
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med., 04 October 2021
Sec. Cardiovascular Metabolism
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.742178
This article is part of the Research TopicMetabolic Related Cardiomyopathy in Hyperglycemic PatientsView all 9 articles
 Florian A. Wenzl1
Florian A. Wenzl1 Samuele Ambrosini1
Samuele Ambrosini1 Shafeeq A. Mohammed1
Shafeeq A. Mohammed1 Simon Kraler1
Simon Kraler1 Thomas F. Lüscher1,2
Thomas F. Lüscher1,2 Sarah Costantino1
Sarah Costantino1 Francesco Paneni1,3,4*
Francesco Paneni1,3,4*Overlapping pandemics of lifestyle-related diseases pose a substantial threat to cardiovascular health. Apart from coronary artery disease, metabolic disturbances linked to obesity, insulin resistance and diabetes directly compromise myocardial structure and function through independent and shared mechanisms heavily involving inflammatory signals. Accumulating evidence indicates that metabolic dysregulation causes systemic inflammation, which in turn aggravates cardiovascular disease. Indeed, elevated systemic levels of pro-inflammatory cytokines and metabolic substrates induce an inflammatory state in different cardiac cells and lead to subcellular alterations thereby promoting maladaptive myocardial remodeling. At the cellular level, inflammation-induced oxidative stress, mitochondrial dysfunction, impaired calcium handling, and lipotoxicity contribute to cardiomyocyte hypertrophy and dysfunction, extracellular matrix accumulation and microvascular disease. In cardiometabolic patients, myocardial inflammation is maintained by innate immune cell activation mediated by pattern recognition receptors such as Toll-like receptor 4 (TLR4) and downstream activation of the NLRP3 inflammasome and NF-κB-dependent pathways. Chronic low-grade inflammation progressively alters metabolic processes in the heart, leading to a metabolic cardiomyopathy (MC) phenotype and eventually to heart failure with preserved ejection fraction (HFpEF). In accordance with preclinical data, observational studies consistently showed increased inflammatory markers and cardiometabolic features in patients with HFpEF. Future treatment approaches of MC may target inflammatory mediators as they are closely intertwined with cardiac nutrient metabolism. Here, we review current evidence on inflammatory processes involved in the development of MC and provide an overview of nutrient and cytokine-driven pro-inflammatory effects stratified by cell type.
Lifestyle-related diseases have reached pandemic proportions and contribute greatly to human suffering and excess mortality. By the year 2030, more than 2.1 billion people will be overweight or obese and 0.5 billion will have diabetes worldwide, with cardiovascular disease remaining the leading cause of death in these patients (1–4). While the burden of coronary artery disease and hypertension is declining in high-income countries, glucometabolic perturbations linked to obesity and diabetes have emerged as key determinants of myocardial remodeling and dysfunction in the past two decades (5, 6). It is now recognized that metabolic disturbances induce a systemic inflammatory state, which in turn impacts myocardial structure and function. The pro-inflammatory milieu created by circulating cytokines, excess metabolic substrate availability, and paracrine signals from activated immune cells in the heart triggers maladaptive myocardial remodeling and its clinical sequelae. Indeed, cytokines and nutrient metabolites activate inflammatory programs in different cardiac cell types through shared pathways causing a disruption of cardiac tissue homeostasis. The resulting subcellular alterations progressively lead to a metabolic cardiomyopathy (MC) phenotype which can become clinically evident as heart failure (HF) with preserved ejection fraction (HFpEF).
Collectively, cellular abnormalities in obesity and diabetes overlap considerably with those observed in HFpEF including inflammation-induced oxidative stress, mitochondrial dysfunction, lipotoxicity, cardiomyocyte hypertrophy and impaired calcium handling, extracellular matrix (ECM) accumulation, and microvascular disease (7). Both obesity and type 2 diabetes (T2D) associate with increased inflammatory markers and are present in the majority of patients with HFpEF (7–9). Given the prominent role of obesity and associated comorbidities in HFpEF, systemic inflammation has emerged as major culprit in disease development (7, 10). Randomized controlled trials in obese HFpEF patients with elevated C-reactive protein (CRP) have shown decreased N-terminal pro-B-type natriuretic peptide (NT-proBNP) levels and improved exercise capacity upon interleukin (IL)-1 blockade (11–13). Yet, recent clinical trials with anti-inflammatory agents have failed to demonstrate a benefit in terms of survival or hospitalization in patients with HF, thus highlighting the unmet need for a better understanding of the underlying pathobiology (11–13). In the present review we provide an overview of inflammatory processes involved in the development of MC stratified by cell type.
Along with the growing burden of lifestyle diseases, the term “metabolic cardiomyopathy” has been increasingly used in the literature to reflect deleterious effects of glucometabolic perturbations on the myocardium unrelated to coronary artery disease, hypertension, valvular heart disease and other traditional risk factors for myocardial remodeling (14–18). As a pathophysiological entity, MC embraces the broad spectrum of metabolic disturbances that compromise myocardial structure and function in patients with obesity, insulin resistance and diabetes (14, 17). In fact, these conditions associate with a distinct form of cardiomyopathy marked by early diastolic dysfunction, interstitial fibrosis and myocellular lipid accumulation (17, 19, 20). Beyond traditional causes of myocardial disease, adverse remodeling is mediated by systemic metabolic dysregulation including circulating metabolic substrates [e.g., free fatty acids (FFAs)] and inflammatory cytokines [e.g., tumor necrosis factor-alpha (TNF-α) and IL-6] (14). Importantly, there is substantial overlap in the molecular mechanisms underlying diabetic cardiomyopathy, obesity-related cardiomyopathy and those observed in patients with a metabolic HFpEF phenotype (7). Considering that pathological alterations in the myocardium linked to obesity and diabetes commonly occur before the onset of HF symptoms, MC may represent a precursor of HFpEF (21). In line with experimental evidence, obesity and T2D confer increased risk for incident HF even after adjustment for known risk factors including coronary artery disease (22–24).
A growing body of evidence indicates that alterations in myocardial structure and function in cardiometabolic patients result from a multi-organ disease process involving systemic inflammatory cytokines, circulating metabolic substrates and immune dysregulation (21, 25). As a general model, nutrient overload activates inflammatory responses in extracardiac tissues with release of pro-inflammatory mediators and subsequent systemic and cardiac inflammation (Figure 1) (14, 25, 26). In parallel, circulating inflammatory cytokines (e.g., TNF-α and IL-6) impair systemic and cardiac insulin sensitivity via activation of evolutionary conserved regulators of inflammation such as nuclear factor (NF)-κB (27, 28) and c-Jun N-terminal kinase (JNK) (29, 30). This state of chronic low-grade inflammation—primarily caused by obesity and associated metabolic conditions has been termed metabolic inflammation or “metainflammation” (25). Unlike acute inflammatory responses to cardiac tissue damage, which represent crucial regenerative processes, chronic inflammation leads to metabolic reprogramming of the heart and contributes to adverse remodeling and functional impairment (14).
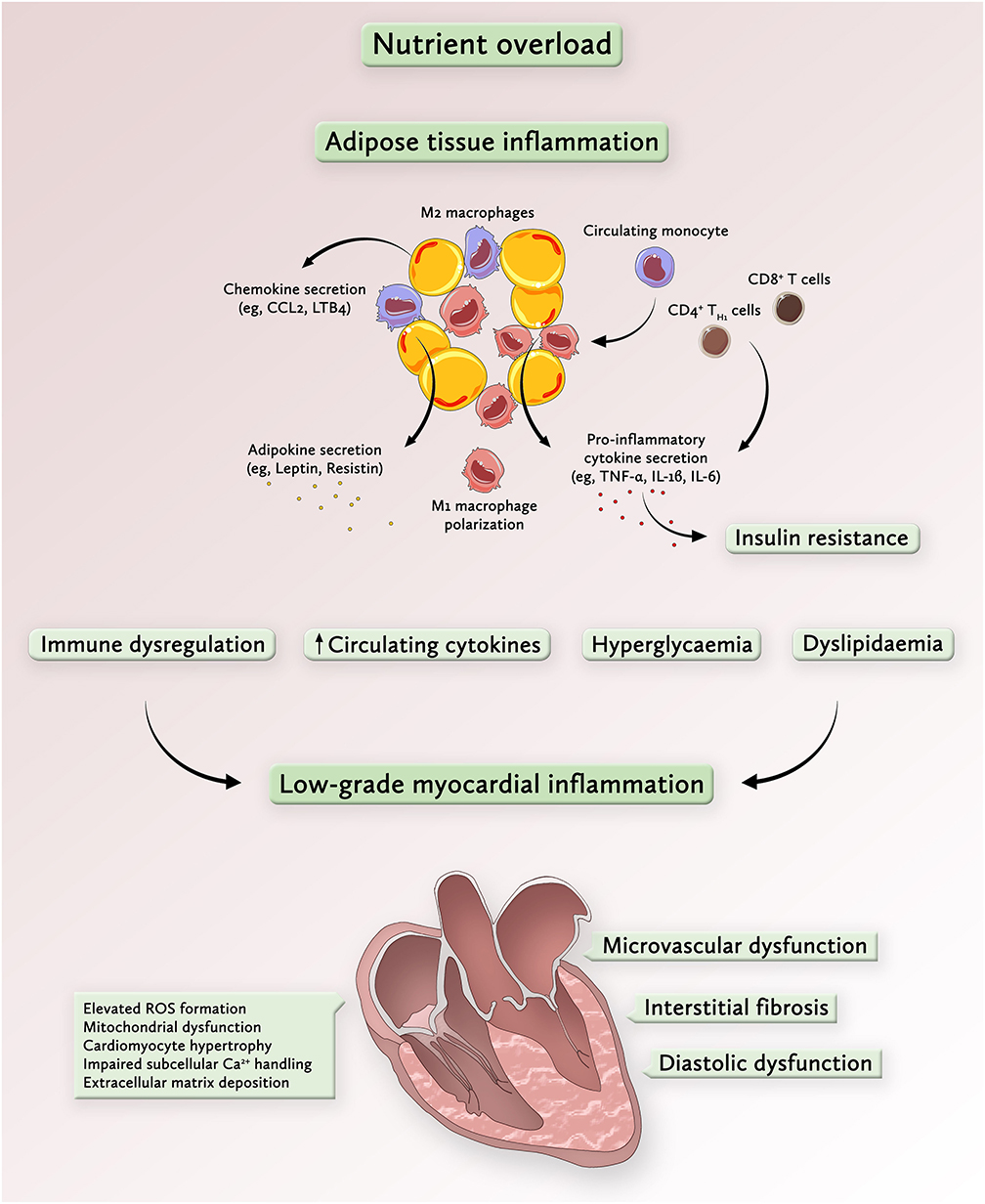
Figure 1. Overnutrition drives metabolic inflammation and promotes a low-grade inflammatory state in the heart. Chronic nutrient overload induces adipose tissue expansion, which enhances the secretion of chemotactic signals, such as chemokine-ligand 2 (CCL2) from enlarging adipocytes. Transmigration of chemokine-ligand receptor 2 (CCR2)+ circulating monocytes into the adipose tissue represents a key event in the development of systemic inflammation in response to nutrient overload. Given the pro-inflammatory milieu, recruited monocytes assume an inflammatory M1 macrophage phenotype, a process that is further accelerated by activated CD8+ T cells and CD4+ TH1 cells. The release of inflammatory cytokines causes insulin resistance, commonly associated with hyperglycemia, dyslipidemia and immune dysregulation. These processes contribute to the activation of inflammatory pathways in the myocardium which are linked to enhanced ROS formation and mitochondrial dysfunction, cardiomyocyte growth and extracellular matrix deposition. Collectively, these alterations on both systemic and myocardial levels drive microvascular dysfunction, interstitial fibrosis and diastolic dysfunction, key features of metabolic cardiomyopathy. CCL2 denotes chemokine ligand 2; IL, interleukin; LB4, leukotriene B4; ROS, reactive-oxygen species; TNF-α, tumor necrosis factor alpha.
The initial event in obesity-induced systemic inflammation is the secretion of specific chemokines such as C-C motif chemokine ligand 2 (CCL2) and leukotriene B4 (LTB4) from adipocytes which promote monocyte trafficking into the adipose tissue (26, 31). Once recruited to adipose tissue via the C-C motif chemokine receptor 2 (CCR2), monocytes polarize toward a pro-inflammatory macrophage phenotype and secrete their own chemotactic and pro-inflammatory cytokines to attract additional monocytes, thus amplifying local and systemic inflammation (26, 32). In particular, visceral adipose tissue has a prominent role in metabolic dysregulation since it recruits more pro-inflammatory macrophages, secretes larger amounts of inflammatory cytokines and causes more pronounced peripheral insulin resistance than subcutaneous white adipose tissue (26, 33, 34). Once a systemic pro-inflammatory state has been initiated, inflammatory triggers (e.g., IL-1β, IL-6, and IL-8) originate from a variety of extracardiac cell types including fibroblasts and vascular cells (7). In the heart, inflammatory cytokines are implicated in several important processes of cardiac remodeling, including cardiomyocyte hypertrophy (35), cardiomyocyte apoptosis (36), microvascular endothelial activation, and myocardial fibrosis (37). Looking beyond the heart, cardiac signs and symptoms in patients with obesity and T2D result from a complex pro-inflammatory inter-organ cross-talk involving the adipose tissue, kidney, lung, spleen, bone marrow, skeletal muscle, and gut (13).
An additional feature of metabolic inflammation is the increased substrate availability. Aside from circulating cytokines, high levels of glucose and saturated FFAs were found to directly promote a pro-inflammatory state in different cardiac cell types (38–40). Importantly, high glucose levels modulate multiple intracellular signaling pathways in cardiomyocytes, fibroblasts and cardiac macrophages that converge toward NF-κB activation and promote the expression of TNF-α and IL-6 (38, 41–46). Although less well-studied, other nutrients such as high fructose corn syrup, contained in a Western diet, may also lead to low-grade myocardial inflammation (suggested by increased expression of macrophage markers) and have recently been included in some animal models for HFpEF (47, 48).
Metabolic inflammation leads to the recruitment of macrophages into the myocardium (25). Animal models for diet-induced obesity (49, 50), pre-diabetes (51), T2D (52–55), and lipotoxic cardiomyopathy (56) conclusively showed upregulation of vascular adhesion molecules [e.g., intercellular adhesion molecule (ICAM)-1 and vascular cell adhesion molecule (VCAM)-1] and infiltration of macrophages into the heart—a phenomenon similarly observed in obese patients with HFpEF (10, 57). In fact, glucometabolic disturbances are tightly coupled with dysregulation of innate immune cells. Saturated fatty acids induce the secretion of inflammatory mediators (e.g., TNF-α, IL-1β, IL-6, and CCL2) by macrophages through mechanisms depending on pattern recognition receptors, such as Toll-like receptor (TLR)4, thus maintaining myocardial inflammation (58–61). In patients with obesity and T2D immune-dysregulation and macrophage recruitment are also promoted by the overproduction of adipocyte-derived aldosterone and neprilysin, leading to accelerated natriuretic peptide degradation (62). In concert, these substances mediate renal sodium reabsorption and contribute to low-grade myocardial inflammation (62, 63). Of note, augmented secretion of aldosterone from the adrenal glands is closely linked to increased body fat mass as it can be directly induced by the adipokine leptin (62).
Next, activation of the renin-angiotensin-aldosterone system, evidenced by pronounced secretion of angiotensinogen by the liver and adipose tissue, contributes to myocardial remodeling and inflammation in cardiometabolic patients (64, 65). Cleavage of circulating Angiotensin (Ang) I by the angiotensin converting enzyme (ACE) yields Ang II, which along with aldosterone, activates NF-κB in cardiac endothelial cells and fibroblasts, thus leading to upregulation of vascular adhesion molecules, recruitment of immune cells, and increased ECM production (65, 66). In the counterregulatory RAAS pathway, ACE2 converts Ang I to Ang-(1-7) which mitigates leukocyte migration, pro-inflammatory cytokine release, fibrosis, and insulin resistance via activation of the Mas receptor (67, 68).
Another mechanism coupling systemic glucometabolic disturbances with myocardial inflammation and hypertrophy is the formation of advanced glycation end products (AGEs) (50). As a result of chronic hyperglycemia AGEs can accumulate in the cardiac ECM and enhance the expression of pro-inflammatory mediators (e.g., TNF-α, IL-6, ICAM-1, and CCL2) via the receptor for AGEs (RAGE) (50). Of note, AGEs also promote myocardial inflammation by direct activation of macrophages via the RAGE/NF-κB pathway (69, 70).
Collectively, systemic cytokines, paracrine signals from recruited immune cells, increased substrate availability and alterations of the ECM all contribute to an inflammatory milieu in the myocardium and disrupt cardiac tissue homeostasis. Maladaptive myocardial remodeling in patients with obesity and T2D therefore can be framed as a chronic inflammatory condition of the heart that is closely intertwined with nutrient metabolism (25).
Under physiological conditions, the myocardium is able to switch between metabolic substrates, mainly fatty acids and carbohydrates, in response to changes in nutrient availability (71). However, systemic low-grade inflammation goes along with cardiac insulin resistance which is accompanied by a shift in substrate utilization toward fatty acid metabolism favoring the accumulation of toxic lipid metabolites (29).
TNF-α causes cardiac insulin resistance by activation of both NF-κB- and the JNK-dependent signaling pathways converging toward serine phosphorylation and proteasomal degradation of the insulin response substrate (IRS)1 (29). Moreover, IL-6 interferes with insulin signal transduction through signal transducer and activator of transcription (STAT)3-dependent suppressor of cytokine signaling (SOCS)3 upregulation, which impairs the coupling of IRS1 with the insulin receptor (72). In line with this notion, genetic knockout of IL-6 attenuates cardiac insulin resistance and inflammation in obese mice (49). At the myocardial level, insulin resistance is further promoted by inflammation-induced oxidative stress (73).
Contrasting reduced cardiac glucose uptake via the insulin-dependent glucose transporter 4 (GLUT4) in insulin resistant states, metabolic stress promotes increased cardiac fatty acid uptake through upregulation of cluster of differentiation (CD)36, the main fatty acid transporter in cardiomyocytes. CD36 is regulated by the peroxisome proliferator-activated receptor (PPAR)-γ/retinoid X receptor (RXR) complex allowing for its enhanced expression in response to nutrient excess (Figure 2) (29, 74). Paired with high levels of circulating FFAs, increased abundance of CD36 on the sarcolemmal membrane raises intracellular fatty acid availability and turnover (75).
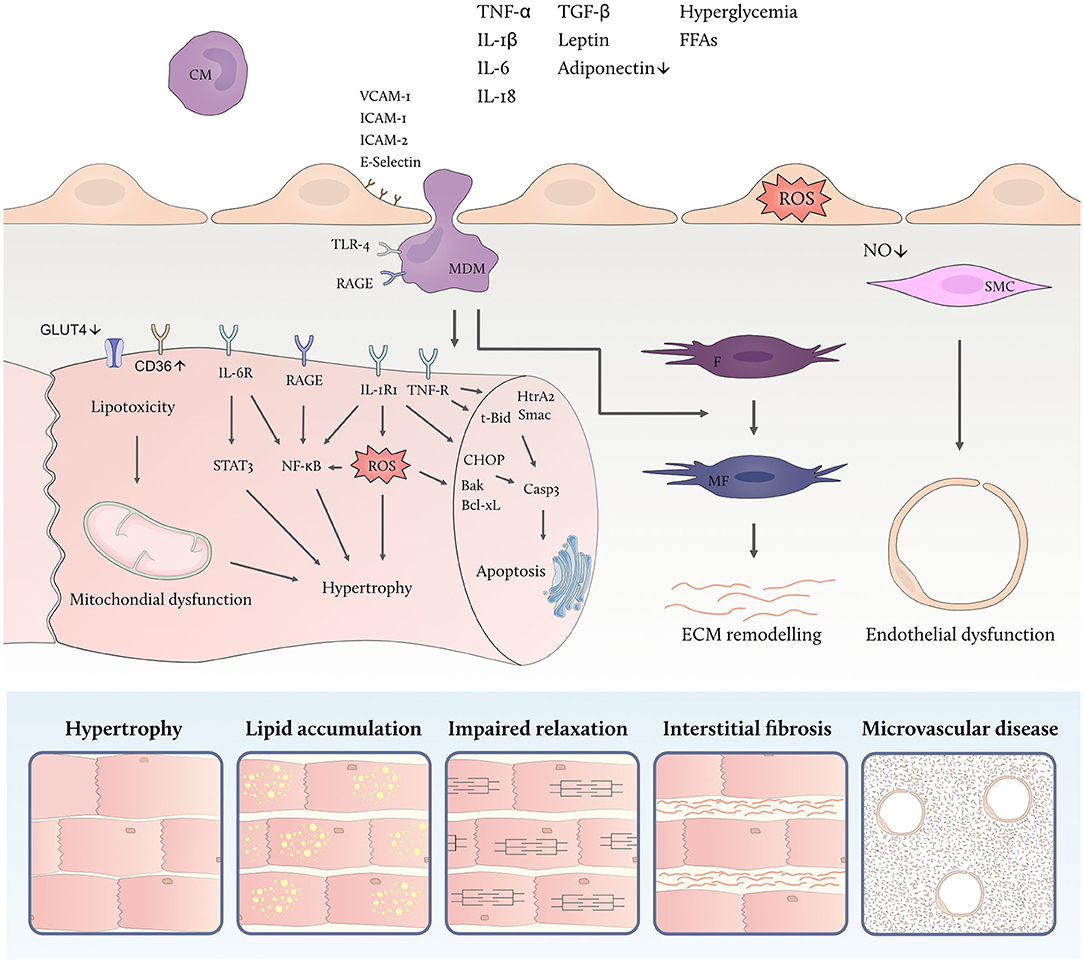
Figure 2. Metabolic inflammation promotes myocardial remodeling. High levels of circulating inflammatory cytokines and metabolic substrates activate inflammatory cascades in different cardiac cell types linked to cellular dysfunction. Endothelial activation facilitates leucocyte adhesion and transmigration into the myocardium thereby aggravating the low-grade inflammatory state. Both free fatty acids (FFAs) and high glucose levels modulate the polarization of monocyte-derived macrophages (MDM) which secrete inflammatory and profibrotic cytokines. Cardiac insulin resistance is promoted by inflammatory cytokines, including tumor necrosis factor alpha (TNF-α), and goes along with down-regulation of the insulin-dependent glucose transporter 4 (GLUT4) and upregulation of the fatty acid transporter cluster of differentiation (CD)36 thus contributing to lipotoxicity, mitochondrial dysfunction and accumulation of reactive oxygen species (ROS). In addition, direct effects of circulating inflammatory mediators lead to endothelial ROS formation and microvascular dysfunction. IL denotes interleukin; IL-1RI, IL-1 receptor type I; IL-6R, IL-6 receptor, TNF-R, TNF receptor; STAT3, signal transducer and activator of transcription 3; NF-κB, nuclear factor kappa-light-chain-enhancer of activated B cells; HtrA2, HtrA serine peptidase 2; Smac, second mitochondria-derived activator of caspase; t-Bid, truncated BH3 interacting domain death agonist; CHOP, C/EBP homologous protein; Bak, BCL2-antagonist/killer; Bcl-xL, BCL-extra-large; Casp3, caspase 3; TGF-β, transforming growth factor beta; VCAM-1, vascular cell adhesion molecule 1; ICAM, intercellular adhesion molecule; RAGE, receptor for advanced glycation end products; IL-R, interleukin receptor; CM, circulating monocyte; MDM, monocyte derived macrophage; F, fibroblast; MF, myofibroblast; SMC, smooth muscle cell; ROS, reactive oxygen species; NO, nitric oxide; ECM, extracellular matrix.
FFA overload leads to mitochondrial dysfunction and uncoupling of fatty acid oxidation from ADP phosphorylation in cardiomyocytes (17). As a result of deranged cardiac lipid metabolism, cardiac triacylglycerols and toxic intermediate products such as diacylglycerols and ceramides are formed (17) and accumulate in the heart of obese and diabetic patients (76–78). Cardiac lipotoxicity has been implicated in the generation of reactive oxygen species (ROS), cell apoptosis, defective insulin signaling, and impaired calcium handling (79–83). While the exact mechanisms underlying cardiac lipotoxicity remain elusive and are subject of ongoing investigations, the combination of myocardial inflammation, insulin resistance and excess supply of FFA emerges as a decisive factor (17).
Nutrient overload activates different inflammatory signaling cascades in cardiomyocytes which contribute to cell hypertrophy, apoptosis, and mechanical dysfunction (38). The regulation of inflammatory programs in cardiomyocytes is closely linked to intracellular ROS accumulation resulting from deranged cardiac substrate utilization in diabetes and obesity (17, 38). Excess availability of lipids and glucose favors the production of ROS (17, 84) which in turn enhances the transcription and functional activity of NF-κB (85–89). Cardiomyocyte-specific inhibition of NF-kB signaling through overexpression of inhibitor of NF-κB (IκB)-α mitigates cardiac alterations in hyperglycemic mice—highlighting the importance of this axis (90).
In addition, high glucose concentrations directly activate a number of pro-inflammatory pathways in cardiomyocytes converging toward NF-κB. Exposure to high glucose levels enhances the expression of high-mobility group box 1 (HMGB1) protein in cardiomyocytes thereby activating mitogen-activated protein kinase (MAPK) and NF-κB which leads to TNF-α and IL-6 secretion (41). High glucose also induces upregulation of TNF-α, IL-1β, IL-6, and IL-12 through activation of JNK and NF-κB (45). Another mechanism linking glucose metabolism to inflammation is histone 3 lysine 9 trimethylation (H3K9me3) at the IL-6 promoter under high glucose conditions favoring its upregulation (91). Moreover, posttranslational modification of the NF-κB p65 subunit by O-linked N-acetylglucosamine (O-GlcNAc) enhancing its transcriptional activity under hyperglycemic conditions may also apply to cardiomyocytes (92). Likewise, hyperglycemia-induced epigenetic changes that increase p65 expression may be of relevance in cardiomyocytes (38, 44, 93).
Excess availability of FFAs contributes to deranged substrate utilization of the heart in high metabolic states leading to lipotoxicity and ROS formation (17). Exposure of human cardiomyocytes to saturated fatty acids enhances NF-κB binding activity and raises nuclear p65 protein levels leading to enhanced expression of TNF-α, IL-6, and CCL-2 (94). Similar findings were reported in hearts from mice fed a high-fat diet (94). Direct activation of the NOD-, LRR- and pyrin domain-containing protein (NLRP) 3 inflammasome by accumulating ceramides has been demonstrated in other cell types including adipocytes and may also be of importance in cardiomyocytes.
Endothelial cells are a central component of the cardiac vasculature forming a barrier between blood and myocardial tissue. Aside from their regulatory function in substrate exchange, endothelial cells control myocardial blood flow, and immune cell recruitment (95–97). Endothelial nitric oxide (NO) production regulates the vascular tone and hinges on functional insulin signaling in endothelial cells (98). In metabolic disorders, such as obesity and T2D, coronary endothelial cell function is markedly impaired by high levels of circulating inflammatory mediators (e.g., TNF-α, IL-1β, and IL-6) contributing to insulin resistance (99). In addition, excess metabolic substrates, namely glucose and FFA, exert a rage of detrimental effects on endothelial cell function linked to ROS formation and inflammatory pathway activation (98).
Exposure of endothelial cells to high glucose levels activates IκB kinase (IKK)β and NF-κB signaling which leads to upregulation of inflammatory cytokine expression, reduced insulin sensitivity and diminished NO production (100, 101). Excess glucose also leads to tight junction disruption—a hallmark of endothelial barrier dysfunction—through activation of the NLRP3 inflammasome (102). In line, high glucose levels associate with increased inflammatory markers in the circulation and in endothelial cells in the setting of acute coronary syndrome (103, 104).
High levels of circulating FFAs disrupt endothelial cell function via induction inflammatory signaling cascades and increased ROS formation (105). FFAs induce vascular inflammation via TLR4-dependent activation of IKKβ and NF-κB which has been linked to endothelial insulin resistance and decreased NO availability (105–108). It has also been reported that FFAs selectively stimulate NF-κB and activator protein (AP)1 transcriptional activation leading to enhanced expression of inflammatory mediators such as TNF-α, CCL-2, and ICAM-1 (105, 109). Conversely, genetic inhibition of NF-κB in endothelial cells blocks ROS formation, improves insulin sensitivity, downregulates vascular adhesion molecules and increases the expression of endothelial NO synthetase (eNOS) in obesity (110). Moreover, palmitic acid, a long-chain saturated fatty acid, activates the NLRP3 inflammasome and increases the expression of IL-1β in endothelial cells thereby contributing to endothelial dysfunction (111).
Fibroblasts are one of the largest non-cardiomyocyte cell populations in the heart and regulate the ECM composition, structure, and turnover (112). Expansion of the cardiac interstitium through accumulation of ECM proteins (i.e., interstitial and perivascular fibrosis) in patients with obesity and diabetes reflects a maladaptive response to glucometabolic disturbances (112, 113). Exposure to high glucose increased the expression of transforming growth factor (TGF)-β, the main fibrogenic cytokine in the heart, and promotes fibroblast proliferation and ECM protein synthesis in vitro (113–118). High glucose levels also activate the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt)/MAPK signaling pathway and leads to upregulation of pro-inflammatory IL-17 synthesis and IL-17 receptor (IL-17R) expression, thus stimulating increased collagen synthesis (119). Apart from the MAPK pathway, these effects may be partially favored by a pro-inflammatory state in cardiac fibroblasts under high glucose conditions manifest from activation of NF-κB and enhanced expression of TNF-α, IL-6, and IL-1β (41, 114, 117, 120). A key event in myocardial remodeling is the conversion of fibroblasts to activated myofibroblasts, the main ECM producing cells (112). Due to differences in study conditions, conflicting data have been reported on the effect of high glucose on myofibroblast transition with the majority of studies pointing toward increased myofibroblast conversion under high glucose conditions (102, 115, 118, 121, 122). Interestingly, obese diabetic (db/db) mice, characterized by increased body weight, hyperglycemia and hyperlipidemia, display cardiac fibrosis in the absence of myofibroblast conversion, suggesting the activation of alternative matrix-synthetic programs in fibroblasts (112, 123).
Macrophages are the predominant immune cell type in the resting heart and have an important role in the regulation of tissue homeostasis (124). In response to chronic nutrient overload, resident macrophages expand and interact with other cardiac cell types via paracrine mechanisms (49–51, 124, 125). In fact, myocardial remodeling observed in patients with obesity or diabetes is largely mediated and amplified by cardiac macrophages (69). Increased levels of circulating nutrients (i.e., glucose and FFAs) alter macrophage function favoring their polarization from a regulatory (M2) toward a pro-inflammatory (M1) phenotype through different mechanisms (60, 126, 127).
First, overnutrition leads to global insulin resistance accompanied by chronic hyperglycemia, which promotes increased glucose uptake by macrophages via the insulin-independent GLUT1. In contrast to cardiomyocytes, macrophages are not sensitive to insulin and maintain glucose uptake in insulin resistant states (69, 128). Elevated intracellular glucose availability shifts the macrophage metabolism toward glycolysis and away from oxidative phosphorylation leading to increased pro-inflammatory gene expression (129). In parallel, the pentose phosphate pathway is activated and generates nicotinamide adenine dinucleotide phosphate (NADPH) which supports the synthesis of inflammatory prostaglandins and leukotrienes, thus activating NF-κB (69, 129).
Second, obesity and diabetes are both associated with elevated circulating and cardiac lipid levels which act as extra- and intracellular pro-inflammatory signaling molecules (17, 69, 130). Saturated fatty acids drive inflammatory responses in macrophages mediated by TLR4 on the cellular surface (14, 59). Multiple studies have demonstrated that long-chain saturated fatty acids (e.g., palmitic acid), but not short-chain saturated fatty acids or long-chain unsaturated fatty acids, induce the expression of inflammatory cytokines in macrophages (e.g., TNF-α) via the JNK signaling pathway in a TLR4-dependent manner (59–61, 126). Mechanistically, it is uncertain whether this effect is mediated by direct binding of FFAs to TLR4 or by an indirect TLR4-dependent mechanism—with a recent systematic study indicating the latter (61). Within the cell, saturated fatty acids also activate the NLRP3 inflammasome via an AMP-activated kinase (AMPK)-dependent pathway hinging on mitochondrial ROS production and cause IL-1β and IL-18 synthesis (131). In addition, excess intracellular fatty acid availability promotes anabolic pathways in macrophages including triacylglycerol, phospholipid, and ceramide synthesis (69, 132). Fatty acid-derived ceramide production activates the NLRP3 inflammasome thereby promoting lipotoxicity and M1 polarization (132, 133). Moreover, oxidized low-density lipoprotein (LDL) induces CD36-dependent mitochondrial ROS production in macrophages which facilitates NF-κB activation and inflammatory cytokine generation (69, 134). In aggregate, inflammatory processes in macrophages are tightly coupled to nutrient metabolism and therefore dictated by the availability of energetic substrates.
Cardiomyocytes are exposed to a broad range of cytokines originating from other cardiomyocytes, non-cardiomyocyte cardiac cells, and extracardiac tissues (135). Inflammatory cytokines such as TNF-α and IL-6, highly abundant in obesity and T2D, bind to receptors on the cardiomyocyte surface which triggers downstream activation of NF-κB and other central regulators of cell metabolism with differential impact on cardiomyocyte function (7–9, 86, 136–138). A number of deleterious effects of inflammatory cytokines on cardiomyocytes have been documented, namely cardiomyocyte hypertrophy, progressive cardiomyocyte loss through apoptosis, activation of fetal gene programs, impaired contractility, and increased passive tension (Table 1) (35, 36, 137, 139–141).
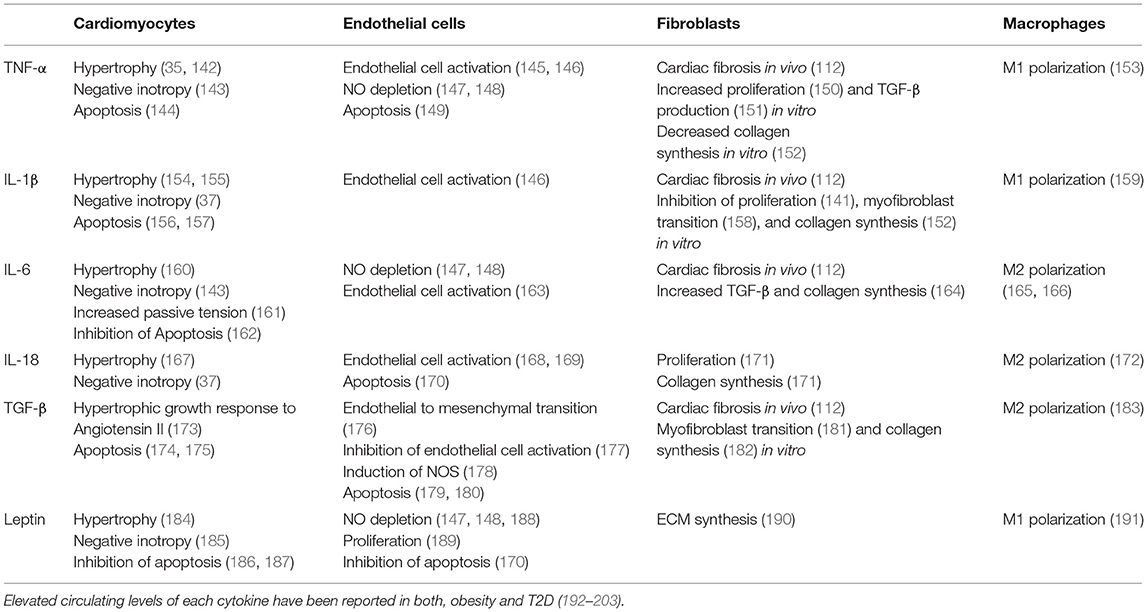
Table 1. Overview of effects on different cardiac cell types mediated by selected cytokines upregulated in cardiometabolic patients.
TNF-α exerts intracellular effects via binding to two different cell surface receptors, TNF receptor (TNFR)1 and TNFR2, both of which are expressed in cardiomyocytes (204). Exposure to TNF-α stimulates protein synthesis and blunts protein degradation in cardiomyocytes leading to cell hypertrophy (35, 142) via Akt/NF-κB and JNK activation (205). IL-1β induces cardiomyocyte hypertrophy through (1) direct interaction with cardiomyocytes (154), and (2) signal transducer and activator of transcription (STAT)3-dependent induction of insulin-like growth factor (IGF)1 by cardiac fibroblasts (155). IL-6 induces cardiomyocyte hypertrophy through Ca2+/calmodulin-dependent protein kinase (CaMK)II-dependent activation of STAT3 (160). IL-18, another upregulated pro-inflammatory cytokine in patients with obesity and T2D, induces cardiomyocyte hypertrophy via PI3K/Akt/GATA binding protein (GATA)4 signaling (167). In line with in vitro results, a number of studies have confirmed the role of pro-inflammatory cytokines in cardiac hypertrophy in vivo. Administration of TNF-α (35, 206) and IL-1β (141, 207) leads to left ventricular (LV) hypertrophy and dysfunction in rodents. Conversely, genetic deletion of TNF-α (208) and IL-1β (155) reduces LV hypertrophy and dysfunction in response to pressure overload. Marked LV hypertrophy and impaired diastolic relaxation and can be induced by infusion of IL-6 (209). Conclusively, IL-6 knockout attenuates myocardial hypertrophy and improves diastolic function in response to pressure overload (160).
Inflammatory cytokines modulate a range of processes controlling cardiomyocyte apoptosis. Sustained TNF signaling induces apoptosis via activation of intrinsic and extrinsic cell death pathways leading to activation of caspases-9 and−3 via cytosolic upregulation of cytochrome c, second mitochondria-derived activator of caspase (Smac), and HtrA serine peptidase (HtrA)2, and to cleavage of BH3 interacting domain death agonist (Bid) to truncated (t-)Bid, respectively (144). IL-1β promotes cardiomyocyte apoptosis (1) by induction of inducible nitric oxide synthase (iNOS) and subsequent generation of oxygen free radicals that alter the cellular balance of BCL2-antagonist/killer (Bak) and BCL-extra-large (Bcl-xL), and (2) by increasing endoplasmatic reticulum stress which promotes interleukin 1 receptor associated kinase (IRAK)2/C/EBP homologous protein (CHOP) signaling (156, 157). In addition, activation of the NLRP3 inflammasome induces cardiomyocyte cell death via caspase-1 (210).
Activation of the NLRP3 inflammasome/caspase-1 has also been liked to LV diastolic dysfunction. In diabetic cardiomyopathy, inhibition of caspase-1 leading to diminished IL-1β and IL-18 synthesis improves diastolic function and reduces myocardial fibrosis (54). Likewise, inhibition of IL-1β and IL-18 synthesis by knockdown of NLRP3 improves diastolic LV function in diabetic rats (211).
Alongside LV diastolic dysfunction, impaired systolic LV function is common in diabetic cardiomyopathy and obese patients with HFpEF (212, 213). In parallel, negative inotropic effects in vivo and in vitro have been reported for TNF-α, IL-1β, IL-6, and IL-18 (37). Cytokines mediate a rapid and reversible reduction of cardiomyocyte contractility by activating myocardial iNOS (143). Moreover, IL-1β and IL-6 decrease the expression of sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA)2a, which in turn may impair cardiomyocyte contractility through altered calcium handling (38).
Structural and functional abnormalities of the coronary microvasculature are propagated by chronic metabolic inflammation and can occur in the absence of macrovascular coronary artery disease (21, 213–217). The combination of systemic inflammation, hyperglycemia, and hyperlipidemia alters the release of vasoactive substances, such as NO, from the vascular endothelium leading to impaired smooth muscle relaxation and decreased myocardial perfusion (21, 147, 213–215, 217). Attenuated vasodilator response irrespective of macrovascular alterations is observed in subjects with diabetes (214, 218, 219), obese subjects with or without diabetes (220–223), subjects at increased risk to develop HFpEF (224) and patients diagnosed with HFpEF (213, 225, 226). Over the past two decades, the strong link between inflammation and microvascular dysfunction has been substantiated by a number of clinical studies (227–230). Notably, reduction in coronary flow reserve correlates with the degree of systemic inflammation assessed by CRP, IL-6, and white blood count (231). Likewise, in obese patients without coronary artery disease high circulating inflammatory markers (e.g., high sensitivity [hs]CRP, TNF-α, IL-6, and Leptin) associate with reduced coronary flow reserve (232, 233).
On a cellular level, TNF-α, IL-6, and leptin activate the NADPH-oxidase in the vessel wall leading to enhanced production of hyperoxide anion which in turn decreases NO availability and impairs vasodilation (147, 148). Moreover, obesity and diabetes-related microangiopathy is accompanied by microvascular rarefaction (222, 234–238). Reduction of coronary capillary density relative to cardiomyocyte surface area in turn promotes cardiomyocyte hypertrophy by decreasing NO-dependent protein kinase (PK)G activity (238). Depressed endothelial NO generation due to systemic inflammation has also been proposed as a leading cause of reduced cGMP-dependent PKG signaling in adjacent cardiomyocytes and impaired diastolic relaxation (7, 215).
Inflammatory processes in the myocardium of patients with the metabolic syndrome are amplified by endothelial activation and recruitment of circulating immune cells (38). Diet-induced obesity and diabetes alike enhance the expression of endothelial transmembrane proteins in the heart, such as VCAM-1 and ICAM-1, which facilitate leucocyte adhesion to the vascular wall and endothelial transmigration (50, 54, 239). Accordingly, both conditions are accompanied by increased abundance of cardiac macrophages and greater propensity of macrophages to assume a pro-inflammatory (M1) phenotype (47, 49, 50, 52, 53, 240). Mechanistically, circulating inflammatory cytokines (e.g., TNF-α, IL-1β) and, in advanced stages of myocardial functional impairment, elevated levels of Ang II induce upregulation of vascular adhesion molecules on the endothelial surface (215, 241–243).
During inflammatory states, cardiac cells secrete inflammatory and profibrotic cytokines which stimulate maladaptive remodeling through direct activation of fibroblasts and indirect effects (38, 112). TNF-α exerts multiple profibrotic effects including fibroblast activation and increased expression of TGF-β, the main profibrotic cytokine in the heart (38, 119, 151). Direct upregulation of collagen production by TNF-α via activation of WNT1 inducible signaling pathway protein (WISP) 1 has been reported (119, 151). Accordingly, pharmacological inhibition of TNF-α by monoclonal antibodies markedly reduces myocardial collagen I and III content and attenuates cardiac fibrosis in diabetic rats (54). Of note, TNF-α also activates matrix-degenerating programs in fibroblasts, such as the expression of matrix metalloproteinases (MMPs), suggesting that TNF-α mediated fibrosis may partly represent a response to ECM degradation (152). Another fibrogenic inflammatory mediator, upregulated under glucometabolic challenge by NF-κB activation, is IL-6 (14, 50). Abundant evidence indicates profibrotic effects of IL-6, mainly attributed to STAT3-dependent induction of collagen synthesis by cardiac fibroblasts and to enhancement of TGF-β expression (112, 164, 244). Genetic deletion of IL-6 mitigates cardiac fibrosis and dysfunction in diabetic mice (164). The inflammatory cytokine IL-1β is released upon activation of NLRP3/caspase 1 and is present in increased abundance in diabetic hearts (55). Il-1β has been implicated in cardiac fibrosis by exerting indirect profibrotic effects on fibroblasts via generation of ECM fragments and by induction of TGF-ß (112). Inhibition of caspase 1 reduces the biologically active form of IL-1β thereby improving cardiac fibrosis and LV function in diabetic rats (54).
Another process linking metabolic inflammation to myocardial fibrosis is endothelial-to-mesenchymal-transition. When exposed to inflammatory cytokines (e.g., TNF-α, IL-1β, IL-6, IL-13) or oxidized LDL endothelial cells adopt a fibroblast-like phenotype displaying mesenchymal cell morphology and function (245, 246). It has been suggested that endothelial-to-mesenchymal transition represents a general response to intracellular inflammation and may have a major role in cardiac ECM remodeling (245, 246).
AGEs are heterogenous molecules formed in a non-enzymatic reaction between the carbonyl group of a reducing sugar and the amino group of proteins, lipids, and nucleic acids. Chronic hyperglycemia leads to enhanced endogenous production and accumulation of AGEs in the cardiac ECM. Binding of AGEs to their cell surface receptor RAGE that is expressed in cardiomyocytes, fibroblasts, endothelial cells, and cardiac immune cells triggers the activation NF-κB via PI3K/Akt/MAPK (247, 248). The resulting pro-inflammatory state associates with enhanced intracellular ROS generation and alteration of cellular protein function (249). Overall, NF-κB activation by AGE-RAGE interaction leads to enhanced transcription and secretion of TNF-α, IL-1β, IL-2, and IL-6 contributing to the inflammatory milieu in the myocardium of hyperglycemic patients (250).
Several lines of evidence suggests that endocrine actions of pro- and anti-inflammatory adipokines in the systemic circulation along with paracrine effects of the epicardial adipose tissue contribute to myocardial inflammation (251, 252). Adipocyte hypertrophy promotes the secretion of leptin which has been linked to inflammatory effects in the myocardium and cardiomyocyte hypertrophy (14, 30, 253–255). In contrast, plasma levels of anti-inflammatory adiponectin are inversely correlated with body fat mass leading to reduced antagonism of inflammatory pathways in cardiometabolic patients (256). Adiponectin blocks TNF-α mediated activation of NF-κB through a protein kinase (PK)A-dependent mechanism (257). In addition, adiponectin potently stimulates ceramidase activity in cardiomyocytes and enhances ceramide catabolism thereby protecting from lipotoxic damage (258).
Given its anatomical intimacy with the underlying heart muscle and a shared microcirculation, the epicardial adipose tissue (EAT) is a pivotal regulator of myocardial inflammation (255). Unhindered passage of pro- and anti-inflammatory cytokines secreted by the EAT to the neighboring myocardium allows for paracrine interactions (251). At baseline, the EAT protects the myocardium form pro-inflammatory and hypertrophic stimuli through secretion of adiponectin (257–259). Along with growing body fat mass, macrophages are recruited to EAT where they foster local adipose tissue inflammation through upregulation of TNF-α, IL-6, IL-1β, and leptin while blunting the secretion of adiponectin (251, 260). Owed to its close proximity to the heart, EAT amplifies the effects of systemic metabolic disturbances on the myocardium (261). Clinically, EAT expansion correlates with elevated systemic inflammatory markers, increased LV mass index, abnormal coronary microcirculation (262), worsened parameters of diastolic function, and left atrial dilation—all features of the metabolic HFpEF phenotype (263–265). Besides, inflammation-induced invasion of pluripotent stem cells from the EAT to the outer myocardial layer and subsequent conversion to fibroblasts has also been proposed as mechanism of maladaptive myocardial remodeling (251).
Abundant observational data support the clinical relevance of inflammation in myocardial remodeling and HF development in cardiometabolic patients. Obesity and T2D associate with elevated biomarkers of inflammation including hsCRP (136), IL-6 (136), TNF-α (7–9), and other markers of metabolic inflammation such as Leptin (192) and TGF-β (193, 194). High hsCRP, TNF-α, and TGF-β levels increase the susceptibility to cardiac damage in hypertensive patients with the metabolic syndrome, in whom they are independently related to the LV mass index and diastolic LV dysfunction (65). In accordance, elevated TNF-α and IL-6 independently predict incident HFpEF, the predominant type of HF in obesity and diabetes, but not HF with reduced ejection fraction (HFrEF) (266, 267). Subjects with obesity or diabetes account for the majority of the HFpEF patient population in which pathophysiological pathway analyses demonstrated a close link to vascular cell adhesion, leucocyte migration and inflammation (268). In line, among patients with established HF, subjects with HFpEF display higher levels of inflammatory markers than those with HFrEF (268, 269). While the majority of clinical trials on direct anti-inflammatory agents were performed in HFrEF and have had neutral results, two randomized controlled trials on IL-1 blockade in HFpEF patients with high hsCRP levels showed a decrease in NT-proBNP levels and improved exercise performance—holding promise for individualized anti-inflammatory treatment approaches (11–13).
Myocardial remodeling in the setting of obesity and diabetes results from a multifaceted disease process involving metabolic dysregulation and systemic inflammation. While the underlying cellular crosstalk within and beyond the heart remains poorly understood, remarkable overlap in subcellular alterations within the spectrum of glucometabolic disturbances has been reported. Nutrients and pro-inflammatory cytokines are intricately linked to the regulation of inflammatory processes in the heart through conserved signal transduction pathways. Recent advances in the field are shedding light on the interplay between lipid metabolites and immune dysregulation underlining their role as key modulators of myocardial hypertrophy and fibrosis. Disentangling the inflammatory programs involved in adverse myocardial remodeling in cardiometabolic patients and their regulation by systemic mediators may help to identify potential drug targets and personalized approaches in this setting. Overnutrition is on the rise worldwide (270) calling for dedicated research on the myocardial sequelae to decipher molecular pathways and improve clinical outcomes.
FW conceptualized and wrote the manuscript. SA, SM, and SK assisted in drafting the manuscript. TL, SC, and FP revised the manuscript critically and provided important intellectual content. FP conceptualized the manuscript and guided the writing process. All authors have contributed significantly.
This work was supported by the Swiss National Science Foundation (n. 310030_197557), the Swiss Heart Foundation (n. FF19045), the Stiftung für wissenschaftliche Forschung, the Olga Mayenfisch Foundation, the Swiss Life Foundation, the Kurt und Senta-Hermann Stiftung, the EMDO Stiftung and the Schweizerische Diabetes-Stiftung (to FP); the Holcim Foundation and the Swiss Heart Foundation (to SC). SA and SM are the recipients of a Forschungskredit Candoc grant from the University of Zürich. Research of SK and TL was supported by the Swiss Heart Foundation (FF20094, FF19056) and the Foundation of Cardiovascular Research – Zurich Heart House (Donation of H.H. Sheikh Khalifa bin Hamad Al-Thani). SK received funding from the Theodor und Ida Herzog-Egli-Stiftung.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, et al. Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Res Clin Pract. (2019) 157:107843. doi: 10.1016/j.diabres.2019.107843
2. Kelly T, Yang W, Chen CS, Reynolds K, He J. Global burden of obesity in 2005 and projections to 2030. Int J Obes. (2008) 32:1431–7. doi: 10.1038/ijo.2008.102
3. Cavallari I, Bhatt DL, Steg PG, Leiter LA, McGuire DK, Mosenzon O, et al. Causes and risk factors for death in diabetes: a competing-risk analysis from the SAVOR-TIMI 53 trial. J Am Coll Cardiol. (2021) 77:1837–40. doi: 10.1016/j.jacc.2021.02.030
4. Bhaskaran K, Dos-Santos-Silva I, Leon DA, Douglas IJ, Smeeth L. Association of BMI with overall and cause-specific mortality: a population-based cohort study of 3.6 million adults in the UK. Lancet Diabetes Endocrinol. (2018) 6:944–53. doi: 10.1016/S2213-8587(18)30288-2
5. Sanchis-Gomar F, Perez-Quilis C, Leischik R, Lucia A. Epidemiology of coronary heart disease and acute coronary syndrome. Ann Transl Med. (2016) 4:256. doi: 10.21037/atm.2016.06.33
6. Mills KT, Stefanescu A, He J. The global epidemiology of hypertension. Nat Rev Nephrol. (2020) 16:223–37. doi: 10.1038/s41581-019-0244-2
7. Mishra S, Kass DA. Cellular and molecular pathobiology of heart failure with preserved ejection fraction. Nat Rev Cardiol. (2021) 18:400–23. doi: 10.1038/s41569-020-00480-6
8. Nabipour I, Vahdat K, Jafari SM, Beigi S, Assadi M, Azizi F, et al. Elevated high sensitivity C-reactive protein is associated with type 2 diabetes mellitus: the Persian Gulf Healthy Heart Study. Endocr J. (2008) 55:717–22. doi: 10.1507/endocrj.K08E-026
9. Visser M, Bouter LM, McQuillan GM, Wener MH, Harris TB. Elevated C-reactive protein levels in overweight and obese adults. J Am Med Assoc. (1999) 282:2131–5. doi: 10.1001/jama.282.22.2131
10. Hahn VS, Knutsdottir H, Luo X, Bedi K, Margulies KB, Haldar SM, et al. Myocardial gene expression signatures in human heart failure with preserved ejection fraction. Circulation. (2021) 143:120–34. doi: 10.1161/CIRCULATIONAHA.120.050498
11. Van Tassell BW, Arena R, Biondi-Zoccai G, Canada JM, Oddi C, Abouzaki NA, et al. Effects of interleukin-1 blockade with anakinra on aerobic exercise capacity in patients with heart failure and preserved ejection fraction (from the D-HART pilot study). Am J Cardiol. (2014) 113:321–7. doi: 10.1016/j.amjcard.2013.08.047
12. Van Tassell BW, Trankle CR, Canada JM, Carbone S, Buckley L, Kadariya D, et al. IL-1 blockade in patients with heart failure with preserved ejection fraction. Circ Heart Fail. (2018) 11:e005036. doi: 10.1161/CIRCHEARTFAILURE.118.005036
13. Murphy SP, Kakkar R, McCarthy CP, Januzzi JL Jr. Inflammation in heart failure: JACC state-of-the-art review. J Am Coll Cardiol. (2020) 75:1324–40. doi: 10.1016/j.jacc.2020.01.014
14. Nishida K, Otsu K. Inflammation and metabolic cardiomyopathy. Cardiovasc Res. (2017) 113:389–98. doi: 10.1093/cvr/cvx012
15. Murphy E, Amanakis G, Fillmore N, Parks RJ, Sun J. Sex differences in metabolic cardiomyopathy. Cardiovasc Res. (2017) 113:370–7. doi: 10.1093/cvr/cvx008
16. Costantino S, Akhmedov A, Melina G, Mohammed SA, Othman A, Ambrosini S, et al. Obesity-induced activation of JunD promotes myocardial lipid accumulation and metabolic cardiomyopathy. Eur Heart J. (2019) 40:997–1008. doi: 10.1093/eurheartj/ehy903
17. Schulze PC, Drosatos K, Goldberg IJ. Lipid use and misuse by the heart. Circ Res. (2016) 118:1736–51. doi: 10.1161/CIRCRESAHA.116.306842
18. Kosmala W, Sanders P, Marwick TH. Subclinical myocardial impairment in metabolic diseases. JACC Cardiovasc Imaging. (2017) 10:692–703. doi: 10.1016/j.jcmg.2017.04.001
19. Grundy SM. Pre-diabetes, metabolic syndrome, and cardiovascular risk. J Am Coll Cardiol. (2012) 59:635–43. doi: 10.1016/j.jacc.2011.08.080
20. Schannwell CM, Schneppenheim M, Perings S, Plehn G, Strauer BE. Left ventricular diastolic dysfunction as an early manifestation of diabetic cardiomyopathy. Cardiology. (2002) 98:33–9. doi: 10.1159/000064682
21. Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res. (2018) 122:624–38. doi: 10.1161/CIRCRESAHA.117.311586
22. McKee PA, Castelli WP, McNamara PM, Kannel WB. The natural history of congestive heart failure: the Framingham study. N Engl J Med. (1971) 285:1441–6. doi: 10.1056/NEJM197112232852601
23. Ho KK, Pinsky JL, Kannel WB, Levy D. The epidemiology of heart failure: the Framingham Study. J Am Coll Cardiol. (1993) 22(4Suppl.A):6A–13A. doi: 10.1016/0735-1097(93)90455-A
24. Kenchaiah S, Evans JC, Levy D, Wilson PW, Benjamin EJ, Larson MG, et al. Obesity and the risk of heart failure. N Engl J Med. (2002) 347:305–13. doi: 10.1056/NEJMoa020245
25. Schiattarella GG, Rodolico D, Hill JA. Metabolic inflammation in heart failure with preserved ejection fraction. Cardiovasc Res. (2021) 117:423–34. doi: 10.1093/cvr/cvaa217
26. Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med. (2012) 18:363–74. doi: 10.1038/nm.2627
27. Yin MJ, Yamamoto Y, Gaynor RB. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature. (1998) 396:77–80. doi: 10.1038/23948
28. Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, et al. Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science. (2001) 293:1673–7. doi: 10.1126/science.1061620
29. Mandavia CH, Aroor AR, Demarco VG, Sowers JR. Molecular and metabolic mechanisms of cardiac dysfunction in diabetes. Life Sci. (2013) 92:601–8. doi: 10.1016/j.lfs.2012.10.028
30. Blüher M. Adipose tissue inflammation: a cause or consequence of obesity-related insulin resistance? Clin Sci (Lond). (2016) 130:1603–14. doi: 10.1042/CS20160005
31. Li P, Oh DY, Bandyopadhyay G, Lagakos WS, Talukdar S, Osborn O, et al. LTB4 promotes insulin resistance in obese mice by acting on macrophages, hepatocytes and myocytes. Nat Med. (2015) 21:239–47. doi: 10.1038/nm.3800
32. Castoldi A, Naffah de Souza C, Câmara NO, Moraes-Vieira PM. The macrophage switch in obesity development. Front Immunol. (2016) 6:637. doi: 10.3389/fimmu.2015.00637
33. O'Rourke RW, Metcalf MD, White AE, Madala A, Winters BR, Maizlin II, et al. Depot-specific differences in inflammatory mediators and a role for NK cells and IFN-gamma in inflammation in human adipose tissue. Int J Obes. (2009) 33:978–90. doi: 10.1038/ijo.2009.133
34. Gastaldelli A, Miyazaki Y, Pettiti M, Matsuda M, Mahankali S, Santini E, et al. Metabolic effects of visceral fat accumulation in type 2 diabetes. J Clin Endocrinol Metab. (2002) 87:5098–103. doi: 10.1210/jc.2002-020696
35. Yokoyama T, Nakano M, Bednarczyk JL, McIntyre BW, Entman M, Mann DL. Tumor necrosis factor-alpha provokes a hypertrophic growth response in adult cardiac myocytes. Circulation. (1997) 95:1247–52. doi: 10.1161/01.CIR.95.5.1247
36. Krown KA, Page MT, Nguyen C, Zechner D, Gutierrez V, Comstock KL, et al. Tumor necrosis factor alpha-induced apoptosis in cardiac myocytes. Involvement of the sphingolipid signaling cascade in cardiac cell death. J Clin Invest. (1996) 98:2854–65. doi: 10.1172/JCI119114
37. Mann DL. Innate immunity and the failing heart: the cytokine hypothesis revisited. Circ Res. (2015) 116:1254–68. doi: 10.1161/CIRCRESAHA.116.302317
38. Frati G, Schirone L, Chimenti I, Yee D, Biondi-Zoccai G, Volpe M, et al. An overview of the inflammatory signalling mechanisms in the myocardium underlying the development of diabetic cardiomyopathy. Cardiovasc Res. (2017) 113:378–88. doi: 10.1093/cvr/cvx011
39. Nan WQ, Shan TQ, Qian X, Ping W, Bing GA, Ying LL. PPARα agonist prevented the apoptosis induced by glucose and fatty acid in neonatal cardiomyocytes. J Endocrinol Invest. (2011) 34:271–5. doi: 10.1007/BF03347084
40. Min W, Bin ZW, Quan ZB, Hui ZJ, Sheng FG. The signal transduction pathway of PKC/NF-kappa B/c-fos may be involved in the influence of high glucose on the cardiomyocytes of neonatal rats. Cardiovasc Diabetol. (2009) 8:8. doi: 10.1186/1475-2840-8-8
41. Volz HC, Seidel C, Laohachewin D, Kaya Z, Müller OJ, Pleger ST, et al. HMGB1: the missing link between diabetes mellitus and heart failure. Basic Res Cardiol. (2010) 105:805–20. doi: 10.1007/s00395-010-0114-3
42. Zachara NE. The roles of O-linked β-N-acetylglucosamine in cardiovascular physiology and disease. Am J Physiol Heart Circ Physiol. (2012) 302:H1905–18. doi: 10.1152/ajpheart.00445.2011
43. Sulaiman M, Matta MJ, Sunderesan NR, Gupta MP, Periasamy M, Gupta M. Resveratrol, an activator of SIRT1, upregulates sarcoplasmic calcium ATPase and improves cardiac function in diabetic cardiomyopathy. Am J Physiol Heart Circ Physiol. (2010) 298:H833–43. doi: 10.1152/ajpheart.00418.2009
44. Planavila A, Iglesias R, Giralt M, Villarroya F. Sirt1 acts in association with PPARα to protect the heart from hypertrophy, metabolic dysregulation, and inflammation. Cardiovasc Res. (2011) 90:276–84. doi: 10.1093/cvr/cvq376
45. Pan Y, Wang Y, Zhao Y, Peng K, Li W, Wang Y, et al. Inhibition of JNK phosphorylation by a novel curcumin analog prevents high glucose-induced inflammation and apoptosis in cardiomyocytes and the development of diabetic cardiomyopathy. Diabetes. (2014) 63:3497–511. doi: 10.2337/db13-1577
46. Wang Y, Feng W, Xue W, Tan Y, Hein DW Li XK, Cai L. Inactivation of GSK-3beta by metallothionein prevents diabetes-related changes in cardiac energy metabolism, inflammation, nitrosative damage, and remodeling. Diabetes. (2009) 58:1391–402. doi: 10.2337/db08-1697
47. Jia G, Habibi J, Bostick BP, Ma L, DeMarco VG, Aroor AR, et al. Uric acid promotes left ventricular diastolic dysfunction in mice fed a Western diet. Hypertension. (2015) 65:531–9. doi: 10.1161/HYPERTENSIONAHA.114.04737
48. Sharp TE 3rd, Scarborough AL, Li Z, Polhemus DJ, Hidalgo HA, Schumacher JD, et al. Novel Göttingen Miniswine model of heart failure with preserved ejection fraction integrating multiple comorbidities. JACC Basic Transl Sci. (2021) 6:154–70. doi: 10.1016/j.jacbts.2020.11.012
49. Ko HJ, Zhang Z, Jung DY, Jun JY, Ma Z, Jones KE, et al. Nutrient stress activates inflammation and reduces glucose metabolism by suppressing AMP-activated protein kinase in the heart. Diabetes. (2009) 58:2536–46. doi: 10.2337/db08-1361
50. Tikellis C, Thomas MC, Harcourt BE, Coughlan MT, Pete J, Bialkowski K, et al. Cardiac inflammation associated with a Western diet is mediated via activation of RAGE by AGEs. Am J Physiol Endocrinol Metab. (2008) 295:E323–30. doi: 10.1152/ajpendo.00024.2008
51. Urbina P, Singla DK. BMP-7 attenuates adverse cardiac remodeling mediated through M2 macrophages in prediabetic cardiomyopathy. Am J Physiol Heart Circ Physiol. (2014) 307:H762–72. doi: 10.1152/ajpheart.00367.2014
52. Fukuda M, Nakamura T, Kataoka K, Nako H, Tokutomi Y, Dong YF, et al. Potentiation by candesartan of protective effects of pioglitazone against type 2 diabetic cardiovascular and renal complications in obese mice. J Hypertens. (2010) 28:340–52. doi: 10.1097/HJH.0b013e32833366cd
53. Fukuda M, Nakamura T, Kataoka K, Nako H, Tokutomi Y, Dong YF, et al. Ezetimibe ameliorates cardiovascular complications and hepatic steatosis in obese and type 2 diabetic db/db mice. J Pharmacol Exp Ther. (2010) 335:70–5. doi: 10.1124/jpet.110.170373
54. Westermann D, Van Linthout S, Dhayat S, Dhayat N, Escher F, Bücker-Gärtner C, et al. Cardioprotective and anti-inflammatory effects of interleukin converting enzyme inhibition in experimental diabetic cardiomyopathy. Diabetes. (2007) 56:1834–41. doi: 10.2337/db06-1662
55. Tschöpe C, Walther T, Escher F, Spillmann F, Du J, Altmann C, et al. Transgenic activation of the kallikrein-kinin system inhibits intramyocardial inflammation, endothelial dysfunction and oxidative stress in experimental diabetic cardiomyopathy. FASEB J. (2005) 19:2057–9. doi: 10.1096/fj.05-4095fje
56. Schilling JD, Machkovech HM, Kim AH, Schwendener R, Schaffer JE. Macrophages modulate cardiac function in lipotoxic cardiomyopathy. Am J Physiol Heart Circ Physiol. (2012) 303:H1366–73. doi: 10.1152/ajpheart.00111.2012
57. Franssen C, Chen S, Unger A, Korkmaz HI, De Keulenaer GW, Tschöpe C, et al. Myocardial microvascular inflammatory endothelial activation in heart failure with preserved ejection fraction. JACC Heart Fail. (2016) 4:312–24. doi: 10.1016/j.jchf.2015.10.007
58. McArdle MA, Finucane OM, Connaughton RM, McMorrow AM, Roche HM. Mechanisms of obesity-induced inflammation and insulin resistance: insights into the emerging role of nutritional strategies. Front Endocrinol (Lausanne). (2013) 4:52. doi: 10.3389/fendo.2013.00052
59. Lee JY, Sohn KH, Rhee SH, Hwang D. Saturated fatty acids, but not unsaturated fatty acids, induce the expression of cyclooxygenase-2 mediated through Toll-like receptor 4. J Biol Chem. (2001) 276:16683–9. doi: 10.1074/jbc.M011695200
60. Nguyen MT, Favelyukis S, Nguyen AK, Reichart D, Scott PA, Jenn A, et al. A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J Biol Chem. (2007) 282:35279–92. doi: 10.1074/jbc.M706762200
61. Lancaster GI, Langley KG, Berglund NA, Kammoun HL, Reibe S, Estevez E, et al. Evidence that TLR4 is not a receptor for saturated fatty acids but mediates lipid-induced inflammation by reprogramming macrophage metabolism. Cell Metab. (2018) 27:1096–110.e5. doi: 10.1016/j.cmet.2018.03.014
62. Packer M, Kitzman DW. Obesity-related heart failure with a preserved ejection fraction: the mechanistic rationale for combining inhibitors of aldosterone, neprilysin, and sodium-glucose cotransporter-2. JACC Heart Fail. (2018) 6:633–9. doi: 10.1016/j.jchf.2018.01.009
63. Brown NJ. Contribution of aldosterone to cardiovascular and renal inflammation and fibrosis. Nat Rev Nephrol. (2013) 9:459–69. doi: 10.1038/nrneph.2013.110
64. Cassis LA, Police SB, Yiannikouris F, Thatcher SE. Local adipose tissue renin-angiotensin system. Curr Hypertens Rep. (2008) 10:93–8. doi: 10.1007/s11906-008-0019-9
65. Sciarretta S, Paneni F, Palano F, Chin D, Tocci G, Rubattu S, et al. Role of the renin-angiotensin-aldosterone system and inflammatory processes in the development and progression of diastolic dysfunction. Clin Sci. (2009) 116:467–77. doi: 10.1042/CS20080390
66. Pueyo ME, Gonzalez W, Nicoletti A, Savoie F, Arnal JF, Michel JB. Angiotensin II stimulates endothelial vascular cell adhesion molecule-1 via nuclear factor-kappaB activation induced by intracellular oxidative stress. Arterioscler Thromb Vasc Biol. (2000) 20:645–51. doi: 10.1161/01.ATV.20.3.645
67. Schinzari F, Tesauro M, Veneziani A, Mores N, Di Daniele N, Cardillo C. Favorable vascular actions of angiotensin-(1-7) in human obesity. Hypertension. (2018) 71:185–91. doi: 10.1161/HYPERTENSIONAHA.117.10280
68. Simões e Silva AC, Silveira KD, Ferreira AJ, Teixeira MM. ACE2, angiotensin-(1-7) and Mas receptor axis in inflammation and fibrosis. Br J Pharmacol. (2013) 169:477–92. doi: 10.1111/bph.12159
69. Mouton AJ Li X, Hall ME, Hall JE. Obesity, hypertension, and cardiac dysfunction: novel roles of immunometabolism in macrophage activation and inflammation. Circ Res. (2020) 126:789–806. doi: 10.1161/CIRCRESAHA.119.312321
70. Jin X, Yao T, Zhou Z, Zhu J, Zhang S, Hu W, et al. Advanced glycation end products enhance macrophages polarization into M1 phenotype through activating RAGE/NF-κB pathway. Biomed Res Int. (2015) 2015:732450. doi: 10.1155/2015/732450
71. Hue L, Taegtmeyer H. The Randle cycle revisited: a new head for an old hat. Am J Physiol Endocrinol Metab. (2009) 297:E578–91. doi: 10.1152/ajpendo.00093.2009
72. Serrano-Marco L, Rodríguez-Calvo R, El Kochairi I, Palomer X, Michalik L, Wahli W, et al. Activation of peroxisome proliferator-activated receptor-β/-δ (PPAR-β/-δ) ameliorates insulin signaling and reduces SOCS3 levels by inhibiting STAT3 in interleukin-6-stimulated adipocytes. Diabetes. (2011) 60:1990–9. doi: 10.2337/db10-0704
73. Pulakat L, DeMarco VG, Ardhanari S, Chockalingam A, Gul R, Whaley-Connell A, et al. Adaptive mechanisms to compensate for overnutrition-induced cardiovascular abnormalities. Am J Physiol Regul Integr Comp Physiol. (2011) 301:R885–95. doi: 10.1152/ajpregu.00316.2011
74. Han S, Sidell N. Peroxisome-proliferator-activated-receptor gamma (PPARgamma) independent induction of CD36 in THP-1 monocytes by retinoic acid. Immunology. (2002) 106:53–9. doi: 10.1046/j.1365-2567.2002.01404.x
75. Herrero P, Peterson LR, McGill JB, Matthew S, Lesniak D, Dence C, et al. Increased myocardial fatty acid metabolism in patients with type 1 diabetes mellitus. J Am Coll Cardiol. (2006) 47:598–604. doi: 10.1016/j.jacc.2005.09.030
76. McGavock JM, Lingvay I, Zib I, Tillery T, Salas N, Unger R, et al. Cardiac steatosis in diabetes mellitus: a 1H-magnetic resonance spectroscopy study. Circulation. (2007) 116:1170–5. doi: 10.1161/CIRCULATIONAHA.106.645614
77. Alavaikko M, Elfving R, Hirvonen J, Järvi J. Triglycerides, cholesterol, and phospholipids in normal heart papillary muscle and in patients suffering from diabetes, cholelithiasis, hypertension, and coronary atheroma. J Clin Pathol. (1973) 26:285–93. doi: 10.1136/jcp.26.4.285
78. Rijzewijk LJ, van der Meer RW, Smit JW, Diamant M, Bax JJ, Hammer S, et al. Myocardial steatosis is an independent predictor of diastolic dysfunction in type 2 diabetes mellitus. J Am Coll Cardiol. (2008) 52:1793–9. doi: 10.1016/j.jacc.2008.07.062
79. Listenberger LL, Ory DS, Schaffer JE. Palmitate-induced apoptosis can occur through a ceramide-independent pathway. J Biol Chem. (2001) 276:14890–5. doi: 10.1074/jbc.M010286200
80. Marfella R, Di Filippo C, Portoghese M, Barbieri M, Ferraraccio F, Siniscalchi M, et al. Myocardial lipid accumulation in patients with pressure-overloaded heart and metabolic syndrome. J Lipid Res. (2009) 50:2314–23. doi: 10.1194/jlr.P900032-JLR200
81. Hickson-Bick DLM, Buja ML, McMillin JB. Palmitate-mediated alterations in the fatty acid metabolism of rat neonatal cardiac myocytes. J Mol Cell Cardiol. (2000) 32:511–9. doi: 10.1006/jmcc.1999.1098
82. Sparagna GC, Hickson-Bick DL, Buja LM, McMillin JB A. metabolic role for mitochondria in palmitate-induced cardiac myocyte apoptosis. Am J Physiol Heart Circ Physiol. (2000) 279:H2124–32. doi: 10.1152/ajpheart.2000.279.5.H2124
83. Park SY, Cho YR, Kim HJ, Higashimori T, Danton C, Lee MK, et al. Unraveling the temporal pattern of diet-induced insulin resistance in individual organs and cardiac dysfunction in C57BL/6 mice. Diabetes. (2005) 54:3530–40. doi: 10.2337/diabetes.54.12.3530
84. Kaludercic N, Di Lisa F. Mitochondrial ROS formation in the pathogenesis of diabetic cardiomyopathy. Front Cardiovasc Med. (2020) 7:12. doi: 10.3389/fcvm.2020.00012
85. Baker RG, Hayden MS, Ghosh S. NF-κB, inflammation, and metabolic disease. Cell Metab. (2011) 13:11–22. doi: 10.1016/j.cmet.2010.12.008
86. Gordon JW, Shaw JA, Kirshenbaum LA. Multiple facets of NF-κB in the heart: to be or not to NF-κB. Circ Res. (2011) 108:1122–32. doi: 10.1161/CIRCRESAHA.110.226928
87. Shah MS, Brownlee M. Molecular and cellular mechanisms of cardiovascular disorders in diabetes. Circ Res. (2016) 118:1808–29. doi: 10.1161/CIRCRESAHA.116.306923
88. Tan Y, Ichikawa T, Li J, Si Q, Yang H, Chen X, et al. Diabetic downregulation of Nrf2 activity via ERK contributes to oxidative stress-induced insulin resistance in cardiac cells in vitro and in vivo. Diabetes. (2011) 60:625–33. doi: 10.2337/db10-1164
89. Cong W, Ruan D, Xuan Y, Niu C, Tao Y, Wang Y, et al. Cardiac-specific overexpression of catalase prevents diabetes-induced pathological changes by inhibiting NF-κB signaling activation in the heart. J Mol Cell Cardiol. (2015) 89:314–25. doi: 10.1016/j.yjmcc.2015.10.010
90. Thomas CM, Yong QC, Rosa RM, Seqqat R, Gopal S, Casarini DE, et al. Cardiac-specific suppression of NF-κB signaling prevents diabetic cardiomyopathy via inhibition of the renin-angiotensin system. Am J Physiol Heart Circ Physiol. (2014) 307:H1036–45. doi: 10.1152/ajpheart.00340.2014
91. Yu XY, Geng YJ, Liang JL, Zhang S, Lei HP, Zhong SL, et al. High levels of glucose induce “metabolic memory” in cardiomyocyte via epigenetic histone H3 lysine 9 methylation. Mol Biol Rep. (2012) 39:8891–8. doi: 10.1007/s11033-012-1756-z
92. Yang WH, Park SY, Nam HW, Kim DH, Kang JG, Kang ES, et al. NFkappaB activation is associated with its O-GlcNAcylation state under hyperglycemic conditions. Proc Natl Acad Sci USA. (2008) 105:17345–50. doi: 10.1073/pnas.0806198105
93. El-Osta A, Brasacchio D, Yao D, Pocai A, Jones PL, Roeder RG, et al. Transient high glucose causes persistent epigenetic changes and altered gene expression during subsequent normoglycemia. J Exp Med. (2008) 205:2409–17. doi: 10.1084/jem.20081188
94. Alvarez-Guardia D, Palomer X, Coll T, Serrano L, Rodríguez-Calvo R, Davidson MM, et al. PPARβ/δ activation blocks lipid-induced inflammatory pathways in mouse heart and human cardiac cells. Biochim Biophys Acta. (2011) 1811:59–67. doi: 10.1016/j.bbalip.2010.11.002
95. Li M, van Esch BCAM, Wagenaar GTM, Garssen J, Folkerts G, Henricks PAJ. Pro- and anti-inflammatory effects of short chain fatty acids on immune and endothelial cells. Eur J Pharmacol. (2018) 831:52–9. doi: 10.1016/j.ejphar.2018.05.003
96. Dalal PJ, Muller WA, Sullivan DP. Endothelial cell calcium signaling during barrier function and inflammation. Am J Pathol. (2020) 190:535–42. doi: 10.1016/j.ajpath.2019.11.004
97. Busse R, Fleming I. Vascular endothelium and blood flow. Handb Exp Pharmacol. (2006) 176:43–78. doi: 10.1007/3-540-36028-X_2
98. Janus A, Szahidewicz-Krupska E, Mazur G, Doroszko A. Insulin resistance and endothelial dysfunction constitute a common therapeutic target in cardiometabolic disorders. Mediators Inflamm. (2016) 2016:3634948. doi: 10.1155/2016/3634948
99. van den Oever IA, Raterman HG, Nurmohamed MT, Simsek S. Endothelial dysfunction, inflammation, and apoptosis in diabetes mellitus. Mediators Inflamm. (2010) 2010:792393. doi: 10.1155/2010/792393
100. Kim F, Tysseling KA, Rice J, Gallis B, Haji L, Giachelli CM, et al. Activation of IKKbeta by glucose is necessary and sufficient to impair insulin signaling and nitric oxide production in endothelial cells. J Mol Cell Cardiol. (2005) 39:327–34. doi: 10.1016/j.yjmcc.2005.05.009
101. Sweet IR, Gilbert M, Maloney E, Hockenbery DM, Schwartz MW, Kim F. Endothelial inflammation induced by excess glucose is associated with cytosolic glucose 6-phosphate but not increased mitochondrial respiration. Diabetologia. (2009) 52:921–31. doi: 10.1007/s00125-009-1272-4
102. Chen Y, Wang L, Pitzer AL, Li X, Li PL, Zhang Y. Contribution of redox-dependent activation of endothelial Nlrp3 inflammasomes to hyperglycemia-induced endothelial dysfunction. J Mol Med (Berl). (2016) 94:1335–47. doi: 10.1007/s00109-016-1481-5
103. Paolisso P, Foà A, Bergamaschi L, Donati F, Fabrizio M, Chiti C, et al. Hyperglycemia, inflammatory response and infarct size in obstructive acute myocardial infarction and MINOCA. Cardiovasc Diabetol. (2021) 20:33. doi: 10.1186/s12933-021-01222-9
104. D'Onofrio N, Sardu C, Paolisso P, Minicucci F, Gragnano F, Ferraraccio F, et al. MicroRNA-33 and SIRT1 influence the coronary thrombus burden in hyperglycemic STEMI patients. J Cell Physiol. (2020) 235:1438–52. doi: 10.1002/jcp.29064
105. Ghosh A, Gao L, Thakur A, Siu PM, Lai CWK. Role of free fatty acids in endothelial dysfunction. J Biomed Sci. (2017) 24:50. doi: 10.1186/s12929-017-0357-5
106. Kim F, Pham M, Luttrell I, Bannerman DD, Tupper J, Thaler J, et al. Toll-like receptor-4 mediates vascular inflammation and insulin resistance in diet-induced obesity. Circ Res. (2007) 100:1589–96. doi: 10.1161/CIRCRESAHA.106.142851
107. Li H, Li H, Bao Y, Zhang X, Yu Y. Free fatty acids induce endothelial dysfunction and activate protein kinase C and nuclear factor-κB pathway in rat aorta. Int J Cardiol. (2011) 152:218–24. doi: 10.1016/j.ijcard.2010.07.019
108. Iwata NG, Pham M, Rizzo NO, Cheng AM, Maloney E, Kim F. Trans fatty acids induce vascular inflammation and reduce vascular nitric oxide production in endothelial cells. PLoS ONE. (2011) 6:e29600. doi: 10.1371/journal.pone.0029600
109. Toborek M, Lee YW, Garrido R, Kaiser S, Hennig B. Unsaturated fatty acids selectively induce an inflammatory environment in human endothelial cells. Am J Clin Nutr. (2002) 75:119–25. doi: 10.1093/ajcn/75.1.119
110. Hasegawa Y, Saito T, Ogihara T, Ishigaki Y, Yamada T, Imai J, et al. Blockade of the nuclear factor-κB pathway in the endothelium prevents insulin resistance and prolongs life spans. Circulation. (2012) 125:1122–33. doi: 10.1161/CIRCULATIONAHA.111.054346
111. Xing JH Li R, Gao YQ, Wang MY, Liu YZ, Hong J, et al. NLRP3 inflammasome mediate palmitate-induced endothelial dysfunction. Life Sci. (2019) 239:116882. doi: 10.1016/j.lfs.2019.116882
112. Frangogiannis NG. Cardiac fibrosis. Cardiovasc Res. (2021) 117:1450–88. doi: 10.1093/cvr/cvaa324
113. Russo I, Frangogiannis NG. Diabetes-associated cardiac fibrosis: cellular effectors, molecular mechanisms and therapeutic opportunities. J Mol Cell Cardiol. (2016) 90:84–93. doi: 10.1016/j.yjmcc.2015.12.011
114. Han DC, Isono M, Hoffman BB, Ziyadeh FN. High glucose stimulates proliferation and collagen type I synthesis in renal cortical fibroblasts: mediation by autocrine activation of TGF-beta. J Am Soc Nephrol. (1999) 10:1891–9. doi: 10.1681/ASN.V1091891
115. Shamhart PE, Luther DJ, Adapala RK, Bryant JE, Petersen KA, Meszaros JG, et al. Hyperglycemia enhances function and differentiation of adult rat cardiac fibroblasts. Can J Physiol Pharmacol. (2014)92:598–604.
116. Aguilar H, Fricovsky E, Ihm S, Schimke M, Maya-Ramos L, Aroonsakool N, et al. Role for high-glucose-induced protein O-GlcNAcylation in stimulating cardiac fibroblast collagen synthesis. Am J Physiol Cell Physiol. (2014) 306:C794–804. doi: 10.1152/ajpcell.00251.2013
117. Che H, Wang Y, Li Y, Lv J, Li H, Liu Y, et al. Inhibition of microRNA-150-5p alleviates cardiac inflammation and fibrosis via targeting Smad7 in high glucose-treated cardiac fibroblasts. J Cell Physiol. (2020) 235:7769–79. doi: 10.1002/jcp.29386
118. Levick SP, Widiapradja A. The diabetic cardiac fibroblast: mechanisms underlying phenotype and function. Int J Mol Sci. (2020) 21:970. doi: 10.3390/ijms21030970
119. Venkatachalam K, Mummidi S, Cortez DM, Prabhu SD, Valente AJ, Chandrasekar B. Resveratrol inhibits high glucose-induced PI3K/Akt/ERK-dependent interleukin-17 expression in primary mouse cardiac fibroblasts. Am J Physiol Heart Circ Physiol. (2008) 294:H2078–87. doi: 10.1152/ajpheart.01363.2007
120. Tang M, Zhang W, Lin H, Jiang H, Dai H, Zhang Y. High glucose promotes the production of collagen types I and III by cardiac fibroblasts through a pathway dependent on extracellular-signal-regulated kinase 1/2. Mol Cell Biochem. (2007) 301:109–14. doi: 10.1007/s11010-006-9401-6
121. Zhang X, Stewart JA Jr, Kane ID, Massey EP, Cashatt DO, Carver WE. Effects of elevated glucose levels on interactions of cardiac fibroblasts with the extracellular matrix. In vitro Cell Dev Biol Anim. (2007) 43:297–305. doi: 10.1007/s11626-007-9052-2
122. Wu H, Li GN, Xie J, Li R, Chen QH, Chen JZ, et al. Resveratrol ameliorates myocardial fibrosis by inhibiting ROS/ERK/TGF-β/periostin pathway in STZ-induced diabetic mice. BMC Cardiovasc Disord. (2016) 16:5. doi: 10.1186/s12872-015-0169-z
123. Alex L, Russo I, Holoborodko V, Frangogiannis NG. Characterization of a mouse model of obesity-related fibrotic cardiomyopathy that recapitulates features of human heart failure with preserved ejection fraction. Am J Physiol Heart Circ Physiol. (2018) 315:H934–49. doi: 10.1152/ajpheart.00238.2018
124. Lafuse WP, Wozniak DJ, Rajaram MVS. Role of cardiac macrophages on cardiac inflammation, fibrosis and tissue repair. Cells. (2020) 10:51. doi: 10.3390/cells10010051
125. Hulsmans M, Sam F, Nahrendorf M. Monocyte and macrophage contributions to cardiac remodeling. J Mol Cell Cardiol. (2016) 93:149–55. doi: 10.1016/j.yjmcc.2015.11.015
126. Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. (2006) 116:3015–25. doi: 10.1172/JCI28898
127. reemerman AJ, Johnson AR, Sacks GN, Milner JJ, Kirk EL, Troester MA, et al. Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J Biol Chem. (2014) 289:7884–96. doi: 10.1074/jbc.M113.522037
128. Watanabe R, Hilhorst M, Zhang H, Zeisbrich M, Berry GJ, Wallis BB, et al. Glucose metabolism controls disease-specific signatures of macrophage effector functions. JCI Insight. (2018) 3:e123047. doi: 10.1172/jci.insight.123047
129. Geeraerts X, Bolli E, Fendt SM, Van Ginderachter JA. Macrophage metabolism as therapeutic target for cancer, atherosclerosis, and obesity. Front Immunol. (2017) 8:289. doi: 10.3389/fimmu.2017.00289
130. Boden G. Obesity and free fatty acids. Endocrinol Metab Clin North Am. (2008) 37:635–46. doi: 10.1016/j.ecl.2008.06.007
131. Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, et al. Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol. (2011) 12:408–15. doi: 10.1038/ni.2022
132. Namgaladze D, Brüne B. Macrophage fatty acid oxidation and its roles in macrophage polarization and fatty acid-induced inflammation. Biochim Biophys Acta. (2016) 1861:1796–807. doi: 10.1016/j.bbalip.2016.09.002
133. Camell CD, Nguyen KY, Jurczak MJ, Christian BE, Shulman GI, Shadel GS, et al. Macrophage-specific de novo synthesis of ceramide is dispensable for inflammasome-driven inflammation and insulin resistance in obesity. J Biol Chem. (2015) 290:29402–13. doi: 10.1074/jbc.M115.680199
134. Chen L, Chen R, Wang H, Liang F. Mechanisms linking inflammation to insulin resistance. Int J Endocrinol. (2015) 2015:508409. doi: 10.1155/2015/508409
135. Frieler RA, Mortensen RM. Immune cell and other noncardiomyocyte regulation of cardiac hypertrophy and remodeling. Circulation. (2015) 131:1019–30. doi: 10.1161/CIRCULATIONAHA.114.008788
136. Fantuzzi G. Adipose tissue, adipokines, and inflammation. J Allergy Clin Immunol. (2005) 115:911–9. doi: 10.1016/j.jaci.2005.02.023
137. Dhingra S, Sharma AK, Arora RC, Slezak J, Singal PK. IL-10 attenuates TNF-alpha-induced NF kappaB pathway activation and cardiomyocyte apoptosis. Cardiovasc Res. (2009) 82:59–66. doi: 10.1093/cvr/cvp040
138. Freund C, Schmidt-Ullrich R, Baurand A, Dunger S, Schneider W, Loser P, et al. Requirement of nuclear factor-kappaB in angiotensin II- and isoproterenol-induced cardiac hypertrophy in vivo. Circulation. (2005) 111:2319–25. doi: 10.1161/01.CIR.0000164237.58200.5A
139. Mann DL. Inflammatory mediators and the failing heart: past, present, and the foreseeable future. Circ Res. (2002) 91:988–98. doi: 10.1161/01.RES.0000043825.01705.1B
140. Kubota T, McTiernan CF, Frye CS, Slawson SE, Lemster BH, Koretsky AP, et al. Dilated cardiomyopathy in transgenic mice with cardiac-specific overexpression of tumor necrosis factor-alpha. Circ Res. (1997) 81:627–35. doi: 10.1161/01.RES.81.4.627
141. Palmer JN, Hartogensis WE, Patten M, Fortuin FD, Long CS. Interleukin-1 beta induces cardiac myocyte growth but inhibits cardiac fibroblast proliferation in culture. J Clin Invest. (1995) 95:2555–64. doi: 10.1172/JCI117956
142. Nakamura K, Fushimi K, Kouchi H, Mihara K, Miyazaki M, Ohe T, et al. Inhibitory effects of antioxidants on neonatal rat cardiac myocyte hypertrophy induced by tumor necrosis factor-alpha and angiotensin II. Circulation. (1998) 98:794–9. doi: 10.1161/01.CIR.98.8.794
143. Finkel MS, Oddis CV, Jacob TD, Watkins SC, Hattler BG, Simmons RL. Negative inotropic effects of cytokines on the heart mediated by nitric oxide. Science. (1992) 257:387–9. doi: 10.1126/science.1631560
144. Haudek SB, Taffet GE, Schneider MD, Mann DL, TNF. provokes cardiomyocyte apoptosis and cardiac remodeling through activation of multiple cell death pathways. J Clin Invest. (2007) 117:2692–701. doi: 10.1172/JCI29134
145. Kindle L, Rothe L, Kriss M, Osdoby P, Collin-Osdoby P. Human microvascular endothelial cell activation by IL-1 and TNF-alpha stimulates the adhesion and transendothelial migration of circulating human CD14+ monocytes that develop with RANKL into functional osteoclasts. J Bone Miner Res. (2006) 21:193–206. doi: 10.1359/JBMR.051027
146. Myers CL, Wertheimer SJ, Schembri-King J, Parks T, Wallace RW. Induction of ICAM-1 by TNF-alpha, IL-1 beta, and LPS in human endothelial cells after downregulation of PKC. Am J Physiol. (1992) 263:C767–72. doi: 10.1152/ajpcell.1992.263.4.C767
147. Bagi Z. Mechanisms of coronary microvascular adaptation to obesity. Am J Physiol Regul Integr Comp Physiol. (2009) 297:R556–67. doi: 10.1152/ajpregu.90817.2008
148. Selthofer-Relatić K, Bošnjak I, Kibel A. Obesity related coronary microvascular dysfunction: from basic to clinical practice. Cardiol Res Pract. (2016) 2016:8173816. doi: 10.1155/2016/8173816
149. Deshpande SS, Angkeow P, Huang J, Ozaki M, Irani K. Rac1 inhibits TNF-alpha-induced endothelial cell apoptosis: dual regulation by reactive oxygen species. FASEB J. (2000) 14:1705–14. doi: 10.1096/fj.99-0910com
150. Venkatachalam K, Venkatesan B, Valente AJ, Melby PC, Nandish S, Reusch JE, et al. WISP1, a pro-mitogenic, pro-survival factor, mediates tumor necrosis factor-alpha (TNF-alpha)-stimulated cardiac fibroblast proliferation but inhibits TNF-alpha-induced cardiomyocyte death. J Biol Chem. (2009) 284:14414–27. doi: 10.1074/jbc.M809757200
151. Voloshenyuk TG, Hart AD, Khoutorova E, Gardner JD. TNF-α increases cardiac fibroblast lysyl oxidase expression through TGF-β and PI3Kinase signaling pathways. Biochem Biophys Res Commun. (2011) 413:370–5. doi: 10.1016/j.bbrc.2011.08.109
152. Siwik DA, Chang DL, Colucci WS. Interleukin-1beta and tumor necrosis factor-alpha decrease collagen synthesis and increase matrix metalloproteinase activity in cardiac fibroblasts in vitro. Circ Res. (2000) 86:1259–65. doi: 10.1161/01.RES.86.12.1259
153. Murray PJ. Macrophage polarization. Annu Rev Physiol. (2017) 79:541–66. doi: 10.1146/annurev-physiol-022516-034339
154. Thaik CM, Calderone A, Takahashi N, Colucci WS. Interleukin-1 beta modulates the growth and phenotype of neonatal rat cardiac myocytes. J Clin Invest. (1995) 96:1093–9. doi: 10.1172/JCI118095
155. Honsho S, Nishikawa S, Amano K, Zen K, Adachi Y, Kishita E, et al. Pressure-mediated hypertrophy and mechanical stretch induces IL-1 release and subsequent IGF-1 generation to maintain compensative hypertrophy by affecting Akt and JNK pathways. Circ Res. (2009) 105:1149–58. doi: 10.1161/CIRCRESAHA.109.208199
156. Ing DJ, Zang J, Dzau VJ, Webster KA, Bishopric NH. Modulation of cytokine-induced cardiac myocyte apoptosis by nitric oxide, Bak, and Bcl-x. Circ Res. (1999) 84:21–33. doi: 10.1161/01.RES.84.1.21
157. Liu Z, Zhao N, Zhu H, Zhu S, Pan S, Xu J, et al. Circulating interleukin-1β promotes endoplasmic reticulum stress-induced myocytes apoptosis in diabetic cardiomyopathy via interleukin-1 receptor-associated kinase-2. Cardiovasc Diabetol. (2015) 14:125. doi: 10.1186/s12933-015-0288-y
158. Brønnum H, Eskildsen T, Andersen DC, Schneider M, Sheikh SP. IL-1β suppresses TGF-β-mediated myofibroblast differentiation in cardiac fibroblasts. Growth Factors. (2013) 31:81–9. doi: 10.3109/08977194.2013.787994
159. Moratal C, Raffort J, Arrighi N, Rekima S, Schaub S, Dechesne CA, et al. IL-1β- and IL-4-polarized macrophages have opposite effects on adipogenesis of intramuscular fibro-adipogenic progenitors in humans. Sci Rep. (2018) 8:17005. doi: 10.1038/s41598-018-35429-w
160. Zhao L, Cheng G, Jin R, Afzal MR, Samanta A, Xuan YT, et al. Deletion of interleukin-6 attenuates pressure overload-induced left ventricular hypertrophy and dysfunction. Circ Res. (2016) 118:1918–29. doi: 10.1161/CIRCRESAHA.116.308688
161. Kötter S, Kazmierowska M, Andresen C, Bottermann K, Grandoch M, Gorressen S, et al. Titin-based cardiac myocyte stiffening contributes to early adaptive ventricular remodeling after myocardial infarction. Circ Res. (2016) 119:1017–29. doi: 10.1161/CIRCRESAHA.116.309685
162. Wollert KC, Drexler H. The role of interleukin-6 in the failing heart. Heart Fail Rev. (2001) 6:95–103. doi: 10.1023/A:1011401825680
163. Watson C, Whittaker S, Smith N, Vora AJ, Dumonde DC, Brown KA. IL-6 acts on endothelial cells to preferentially increase their adherence for lymphocytes. Clin Exp Immunol. (1996) 105:112–9. doi: 10.1046/j.1365-2249.1996.d01-717.x
164. Zhang Y, Wang JH, Zhang YY, Wang YZ, Wang J, Zhao Y, et al. Deletion of interleukin-6 alleviated interstitial fibrosis in streptozotocin-induced diabetic cardiomyopathy of mice through affecting TGFβ1 and miR-29 pathways. Sci Rep. (2016) 6:23010. doi: 10.1038/srep23010
165. Braune J, Weyer U, Hobusch C, Mauer J, Brüning JC, Bechmann I, et al. IL-6 regulates M2 polarization and local proliferation of adipose tissue macrophages in obesity. J Immunol. (2017) 198:2927–34. doi: 10.4049/jimmunol.1600476
166. Sanmarco LM, Ponce NE, Visconti LM, Eberhardt N, Theumer MG, Minguez ÁR, et al. IL-6 promotes M2 macrophage polarization by modulating purinergic signaling and regulates the lethal release of nitric oxide during Trypanosoma cruzi infection. Biochim Biophys Acta Mol Basis Dis. (2017) 1863:857–69. doi: 10.1016/j.bbadis.2017.01.006
167. Yoshida T, Friehs I, Mummidi S, del Nido PJ, Addulnour-Nakhoul S, Delafontaine P, et al. Pressure overload induces IL-18 and IL-18R expression, but markedly suppresses IL-18BP expression in a rabbit model IL-18 potentiates TNF-α-induced cardiomyocyte death. J Mol Cell Cardiol. (2014) 75:141–51. doi: 10.1016/j.yjmcc.2014.07.007
168. Morel JC, Park CC, Woods JM, Koch AE. A novel role for interleukin-18 in adhesion molecule induction through NF kappa B and phosphatidylinositol (PI) 3-kinase-dependent signal transduction pathways. J Biol Chem. (2001) 276:37069–75. doi: 10.1074/jbc.M103574200
169. Gerdes N, Sukhova GK, Libby P, Reynolds RS, Young JL, Schönbeck U. Expression of interleukin (IL)-18 and functional IL-18 receptor on human vascular endothelial cells, smooth muscle cells, and macrophages: implications for atherogenesis. J Exp Med. (2002) 195:245–57. doi: 10.1084/jem.20011022
170. handrasekar B, Valente AJ, Freeman GL, Mahimainathan L, Mummidi S. Interleukin-18 induces human cardiac endothelial cell death via a novel signaling pathway involving NF-kappaB-dependent PTEN activation. Biochem Biophys Res Commun. (2006) 339:956–63. doi: 10.1016/j.bbrc.2005.11.100
171. Fix C, Bingham K, Carver W. Effects of interleukin-18 on cardiac fibroblast function and gene expression. Cytokine. (2011) 53:19–28. doi: 10.1016/j.cyto.2010.10.002
172. Kobori T, Hamasaki S, Kitaura A, Yamazaki Y, Nishinaka T, Niwa A, et al. Interleukin-18 amplifies macrophage polarization and morphological alteration, leading to excessive angiogenesis. Front Immunol. (2018) 9:334. doi: 10.3389/fimmu.2018.00334
173. Schultz Jel J, Witt SA, Glascock BJ, Nieman ML, Reiser PJ, Nix SL, et al. TGF-beta1 mediates the hypertrophic cardiomyocyte growth induced by angiotensin II. J Clin Invest. (2002) 109:787–96. doi: 10.1172/JCI0214190
174. Schneiders D, Heger J, Best P, Michael Piper H, Taimor G. SMAD proteins are involved in apoptosis induction in ventricular cardiomyocytes. Cardiovasc Res. (2005) 67:87–96. doi: 10.1016/j.cardiores.2005.02.021
175. Heger J, Warga B, Meyering B, Abdallah Y, Schlüter KD, Piper HM, et al. TGFβ receptor activation enhances cardiac apoptosis via SMAD activation and concomitant NO release. J Cell Physiol. (2011) 226:2683–90. doi: 10.1002/jcp.22619
176. Ma J, Sanchez-Duffhues G, Goumans MJ, Ten Dijke P. TGF-β-induced endothelial to mesenchymal transition in disease and tissue engineering. Front Cell Dev Biol. (2020) 8:260. doi: 10.3389/fcell.2020.00260
177. Pintavorn P, Ballermann BJ. TGF-beta and the endothelium during immune injury. Kidney Int. (1997) 51:1401–12. doi: 10.1038/ki.1997.192
178. Inoue N, Venema RC, Sayegh HS, Ohara Y, Murphy TJ, Harrison DG. Molecular regulation of the bovine endothelial cell nitric oxide synthase by transforming growth factor-beta 1. Arterioscler Thromb Vasc Biol. (1995) 15:1255–61. doi: 10.1161/01.ATV.15.8.1255
179. Ferrari G, Terushkin V, Wolff MJ, Zhang X, Valacca C, Poggio P, et al. TGF-β1 induces endothelial cell apoptosis by shifting VEGF activation of p38(MAPK) from the prosurvival p38β to proapoptotic p38α. Mol Cancer Res. (2012) 10:605–14. doi: 10.1158/1541-7786.MCR-11-0507
180. Leksa V, Godar S, Schiller HB, Fuertbauer E, Muhammad A, Slezakova K, et al. TGF-beta-induced apoptosis in endothelial cells mediated by M6P/IGFII-R and mini-plasminogen. J Cell Sci. (2005) 118:4577–86. doi: 10.1242/jcs.02587
181. Desmoulière A, Geinoz A, Gabbiani F, Gabbiani G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol. (1993) 122:103–11. doi: 10.1083/jcb.122.1.103
182. Dobaczewski M, Bujak M, Li N, Gonzalez-Quesada C, Mendoza LH, Wang XF, et al. Smad3 signaling critically regulates fibroblast phenotype and function in healing myocardial infarction. Circ Res. (2010) 107:418–28. doi: 10.1161/CIRCRESAHA.109.216101
183. Zhang F, Wang H, Wang X, Jiang G, Liu H, Zhang G, et al. TGF-β induces M2-like macrophage polarization via SNAIL-mediated suppression of a pro-inflammatory phenotype. Oncotarget. (2016) 7:52294–306. doi: 10.18632/oncotarget.10561
184. Xu FP, Chen MS, Wang YZ Yi Q, Lin SB, Chen AF, Luo JD. Leptin induces hypertrophy via endothelin-1-reactive oxygen species pathway in cultured neonatal rat cardiomyocytes. Circulation. (2004) 110:1269–75. doi: 10.1161/01.CIR.0000140766.52771.6D
185. Dong F, Zhang X, Ren J. Leptin regulates cardiomyocyte contractile function through endothelin-1 receptor-NADPH oxidase pathway. Hypertension. (2006) 47:222–9. doi: 10.1161/01.HYP.0000198555.51645.f1
186. McGaffin KR, Zou B, McTiernan CF, O'Donnell CP. Leptin attenuates cardiac apoptosis after chronic ischaemic injury. Cardiovasc Res. (2009) 83:313–24. doi: 10.1093/cvr/cvp071
187. Eguchi M, Liu Y, Shin EJ, Sweeney G. Leptin protects H9c2 rat cardiomyocytes from H2O2-induced apoptosis. FEBS J. (2008) 275:3136–44. doi: 10.1111/j.1742-4658.2008.06465.x
188. Korda M, Kubant R, Patton S, Malinski T. Leptin-induced endothelial dysfunction in obesity. Am J Physiol Heart Circ Physiol. (2008) 295:H1514–21. doi: 10.1152/ajpheart.00479.2008
189. Park HY, Kwon HM, Lim HJ, Hong BK, Lee JY, Park BE, et al. Potential role of leptin in angiogenesis: leptin induces endothelial cell proliferation and expression of matrix metalloproteinases in vivo and in vitro. Exp Mol Med. (2001) 33:95–102. doi: 10.1038/emm.2001.17
190. Ezure T, Amano S. Adiponectin and leptin up-regulate extracellular matrix production by dermal fibroblasts. Biofactors. (2007) 31:229–36. doi: 10.1002/biof.5520310310
191. Raso GM, Pacilio M, Esposito E, Coppola A, Di Carlo R, Meli R. Leptin potentiates IFN-gamma-induced expression of nitric oxide synthase and cyclo-oxygenase-2 in murine macrophage J774A1. Br J Pharmacol. (2002) 137:799–804. doi: 10.1038/sj.bjp.0704903
192. Ostlund RE Jr, Yang JW, Klein S, Gingerich R. Relation between plasma leptin concentration and body fat, gender, diet, age, and metabolic covariates. J Clin Endocrinol Metab. (1996) 81:3909–13. doi: 10.1210/jc.81.11.3909
193. Wang YC Li Y, Wang XY, Zhang D, Zhang H, Wu Q, et al. Circulating miR-130b mediates metabolic crosstalk between fat and muscle in overweight/obesity. Diabetologia. (2013) 56:2275–85. doi: 10.1007/s00125-013-2996-8
194. Fain JN, Tichansky DS, Madan AK. Transforming growth factor beta1 release by human adipose tissue is enhanced in obesity. Metabolism. (2005) 54:1546–51. doi: 10.1016/j.metabol.2005.05.024
195. Di Renzo L, Bigioni M, Del Gobbo V, Premrov MG, Barbini U, Di Lorenzo N, et al. Interleukin-1 (IL-1) receptor antagonist gene polymorphism in normal weight obese syndrome: relationship to body composition and IL-1 alpha and beta plasma levels. Pharmacol Res. (2007) 55:131–8. doi: 10.1016/j.phrs.2006.11.002
196. Ballak DB, Stienstra R, Tack CJ, Dinarello CA, van Diepen JA. IL-1 family members in the pathogenesis and treatment of metabolic disease: focus on adipose tissue inflammation and insulin resistance. Cytokine. (2015) 75:280–90. doi: 10.1016/j.cyto.2015.05.005
197. Esposito K, Pontillo A, Ciotola M, Di Palo C, Grella E, Nicoletti G, et al. Weight loss reduces interleukin-18 levels in obese women. J Clin Endocrinol Metab. (2002) 87:3864–6. doi: 10.1210/jcem.87.8.8781
198. Chen YL, Qiao YC, Xu Y, Ling W, Pan YH, Huang YC, et al. Serum TNF-α concentrations in type 2 diabetes mellitus patients and diabetic nephropathy patients: a systematic review and meta-analysis. Immunol Lett. (2017) 186:52–8. doi: 10.1016/j.imlet.2017.04.003
199. Amitani M, Asakawa A, Amitani H, Inui A. The role of leptin in the control of insulin-glucose axis. Front Neurosci. (2013) 7:51. doi: 10.3389/fnins.2013.00051
200. Ehnert S, Freude T, Ihle C, Mayer L, Braun B, Graeser J, et al. Factors circulating in the blood of type 2 diabetes mellitus patients affect osteoblast maturation - description of a novel in vitro model. Exp Cell Res. (2015) 332:247–58. doi: 10.1016/j.yexcr.2014.12.011
201. Esposito K, Nappo F, Marfella R, Giugliano G, Giugliano F, Ciotola M, et al. Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans: role of oxidative stress. Circulation. (2002) 106:2067–72. doi: 10.1161/01.CIR.0000034509.14906.AE
202. Jagannathan-Bogdan M, McDonnell ME, Shin H, Rehman Q, Hasturk H, Apovian CM, et al. Elevated proinflammatory cytokine production by a skewed T cell compartment requires monocytes and promotes inflammation in type 2 diabetes. J Immunol. (2011) 186:1162–72. doi: 10.4049/jimmunol.1002615
203. Trøseid M, Seljeflot I, Arnesen H. The role of interleukin-18 in the metabolic syndrome. Cardiovasc Diabetol. (2010) 9:11. doi: 10.1186/1475-2840-9-11
204. Schulz R, Heusch G. Tumor necrosis factor-alpha and its receptors 1 and 2: Yin and Yang in myocardial infarction? Circulation. (2009) 119:1355–7. doi: 10.1161/CIRCULATIONAHA.108.846105
205. Condorelli G, Morisco C, Latronico MV, Claudio PP, Dent P, Tsichlis P, et al. TNF-alpha signal transduction in rat neonatal cardiac myocytes: definition of pathways generating from the TNF-alpha receptor. FASEB J. (2002) 16:1732–7. doi: 10.1096/fj.02-0419com
206. Bozkurt B, Kribbs SB, Clubb FJ Jr, Michael LH, Didenko VV, Hornsby PJ, et al. Pathophysiologically relevant concentrations of tumor necrosis factor-alpha promote progressive left ventricular dysfunction and remodeling in rats. Circulation. (1998) 97:1382–91. doi: 10.1161/01.CIR.97.14.1382
207. Van Tassell BW, Arena RA, Toldo S, Mezzaroma E, Azam T, Seropian IM, et al. Enhanced interleukin-1 activity contributes to exercise intolerance in patients with systolic heart failure. PLoS ONE. (2012) 7:e33438. doi: 10.1371/journal.pone.0033438
208. Sun M, Chen M, Dawood F, Zurawska U, Li JY, Parker T, et al. Tumor necrosis factor-alpha mediates cardiac remodeling and ventricular dysfunction after pressure overload state. Circulation. (2007) 115:1398–407. doi: 10.1161/CIRCULATIONAHA.106.643585
209. Meléndez GC, McLarty JL, Levick SP, Du Y, Janicki JS, Brower GL. Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension. (2010) 56:225–31. doi: 10.1161/HYPERTENSIONAHA.109.148635
210. Mezzaroma E, Toldo S, Farkas D, Seropian IM, Van Tassell BW, Salloum FN, et al. The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc Natl Acad Sci USA. (2011) 108:19725–30. doi: 10.1073/pnas.1108586108
211. Luo B, Li B, Wang W, Liu X, Xia Y, Zhang C, et al. NLRP3 gene silencing ameliorates diabetic cardiomyopathy in a type 2 diabetes rat model. PLoS ONE. (2014) 9:e104771. doi: 10.1371/journal.pone.0104771
212. Kraigher-Krainer E, Shah AM, Gupta DK, Santos A, Claggett B, Pieske B, et al. Impaired systolic function by strain imaging in heart failure with preserved ejection fraction. J Am Coll Cardiol. (2014) 63:447–56. doi: 10.1016/j.jacc.2013.09.052
213. Taqueti VR, Di Carli MF. Coronary microvascular disease pathogenic mechanisms and therapeutic options: JACC state-of-the-art review. J Am Coll Cardiol. (2018) 72:2625–41. doi: 10.1016/j.jacc.2018.09.042
214. Di Carli MF, Janisse J, Grunberger G, Ager J. Role of chronic hyperglycemia in the pathogenesis of coronary microvascular dysfunction in diabetes. J Am Coll Cardiol. (2003) 41:1387–93. doi: 10.1016/S0735-1097(03)00166-9
215. Paulus WJ, Tschöpe C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. (2013) 62:263–71. doi: 10.1016/j.jacc.2013.02.092
216. Paulus WJ, van Heerebeek L. Ancient gunpowder and novel insights team up against heart failure with preserved ejection fraction. J Am Coll Cardiol. (2015) 66:1683–6. doi: 10.1016/j.jacc.2015.08.028
217. Kibel A, Selthofer-Relatic K, Drenjancevic I, Bacun T, Bosnjak I, Kibel D, et al. Coronary microvascular dysfunction in diabetes mellitus. J Int Med Res. (2017) 45:1901–29. doi: 10.1177/0300060516675504
218. Nahser PJ Jr, Brown RE, Oskarsson H, Winniford MD, Rossen JD. Maximal coronary flow reserve and metabolic coronary vasodilation in patients with diabetes mellitus. Circulation. (1995) 91:635–40. doi: 10.1161/01.CIR.91.3.635
219. Galderisi M, Capaldo B, Sidiropulos M, D'Errico A, Ferrara L, Turco A, et al. Determinants of reduction of coronary flow reserve in patients with type 2 diabetes mellitus or arterial hypertension without angiographically determined epicardial coronary stenosis. Am J Hypertens. (2007) 20:1283–90. doi: 10.1016/j.amjhyper.2007.08.005
220. Bajaj NS, Osborne MT, Gupta A, Tavakkoli A, Bravo PE, Vita T, et al. Coronary microvascular dysfunction and cardiovascular risk in obese patients. J Am Coll Cardiol. (2018) 72:707–17. doi: 10.1016/j.jacc.2018.05.049
221. Eroglu S, Sade LE, Bozbaş H, Müderrisoglu H. Decreased coronary flow reserve in obese women. Turk Kardiyol Dern Ars. (2009) 37:391–6.
222. de Jongh RT, Serné EH, IJzerman RG, de Vries G, Stehouwer CD. Impaired microvascular function in obesity: implications for obesity-associated microangiopathy, hypertension, and insulin resistance. Circulation. (2004) 109:2529–35. doi: 10.1161/01.CIR.0000129772.26647.6F
223. Al Suwaidi J, Higano ST, Holmes DR Jr, Lennon R, Lerman A. Obesity is independently associated with coronary endothelial dysfunction in patients with normal or mildly diseased coronary arteries. J Am Coll Cardiol. (2001) 37:1523–8. doi: 10.1016/S0735-1097(01)01212-8
224. Taqueti VR, Solomon SD, Shah AM, Desai AS, Groarke JD, Osborne MT, et al. Coronary microvascular dysfunction and future risk of heart failure with preserved ejection fraction. Eur Heart J. (2018) 39:840–9. doi: 10.1093/eurheartj/ehx721
225. Shah SJ, Lam CSP, Svedlund S, Saraste A, Hage C, Tan RS, et al. Prevalence and correlates of coronary microvascular dysfunction in heart failure with preserved ejection fraction: PROMIS-HFpEF. Eur Heart J. (2018) 39:3439–50. doi: 10.1093/eurheartj/ehy531
226. Srivaratharajah K, Coutinho T, deKemp R, Liu P, Haddad H, Stadnick E, et al. Reduced myocardial flow in heart failure patients with preserved ejection fraction. Circ Heart Fail. (2016) 9:e002562. doi: 10.1161/CIRCHEARTFAILURE.115.002562
227. Tomai F, Ribichini F, Ghini AS, Ferrero V, Andò G, Vassanelli C. Elevated C-reactive protein levels and coronary microvascular dysfunction in patients with coronary artery disease. Eur Heart J. (2005) 26:2099–105. doi: 10.1093/eurheartj/ehi356
228. Faccini A, Kaski JC, Camici PG. Coronary microvascular dysfunction in chronic inflammatory rheumatoid diseases. Eur Heart J. (2016) 37:1799–806. doi: 10.1093/eurheartj/ehw018
229. Taqueti VR, Ridker PM. Inflammation, coronary flow reserve, and microvascular dysfunction: moving beyond cardiac syndrome X. JACC Cardiovasc Imaging. (2013) 6:668–71. doi: 10.1016/j.jcmg.2013.02.005
230. Ong P, Athanasiadis A, Borgulya G, Mahrholdt H, Kaski JC, Sechtem U. High prevalence of a pathological response to acetylcholine testing in patients with stable angina pectoris and unobstructed coronary arteries. The ACOVA Study (Abnormal COronary VAsomotion in patients with stable angina and unobstructed coronary arteries). J Am Coll Cardiol. (2012) 59:655–62. doi: 10.1016/j.jacc.2011.11.015
231. Vaccarino V, Khan D, Votaw J, Faber T, Veledar E, Jones DP, et al. Inflammation is related to coronary flow reserve detected by positron emission tomography in asymptomatic male twins. J Am Coll Cardiol. (2011) 57:1271–9. doi: 10.1016/j.jacc.2010.09.074
232. Tona F, Serra R, Di Ascenzo L, Osto E, Scarda A, Fabris R, et al. Systemic inflammation is related to coronary microvascular dysfunction in obese patients without obstructive coronary disease. Nutr Metab Cardiovasc Dis. (2014) 24:447–53. doi: 10.1016/j.numecd.2013.09.021
233. Quercioli A, Pataky Z, Montecucco F, Carballo S, Thomas A, Staub C, et al. Coronary vasomotor control in obesity and morbid obesity: contrasting flow responses with endocannabinoids, leptin, and inflammation. JACC Cardiovasc Imaging. (2012) 5:805–15. doi: 10.1016/j.jcmg.2012.01.020
234. Campbell DJ, Somaratne JB, Prior DL Yii M, Kenny JF, Newcomb AE, Kelly DJ, et al. Obesity is associated with lower coronary microvascular density. PLoS ONE. (2013) 8:e81798. doi: 10.1371/journal.pone.0081798
235. Machado MV, Vieira AB, da Conceição FG, Nascimento AR, da Nóbrega ACL, Tibirica E. Exercise training dose differentially alters muscle and heart capillary density and metabolic functions in an obese rat with metabolic syndrome. Exp Physiol. (2017) 102:1716–28. doi: 10.1113/EP086416
236. Gonzalez-Quesada C, Cavalera M, Biernacka A, Kong P, Lee DW, Saxena A, et al. Thrombospondin-1 induction in the diabetic myocardium stabilizes the cardiac matrix in addition to promoting vascular rarefaction through angiopoietin-2 upregulation. Circ Res. (2013) 113:1331–44. doi: 10.1161/CIRCRESAHA.113.302593
237. Toblli JE, Cao G, DeRosa G, Di Gennaro F, Forcada P. Angiotensin-converting enzyme inhibition and angiogenesis in myocardium of obese Zucker rats. Am J Hypertens. (2004) 17:172–80. doi: 10.1016/j.amjhyper.2003.10.006
238. Seferović PM, Paulus WJ. Clinical diabetic cardiomyopathy: a two-faced disease with restrictive and dilated phenotypes. Eur Heart J. (2015) 36:1718–27. doi: 10.1093/eurheartj/ehv134
239. Othman AI, El-Sawi MR, El-Missiry MA, Abukhalil MH. Epigallocatechin-3-gallate protects against diabetic cardiomyopathy through modulating the cardiometabolic risk factors, oxidative stress, inflammation, cell death and fibrosis in streptozotocin-nicotinamide-induced diabetic rats. Biomed Pharmacother. (2017) 94:362–73. doi: 10.1016/j.biopha.2017.07.129
240. Jia G, DeMarco VG, Sowers JR. Insulin resistance and hyperinsulinaemia in diabetic cardiomyopathy. Nat Rev Endocrinol. (2016) 12:144–53. doi: 10.1038/nrendo.2015.216
241. Singh RJ, Mason JC, Lidington EA, Edwards DR, Nuttall RK, Khokha R, et al. Cytokine stimulated vascular cell adhesion molecule-1 (VCAM-1) ectodomain release is regulated by TIMP-3. Cardiovasc Res. (2005) 67:39–49. doi: 10.1016/j.cardiores.2005.02.020
242. Osborn L, Hession C, Tizard R, Vassallo C, Luhowskyj S, Chi-Rosso G, et al. Direct expression cloning of vascular cell adhesion molecule 1, a cytokine-induced endothelial protein that binds to lymphocytes. Cell. (1989) 59:1203–11. doi: 10.1016/0092-8674(89)90775-7
243. Ruiz-Ortega M, Lorenzo O, Rupérez M, Esteban V, Suzuki Y, Mezzano S, et al. Role of the renin-angiotensin system in vascular diseases: expanding the field. Hypertension. (2001) 38:1382–7. doi: 10.1161/hy1201.100589
244. Datta R, Bansal T, Rana S, Datta K, Datta Chaudhuri R, Chawla-Sarkar M, et al. Myocyte-derived Hsp90 modulates collagen upregulation via biphasic activation of STAT-3 in fibroblasts during cardiac hypertrophy. Mol Cell Biol. (2017) 37:e00611–16. doi: 10.1128/MCB.00611-16
245. Tschöpe C, Van Linthout S. New insights in (inter)cellular mechanisms by heart failure with preserved ejection fraction. Curr Heart Fail Rep. (2014) 11:436–44. doi: 10.1007/s11897-014-0219-3
246. Piera-Velazquez S, Jimenez SA. Endothelial to mesenchymal transition: role in physiology and in the pathogenesis of human diseases. Physiol Rev. (2019) 99:1281–324. doi: 10.1152/physrev.00021.2018
247. Chaudhuri J, Bains Y, Guha S, Kahn A, Hall D, Bose N, et al. The role of advanced glycation end products in aging and metabolic diseases: bridging association and causality. Cell Metab. (2018) 28:337–52. doi: 10.1016/j.cmet.2018.08.014
248. Ramasamy R, Schmidt AM. Receptor for advanced glycation end products (RAGE) and implications for the pathophysiology of heart failure. Curr Heart Fail Rep. (2012) 9:107–16. doi: 10.1007/s11897-012-0089-5
249. Rungratanawanich W, Qu Y, Wang X, Essa MM, Song BJ. Advanced glycation end products (AGEs) and other adducts in aging-related diseases and alcohol-mediated tissue injury. Exp Mol Med. (2021) 53:168–88. doi: 10.1038/s12276-021-00561-7
250. Prasad K, Mishra M. AGE-RAGE stress, stressors, and antistressors in health and disease. Int J Angiol. (2018) 27:1–12. doi: 10.1055/s-0037-1613678
251. Packer M. Epicardial adipose tissue may mediate deleterious effects of obesity and inflammation on the myocardium. J Am Coll Cardiol. (2018) 71:2360–72. doi: 10.1016/j.jacc.2018.03.509
252. Turer AT, Hill JA, Elmquist JK, Scherer PE. Adipose tissue biology and cardiomyopathy: translational implications. Circ Res. (2012) 111:1565–77. doi: 10.1161/CIRCRESAHA.111.262493
253. Blüher M. Obesity: global epidemiology and pathogenesis. Nat Rev Endocrinol. (2019) 15:288–98. doi: 10.1038/s41574-019-0176-8
254. Rajapurohitam V, Javadov S, Purdham DM, Kirshenbaum LA, Karmazyn M. An autocrine role for leptin in mediating the cardiomyocyte hypertrophic effects of angiotensin II and endothelin-1. J Mol Cell Cardiol. (2006) 41:265–74. doi: 10.1016/j.yjmcc.2006.05.001
255. Moro C, Grauzam S, Ormezzano O, Toufektsian MC, Tanguy S, Calabrese P, et al. Inhibition of cardiac leptin expression after infarction reduces subsequent dysfunction. J Cell Mol Med. (2011) 15:1688–94. doi: 10.1111/j.1582-4934.2010.01154.x
256. Matsubara M, Maruoka S, Katayose S. Inverse relationship between plasma adiponectin and leptin concentrations in normal-weight and obese women. Eur J Endocrinol. (2002) 147:173–80. doi: 10.1530/eje.0.1470173
257. Ouchi N, Kihara S, Arita Y, Okamoto Y, Maeda K, Kuriyama H, et al. Adiponectin, an adipocyte-derived plasma protein, inhibits endothelial NF-kappaB signaling through a cAMP-dependent pathway. Circulation. (2000) 102:1296–301. doi: 10.1161/01.CIR.102.11.1296
258. Holland WL, Miller RA, Wang ZV, Sun K, Barth BM, Bui HH, et al. Receptor-mediated activation of ceramidase activity initiates the pleiotropic actions of adiponectin. Nat Med. (2011) 17:55–63. doi: 10.1038/nm.2277
259. Antonopoulos AS, Margaritis M, Verheule S, Recalde A, Sanna F, Herdman L, et al. Mutual regulation of epicardial adipose tissue and myocardial redox state by PPAR-γ/adiponectin signalling. Circ Res. (2016) 118:842–55. doi: 10.1161/CIRCRESAHA.115.307856
260. Gruzdeva OV, Akbasheva OE, Dyleva YA, Antonova LV, Matveeva VG, Uchasova EG, et al. Adipokine and cytokine profiles of epicardial and subcutaneous adipose tissue in patients with coronary heart disease. Bull Exp Biol Med. (2017) 163:608–11. doi: 10.1007/s10517-017-3860-5
261. Greulich S, Maxhera B, Vandenplas G, de Wiza DH, Smiris K, Mueller H, et al. Secretory products from epicardial adipose tissue of patients with type 2 diabetes mellitus induce cardiomyocyte dysfunction. Circulation. (2012) 126:2324–34. doi: 10.1161/CIRCULATIONAHA.111.039586
262. Nakanishi K, Fukuda S, Tanaka A, Otsuka K, Taguchi H, Shimada K. Relationships between periventricular epicardial adipose tissue accumulation, coronary microcirculation, and left ventricular diastolic dysfunction. Can J Cardiol. (2017) 33:1489–97. doi: 10.1016/j.cjca.2017.08.001
263. Lai YH, Yun CH, Yang FS, Liu CC, Wu YJ, Kuo JY, et al. Epicardial adipose tissue relating to anthropometrics, metabolic derangements and fatty liver disease independently contributes to serum high-sensitivity C-reactive protein beyond body fat composition: a study validated with computed tomography. J Am Soc Echocardiogr. (2012) 25:234–41. doi: 10.1016/j.echo.2011.09.018
264. Patel VB, Shah S, Verma S, Oudit GY. Epicardial adipose tissue as a metabolic transducer: role in heart failure and coronary artery disease. Heart Fail Rev. (2017) 22:889–902. doi: 10.1007/s10741-017-9644-1
265. Fontes-Carvalho R, Fontes-Oliveira M, Sampaio F, Mancio J, Bettencourt N, Teixeira M, et al. Influence of epicardial and visceral fat on left ventricular diastolic and systolic functions in patients after myocardial infarction. Am J Cardiol. (2014) 114:1663–9. doi: 10.1016/j.amjcard.2014.08.037
266. Kalogeropoulos A, Georgiopoulou V, Psaty BM, Rodondi N, Smith AL, Harrison DG, et al. Inflammatory markers and incident heart failure risk in older adults: the Health ABC (Health, Aging, and Body Composition) study. J Am Coll Cardiol. (2010) 55:2129–37. doi: 10.1016/j.jacc.2009.12.045
267. Chia YC, Kieneker LM, van Hassel G, Binnenmars SH, Nolte IM, van Zanden JJ, et al. Interleukin 6 and development of heart failure with preserved ejection fraction in the general population. J Am Heart Assoc. (2021) 10:e018549. doi: 10.1161/JAHA.120.018549
268. Tromp J, Westenbrink BD, Ouwerkerk W, van Veldhuisen DJ, Samani NJ, Ponikowski P, et al. Identifying pathophysiological mechanisms in heart failure with reduced versus preserved ejection fraction. J Am Coll Cardiol. (2018) 72:1081–90. doi: 10.1016/j.jacc.2018.06.050
269. Tromp J, Khan MA, Klip IT, Meyer S, de Boer RA, Jaarsma T. Biomarker profiles in heart failure patients with preserved and reduced ejection fraction. J Am Heart Assoc. (2017) 6:e003989. doi: 10.1161/JAHA.116.003989
Keywords: obesity, inflammation, lipotoxicity, HFpEF, cardiometabolic disease
Citation: Wenzl FA, Ambrosini S, Mohammed SA, Kraler S, Lüscher TF, Costantino S and Paneni F (2021) Inflammation in Metabolic Cardiomyopathy. Front. Cardiovasc. Med. 8:742178. doi: 10.3389/fcvm.2021.742178
Received: 15 July 2021; Accepted: 31 August 2021;
Published: 04 October 2021.
Edited by:
Annalisa Capuano, University of Campania Luigi Vanvitelli, ItalyReviewed by:
Michelangela Barbieri, University of Campania Luigi Vanvitelli, ItalyCopyright © 2021 Wenzl, Ambrosini, Mohammed, Kraler, Lüscher, Costantino and Paneni. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Francesco Paneni, ZnJhbmNlc2NvLnBhbmVuaUB1emguY2g=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.