 Mohapradeep Mohan
Mohapradeep Mohan Adel Dihoum
Adel Dihoum Ify R. Mordi
Ify R. Mordi Anna-Maria Choy
Anna-Maria Choy Graham Rena
Graham Rena Chim C. Lang
Chim C. Lang
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Cardiovasc. Med. , 29 September 2021
Sec. Cardiovascular Biologics and Regenerative Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.746382
This article is part of the Research Topic Understanding Molecular Mechanisms in Diabetic Cardiomyopathy (DCM) View all 13 articles
Heart failure is an important manifestation of diabetic heart disease. Before the development of symptomatic heart failure, as much as 50% of patients with type 2 diabetes mellitus (T2DM) develop asymptomatic left ventricular dysfunction including left ventricular hypertrophy (LVH). Left ventricular hypertrophy (LVH) is highly prevalent in patients with T2DM and is a strong predictor of adverse cardiovascular outcomes including heart failure. Importantly regression of LVH with antihypertensive treatment especially renin angiotensin system blockers reduces cardiovascular morbidity and mortality. However, this approach is only partially effective since LVH persists in 20% of patients with hypertension who attain target blood pressure, implicating the role of other potential mechanisms in the development of LVH. Moreover, the pathophysiology of LVH in T2DM remains unclear and is not fully explained by the hyperglycemia-associated cellular alterations. There is a growing body of evidence that supports the role of inflammation, oxidative stress, AMP-activated kinase (AMPK) and insulin resistance in mediating the development of LVH. The recognition of asymptomatic LVH may offer an opportune target for intervention with cardio-protective therapy in these at-risk patients. In this article, we provide a review of some of the key clinical studies that evaluated the effects of allopurinol, SGLT2 inhibitor and metformin in regressing LVH in patients with and without T2DM.
Diabetic cardiomyopathy (DCM) is defined as cardiac dysfunction, characterised by abnormal structural, functional and metabolic changes in the myocardium, that occurs in the absence of significant coronary, valvular or hypertensive diseases in individuals with diabetes (1). Diabetic cardiomyopathy was first described five decades ago (2), and the higher incidence of heart failure (HF) in patients with type 2 diabetes mellitus (T2DM) was further confirmed in several epidemiological studies, including the Framingham Heart Study (3–5). The aetiology of DCM in the diabetic heart is complex and is likely to multifactorial (Figure 1). In the early stages of its progression, DCM is usually asymptomatic and is characterised by subclinical structural and functional abnormalities including left ventricular hypertrophy (LVH), reduced LV compliance, myocardial fibrosis and stiffness (6). These pathophysiological changes associated with subclinical cardiac dysfunction can be progressive and lead to HF symptoms and to the clinical syndrome of HF (7). Despite the success of various antihyperglycemic agents in the intensive management of hyperglycemia in people with T2DM in reducing the risk of cardiovascular (CV) complications, the high prevalence of HF persists (8–10), that might suggest the role of other non-hyperglycemia associated pathophysiological mechanisms that might contribute to the development of DCM and consequent higher risk of HF in T2DM.
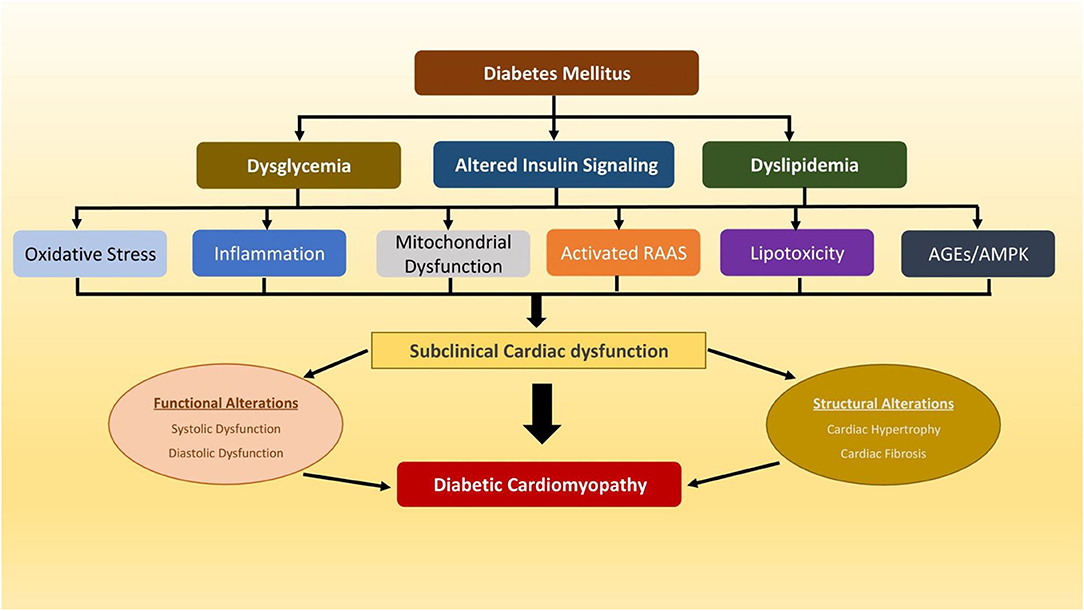
Figure 1. A schematic diagram depicting multiple potential mechanisms that have been implicated in pathophysiology of diabetic cardiomyopathy. AGEs, advanced glycation end products; AMPK, AMP-activated protein kinase; RAAS, renin-angiotensin-aldosterone system.
LVH is defined by an elevated left ventricular mass (LVM), either due to an increase in wall thickness or due to left ventricular cavity enlargement, or both. Typically, the LV wall thickening manifests in response to hemodynamic pressure overload, and chamber dilatation occurs in response to volumetric burden, with the caveat that other molecular and metabolic mechanisms may play a synergistic and potentially independent role. Importantly, LVH is strong predictor of CV events (11, 12), and is a common feature in the diabetic myocardium (7, 13). The reason why LVH is predictive of CV events is because it precedes many potentially fatal CV sequelae of events. For instance, the presence of LVH can lead to adverse cardiovascular (CV) outcome—LVH impedes left ventricular (LV) filling and can lead to diastolic heart failure; it reduces coronary perfusion reserve and can induce ischemia; it can lead to left atrial enlargement and subsequent atrial fibrillation and it is intrinsically arrhythmogenic and can cause sudden cardiac death (14). The development of LVH in T2DM is not fully explained by the hyperglycemia-associated cellular alterations alone (15). Specifically, there is extensive body of clinical and experimental underpinnings that supports the role of inflammation, oxidative stress, insulin resistance and AMP-activated kinase (AMPK) in mediating the development of LVH in T2DM (15–18). While the pathophysiology of LVH in diabetes is not yet fully elucidated, the increasing recognition that LVH is an exquisite orchestration of a wide array of pathophysiological process, that are not limited to hyperglycemia, hypertension and valvular disease, provided new opportunities to examine new pharmacological therapies in LVH regression.
As defined by the American College of Cardiology/American Heart Association (ACC/AHA) guidelines, HF progresses as a clinical continuum of four stages (19). Stage B HF represents patients with structural heart disease including LVH, but with no current or prior symptoms of HF (19). To prevent progression to symptomatic HF (Stage C HF), it is important to identify the presence of DCM at the early stages of its development. While the electrocardiogram (ECG) is a widely used method to diagnose LVH, its diagnostic accuracy is limited due to its poor sensitivity in detecting LVH (20, 21). Echocardiography has been conventionally considered a test of choice to evaluate the presence of cardiac dysfunction and early structural changes of the heart including LVH (22). Its sensitivity is significantly higher than ECG in detecting LVH (23), and can also diagnosis other subclinical cardiac abnormalities and valvular heart disease. However, cardiac magnetic resonance imaging is considered the gold standard due to high precision and reproducibility in estimating LV mass (24) and other structural cardiac abnormalities, albeit its use is limited due to its high costs and limited availability.
In addition to multiple comorbidities and several molecular and metabolic mechanisms, several studies have reported that LVH is also influenced by various other factors such as age, gender, ethnicity and genetic factors. For instance, LVH is more prevalent in blacks compared with other race/ethnic groups (25, 26). The prevalence of LVH is not reported to be dissimilar between men and women, irrespective of diagnostic criteria applied (27). The prevalence of LVH also increased significantly with age, occurring in 33% of men and 49% of women over 70 (28). Furthermore, genome wide association studies (GWAS) have also reported substantial heritability for LVH in diverse populations including diabetic patients (29, 30).
In this mini-review, we summarise the epidemiology of LVH in T2DM and the key pathophysiological mechanisms and provide a review of some of the key studies that evaluated the effects of different classes drugs on LVH regression.
It is becoming increasingly recognised that T2DM is an independent risk factor for LVH, even in the absence of overt cardiovascular diseases (CVD) (31, 32). T2DM is often associated with cardiomyopathy, manifested by LVH, and the reported prevalence of LVH in patients with T2DM ranges from 32 to 71% in different studies, depending on the criteria used for defining LVH (12, 33–36). Importantly, previous research has demonstrated that LVH regression per se independently reduces CV mortality and events (37, 38). Thus, routine screening of LVH, followed by targeted intervention could be a promising way of reducing CV events and mortality in people with T2DM. Furthermore, disability caused by CVD carries major health economic consequences at both individual levels, and the society as a whole, and this burden is expected to increase in the future. Therefore, routine screening of LVH and targeted treatment may have the potential to reduce the economic burden of CVD. While it is difficult to evaluate the economic value of saving life, the costs of screening for and treating LVH is anticipated to be low, primarily due to the high prevalence of the condition and the low unit cost of echocardiography (39).
The efficacy and cost effectiveness of early screening for traditional CV risk factors has already been proven in the general population (40–42). However, an ongoing challenge is to develop adequate screening strategies that are cost-effective that can identify individuals at risk for future CV events, prior to developing overt CVD. Since people with T2DM are at a higher risk for future CV events than non-diabetic people, it may be more cost-effective to target subclinical cardiac abnormalities including LVH in diabetic population. Whilst this approach may initially increase healthcare costs, especially with the costs associated with CV imaging and treatment, such an approach may in the longer term reduce CV related mortality and morbidity in patients with T2DM. Witham et al. in a hypothetical analysis of the cost-effectiveness of screening LVH, suggested that this may indeed be a very cost-effective strategy to reduce CV events in high-risk people including those with T2DM (39). Biomarkers may prove to be an alternate tool that is even more cost-effective, when used in CVD risk prognostication (43). In this context, previous studies have utilised a biomarker-based approach for the identification of subclinical LV abnormalities in patients without CVD. For instance, in patients with T2DM and without HF, higher levels of B-type natriuretic peptide (BNP) associated with LVH and lower left ventricular ejection fraction (44). Another study of non-diabetic individuals without overt CVD, found that high sensitivity cardiac troponin T (hs-cTnT) and BNP may be suitable for (the c-statistic for screening with BNP ± hs-cTnT was 0·81) identifying asymptomatic cardiac diseases including LVH (45). If the results of these studies can be confirmed in future large studies, pre-screening of people with T2DM using the biomarker approach may offer a cost-effective modality in screening LVH. Further systematic health economics studies are warranted to assess the cost-effectiveness of screening T2DM patients for LVH, and then pre-emptive treatment.
Inflammation is a prominent hallmark of LVH. Pro-inflammatory cytokines are found to modulate left ventricle LV structure and play a critical role in LV remodelling (46). Inflammatory mediators can result in changes in cardiac size, shapes, and composition, including myocyte hypertrophy, alterations in expression of foetal gene, and progressive myocyte loss through apoptosis (46). Pro-inflammatory cytokines such as IL-1, IL-6 and tumour necrosis factor-alpha (TNF-α) are normally not expressed in the heart, but they are activated and up regulated as response to myocardial injury or mechanical stress, contributing to cardiac remodelling (46). Soon after ischemic myocardial injury, TNF-α and IL-6 are elaborated and trigger additional cellular inflammatory responses (47). Chronically, to repair and remodelling, these cytokines activate matrix metalloproteinases and collagen formation, and regulate angiogenesis and progenitor cell mobilisation, resulting in development of myocardial hypertrophy, collagen deposition, and fibrosis (47). In macrophages, lipopolysaccharide (LPS), tumour necrosis factor-α, and interferon-γ, has been shown to be implicated in cardiac hypertrophy via their effects on the expression of microRNA-155 (miR-155) expression, a powerful mediator of cardiac hypertrophy (48). Notably, variable myocyte hypertrophy and inflammatory cell infiltration with activation of nuclear factor kappa B (NF-κB) have been identified in endomyocardial tissue of patients with Hypertrophic cardiomyopathy (HCM) while the levels of in interleukins (IL-1β, IL-1RA, IL-6, IL-10) and high-sensitivity C-reactive protein (hsCRP) have been found to be significantly higher in patients with HCM than in control subjects (49). These suggest that a low-grade inflammatory response may play an important role in the development of cardiac hypertrophy in patients with HCM and support the causative significance of inflammatory signalling in hypertrophic heart disease, demonstrating the feasibility of therapeutic targeting of inflammation in heart failure.
Growing evidence points to the potential involvement of oxidative stress in the pathophysiology of cardiac hypertrophy via different possible mechanisms. Dysregulated expression of nitrogen oxides (NOX) proteins, which are predominant isoforms expressed in cardiac tissue, has been proposed to contribute to the development of cardiac hypertrophy and is believed to driven by eroxisome proliferator-activated receptor α (PPARα) downregulation (50). Upregulation of NOX4 by angiotensin II, which is primary in mitochondria of cardiomyocytes, can also result in inducing nuclear export of histone deacetylase 4 (HDAC4), a crucial precursor of cardiac hypertrophy (51). NOX2 and NOX4 have also been found to be associated with cardiac hypertrophy and fibrosis in diabetic rats, and the elevation of NOX2 has been shown to be associated with an increase in cardiomyocytes size in mice subjected to high fat diet (50). Furthermore, In an experimental guinea pig model of pressure-overload induced cardiac hypertrophy, increased oxidative stress can result in an increase in cardiac ROS production, which induces cardiac hypertrophy through activation of redox-sensitive protein kinases such as mitogen activated protein kinase (MAPK) (52). In neonatal cardiomyocytes, however, ROS has been found to active a wide variety of hypertrophic signalling kinases and transcription factors, including the tyrosine kinase Src, GTP-binding protein Ras, protein kinase C, mitogen-activated protein kinases (extracellular response kinase and extracellular signal–regulated kinase), and Jun-nuclear kinase, NK-KB and Phosphoinositol 3-kinase, which is required for H2O2-induced hypertrophy (53). Finally, in diabetic patients, it is hypothesised that myocardial kinases β isoform of protein kinase C (PKCβ), which is preferentially overexpressed in diabetic myocardium accompanied with increased upregulation of pro-oxidant enzyme NAPH oxidase, that is a major upstream moderator of oxidative stress and that inhibition of PKCβ can attenuate myocardial hypertrophy (54). All these observations support the role of oxidative stress in the pathophysiology of cardiac hypertrophy.
Insulin resistance has long been recognised as a common factor in and contributing towards the intersection between diagnosis of heart disease and diabetes in the same patient (55). In brief, development of insulin resistance over time, initially compensated by hyperinsulinemia, but ultimately resulting in beta cell failure, likely results in successive periods where there is first overstimulation and then under stimulation of anabolic insulin-sensitive signalling pathways in cardiomyocytes and other tissues, any of which, as well as the metabolic flexibility inherent in insulin resistance, may contribute to pathology. Many of the proteins mediating insulin signalling are encoded by more than one gene, which has complicated mechanistic genetic analysis. Consistent with the likely complex role of insulin signalling in heart disease, deletion of IRS1 and IRS2 docking proteins in liver results in heart failure in mice, whereas deletion of the same genes in cardiac tissue resulted in smaller ventricular mass (56). These findings also illustrate that insulin signalling in cardiac tissue and in non-cardiac tissue, are both likely to have an important impact on progression of heart disease. More work is required to determine the critical metabolic regulatory pathways underlying these effects.
One of the metabolic signalling pathways most studied in cardiac dysfunction is mammalian target of rapamycin (mTOR) signalling. The mTOR signalling pathways are at the crossroads of anabolic and catabolic cellular process (57). mammalian target of rapamycin complex 1 (mTORC1) promotes anabolic metabolism in response to growth factor signalling, nutrients (particularly amino acids), and increased energy supply. mTORC1 also suppresses autophagy (58). mTORC1 phosphorylates Unc-51 like autophagy activating kinase (ULK1), a kinase that forms a complex with ATG13, FIP2000, and ATG101 and drives autophagosome formation, disrupting the interaction between Ulk1 and AMPK and prevent Ulk1 activation by AMPK (59). At transcriptional level, mTORC1 can also regulate autophagy. It prevents nuclear transport of the transcription factor EB (TFEB), which drives genes expression for lysosomes biogenesis and the autophagy machinery (60). In these ways mTORC1 is understood largely to determine the extent of cellular anabolic and catabolic processes, whilst in contrast mTORC2 seems to function mainly as an effector of insulin/IGF-1 signalling ref 8. mTORC2 activates and phosphorylates protein kinase B (PKB), a key effector of insulin/PI3K signalling. Phosphorylation of PKB results in cell promotion and proliferation via activation and suppression of many key substrates including the transcription factors of FoxO1/3a, the metabolic regulator (GSK3β), and the mTORC1 inhibitor (TSC2) (58). mTORC2 is also regulated by mTORC1 through a negative feedback loop between mTORC1 and insulin PI3K signalling. mTORC1 activates (Grb10), a negative regulator of insulin/IGF-1 receptor signalling upstream of Akt and mTORC2 (58).
In the cardiovascular system, mTOR pathways are understood to be important regulators of the hypertrophic response (61). Loss of function genetic studies indicate that mTORC1 activation is indispensable for development of cardiac hypertrophy in response to pressure overload. An animal study showed that the deletion of Rheb 1, which mediates mTORC1 activation, in cardiomyocytes confers cardioprotection against pathological remodelling in pressure overload (62). The mTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. A study showed a significant improvement in apoptosis and heart function when Mtor and the gene encoding 4E-BP1, an mTOR-containing multiprotein complex-1 (mTORC1) substrate that inhibits translation initiation, was deleted together (63).
Furthermore, in mice, ablation of cardiac raptor, a gene required for normal cardiac physiological function and for heart adaptation to increased workload, is shown to impair adaptive hypertrophy, change metabolic gene expression, and causing heart failure (64). In raptor-cKO mice, pressure overload causes a significant cardiac dilatation and immediate reduction in ejection fraction (64).
Consistent with this, pharmacological inhibition of mTORC1 also suppresses hypertrophy. Rapamycin, a specific inhibitor of mTOR, inhibits established cardiac hypertrophy via a complete suppression the phosphorylation of S6K1 and S6 phosphorylation in response to pressure overload (65). This can explain the attenuation of the increase in myocyte cell size induced by aortic constriction after administering Rapamycin to mice (66). Rapamycin is also shown to extend lifespan of mice (67). However, although it is shown that rapamycin can reverse age-dependent defects in cardiac function (68) and can protect against the changes of atherosclerosis (69), it is still unclear if an increase in lifespan owes to effects on cardiovascular system (67).
AMPK is another protein kinase that regulates many aspects of cellular energy balance and is seen as a promising target for anti-hypertrophic drugs. AMPK activation reverses increased protein O-GlcNAcylation, which is associated with cardiac hypertrophy, mainly through controlling the glutamine: fructose-6-phosphate aminotransferase (GFAT) phosphorylation, resulting in a decrease in O-GlcNAcylation of proteins such as troponin T (70). In line with this, an increase in cellular O-GlcNAc levels in response to O-linked N-acetylglucosamine (O-GlcNAc) inducers completely supresses the anti-hypertrophic effect of AMPK (70).
Metformin and AICAR have each been shown in preclinical studies to inhibit cardiac hypertrophy. Long-term metformin treatment significantly increases AMPK phosphorylation and attenuates cardiac hypertrophy induced by Transverse aortic constriction (TAC) (71). Interestingly, the antihypertrophic effects of metformin were not observed in AMPKα2–/– mice (71), suggesting that the chronic activation of AMPK during the development of cardiac hypertrophy is an important mechanism that mediates the beneficial effect of metformin. Moreover, long-term activation of AMPK by AICAR has been shown to block load-induced calcineurin–NFAT pathway as calcineurin is also regulated by MAPK pathway. Thus, the activation of AMPK may counteract MAPK pathway though blocking calcineurin–NFAT pathway (72).
However, counterbalancing these promising findings is recent evidence that a pan-AMPK activator improves glucose homeostasis, but at the cost of increased hypertrophy. Chronic activation of MK-8722, pan-AMPK activator, increases glucose uptake into skeletal muscle, but it results in cardiac hypertrophy associated with increased cardiac glycogen contents (73). To determine which of these beneficial and less beneficial aspects of the drugs really do owe to AMPK, further genetic studies are required.
Due to the high attrition rates, significant economic burden and lengthy new drug discovery process, repurposing of ‘old’ drugs (also known as drug repositioning, reprofiling or re-tasking) to treat both common and rare diseases is increasingly becoming a promising field in drug discovery. Drug repurposing identifies new therapeutic uses for already approved drugs that are outside the scope of the original use (74). This is an attractive proposition because it involves the use of existing compounds that have undergone rigorous testing with potentially lower overall development costs and shorter development timelines.
Hypertension is the most common causes of LVH and therefore, controlling blood pressure (BP) especially with drugs that blocks the renin-angiotensin system (RAS) is the standard approach to the management of LVH. However, this approach is only partially effective since LVH persists in 20% of hypertensives that attain target BP (75). Thus “normotensive LVH” is very common (12). Indeed, BP only contributes 25% to the variability in LV mass seen in a population (76). Despite a “normal” BP, normotensive LVH is just as risky as is hypertensive LVH (77). Nevertheless, regressing LVH irrespective of BP changes is an effective way to reduce the incidence of all major CV events including specifically sudden deaths, heart failure hospitalisations, new onset AF and strokes (37, 38, 78–83). The LIFE study demonstrates that LVH regression per se reduces future cardiovascular events irrespective of BP (84). Since controlling BP and using a RAS blocker is only partially effective at regressing LVH, we now need additional ways of regressing LVH. In addition to BP, as we have discussed above, the pathophysiology of LVH may involve a complex cocktail of various non-hemodynamic disease processes including inflammation, oxidative stress, obesity, and insulin resistance (15, 29, 31, 53, 85–90).
Below, we provide a narrative review of some of the key clinical trials that evaluated the effects of interventions targeting LVH in patients with and without T2DM, in the context of normal BP.
Allopurinol, a xanthine oxidase (XO) inhibitor, has been the mainstay of treatment for patients with gout associated with hyperuricemia for several decades. In addition, there is mounting evidence to suggest cardioprotective effects of allopurinol (91–93). Allopurinol has been shown to exert its cardioprotective effects through three key mechanisms: (i) reduction of uric acid concentrations which has pro-inflammatory effects; (ii) inhibiting xanthine oxidase mediated production of reactive oxygen species (ROS) which aggravate endothelial dysfunction and atherosclerosis plaque instability; and (iii) increasing local tissue availability of adenosine triphosphate and oxygen by inhibiting purine metabolism (94).
Allopurinol has been shown to regress LVM in different cohorts along the cardiovascular spectrum with significant pre-existing disease, oxidative stress and inflammation. In a RCT of people with T2DM, 9 months treatment of allopurinol treatment significantly reduced absolute left ventricular mass (LVM) (allopurinol −2.65 ± 5.91 g vs. placebo group +1.21 ± 5.10 g; p = 0.012) and LVM indexed (LVMI) to body surface area (allopurinol −1.32 ± 2.84 g/m(2) vs. placebo group +0.65 ± 3.07 g/m(2); p = 0.017) (18). In another RCT of people with ischemic heart disease, allopurinol treatment significantly reduced LVM (allopurinol −5.2 ± 5.8 g vs. placebo −1.3 ± 4.48 g; p = 0.007) and LVMI (allopurinol −2.2 ± 2.78 g/m(2) vs. placebo −0.53 ± 2.5 g/m(2); p = 0.023) (17). Furthermore, Kao et al., in another RCT of patients with severe renal disease (CKD 3) reported significant reduction in LVM with 9 months allopurinol treatment (95). In contrast, a recent study by Gingles et al., reported that LVM regression was significantly reduced with allopurinol treatment than placebo, suggestive of a potential adverse effect (96). However, unlike other studies, in this RCT, the study population had well-controlled BP and were normouricemic with low oxidative stress. This may have negated any direct effect of allopurinol, who rely on urate for antioxidant defence, in reducing ROS generation by XO inhibition, and consequent null effect on LVH. Therefore, the regressive effect of allopurinol may not be observed universally in all study populations.
Metformin is an oral antihyperglycemic agent that has been used widely for the treatment of T2DM for over many decades. Beyond its antihyperglycemic effects, there is now accumulating evidence to suggest that metformin is cardioprotective (97). While the exact mechanism of cardioprotective actions of metformin is not fully understood, several ancillary mechanisms have been proposed to explain the metformin induced LVH regression. First, as stated already, insulin resistance, inflammation, oxidative stress, endothelial dysfunction and obesity is understood to contribute to the development of LVH (15, 53, 85–89), and metformin has been shown to reduce insulin resistance (98), inflammation (99), oxidative stress (100–104), central obesity (105) and endothelial dysfunction (106), albeit the latter is not a consistent finding (107). Second, in vivo studies have reported activation of AMP-activated protein kinase (AMPK) as one of the putative mechanisms for the anti-hypertrophic effect of metformin (15), leading to the view that AMPK stimulation is a promising new strategy to prevent or reduce LVH (108–110).
For clinical trials, to date, only one study: The MET-REMODEL trial, has explicitly investigated the effect of metformin on LVH in non-diabetic CAD patients identified to have IR and/or diabetes. This study demonstrated that 12 months metformin treatment (2 g/day) significantly reduced LVMI (absolute mean difference −1.37 (95% CI: −2.63 to −0.12, P = 0.033) (111). In this study, metformin also significantly reduced LVM, systolic blood pressure, and oxidative stress. In line with these findings, few other clinical studies and a network analysis also reported anti-hypertrophic effects of metformin (112–114).
The sodium-glucose linked cotransporter type 2 (SGLT2) class of inhibitors was developed as a novel anti-diabetic agent that acts independent of the insulin-incretin pathway to lower blood sugar. Various classes of SGLT2 inhibitors such as empagliflozin, canagliflozin and dapagliflozin have been shown to reduce cardiovascular mortality in patients with diabetes mellitus (115–118), but its cardioprotective mechanism remains elusive. More recent evidence from RCTs, suggest the potential of dapagliflozin in reducing the risk of worsening heart failure or CV mortality, even in non-diabetic population (119, 120). However, it is not clear whether the cardioprotective effects of SGLT2 inhibition in non-diabetic patients is class effect or drug-specific effect (121–124). Unlike other antidiabetic agents that are dependent on pancreatic beta-cell function and insulin sensitivity for the glucose lowering effect, the principal mechanism by which SGLT2 inhibitors lower blood glucose is by excreting excess glucose by enhancing urinary glucose excretion. Part of the off-target effects that have been observed with this class of drugs include weight loss, improved glycemia/lipid profile, arterial stiffness, reduce preload and afterload (blood pressure) and diuresis (125, 126)—all of which are key risk factors implicated in the development of LVH (15).
The DAPA-LVH study was the first placebo controlled RCT to investigate the efficacy of dapagliflozin in regressing LVH in normotensive patients with T2DM, without pre-existing CVD (127). At 12 months, dapagliflozin treatment significantly reduced LVM in people with T2DM, as assessed by cardiac magnetic resonance imaging (128). In this study, dapagliflozin was also shown to significantly reduce systolic BP, body weight, abdominal obesity (both visceral and subcutaneous), insulin resistance, and hsCRP. A similar finding was observed in the EMPA-HEART study that reported anti-hypertrophic effect of empagliflozin (129). In the DAPA-LVH trial, LVH regression was greater in those with higher baseline LVM (128). It is to be noted that a recent subgroup analysis of the EMPA-REG OUTCOME trial reported lower incidence of CVD in patients with LVH compared with those without LVH (130). Furthermore, post-hoc exploratory analysis from DAPA-LVH trial suggest dapagliflozin may improve subclinical dysfunction, as evidenced by improved myocardial longitudinal function (131). Taken together, there is compelling evidence to suggest that SGLT2 have the potential to promote reverse LV remodelling in patients with diabetes, which may, at least in part, explain the cardioprotective effects observed in large outcome trials of SGLT2.
In this mini-review, we have argued that LVH is a good surrogate marker of diabetic cardiomyopathy and discussed the trials targeting LVH as a manifestation of Stage B Cardiomyopathy, and potential mechanisms behind LVH regression (Figure 2) in patients with T2DM or in insulin-resistant individuals. We believe that cardiovascular outcome trials are still needed to provide definitive evidence for the cardio-protective role of the proposed repurposed drugs. With respect to metformin, the results of the on-going outcome trials such as the VA IMPACT trial (VA IMPACTNCT02915198) and Glucose Lowering in Non-diabetic hyperglycaemia Trial (GLINT; ISRCTN34875079), will be informative and might provide the needed evidence for recommending metformin in these at risk patients.
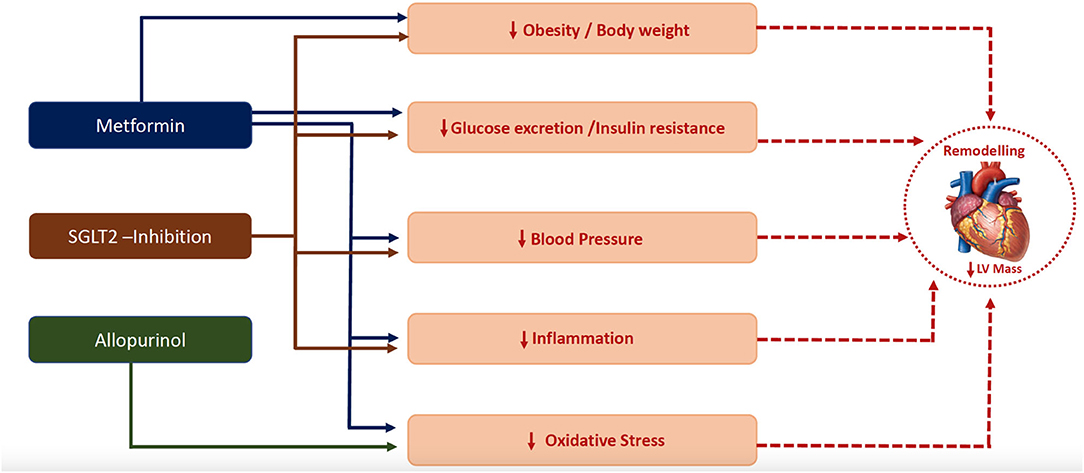
Figure 2. Plausible mechanisms by which metformin (111), SGLT-2 inhibitor (128) and allopurinol (17, 18) regressed left ventricular hypertrophy.
All the clinical studies discussed in this manuscript were “proof of concept” studies, conducted in small sample size, to evaluate the possible mechanisms behind the purported cardio-protective effects of each drugs. Unlike, the LVH regression observed in previous hypertension trials, the magnitude of the LVH regression observed in these trials were small, which may be, at least in part due to the shorter follow-up period and treatment duration. The results of these proof-of-concept trials are encouraging and help underpin.future large cardiovascular outcome trials, with longer follow-up period, incorporating better hard end points. One such trial is the VA-IMPACT trial (VA IMPACT NCT−02915198) of metformin. Such trials will be informative and help provide the medical evidence to support the use these drugs in diabetic cardiomyopathy.
CCL and MM conceived the idea. MM designed the figure. MM and AD wrote the first draft of the manuscript and was responsible for synthesizing the evidence, the search strategy, and conducted the literature search. IM, AMC, GR, and CCL critically reviewed and revised the manuscript. All authors read and approved the final manuscript.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
MM is supported by the National Institute of Health Research (NIHR) (grant no: 16/137/107). CCL has received funding by a Distinguished Professorship from the University Kebangsaan Malaysia.
1. Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res. (2018) 122:624–38. doi: 10.1161/CIRCRESAHA.117.311586
2. Rubler S, Dlugash J, Yuceoglu YZ, Kumral T, Branwood AW, Grishman A. New type of cardiomyopathy associated with diabetic glomerulosclerosis. Am J Cardiol. (1972) 30:595–602. doi: 10.1016/0002-9149(72)90595-4
3. Cas D, Khan SS, Butler J, Mentz RJ, Bonow RO, Avogaro A, et al. Impact of diabetes on epidemiology, treatment, and outcomes of patients with heart failure. JACC Heart Fail. (2015) 3:136–45. doi: 10.1016/j.jchf.2014.08.004
4. Tousoulis D, Oikonomou E, Siasos G, Stefanadis C. Diabetes mellitus and heart failure. Eur Cardiol. (2014) 9:37–42. doi: 10.15420/ecr.2014.9.1.37
5. Aune D, Schlesinger S, Neuenschwander M, Feng T, Janszky I, Norat T, et al. Diabetes mellitus, blood glucose and the risk of heart failure: a systematic review and meta-analysis of prospective studies. Nutr Metab Cardiovasc Dis. (2018) 28:1081–91. doi: 10.1016/j.numecd.2018.07.005
6. Jia G, DeMarco VG, Sowers JR. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol. (2016) 12:144–53. doi: 10.1038/nrendo.2015.216
7. Lee M, Gardin JM, Lynch JC, Smith VE, Tracy RP, Savage PJ, et al. Diabetes mellitus and echocardiographic left ventricular function in free-living elderly men and women: the cardiovascular health study. Am Heart J. (1997) 133:36–43. doi: 10.1016/S0002-8703(97)70245-X
8. Moritz T, Duckworth W, Abraira C. Veterans Affairs diabetes trial—corrections. N Engl J Med. (2009) 361:1024–5. doi: 10.1056/NEJMc096250
9. Patel A, MacMahon S, Chalmers J, Neal B, Billot L, Woodward M, et al. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N Engl J Med. (2008) 358:2560–72. doi: 10.1056/NEJMoa0802987
10. Stratton IM, Adler AI, Neil HA, Matthews DR, Manley SE, Cull CA, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ. (2000) 321:405–12. doi: 10.1136/bmj.321.7258.405
11. Gosse PS. Left ventricular hypertrophy–the problem and possible solutions. J Int Med Res. (2005) 33 (Suppl 1):3A−11A. doi: 10.1177/14732300050330S102
12. Dawson A, Morris AD, Struthers AD. The epidemiology of left ventricular hypertrophy in type 2 diabetes mellitus. Diabetologia. (2005) 48:1971–9. doi: 10.1007/s00125-005-1896-y
13. Devereux RB, Roman MJ, Paranicas M, O'Grady MJ, Lee ET, Welty TK, et al. Impact of diabetes on cardiac structure and function: the strong heart study. Circulation. (2000) 101:2271–6. doi: 10.1161/01.CIR.101.19.2271
14. Artham SM, Lavie CJ, Milani RV, Patel DA, Verma A, Ventura HO. Clinical impact of left ventricular hypertrophy and implications for regression. Prog Cardiovasc Dis. (2009) 52:153–67. doi: 10.1016/j.pcad.2009.05.002
15. Mohan M, McSwiggan S, Baig F, Rutherford L, Lang CC. Metformin and its effects on myocardial dimension and left ventricular hypertrophy in normotensive patients with coronary heart disease (the MET-REMODEL study): rationale and design of the MET-REMODEL study. Cardiovasc Ther. (2015) 33:1–8. doi: 10.1111/1755-5922.12101
16. Öncel CR. Left ventricular hypertrophy, inflammation, insulin resistance. Anatol J Cardiol. (2016) 16:142. doi: 10.14744/AnatolJCardiol.2015.6857
17. Rekhraj S, Gandy SJ, Szwejkowski BR, Nadir MA, Noman A, Houston JG, et al. High-dose allopurinol reduces left ventricular mass in patients with ischemic heart disease. J Am Coll Cardiol. (2013) 61:92632. doi: 10.1016/j.jacc.2012.09.066
18. Szwejkowski BR, Gandy SJ, Rekhraj S, Houston JG, Lang CC, Morris AD, et al. Allopurinol reduces left ventricular mass in patients with type 2 diabetes and left ventricular hypertrophy. J Am Coll Cardiol. (2013) 62:2284–93. doi: 10.1016/j.jacc.2013.07.074
19. Goldberg LR, Jessup M. Stage B heart failure: management of asymptomatic left ventricular systolic dysfunction. Circulation. (2006) 113:2851–60. doi: 10.1161/CIRCULATIONAHA.105.600437
20. Pewsner D, Jüni P, Egger M, Battaglia M, Sundström J, Bachmann LM. Accuracy of electrocardiography in diagnosis of left ventricular hypertrophy in arterial hypertension: systematic review. BMJ. (2007) 335:711. doi: 10.1136/bmj.39276.636354.AE
21. Burgos PF, Luna Filho B, Costa FA, Bombig MT, Souza D, Bianco HT, et al. Electrocardiogram performance in the diagnosis of left ventricular hypertrophy in hypertensive patients with left bundle branch block. Arq Bras Cardiol. (2017) 108:47–52. doi: 10.5935/abc.20160187
22. Foppa M, Duncan BB, Rohde LE. Echocardiography-based left ventricular mass estimation. How should we define hypertrophy? Cardiovasc Ultrasound. (2005) 3:17. doi: 10.1186/1476-7120-3-17
23. Levy D, Labib SB, Anderson KM, Christiansen JC, Kannel WB, Castelli WP. Determinants of sensitivity and specificity of electrocardiographic criteria for left ventricular hypertrophy. Circulation. (1990) 81:815–20. doi: 10.1161/01.CIR.81.3.815
24. Pennell DJ, Sechtem UP, Higgins CB, Manning WJ, Pohost GM, Rademakers FE, et al. Cardiology. Clinical indications for cardiovascular magnetic resonance (CMR): Consensus Panel report. Eur Heart J. (2004) 25:1940–65. doi: 10.1016/j.ehj.2004.06.040
25. Drazner MH, Dries DL, Peshock RM, Cooper RS, Klassen C, Kazi F, et al. Left ventricular hypertrophy is more prevalent in blacks than whites in the general population: the Dallas Heart Study. Hypertension. (2005) 46:124–9. doi: 10.1161/01.HYP.0000169972.96201.8e
26. Lewis AA, Ayers CR, Selvin E, Neeland I, Ballantyne CM, Nambi V, et al. Racial differences in malignant left ventricular hypertrophy and incidence of heart failure: a multicohort study. Circulation. (2020) 141:957–67. doi: 10.1161/CIRCULATIONAHA.119.043628
27. Cuspidi C, Sala C, Negri F, Mancia G, Morganti A, Italian Society of Hypertension. Prevalence of left-ventricular hypertrophy in hypertension: an updated review of echocardiographic studies. J Hum Hypertens. (2012) 26:343–9. doi: 10.1038/jhh.2011.104
28. Levy D, Anderson KM, Savage DD, Kannel WB, Christiansen JC, Castelli WP. Echocardiographically detected left ventricular hypertrophy: prevalence and risk factors. The Framingham Heart Study. Ann Intern Med. (1988) 108:7–13. doi: 10.7326/0003-4819-108-1-7
29. Bella JN, Göring HH. Genetic epidemiology of left ventricular hypertrophy. Am J Cardiovasc Dis. (2012) 2:267–78.
30. Parry HM, Donnelly LA, Van Zuydam N, Doney AS, Elder DH, Morris AD, et al. Genetic variants predicting left ventricular hypertrophy in a diabetic population: a Go-DARTS study including meta-analysis. Cardiovasc Diabetol. (2013) 12:109. doi: 10.1186/1475-2840-12-109
31. Palmieri V, Tracy RP, Roman MJ, Liu JE, Best LG, Bella JN, et al. Relation of left ventricular hypertrophy to inflammation and albuminuria in adults with type 2 diabetes: the strong heart study. Diabetes Care. (2003) 26:2764–9. doi: 10.2337/diacare.26.10.2764
32. Fang ZY, Prins JB, Marwick TH. Diabetic cardiomyopathy: evidence, mechanisms, therapeutic implications. Endocr Rev. (2004) 25:543–67. doi: 10.1210/er.2003-0012
33. Muddu M, Mutebi E, Mondo C. Prevalence, types and factors associated with echocardiographic abnormalities among newly diagnosed diabetic patients at Mulago Hospital. Afr Health Sci. (2016) 16:183–93. doi: 10.4314/ahs.v16i1.25
34. Srivastava PM, Calafiore P, Macisaac RJ, Patel SK, Thomas MC, Jerums G, et al. Prevalence and predictors of cardiac hypertrophy and dysfunction in patients with Type 2 diabetes. Clin Sci. (2008) 114:313–20. doi: 10.1042/CS20070261
35. Somaratne JB, Whalley GA, Poppe KK, ter Bals MM, Wadams G, Pearl A, et al. Screening for left ventricular hypertrophy in patients with type 2 diabetes mellitus in the community. Cardiovasc Diabetol. (2011) 10:29. doi: 10.1186/1475-2840-10-29
36. Wu PY, Huang JC, Chen SC, Chen LI. Type 2 diabetes mellitus-related changes in left ventricular structure and function in patients with chronic kidney disease. Oncotarget. (2018) 9:14661–8. doi: 10.18632/oncotarget.24482
37. Okin PM, Devereux RB, Harris KE, Jern S, Kjeldsen SE, Julius S, et al. Regression of Electrocardiographic left ventricular hypertrophy is associated with less hospitalization for heart failure in hypertensive patients. Annals of Internal Medicine. (2007) 147:311–9. doi: 10.7326/0003-4819-147-5-200709040-00006
38. Wachtell K, Okin PM, Olsen MH, Dahlof B, Devereux RB, Ibsen H, et al. Regression of electrocardiographic left ventricular hypertrophy during antihypertensive therapy and reduction in sudden cardiac death: the LIFE Study. Circulation. (2007) 116:700–5. doi: 10.1161/CIRCULATIONAHA.106.666594
39. Witham MD, Davies JI, Dawson A, Davey PG, Struthers AD. Hypothetical economic analysis of screening for left ventricular hypertrophy in high-risk normotensive populations. QJM. (2004) 97:87–93. doi: 10.1093/qjmed/hch016
40. Selvarajah S, Haniff J, Kaur G, Guat Hiong T, Bujang A, Chee Cheong K, et al. Identification of effective screening strategies for cardiovascular disease prevention in a developing country: using cardiovascular risk-estimation and risk-reduction tools for policy recommendations. BMC Cardiovasc Disord. (2013) 13:10. doi: 10.1186/1471-2261-13-10
41. Barton P, Andronis L, Briggs A, McPherson K, Capewell S. Effectiveness and cost effectiveness of cardiovascular disease prevention in whole populations: modelling study. BMJ. (2011) 343:d4044. doi: 10.1136/bmj.d4044
42. Chamnan P, Simmons RK, Khaw KT, Wareham NJ, Griffin SJ. Estimating the population impact of screening strategies for identifying and treating people at high risk of cardiovascular disease: modelling study. BMJ. (2010) 340:c1693. doi: 10.1136/bmj.c1693
43. Vasan RS. Biomarkers of cardiovascular disease: molecular basis and practical considerations. Circulation. (2006) 113:2335–62. doi: 10.1161/CIRCULATIONAHA.104.482570
44. Dawson A, Davies JI, Morris AD, Struthers AD. B-type natriuretic Peptide is associated with both augmentation index and left ventricular mass in diabetic patients without heart failure. Am J Hypertens. (2005) 18(12 Pt 1):1586–91. doi: 10.1016/j.amjhyper.2005.06.016
45. Nadir MA, Rekhraj S, Wei L, Lim TK, Davidson J, MacDonald TM, et al. Improving the primary prevention of cardiovascular events by using biomarkers to identify individuals with silent heart disease. J Am Coll Cardiol. (2012) 60:960–8. doi: 10.1016/j.jacc.2012.04.049
46. Mann DL. Inflammatory mediators and the failing heart: past, present, and the foreseeable future. Circ Res. (2002) 91:988–98. doi: 10.1161/01.RES.0000043825.01705.1B
47. Nian M, Lee P, Khaper N, Liu P. Inflammatory cytokines and postmyocardial infarction remodeling. Circ Res. (2004) 94:1543–53. doi: 10.1161/01.RES.0000130526.20854.fa
48. Heymans S, Corsten MF, Verhesen W, Carai P, van Leeuwen RE, Custers K, et al. Macrophage microRNA-155 promotes cardiac hypertrophy and failure. Circulation. (2013) 128:1420–32. doi: 10.1161/CIRCULATIONAHA.112.001357
49. Kuusisto J, Karja V, Sipola P, Kholova I, Peuhkurinen K, Jaaskelainen P, et al. Low-grade inflammation and the phenotypic expression of myocardial fibrosis in hypertrophic cardiomyopathy. Heart. (2012) 98:1007–13. doi: 10.1136/heartjnl-2011-300960
50. Ramachandra CJA, Cong S, Chan X, Yap EP, Yu F, Hausenloy DJ. Oxidative stress in cardiac hypertrophy: From molecular mechanisms to novel therapeutic targets. Free Radic Biol Med. (2021) 166:297–312. doi: 10.1016/j.freeradbiomed.2021.02.040
51. Okabe K, Matsushima S, Ikeda S, Ikeda M, Ishikita A, Tadokoro T, et al. DPP (Dipeptidyl Peptidase)-4 Inhibitor Attenuates Ang II (Angiotensin II)-Induced Cardiac Hypertrophy via GLP (Glucagon-Like Peptide)-1-Dependent Suppression of Nox (Nicotinamide Adenine Dinucleotide Phosphate Oxidase) 4-HDAC (Histone Deacetylase) 4 Pathway. Hypertension. (2020) 75:991–1001. doi: 10.1161/HYPERTENSIONAHA.119.14400
52. Li JM, Gall NP, Grieve DJ, Chen M, Shah AM. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension. (2002) 40:477–84. doi: 10.1161/01.HYP.0000032031.30374.32
53. Takimoto E, Kass DA. Role of oxidative stress in cardiac hypertrophy and remodeling. Hypertension. (2007) 49:241–8. doi: 10.1161/01.HYP.0000254415.31362.a7
54. Liu Y, Lei S, Gao X, Mao X, Wang T, Wong GT, et al. PKCbeta inhibition with ruboxistaurin reduces oxidative stress and attenuates left ventricular hypertrophy and dysfunction in rats with streptozotocin-induced diabetes. Clin Sci. (2012) 122:161–73. doi: 10.1042/CS20110176
55. MacDonald MR, Petrie MC, Hawkins NM, Petrie JR, Fisher M, McKelvie R, et al. Diabetes, left ventricular systolic dysfunction, and chronic heart failure. Eur Heart J. (2008) 29:1224–40. doi: 10.1093/eurheartj/ehn156
56. Qi Y, Xu Z, Zhu Q, Thomas C, Kumar R, Feng H, et al. Myocardial loss of IRS1 and IRS2 causes heart failure and is controlled by p38alpha MAPK during insulin resistance. Diabetes. (2013) 62:3887–900. doi: 10.2337/db13-0095
57. Eichner R, Fernandez-Saiz V, Targosz BS, Bassermann F. Cross talk networks of mammalian target of rapamycin signaling with the ubiquitin proteasome system and their clinical implications in multiple myeloma. Int Rev Cell Mol Biol. (2019) 343, 219–97. doi: 10.1016/bs.ircmb.2018.06.001
58. Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. (2017) 168:960–76. doi: 10.1016/j.cell.2017.02.004
59. Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. (2011) 13:132–41. doi: 10.1038/ncb2152
60. Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFE Autophagy B. (2012) 8:903–14. doi: 10.4161/auto.19653
61. Sciarretta S, Forte M, Frati G, Sadoshima J. New insights into the role of mTOR signaling in the cardiovascular system. Circ Res. (2018) 122:489–505. doi: 10.1161/CIRCRESAHA.117.311147
62. Wu X, Cao Y, Nie J, Liu H, Lu S, Hu X, et al. Genetic and pharmacological inhibition of Rheb1-mTORC1 signaling exerts cardioprotection against adverse cardiac remodeling in mice. Am J Pathol. (2013) 182:2005–14. doi: 10.1016/j.ajpath.2013.02.012
63. Zhang D, Contu R, Latronico MV, Zhang J, Rizzi R, Catalucci D, et al. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J Clin Invest. (2010) 120:2805–16. doi: 10.1172/JCI43008
64. Shende P, Plaisance I, Morandi C, Pellieux C, Berthonneche C, Zorzato F, et al. Cardiac raptor ablation impairs adaptive hypertrophy, alters metabolic gene expression, and causes heart failure in mice. Circulation. (2011) 123:1073–82. doi: 10.1161/CIRCULATIONAHA.110.977066
65. McMullen JR, Sherwood MC, Tarnavski O, Zhang L, Dorfman AL, Shioi T, et al. Inhibition of mTOR signaling with rapamycin regresses established cardiac hypertrophy induced by pressure overload. Circulation. (2004) 109:3050–5. doi: 10.1161/01.CIR.0000130641.08705.45
66. Shioi T, McMullen JR, Tarnavski O, Converso K, Sherwood MC, Manning WJ, et al. Rapamycin attenuates load-induced cardiac hypertrophy in mice. Circulation. (2003) 107:1664–70. doi: 10.1161/01.CIR.0000057979.36322.88
67. Miller RA, Harrison DE, Astle CM, Fernandez E, Flurkey K, Han M, et al. Rapamycin-mediated lifespan increase in mice is dose and sex dependent and metabolically distinct from dietary restriction. Aging Cell. (2014) 13:468–77. doi: 10.1111/acel.12194
68. Flynn JM, O'Leary MN, Zambataro CA, Academia EC, Presley MP, Garrett BJ, et al. Late-life rapamycin treatment reverses age-related heart dysfunction. Aging Cell. (2013) 12:851–62. doi: 10.1111/acel.12109
69. Mueller MA, Beutner F, Teupser D, Ceglarek U, Thiery J. Prevention of atherosclerosis by the mTOR inhibitor everolimus in LDLR-/- mice despite severe hypercholesterolemia. Atherosclerosis. (2008) 198:39–48. doi: 10.1016/j.atherosclerosis.2007.09.019
70. Gelinas R, Mailleux F, Dontaine J, Bultot L, Demeulder B, Ginion A, et al. AMPK activation counteracts cardiac hypertrophy by reducing O-GlcNAcylation. Nat Commun. (2018) 9:374. doi: 10.1038/s41467-017-02795-4
71. Fu YN, Xiao H, Ma XW, Jiang SY, Xu M, Zhang YY. Metformin attenuates pressure overload-induced cardiac hypertrophy via AMPK activation. Acta Pharmacol Sin. (2011) 32:879–87. doi: 10.1038/aps.2010.229
72. Li HL, Yin R, Chen D, Liu D, Wang D, Yang Q, et al. Long-term activation of adenosine monophosphate-activated protein kinase attenuates pressure-overload-induced cardiac hypertrophy. J Cell Biochem. (2007) 100:1086–99. doi: 10.1002/jcb.21197
73. Myers RW, Guan HP, Ehrhart J, Petrov A, Prahalada S, Tozzo E, et al. Systemic pan-AMPK activator MK-8722 improves glucose homeostasis but induces cardiac hypertrophy. Science. (2017) 357:507–11. doi: 10.1126/science.aah5582
74. Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov. (2004) 3:673–83. doi: 10.1038/nrd1468
75. Mancia G, Carugo S, Grassi G, Lanzarotti A, Schiavina R, Cesana G, et al. Prevalence of left ventricular hypertrophy in hypertensive patients without and with blood pressure control: data from the PAMELA population. Pressioni Arteriose Monitorate E Loro Associazioni. Hypertension. (2002) 39:744–9. doi: 10.1161/hy0302.104669
76. Fraser R. Studying genes and the development of cardiac hypertrophy: convenient intermediate phenotypes in man. J Hypertens. (2003) 21:873–4. doi: 10.1097/00004872-200305000-00010
77. Brown DW, Giles WH, Croft JB. Left ventricular hypertrophy as a predictor of coronary heart disease mortality and the effect of hypertension. Am Heart J. (2000) 140:848–56. doi: 10.1067/mhj.2000.111112
78. Verdecchia P, Schillaci G, Borgioni C, Ciucci A, Gattobigio R, Zampi I, et al. Prognostic significance of serial changes in left ventricular mass in essential hypertension. Circulation. (1998) 97:48–54. doi: 10.1161/01.CIR.97.1.48
79. Verdecchia P, Angeli F, Borgioni C, Gattobigio R, de Simone G, Devereux RB, et al. Changes in cardiovascular risk by reduction of left ventricular mass in hypertension: a meta-analysis. Am J Hypertens. (2003) 16(11 Pt 1):895–9. doi: 10.1016/S0895-7061(03)01018-5
80. Verdecchia P, Angeli F, Gattobigio R, Sardone M, Pede S, Reboldi GP. Regression of left ventricular hypertrophy and prevention of stroke in hypertensive subjects. Am J Hypertens. (2006) 19:493–9. doi: 10.1016/j.amjhyper.2005.10.018
81. Okin PM, Devereux RB, Jern S, Kjeldsen SE, Julius S, Nieminen MS, et al. Regression of electrocardiographic left ventricular hypertrophy during antihypertensive treatment and the prediction of major cardiovascular events. JAMA. (2004) 292:2343–9. doi: 10.1001/jama.292.19.2343
82. Okin PM, Wachtell K, Devereux RB, Harris KE, Jern S, Kjeldsen SE, et al. Regression of electrocardiographic left ventricular hypertrophy and decreased incidence of new-onset atrial fibrillation in patients with hypertension. JAMA. (2006) 296:1242–8. doi: 10.1001/jama.296.10.1242
83. Koren MJ, Ulin RJ, Koren AT, Laragh JH, Devereux RB. Left ventricular mass change during treatment and outcome in patients with essential hypertension. Am J Hypertens. (2002) 15:1021–8. doi: 10.1016/S0895-7061(02)03061-3
84. Dahlöf B, Devereux RB, Kjeldsen SE, Julius S, Beevers G, de Faire U, et al. Cardiovascular morbidity and mortality in the Losartan Intervention For Endpoint reduction in hypertension study (LIFE): a randomised trial against atenolol. Lancet. (2002) 359:995–1003. doi: 10.1016/S0140-6736(02)08089-3
85. Rababa'h AM, Guillory AN, Mustafa R, Hijjawi T. Oxidative stress and cardiac remodeling: an updated edge. Curr Cardiol Rev. (2018) 14:53–9. doi: 10.2174/1573403X14666180111145207
86. de Simone G, Izzo R, De Luca N, Gerdts E. Left ventricular geometry in obesity: is it what we expect? Nutr Metab Cardiovasc Dis. (2013) 23:905–12. doi: 10.1016/j.numecd.2013.06.012
87. Cuspidi C, Rescaldani M, Sala C, Grassi G. Left-ventricular hypertrophy and obesity: a systematic review and meta-analysis of echocardiographic studies. J Hypertens. (2014) 32:16–25. doi: 10.1097/HJH.0b013e328364fb58
88. Masiha S, Sundström J, Lind L. Inflammatory markers are associated with left ventricular hypertrophy and diastolic dysfunction in a population-based sample of elderly men and women. J Hum Hypertens. (2013) 27:13–7. doi: 10.1038/jhh.2011.113
89. Gamrat A, Surdacki MA, Chyrchel B, Surdacki A. Endothelial dysfunction: a contributor to adverse cardiovascular remodeling and heart failure development in type 2 diabetes beyond accelerated atherogenesis. J Clin Med. (2020) 9:2090. doi: 10.3390/jcm9072090
90. Gottdiener JS, Reda DJ, Massie BM, Materson BJ, Williams DW, Anderson RJ. Effect of single-drug therapy on reduction of left ventricular mass in mild to moderate hypertension: comparison of six antihypertensive agents. The department of veterans affairs cooperative study group on antihypertensive agents. Circulation. (1997) 95:2007–14. doi: 10.1161/01.CIR.95.8.2007
91. Higgins P, Dawson J, Walters M. The potential for xanthine oxidase inhibition in the prevention and treatment of cardiovascular and cerebrovascular disease. Cardiovasc Psychiatry Neurol. (2009) 2009:282059. doi: 10.1155/2009/282059
92. Higgins P, Dawson J, Lees KR, McArthur K, Quinn TJ, Walters MR. Xanthine oxidase inhibition for the treatment of cardiovascular disease: a systematic review and meta-analysis. Cardiovasc Ther. (2012) 30:217–26. doi: 10.1111/j.1755-5922.2011.00277.x
93. Guedes M, Esperança A, Pereira AC, Rego C. What is the effect on cardiovascular events of reducing hyperuricemia with allopurinol? An evidence-based review. Rev Port Cardiol. (2014) 33:727–32. doi: 10.1016/j.repce.2014.06.003
94. Singh TP, Skalina T, Nour D, Murali A, Morrison S, Moxon JV, et al. A meta-analysis of the efficacy of allopurinol in reducing the incidence of myocardial infarction following coronary artery bypass grafting. BMC Cardiovasc Disord. (2018) 18:143. doi: 10.1186/s12872-018-0881-6
95. Kao MP, Ang DS, Gandy SJ, Nadir MA, Houston JG, Lang CC, et al. Allopurinol benefits left ventricular mass and endothelial dysfunction in chronic kidney disease. J Am Soc Nephrol. (2011) 22:1382–9. doi: 10.1681/ASN.2010111185
96. Gingles CR, Symon R, Gandy SJ, Struthers AD, Houston G, MacDonald TM, et al. Allopurinol treatment adversely impacts left ventricular mass regression in patients with well-controlled hypertension. J Hypertens. (2019) 37:2481–9. doi: 10.1097/HJH.0000000000002189
97. Rena G, Lang CC. Repurposing metformin for cardiovascular disease. Circulation. (2018) 137:422–4. doi: 10.1161/CIRCULATIONAHA.117.031735
98. Kirpichnikov D, McFarlane SI, Sowers JR. Metformin: an update. Ann Intern Med. (2002) 137:25–33. doi: 10.7326/0003-4819-137-1-200207020-00009
99. Cameron AR, Morrison VL, Levin D, Mohan M, Forteath C, Beall C, et al. Anti-inflammatory effects of metformin irrespective of diabetes status. Circ Res. (2016) 119:652–65. doi: 10.1161/CIRCRESAHA.116.308445
100. Esteghamati A, Eskandari D, Mirmiranpour H, Noshad S, Mousavizadeh M, Hedayati M, et al. Effects of metformin on markers of oxidative stress and antioxidant reserve in patients with newly diagnosed type 2 diabetes: a randomized clinical trial. Clin Nutr. (2013) 32:179–85. doi: 10.1016/j.clnu.2012.08.006
101. Chakraborty A, Chowdhury S, Bhattacharyya M. Effect of metformin on oxidative stress, nitrosative stress and inflammatory biomarkers in type 2 diabetes patients. Diabetes Res Clin Pract. (2011) 93:56–62. doi: 10.1016/j.diabres.2010.11.030
102. Formoso G, De Filippis EA, Michetti N, Di Fulvio P, Pandolfi A, Bucciarelli T, et al. Decreased in vivo oxidative stress and decreased platelet activation following metformin treatment in newly diagnosed type 2 diabetic subjects. Diabetes Metab Res Rev. (2008) 24:231–7. doi: 10.1002/dmrr.794
103. Skrha J, Prazny M, Hilgertova J, Kvasnicka J, Kalousova M, Zima T. Oxidative stress and endothelium influenced by metformin in type 2 diabetes mellitus. Eur J Clin Pharmacol. (2007) 63:1107–14. doi: 10.1007/s00228-007-0378-1
104. Pavlovic D, Kocic R, Kocic G, Jevtovic T, Radenkovic S, Mikic D, et al. Effect of four-week metformin treatment on plasma and erythrocyte antioxidative defense enzymes in newly diagnosed obese patients with type 2 diabetes. Diabetes Obes Metab. (2000) 2:251–6. doi: 10.1046/j.1463-1326.2000.00089.x
105. Srinivasan S, Ambler GR, Baur LA, Garnett SP, Tepsa M, Yap F, et al. Controlled trial of metformin for obesity and insulin resistance in children and adolescents: improvement in body composition and fasting insulin. J Clin Endocrinol Metab. (2006) 91:2074–80. doi: 10.1210/jc.2006-0241
106. Mather KJ, Verma S, Anderson TJ. Improved endothelial function with metformin in type 2 diabetes mellitus. J Ame Coll Cardiol. (2001) 37:1344–50. doi: 10.1016/S0735-1097(01)01129-9
107. Wong AK, Symon R, AlZadjali MA, Ang DS, Ogston S, Choy A, et al. The effect of metformin on insulin resistance and exercise parameters in patients with heart failure. Eur J Heart Fail. (2012) 14:1303–10. doi: 10.1093/eurjhf/hfs106
108. Chan AY, Dyck JR. Activation of AMP-activated protein kinase (AMPK) inhibits protein synthesis: a potential strategy to prevent the development of cardiac hypertrophy. Can J Physiol Pharmacol. (2005) 83:24–8. doi: 10.1139/y04-107
109. Dolinsky VW, Dyck JR. Role of AMP-activated protein kinase in healthy and diseased hearts. Am J Physiol Heart Circ Physiol. (2006) 291: H2557–69. doi: 10.1152/ajpheart.00329.2006
110. Wong AKF, Howie J, Petrie JR, Lang CC. AMP-activated protein kinase pathway: a potential therapeutic target in cardiometabolic disease. Clin Sci. (2009) 116:607–20. doi: 10.1042/CS20080066
111. Mohan M, Al-Talabany S, McKinnie A, Mordi IR, Singh JSS, Gandy SJ, et al. A randomized controlled trial of metformin on left ventricular hypertrophy in patients with coronary artery disease without diabetes: the MET-REMODEL trial. Eur Heart J. (2019) 40:3409–17. doi: 10.1136/heartjnl-2018-BCS.6
112. Hugo Velázquez, Meaney A, Galeana C, Zempoalteca JC, Gutiérrez-Salmeán G, Nájera N, et al. Metformin enhances left ventricular function in patients with metabolic syndrome. Rev Colomb Cardiol. (2016) 27:16–25.
113. Al Ali L, Hartman MT, Lexis CP, Hummel YM, Lipsic E, van Melle JP, et al. The effect of metformin on diastolic function in patients presenting with st-elevation myocardial infarction. PLoS ONE. (2016) 11:e0168340. doi: 10.1371/journal.pone.0168340
114. Ida S, Kaneko R, Murata K. Effects of oral antidiabetic drugs on left ventricular mass in patients with type 2 diabetes mellitus: a network meta-analysis. Cardiovasc Diabetol. (2018) 17:129. doi: 10.1186/s12933-018-0773-1
115. Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. (2015) 373:2117–28. doi: 10.1056/NEJMoa1504720
116. Neal B, Perkovic V, Mahaffey KW, de Zeeuw D, Fulcher G, Erondu N, et al. Canagliflozin and cardiovascular and renal events in type 2 diabetes. N Engl J Med. (2017) 377:644–57. doi: 10.1056/NEJMoa1611925
117. Wiviott SD, Raz I, Bonaca MP, Mosenzon O, Kato ET, Cahn A, et al. Dapagliflozin and cardiovascular outcomes in type 2 diabetes. N Engl J Med. (2019) 380:347–57. doi: 10.1056/NEJMoa1812389
118. Zelniker TA, Wiviott SD, Raz I, Im K, Goodrich EL, Bonaca MP, et al. SGLT2 inhibitors for primary and secondary prevention of cardiovascular and renal outcomes in type 2 diabetes: a systematic review and meta-analysis of cardiovascular outcome trials. Lancet. (2019) 393:31–9. doi: 10.1016/S0140-6736(18)32590-X
119. McMurray JJV, Solomon SD, Inzucchi SE, Køber L, Kosiborod MN, Martinez FA. Dapagliflozin in patients with heart failure and reduced ejection fraction. N Engl J Med. (2019) 381:1995–2008. doi: 10.1056/NEJMoa1911303
120. Nassif ME, Windsor SL, Tang F, Khariton Y, Husain M, Inzucchi SE, et al. Dapagliflozin effects on biomarkers, symptoms, and functional status in patients with heart failure with reduced ejection fraction: the DEFINE-HF trial. Circulation. (2019) 140:1463–76. doi: 10.1161/CIRCULATIONAHA.119.042929
121. Packer M, Anker SD, Butler J, Filippatos G, Zannad F. Effects of sodium-glucose cotransporter 2 inhibitors for the treatment of patients with heart failure: proposal of a novel mechanism of action. JAMA Cardiol. (2017) 2:1025–9. doi: 10.1001/jamacardio.2017.2275
122. Maack C, Lehrke M, Backs J, Heinzel FR, Hulot JS, Marx N, et al. Heart failure and diabetes: metabolic alterations and therapeutic interventions: a state-of-the-art review from the Translational Research Committee of the Heart Failure Association-European Society of Cardiology. Eur Heart J. (2018) 39:4243–54. doi: 10.1093/eurheartj/ehy596
123. Santos-Gallego CG, Requena-Ibanez JA, San Antonio R, Ishikawa K, Watanabe S, Picatoste B, et al. Empagliflozin ameliorates adverse left ventricular remodeling in nondiabetic heart failure by enhancing myocardial energetics. J Am Coll Cardiol. (2019) 73:1931–44. doi: 10.1016/j.jacc.2019.01.056
124. Yurista SR, Silljé HHW, Oberdorf-Maass SU, Schouten EM, Pavez Giani MG, Hillebrands JL, et al. Sodium-glucose co-transporter 2 inhibition with empagliflozin improves cardiac function in non-diabetic rats with left ventricular dysfunction after myocardial infarction. Eur J Heart Fail. (2019) 21:862–73. doi: 10.1002/ejhf.1473
125. Packer M. Cardioprotective effects of sirtuin-1 and its downstream effectors: potential role in mediating the heart failure benefits of SGLT2 (Sodium-Glucose Cotransporter 2) Inhibitors. Circ Heart Fail. (2020) 13:e007197. doi: 10.1161/CIRCHEARTFAILURE.120.007197
126. Tentolouris A, Vlachakis P, Tzeravini E, Eleftheriadou I, Tentolouris N. SGLT2 inhibitors: a review of their antidiabetic and cardioprotective effects. Int J Environ Res Public Health. (2019) 16:2965. doi: 10.3390/ijerph16162965
127. Brown AJM, Lang C, McCrimmon R, Struthers A. Does dapagliflozin regress left ventricular hypertrophy in patients with type 2 diabetes? A prospective, double-blind, randomised, placebo-controlled study. BMC Cardiovasc Disord. (2017) 17:229. doi: 10.1186/s12872-017-0663-6
128. Brown AJM, Gandy S, McCrimmon R, Houston JG, Struthers AD, Lang CC. A randomized controlled trial of dapagliflozin on left ventricular hypertrophy in people with type two diabetes: the DAPA-LVH trial. Eur Heart J. (2020) 41:3421–32. doi: 10.1093/eurheartj/ehaa419
129. Verma S, Mazer CD, Yan AT, Mason T, Garg V, Teoh H, et al. Effect of empagliflozin on left ventricular mass in patients with type 2 diabetes mellitus and coronary artery disease: the EMPA-HEART cardiolink-6 randomized clinical trial. Circulation. (2019) 140:1693–702. doi: 10.1161/CIRCULATIONAHA.119.042375
130. Verma S, Mazer CD, Bhatt DL, Raj SR, Yan AT, Verma A, et al. Empagliflozin and cardiovascular outcomes in patients with type 2 diabetes and left ventricular hypertrophy: a subanalysis of the EMPA-REG OUTCOME trial. Diabetes Care. (2019) 42: e42–4. doi: 10.2337/dc18-1959
Keywords: diabetic cardiomyopathy (DCM), heart failure, type 2 diabetes mellitus, metformin, allopurinol, SGLT2 inhibitors, left ventricular hypertrophy (LVH)
Citation: Mohan M, Dihoum A, Mordi IR, Choy A-M, Rena G and Lang CC (2021) Left Ventricular Hypertrophy in Diabetic Cardiomyopathy: A Target for Intervention. Front. Cardiovasc. Med. 8:746382. doi: 10.3389/fcvm.2021.746382
Received: 23 July 2021; Accepted: 02 September 2021;
Published: 29 September 2021.
Edited by:
VijayaKumar Sukumaran, Qatar University, QatarReviewed by:
Rizwan Ahmed, Johns Hopkins University, United StatesCopyright © 2021 Mohan, Dihoum, Mordi, Choy, Rena and Lang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mohapradeep Mohan, bW9oYXByYWRlZXAubW9oYW5Ad2Fyd2ljay5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.