion are the main hemodynamic profile. The exact mechanism of WRF induced by renal congestion has not been well clarified. Increased renal vessel pressure caused by renal congestion may trigger interstitial edema, tubular dysfunction, and reversible azotemia (25, 26). The phenomenon that increased central venous pressure (CVP) and/or raised intra-abdominal pressure (IAP) positively correlate with WRF in AHF also provides more testimonies. Previous literature has indicated that the increase in CVP has a strong association with declined GFR and WRF in patients with AHF (27). Mullens et al. demonstrated that it is the elevation of the admission baseline CVP, mean CVP, and discharge CVP rather than the cardiac index (CI) or other hemodynamic parameters that are strongly correlated with the increased risk of WRF for patients with acute decompensated heart failure (ADHF) (28). The role of CVP in maintaining renal perfusion pressure and trans-renal perfusion is decisive. For patients with systemic congestion, CVP elevates dramatically. Renal interstitial pressure can escalate along with increased CVP which may cause the congestion of renal vasculature, thus congestive kidney failure is developed. Analogous to mechanisms of liver failure caused by hepatic congestion, the congestive kidney injuries, manifesting as ischemic injury of renal parenchyma and hypoxia damage of renal cortex, will emerge under the increased renal interstitial pressure (12, 29). Raised IAP also contributes to the development of WRF. Although distinct ascites can be only detected in a small population of patients with acute CRS, symptoms of visceral/tissue edema are prevalent for patients with systemic congestion and the prevalence of raised IAP can reach up to 60% in patients ADHF. On one hand, elevated IAP indirectly increases CVP and may further induce congestive renal failure. On the other hand, the kidney is compressed when exposed to raised IAP and renal infusion displays a precipitous decline. Hence ischemic injuries will develop due to the reduced renal blood flow. Mullens et al. also illustrated that elevated IAP was associated with WRF for patients with ADHF and reductions of IAP were associated with improved renal function (30).
Neurohormonal Activation
The renin-angiotensin-aldosterone system (RAAS) is crucial in maintaining the hemodynamic homeostasis and plays a major role in the cardiorenal physiological/pathophysiologic bidirectional interaction in CRS-1 (31). The persistent renal hypoperfusion results in RAAS overactivation. Besides, sympathetic nerve activity (SNA) and the secretion and release of arginine vasopressin (AVP) will also be increased to tackle systemic hypovolemia and to maintain kidney blood flow.
The activation of RAAS plays a vital role in maintaining systemic and renal hemodynamic hemostasis and contributes to cellular hypertrophy, apoptosis, and fibrosis in both the heart and the kidney. At the cardiac level, it can reflect the degree of cardiac remodeling, ventricular hypertrophy, and fibrosis. What is more, it promotes sodium and water retention, mediates the redistribution of intrarenal perfusion which refers to the reduced medullary blood flow and increased cortical blood flow, to compensate for the hemodynamic changes in CRS-1 (32). In detail, angiotensin II, renin, as well as numerous other vasoactive agents, including vasodilators such as prostaglandin I2 and nitric oxide, are subsequently generated in response to the activation of RAAS. The former, vasoconstrictors, can constrict efferent arterioles, and the latter, vasodilators, can dilate afferent arterioles. Thus the glomerular hydrostatic pressure and filtration are preserved (22). In this compensated stage, the kidney function is approximately normal. With the progression of WRF, both afferent and efferent arterioles are both constricted, thus the vasoconstriction effect of angiotensin II dominates renal hemodynamic physiopathologic changes. In the decompensated phase, GFR falls dramatically (33, 34).
Overactivation of RAAS accounts for approximately 50% of the over-production of aldosterone (35). The mineralocorticoid, aldosterone, is of great importance in increasing sodium and water retention and the regulation of blood pressure and fluid homeostasis. Furthermore, Angiotensin II and aldosterone are tightly associated with the hypertrophy, apoptosis, and fibrosis of renal tubular cells (36). On one hand, Angiotensin II and aldosterone promotes the expression of numerous profibrotic molecules, such as plasminogen activator inhibitor 1, osteopontin, and galectin-3, which promote collagen and matrix production. On the other hand, angiotensin II and aldosterone also stimulates mitochondrial production of reactive oxygen species (ROS) and thus exacerbates renal tubulointerstitial fibrosis and endothelial dysfunction through the inflammatory pathway (37).
The SNA will also be potentiated for hypovolemia and decreased artery pressure (38, 39). Angiotensin is the stimulator of the sympathetic nervous system (SNS) and can also contribute to the activation of SNS via numerous biological effects, such as the direct activation of critical sympathoexcitatory neurons in the paraventricular nucleus of the hypothalamus (40). At the cardiac level, it increases cardiac output while worsens cardiac fibrosis and hypertrophy. For the kidney, over-activated SNS stimulates not only α1-adrenergic receptors on vascular smooth muscle cells to enhance vasoconstriction but also β1-adrenergic receptors on juxtaglomerular cells to increase the secretion of renin, which can activate RAAS in reverse (41). Hence, enhanced SNA results in vessel constriction, increased systemic vascular resistance, and sodium retention.
Arginine vasopressin (AVP), the antidiuretic hormone secreted by the posterior pituitary gland receiving the stimulation of hypovolemia and increased osmolality, also takes on pivotal importance in sustaining the renal filtration fraction (42). AVP can activate V1 receptors on the vasculature, which results in renal vessels constriction and elevated vascular resistance. V2 receptors on principal cells can also be activated thus water and sodium reabsorption, as well as hyponatremia, is aggravated (43). Both water and sodium-retaining neurohormones work together to maintain hemodynamic homeostasis and sufficient kidney perfusion pressure. Hypervolemia induced by water and sodium retention also triggers congestive renal injuries.
Inflammation and Oxidative Stress
The increase of proinflammatory cytokines and oxidative stress is the vital factor for the pathogenesis of WRF, which have been validated by numerous in vivo and in vitro experimental studies (44). The underlying mechanisms may incorporate oxidative stress and inflammation leading to monocyte phenotype transition, kidney interstitial fibrosis, and renal cellular apoptosis. Inflammatory and apoptosis pathway plays a crucial role in the pathogenesis of WRF in CRS-1. Inflammatory cytokines can induce AKI via activating death signaling receptors and other various signaling pathways. It can cause endothelial function impairment, renal vasoconstriction, and capillary obstruction (45, 46). The oxidative stress pathway is a classic pathway inducing cellular injury, interstitial fibrosis, and organ dysfunction. An increase of oxidative stress leads to the over-generation of ROS and reactive nitrogen species (RNS), which facilitates the formation of proinflammatory and profibrotic milieu. The over-activation of RAAS can also stimulate the over-production of ROS and RNS in the kidney (47). Furthermore, over-production of ROS and RNS exacerbates renal and cardiovascular structural and functional abnormalities through inactivating mitochondrial enzymes, accelerating DNA damage, and promoting base hydroxylation (46). Both cardiac and renal injuries are thus developed.
The inflammatory and oxidative stress activities are increased in acute CRS. Vitro experiments conducted by Pastori et al. revealed that when adding the plasm of patients with acute CRS, the apoptosis of monocyte cell lines was obviously upregulated vs. those incubated with plasm from patients with AHF or healthy individuals. Besides, the expression levels of caspase-3 and-8 of monocytes were in line with apoptosis rates. Plasm concentrations of inflammatory cytokines including Interleukin (IL) −6 and −18 were also apparently higher in patients with acute CRS than which in patients with AHF (48). A similar phenomenon, which refers to the more obvious elevation of plasm levels of inflammatory cytokine and markers of oxidative stress including IL-6 and myeloperoxidase (MPO) in acute CRS patients compared with patients with AHF, was also observed in the study of Virzì et al. (46, 49). Vivo experiments also showed significant elevation of serum concentration of cytokines and MPO in patients with CRS-1 vs. patients with AHF without renal insufficiency (50). An observational pilot study reflected that the activation of peripheral blood monocytes may be the stimulator of inflammatory pathway way in acute CRS patients. Monocyte phenotype transition and apoptosis are crucial in the pathogenesis of WRF (51).
Markers for WRF
Creatinine
Creatinine is the most widely used biomarker to evaluate GFR. As the pioneer of biomarkers estimating renal function, creatinine negatively correlates with GFR. Creatinine is a 113kDa end-product of creatine phosphate metabolism. Creatine is a nitrogenous organic acid that is predominantly generated in the kidney and liver and is mainly transported to and metabolized in skeletal and heart muscles tissues for energy generation. Serum creatinine is generated during the spontaneous, non-enzymatic anhydration of creatine in muscle cells (62, 63). Creatinine is filtered by the glomerulus. The highlighted peculiarity of creatinine is that it will not be reabsorbed by the renal tubules (64). Since creatinine was discovered, it has been wildly used as the “golden standard” to estimate GFR in clinical practice.
There are blemishes of creatinine in assessing renal function. Above all, the synthesis and secretion of creatinine are heterogeneous across individuals. The production of creatine principally depends on muscle mass, physical activity, dietary meat consumption, ethnic and age factors. Chronic illness, including inflammatory disease and malignant tumors, can also decrease creatinine generation. For another, the clearance of creatinine is also influenced by diversified factors such as pharmacotherapy including cimetidine and trimethoprim (65). Moreover, the relationship between creatinine and GFR is non-linear and exponential (66). On some occasions, creatinine is impotent to detect a mild alternation in GFR and will underestimate the degree of kidney impairment. Considerable reduction of GFR may accompany the weeny elevation of serum creatinine concentration, especially in patients with advanced age and low muscle mass (8). In addition, the uplift of serum creatinine is comparatively lagged behind AKI and is insensitive to tubular impairment (67). These characteristics impose restrictions on the utilization of creatinine.
Cystatin C
Cystatin C (CysC), a non-glycosylated 13kDa protein, is a member of endogenous cysteine proteinase inhibitors (68). Cystatin C is synthesized in all human nucleated cells and is encoded by the CST3 gene, a housekeeping gene located in chromosome 20 (69). For this reason, the generation rate of CysC is relatively constant. Similar to creatinine, CysC is freely filtered by glomeruli (70). CysC is completely reabsorbed and metabolized by renal tubular epithelial cells. The changing trend of serum concentration of CysC is in keeping with which of GFR, thus CysC is considered as a novel and dependable biomarker in identifying kidney disease (71). For that, CysC is insensitive to various confound factors, numerous studies have indicated that the performance of CysC in diagnosis renal insufficiency is comparably superior to creatinine (68). Rafouli-Stergiou et al. revealed that a rise in cystatin C during the admission of patients with ADHF was an independent predictor of short-term prognosis. Besides, the combination of cystatin C and NT-proBNP could provide additional prognostic information for WRF in CRS-1 (66). One multicenter clinical study revealed that CysC could well predict AKI in the admission patients with AHF and a rise in cystatin C > >0.3 mg/L within 48h from admission positively correlated with higher in-hospital mortality and longer hospital stay (Table 2) (52).
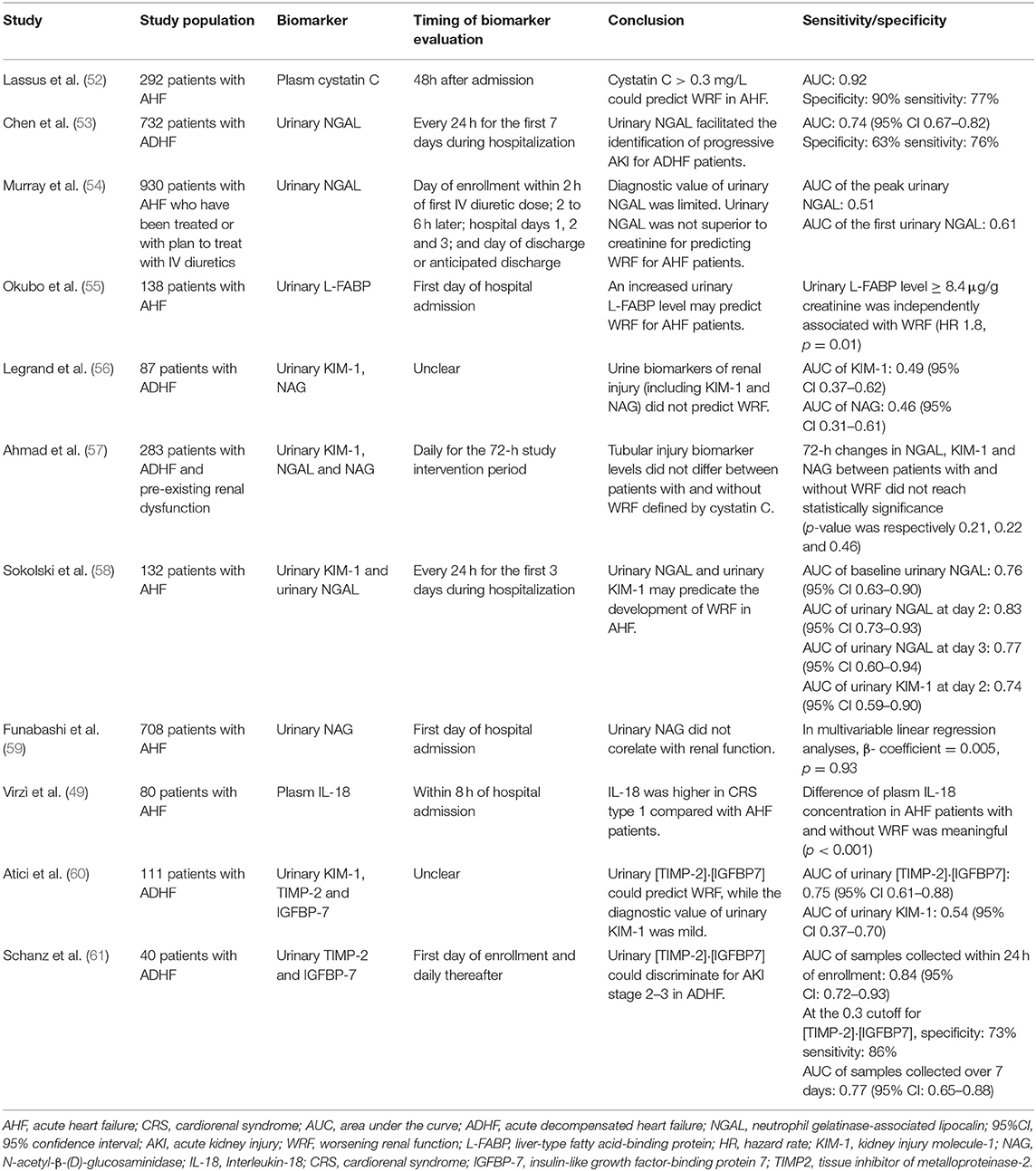
Table 2. Summary of studies on biomarkers of WRF in CRS-1 in this review.
Neutrophil Gelatinase-Associated Lipocalin
As a member of the lipocalin family, neutrophil gelatinase-associated lipocalin (NGAL) is a 25kDa protein, which is filtered by the glomerulus and is immediately reabsorbed in proximal tubule through a megalin-dependent pathway (72). The serum concentration of NGAL is relatively low in healthy individuals, whereas its plasm level significantly elevates (about 300-folds) in response to tubular epithelial damage (73). The expression of NGAL is upregulated in kidney tissue when AKI is developed, which may result in the increase of NGLA plasm concentrations. Furthermore, impaired reabsorption function of the proximal tubule may contribute to the increased levels in urine (74). Previous studies have validated the efficiency and accuracy of urinary NGAL in the diagnosis of AKI and WRF in patients with AHF, while the sensitivity and specificity of plasm NGAL are not so excellent (53). Interestingly, the Acute Kidney Injury N-gal Evaluation of Symptomatic heart failure Study (AKINESIS) study confirmed that the diagnostic value and prognostic predicting value of plasm NGAL for WRF in patients with AHF was limited and was not superior to creatinine. Similarly, the performance of urine NGAL in predicting WRF and prognosis did not overmatch creatinine (54).
Liver-Type Fatty Acid-Binding Protein
Fatty acid-binding protein (FABP) is a 14KDa molecule protein and belongs to the superfamily of lipid-binding proteins. FABP plays a significantly important role in the metabolism and transportation of fatty acids (75). FABP consists of nine tissue-specific subtypes, including liver (L), intestinal (I), muscle and heart (H), adipocyte (A), epidermal (E), ileal (IL), brain (B), myelin (M), and testis (T) (76). For liver-type fatty acid-binding protein (L-FABP), it is expressed in both hepatocytes and tubular epithelial cells. It participates in the transportation of fatty acids and is essential to fatty acid β-oxidation and power supply of tubular epithelial cells. For patients with AKI, increased oxidative stress promotes the over-dosed production of ROS. For the peroxidation of plasma membrane and cytoplasmic membrane mediated by ROS, the cytotoxic lipid peroxidation products, which are bound and cleared by L-FABP, are excessively generated and accumulated in proximal tubules. Thus L-FABP was excreted from the proximal tubules into urine together with cytotoxic lipids. Urinary L-FABP has been proposed as an emerging acute proximal tubules injury biomarker for WRF in CRS-1 despites few related studies (77, 78). Yousaku Okubo et al. revealed that urinary L-FABP can predict WRF in patients with AHF and patients with higher L-FABP levels were more likely to have rehospitalization (55).
Kidney Injury Molecule-1
Kidney injury molecule-1 (KIM-1) is a 38.7kDa transmembrane glycoprotein, which is a member of type I transmembrane glycoproteins (79). KIM-1 is principally expressed at the apical membrane of proximal tubular epithelial cells and its expression maintains a low state under normal circumstances. The expression of KIM-1 is dramatically upregulated when the renal tubule undergoes different injuries including ischemia-reperfusion injury (80, 81). KIM-1 is regarded as a promising biomarker for detecting renal tubular injuries, while its sensitivity and accuracy in the diagnosis of AKI have not been validated (82). Studies engaged in the association between KIM-1 and WRF in patients with CRS-1 are rare. Legrand et al. revealed a slight but non-statistically significant association between KIM-1 and increased risk of WRF for AHF patients in the Biomonitoring and Cardiorenal Syndrome in Heart Failure Trial (BIONICS) trial population (56). A similar result was drawn in the Renal Optimization Strategies Evaluation AHF (ROSE-AHF) trial, which exhibited no relationship between the change of plasm KIM-1 level and the occurrence of WRF in patients with AHF (57). On the contrary, urinary KIM-1 is a meaningful biomarker for predicting WRF. Sokolski et al. exhibited the excellent diagnostic value of urinary KIM-1 for WRF (58).
N-Acetyl-β-(D)-Glucosaminidase
N-acetyl-β-(D)-glucosaminidase (NAG) is a brush-border lysosomal enzyme found in several human cells including proximal tubule cells (83). On the grounds that NAG is a large molecular weight compound (>>130kDa), NAG obviates the elimination through glomerular filtration and thus, the elevation of urinary NAG levels can be considered as a tubular origin (84). Urinary NAG is regarded as an outstanding predictor reflecting the impairment of tubular injury. Funabashi et al. demonstrated that for inpatients with AHF recruited from the National Cerebral and Cardiovascular Center Acute Decompensated Heart Failure (NaDEF) registry study, those with higher urinary NAG tended to have lower eGFR than those with low urinary NAG levels, while multivariable linear regression analyses showed no significant relation between renal function and urinary NAG concentrations. Besides, elevated urinary NAG levels correlated with long-term adverse events (59). In contrast, the ROSE-AHF trial indicated no relationship between the development of WRF and the increase of urinary NAG (57). Related clinical studies are relatively limited and further studies should be conducted.
Interleukin-18
As a member of the IL-1 family, Interleukin-18 (IL-18), which was first described as an “interferon (INF) γ-inducing factor,” is an 18kDa biologically active proinflammatory cytokine (85). IL-18 is involved in numerous renal pathogenic processes such as apoptosis, ischemia-reperfusion injuries, allograft rejection, and malignancy. IL-18 is generated by proximal tubules and is excreted into the urine after acute ischemic injuries, which have been verified by several in vivo studies (86, 87). As mentioned above, inflammation and oxidative stress factors are crucial for the pathogenesis process of WRF. Hence inflammation cytokines (such as IL-18) may be potential candidates for the identification of WRF in the early stages of acute renal insufficiency. Parikh et al. revealed that urinary IL-18 could accurately predict WRF even in the first 24 h of onset of deterioration of renal function, which was obviously earlier than the elevation of serum creatinine (88). The Systolic Blood Pressure Intervention Trial (SPRINT) trial also indicated that IL-18 was an independent predictor of future risk of AKI with high sensitivity and accuracy (89). Virzì et al. indicated that plasma levels of proinflammatory cytokines including IL-18 were higher in CRS-1 patients compared with AHF patients, which hinted at the potential of IL-18 in the diagnosis of WRF in CRS-1 (49).
Insulin-Like Growth Factor-Binding Protein 7 (IGFBP-7) and Tissue Inhibitors of Metalloproteinase-2 (TIMP2)
Insulin-like growth factor-binding protein 7 (IGFBP-7) and TIMP2 are both small molecular weight proteins (29 kDa and 24 kDa, respectively) and stimulators of G1 cell cycle arrest which involves in the pathogenesis of AKI (90, 91). Both biomarkers prevent the division of injured cells with damaged DNA. The normal cell cycle will re-initiate until the repair is accomplished. However, pathological changes such as senescence and fibrosis will emerge if the arrested cell cycle lasts for a too long period (92). These two biomarkers are both expressed, generated, and secreted in renal tubular cells at the early phase of AKI/WRF. Related studies have validated that the combination of these two biomarkers, which refers to urinary [TIMP-2]·[IGFBP7], had a significantly superior performance in the early identification and diagnosis of AKI compared with classic renal injury biomarkers such as KIM-1 (93, 94). When focused on its application in the diagnosis of WRF in CRS-1, associated studies are limited. In Atici et al.'s study, the levels of urinary [TIMP-2]·[IGFBP7] were significantly elevated in patients with acute CRS compared with those with simple AHF (60). Schanz et al. also revealed that urinary [TIMP-2]·[IGFBP7] was a promising biomarker in the early discrimination of WRF in patients with ADHF with considerable sensitivity and specificity. Furthermore, the urinary [TIMP-2]·[IGFBP7] also well predicted all-cause mortality at 1 year after discharge (61).
B-Type Natriuretic Peptide (BNP)
B-type natriuretic peptide (BNP) and N-terminal proB-type natriuretic peptide (NT-proBNP) are well-established biomarkers for HF and have been universally manipulated in estimating the presence, severity, and prognosis of HF (95). The serum concentration of NT-proBNP can be influenced by a variety of factors, such as renal function, advanced age, blood pressure, severe infection, gender, and obesity (5). Hence BNP and NT-proBNP are not regarded as eligible and reliable biomarkers for the identification of WRF. Based on that, the term “the estimated mature BNP” (emBNP), which was calculated by subtracting proBNP from total BNP, has been proposed. It may contribute to the identification of WRF in CRS-1 patients for its peculiar clearing mechanisms (96). The emBNP is cleared through membrane-bound natriuretic peptide receptors A and C rather than kidney, whereas NT-proBNP was mainly cleared by renal excretion (97). Previous literature validated that patients with AHF and WRF had lower emBNP levels and higher NT-proBNP/emBNP ratios. Besides, NT-proBNP/emBNP ratios were associated with composite clinical events, including all causes of death and rehospitalization for HF (96).
Treatment
Diuretics
Congestion is the hallmark of AHF and decongestion therapy, especially diuresis pharmacotherapy, is the cornerstone for patients with AHF. However, controversies concerning the utilization of diuretics in patients with CRS-1 have never been eliminated for the reason that prior studies have identified the association between aggressive diuresis (loop diuretics in especial) and increased risk of WRF. The possibility that administration of diuretics may induce or aggravate WRF in AHF patients astricts the appropriate use of diuretics. The optimal diuretics therapeutic regimen in CRS-1 patients is still unclear.
It is a conundrum whether aggressive fluid removal via escalating doses of diuretics can be safely and effectively applied in the setting of AHF. It deserves the deliberateness to weigh up the pros and cons. From pathogenesis, diuretics therapy will rapidly mitigate congestion and reduce increased CVP, which may ameliorate the kidney dysfunction caused by congestive kidney damages. While hypovolemia and impaired renal preload will induce hypo-infusion renal injuries. Hence different viewpoints of rational diuretics use have been concluded in different clinical researches. The Description de la Filière de Soins dans les Syndromes d'Insuffisance Cardiaque Aigue (DeFSSICA) study indicated no limitation of loop diuretics in patients with AHF with renal insufficiency for the reason that the prognosis of patients with acute CRS on diuretics therapy was indiscriminate compared with that of patients with AHF (98). Conversely, the Randomized Evaluation of Heart Failure with Preserved Ejection Fraction Patients with Acute Heart Failure and Dopamine (ROPA-DOP) study and the DOSE-AHF study clarified the association between the utilization of diuretics and WRF in patients with AHF (99, 100). The DOSE-AHF study illustrated that patients with AHF receiving high-dose furosemide treatment were more vulnerable to transient WRF than those accepting low-dose furosemide (99). The ROPA-DOP study also revealed that a continuous infusion diuretic strategy was associated with a higher incidence of WRF in patients with heart failure with preserved ejection fraction (HFpEF) hospitalized for AHF treatment (100).
Given this evidence, how to suitably and optimally use diuretics is still a predicament. The dosage and mode of administration of diuretics are relatively crucial to balance the therapeutic effect of decongestion and the possibility of WRF. For the issue of administration mode, in Palazzuoli et al.'s study, patients receiving continuous furosemide infusion therapy were more vulnerable to WRF than those receiving bolus injections of furosemide, while the efficiency of diuresis was better in the former (101, 102). For the question of diuretics dosage, the DOSE-AHF study demonstrated that AHF patients on high-dose furosemide therapy had an increased risk of transient WRF vs. those on low-dose furosemide, and the primary endpoints (the patient's global assessment of symptoms) of two groups had non-significant difference (99). Carbohydrate antigen 125 (CA-125) diuretic-guided treatment proposed by Núñez et al. perhaps can solve the dilemma for that CA-125 is an excellent marker reflecting congestion. In this study, during the diuretic therapy process, serum CA-125 was detected and the dosage of diuretics would be determined and adjusted according to the plasm CA-125 concentration stratification. Individualized decongestion treatment may be realized when CA-125 diuretic-guided treatment is utilized.
Vasopressin Antagonists
In the pathogenetic process of WRF in CRS-1, as described above, AVP is over-generated and over-secreted in response to neurohormonal activation. AVP can activate both V1 receptors on the vasculature and V2 receptors on principal cells resulting in renal vasculature vasoconstriction and the enhancement of water and sodium reabsorption (103). Tolvaptan, a selective V2 receptor antagonist of AVP, can act on the distal portion of the nephron and competitively blocks the bond of AVP and V2 receptors, which facilitates the activation of the aquaporin system and the prevention of water and sodium reabsorption (104).
The Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan (EVEREST) trial has ascertained the efficacy and safety of tolvaptan in AHF treatment while the renal function was not included as observation criteria or endpoint criteria in this trial (105). Subsequent studies supplemented related evidence. The Clinical Effectiveness of Tolvaptan in Patients with Acute Heart Failure and Renal Dysfunction (AQUAMARINE) study validated that for patients with CRS-1, tolvaptan had better diuresis effectiveness compared with conventional diuretic therapy while the incidence of WRF was comparable (106). A secondary analysis of the AQUAMARINE study also revealed that patients with CRS-1 were well-tolerated to tolvaptan and tolvaptan can improve diuretic response (107). Another study conducted by Matsue et al. illustrated that for patients with AHF with renal dysfunction, tolvaptan could improve the prognosis and reduce the risk of all-cause death and HF readmission in specific individuals whose eGFR was 30mL/min/1.73m2 or above (108). All in all, vasopressin antagonist is an effective decongestion therapy with less influence on renal function.
Vasodilators
Vasodilators are another fundamental therapeutic regimen and are the second most commonly used drugs for AHF treatment, which has been validated by current clinical practice guidelines. Vasodilators can rapidly reduce ventricular filling pressure and central venous tone, thus reducing both cardiac preload and afterload, and decreasing the myocardial oxygen consumption (109). In hypertensive AHF, vasodilators are widely used and their efficacy is considerable, while for patients with low systolic pressure and/or systemic hypotension, vasodilators should be avoided. For hemodynamic influences of vasodilators, which may induce WRF, the utilization of vasodilators in patients with AHF with renal dysfunction should be cautious.
Nesiritide, or recombinant human BNP, is a type of vasodilator with both natriuretic and diuretic effects. Nesiritide has the same amino acid sequence and pharmacological effect on endogenous BNP. It can interact with natriuretic peptide receptor A on vascular smooth muscle cells and endothelial cells and can activate the guanylyl cyclase pathway, resulting in an increase of intracellular cyclic guanosine monophosphate (110). Besides, it can antagonize the effect of RAAS. Nesiritide has been approved for the treatment of AHF and previous literature indicated that nesiritide can mitigate congestion with no influence on renal function. The Acute Study of Clinical Effectiveness of Nesiritide in Decompensated Heart Failure (ASCEND-HF) study revealed that compared with placebo, nesiritide did not improve clinical outcomes and did not worsen renal function (111, 112). The ROSE-AHF study also showed similar results. These randomized clinical trials validated the renal safety of nesiritide despite its feeblish effects on decongestion and improvement of clinical outcomes and prognosis.
Relaxin, a 6kDa hormone of pregnancy mainly secreted by the corpus luteum of the ovary, plays an important role in maintaining the homeostasis of cardiovascular and hemodynamic during pregnancy. Relaxin can cause systemic and renal vasodilation via stimulating the relaxin family peptide receptors (RXFP), which are widely distributed in the heart, skeletal muscle, kidney, arteries, veins, and various tissues and organs. The activation of RXFP will increase the generation of second messengers, thus activating the nitric oxide pathway and cyclic adenosine monophosphate (cAMP) pathway, promoting the production of vasodilator substances, resulting in decreased systemic and renal vascular tone (113, 114). Serelaxin, the recombinant human relaxin-2, has similar pharmacologic action to relaxin and is considered as a potential therapeutic agent for AHF (115). The RELAX-AHF study recruited AHF participants with mild-to-moderate renal dysfunction (with eGFR of 30–75 mL/min/1·73 m2) who would be randomly assigned to the serelaxin treatment group and placebo treatment group. The study revealed that serelaxin was associated with greater dyspnoea relief, reduced worsening HF events, reduced cardiovascular and all-cause mortality compared to placebo, while lower proportions of patients on serelaxin therapy had development of WRF and adverse events related to renal function impairment compared with the placebo group (116). Additional studies concerning relaxin/ serelaxin have been ongoing. In the Pre-relaxin for the treatment of patients with acute heart failure (RELAX-AHF) study, patients with AHF receiving intravenous relaxin therapy tended to have a similar risk of WRF vs. those on placebo therapy (117, 118). The RELAX-AHF-EU study explored the effect of serelaxin when standard-of-care (SoC) therapy was added in AHF patients with mild-to-moderate renal dysfunction (119). Patients receiving serelaxin+SoC therapy were less likely to suffer renal deterioration compared to those with SoC therapy alone. The RELAX-AHF-2 study also affirmed the renal safety of serelaxin (120). To sum up, serelaxin is beneficial to ameliorate congestion symptoms and signs and improve the prognosis of CRS-1 patients with potential kidney protective effects.
Inotropes
For patients with AHF with low cardiac output and hypotension, especially for those with low systolic blood pressure, inadequate peripheral infusion, and poor response to strand therapy, inotropes are still effective remedies to maintain vital organs (including kidney) perfusion and function (121). Various studies have reported the renal protective effect of inotropes for AHF patients.
Dopamine, the most commonly used inotropes, can increase cardiac output by reducing cardiac afterload and may improve renal perfusion and GFR through dilating both afferent and efferent arterioles. However, only small-scale studies have validated the potential mechanism (122). The Dopamine in Acute Decompensated Heart Failure (DAD-HF) trial confirmed for AHF patients on high dose furosemide (20mg/h continuous for 8 h), the incidence of WRF was obviously higher than those on low dose furosemide combined with low dose dopamine (furosemide 5 mg/h plus dopamine 5 mg·kg−1·min−1 continuous infusion for 8 h). while there was no obvious difference in the 60-day mortality and/or rehospitalization rates between the two groups (123). In the DAD-HFIItrail, compared with the DAD-HFItrail, an isolated low dose furosemide (furosemide 5 mg/h continuous for 8 h) treatment group was added. Similar to the DAD-HFItrail, the incidence of WRF was higher in the high-dose furosemide group than the low-dose furosemide plus low-dose dopamine group and low-dose furosemide group at 24 h after initiation of treatment, no better prognosis was detected in latter groups during 1-year follow up period (124). Besides, controversial outcomes were concluded in other clinical trials including the ROSE-AHF study, the adrenergic inotropes exhibited more adverse events and no renoprotective effect compared with other types of inotropes such as levosimendan.
Levosimendan exerts its inotropic action through increasing sensitivity of troponin C to calcium in cardiomyocytes via c-AMP independent effect. Besides, it owns vasodilation effect through acting on adenosine triphosphate-sensitive potassium channels (KATP channels) in the smooth muscle cell. Based on these properties, levosimendan can dilate afferent arterioles of the kidney and ameliorate renal perfusion, thus increasing GFR. Some studies also indicated can selectively dilate afferent arterioles and own organ-specific effects (125). Fedele et al. showed that based on standard HF therapy, levosimendan could significantly increase GFR compared with placebo for CRS-1 patients with moderate renal impairment (126). Similar favorable results were also observed in Yilmaz et al.'s study. For CRS-1 patients requiring inotropic therapy, levosimendan could provide additional renal protective effects compared with dobutamine (127).
Adenosine Antagonists
Adenosine is a nucleoside that is generated by phosphohydrolase of precursor molecules, including adenosine triphosphate and adenosine monophosphate. Adenosine is an important compound in renal hemodynamic regulation and can induce renal vasoconstriction and reduce GFR via activating adenosine A1 receptors expressed on afferent arteriole. The secretion of adenosine will dramatically increase in response to renal hypoxic and ischemic injuries (128, 129). Besides, it can also enhance water and sodium reabsorption by stimulating adenosine A1 receptors on the proximal tubules. Based on this evidence, for patients with AHF with renal dysfunction, adenosine A1 receptor antagonists are thought to be novel pharmacologic agents for their antagonistic effects of renal vasoconstriction and favorable effects of diuresis, natriuresis, and amelioration of renal infusion (130).
In Pilot Effects of Rolofylline, a New Adenosine A1 Receptor Antagonist on Symptoms, Renal Function, and Outcomes in Patients with Acute Heart Failure (PROTECT) study, a total of 301 patients with AHF with impaired kidney function (estimated creatinine clearance between 20 and 80 ml/min) were enrolled and were randomized to the rolofylline (a type of adenosine A1 receptor antagonists) therapy group with different dosage administration and the placebo group. The serum creatinine concentration increased in patients on placebo therapy and remained stable or tended to decrease in those receiving rolofylline therapy. Besides, treatment with 30 mg rolofylline was associated with reduced 60-day mortality or readmission for cardiovascular or renal causes. The kidney protective effects of rolofylline were validated in this trial (131).
Based on the phenomenon that rolofylline has a renal protective effect, the Placebo-Controlled Randomized Study of the Selective A1 Adenosine Receptor Antagonist Rolofylline for Patients Hospitalized With Acute Decompensated Heart Failure and Volume Overload to Assess Treatment Effect on Congestion and Renal Function (PROTECT) trial, which recruited more participants (amount to 2,033 patients), was designed and conducted to provide additional details about the adenosine A1 receptor antagonist therapy. Interestingly, inconsistent with the prior results, there were no statistically significant differences in short-term renal function changes and persistent WRF incidence between the two groups, which indicated no renal protective effect of rolofylline in patients with CRS-1 (132). No clear renal protective effect of rolofylline was also found in a multicenter, randomized, double-blind, Placebo-Controlled Study of the Effects of KW-3902 Injectable Emulsion on Heart Failure Signs and Symptoms, Diuresis, Renal Function, and Clinical Outcomes in Subjects Hospitalized With Worsening Renal Function and Heart Failure Requiring Intravenous Therapy (REACH UP) study (133). Until now, for rolofylline, the beneficial effect of preservation of renal function is imprecise.
Ultrafiltration
Ultrafiltration is a mechanically therapeutic method to remove excess fluid through a semipermeable membrane by the actuation of transmembrane pressure gradient (134). Current studies and clinical practice guidelines have not recommended ultrafiltration as a routine strategy to tackle over-congestion in CRS-1, even in those who have a poor response to diuretics. However, for patients with refractory volume overload, obstinate DR, renal deficiency, severe hyperkalemia, and acidosis, ultrafiltration is recommended and can serve as an effective therapeutic regimen (122).
In the Aquapheresis vs. Intravenous Diuretics and Hospitalization for Heart Failure (AVOID-HF) trial, compared with patients with AHF on adjustable intravenous loop diuretics therapy, those receiving adjustable ultrafiltration therapy had a better decongestive effect and reduced risk of recurrent HF and cardiovascular events within 90 days of discharge from the index hospitalization. It is remarkable that adjustable ultrafiltration therapy would not worsen renal function (135). In previous randomized controlled trials, including the Early ultrafiltration in patients with decompensated heart failure and diuretic resistance (EUPHORIA) trial and the Ultrafiltration vs. Intravenous Diuretics for Patients Hospitalized for Acute Decompensated Congestive Heart Failure (UNLOAD) trial, no WRF was detected in patients with AHF receiving ultrafiltration therapy (136, 137). Suspicion of renal safety of ultrafiltration is proposed in the Cardiorenal Rescue Study in Acute Decompensated Heart Failure (CARRESS-HF) trial which indicated the correlation of ultrafiltration therapy and increased risk of WRF in patients with acute CRS (138). The opposite results of the CARRESS-HF trial were further evaluated. Although the deterioration of renal function in the ultrafiltration group was statistically significant but may not be clinically significant. Besides, the serum creatinine concentration (0.23 ± 0.70 mg/dL) did not reach the acknowledged criteria of WRF (0.30–0.50 mg/dL or greater), which has been described above (134).
Conclusion
CRS-1 is a major clinical problem and WRF predicts a poor prognosis. Despite no fully explicit mechanism, multiple factors, including hemodynamic imbalance, neurohormonal activation, sympathetic activity, pharmacological interventions, inflammation along with oxidative stress, are involved in pathogenesis. Numerous biomarkers have a good performance in the early identification of WRF in CRS-1 while some are not so excellent. Effective treatment strategies with good renal safety have been explored and further studies should be conducted.
Author Contributions
All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
Funding
This study was supported by the National Natural Science Foundation of China (81873516, 81873522, and 81900444), the National Key Research and Development Program of Shandong Province (2017YFC1308303), the Shandong Provincial Natural Science Foundation of China (ZR2019PH030), and the Clinical Research Center of Shandong University (No. 2020SDUCRCA009).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Harjola VP, Mullens W, Banaszewski M, Bauersachs J, Brunner-La Rocca HP, Chioncel O, et al. Organ dysfunction, injury and failure in acute heart failure: from pathophysiology to diagnosis and management. A review on behalf of the Acute Heart Failure Committee of the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). Eur J Heart Fail. (2017) 19:821–36. doi: 10.1002/ejhf.872
2. Cheema B, Ambrosy AP, Kaplan RM, Senni M, Fonarow GC, Chioncel O, et al. Lessons learned in acute heart failure. Eur J Heart Fail. (2018) 20:630–41. doi: 10.1002/ejhf.1042
3. Zannad F, Rossignol P. Cardiorenal syndrome revisited. Circulation. (2018) 138:929–44. doi: 10.1161/CIRCULATIONAHA.117.028814
4. Ronco C, Bellasi A, Di Lullo L. Cardiorenal syndrome: an overview. Adv Chronic Kidney Dis. (2018) 25:382–90. doi: 10.1053/j.ackd.2018.08.004
5. Rangaswami J, Bhalla V, Blair JEA, Chang TI, Costa S, Lentine KL, et al. Cardiorenal syndrome: classification, pathophysiology, diagnosis, and treatment strategies: a scientific statement from the American Heart Association. Circulation. (2019) 139:e840–78. doi: 10.1161/CIR.0000000000000664
6. Metra M, Nodari S, Parrinello G, Bordonali T, Bugatti S, Danesi R, et al. Worsening renal function in patients hospitalised for acute heart failure: clinical implications and prognostic significance. Eur J Heart Fail. (2008) 10:188–95. doi: 10.1016/j.ejheart.2008.01.011
7. Ronco C, McCullough P, Anker SD, Anand I, Aspromonte N, Bagshaw SM, et al. Cardio-renal syndromes: report from the consensus conference of the acute dialysis quality initiative. Eur Heart J. (2010) 31:703–11. doi: 10.1093/eurheartj/ehp507
8. Logeart D, Tabet JY, Hittinger L, Thabut G, Jourdain P, Maison P, et al. Transient worsening of renal function during hospitalization for acute heart failure alters outcome. Int J Cardiol. (2008) 127:228–32. doi: 10.1016/j.ijcard.2007.06.007
9. Damman K, Testani JM. The kidney in heart failure: an update. Eur Heart J. (2015) 36:1437–44. doi: 10.1093/eurheartj/ehv010
10. Sheerin NJ, Newton PJ, Macdonald PS, Leung DY, Sibbritt D, Spicer ST, et al. Worsening renal function in heart failure: the need for a consensus definition. Int J Cardiol. (2014) 174:484–91. doi: 10.1016/j.ijcard.2014.04.162
11. Triposkiadis F, Starling RC, Boudoulas H, Giamouzis G, Butler J. The cardiorenal syndrome in heart failure: cardiac? renal? syndrome? Heart Fail Rev. (2012) 17:355–66. doi: 10.1007/s10741-011-9291-x
12. Tang WH, Mullens W. Cardiorenal syndrome in decompensated heart failure. Heart. (2010) 96:255–60. doi: 10.1136/hrt.2009.166256
13. Shirakabe A, Hata N, Kobayashi N, Okazaki H, Matsushita M, Shibata Y, et al. Worsening renal function definition is insufficient for evaluating acute renal failure in acute heart failure. ESC Heart Fail. (2018) 5:322–31. doi: 10.1002/ehf2.12264
14. Kidney Disease: Improving Global Outcomes (KDIGO). KDIGO clinical practice guidelines for the prevention, diagnosis, evaluation, and treatment of hepatitis C in chronic kidney disease. Kidney Int Suppl. (2008) 109:S1–99. doi: 10.1038/ki.2008.81
15. Mehta RL, Kellum JA, Shah SV, Molitoris BA, Ronco C, Warnock DG, et al. Acute Kidney Injury Network: report of an initiative to improve outcomes in acute kidney injury. Crit Care. (2007) 11:R31. doi: 10.1186/cc5713
16. Kellum JA, Bellomo R, Ronco C, Mehta R, Clark W, Levin NW. The 3rd International Consensus Conference of the Acute Dialysis Quality Initiative (ADQI). Int J Artif Organs. (2005) 28:441–4. doi: 10.1177/039139880502800503
17. Ronco C, Ronco F. Cardio-renal syndromes: a systematic approach for consensus definition and classification. Heart Fail Rev. (2012) 17:151–60. doi: 10.1007/s10741-010-9224-0
18. Palazzuoli A, Ruocco G. Heart-kidney interactions in cardiorenal syndrome type 1. Adv Chronic Kidney Dis. (2018) 25:408–17. doi: 10.1053/j.ackd.2018.08.013
19. Bongartz LG, Cramer MJ, Doevendans PA, Joles JA, Braam B. The severe cardiorenal syndrome: 'Guyton revisited'. Eur Heart J. (2005) 26:11–7. doi: 10.1093/eurheartj/ehi020
20. Felker GM, Mentz RJ. Diuretics and ultrafiltration in acute decompensated heart failure. J Am Coll Cardiol. (2012) 59:2145–53. doi: 10.1016/j.jacc.2011.10.910
21. Testani JM, Coca SG, McCauley BD, Shannon RP, Kimmel SE. Impact of changes in blood pressure during the treatment of acute decompensated heart failure on renal and clinical outcomes. Eur J Heart Fail. (2011) 13:877–84. doi: 10.1093/eurjhf/hfr070
22. Yancy CW, Lopatin M, Stevenson LW, De Marco T, Fonarow GC. ADHERE Scientific Advisory Committee and Investigators. Clinical presentation, management, and in-hospital outcomes of patients admitted with acute decompensated heart failure with preserved systolic function: a report from the Acute Decompensated Heart Failure National Registry (ADHERE) Database. J Am Coll Cardiol. (2006) 47:76–84. doi: 10.1016/j.jacc.2005.09.022
23. Nohria A, Hasselblad V, Stebbins A, Pauly DF, Fonarow GC, Shah M, et al. Cardiorenal interactions: insights from the ESCAPE trial. J Am Coll Cardiol. (2008) 51:1268–74. doi: 10.1016/j.jacc.2007.08.072
24. McCallum W, Tighiouart H, Testani JM, Griffin M, Konstam MA, Udelson JE, et al. Acute kidney function declines in the context of decongestion in acute decompensated heart failure. JACC Heart Fail. (2020) 8:537–47. doi: 10.1016/j.jchf.2020.03.009
25. Shamseddin MK, Parfrey PS. Mechanisms of the cardiorenal syndromes. Nat Rev Nephrol. (2009) 5:641–9. doi: 10.1038/nrneph.2009.156
26. Wencker D. Acute cardio-renal syndrome: progression from congestive heart failure to congestive kidney failure. Curr Heart Fail Rep. (2007) 4:134–8. doi: 10.1007/s11897-007-0031-4
27. Uthoff H, Breidthardt T, Klima T, Aschwanden M, Arenja N, Socrates T, et al. Central venous pressure and impaired renal function in patients with acute heart failure. Eur J Heart Fail. (2011) 13:432–9. doi: 10.1093/eurjhf/hfq195
28. Mullens W, Abrahams Z, Francis GS, Sokos G, Taylor DO, Starling RC, et al. Importance of venous congestion for worsening of renal function in advanced decompensated heart failure. J Am Coll Cardiol. (2009) 53:589–96. doi: 10.1016/j.jacc.2008.05.068
29. Textor SC. Renal Arterial Disease and Hypertension. Med Clin North Am. (2017) 101:65–79. doi: 10.1016/j.mcna.2016.08.010
30. Mullens W, Abrahams Z, Skouri HN, Francis GS, Taylor DO, Starling RC, et al. Elevated intra-abdominal pressure in acute decompensated heart failure: a potential contributor to worsening renal function? J Am Coll Cardiol. (2008) 51:300–6. doi: 10.1016/j.jacc.2007.09.043
31. Yacoub R, Patel N, Lohr JW, Rajagopalan S, Nader N, Arora P. Acute kidney injury and death associated with renin angiotensin system blockade in cardiothoracic surgery: a meta-analysis of observational studies. Am J Kidney Dis. (2013) 62:1077–86. doi: 10.1053/j.ajkd.2013.04.018
32. Palazzuoli A, Ruocco G, Pellicori P, Incampo E, Di Tommaso C, Favilli R, et al. The prognostic role of different renal function phenotypes in patients with acute heart failure. Int J Cardiol. (2019) 276:198–203. doi: 10.1016/j.ijcard.2018.11.108
33. Ruocco G, Palazzuoli A, Ter Maaten JM. The role of the kidney in acute and chronic heart failure. Heart Fail Rev. (2020) 25:107–18. doi: 10.1007/s10741-019-09870-6
34. Palazzuoli A, Lombardi C, Ruocco G, Padeletti M, Nuti R, Metra M, et al. Chronic kidney disease and worsening renal function in acute heart failure: different phenotypes with similar prognostic impact? Eur Heart J Acute Cardiovasc Care. (2016) 5:534–48. doi: 10.1177/2048872615589511
35. Ronco C, Cicoira M, McCullough PA. Cardiorenal syndrome type 1: pathophysiological crosstalk leading to combined heart and kidney dysfunction in the setting of acutely decompensated heart failure. J Am Coll Cardiol. (2012) 60:1031–42. doi: 10.1016/j.jacc.2012.01.077
36. Pan X, Shao Y, Wu F, Wang Y, Xiong R, Zheng J, et al. FGF21 prevents angiotensin II-induced hypertension and vascular dysfunction by activation of ACE2/Angiotensin-(1-7) axis in mice. Cell Metab. (2018) 27:1323–37.e5. doi: 10.1016/j.cmet.2018.04.002
37. Brown NJ. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nat Rev Nephrol. (2013) 9:459–69. doi: 10.1038/nrneph.2013.110
38. Wang Y, Seto SW, Golledge J. Angiotensin II, sympathetic nerve activity and chronic heart failure. Heart Fail Rev. (2014) 19:187–98. doi: 10.1007/s10741-012-9368-1
39. Jönsson S, Agic MB, Narfström F, Melville JM, Hultström M. Renal neurohormonal regulation in heart failure decompensation. Am J Physiol Regul Integr Comp Physiol. (2014) 307:R493–7. doi: 10.1152/ajpregu.00178.2014
40. Mendelowitz D. How does angiotensin activate hypothalamic neurons essential for controlling sympathetic activity and blood pressure? Hypertension. (2016) 68:1340–1. doi: 10.1161/HYPERTENSIONAHA.116.07914
41. Cole RT, Masoumi A, Triposkiadis F, Giamouzis G, Georgiopoulou V, Kalogeropoulos A, et al. Renal dysfunction in heart failure. Med Clin North Am. (2012) 96:955–74. doi: 10.1016/j.mcna.2012.07.005
42. Bankir L, Bichet DG, Morgenthaler NG. Vasopressin: physiology, assessment and osmosensation. J Intern Med. (2017) 282:284–97. doi: 10.1111/joim.12645
43. Sarraf M, Masoumi A, Schrier RW. Cardiorenal syndrome in acute decompensated heart failure. Clin J Am Soc Nephrol. (2009) 4:2013–26. doi: 10.2215/CJN.03150509
44. Pugliese NR, Fabiani I, Conte L, Nesti L, Masi S, Natali A, et al. Persistent congestion, renal dysfunction and inflammatory cytokines in acute heart failure: a prognosis study. J Cardiovasc Med (Hagerstown). (2020) 21:494–502. doi: 10.2459/JCM.0000000000000974
45. Clementi A, Virzì GM, Battaglia GG, Ronco C. Neurohormonal, endocrine, and immune dysregulation and inflammation in cardiorenal syndrome. Cardiorenal Med. (2019) 9:265–73. doi: 10.1159/000500715
46. Virzì GM, Clementi A, de Cal M, Brocca A, Day S, Pastori S, et al. Oxidative stress: dual pathway induction in cardiorenal syndrome type 1 pathogenesis. Oxid Med Cell Longev. (2015) 2015:391790. doi: 10.1155/2015/391790
47. Rubattu S, Mennuni S, Testa M, Mennuni M, Pierelli G, Pagliaro B, et al. Pathogenesis of chronic cardiorenal syndrome: is there a role for oxidative stress? Int J Mol Sci. (2013) 14:23011–32. doi: 10.3390/ijms141123011
48. Pastori S, Virzì GM, Brocca A, de Cal M, Clementi A, Vescovo G, et al. Cardiorenal syndrome type 1: a defective regulation of monocyte apoptosis induced by proinflammatory and proapoptotic factors. Cardiorenal Med. (2015) 5:105–15. doi: 10.1159/000371898
49. Virzì GM, Breglia A, Brocca A, de Cal M, Bolin C, Vescovo G, et al. Levels of proinflammatory cytokines, oxidative stress, and tissue damage markers in patients with acute heart failure with and without cardiorenal syndrome type 1. Cardiorenal Med. (2018) 8:321–31. doi: 10.1159/000492602
50. Virzì GM, Breglia A, Castellani C, Ankawi G, Bolin C, de Cal M, et al. Lipopolysaccharide in systemic circulation induces activation of inflammatory response and oxidative stress in cardiorenal syndrome type 1. J Nephrol. (2019) 32:803–10. doi: 10.1007/s40620-019-00613-2
51. Linhart C, Ulrich C, Greinert D, Dambeck S, Wienke A, Girndt M, et al. Systemic inflammation in acute cardiorenal syndrome: an observational pilot study. ESC Heart Fail. (2018) 5:920–30. doi: 10.1002/ehf2.12327
52. Lassus JP, Nieminen MS, Peuhkurinen K, Pulkki K, Siirilä-Waris K, Sund R, et al. Markers of renal function and acute kidney injury in acute heart failure: definitions and impact on outcomes of the cardiorenal syndrome. Eur Heart J. (2010) 31:2791–8. doi: 10.1093/eurheartj/ehq293
53. Chen C, Yang X, Lei Y, Zha Y, Liu H, Ma C, et al. Urinary Biomarkers at the Time of AKI Diagnosis as Predictors of Progression of AKI among Patients with Acute Cardiorenal Syndrome. Clin J Am Soc Nephrol. (2016) 11:1536–44. doi: 10.2215/CJN.00910116
54. Murray PT, Wettersten N, van Veldhuisen DJ, Mueller C, Filippatos G, Nowak R, et al. Utility of urine neutrophil gelatinase-associated lipocalin for worsening renal function during hospitalization for acute heart failure: primary findings of the urine N-gal acute kidney injury N-gal evaluation of symptomatic heart failure study (AKINESIS). J Card Fail. (2019) 25:654–65. doi: 10.1016/j.cardfail.2019.05.009
55. Okubo Y, Sairaku A, Morishima N, Ogi H, Matsumoto T, Kinoshita H, et al. Increased urinary liver-type fatty acid-binding protein level predicts worsening renal function in patients with acute heart failure. J Card Fail. (2018) 24:520–4. doi: 10.1016/j.cardfail.2018.07.003
56. Legrand M, De Berardinis B, Gaggin HK, Magrini L, Belcher A, Zancla B, et al. Evidence of uncoupling between renal dysfunction and injury in cardiorenal syndrome: insights from the BIONICS study. PLoS One. (2014) 9:e112313. doi: 10.1371/journal.pone.0112313
57. Ahmad T, Jackson K, Rao VS, Tang WHW, Brisco-Bacik MA, Chen HH, et al. Worsening renal function in patients with acute heart failure undergoing aggressive diuresis is not associated with tubular injury. Circulation. (2018) 137:2016–28. doi: 10.1161/CIRCULATIONAHA.117.030112
58. Sokolski M, Zymliński R, Biegus J, Siwołowski P, Nawrocka-Millward S, Todd J, et al. Urinary levels of novel kidney biomarkers and risk of true worsening renal function and mortality in patients with acute heart failure. Eur J Heart Fail. (2017) 19:760–7. doi: 10.1002/ejhf.746
59. Funabashi S, Omote K, Nagai T, Honda Y, Nakano H, Honda S, et al. Elevated admission urinary N-acetyl-β-D-glucosamidase level is associated with worse long-term clinical outcomes in patients with acute heart failure. Eur Heart J Acute Cardiovasc Care. (2020) 9:429–36. doi: 10.1177/2048872620901986
60. Atici A, Emet S, Cakmak R, Yuruyen G, Alibeyoglu A, Akarsu M, et al. Type I cardiorenal syndrome in patients with acutely decompensated heart failure: the importance of new renal biomarkers. Eur Rev Med Pharmacol Sci. (2018) 22:3534–43. doi: 10.26355/eurrev_201806_15180
61. Schanz M, Shi J, Wasser C, Alscher MD, Kimmel M. Urinary [TIMP-2] × [IGFBP7] for risk prediction of acute kidney injury in decompensated heart failure. Clin Cardiol. (2017) 40:485–91. doi: 10.1002/clc.22683
62. Kashani K, Rosner MH, Ostermann M. Creatinine: From physiology to clinical application. Eur J Intern Med. (2020) 72:9–14. doi: 10.1016/j.ejim.2019.10.025
63. Wyss M, Kaddurah-Daouk R. Creatine and creatinine metabolism. Physiol Rev. (2000) 80:1107–213. doi: 10.1152/physrev.2000.80.3.1107
64. Ferguson MA, Waikar SS. Established and emerging markers of kidney function. Clin Chem. (2012) 58:680–9. doi: 10.1373/clinchem.2011.167494
65. Stevens LA, Coresh J, Greene T, Levey AS. Assessing kidney function–measured and estimated glomerular filtration rate. N Engl J Med. (2006) 354:2473–83. doi: 10.1056/NEJMra054415
66. Rafouli-Stergiou P, Parissis J, Farmakis D, Bistola V, Nikolaou M, Vasiliadis K, et al. Prognostic value of in-hospital change in cystatin C in patients with acutely decompensated heart failure and renal dysfunction. Int J Cardiol. (2015) 182:74–6. doi: 10.1016/j.ijcard.2014.12.135
67. Metra M, Voors AA. The puzzle of kidney dysfunction in heart failure: an introduction. Heart Fail Rev. (2012) 17:129–31. doi: 10.1007/s10741-012-9304-4
68. Zhang Z, Lu B, Sheng X, Jin N. Cystatin C in prediction of acute kidney injury: a systemic review and meta-analysis. Am J Kidney Dis. (2011) 58:356–65. doi: 10.1053/j.ajkd.2011.02.389
69. Porrini E, Ruggenenti P, Luis-Lima S, Carrara F, Jiménez A, de Vries APJ, et al. Estimated GFR: time for a critical appraisal. Nat Rev Nephrol. (2019) 15:177–90. doi: 10.1038/s41581-018-0080-9
70. Ferguson TW, Komenda P, Tangri N. Cystatin C as a biomarker for estimating glomerular filtration rate. Curr Opin Nephrol Hypertens. (2015) 24:295–300. doi: 10.1097/MNH.0000000000000115
71. Shlipak MG, Mattes MD, Peralta CA. Update on cystatin C: incorporation into clinical practice. Am J Kidney Dis. (2013) 62:595–603. doi: 10.1053/j.ajkd.2013.03.027
72. George JA, Gounden V. Novel glomerular filtration markers. Adv Clin Chem. (2019) 88:91–119. doi: 10.1016/bs.acc.2018.10.005
73. Buonafine M, Martinez-Martinez E, Jaisser F. More than a simple biomarker: the role of NGAL in cardiovascular and renal diseases. Clin Sci (Lond). (2018) 132:909–23. doi: 10.1042/CS20171592
74. Singer E, Markó L, Paragas N, Barasch J, Dragun D, Müller DN, et al. Neutrophil gelatinase-associated lipocalin: pathophysiology and clinical applications. Acta Physiol (Oxf). (2013) 207:663–72. doi: 10.1111/apha.12054
75. Yamamoto T, Noiri E, Ono Y, Doi K, Negishi K, Kamijo A, et al. Renal L-type fatty acid–binding protein in acute ischemic injury. J Am Soc Nephrol. (2007) 18:2894–902. doi: 10.1681/ASN.2007010097
76. Li B, Hao J, Zeng J, Sauter ER. SnapShot: FABP functions. Cell. (2020) 182:1066–1066.e1. doi: 10.1016/j.cell.2020.07.027
77. Xu Y, Xie Y, Shao X, Ni Z, Mou S. L-FABP: A novel biomarker of kidney disease. Clin Chim Acta. (2015) 445:85–90. doi: 10.1016/j.cca.2015.03.017
78. Holzscheiter L, Beck C, Rutz S, Manuilova E, Domke I, Guder WG, et al. L-FABP, and KIM-1 in comparison to established markers of renal dysfunction. Clin Chem Lab Med. (2014) 52:537–46. doi: 10.1515/cclm-2013-0693
79. Geng J, Qiu Y, Qin Z, Su B. The value of kidney injury molecule 1 in predicting acute kidney injury in adult patients: a systematic review and Bayesian meta-analysis. J Transl Med. (2021) 19:105. doi: 10.1186/s12967-021-02776-8
80. Lim AI, Tang SC, Lai KN, Leung JC. Kidney injury molecule-1: more than just an injury marker of tubular epithelial cells? J Cell Physiol. (2013) 228:917–24. doi: 10.1002/jcp.24267
81. Coca SG, Yalavarthy R, Concato J, Parikh CR. Biomarkers for the diagnosis and risk stratification of acute kidney injury: a systematic review. Kidney Int. (2008) 73:1008–16. doi: 10.1038/sj.ki.5002729
82. Bonventre JV, Yang L. Kidney injury molecule-1. Curr Opin Crit Care. (2010) 16:556–61. doi: 10.1097/MCC.0b013e32834008d3
83. Comnick M, Ishani A. Renal biomarkers of kidney injury in cardiorenal syndrome. Curr Heart Fail Rep. (2011) 8:99–105. doi: 10.1007/s11897-011-0052-x
84. Mårtensson J, Martling CR, Bell M. Novel biomarkers of acute kidney injury and failure: clinical applicability. Br J Anaesth. (2012) 109:843–50. doi: 10.1093/bja/aes357
85. Kaplanski G. Interleukin-18: Biological properties and role in disease pathogenesis. Immunol Rev. (2018) 281:138–53. doi: 10.1111/imr.12616
86. Liu Y, Guo W, Zhang J, Xu C, Yu S, Mao Z, et al. Urinary interleukin 18 for detection of acute kidney injury: a meta-analysis. Am J Kidney Dis. (2013) 62:1058–67. doi: 10.1053/j.ajkd.2013.05.014
87. Melnikov VY, Ecder T, Fantuzzi G, Siegmund B, Lucia MS, Dinarello CA, et al. Impaired IL-18 processing protects caspase-1-deficient mice from ischemic acute renal failure. J Clin Invest. (2001) 107:1145–52. doi: 10.1172/JCI12089
88. Parikh CR, Abraham E, Ancukiewicz M, Edelstein CL. Urine IL-18 is an early diagnostic marker for acute kidney injury and predicts mortality in the intensive care unit. J Am Soc Nephrol. (2005) 16:3046–52. doi: 10.1681/ASN.2005030236
89. Bullen AL, Katz R, Jotwani V, Garimella PS, Lee AK, Estrella MM, et al. Biomarkers of kidney tubule health, CKD progression, and acute kidney injury in SPRINT (Systolic Blood Pressure Intervention Trial) participants. Am J Kidney Dis. (2021) 78:361–8.e1. doi: 10.1053/j.ajkd.2021.01.021
90. Fan W, Ankawi G, Zhang J, Digvijay K, Giavarina D, Yin Y, et al. Current understanding and future directions in the application of TIMP-2 and IGFBP7 in AKI clinical practice. Clin Chem Lab Med. (2019) 57:567–76. doi: 10.1515/cclm-2018-0776
91. Srisawat N, Kellum JA. The role of biomarkers in acute kidney injury. Crit Care Clin. (2020) 36:125–40. doi: 10.1016/j.ccc.2019.08.010
92. Meersch M, Schmidt C, Van Aken H, Martens S, Rossaint J, Singbartl K, et al. Urinary TIMP-2 and IGFBP7 as early biomarkers of acute kidney injury and renal recovery following cardiac surgery. PLoS ONE. (2014) 9:e93460. doi: 10.1371/journal.pone.0093460
93. Kellum JA, Chawla LS. Cell-cycle arrest and acute kidney injury: the light and the dark sides. Nephrol Dial Transplant. (2016) 31:16–22. doi: 10.1093/ndt/gfv130
94. Jia HM, Huang LF, Zheng Y, Li WX. Diagnostic value of urinary tissue inhibitor of metalloproteinase-2 and insulin-like growth factor binding protein 7 for acute kidney injury: a meta-analysis. Crit Care. (2017) 21:77. doi: 10.1186/s13054-017-1660-y
95. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE Jr, Colvin MM, et al. 2017 ACC/AHA/HFSA Focused Update of the 2013 ACCF/AHA Guideline for the Management of Heart Failure: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Failure Society of America. J Am Coll Cardiol. (2017) 70:776–803. doi: 10.1161/CIR.0000000000000509
96. Takahama H, Nishikimi T, Takashio S, Hayashi T, Nagai-Okatani C, Asada T, et al. Change in the NT-proBNP/Mature BNP molar ratio precedes worsening renal function in patients with acute heart failure: a novel predictor candidate for cardiorenal syndrome. J Am Heart Assoc. (2019) 8:e011468. doi: 10.1161/JAHA.118.011468
97. Weber M, Hamm C. Role of B-type natriuretic peptide (BNP) and NT-proBNP in clinical routine. Heart. (2006) 92:843–9. doi: 10.1136/hrt.2005.071233
98. Dos Reis D, Fraticelli L, Bassand A, Manzo-Silberman S, Peschanski N, Charpentier S, et al. Impact of renal dysfunction on the management and outcome of acute heart failure: results from the French prospective, multicentre, DeFSSICA survey. BMJ Open. (2019) 9:e022776. doi: 10.1136/bmjopen-2018-022776
99. Felker GM, Lee KL, Bull DA, Redfield MM, Stevenson LW, Goldsmith SR, et al. Diuretic strategies in patients with acute decompensated heart failure. N Engl J Med. (2011) 364:797–805. doi: 10.1056/NEJMoa1005419
100. Sharma K, Vaishnav J, Kalathiya R, Hu JR, Miller J, Shah N, et al. Randomized evaluation of heart failure with preserved ejection fraction patients with acute heart failure and dopamine: the ROPA-DOP trial. JACC Heart Fail. (2018) 6:859–70. doi: 10.1016/j.jchf.2018.04.008
101. Palazzuoli A, Pellegrini M, Franci B, Beltrami M, Ruocco G, Gonnelli S, et al. Short and long-term effects of continuous versus intermittent loop diuretics treatment in acute heart failure with renal dysfunction. Intern Emerg Med. (2015) 10:41–9. doi: 10.1007/s11739-014-1112-5
102. Palazzuoli A, Pellegrini M, Ruocco G, Martini G, Franci B, Campagna MS, et al. Continuous versus bolus intermittent loop diuretic infusion in acutely decompensated heart failure: a prospective randomized trial. Crit Care. (2014) 18:R134. doi: 10.1186/cc13952
103. Christ-Crain M. Vasopressin and Copeptin in health and disease. Rev Endocr Metab Disord. (2019) 20:283–94. doi: 10.1007/s11154-019-09509-9
105. Konstam MA, Gheorghiade M, Burnett JC Jr, Grinfeld L, Maggioni AP, Swedberg K, et al. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST outcome trial. JAMA. (2007) 297:1319–31. doi: 10.1001/jama.297.12.1319
106. Matsue Y, Suzuki M, Torii S, Yamaguchi S, Fukamizu S, Ono Y, et al. Clinical effectiveness of tolvaptan in patients with acute heart failure and renal dysfunction. J Card Fail. (2016) 22:423–32. doi: 10.1016/j.cardfail.2016.02.007
107. Matsue Y, Ter Maaten JM, Suzuki M, Torii S, Yamaguchi S, Fukamizu S, et al. Early treatment with tolvaptan improves diuretic response in acute heart failure with renal dysfunction. Clin Res Cardiol. (2017) 106:802–12. doi: 10.1007/s00392-017-1122-1
108. Matsue Y, Suzuki M, Torii S, Yamaguchi S, Fukamizu S, Ono Y, et al. Prognostic impact of early treatment with tolvaptan in patients with acute heart failure and renal dysfunction. Int J Cardiol. (2016) 221:188–93. doi: 10.1016/j.ijcard.2016.07.063
109. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JGF, Coats AJS, et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: the Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC) Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur Heart J. (2016) 37:2129–200. doi: 10.1093/eurheartj/ehw128
110. Keating GM, Goa KL. Nesiritide: a review of its use in acute decompensated heart failure. Drugs. (2003) 63:47–70. doi: 10.2165/00003495-200363010-00004
111. O'Connor CM, Starling RC, Hernandez AF, Armstrong PW, Dickstein K, Hasselblad V, et al. Effect of nesiritide in patients with acute decompensated heart failure. N Engl J Med. (2011) 365:32–43. doi: 10.1056/NEJMx110061
112. van Deursen VM, Hernandez AF, Stebbins A, Hasselblad V, Ezekowitz JA, Califf RM, et al. Nesiritide, renal function, and associated outcomes during hospitalization for acute decompensated heart failure: results from the Acute Study of Clinical Effectiveness of Nesiritide and Decompensated Heart Failure (ASCEND-HF). Circulation. (2014) 130:958–65. doi: 10.1161/CIRCULATIONAHA.113.003046
113. Feijóo-Bandín S, Aragón-Herrera A, Rodríguez-Penas D, Portolés M, Roselló-Lletí E, Rivera M, et al. Relaxin-2 in cardiometabolic diseases: mechanisms of action and future perspectives. Front Physiol. (2017) 8:599. doi: 10.3389/fphys.2017.00599
114. Schmieder RE, Mitrovic V, Hengstenberg C. Renal impairment and worsening of renal function in acute heart failure: can new therapies help? The potential role of serelaxin. Clin Res Cardiol. (2015) 104:621–31. doi: 10.1007/s00392-015-0839-y
115. Unemori E. Serelaxin in clinical development: past, present and future. Br J Pharmacol. (2017) 174:921–32. doi: 10.1111/bph.13695
116. Teerlink JR, Cotter G, Davison BA, Felker GM, Filippatos G, Greenberg BH, et al. Serelaxin, recombinant human relaxin-2, for treatment of acute heart failure (RELAX-AHF): a randomised, placebo-controlled trial. Lancet. (2013) 381:29–39. doi: 10.1016/S0140-6736(12)61855-8
117. Teerlink JR, Metra M, Felker GM, Ponikowski P, Voors AA, Weatherley BD, et al. Relaxin for the treatment of patients with acute heart failure (Pre-RELAX-AHF): a multicentre, randomised, placebo-controlled, parallel-group, dose-finding phase IIb study. Lancet. (2009) 373:1429–39. doi: 10.1016/S0140-6736(09)60622-X
118. Voors AA, Davison BA, Felker GM, Ponikowski P, Unemori E, Cotter G, et al. Early drop in systolic blood pressure and worsening renal function in acute heart failure: renal results of Pre-RELAX-AHF. Eur J Heart Fail. (2011) 13:961–7. doi: 10.1093/eurjhf/hfr060
119. Maggioni AP, López-Sendón J, Nielsen OW, Hallén J, Aalamian-Mattheis M, Wang Y, et al. Efficacy and safety of serelaxin when added to standard of care in patients with acute heart failure: results from a PROBE study, RELAX-AHF-EU. Eur J Heart Fail. (2019) 21:322–33. doi: 10.1002/ejhf.1368
120. Metra M, Teerlink JR, Cotter G, Davison BA, Felker GM, Filippatos G, et al. Effects of serelaxin in patients with acute heart failure. N Engl J Med. (2019) 381:716–26. doi: 10.1056/NEJMoa1801291
121. McDonagh TA, Metra M, Adamo M, Gardner RS, Baumbach A, Böhm M, et al. 2021 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure. Eur Heart J. (2021) 42:3599–726. doi: 10.1093/eurheartj/ehab368
122. Zima E, Farmakis D, Pollesello P, Parissis JT. Differential effects of inotropes and inodilators on renal function in acute cardiac care. Eur Heart J Suppl. (2020) 22:D12–9. doi: 10.1093/eurheartj/suaa091
123. Giamouzis G, Butler J, Starling RC, Karayannis G, Nastas J, Parisis C, et al. Impact of dopamine infusion on renal function in hospitalized heart failure patients: results of the Dopamine in Acute Decompensated Heart Failure (DAD-HF) Trial. J Card Fail. (2010) 16:922–30. doi: 10.1016/j.cardfail.2010.07.246
124. Triposkiadis FK, Butler J, Karayannis G, Starling RC, Filippatos G, Wolski K, et al. Efficacy and safety of high dose versus low dose furosemide with or without dopamine infusion: the Dopamine in Acute Decompensated Heart Failure II (DAD-HF II) trial. Int J Cardiol. (2014) 172:115–21. doi: 10.1016/j.ijcard.2013.12.276
125. Bouchez S, Fedele F, Giannakoulas G, Gustafsson F, Harjola VP, Karason K, et al. Levosimendan in acute and advanced heart failure: an expert perspective on posology and therapeutic application. Cardiovasc Drugs Ther. (2018) 32:617–24. doi: 10.1007/s10557-018-6838-2
126. Fedele F, Bruno N, Brasolin B, Caira C, D'Ambrosi A, Mancone M. Levosimendan improves renal function in acute decompensated heart failure: possible underlying mechanisms. Eur J Heart Fail. (2014) 16:281–8. doi: 10.1002/ejhf.9
127. Yilmaz MB, Yalta K, Yontar C, Karadas F, Erdem A, Turgut OO, et al. Levosimendan improves renal function in patients with acute decompensated heart failure: comparison with dobutamine. Cardiovasc Drugs Ther. (2007) 21:431–5. doi: 10.1007/s10557-007-6066-7
128. Bauerle JD, Grenz A, Kim JH, Lee HT, Eltzschig HK. Adenosine generation and signaling during acute kidney injury. J Am Soc Nephrol. (2011) 22:14–20. doi: 10.1681/ASN.2009121217
129. Oyarzún C, Garrido W, Alarcón S, Yáñez A, Sobrevia L, Quezada C, et al. Adenosine contribution to normal renal physiology and chronic kidney disease. Mol Aspects Med. (2017) 55:75–89. doi: 10.1016/j.mam.2017.01.004
130. Hocher B. Adenosine A1 receptor antagonists in clinical research and development. Kidney Int. (2010) 78:438–45. doi: 10.1038/ki.2010.204
131. Cotter G, Dittrich HC, Weatherley BD, Bloomfield DM, O'Connor CM, Metra M, et al. The PROTECT pilot study: a randomized, placebo-controlled, dose-finding study of the adenosine A1 receptor antagonist rolofylline in patients with acute heart failure and renal impairment. J Card Fail. (2008) 14:631–40. doi: 10.1016/j.cardfail.2008.08.010
132. Voors AA, Dittrich HC, Massie BM, DeLucca P, Mansoor GA, Metra M, et al. Effects of the adenosine A1 receptor antagonist rolofylline on renal function in patients with acute heart failure and renal dysfunction: results from PROTECT (Placebo-Controlled Randomized Study of the Selective Adenosine A1 Receptor Antagonist Rolofylline for Patients Hospitalized with Acute Decompensated Heart Failure and Volume Overload to Assess Treatment Effect on Congestion and Renal Function). J Am Coll Cardiol. (2011) 57:1899–907. doi: 10.1016/j.jacc.2010.11.057
133. Gottlieb SS, Givertz MM, Metra M, Gergich K, Bird S, Jones-Burton C, et al. The effects of adenosine A(1) receptor antagonism in patients with acute decompensated heart failure and worsening renal function: the REACH UP study. J Card Fail. (2010) 16:714–9. doi: 10.1016/j.cardfail.2010.04.006
134. Emani S. Ultrafiltration for the treatment of acute heart failure. Heart Fail Clin. (2018) 14:517–24. doi: 10.1016/j.hfc.2018.06.013
135. Costanzo MR, Negoianu D, Jaski BE, Bart BA, Heywood JT, Anand IS, et al. Aquapheresis versus intravenous diuretics and hospitalizations for heart failure. JACC Heart Fail. (2016) 4:95–105. doi: 10.1016/j.jchf.2015.08.005
136. Costanzo MR, Saltzberg M, O'Sullivan J, Sobotka P. Early ultrafiltration in patients with decompensated heart failure and diuretic resistance. J Am Coll Cardiol. (2005) 46:2047–51. doi: 10.1016/j.jacc.2005.05.099
137. Costanzo MR, Guglin ME, Saltzberg MT, Jessup ML, Bart BA, Teerlink JR, et al. Ultrafiltration versus intravenous diuretics for patients hospitalized for acute decompensated heart failure. J Am Coll Cardiol. (2007) 49:675–83. doi: 10.1016/j.jacc.2006.07.073
Keywords: worsening renal function, type 1 cardiorenal syndrome, acute heart failure, pathogenesis, biomarker, treatment
Citation: Fu K, Hu Y, Zhang H, Wang C, Lin Z, Lu H and Ji X (2021) Insights of Worsening Renal Function in Type 1 Cardiorenal Syndrome: From the Pathogenesis, Biomarkers to Treatment. Front. Cardiovasc. Med. 8:760152. doi: 10.3389/fcvm.2021.760152
Received: 17 August 2021; Accepted: 11 November 2021;
Published: 14 December 2021.
Edited by:
Gaetano Ruocco, Regina Montis Regalis Hospital, ItalyReviewed by:
Mrudula Reddy Munagala, Jackson Health System, United StatesAlberto Palazzuoli, University of Siena, Italy
Copyright © 2021 Fu, Hu, Zhang, Wang, Lin, Lu and Ji. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoping Ji, aml4aWFvcGluZ0BzZHUuZWR1LmNu; Huixia Lu, bHVodWl4aWFAc2R1LmVkdS5jbg==