 Shugang Zhang
Shugang Zhang Weigang Lu1,2
Weigang Lu1,2 Zhiqiang Wei
Zhiqiang Wei Henggui Zhang
Henggui Zhang- 1Computational Cardiology Group, College of Computer Science and Technology, Ocean University of China, Qingdao, China
- 2Biological Physics Group, School of Physics and Astronomy, University of Manchester, Manchester, United Kingdom
Cardiovascular disease is the leading cause of death worldwide and kills over 17 million people per year. In the recent decade, growing epidemiological evidence links air pollution and cardiac arrhythmias, suggesting a detrimental influence of air pollution on cardiac electrophysiological functionality. However, the proarrhythmic mechanisms underlying the air pollution-induced cardiac arrhythmias are not fully understood. The purpose of this work is to provide recent advances in air pollution-induced arrhythmias with a comprehensive review of the literature on the common air pollutants and arrhythmias. Six common air pollutants of widespread concern are discussed, namely particulate matter, carbon monoxide, hydrogen sulfide, sulfur dioxide, nitrogen dioxide, and ozone. The epidemiological and clinical reports in recent years are reviewed by pollutant type, and the recently identified mechanisms including both the general pathways and the direct influences of air pollutants on the cellular electrophysiology are summarized. Particularly, this review focuses on the impaired ion channel functionality underlying the air pollution-induced arrhythmias. Alterations of ionic currents directly by the air pollutants, as well as the alterations mediated by intracellular signaling or other more general pathways are reviewed in this work. Finally, areas for future research are suggested to address several remaining scientific questions.
Introduction
Cardiovascular disease (CVD) is the number one cause of death globally and accounts for more than 17 million deaths annually (1, 2). Air pollution is among the leading risk factors for cardiovascular disease and responsible for ~19% of the total CVD deaths (3, 4). Cardiac arrhythmias, which refer to any types of irregular heartbeats or abnormal heart rates, are a major cause of morbidity and mortality in cardiovascular diseases. Common types of cardiac arrhythmias include sinus node dysfunction, supraventricular tachycardia, atrial fibrillation (AF), conduction disorders, ventricular tachycardia (VT), and ventricular fibrillation (VF). Epidemiological studies have substantiated the association of air pollution with a variety of arrhythmia types (5–7).
Toxicity of pollutants can be significantly modified with their components, which suggest disproportionate contributions of different pollutants (8). Therefore, this review is organized with regard to each pollutant separately. Six common air pollutants, including particulate matter (PM), carbon monoxide (CO), hydrogen sulfide (H2S), sulfur dioxide (SO2), nitrogen dioxide (NO2), and ozone (O3). The independent role of each pollutant as well as their potential pathological pathways were explored and emphasized regarding the mechanisms of air pollution-induced arrhythmias.
Systemic inflammatory response, cardiac autonomic dysfunction, cardiac structural remodeling, and translocation of particles into cardiovascular systems are considered the dominant mechanisms underlying air pollution-induced arrhythmias (9). The systemic inflammation and the altered autonomic nervous system (ANS) indirectly cause arrhythmias by a series of actions comprising the progression of atherosclerosis, the increased cytokine levels, and the decreased heart rate variability (HRV), etc. The structural remodeling results in myocardial fibrosis and decreased conduction velocity (CV), and provides substrates for arrhythmias, while the translocated particles can affect ion channel functions through elevating the intracellular levels of reactive oxygen species (ROS). Different from previous surveys that introduced more generalized mechanisms, the present review particularly emphasizes the alterations of cardiac ion channels. This is because most arrhythmia types originate from the dysfunction of ion channels, and minor changes of ionic currents can result in severe cardiac disorder. In the present work, we review both the effects of air pollutants on ion channels that have been proved in experiments and the electrical remodeling that arises as secondary effects of systemic inflammation and other general mechanisms (Figure 1).
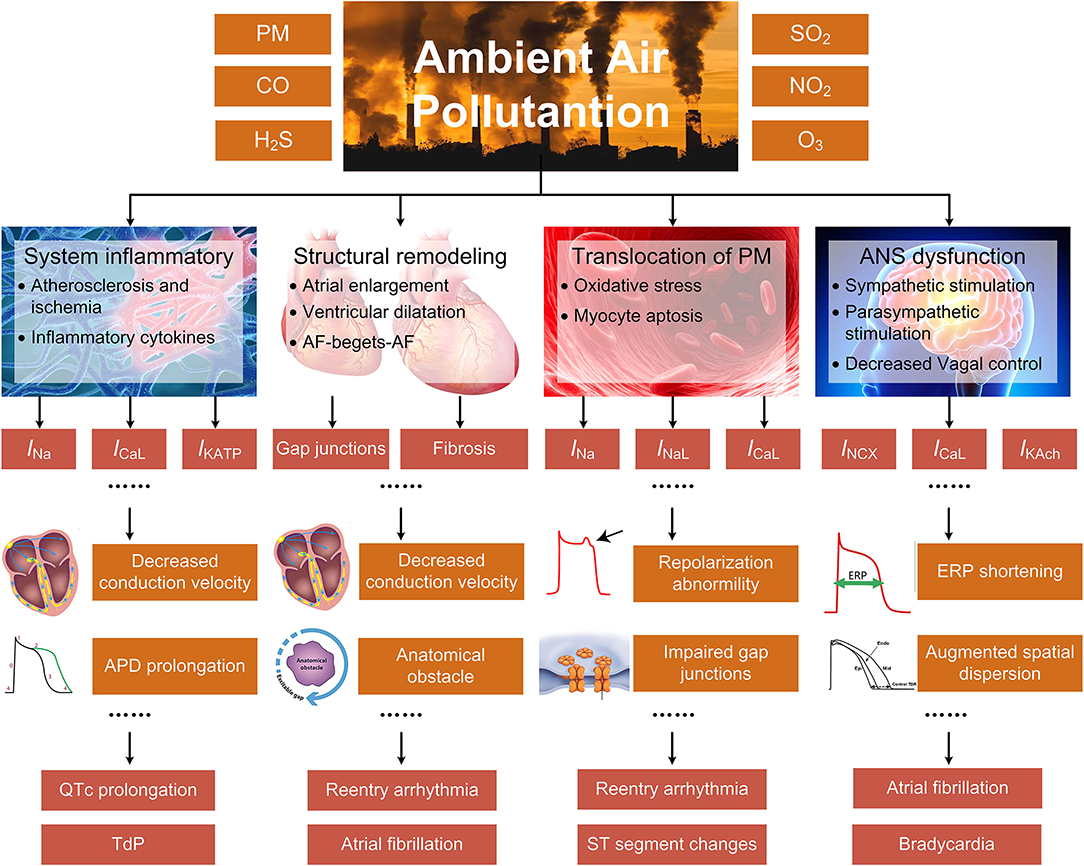
Figure 1. The indirect mechanisms underlying air pollution-induced arrhythmias, including systemic inflammatory response, cardiac structural remodeling, translocation of particles into cardiovascular systems, and ANS dysfunction. The specific pathways are closely linked or overlapped, and only the primary and representative pathways are illustrated for each process.
Particulate Matter
Particulate matters (PM) are the materials suspended in Earth's atmosphere in the form of solid particles and liquid drops. There is no strict size boundary within which the particles were considered hazardous. But due to that the larger particles can be filtered by cilia and mucus in the nose and throat, the particles that are <10 μm (PM10) in diameter are thought to influence human health as they can be inhaled and get deep into lungs or bloodstream. PM10 can be subclassified into coarse PM (PM2.5−10), fine PM (PM2.5), and ultrafine PM (PM0.1) according to the aerodynamic diameter (10). Fine and ultrafine particles derive primarily from the combustion of fossil fuels in industry and traffic (11, 12). Instead, PM2.5−10 comes primarily from mechanical processes and is associated with surface or fugitive releases by various human and natural activities (11).
Epidemiological and Clinical Reports
A growing body of epidemiological evidence demonstrates the associations between PM and cardiovascular diseases (13–15). Among the three size categories of PM, the PM2.5 has received the most attention in the past decade. A nationwide study in China suggested a concentration-dependent influence of long-term exposure to PM2.5 on cardiovascular health (15). Seniors, rural residents, and non-smokers were shown to be more prone to the adverse effects of PM2.5 exposure. It was estimated that each 10 μg/m3 decrease in the concentration of PM2.5 would avoid over 1.5 million cardiovascular incidents and saved more than 433 thousand lives from cardiovascular diseases annually in mainland China (16). The association between PM2.5 and cardiovascular disease is still evident in developed countries where the level of PM2.5 is well below the present standard. An investigation conducted in the United States demonstrated that each increase of 10 μg/m3 PM2.5 was correlated with a 16% increase in mortality from ischemic heart disease, despite the low concentration of PM2.5 that below the present 12 μg/m3 annual average standard in the United States (17). Though PM2.5 is considered to pose the greatest risk to health, there is emerging evidence that coarse particles can exert adverse health influences that are competitive to PM2.5 (18). Wang et al. reported that each 10 μg/m3 increase of daily PM10 was associated with a 1.63% increase in cardiovascular mortality (19). In addition to the size of particles, recent studies confirmed that the toxicity of PM is also related to their constituents, and some components of PM may be more harmful than others. Generally, most studies proved the dominant role of combustion-associated pollutants (20–23). Bell et al. reported that black carbon was one of the most harmful particle types (21). Similarly, a meta-analysis also found a significant association between the black carbon and adverse cardiovascular effects (22). An investigation in Pakistan indicated that exposure to nickel was closely associated with a substantial increase in the risk of cardiovascular diseases (23). As black carbon and nickel were known to be indicators for fossil fuel combustions and industrial emissions, these studies together showed the significant cardiovascular toxicity of combustion-related PM2.5. In contrast, constituents related to sea salt showed weak or no association (22, 24).
Cardiac arrhythmia is one of the various cardiovascular events induced by PM. A significant association exists between PM exposure and incidences of various arrhythmia subtypes (25, 26). Atrial fibrillation (AF) is the most common type of arrhythmia induced by PM exposure. A nationwide cohort study concerning the proarrhythmic effects of long-term exposure to PM reported that 10 μg/m3 increments of PM2.5 were associated with a 17.9% increase of AF, and 10 μg/m3 increments of PM10 was associated with a 3.4% increase (27). A recent epidemiological study conducted in Canada demonstrated that long-term exposure to PM2.5 was in association with the occurrence of AF even at a very low concentration level (28). A correlation was also found between short-term PM exposure and increased risks of AF. An investigation based on patients with dual-chamber implantable cardioverter-defibrillators (ICDs) suggested significant effects of PM2.5 on triggering AF. The odds of AF increased by 26% for each 6 μg/m3 increase in PM2.5 in the 2 h prior to the onset of AF (29). For healthy individuals, Lee et al. demonstrated that short-term exposure to PM2.5 was associated with AF in the general population with no AF history (30). Similar findings were also reported in other investigations (5, 31). Besides atrial arrhythmias, PM was also associated with ventricular arrhythmias. An investigation based on high-risk populations with implantable cardioverter-defibrillators (ICDs) or cardiac resynchronization therapy defibrillators (ICD-CRT) showed a positive and significant correlation between short-term exposure to PM and ventricular arrhythmias including VT and VF (32). The study also showed that patients who had previous myocardial infarction were more prone to PM-induced ventricular arrhythmias (32). Tsai et al. reported that PM2.5 exposure was associated with high ventricular premature complex (VPC) burden in patients without structural heart disease (33). The VPC burden is one of the clinical indicators of decreased cardiac function in continuous ECG monitoring (34, 35). It measures the percentage of ectopic beats in all heartbeats, and a higher VPC burden is associated with increased risks of cardiovascular mortality (36).
Potential Proarrhythmic Mechanisms
It is generally considered that PM-induced cardiac arrhythmias are triggered by several major mechanisms, including inflammatory responses, ANS dysfunctions, direct effects of translocated PM into circulation, and cardiac structural remodeling (9, 37) (Figure 2).
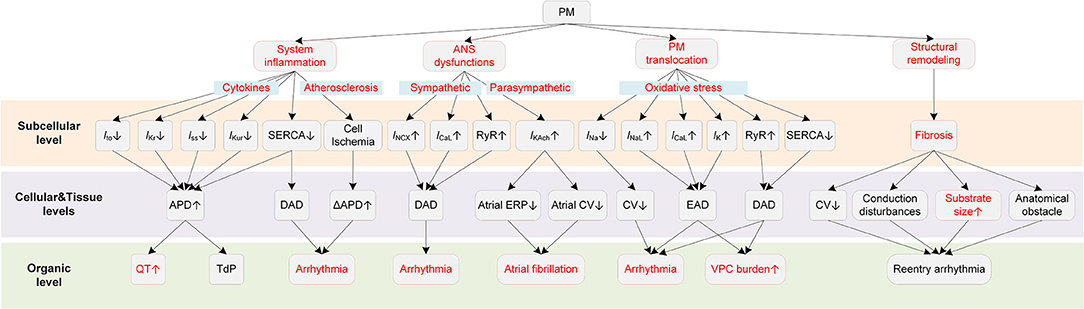
Figure 2. Potential mechanisms underlying PM-induced arrhythmias. Red boxes indicate effects of PM that have been explicitly demonstrated.
PM can exert proarrhythmic effects indirectly that are mediated through the systemic inflammatory responses (38–40). Systemic inflammation is known to contribute to the progression of atherosclerosis (41), resulting in chronic and acute ischemia. Ischemic heart disease leads to ion channel remodeling, augments spatial dispersion of depolarization, thereby providing substrates for cardiac arrhythmias (42, 43). Second, in some severe inflammatory states such as sepsis and septic shock, the increased cytokine levels cause myocardial dysfunction that might contribute to the occurrence of various types of arrhythmias. Besides, the inflammation-related coagulation response might also act as an indirect factor for arrhythmogenesis (38). Inflammatory cytokines can also directly modulate the function of ion channels and calcium homeostasis (44). For example, tumor necrosis factor (TNF), as one of the inflammatory cytokines, could reduce the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA), leading to increases in intracellular Ca2+ levels and risks of arrhythmogenesis (45). Recent evidence from clinical studies proved the direct influences of inflammatory cytokines on potassium currents, suggesting inflammation as a novel risk factor for QT-syndrome and Torsade de Pointes (TdP) (46, 47). Lazzerini et al. showed that systemic inflammation could directly prolong the corrected QT (QTc) interval via the cytokine-mediated ventricular electrical remodeling effects (48). Overall, these findings together reveal the role of inflammation in arrhythmia pathogenesis and point out one of the potential explanations for the PM2.5-induced cardiac arrhythmias.
Another indirect proarrhythmic pathway is the ANS dysfunction (49). PM is documented to be in relation with a decreased HRV (50, 51), which is an indicator of cardiac autonomic dysfunction and is a risk factor for fatal ventricular arrhythmias (52). Liao et al. observed that the magnitude of association between PM2.5 and AF was greatly attenuated after adjusting for cardiac autonomic modulation among healthy people, suggesting that ANS was partly responsible for the PM-induced cardiac electrophysiological changes (53). ANS can be subdivided into two antagonistic sets of nerves, i.e., the sympathetic nervous system, and the parasympathetic nervous system. The sympathetic stimulation in normal hearts increases the heart rate by enhancing the funny current (If), shortens APD, and reduces transmural dispersion of repolarization (54, 55). In contrast, in pathological states induced by PM, the dysregulated sympathetic stimulation might enhance the dispersion of repolarization (56, 57) and promotes afterdepolarization (58, 59). Specifically, the β-adrenergic receptor stimulation activates the stimulatory Gs proteins, which stimulates the adenylyl cyclase that catalyzes the conversion of adenosine triphosphate (ATP) to cyclic adenosine monophosphate (cAMP). The cAMP then activates the protein kinase A (PKA) and the latter causes the phosphorylation of the L-type calcium current (ICaL) and ryanodine receptors, leading to excessive calcium influx and sarcoplasmic reticulum (SR) Ca2+ release. Elevated intracellular Ca2+ activates the Na+/Ca2+ exchanger current (INCX) and finally induces delayed afterdepolarization (DAD) in myocytes (60, 61). Computer simulations further revealed that the SR Ca2+ overload and release can be produced synchronously and finally lead to ectopic beats (62). While it seems apparent that the vagal stimulation, as opposed to sympathetic stimulation, is antiarrhythmic by mitigating the influences of its counterpart, it can be proarrhythmic in the atria due to its different effects in atria and ventricles (54). In detail, the vagal activation prolongs APD and ERP in the ventricle (63, 64), whereas it reduces the atrial ERP, augments the spatial electrophysiological heterogeneity, and promotes EAD (54). Such difference is partly attributable to an inwardly rectifying potassium current abundant in the atria, i.e., IKAch. IKAch is activated by the parasympathetic neurotransmitter acetylcholine (Ach), and the activation of IKAch under the condition of vagal stimulation substantially shortens APD. Besides, simulations show that the IKAch can result in the hyperpolarization of the resting membrane potential, which in turn slows the conduction velocity in the atria (65). The effect of conduction slowing can be further enhanced by fibrosis—a condition that widely occurred in AF patients. The shortened APD and decreased CV induced by IKAch together contribute to a reduced wavelength for reentry and therefore increase the susceptibility to atrial arrhythmias (65). Above all, either sympathetic or parasympathetic dysfunctions can be proarrhythmic, with afterdepolarization acting as triggers while shortened refractory period and augmented spatial dispersion of repolarization acting as substrates for arrhythmogenesis.
Though the proarrhythmic effect of PM on cardiac electrical activity can be secondary to the systemic inflammatory process, in some instances, some small PM such as ultrafine particles can cross the pulmonary epithelium and penetrate into the circulation, leading to direct influences on the cardiovascular system (66, 67). Miller et al. observed that nanoparticles could translocate from the lung to the circulation, and these nanoparticles were found accumulated at sites of vascular inflammation in both mice and humans (68). These translocated particles can directly alter the membrane ion channels and the cellular electrophysiology of myocytes. Savi et al. reported that the nanoparticles directly entered ventricular cardiomyocytes, resulting in shortened action potential duration (APD) and effective refractory period (ERP), and also an increased membrane excitability (69). These effects together provided substrates for electrical alternans and increased the propensity to arrhythmias. It was also reported that the intracellular ROS formation was increased by 16% after only 1-h exposure, which partially accounted for the impaired channel functions (69). Indeed, oxidative stress was proved to be associated with arrhythmias via various actions (70, 71). In terms of ionic effects, oxidative stress was recorded to affect all major ionic currents (71). For sodium channels, the elevated ROS enhanced the late sodium current (INaL) (72) and decreased the sodium current (INa) (73). Such effects resulted in the occurrence of early afterdepolarization (EAD) and the decreased CV, which facilitated the formation of reentry arrhythmia by providing both the trigger and the substrate. Besides, ROS can also stimulate ICaL (74) and modulate intracellular Ca2+ handling through influencing ryanodine receptors and SERCA (75–78). These effects can lead to abnormal depolarization and induce triggered activity. Specifically, ROS decreases the repolarization reserve by stimulating ICaL, which facilitates the formation of EAD (74). Besides, the elevated ROS oxidases and enhances the Ca2+/calmodulin-dependent kinase II (CaMKII), and the latter then phosphorylates and activates the ryanodine receptor (79, 80). The ryanodine receptor is known to act a physiological role in a biological process named “calcium-induced calcium release (CICR)”, whereas the oxidative stress-induced activation of the ryanodine receptor may lead to Ca2+ leak (79). ROS can also inhibit SERCA. SERCA is responsible for the calcium transportation from the cytosol back into the SR during diastole, and the inhibition of SERCA hinders the SR Ca2+ reuptake (81). Above two effects of ROS together contribute to overloaded intracellular Ca2+. The overloaded Ca2+ is expelled in exchange for Na+ via Na+/Ca2+ exchanger, thereby forming INCX and may triggering delayed afterdepolarizations (DADs). For potassium channels, ROS was reported to suppress the ATP-sensitive potassium current (IKATP) (82), the transient outward potassium current (Ito) (83), the rapid delayed rectifier potassium current (IKr) (84), and the slow delayed rectifier potassium current (IKs) (84), which led to a prolonged APD and the occurrence of EAD. Besides the ionic currents, ROS can also change the cell coupling by promoting myocardial fibrosis (85) or impairing gap junctions (86). Taken together, translocated particles can directly influence cardiac electrophysiological activities by affecting various ion channels, and ROS might be an important mediator in these actions.
Finally, PM2.5 can also exert proarrhythmic effects by promoting cardiac structural remodeling. An epidemiological study reported that exposure to PM2.5 was significantly associated with cardiac ventricular dilatation including larger left ventricular end-diastolic and end-systolic volumes, and right ventricular end-diastolic volume (87). The remodeling effect of PM2.5 was also confirmed in experimental studies (88–90). The structural remodeling induced by air pollutants can promote the development of the AF and the lethal VF. In detail, tissue fibrosis slows the conduction velocity while increases the heart dimension (91), resulting in an increased substrate size for reentry arrhythmias (92). Fibrosis also promotes arrhythmias by interrupting the fiber bundle continuity, leading to local conduction disturbances or anatomical reentry (93). The induced arrhythmias in turn cause more serious cardiac remodeling and abnormalities, forming an auto-reinforcing process such as ‘AF-begets-AF' (91, 94, 95). Computer simulations substantiate the association between structural remodeling caused by PM exposure and cardiac arrhythmias and provide insight into the mechanisms. Most recently, Palacio et al. modeled the diffuse myocardial fibrosis, a type of structural remodeling caused by exposure to ambient PM, in a 3D model of human atria (96). The simulation results suggested that the PM-induced diffuse fibrosis reduced the CV and increased the reentrant fibrillary dynamics with the increase of the fibrosis density.
Carbon Monoxide
Carbon monoxide (CO) is a hazardous air pollutant presented particularly in heavily polluted urban areas. It is colorless and odorless, and primarily arises from incomplete combustion of hydrocarbon sources. Exposure to environmental CO such as road traffic has shown to be associated with poor cardiovascular outcomes (97).
Epidemiological and Clinical Reports
CO is among the most frequently studied gaseous pollutants (98). A meta-analysis based on 9 million people in the United States established the correlation between the ambient CO exposure and heart failure that 1 ppm increment of CO was associated with a 3.52% increase in heart failure hospitalizations or mortality (98). A recent study reported that ischemic heart disease deaths increased 21.1% for every 10 μg/m3 increase in CO (99). Accidental exposure to higher levels of CO is not rare in industrial settings or common road traffic environments, which could lead to ECG changes and arrhythmias. A case-control study by Hanci et al. reported that 5 of 30 acute CO poisoning patients had arrhythmias, with one of them had ventricular arrhythmias and the other four had atrial arrhythmias (100). Among those patients of atrial arrhythmias, three of them were supraventricular tachycardia and the other one had atrial extrasystoles. Akdemir et al. reported that a patient with no history of heart diseases and arrhythmias developed AF after CO exposure (101). Gedela et al. reported a similar case for CO-induced AF (102). Effects of CO on cardiac electrophysiology can be manifested on the ECG as ST-T changes, prolonged QT intervals, increased QT dispersion durations, and increased P-wave dispersion durations. ST-segment changes are among the most frequently observed ECG manifestations of CO poisoning, and various abnormal ST-T morphologies including ST-segment elevation, ST depression, and T-wave inversion have been reported in CO poisoning patients (103–105). Frequently observed ST-segment changes indicate that the myocardium is prone to be injured in CO poisoning, and a previous clinical investigation showed that up to 37% of the enrolled patients developed myocardial injury after CO exposure (103). As for the QT changes, Hanci et al. reported an average QTc interval of 429.6 ms in the CO poisoning group among which 30% had QTc intervals above 440 ms. The average QTc interval in the control group, in contrast, was only 385.6 ms and none of them had extended QTc (100). In a recent clinical case, a QTc interval of up to 622 ms was observed (106). In addition to the QT prolongation effect, CO was also reported to significantly increase the dispersions of both QT intervals (QTd) and P-waves (Pwd), and the Pwd was almost doubled in the CO group (106).
Potential Proarrhythmic Mechanisms
Potential mechanisms of CO-induced arrhythmias are summarized in Figure 3. CO has been proven to have mitochondrial toxicity. The high binding affinity of CO to intracellular myoglobin in the myocardium would impair the oxygen delivery to the mitochondria, causing subsequent energy crisis and cardiac contractile decrease (107). CO could also impair the mitochondrial respiratory chain via inhibiting cytochrome C oxidase, causing decreased glutathione concentrations and declined ATP generation (107). In addition to the intracellular influences, CO also affects multiple membrane channels. For the calcium channel, Scragg et al. found that CO suppressed the ICaL (108). The inhibition effects occurred via an increase in ROS production specifically from mitochondria. In settings of chronic exposure to environmentally relevant CO levels, the alterations of ICaL were not consistent with above findings. Specifically, CO was observed to increase ICaL, and such influences were only observed in epicardial cells (109). Though the uneven changes of the ICaL might alter the transmural repolarization dispersion and potentially provide a substrate for ventricular arrhythmias, this study considered it to have limited impact (109). Instead, the study attributed the arrhythmogenic effect of chronic CO exposure to Ca2+ overload. Particularly, the intracellular Ca2+ overload results from the reduced SERCA expression and the PKA-phosphorylation of ryanodine receptors. The impairment in the Ca2+ reuptake by SR, and the Ca2+ leak in SR, together contribute to the Ca2+ overload. The overloaded Ca2+ in turn activates INCX and leads to DAD (109). In addition to calcium currents, sodium currents including INa and INaL could also be affected by CO. Dallas et al. showed that CO augmented INaL through the NO-mediated pathway (110). CO was able to activate the synthesis of NO, and the elevated NO level consequently led to nitrosylation of the Nav1.5 channel protein, causing the augmentation of INaL. The above effect of CO led to EAD-like Ca2+ transients. In the circumstances of additional stress (mimicked by injecting β-adrenergic agonist isoprenaline), CO-induced EADs could easily lead to VF and sudden death (110). In contrast to the enhanced INaL, CO was reported to inhibit the INa, and the inhibition was also NO-dependent (111). The inhibition effect was associated with a hyperpolarization shift of steady-state inactivation properties of channels. Given the fact that the inhibition of INa can lead to Brugada syndrome-like arrhythmias (112), the study suggested a new potential proarrhythmic mechanism of CO (111). Finally, for potassium channels, CO decreases the rapidly activating delayed rectifier potassium current (IKr) (113) and the inwardly rectifying potassium channel current (IK1) (114). The former was observed in guinea pigs and HEK293 cells and was mediated by the formation of NO and peroxynitrite (113). Liang et al. suggested that the prolonged APD of rat myocytes arouse from the combined effects of CO on INaL and IK1, and the ratio of APD prolonging by INaL and IK1 was about 4:3. In addition to above experimental observations about the alteration in ionic currents upon exposure to CO, it should be noted that CO can also indirectly contribute to the cellular electrophysiological remodeling via myocardial ischemia. Under the ischemic condition, intracellular acidosis activates the Na+/H+ exchanger to exclude hydrogen ions in exchange for sodium ions, and the elevated sodium is then exchanged for calcium through the Na+/Ca2+ exchanger, which finally leads to intracellular Ca2+ overload (115, 116). Acidosis also inhibits ICaL, and this effect results in APD shortening together with the reduced IKATP by hypoxia (115). Besides, the ischemia is also accompanied by hyperkalemia. The elevated extracellular potassium results in depolarization of the resting potential, which then partially or completely inactivates sodium channels but activates the late sodium channel (115, 117).
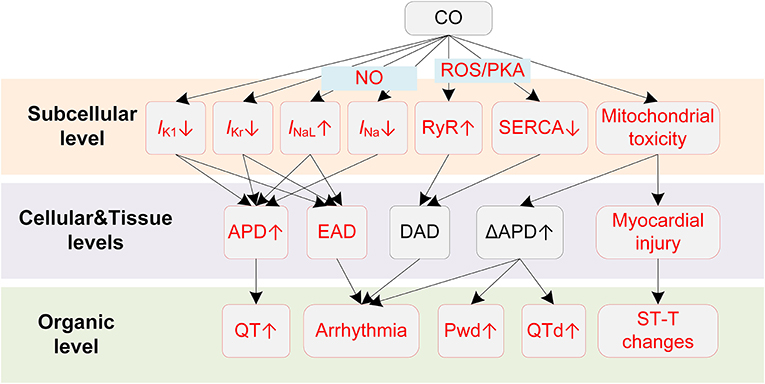
Figure 3. Potential mechanisms underlying CO-induced arrhythmias. Red boxes indicate effects of CO that have been explicitly demonstrated.
Above experiments regarding the alterations of ion channels by CO provided the cellular basis for the clinically observed ECG changes and CO-induced arrhythmias. The myocardial injury that comprises myocardial ischemia and infarction is the direct cause of the ST-T change, and it is also the primary factor responsible for the CO-induced arrhythmias (97). Depending on the injured location and the time of injury, the MI-induced ST-T changes can be very different. In the case of subendocardial infarction, as a common clinical type, the phase-2 of the endocardial action potential is lower than the healthy epicardial myocytes, which is manifested as the depressed ST-segment on the ECG. The involvement of IKATP and the decreased ICaL by acidosis under ischemia condition shortens the endocardial APD, and therefore changes the transmural repolarization sequence, causing an inverted T-wave. In contrast, the transmural or subepicardial infarction types are manifested as enhanced ST-segment. Arrhythmogenesis substrates can be formed in either case, as the spatial dispersion of repolarization increases the risk of developing unidirectional block when the premature stimulus (i.e., EAD, DAD) occurs. Furthermore, the ischemia-induced cellular electrical remodeling also contributes to repolarization heterogeneities, especially near the ‘border zone' of infarction (118).
In addition to myocardial injury-related ST-T changes, CO also results in QT changes that might be achieved through distinct pathways other than ischemia-induced ion channel remodeling. The QT interval on the ECG corresponds to the time it takes for heart ventricles to depolarize and repolarize, and as the depolarization is quite fast, the repolarization phase accounts for most of the QT duration. A prolonged QT interval on the ECG indicates that the ventricle repolarization is delayed, or from a cellular perspective, the duration of AP repolarization is prolonged. In this regard, the observed QT prolongation in patients with CO poisoning can be attributed to the impaired repolarization reserve resulted from several altered ion channels, i.e., increased INaL (110), decreased IKr (113), and decreased IK1 (114). Prolonged QT or QTc durations increase the risk for polymorphic VT, VF, TdP, and even sudden death (119, 120). In this regard, Hanci et al. found a positive correlation between COHb level and QTc duration, providing a rationale between acute CO poisoning and the development of ventricular arrhythmia (100). Noted that the prolonged QTc was also observed when people were exposed to traffic-related air pollutants (121). Therefore, CO might be a major role in traffic-related pollution-induced heart diseases, exerting independent detrimental influences on cardiovascular systems. Another observed manifestation of QT changes by CO is the increased QTd. The QTd reflects the transmural heterogeneities of repolarization time within ventricles, and the heterogeneities are secondary to regional differences in APD and activation time (122). Recent simulation studies suggested that the increased transmural dispersion of repolarization (TDR) by CO was attributed to the more prolonged APD in midmyocardial cells comparing to epicardial or endocardial cells (123, 124), and the different APD changes may arise from the heterogeneous current densities in different cell types (125). The increased TDR then contributes to higher risks of developing unidirectional conduction block (measured as the vulnerable window), and finally leads to TdP, reentry arrhythmias, and VF (126). Extensive investigations have reported that QTd was significantly increased in CO intoxicated patients (100, 127). Therefore, the enhanced repolarization dispersion is another potential proarrhythmic factor of CO.
The CO-induced ECG manifestations and arrhythmias related to the atria also have their cellular basis. CO was observed to increase the P-wave dispersion (Pwd), which is a non-invasive indicator of AF. The Pwd is measured as the difference between the maximum and minimum P-wave durations on the ECG, and it reflects the inhomogeneous and discontinuous propagation of sinus impulses through the atria. Similar to QTd, the increased Pwd is secondary to the increased heterogeneity of electrophysiological properties within the atrial myocardium (128). In the case of CO, the clinically observed Pwd may originate from several ionic remodeling effects. First, CO could decrease the conduction velocity of excitation wave (123, 124); therefore the intra- and interatrial conduction times of sinus node impulses can be prolonged. Such assumption is based on the fact that CO could inhibit INa (111). As INa is a prominent factor in determining the conduction velocity in cardiac tissues, its inhibition will delay the propagation of excitation in the atria. In addition to the conduction properties, CO may also contribute to the regional heterogeneity by differently prolonging the APD in a dose-dependent way (124). The increased heterogeneity provides substrates for atrial arrhythmias by increasing the likelihood of unidirectional conduction block, while the CO-induced EAD (via increased INaL) (110) and DAD (due to the Ca2+ overload) (109) act as triggers in initiating atrial arrhythmias, providing that the afterdepolarization activities were able to overcome the source-sink effect and form an ectopic beat (129, 130). These cellular mechanisms provide insights of the clinical observed Pwd changes and atrial arrhythmias.
Hydrogen Sulfide
Hydrogen sulfide (H2S) is a colorless air pollutant with a strong odor of rotten eggs. It can be largely produced in industrial activities, such as food processing, coke ovens, and petroleum refineries.
Epidemiological and Clinical Reports
Acute H2S poisoning often occurs in occupational exposures. H2S is immediately fatal when it reaches concentrations of over 500–1,000 ppm (131). For environmentally relevant H2S concentration, an investigation based on the naturally H2S-exposed population showed that cardiovascular was one of the adverse health effects of chronic H2S exposure (132). Another study that focused on the short-term effects of low-level H2S exposure showed that H2S exceeding 7 μg/m3 was associated with admission and emergency department visits with heart disease (133). The study also concluded that males and old people were more susceptible to H2S. Available clinical reports of H2S poisoning demonstrated consistent cardiac changes, i.e., ST-segment elevation and malignant arrhythmias. Specifically, in an acute H2S exposure case, the patient was reported to have episodes of VT two days after exposure, and the course was complicated by other observations including ST-segment elevation, hyperkalemia, acidosis, etc. The patient died on the third day in the hospital (134). The ST-segment elevation was also reported in another clinical study (135). The patient was exposed to 20.3~25.6 mg/m3 (20.3~25.6 ppb) H2S but showed no obvious ECG changes on the first day of admission; however, subsequent significant ST-segment elevation along with changes in markers of myocardial injury was observed on the fourth day of hospitalization (135). Another recent case presented identical cardiac changes and clinical course, where the patient was free of ECG abnormalities on the first day of exposure, but showed extensive elevation of ST-segment and dramatic increase of myocardial enzyme index two days after exposure. The patient then developed VF on the next day (136). It can be easily observed from above cases that patients with H2S poisoning usually presented no clinical symptoms or ECG changes immediately after exposure, but could experience ST-segment elevation and develop lethal arrhythmias after 2~4 days. The delayed and abrupt onset of cardiac changes are rather dangerous and suggest the insidious effects of H2S on the myocardium.
Potential Proarrhythmic Mechanisms
Potential mechanisms of H2S-induced arrhythmias are illustrated in Figure 4. The clinically observed ST-segment changes on the ECG suggest the occurrence of myocardial injury when exposed to H2S. Similar to CO, H2S causes myocardial damages due to its mitochondria toxicity. Specifically, H2S was reported to inhibit cytochrome C oxidase (137), leading to the blockade of the electron transport in the respiratory chain and the utilization of molecular oxygen (135, 138). The damaged respiratory chain could subsequently result in disrupted aerobic metabolism of cells and impaired ATP production, and ultimately leads to reduced oxygen use of cardiomyocytes and myocardial damage (136). Large dispersion in repolarization can be formed under myocardial injury conditions, e.g., in the border zone of the regionally ischemic heart or near the infarcted tissue area (139), and thereby increases the likelihood of induction of unidirectional block and arrhythmias.
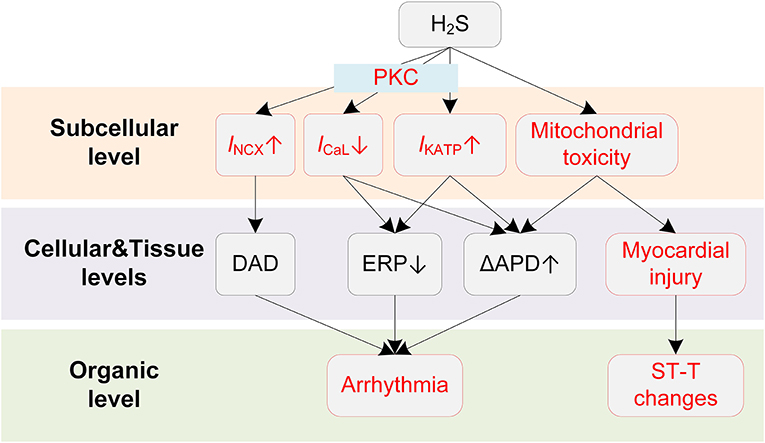
Figure 4. Potential mechanisms underlying H2S-induced arrhythmias. Red boxes indicate effects of H2S that have been explicitly demonstrated.
While the repolarization dispersion provides a plausible explanation for the frequently observed malignant arrhythmias, the proarrhythmic effects of H2S can be further exaggerated by its actions on cardiac cellular electrophysiology. Specifically, hypoxia is known to activate the IKATP due to a dropped ratio of adenosine triphosphate (ATP) and adenosine diphosphate (ADP), and H2S is also proved to be a KATP channel opener (140). The activation of IKATP can be proarrhythmic as it accelerates the phase 3 repolarization and reduces APD and ERP, leading to shortened critical length for initiating reentry arrhythmias (141). H2S was also reported to inhibit the ICaL in a concentration-dependent manner (142, 143), and the involvement of ICaL was further evidenced by the observation that blockage of IKATP did not completely eliminate the effect of H2S (144). In our previous simulation study, we investigated the proarrhythmic effects of H2S using a multi-scale virtual heart (43). Briefly, the effects of H2S on ionic currents were incorporated into a mathematical myocyte model, and were further expanded to tissue levels. The simulation results suggested that H2S could decrease the spatial critical length of initiating arrhythmias, augment transmural repolarization dispersion, and depress cell excitability. These actions together lead to increased tissue susceptibility for initiation and maintenance of reentry arrhythmia. In addition, the vulnerability to reentry arrhythmia could be further exacerbated in mild ischemia due to the involvement of IKATP (43).
The mechanism of proarrhythmic effects of H2S can be more complicated in the atria. Recently, Chan et al. performed a comprehensive investigation on the influences of H2S on the atria and provided novel insights into the proarrhythmic effects of H2S (145). The study reported that NaHS (H2S donor) significantly reduced sinoatrial node (SAN) beating rates, causing SAN dysfunction and potentially the AF. In pulmonary vein myocytes, NaHS increased IKATP and INCX. The activation of INCX could potentially lead to DADs, acting as triggers for arrhythmogenesis. These effects were mediated by the protein kinase C (PKC) as they were attenuated by PKC inhibitors. The PKC-mediated increases in IKATP and INCX were also observed in atrial myocytes; however, such effects were only presented in left atrial myocytes but not right atrial myocytes. This interesting observation demonstrates that H2S can lead to increased interatrial dispersion, and therefore increases the risk of supraventricular arrhythmias (145). Noted that PKC may also regulate other channels in addition to IKATP and INCX, such as Ito (146), INaL (147), IKs (148), and their potential roles in H2S-induced arrhythmias warrants further investigation.
Sulfur Dioxide
Sulfur dioxide (SO2) is a colorless gaseous pollutant with a pungent odor. SO2 in the air comes mainly from the burning of fossil fuels contaminated with sulfur compounds or from copper smelting. It can also be released naturally from volcanic eruptions.
Epidemiological and Clinical Reports
Epidemiological studies have demonstrated the correlation between SO2 and cardiovascular events. A nationwide study in China demonstrated that SO2 was associated with increased total and cardiorespiratory mortality (149). Amsalu et al. reported that the increased SO2 concentration was associated with multiple cardiovascular diseases, including coronary heart disease, AF, and heart failure (150). Specifically for arrhythmias, Zhao et al. reported that a 10 μg/m3 increase of daily SO2 led to a 2.07% increase of outpatient arrhythmia visits, and the influences were stronger in older people and in females (151). A recent epidemiological study showed that SO2 was significantly associated with arrhythmias (152). The proarrhythmic effect of SO2 was significant in the middle-aged population and was primarily observed in the cold season (152). The cardiovascular influences were also observed after long-term SO2 exposure. An investigation conducted in South Korea suggested that long-term exposure to a high level of SO2 was consistently and significantly associated with an increased ischemic heart disease mortality (153).
Potential Proarrhythmic Mechanisms
SO2 can modulate various ion channels in cardiac myocytes, causing electrophysiological remodeling that predisposes to cardiac arrhythmias (Figure 5). Nie and Meng presented a series of experiments about the effects of SO2 derivations on ion channels in rat cardiac myocytes (154–156). In (154), the authors reported that the SO2 derivatives dose-dependently enhanced INa through a depolarizing shift of the inactivation curve. At a concentration of 10 μM, the half inactivation potential was shifted by 7 mV, causing an increase of ~42% in peak amplitude of INa. Besides, the time courses of activation, inactivation, and recovery from inactivation were all accelerated (154). SO2 derivatives were also found to increase ICaL in a dose-dependent manner (156). The activation and inactivation curves of ICaL were shifted by 10 and 5 mV, respectively, toward depolarization direction, and the channel inactivation and recovery were accelerated (156). Consistent with above two studies, the INCX was inhibited by SO2 derivatives, and this was accompanied by the intracellular Ca2+ overload (157). To be more specific, the enhanced ICaL leads to an increased intracellular Ca2+, which should be expelled via Na+/Ca2+ exchanger under physiological conditions. However, the expulsion is attenuated by the increased intracellular Na+ because of the SO2-induced increase in INa. The accumulated Ca2+ eventually leads to irreversible myocardial damages, providing a possible mechanism for SO2-induced myocardial injury. For potassium channels, Nie and Meng reported that SO2 derivatives at 10 μM increased Ito and IK1 by 37.4% and 26.2, respectively. The enhancement was accompanied by hyperpolarizing shifted activation curve and depolarizing shifted inactivation curve and the shortened time courses (155). Similar to H2S, we investigated the proarrhythmic effects of SO2 using simulation approaches (158). Simulation results revealed that SO2-induced arrhythmogenesis may arise from several aspects. First, SO2 decreased APD and ERP, which generally facilitated the formulation of triggered activities and increased susceptibility to arrhythmias. As another consequence of the decreased ERP, the spatial vulnerability to reentry arrhythmia was increased in the SO2 affected tissue. The critical size for initiating reentry arrhythmia was shortened due to the decreased wavelength of spiral waves, which provided substrates for arrhythmogenesis. Third, the transmural repolarization dispersion was remodeled, and the decreased ΔERP attenuated the drift of the spiral wave and prolonged its lifespan (158).
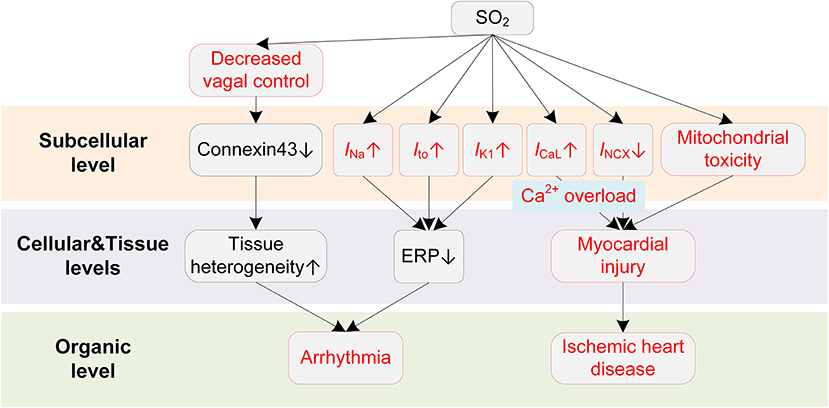
Figure 5. Potential mechanisms underlying SO2-induced arrhythmias. Red boxes indicate effects of SO2 that have been explicitly demonstrated.
SO2 also causes other cardiac dysfunctions that indirectly lead to arrhythmias. For example, a human challenge study presented direct evidence that SO2 could cause a decrease in cardiac vagal control (159). The vagal control reflects the input of the parasympathetic branch of the ANS to cardiac regulation (160), and it is classically thought to be antiarrhythmic by balancing the sympathetic stimulation and maintaining the gap junction function (i.e., preservation of phosphorylated connexin 43) in structural remodeled hearts (161). The decreased vagal activities, consequently, lead to the occurrence of arrhythmias. The systemic inflammatory response was excluded since no systemic acute phase or coagulant response was observed during exposure (159). Besides, several studies have suggested that SO2 could cause mitochondrial dysfunction and induce ROS production (162, 163), which provide possible underlying mechanisms for the significant association of SO2 to ischemic heart diseases. Above two pathological pathways for promoting arrhythmias have been discussed in previous sections and are not discussed here.
Ozone
Ozone (O3) is a pale blue gas and has a very pungent odor. Ground-level O3 is formed primarily from photochemical reactions between volatile organic compounds and nitrogen oxides (164).
Epidemiological and Clinical Reports
Epidemiological reports have provided substantial evidence on the associations of both short-term and long-term exposures to ambient O3 with adverse cardiovascular effects. For short-term effects, Buadong et al. reported that a 10 μg/m3 increase of O3 contributed to a 0.5% increase in the numbers of daily emergency hospital visits for cardiovascular diseases among aged individuals (165). Ensor et al. reported that the increase of O3 in the previous 1–3 h was associated with an increased cardiac arrest risk (166). An investigation conducted in Italy reported a direct and significant correlation between the number of daily ST-elevation myocardial infarction patients and the O3 concentration of the same day (167). On the other hand, Turner et al. reported significant positive associations between the long-term O3 exposure and cardiovascular mortality (168). The long-term influences were also observed in a recent cohort study in the United States, which reported significant associations of the long-term O3 exposure to cardiovascular diseases and ischemic heart diseases (169). For heart rhythm disorders, Rich et al. found a statistically significant association between ventricular arrhythmia and O3 exposure in 24 h before the arrhythmia (170). They also reported a positive association between episodes of paroxysmal AF and 1-h O3 exposure (171). The controlled human exposure to O3 reported three changes in cardiovascular systems, including an increase of inflammatory markers, changes in fibrinolytic markers, and changes in autonomic control of heart rate (172, 173). Prolonged QT intervals were also reported in both prospective follow-up studies and human exposure experiments, and such effect was immediately observed after exposure (121, 172).
Potential Proarrhythmic Mechanisms
Animal exposure studies have revealed that O3 could result in a series of changes in cardiac electrophysiology (Figure 6). Farraj et al. reported that exposure to O3 of 0.2 ppm was already able to increase myocardial susceptibility to arrhythmias, and a higher concentration (0.8 ppm) resulted in more rhythm abnormalities, including bradycardia, prolonged PR intervals, depressed ST segments, and frequent atrial premature beats and conduction blocks (174). The mechanistic investigation using a rat exposure model suggested that the inflammation played a major role in O3-induced arrhythmias (175). It was observed that acute O3 exposure led to paroxysmal VT in rats, and the changes of inflammatory cytokines such as tumor necrosis factor α (TNF-α) and interleukin 6 (IL-6) were greater than those of oxidative stress, e.g., superoxide dismutase (SOD) and serum malondialdehyde (MDA) (175). A recent clinical study demonstrated that systemic inflammation could lead to QTc prolongation via cytokine-mediated effects (48), and this finding provides new possible mechanisms for the O3-induced QT-prolonging observed in O3 exposure experiments. From a cellular perspective, the inflammation-induced QT prolongation arises from the alterations of multiple ion channels by cytokines. Specifically, TNF-α at physiologically relevant concentrations was recorded to reduce SERCA (45) and downregulate multiple repolarization currents in cardiomyocytes of rodent animals, including the transient outward potassium current (Ito) (45, 176, 177), the steady state current (Iss) (45), and the ultra-rapid outward current (IKur) (177). TNF-α was also reported to decrease IKr in the HEK293 cell (178). In addition to the TNF-α, other inflammatory cytokines, e.g., IL-1β and IL-6, could also lead to APD prolongation via decreasing Ito (179) or increasing ICaL (180). Above alterations of ionic currents in the inflammatory condition together contribute to delayed repolarization of AP and manifest as the prolonged QT interval on the ECG.
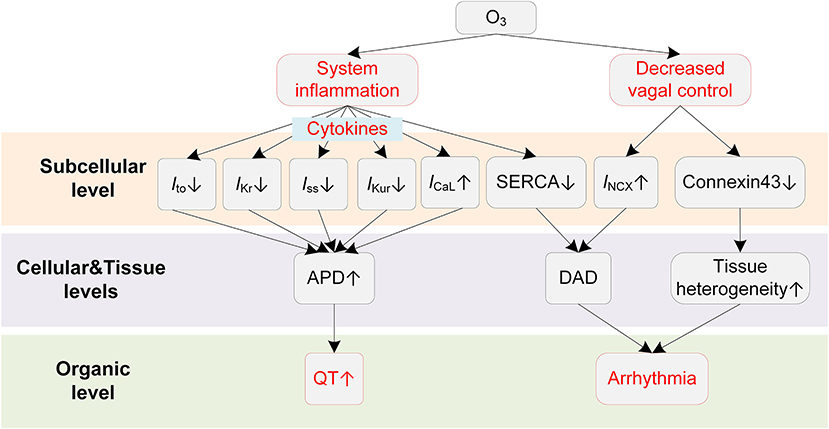
Figure 6. Potential mechanisms underlying O3-induced arrhythmias. Red boxes indicate effects of O3 that have been explicitly demonstrated.
ANS is also involved in the proarrhythmic effects of O3. The decreased HRV has been observed in both animal (175) and human exposure studies (172). As stated in previous sections, the decreased HRV implies weakened antiarrhythmic effects by the parasympathetic nervous system; it therefore acts as another proarrhythmic pathway after exposure to O3.
Currently, there is no experimental investigation showing the influence of O3 on cardiac ion channels. Therefore, the O3-induced subcellular electrophysiological changes are worth further study in the future.
Nitrogen Dioxide
Nitrogen dioxide (NO2) is a reddish-brown gaseous pollutant with a pungent and irritating odor. It is formed primarily from high temperature combustion, and the prominent sources of NO2 include power plants, industrial furnaces and boilers, and motor vehicles (181).
Epidemiological and Clinical Reports
Epidemiological studies have identified significant associations between short-term or long-term exposures to NO2 and hospital admissions for heart rhythm disorders and other cardiovascular diseases. An investigation conducted in the UK showed positive and statistically significant associations between short-term NO2 exposure and cardiovascular events, including an increase of 2.9% for arrhythmias (182). Similar results were reported worldwide, for example, in Spain (183), Belgium (184), and China (151, 152, 185). For the long-term NO2 exposures, an early epidemiological study showed that long-term exposure to NO2 had a significant effect on emergency room visits for arrhythmias (186). Such proarrhythmic effect of long-exposure to NO2 was supported by a recent investigation that traffic-relevant NO2 was in association with a higher risk of AF in both the young and middle-aged population (187). In addition, long-term NO2 exposure was also strongly associated with the risk of myocardial infarction (188).
Noted that an increasing amount of investigations propose the hypothesis that NO2 might be a surrogate for other pollutants (PM2.5) rather than directly influencing cardiovascular health (184, 189). The hypothesis is raised due to the same combustion emission sources of NO2 and other pollutants, but it is still disputable and not consistent in different studies. Seaton et al. first suggested a surrogate role of NO2, indicating that the toxicity of low concentration NO2 was just the result of confounding by PM (190). Scaife et al. also reported that NO2 failed to affect HRV in the absence of other pollutants (191). On the other hand, Samoli et al. reported an independent effect of NO2 on cardiovascular mortality (192). Besides, a national coverage study in the UK reported a strong NO2 effect with arrhythmias including AF and heart failure, and this strong effect persisted after adjusting PM2.5. In contrast, the PM2.5 effect was somewhat reduced when NO2 was adjusted (182). In summary, the underlying mechanisms for NO2-induced cardiovascular diseases, and the joint effects of NO2 and other pollutants deserve more in-depth investigations.
Potential Proarrhythmic Mechanisms
Potential mechanisms of NO2-induced arrhythmias are summarized in Figure 7. Bradycardia is the major ECG abnormality after acute exposure to NO2, according to previous animal exposure experiments (193, 194). More severe arrhythmias were observed after 90 min to 3 h exposure, which were accompanied by atrioventricular blocks, premature beats, and wandering pacemakers. The severity of abnormalities was dependent on the NO2 concentration and exposure time (193).
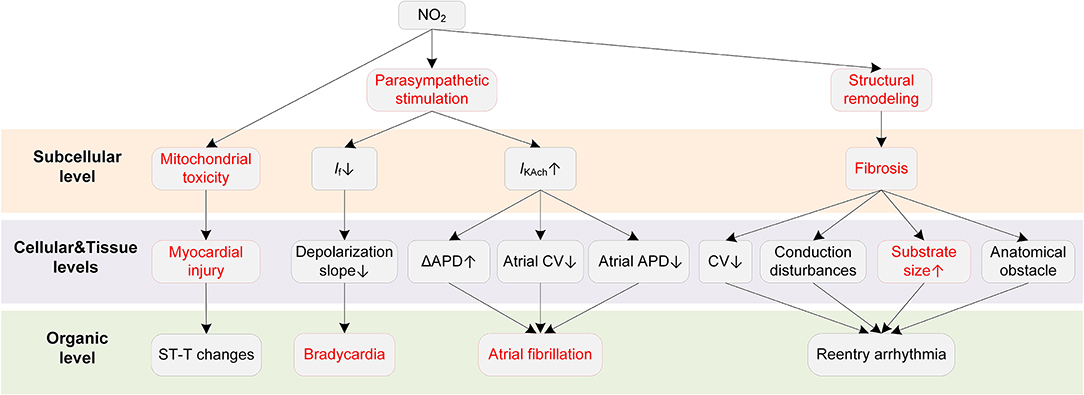
Figure 7. Potential mechanisms underlying NO2-induced arrhythmias. Red boxes indicate effects of NO2 that have been explicitly demonstrated.
The ANS is a critical pathway for NO2 to exert adverse cardiovascular effects. In an early animal investigation, exposure to NO2 led to bradycardia arrhythmias in healthy rats (193). However, the bradycardia was not observed in rats administered with the nitrite solution. The experimental results suggested that the elevated nitrite (NO2−) and nitrate (NO3−) in blood, despite being one of the consequences of the exposure to NO2, had no association with the arrhythmias caused by NO2. The study concluded that the changes in parasympathetic nervous activity were responsible for the proarrhythmic effects of NO2 (193). The involvement of ANS in the effects of NO2 was supported by some following studies (195, 196). From a cellular perspective, the NO2-induced parasympathetic stimulation decreases the If and the slope of phase 4 depolarization in the SAN and atrioventricular node (AVN), and finally slows the heart rate (197). The parasympathetic activation by NO2 may also contribute to atrial arrhythmia. It has been demonstrated that whether the parasympathetic stimulation is proarrhythmic or antiarrhythmic depends on the heart region (54). Specifically, the parasympathetic stimulation reduces ERP, augments the spatial electrophysiological heterogeneity, and promotes EAD particularly in the atria (54), whereas it prolongs APD and ERP in the ventricle (63, 64). These atria-specific effects provide a possible basis for the associations between NO2 and atrial arrhythmias that are frequently reported in epidemiological studies. In addition to the ANS, structural remodeling is another proarrhythmic factor of NO2. Exposure to a higher NO2 concentration was reported to be associated with larger biventricular volume in healthy individuals (87, 198). There were also studies considering NO2 and PM together as a joint proarrhythmic factor (182, 183).
As far as we are concerned, there are no studies investigating the alterations in ionic currents upon exposure to NO2. However, a recent study suggested that NO2 could impair the mitochondrial function under conditions of repeated exposures to NO2 at air pollution relevant levels (199). Mitochondrial dysfunctions including alterations of ATP synthesis and oxidative phosphorylation, and an increase in mitochondrial ROS production was observed after repeated NO2 exposures (15 h per week that lasted three weeks). In contrast, one-time acute exposure only induced moderate and reversible mitochondrial ROS production, and the ROS increase was not accompanied by mitochondrial alterations (199). The mitochondrial toxicity provides possible reasons for the myocardial infarction induced by the long-term exposure to NO2.
Discussion
Air pollution is among the main factors in triggering cardiac arrhythmias. In this review, we focused on six common air pollutants, namely PM, CO, H2S, SO2, O3, and NO2, presented the epidemiological and clinical evidence of air pollution-induced arrhythmias in recent years, and analyzed the underlying mechanisms for each of the air pollutants. Based on the literature in this review, we summarized several areas that warrant further studies in the future.
First, confounding effects among pollutants should be examined at molecular and cellular levels, while epidemiological or exposure studies concerning individual air pollutant are needed. In detail, epidemiological studies have proved that some combinations of pollutants might enhance the toxicity of each other and lead to unexpected biological alterations; however, the corresponding cellular mechanisms remain unclear, with most basic medical research still focusing on a single factor, i.e., an individual pollutant or a specific component of PM. On the other hand, epidemiological or exposure studies with regard to a specific pollutant or PM component are relatively rare, which aggregates the mismatch of epidemiological evidence and mechanical findings at the cellular level. Since most epidemiological studies present the integral influence of various air pollutants, confounding research can be more persuasive in explaining complex mechanisms of pollution-induced arrhythmias.
Next, as illustrated in Figure 1, the pathological pathways for air pollution-induced arrhythmias can be interrelated or overlapped. For example, prolonged QT interval can be an integral effect of CO by enhancing INaL, suppressing IK1, and subsequently prolonging the APD (114). However, such an effect may also arise as a consequence of systemic inflammation induced by O3, as the produced inflammatory cytokines contribute to electrical remodeling and prolong the QTc (48). Another example that has been frequently reported in relevant studies is the ST-T changes, including ST-segment elevation, ST-segment depression, and T wave inversion. The altered ST indicates impaired repolarization. It may arise from the tissue ischemia directly by some gaseous air pollutants, i.e., H2S and CO, but it can also result from the aggravated atherosclerosis by the PM2.5-induced systemic inflammation. In addition, the elevated ROS triggered by translocated PM can lead to myocyte apoptosis. These factors together lead to myocardial injury, generating an abnormal ST morphology. Due to the interrelated mechanisms, thorough investigations are always necessary before concluding proarrhythmic effects for a specific air pollutant. In contrast to the complicated and interrelated pathways, there are only a handful of experimental studies presenting the specific alterations in ionic currents upon exposure to air pollutants, which limits the summarized mechanisms in this review. Therefore, more fundamental studies are expected to elucidate the cellular electrophysiological effects of air pollutants.
Third, the computer simulation supplies an effective tool for integrating multiscale experimental data and for investigating the mechanisms underlying air pollution-induced arrhythmias. Mathematical models from the cell level to the tissue and organ levels have been extensively developed in past decades and have been successfully applied to investigate the mechanisms underlying congenital heart diseases (200), coronary heart diseases (42), and malignant ventricular arrhythmias (201), etc. Despite the sophisticated simulation measurements, the number of simulation studies regarding air pollution-induced cardiac arrhythmias is still quite scarce. In fact, the multiscale heart model has been proved to be a promising tool in investigating air pollution-induced arrhythmia. For instance, the proarrhythmic effects of CO, including the APD prolongation and the occurrence of EAD, were simulated using cardiac cell models by two separate groups (124, 202). Palacio et al. modeled the myocardial fibrosis caused by the PM2.5 in the human atria, which suggested the involvement of structural remodeling in the detrimental influences of PM2.5 (96). Recently, we conducted simulation studies regarding the proarrhythmic effects of H2S (43) and SO2 (158), and these two parallel studies explained important commonalities and differences of the adverse effects of sulfur-containing pollutants. Based on the physiological and the pathophysiological characteristics reflected by different levels of the multiscale heart model, these studies revealed the individual role of air pollutants in promoting arrhythmias and provided insightful mechanisms underlying electrophysiological dysfunctions.
In summary, although there has been a great deal of research focusing on air pollution-induced arrhythmias, studies regarding the effects of pollutants at the cellular level, especially the confounding effects, are relatively few. This significantly hinders the development in this area. Future investigations are encouraged to focus on the basic experimental research before associating pollutants with arrhythmia. The cardiac simulation is also a promising tool for investigating the air pollution-induced arrhythmia, as it provides multiscale insights into the pathological changes of electrophysiology at different levels. As a growing amount of data regarding interactions between air pollutants and cardiac ion channels become available, mechanisms underlying air pollution-induced arrhythmias are expected to be further revealed in computational simulations.
Author Contributions
ZW and HZ conceived this study. SZ and WL drafted and edited the manuscript. All authors reviewed the final version of the manuscript.
Funding
This work was supported by the National Key Research and Development Program of China (NO. 2018YFB0204204) and Shandong Postdoctoral Program for Innovative Talents (grantee SZ).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abbreviations
ΔAPD, Dispersion of repolarization; ADP, Adenosine diphosphate; AF, Atrial fibrillation; ANS, Autonomic nervous system; APD, Action potential duration; ATP, Adenosine triphosphate; AVN, Atrioventricular node; CaMKII, Ca2+/calmodulin-dependent kinase II; CICR, Calcium-induced calcium release; CO, Carbon monoxide; CV, Conduction velocity; CVD, Cardiovascular disease; DAD, Delayed afterdepolarization; EAD, Early afterdepolarization; ERP, Effective refractory period; H2S, Hydrogen sulfide; HEK293, Human embryonic kidney 293 cell; HRV, Heart rate variability; ICaL, L-type calcium current; If, Funny channel current; IK1, Inwardly rectifying potassium channel; IKATP, ATP-sensitive potassium current; IKr, Rapid delayed rectifier potassium current; IKs, Slow delayed rectifier potassium current; IKur, Ultra-repid outward current; IL-6, Interleukin 6; INa, Sodium channel current; INaL, Late sodium channel current; INCX, Na+/Ca2+ exchanger current; Ito, Transient outward potassium current; LQT, Long QT syndrome; MDA, Serum malondialdehyde; MI, Myocardial injury; NO2, Nitrogen dioxide; NO2−, Nitrite; NO3−, Nitrate; O3, Ozone; PKC, Protein kinase C; PM, Particulate matter; Pwd, P-wave dispersion; QTc, Corrected QT interval; QTd, QT dispersion; ROS, Reactive oxygen species; SAN, Sinoatrial node; SERCA, Sarco/endoplasimic reticulum Ca2+ ATPase; SO2, Sulfur dioxide; SOD, Superoxide dismutase; TdP, Torsade de Pointes; TDR, Transmural dispersion of repolarization; TNF, Tumor necrosis factor; VF, Ventricular fibrillation; VPC, Ventricular premature complex; VT, Ventricular tachycardia.
References
1. Sun D, Chen J, Wang Y, Ji H, Peng R, Jin L, et al. Advances in refunctionalization of erythrocyte-based nanomedicine for enhancing cancer-targeted drug delivery. Theranostics. (2019) 9:6885. doi: 10.7150/thno.36510
2. Yang L, Li L, Lewington S, Guo Y, Sherliker P, Bian Z, et al. Outdoor temperature, blood pressure, and cardiovascular disease mortality among 23 000 individuals with diagnosed cardiovascular diseases from China. Eur Heart J. (2015) 36:1178–85. doi: 10.1093/eurheartj/ehv023
3. Gakidou E, Afshin A, Abajobir AA, Abate KH, Abbafati C, Abbas KM, et al. Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet. (2017) 390:1345–422. doi: 10.1016/S0140-6736(17)32366-8
4. Hadley MB, Vedanthan R, Fuster V. Air pollution and cardiovascular disease: a window of opportunity. Nat Rev Cardiol. (2018) 15:193–94. doi: 10.1038/nrcardio.2017.207
5. Kwon OK, Kim S-YS-H, Kang S-H, Cho Y, Oh I-Y, Yoon C-H, et al. Association of short-and long-term exposure to air pollution with atrial fibrillation. Eur J Prev Cardiol. (2019) 26:1208–16. doi: 10.1177/2047487319835984
6. Peralta AA, Link MS, Schwartz J, Luttmann-Gibson H, Dockery DW, Blomberg A, et al. Exposure to air pollution and particle radioactivity with the risk of ventricular arrhythmias. Circulation. (2020) 142:858–67. doi: 10.1161/CIRCULATIONAHA.120.046321
7. Yue C, Yang F, Li F, Chen Y. Association between air pollutants and atrial fibrillation in general population: a systematic review and meta-analysis. Ecotoxicol Environ Saf. (2020) 208:111508. doi: 10.1016/j.ecoenv.2020.111508
8. Park M, Joo HS, Lee K, Jang M, Kim SD, Kim I, et al. Differential toxicities of fine particulate matters from various sources. Sci Rep. (2018) 8:1–11. doi: 10.1038/s41598-018-35398-0
9. Fiordelisi A, Piscitelli P, Trimarco B, Coscioni E, Iaccarino G, Sorriento D. The mechanisms of air pollution and particulate matter in cardiovascular diseases. Heart Fail Rev. (2017) 22:337–47. doi: 10.1007/s10741-017-9606-7
10. Hamanaka RB, Mutlu GM. Particulate matter air pollution: effects on the cardiovascular system. Front Endocrinol. (2018) 9:680. doi: 10.3389/fendo.2018.00680
11. Martinelli N, Olivieri O, Girelli D. Air particulate matter and cardiovascular disease: a narrative review. Eur J Intern Med. (2013) 24:295–302. doi: 10.1016/j.ejim.2013.04.001
12. Wu Q, Han X, Wang D, Zhao F, Wang D. Coal combustion related fine particulate matter (PM2. 5) induces toxicity in Caenorhabditis elegans by dysregulating microRNA expression. Toxicol Res. (2017) 6:432–41. doi: 10.1039/C7TX00107J
13. Amsalu E, Wang T, Li H, Liu Y, Wang A, Liu X, et al. Acute effects of fine particulate matter (PM 2.5) on hospital admissions for cardiovascular disease in Beijing, China: a time-series study. Environ Heal. (2019) 18:1–12. doi: 10.1186/s12940-019-0506-2
14. Wu T, Ma Y, Wu X, Bai M, Peng Y, Cai W, et al. Association between particulate matter air pollution and cardiovascular disease mortality in Lanzhou, China. Environ Sci Pollut Res. (2019) 26:15262–72. doi: 10.1007/s11356-019-04742-w
15. Huang K, Yang X, Liang F, Liu F, Li J, Xiao Q, et al. Long-term exposure to fine particulate matter and hypertension incidence in China: the China-PAR cohort study. Hypertension. (2019) 73:1195–201. doi: 10.1161/HYPERTENSIONAHA.119.12666
16. Liang F, Liu F, Huang K, Yang X, Li J, Xiao Q, et al. Long-term exposure to fine particulate matter and cardiovascular disease in China. J Am Coll Cardiol. (2020) 75:707–17. doi: 10.1016/j.jacc.2019.12.031
17. Hayes RB, Lim C, Zhang Y, Cromar K, Shao Y, Reynolds HR, et al. PM2. 5 air pollution and cause-specific cardiovascular disease mortality. Int J Epidemiol. (2020) 49:25–35. doi: 10.1093/ije/dyz114
18. Deng Q, Deng L, Miao Y, Guo X, Li Y. Particle deposition in the human lung: health implications of particulate matter from different sources. Environ Res. (2019) 169:237–45. doi: 10.1016/j.envres.2018.11.014
19. Wang X, Zhang L, Yao Z, Ai S, Qian ZM, Wang H, et al. Ambient coarse particulate pollution and mortality in three Chinese cities: association and attributable mortality burden. Sci Total Environ. (2018) 628:1037–42. doi: 10.1016/j.scitotenv.2018.02.100
20. Feng B, Song X, Dan M, Yu J, Wang Q, Shu M, et al. High level of source-specific particulate matter air pollution associated with cardiac arrhythmias. Sci Total Environ. (2019) 657:1285–93. doi: 10.1016/j.scitotenv.2018.12.178
21. Bell ML, Ebisu K, Leaderer BP, Gent JF, Lee HJ, Koutrakis P, et al. Associations of PM2. 5 constituents and sources with hospital admissions: analysis of four counties in Connecticut and Massachusetts (USA) for persons≥ 65 years of age. Environ Health Perspect. (2014) 122:138–44. doi: 10.1289/ehp.1306656
22. Yang Y, Ruan Z, Wang X, Yang Y, Mason TG, Lin H, et al. Short-term and long-term exposures to fine particulate matter constituents and health: a systematic review and meta-analysis. Environ Pollut. (2019) 247:874–82. doi: 10.1016/j.envpol.2018.12.060
23. Lu Y, Lin S, Fatmi Z, Malashock D, Hussain MM, Siddique A, et al. Assessing the association between fine particulate matter (PM2. 5) constituents and cardiovascular diseases in a mega-city of Pakistan. Environ Pollut. (2019) 252:1412–22. doi: 10.1016/j.envpol.2019.06.078
24. Hvidtfeldt UA, Geels C, Sørensen M, Ketzel M, Khan J, Tjønneland A, et al. Long-term residential exposure to PM2. 5 constituents and mortality in a Danish cohort. Environ Int. (2019) 133:105268. doi: 10.1016/j.envint.2019.105268
25. Zhang Z, Kang J, Hong YS, Chang Y, Ryu S, Park J, et al. Long-term particulate matter exposure and incidence of arrhythmias: a cohort study. J Am Heart Assoc. (2020) 9:e016885. doi: 10.1161/JAHA.120.016885
26. Rivera-Caravaca JM, Roldán V, Vicente V, Lip GYH, Marín F. Particulate matter and temperature: increased risk of adverse clinical outcomes in patients with atrial fibrillation. Mayo Clin Proc. (2020) 95:2360–9. doi: 10.1016/j.mayocp.2020.05.046
27. Kim I-S, Yang P-S, Lee J, Yu HT, Kim T-H, Uhm J-S, et al. Long-term exposure of fine particulate matter air pollution and incident atrial fibrillation in the general population: a nationwide cohort study. Int J Cardiol. (2019) 283:178–83. doi: 10.1016/j.ijcard.2018.12.048
28. Shin S, Burnett RT, Kwong JC, Hystad P, van Donkelaar A, Brook JR, et al. Ambient air pollution and the risk of atrial fibrillation and stroke: a population-based cohort study. Environ Health Perspect. (2019) 127:87009. doi: 10.1289/EHP4883
29. Link MS, Luttmann-Gibson H, Schwartz J, Mittleman MA, Wessler B, Gold DR, et al. Acute exposure to air pollution triggers atrial fibrillation. J Am Coll Cardiol. (2013) 62:816–25. doi: 10.1016/j.jacc.2013.05.043
30. Lee HH, Pan SC, Chen BY, Lo SH, Guo YL. Atrial fibrillation hospitalization is associated with exposure to fine particulate air pollutants. Environ Heal. (2019) 18:1–8. doi: 10.1186/s12940-019-0554-7
31. Solimini AG, Renzi M. Association between air pollution and emergency room visits for atrial fibrillation. Int J Environ Res Public Health. (2017) 14:661. doi: 10.3390/ijerph14060661
32. Folino F, Buja G, Zanotto G, Marras E, Allocca G, Vaccari D, et al. Association between air pollution and ventricular arrhythmias in high-risk patients (ARIA study): a multicentre longitudinal study. Lancet Planet Heal. (2017) 1:e58–64. doi: 10.1016/S2542-5196(17)30020-7
33. Tsai T-Y, Lo L-W, Liu S-H, Cheng W-H, Chou Y-H, Lin W-L, et al. Ambient fine particulate matter (PM2.5) exposure is associated with idiopathic ventricular premature complexes burden: a cohort study with consecutive Holter recordings. J Cardiovasc Electrophysiol. (2019) 30:487–92. doi: 10.1111/jce.13829
34. Kang JW, Yang WH, Chi JE, Chen WT. Higher ventricular premature complex burden is associated with lower systolic blood pressure response. Acta Cardiol Sin. (2018) 34:152–8. doi: 10.6515/ACS.201803_34(2).20171117A
35. Guerrier K, Anderson JB, Czosek RJ, Mays WA, Statile C, Knilans TK, et al. Usefulness of ventricular premature complexes in asymptomatic patients ≤ 21 years as predictors of poor left ventricular function. Am J Cardiol. (2015) 115:652–5. doi: 10.1016/j.amjcard.2014.12.020
36. Nabil Ali A, Abd Elfattah Badran H, Ahmed El-Alfy M. Complex ventricular premature beats detected by Holter monitoring as a predictor of sudden cardiac death in patients with cardiomyopathy. QJM An Int J Med. (2018) 111:hcy200.026. doi: 10.1093/qjmed/hcy200.026
37. Yue C, Yang F, Wang L, Li F, Chen Y. Association between fine particulate matter and atrial fibrillation in implantable cardioverter defibrillator patients: a systematic review and meta-analysis. J Interv Card Electrophysiol. (2020) 59:595–601. doi: 10.1007/s10840-020-00864-1
38. Yalta T, Yalta K. Systemic inflammation and arrhythmogenesis: a review of mechanistic and clinical perspectives. Angiology. (2018) 69:288–96. doi: 10.1177/0003319717709380
39. Dabass A, Talbott EO, Rager JR, Marsh GM, Venkat A, Holguin F, et al. Systemic inflammatory markers associated with cardiovascular disease and acute and chronic exposure to fine particulate matter air pollution (PM2.5) among US NHANES adults with metabolic syndrome. Environ Res. (2018) 161:485–91. doi: 10.1016/j.envres.2017.11.042
40. Liu Q, Gu X, Deng F, Mu L, Baccarelli AA, Guo X, et al. Ambient particulate air pollution and circulating C-reactive protein level: a systematic review and meta-analysis. Int J Hyg Environ Health. (2019) 222:756–64. doi: 10.1016/j.ijheh.2019.05.005
41. Liang S, Zhang J, Ning R, Du Z, Liu J, Batibawa JW, et al. The critical role of endothelial function in fine particulate matter-induced atherosclerosis. Part Fibre Toxicol. (2020) 17:1–24. doi: 10.1186/s12989-020-00391-x
42. Lu W, Li J, Yang F, Luo C, Wang K, Adeniran I, et al. Effects of acute global ischemia on re-entrant arrhythmogenesis: a simulation study. J Biol Syst. (2015) 23:213–30. doi: 10.1142/S0218339015500114
43. Zhang S, Fan X, Wang W, Li Z, Jia D, Wei Z, et al. Pro-arrhythmic effects of hydrogen sulfide in healthy and ischemic cardiac tissue: insight from a simulation study. Front Physiol. (2019) 10:1482. doi: 10.3389/fphys.2019.01482
44. Hu Y-F, Chen Y-J, Lin Y-J, Chen S-A. Inflammation and the pathogenesis of atrial fibrillation. Nat Rev Cardiol. (2015) 12:230. doi: 10.1038/nrcardio.2015.2
45. Lee SH, Chen YC, Chen YJ, Chang SL, Tai CT, Wongcharoen W, et al. Tumor necrosis factor-α alters calcium handling and increases arrhythmogenesis of pulmonary vein cardiomyocytes. Life Sci. (2007) 80:1806–15. doi: 10.1016/j.lfs.2007.02.029
46. Lazzerini PE, Capecchi PL, Laghi-Pasini F. Systemic inflammation and arrhythmic risk: lessons from rheumatoid arthritis. Eur Heart J. (2017) 38:1717–27. doi: 10.1093/eurheartj/ehw208
47. Lazzerini PE, Boutjdir M, Capecchi PL. COVID-19, arrhythmic risk, and inflammation: mind the gap! Circulation. (2020) 142:7–9. doi: 10.1161/CIRCULATIONAHA.120.047293
48. Lazzerini PE, Acampa M, Laghi-Pasini F, Bertolozzi I, Finizola F, Vanni F, et al. Cardiac arrest risk during acute infections: systemic inflammation directly prolongs QTc interval via cytokine-mediated effects on potassium channel expression. Circ Arrhythmia Electrophysiol. (2020) 13:e008627. doi: 10.1161/CIRCEP.120.008627
49. Taylor-Clark TE. Air pollution-induced autonomic modulation. Physiology. (2020) 35:363–74. doi: 10.1152/physiol.00017.2020
50. Riediker M, Franc Y, Bochud M, Meier R, Rousson V. Exposure to fine particulate matter leads to rapid heart rate variability changes. Front Environ Sci. (2018) 6:2. doi: 10.3389/fenvs.2018.00002
51. Ito K, Mathes R, Ross Z, Nádas A, Thurston G, Matte T. Fine particulate matter constituents associated with cardiovascular hospitalizations and mortality in New York City. Environ Health Perspect. (2011) 119:467. doi: 10.1289/ehp.1002667
52. Franchini M, Mannucci PM. Thrombogenicity and cardiovascular effects of ambient air pollution. Blood. (2011) 118:2405–12. doi: 10.1182/blood-2011-04-343111
53. Liao D, Shaffer ML, He F, Rodriguez-Colon S, Wu R, Whitsel EA, et al. Fine particulate air pollution is associated with higher vulnerability to atrial fibrillation — the APACR study. J Toxicol Environ Heal Part A. (2011) 74:693–705. doi: 10.1080/15287394.2011.556056
54. Shen MJ, Zipes DP. Role of the autonomic nervous system in modulating cardiac arrhythmias. Circ Res. (2014) 114:1004–21. doi: 10.1161/CIRCRESAHA.113.302549
55. Wu P, Vaseghi M. The autonomic nervous system and ventricular arrhythmias in myocardial infarction and heart failure. Pacing Clin Electrophysiol. (2020) 43:172–80. doi: 10.1111/pace.13856
56. Kolettis TM, La Rocca V, Psychalakis N, Karampela E, Kontonika M, Tourmousoglou C, et al. Effects of central sympathetic activation on repolarization-dispersion during short-term myocardial ischemia in anesthetized rats. Life Sci. (2016) 144:170–7. doi: 10.1016/j.lfs.2015.12.019
57. Vaseghi M, Lux RL, Mahajan A, Shivkumar K. Sympathetic stimulation increases dispersion of repolarization in humans with myocardial infarction. Am J Physiol Hear Circ Physiol. (2012) 302:H1838–46. doi: 10.1152/ajpheart.01106.2011
58. Amoni M, Claus P, Dries E, Nagaraju C, De Buck S, Vandenberk B, et al. Discrete sites of frequent premature ventricular complexes cluster within the infarct border zone and coincide with high frequency of delayed afterdepolarizations under adrenergic stimulation. Hear Rhythm. (2021) doi: 10.1016/j.hrthm.2021.07.067
59. Xie Y, Grandi E, Puglisi JL, Sato D, Bers DM. β-adrenergic stimulation activates early afterdepolarizations transiently via kinetic mismatch of PKA targets. J Mol Cell Cardiol. (2013) 58:153–61. doi: 10.1016/j.yjmcc.2013.02.009
60. Liu C, Jiang H, Yu L, Po SS. Vagal stimulation and Arrhythmias. J Atr Fibrillation. (2020) 13:2398. doi: 10.4022/jafib.2398
61. Doste R, Bueno-Orovio A. Multiscale modelling of β-adrenergic stimulation in cardiac electromechanical function. Mathematics. (2021) 9:1785. doi: 10.3390/math9151785
62. Myles RC, Wang L, Kang C, Bers DM, Ripplinger CM. Local β-adrenergic stimulation overcomes source-sink mismatch to generate focal arrhythmia. Circ Res. (2012) 110:1454–64. doi: 10.1161/CIRCRESAHA.111.262345
63. Yamakawa K, So EL, Rajendran PS, Hoang JD, Makkar N, Mahajan A, et al. Electrophysiological effects of right and left vagal nerve stimulation on the ventricular myocardium. Am J Physiol Circ Physiol. (2014) 307:H722–31. doi: 10.1152/ajpheart.00279.2014
64. Yamakawa K, Rajendran PS, Takamiya T, Yagishita D, So EL, Mahajan A, et al. Vagal nerve stimulation activates vagal afferent fibers that reduce cardiac efferent parasympathetic effects. Am J Physiol Circ Physiol. (2015) 309:H1579–90. doi: 10.1152/ajpheart.00558.2015
65. Bayer JD, Boukens BJ, Krul SPJ, Roney CH, Driessen AHG, Berger WR, et al. Acetylcholine delays atrial activation to facilitate atrial fibrillation. Front Physiol. (2019) 10:1105. doi: 10.3389/fphys.2019.01105
66. Rajagopalan S, Al-Kindi SG, Brook RD. Air pollution and cardiovascular disease: JACC state-of-the-art review. J Am Coll Cardiol. (2018) 72:2054–70. doi: 10.1016/j.jacc.2018.07.099
67. Baranowska-Wójcik E, Szwajgier D, Oleszczuk P, Winiarska-Mieczan A. Effects of titanium dioxide nanoparticles exposure on human health—a review. Biol Trace Elem Res. (2020) 193:118–29. doi: 10.1007/s12011-019-01706-6
68. Miller MR, Raftis JB, Langrish JP, McLean SG, Samutrtai P, Connell SP, et al. Inhaled nanoparticles accumulate at sites of vascular disease. ACS Nano. (2017) 11:4542–52. doi: 10.1021/acsnano.6b08551
69. Savi M, Rossi S, Bocchi L, Gennaccaro L, Cacciani F, Perotti A, et al. Titanium dioxide nanoparticles promote arrhythmias via a direct interaction with rat cardiac tissue. Part Fibre Toxicol. (2014) 11:63. doi: 10.1186/s12989-014-0063-3
70. Adameova A, Shah AK, Dhalla NS. Role of oxidative stress in the genesis of ventricular arrhythmias. Int J Mol Sci. (2020) 21:4200. doi: 10.3390/ijms21124200
71. Sovari AA. Cellular and molecular mechanisms of arrhythmia by oxidative stress. Cardiol Res Pract. (2016) 2016:9656078. doi: 10.1155/2016/9656078
72. Song Y, Shryock JC, Wagner S, Maier LS, Belardinelli L. Blocking late sodium current reduces hydrogen peroxide-induced arrhythmogenic activity and contractile dysfunction. J Pharmacol Exp Ther. (2006) 318:214–22. doi: 10.1124/jpet.106.101832
73. Shang LL, Sanyal S, Pfahnl AE, Jiao Z, Allen J, Liu H, et al. NF-κB-dependent transcriptional regulation of the cardiac scn5a sodium channel by angiotensin II. Am J Physiol Cell Physiol. (2008) 294:C372–9. doi: 10.1152/ajpcell.00186.2007
74. Thomas GP, Sims SM, Cook MA, Karmazyn M. Hydrogen peroxide-induced stimulation of L-type calcium current in guinea pig ventricular myocytes and its inhibition by adenosine A1 receptor activation. J Pharmacol Exp Ther. (1998) 286:1208–14.
75. Coetzee WA, Ichikawa H, Hearse DJ. Oxidant stress inhibits Na-Ca-exchange current in cardiac myocytes: mediation by sulfhydryl groups? Am J Physiol Circ Physiol. (1994) 266:H909–19. doi: 10.1152/ajpheart.1994.266.3.H909
76. Chiamvimonvat N, O'Rourke B, Kamp TJ, Kallen RG, Hofmann F, Flockerzi V, et al. Functional consequences of sulfhydryl modification in the pore-forming subunits of cardiovascular Ca2+ and Na+ channels. Circ Res. (1995) 76:325–34. doi: 10.1161/01.RES.76.3.325
77. Anzai K, Ogawa K, Kuniyasu A, Ozawa T, Yamamoto H, Nakayama H. Effects of hydroxyl radical and sulfhydryl reagents on the open probability of the purified cardiac ryanodine receptor channel incorporated into planar lipid bilayers. Biochem Biophys Res Commun. (1998) 249:938–42. doi: 10.1006/bbrc.1998.9244
78. Morris TE, Sulakhe P V. Sarcoplasmic reticulum Ca2+-pump dysfunction in rat cardiomyocytes briefly exposed to hydoxyl radicals. Free Radic Biol Med. (1997) 22:37–47. doi: 10.1016/S0891-5849(96)00238-9
79. Feno S, Butera G, Vecellio Reane D, Rizzuto R, Raffaello A. Crosstalk between calcium and ROS in pathophysiological conditions. Oxid Med Cell Longev. (2019) 2019:9324018. doi: 10.1155/2019/9324018
80. Erickson JR, Mei-ling AJ, Guan X, Kutschke W, Yang J, Oddis CV, et al. A dynamic pathway for calcium-independent activation of CaMKII by methionine oxidation. Cell. (2008) 133:462–74. doi: 10.1016/j.cell.2008.02.048
81. Zima A V, Blatter LA. Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res. (2006) 71:310–21. doi: 10.1016/j.cardiores.2006.02.019
82. Yang Y, Shi W, Cui N, Wu Z, Jiang C. Oxidative stress inhibits vascular KATP channels by S-glutathionylation. J Biol Chem. (2010) 285:38641–8. doi: 10.1074/jbc.M110.162578
83. Santos CXC, Anilkumar N, Zhang M, Brewer AC, Shah AM. Redox signaling in cardiac myocytes. Free Radic Biol Med. (2011) 50:777–93. doi: 10.1016/j.freeradbiomed.2011.01.003
84. Zhang Y, Xiao J, Wang H, Luo X, Wang J, Villeneuve LR, et al. Restoring depressed HERG K+ channel function as a mechanism for insulin treatment of abnormal QT prolongation and associated arrhythmias in diabetic rabbits. Am J Physiol Circ Physiol. (2006) 291:H1446–55. doi: 10.1152/ajpheart.01356.2005
85. Karam BS, Chavez-Moreno A, Koh W, Akar JG, Akar FG. Oxidative stress and inflammation as central mediators of atrial fibrillation in obesity and diabetes. Cardiovasc Diabetol. (2017) 16:1–9. doi: 10.1186/s12933-017-0604-9
86. Beckmann A, Grißmer A, Wolf S, Recktenwald J, Meier C. Oxygen-glucose deprivation in mouse astrocytes is associated with ultrastructural changes in connexin 43 gap junctions. Neuroscience. (2019) 397:67–79. doi: 10.1016/j.neuroscience.2018.11.043
87. Aung N, Sanghvi MM, Zemrak F, Lee AM, Cooper JA, Paiva JM, et al. Association between ambient air pollution and cardiac morpho-functional phenotypes: insights from the UK Biobank population imaging study. Circulation. (2018) 138:2175–86. doi: 10.1161/CIRCULATIONAHA.118.034856
88. de Oliveira-Fonoff AM, Mady C, Pessoa FG, Fonseca KCB, Salemi VMC, Fernandes F, et al. The role of air pollution in myocardial remodeling. PLoS ONE. (2017) 12:e0176084. doi: 10.1371/journal.pone.0176084
89. Liu Y, Goodson JM, Zhang B, Chin MT. Air pollution and adverse cardiac remodeling: clinical effects and basic mechanisms. Front Physiol. (2015) 6:162. doi: 10.3389/fphys.2015.00162
90. Su X, Tian J, Li B, Zhou L, Kang H, Pei Z, et al. Ambient PM2. 5 caused cardiac dysfunction through FoxO1-targeted cardiac hypertrophy and macrophage-activated fibrosis in mice. Chemosphere. (2020) 247:125881. doi: 10.1016/j.chemosphere.2020.125881
91. Nattel S, Harada M. Atrial remodeling and atrial fibrillation: recent advances and translational perspectives. J Am Coll Cardiol. (2014) 63:2335–45. doi: 10.1016/j.jacc.2014.02.555
92. Zou R, Kneller J, Leon LJ, Nattel S. Substrate size as a determinant of fibrillatory activity maintenance in a mathematical model of canine atrium. Am J Physiol Hear Circ Physiol. (2005) 289: H1002–12. doi: 10.1152/ajpheart.00252.2005
93. Burstein B, Comtois P, Michael G, Nishida K, Villeneuve L, Yeh YH, et al. Changes in connexin expression and the atrial fibrillation substrate in congestive heart failure. Circ Res. (2009) 105:1213–22. doi: 10.1161/CIRCRESAHA.108.183400
94. Seko Y, Kato T, Haruna T, Izumi T, Miyamoto S, Nakane E, Inoko M. Association between atrial fibrillation, atrial enlargement, and left ventricular geometric remodeling. Sci Rep. (2018) 8:1–8. doi: 10.1038/s41598-018-24875-1
95. Kowallick JT, Staab W, Schuster A, Backhaus SJ, Weber-Krüger M, Bauer L, et al. Reverse left ventricular structural remodeling after catheter ablation of atrial fibrillation in patients with preserved left ventricular function: insights from cardiovascular magnetic resonance native T1 mapping. Hear Rhythm. (2019) 16:424–32. doi: 10.1016/j.hrthm.2018.09.016
96. Palacio LC, Ugarte JP, Saiz J, Tobón C. Genesis of atrial fibrillation under different diffuse fibrosis density related with atmospheric pollution. In-silico study. In: Workshop on Engineering Applications. Bogotá: Springer (2020). p. 291–301. doi: 10.1007/978-3-030-61834-6_25
97. Garg J, Krishnamoorthy P, Palaniswamy C, Khera S, Ahmad H, Jain D, et al. Cardiovascular abnormalities in carbon monoxide poisoning. Am J Ther. (2018) 25:e339–48. doi: 10.1097/MJT.0000000000000016
98. Shah ASV, Langrish JP, Nair H, McAllister DA, Hunter AL, Donaldson K, et al. Global association of air pollution and heart failure: a systematic review and meta-analysis. Lancet. (2013) 382:1039–48. doi: 10.1016/S0140-6736(13)60898-3
99. Dastoorpoor M, Riahi A, Yazdaninejhad H, Borsi SH, Khanjani N, Khodadadi N, et al. Exposure to particulate matter and carbon monoxide and cause-specific Cardiovascular-Respiratory disease mortality in Ahvaz. Toxin Rev. (2020) 1–11. doi: 10.1080/15569543.2020.1716256
100. Hanci V, Ayoglu H, Yurtlu S, Yildirim N, Okyay D, Erdogan G, et al. Effects of acute carbon monoxide poisoning on the P-wave and QT interval dispersions. Anatol J Cardiol Kardiyol Derg. (2011) 11:48–52. doi: 10.5152/akd.2011.009
101. Akdemir HU, Güngörer B, Çalişkan F, Çolak S, Güzel M. Atrial fibrillation related to carbon monoxide poisoning in a female patient. Am J Emerg Med. (2014) 32:1154.e3–5. doi: 10.1016/j.ajem.2014.02.050
102. Gedela M, Weltman NY, Chavvakula NS, Carpenter PL, Sturm T. Atrial fibrillation induced by carbon monoxide poisoning and successful treatment with hyperbaric oxygen. SD Med. (2017) 70:319–21.
103. Satran D, Henry CR, Adkinson C, Nicholson CI, Bracha Y, Henry TD. Cardiovascular manifestations of moderate to severe carbon monoxide poisoning. J Am Coll Cardiol. (2005) 45:1513–6. doi: 10.1016/j.jacc.2005.01.044
104. Kim S, Lim JH, Kim Y, Oh S, Choi WG. A case of acute carbon monoxide poisoning resulting in an ST elevation myocardial infarction. Korean Circ J. (2012) 42:133. doi: 10.4070/kcj.2012.42.2.133
105. Isik T, Tanboga IH, Güvenç TS, Uyarel H, Varol E. ST-elevation myocardial infarction after acute carbon monoxide poisoning. Anatol J Cardiol. (2012) 12:278–9. doi: 10.5152/akd.2012.077
106. Olatunde O, Raj V, Tambe V, Szombathy T. Carbon monoxide poisoning and its effect on QTc prolongation. J Cardiol Cases. (2020) 22:19–21. doi: 10.1016/j.jccase.2020.03.009
107. Hantson P. Mechanisms of toxic cardiomyopathy. Clin Toxicol. (2019) 57:1–9. doi: 10.1080/15563650.2018.1497172
108. Scragg JL, Dallas ML, Wilkinson JA, Varadi G, Peers C. Carbon monoxide inhibits L-type Ca2+ channels via redox modulation of key cysteine residues by mitochondrial reactive oxygen species. J Biol Chem. (2008) 283:24412–9. doi: 10.1074/jbc.M803037200
109. Andre L, Boissière J, Reboul C, Perrier R, Zalvidea S, Meyer G, et al. Carbon monoxide pollution promotes cardiac remodeling and ventricular arrhythmia in healthy rats. Am J Respir Crit Care Med. (2010) 181:587–95. doi: 10.1164/rccm.200905-0794OC
110. Dallas ML, Yang Z, Boyle JP, Boycott HE, Scragg JL, Milligan CJ, et al. Carbon monoxide induces cardiac arrhythmia via induction of the late Na+ current. Am J Respir Crit Care Med. (2012) 186:648–56. doi: 10.1164/rccm.201204-0688OC
111. Elies J, Dallas ML, Boyle JP, Scragg JL, Duke A, Steele DS, et al. Inhibition of the cardiac Na+ channel Nav1. 5 by carbon monoxide. J Biol Chem. (2014) 289:16421–9. doi: 10.1074/jbc.M114.569996
112. Berne P, Brugada J. Brugada syndrome 2012. Circ J. (2012) 76:1563–71. doi: 10.1253/circj.CJ-12-0717
113. Al-Owais MM, Hettiarachchi NT, Kirton HM, Hardy ME, Boyle JP, Scragg JL, et al. A key role for peroxynitrite-mediated inhibition of cardiac ERG (Kv11.1) K+ channels in carbon monoxide-induced proarrhythmic early afterdepolarizations. FASEB J. (2017) 31:4845–54. doi: 10.1096/fj.201700259R
114. Liang S, Wang Q, Zhang W, Zhang H, Tan S, Ahmed A, et al. Carbon monoxide inhibits inward rectifier potassium channels in cardiomyocytes. Nat Commun. (2014) 5:4676. doi: 10.1038/ncomms5676
115. Bhar-Amato J, Davies W, Agarwal S. Ventricular arrhythmia after acute myocardial infarction: “The perfect storm.” Arrhythmia Electrophysiol Rev. (2017) 6:134–9. doi: 10.15420/aer.2017.24.1
116. Ogawa T, Honjo H, Yamazaki M, Kushiyama Y, Sakuma I, Kodama I, et al. Ranolazine facilitates termination of ventricular tachyarrhythmia associated with acute myocardial ischemia through suppression of late INa-mediated focal activity. Circ J. (2017) 81:1411–28. doi: 10.1253/circj.CJ-17-0128
117. Shaw RM, Rudy Y. Electrophysiologic effects of acute myocardial ischemia: a theoretical study of altered cell excitability and action potential duration. Cardiovasc Res. (1997) 35:256–72. doi: 10.1016/S0008-6363(97)00093-X
118. Mendonca Costa C, Plank G, Rinaldi CA, Niederer SA, Bishop MJ. Modeling the electrophysiological properties of the infarct border zone. Front Physiol. (2018) 9:356. doi: 10.3389/fphys.2018.00356
119. Schwartz PJ, Crotti L, Insolia R. Long-QT syndrome: from genetics to management. Circ Arrhythmia Electrophysiol. (2012) 5:868–77. doi: 10.1161/CIRCEP.111.962019
120. John RM, Tedrow UB, Koplan BA, Albert CM, Epstein LM, Sweeney MO, et al. Ventricular arrhythmias and sudden cardiac death. Lancet. (2012) 380:1520–9. doi: 10.1016/S0140-6736(12)61413-5
121. Xu H, Chen J, Zhao Q, Zhang Y, Wang T, Feng B, et al. Ambient air pollution is associated with cardiac repolarization abnormalities in healthy adults. Environ Res. (2019) 171:239–46. doi: 10.1016/j.envres.2019.01.023
122. Antzelevitch C, Shimizu W, Yan G-X, Sicouri S. Cellular basis for QT dispersion. J Electrocardiol. (1998) 30:168–75. doi: 10.1016/S0022-0736(98)80070-8
123. Al-Owais MM, Steele DS, Holden AV, Benson AP. Deterministic and stochastic cellular mechanisms contributing to carbon monoxide induced ventricular arrhythmias. Front Pharmacol. (2021) 12:936. doi: 10.3389/fphar.2021.651050
124. Al-Owais MM, Peers C, Steele DS, Holden AV, Benson AP. Carbon monoxide effects on electrophysiological mechanisms of ventricular arrhythmogenesis. In: 2018 Computing in Cardiology Conference (CinC). Maastricht: IEEE (2018). p. 1–4. doi: 10.22489/CinC.2018.124
125. O'Hara T, Virág L, Varró A, Rudy Y. Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput Biol. (2011) 7:e1002061. doi: 10.1371/journal.pcbi.1002061
126. Hancox JC, Whittaker DG, Du C, Stuart AG, Zhang H. Emerging therapeutic targets in the short QT syndrome. Expert Opin Ther Targets. (2018) 22:439–51. doi: 10.1080/14728222.2018.1470621
127. Atescelik M, Bozdemir MN, Yildiz M, Gurbuz S, Ayranci M, Goktekin MC, et al. QT dispersion in carbon monoxide poisoning. Eur Rev Med Pharmacol Sci. (2012) 16:25–9.
128. Yenerçag M, Arslan U, Seker OO, Dereli S, Kaya A, Dogdus M, et al. Evaluation of P-wave dispersion in patients with newly diagnosed coronavirus disease 2019. J Cardiovasc Med. (2021) 22:197–203. doi: 10.2459/JCM.0000000000001135
129. Zhang Z, Liu MB, Huang X, Song Z, Qu Z. Mechanisms of premature ventricular complexes caused by QT prolongation. Biophys J. (2021) 120:352–69. doi: 10.1016/j.bpj.2020.12.001
130. Zhang Z, Liu MB, Song Z, Qu Z. Can Phase-2 early afterdepolarizations propagate in cardiac tissue? Insights from multiple action potential models. Biophys J. (2020) 118:409a−10. doi: 10.1016/j.bpj.2019.11.2317
131. Doujaiji B, Al-Tawfiq JA. Hydrogen sulfide exposure in an adult male. Ann Saudi Med. (2010) 30:76–80. doi: 10.5144/0256-4947.59379
132. Bates MN, Garrett N, Shoemack P. Investigation of health effects of hydrogen sulfide from a geothermal source. Arch Environ Heal An Int J. (2002) 57:405–11. doi: 10.1080/00039890209601428
133. Finnbjornsdottir RG, Carlsen HK, Thorsteinsson T, Oudin A, Lund SH, Gislason T, et al. Association between daily hydrogen sulfide exposure and incidence of emergency hospital visits: a population-based study. PLoS ONE. (2016) 11:e0154946. doi: 10.1371/journal.pone.0154946
134. Gummin DD, Mowry JB, Spyker DA, Brooks DE, Beuhler MC, Rivers LJ, et al. 2018 Annual report of the American Association of Poison control centers' National Poison Data System (NPDS): 36th annual report. Clin Toxicol. (2019) 57:1220–413. doi: 10.1080/15563650.2019.1677022
135. Mo W, Shen J, Huang X, Zhang Y, Zhang Z. Acute myocardial injury following hydrogen sulfide poisoning. Toxicol Ind Health. (2020) 36:750–8. doi: 10.1177/0748233720945184
136. Jiang J, Jiang L, Chen X. Cardiogenic shock following hydrogen sulfide poisoning. Authorea. (2020). doi: 10.22541/au.160509653.33194077/v1
137. Lindenmann J, Matzi V, Neuboeck N, Ratzenhofer-Komenda B, Maier A, Smolle-Juettner F-M. Severe hydrogen sulphide poisoning treated with 4-dimethylaminophenol and hyperbaric oxygen. Diving Hyperb Med. (2010) 40:213–7.
138. Hirakawa A, Takeyama N, Iwatsuki S, Iwata T, Kano H. Delayed myocardial injury following acute hydrogen sulfide intoxication. Chudoku Kenkyu. (2013) 26:44–8.
139. Luqman N, Sung RJ, Wang C-L, Kuo C-T. Myocardial ischemia and ventricular fibrillation: pathophysiology and clinical implications. Int J Cardiol. (2007) 119:283–90. doi: 10.1016/j.ijcard.2006.09.016
140. Peers C, Bauer CC, Boyle JP, Scragg JL, Dallas ML. Modulation of ion channels by hydrogen sulfide. Antioxid Redox Signal. (2012) 17:95–105. doi: 10.1089/ars.2011.4359
141. Foster MN, Coetzee WA. KATP channels in the cardiovascular system. Physiol Rev. (2016) 96:177–252. doi: 10.1152/physrev.00003.2015
142. Zhang R, Sun Y, Tsai H, Tang C, Jin H, Du J. Hydrogen sulfide inhibits L-type calcium currents depending upon the protein sulfhydryl state in rat cardiomyocytes. PLoS ONE. (2012) 7:e37073. doi: 10.1371/journal.pone.0037073
143. Sun Y-G, Cao Y-X, Wang W-W, Ma S-F, Yao T, Zhu Y-C. Hydrogen sulphide is an inhibitor of L-type calcium channels and mechanical contraction in rat cardiomyocytes. Cardiovasc Res. (2008) 79:632–41. doi: 10.1093/cvr/cvn140
144. Abramochkin DV, Moiseenko LS, Kuzmin VS. The effect of hydrogen sulfide on electrical activity of rat atrial myocardium. Bull Exp Biol Med. (2009) 147:683–6. doi: 10.1007/s10517-009-0607-y
145. Chan C-S, Lin Y-K, Kao Y-H, Chen Y-C, Chen S-A, Chen Y-J. Hydrogen sulphide increases pulmonary veins and atrial arrhythmogenesis with activation of protein kinase C. J Cell Mol Med. (2018) 22:3503–13. doi: 10.1111/jcmm.13627
146. Walsh KB, Zhang J. Neonatal rat cardiac fibroblasts express three types of voltage-gated K+ channels: regulation of a transient outward current by protein kinase C. Am J Physiol Hear Circ Physiol. (2008) 294:H1010–7. doi: 10.1152/ajpheart.01195.2007
147. Fu C, Hao J, Zeng M, Song Y, Jiang W, Zhang P, et al. Modulation of late sodium current by Ca2+-calmodulin-dependent protein kinase II, protein kinase C and Ca2+ during hypoxia in rabbit ventricular myocytes. Exp Physiol. (2017) 102:818–34. doi: 10.1113/EP085990
148. Mi X, Ding WG, Toyoda F, Kojima A, Omatsu-Kanbe M, Matsuura H. Selective activation of adrenoceptors potentiates IKs current in pulmonary vein cardiomyocytes through the protein kinase A and C signaling pathways. J Mol Cell Cardiol. (2021) 161:86–97. doi: 10.1016/j.yjmcc.2021.08.004
149. Wang L, Liu C, Meng X, Niu Y, Lin Z, Liu Y, et al. Associations between short-term exposure to ambient sulfur dioxide and increased cause-specific mortality in 272 Chinese cities. Environ Int. (2018) 117:33–9. doi: 10.1016/j.envint.2018.04.019
150. Amsalu E, Guo Y, Li H, Wang T, Liu Y, Wang A, et al. Short-term effect of ambient sulfur dioxide (SO2) on cause-specific cardiovascular hospital admission in Beijing, China: a time series study. Atmos Environ. (2019) 208:74–81. doi: 10.1016/j.atmosenv.2019.03.015
151. Zhao A, Chen R, Kuang X, Kan H. Ambient air pollution and daily outpatient visits for cardiac arrhythmia in Shanghai, China. J Epidemiol. (2014) 24:321–6. doi: 10.2188/jea.JE20140030
152. Wang M, Chen J, Zhang Z, Yu P, Gan W, Tan Z, Bao J. Associations between air pollution and outpatient visits for arrhythmia in Hangzhou, China. BMC Public Health. (2020) 20:1524. doi: 10.1186/s12889-020-09628-y
153. Hwang J, Kwon J, Yi H, Bae H-J, Jang M, Kim N. Association between long-term exposure to air pollutants and cardiopulmonary mortality rates in South Korea. BMC Public Health. (2020) 20:1402. doi: 10.1186/s12889-020-09521-8
154. Nie A, Meng Z. Study of the interaction of sulfur dioxide derivative with cardiac sodium channel. Biochim Biophys Acta. (2005) 1718:67–73. doi: 10.1016/j.bbamem.2005.09.020
155. Nie A, Meng Z. Sulfur dioxide derivative modulation of potassium channels in rat ventricular myocytes. Arch Biochem Biophys. (2005) 442:187–95. doi: 10.1016/j.abb.2005.08.004
156. Nie A, Meng Z. Modulation of L-type calcium current in rat cardiac myocytes by sulfur dioxide derivatives. Food Chem Toxicol. (2006) 44:355–63. doi: 10.1016/j.fct.2005.08.006
157. Nie A, Meng Z. Sulfur dioxide derivatives modulate Na/Ca exchange currents and cytosolic [Ca2+]i in rat myocytes. Biochem Biophys Res Commun. (2007) 358:879–84. doi: 10.1016/j.bbrc.2007.05.008
158. Zhang S, Lu W, Li Z, Wang S, Jiang M, Yuan Q, et al. Mechanisms underlying sulfur dioxide pollution induced ventricular arrhythmia: a simulation study. In: 2020 IEEE International Conference on Bioinformatics and Biomedicine (BIBM). (2020). p. 381–6. doi: 10.1109/BIBM49941.2020.9313277
159. Routledge HC, Manney S, Harrison RM, Ayres JG, Townend JN. Effect of inhaled sulphur dioxide and carbon particles on heart rate variability and markers of inflammation and coagulation in human subjects. Heart. (2006) 92:220–7. doi: 10.1136/hrt.2004.051672
160. Kimhy D, Crowley OV, McKinley PS, Burg MM, Lachman ME, Tun PA, et al. The association of cardiac vagal control and executive functioning–findings from the MIDUS study. J Psychiatr Res. (2013) 47:628–35. doi: 10.1016/j.jpsychires.2013.01.018
161. Kalla M, Herring N, Paterson DJ. Cardiac sympatho-vagal balance and ventricular arrhythmia. Auton Neurosci. (2016) 199:29–37. doi: 10.1016/j.autneu.2016.08.016
162. Li S, Xu Z, Xia J, Qin G, Sang N. Sulfur dioxide induces apoptosis via reactive oxygen species generation in rat cardiomyocytes. Environ Sci Pollut Res. (2019) 26:8758–67. doi: 10.1007/s11356-019-04319-7
163. Qin G, Wu M, Wang J, Xu Z, Xia J, Sang N. Sulfur dioxide contributes to the cardiac and mitochondrial dysfunction in rats. Toxicol Sci. (2016) 151:334–46. doi: 10.1093/toxsci/kfw048
164. Zhang JJ, Wei Y, Fang Z. Ozone pollution: a major health hazard worldwide. Front Immunol. (2019) 10:2518. doi: 10.3389/fimmu.2019.02518
165. Buadong D, Jinsart W, Funatagawa I, Karita K, Yano E. Association between PM10 and O3 levels and hospital visits for cardiovascular diseases in Bangkok, Thailand. J Epidemiol. (2009) 19:182–8. doi: 10.2188/jea.JE20080047
166. Ensor KB, Raun LH, Persse D. A case-crossover analysis of out-of-hospital cardiac arrest and air pollution. Circulation. (2013) 127:1192–9. doi: 10.1161/CIRCULATIONAHA.113.000027
167. Zuin M, Rigatelli G, dell'Avvocata F, Picariello C, Conte L, Marcantoni L, et al. Air pollution and ST-elevation myocardial infarction treated with primary percutaneous coronary angioplasty: a direct correlation. Int J Cardiol. (2017) 236:49–53. doi: 10.1016/j.ijcard.2017.02.050
168. Turner MC, Jerrett M, Pope CA III, Krewski D, Gapstur SM, Diver WR, et al. Long-term ozone exposure and mortality in a large prospective study. Am J Respir Crit Care Med. (2016) 193:1134–42. doi: 10.1164/rccm.201508-1633OC
169. Lim CC, Hayes RB, Ahn J, Shao Y, Silverman DT, Jones RR, et al. Long-term exposure to ozone and cause-specific mortality risk in the United States. Am J Respir Crit Care Med. (2019) 200:1022–31. doi: 10.1164/rccm.201806-1161OC
170. Rich DQ, Schwartz J, Mittleman MA, Link M, Luttmann-Gibson H, Catalano PJ, et al. Association of short-term ambient air pollution concentrations and ventricular arrhythmias. Am J Epidemiol. (2005) 161:1123–32. doi: 10.1093/aje/kwi143
171. Rich DQ, Mittleman MA, Link MS, Schwartz J, Luttmann-Gibson H, Catalano PJ, et al. Increased risk of paroxysmal atrial fibrillation episodes associated with acute increases in ambient air pollution. Environ Health Perspect. (2006) 114:120. doi: 10.1289/ehp.8371
172. Devlin RB, Duncan KE, Jardim M, Schmitt MT, Rappold AG, Diaz-Sanchez D. Controlled exposure of healthy young volunteers to ozone causes cardiovascular effects. Circulation. (2012) 126:104–11. doi: 10.1161/CIRCULATIONAHA.112.094359
173. Wang C, Lin J, Niu Y, Wang W, Wen J, Lv L, et al. Impact of ozone exposure on heart rate variability and stress hormones: a randomized-crossover study. J Hazard Mater. (2022) 421:126750. doi: 10.1016/j.jhazmat.2021.126750
174. Farraj AK, Hazari MS, Winsett DW, Kulukulualani A, Carll AP, Haykal-Coates N, et al. Overt and latent cardiac effects of ozone inhalation in rats: evidence for autonomic modulation and increased myocardial vulnerability. Environ Health Perspect. (2012) 120:348–54. doi: 10.1289/ehp.1104244
175. Tian L, Chu N, Yang H, Yan J, Lin B, Zhang W, et al. Acute ozone exposure can cause cardiotoxicity: mitochondria play an important role in mediating myocardial apoptosis. Chemosphere. (2020) 268:128838. doi: 10.1016/j.chemosphere.2020.128838
176. Fernández-Velasco M, Ruiz-Hurtado G, Hurtado O, Moro MÁ, Delgado C. TNF-α downregulates transient outward potassium current in rat ventricular myocytes through iNOS overexpression and oxidant species generation. Am J Physiol Hear Circ Physiol. (2007) 293:H238–45. doi: 10.1152/ajpheart.01122.2006
177. Grandy SA, Fiset C. Ventricular K+ currents are reduced in mice with elevated levels of serum TNFα. J Mol Cell Cardiol. (2009) 47:238–46. doi: 10.1016/j.yjmcc.2009.02.025
178. Wang J, Wang H, Zhang Y, Gao H, Nattel S, Wang Z. Impairment of HERG K+ channel function by tumor necrosis factor-α: role of reactive oxygen species as a mediator. J Biol Chem. (2004) 279:13289–92. doi: 10.1074/jbc.C400025200
179. Monnerat G, Alarcón ML, Vasconcellos LR, Hochman-Mendez C, Brasil G, Bassani RA, et al. Macrophage-dependent IL-1β production induces cardiac arrhythmias in diabetic mice. Nat Commun. (2016) 7:1–15. doi: 10.1038/ncomms13344
180. Hagiwara Y, Miyoshi S, Fukuda K, Nishiyama N, Ikegami Y, Tanimoto K, et al. SHP2-mediated signaling cascade through gp130 is essential for LIF-dependent ICaL,[Ca2+] i transient, and APD increase in cardiomyocytes. J Mol Cell Cardiol. (2007) 43:710–6. doi: 10.1016/j.yjmcc.2007.09.004
181. Nitrogen dioxide (NO2) pollution. United States Environ Prot Agency. (2021). Available online at: https://www.epa.gov/no2-pollution (accessed February 7, 2021).
182. Milojevic A, Wilkinson P, Armstrong B, Bhaskaran K, Smeeth L, Hajat S. Short-term effects of air pollution on a range of cardiovascular events in England and Wales: case-crossover analysis of the MINAP database, hospital admissions and mortality. Heart. (2014) 100:1093–8. doi: 10.1136/heartjnl-2013-304963
183. Santurtún A, Sanchez-Lorenzo A, Villar A, Riancho JA, Zarrabeitia MT. The influence of nitrogen dioxide on arrhythmias in Spain and its relationship with atmospheric circulation. Cardiovasc Toxicol. (2017) 17:88–96. doi: 10.1007/s12012-016-9359-x
184. Collart P, Dubourg D, Levêque A, Sierra NB, Coppieters Y. Short-term effects of nitrogen dioxide on hospital admissions for cardiovascular disease in Wallonia, Belgium. Int J Cardiol. (2018) 255:231–6. doi: 10.1016/j.ijcard.2017.12.058
185. Chiu H-F, Tsai S-S, Weng H-H, Yang C-Y. Short-term effects of fine particulate air pollution on emergency room visits for cardiac arrhythmias: a case-crossover study in Taipei. J Toxicol Environ Heal Part A. (2013) 76:614–23. doi: 10.1080/15287394.2013.801763
186. Tsai S-S, Chiu H-F, Wu T-N, Yang C-Y. Air pollution and emergency room visits for cardiac arrhythmia in a subtropical city: Taipei, Taiwan. Inhal Toxicol. (2009) 21:1113–8. doi: 10.3109/08958370902758939
187. Monrad M, Sajadieh A, Christensen JS, Ketzel M, Raaschou-Nielsen O, Tjønneland A, et al. Long-term exposure to traffic-related air pollution and risk of incident atrial fibrillation: a cohort study. Environ Health Perspect. (2017) 125:422–7. doi: 10.1289/EHP392
188. Grazuleviciene R, Maroziene L, Dulskiene V, Malinauskiene V, Azaraviciene A, Laurinaviciene D, et al. Exposure to urban nitrogen dioxide pollution and the risk of myocardial infarction. Scand J Work Environ Health. (2004) 30:293–8. doi: 10.5271/sjweh.797
189. Billionnet C, Sherrill D, Annesi-Maesano I. Estimating the health effects of exposure to multi-pollutant mixture. Ann Epidemiol. (2012) 22:126–41. doi: 10.1016/j.annepidem.2011.11.004
190. Seaton A, Dennekamp M. Hypothesis: ill health associated with low concentrations of nitrogen dioxide — an effect of ultrafine particles? Thorax. (2003) 58:1012–5. doi: 10.1136/thorax.58.12.1012
191. Scaife A, Barclay J, Hillis GS, Srinivasan J, Macdonald DW, Ross JAS, et al. Lack of effect of nitrogen dioxide exposure on heart rate variability in patients with stable coronary heart disease and impaired left ventricular systolic function. Occup Env Med. (2012) 69:587–91. doi: 10.1136/oemed-2011-100126
192. Samoli E, Aga E, Touloumi G, Nisiotis K, Forsberg B, Lefranc A, et al. Short-term effects of nitrogen dioxide on mortality: an analysis within the APHEA project. Eur Respir J. (2006) 27:1129–38. doi: 10.1183/09031936.06.00143905
193. Hirokazu T, Hajime O, Suzuki AK, Kentaro K. Electrocardiographic abnormalities in rats by acute exposure to nitrogen dioxide. Toxicol Lett. (1982) 12:125–9. doi: 10.1016/0378-4274(82)90174-6
194. Tsubone H, Suzuki AK, Sagai M, Sugano S. Changes of cardiac and respiratory rhythm in non-and tracheostomized rats exposed to nitrogen dioxide. Environ Res. (1984) 35:197–203. doi: 10.1016/0013-9351(84)90127-0
195. Chan C-C, Chuang K-J, Su T-C, Lin L-Y. Association between nitrogen dioxide and heart rate variability in a susceptible population. Eur J Prev Cardiol. (2005) 12:580–6. doi: 10.1097/01.hjr.0000185975.44265.2e
196. Sørensen M, Wendelboe Nielsen O, Sajadieh A, Ketzel M, Tjønneland A, Overvad K, et al. Long-term exposure to road traffic noise and nitrogen dioxide and risk of heart failure: a cohort study. Environ Health Perspect. (2017) 125:97021. doi: 10.1289/EHP1272
197. DiFrancesco D. The role of the funny current in pacemaker activity. Circ Res. (2010) 106:434–46. doi: 10.1161/CIRCRESAHA.109.208041
198. Leary PJ, Kaufman JD, Barr RG, Bluemke DA, Curl CL, Hough CL, et al. Traffic-related air pollution and the right ventricle. The multi-ethnic study of atherosclerosis. Am J Respir Crit Care Med. (2014) 189:1093–100. doi: 10.1164/rccm.201312-2298OC
199. Karoui A, Crochemore C, Harouki N, Corbiere C, Preterre D, Vendeville C, et al. nitrogen dioxide inhalation exposures induce cardiac mitochondrial reactive oxygen species production, impair mitochondrial function and promote coronary endothelial dysfunction. Int J Environ Res Public Health. (2020) 17:5526. doi: 10.3390/ijerph17155526
200. Whittaker DG, Colman MA, Ni H, Hancox JC, Zhang H. Human atrial arrhythmogenesis and sinus bradycardia in KCNQ1-linked short QT syndrome: insights from computational modelling. Front Physiol. (2018) 9:1402. doi: 10.3389/fphys.2018.01402
201. Varshneya M, Irurzun-Arana I, Campana C, Dariolli R, Gutierrez A, Pullinger TK, et al. Investigational treatments for COVID-19 may increase ventricular arrhythmia risk through drug interactions. CPT Pharmacometr Syst Pharmacol. (2020) 10:100–7. doi: 10.1101/2020.05.21.20109397
Keywords: air pollution, arrhythmias, cardiac electrophysiology, epidemiology, review
Citation: Zhang S, Lu W, Wei Z and Zhang H (2021) Air Pollution and Cardiac Arrhythmias: From Epidemiological and Clinical Evidences to Cellular Electrophysiological Mechanisms. Front. Cardiovasc. Med. 8:736151. doi: 10.3389/fcvm.2021.736151
Received: 04 July 2021; Accepted: 04 October 2021;
Published: 28 October 2021.
Edited by:
Christopher Huang, University of Cambridge, United KingdomReviewed by:
Bence Hegyi, University of California, Davis, United StatesKamalan Jeevaratnam, University of Surrey, United Kingdom
Copyright © 2021 Zhang, Lu, Wei and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiqiang Wei, d2VpemhpcWlhbmcmI3gwMDA0MDtvdWMuZWR1LmNu; Henggui Zhang, aGVuZ2d1aS56aGFuZyYjeDAwMDQwO21hbmNoc3Rlci5hYy51aw==