
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med. , 22 November 2021
Sec. General Cardiovascular Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.734400
This article is part of the Research Topic Novel Risk Predicting System for Heart Failure View all 16 articles
 Xintao Li1†
Xintao Li1† Shi Peng1†
Shi Peng1† Bo Guan2
Bo Guan2 Songwen Chen1Genqing Zhou1Yong Wei1Chao Gong1Juan Xu1Xiaofeng Lu1Xiaoyu Zhang3,4*Shaowen Liu1*
Songwen Chen1Genqing Zhou1Yong Wei1Chao Gong1Juan Xu1Xiaofeng Lu1Xiaoyu Zhang3,4*Shaowen Liu1*Background: Positive associations between inflammatory biomarkers and the risk of heart failure (HF) have been reported in conventional observational studies. However, the causal effects of inflammatory biomarkers on HF have not been fully elucidated. We conducted a Mendelian randomization (MR) study to examine the possible etiological roles of inflammatory biomarkers in HF.
Methods: Summary statistical data for the associations between single nucleotide polymorphisms (SNPs) and C-reactive protein (CRP), fibrinogen, and components of the interleukin-1 (IL-1)-interleukin-6 (IL-6) inflammatory signaling pathway, namely, interleukin-1β (IL-1β), IL-1 receptor antagonist (IL-1ra), IL-6, and soluble IL-6 receptor (sIL-6r), were obtained from genome-wide association studies (GWASs) for individuals of European descent. The GWAS dataset of 977,323 participants of European ancestry, which included 47,309 HF cases and 930,014 controls, was collected to identify genetic variants underlying HF. A two-sample Mendelian randomization framework was implemented to examine the causality of the association between these inflammatory biomarkers and HF.
Results: Our MR analyses found that genetically determined CRP and fibrinogen were not causally associated with HF risk (odds ratio [OR] = 0.93, 95% confidence interval [CI] = 0.84–1.02, p = 0.15; OR = 0.94, 95% CI = 0.55–1.58, p = 0.80, respectively). These findings remained consistent using different Mendelian randomization methods and in sensitivity analyses. For the IL-1-IL-6 pathway, causal estimates for IL-6 (OR = 0.86, 95% CI 0.81–0.91, p < 0.001), but not for IL-1β, IL-1ra, or sIL-6r, were significant. However, the association between genetically determined IL-6 and HF risk became non-significant after excluding SNPs with potential pleiotropy (OR = 0.89, 95% CI = 0.77–1.03, p = 0.12).
Conclusion: Our study did not identify convincing evidence to support that CRP and fibrinogen, together with their upstream IL-1-IL-6 signaling pathway, were causally associated with HF risk.
Heart failure, a debilitating condition in which the heart fails to respond to increased cardiac output to meet peripheral demands, is a worldwide health burden (1). Currently, ~1–2% of adult populations in developed countries are victims of heart failure (2). Given the prolonged life expectancy of the general population and the vulnerability of the elderly to cardiac dysfunction (3), the prevalence of HF is predicted to have a two-fold increase by 2060 (4). Therefore, exploring underlying pathophysiological mechanisms to identify therapeutic targets for improving HF prognosis is a clinically unmet need.
Inflammation has been implicated in the pathogenesis of heart failure (HF). Previously, observational studies have reported that C-reactive protein (CRP), a representative biomarker of systemic inflammation, can predict the development and prognosis of HF (5–7). Another inflammatory marker, fibrinogen, is a vital determinant of blood viscosity and platelet aggregation, and has also been suggested to be associated with HF risk (8, 9). However, residual confounding and reverse causality may remain alternative explanations for the strong association between CRP and fibrinogen with heart failure because of the inherent limitation of conventional observational studies (10).
“Upstream” proinflammatory cytokines, such as interleukin-1 (IL-1) and interleukin-6 (IL-6), are major initiators for the production of “downstream” biomarkers, such as CRP and fibrinogen, from the liver (11, 12). Established associations between the classic IL-1-IL6-CRP signaling and adverse cardiac remodeling have led to investigations targeting this pathway to reduce inflammation and alleviate cardiac dysfunction (13). However, contradictory results from HF therapy based on anti-inflammation suggested otherwise (14, 15). High serum levels of IL-1 receptor antagonist (IL-1ra) and soluble IL-6 receptor (sIL-6r) have been reported to mimic the effects of endogenous inhibition of IL-1 and IL-6 signaling, respectively (16). However, investigation of these proximal inflammatory mediators is difficult given that their concentrations are subjected to constant fluctuations in the bloodstream.
Mendelian randomization is a form of analysis that uses genetic variants as instrumental variables (IVs) to generate more reliable causal estimates of long-exposure effects of risk factors on disease outcomes (17). This approach takes advantage of the naturally occurring random allocation of alleles at conception (18). Therefore, the level of a specific exposure will usually be independent of other exposures and unaffected by disease status (19). Thus, Mendelian randomization (MR) is capable of overcoming the limitations of residual confounding and reverse causation in observational studies. Here, we perform a two-sample MR analysis to test the hypothesis that genetically determined CRP and fibrinogen and their upstream inflammatory biomarkers, IL-1 and IL-6, are associated with HF risk in a European population.
Mendelian randomization is built upon three main assumptions (Figure 1) (20). First, genetic variants selected as instrumental variables should be robustly associated with the risk factor. Second, no association should exist between genetic variants and confounders. Third, genetic variants should affect the risk of outcome through the risk factor and not via other pathways.
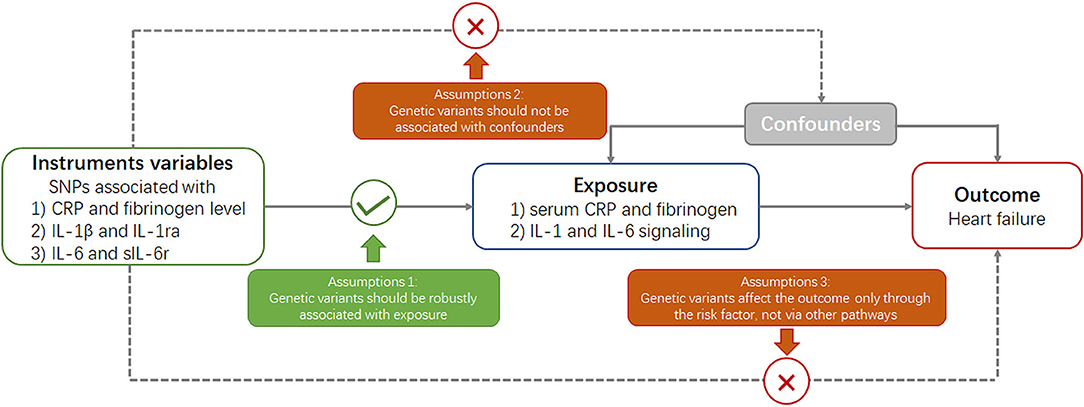
Figure 1. Key assumptions underlying Mendelian randomization study design. CRP, C-reactive protein; SNP, single nucleotide polymorphism.
Circulating CRP-associated variants were collected from the largest genome-wide association study (GWAS) aimed at identifying variants in relation to CRP concentration (involving 204,402 individuals from 88 population-based cohort studies) (21). Genetic variants for fibrinogen were identified from a GWAS meta-analysis, which enrolled >90,000 individuals from 28 studies (22). Genetic instruments for IL-1β were obtained from a GWAS of the Northern Finland Birth Cohort (23), and those for IL-1ra were obtained from a GWAS meta-analysis of 11 cohorts (24). Genetic variants for IL-6 were collected from a GWAS of the SardiNIA project (25), and those for sIL-6r were collected from a collaborative meta-analysis of human genetic and biomarker data (26) (the analytical procedure is presented in Figure 2).
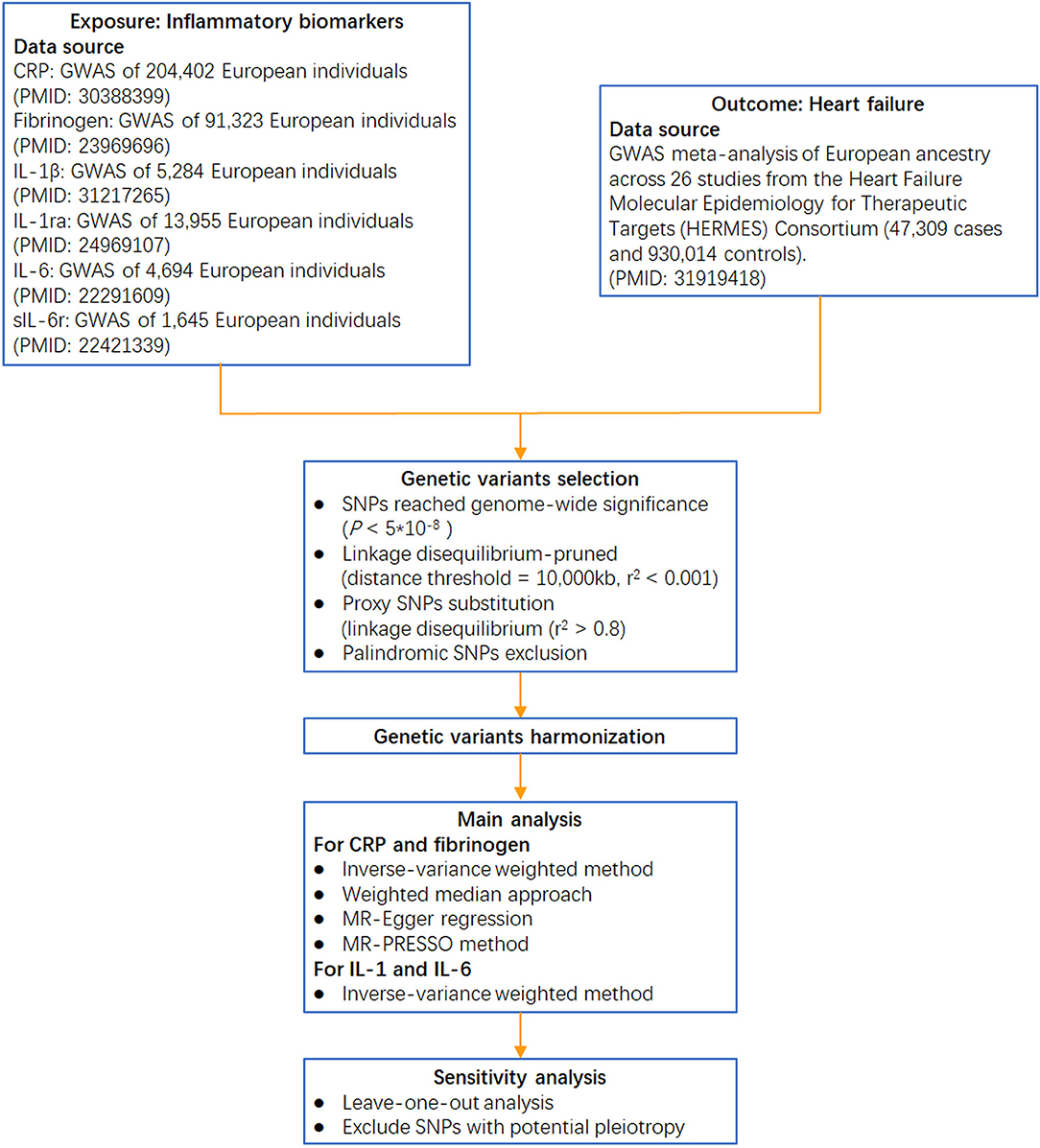
Figure 2. Flow chart of this Mendelian randomization study. IL-1β: interleukin-1β; IL-1ra: interleukin-1 receptor antagonist; sIL-6r: soluble IL-6 receptor; GWAS: genome-wide association study.
Following the identification of the genetic variants, summary statistical data on the association of genetic variants with heart failure were extracted from the published GWAS performed by the Heart Failure Molecular Epidemiology for Therapeutic Targets (HERMES) Consortium on 47,309 cases and 930,014 controls (27). HF cases from 26 cohorts of the HERMES Consortium were identified based on the clinical diagnosis of HF of any etiology with no specific inclusion criteria for left ventricular (LV) ejection fraction (Supplementary Table S1). Details of participant selection can be found elsewhere (27).
The datasets used in our study included individuals of European ancestry to reduce selection bias and to improve the robustness of the analysis. All the data used in this study were derived from GWAS for which ethical approval and patient consent were previously obtained. The study protocols were in accordance with the guidelines of the Helsinki Declaration and approved by the ethics committee of all participating sites.
Single-nucleotide polymorphisms that reached genome-wide significance (p < 5 * 10−8) were selected as instrumental variables. These SNPs were further linkage disequilibrium (LD)-pruned (distance threshold = 10,000 kb, r2 < 0.001) to ensure independence among the genetic variants (28). If the selected SNPs were not collected in the GWAS of HF, proxy SNPs in the linkage disequilibrium (r2 > 0.8) were chosen for substitution. Subsequently, palindromic SNPs were removed to ensure that the effects of the SNPs on the exposure corresponded to the same allele as their effects on HF. Accordingly, for CRP and fibrinogen, 41 and 19 SNPs, respectively, were included in the primary analysis of the association with HF. One SNP for IL-1β (rs6917603), two SNPs for IL-1ra (rs4251961, rs6759676), two SNPs for IL-6 (rs4129267, rs643434), and one SNP for sIL-6r (rs2228145) were also enrolled. The phenotypic variance explained by the selected SNPs was ~6.5% for CRP variation, 3.7% for fibrinogen variation, 1% for IL-1β, 2% for IL-1ra, 0.6% for IL-6, and 4% for sIL-6r.
We first harmonized the summary exposure and outcome data based on a previously described method (Supplementary Table S2) (29). Then, for CRP- and fibrinogen-associated instruments, four different methods of two-sample MR, namely, inverse-variance weighted, weighted median, MR-Egger, and MR-PRESSO, were implemented to calculate estimates and address possible heterogeneity and horizontal pleiotropy across the causal estimates (30). We used a predefined decision tree to select the best statistical estimation from the four methods as previously described (Supplementary Figure S1) (18). For biomarkers of IL-1β, IL-1ra, IL-6, and sIL-6r, the Wald ratio method was used to calculate estimates by dividing the beta coefficient for the SNP-outcome association with the beta coefficient for the SNP-biomarker effect. The inverse-variance weighted method with fixed effects was used to combine the Wald estimates for two SNPs.
To test the reliability of causal effect estimates, several sensitivity analyses were performed. First, a leave-one-out analysis was further conducted by removing a single variant from the analysis each time to determine whether the influence of a single SNP disproportionately affected the association. An additional sensitivity analysis was performed to address horizontal pleiotropic bias by excluding any SNPs significantly associated with potential confounders, such as body mass index (31), type 2 diabetes (32), systolic blood pressure (33), diastolic blood pressure (33), high-density lipoprotein cholesterol (34), low-density lipoprotein cholesterol (34), triglyceride (34), and smoking (35).
All results are presented as odds ratios (ORs) and corresponding 95% confidence intervals (CIs) of the outcomes with per predicted increase in CRP and fibrinogen concentrations (36). A two-sided P < 0.05 was defined as statistically significant. All the analyses were performed with the TwoSampleMR and MR-PRESSO packages with R version 4.0.2.
The causal effect estimates of genetically determined CRP and fibrinogen on the risk of HF are presented in Figures 3, 4, respectively. Significant heterogeneity was detected by the Cochran heterogeneity test among SNPs of CRP (Q = 108.8; p < 0.001) and fibrinogen (Q = 38.2; p = 0.004). The MR-Egger method showed significant directional pleiotropy for the association of CRP with HF [odds (intercept), 0.0076; p = 0.025] but not for fibrinogen [odds (intercept), 0.0074; p = 0.229]. Thus, based on a predefined decision tree (Supplementary Figure S1), no significant causal effects between CRP (MR Egger; OR = 0.93, 95% CI = 0.84–1.02, p = 0.15) and fibrinogen (weighted median; OR = 0.94, 95% CI = 0.55–1.58, p = 0.80) and HF risk were observed (Table 1). The lack of causal association persisted using all of the MR methods employed here. Leave-one-out sensitivity analyses did not reveal any significant change (Supplementary Figure S2). After excluding SNPs with potential pleiotropy, 13 SNPs for CRP and 11 SNPs for fibrinogen remained for analysis, and the results confirmed that neither genetically predicted CRP levels (weighted median; OR = 1.01, 95% CI = 0.93–1.09, p = 0.83) nor fibrinogen concentration (IVW, OR = 1.18, 95% CI = 0.77–1.81, p = 0.46) was associated with HF risk (Supplementary Table S3; Supplementary Figure S3).
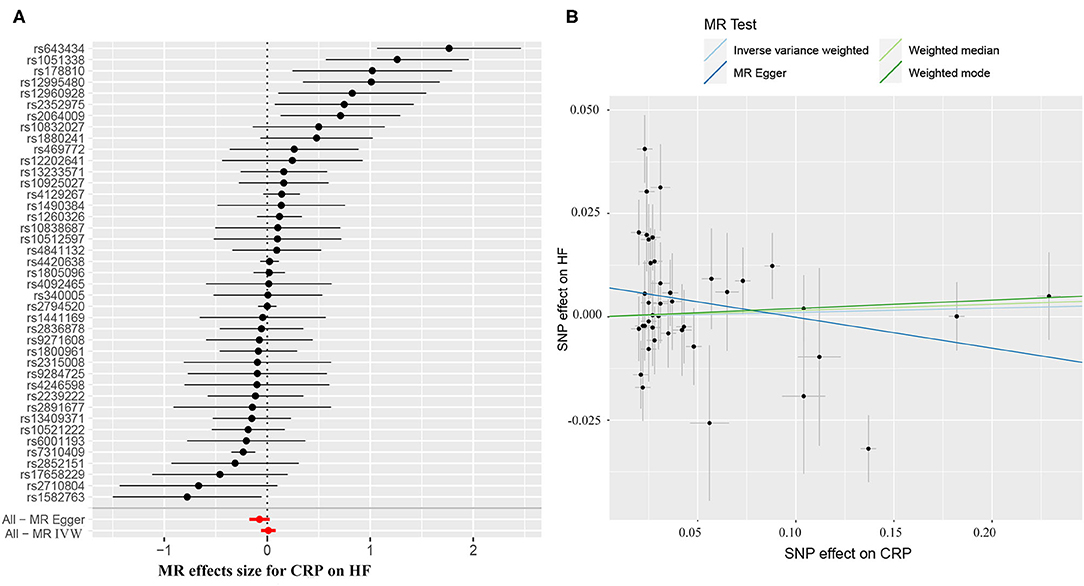
Figure 3. (A) Forest plot and (B) scatter plot of the potential effects of CRP-associated SNPs on heart failure (HF). IVW, inverse variance weighted. MR, Mendelian randomization.
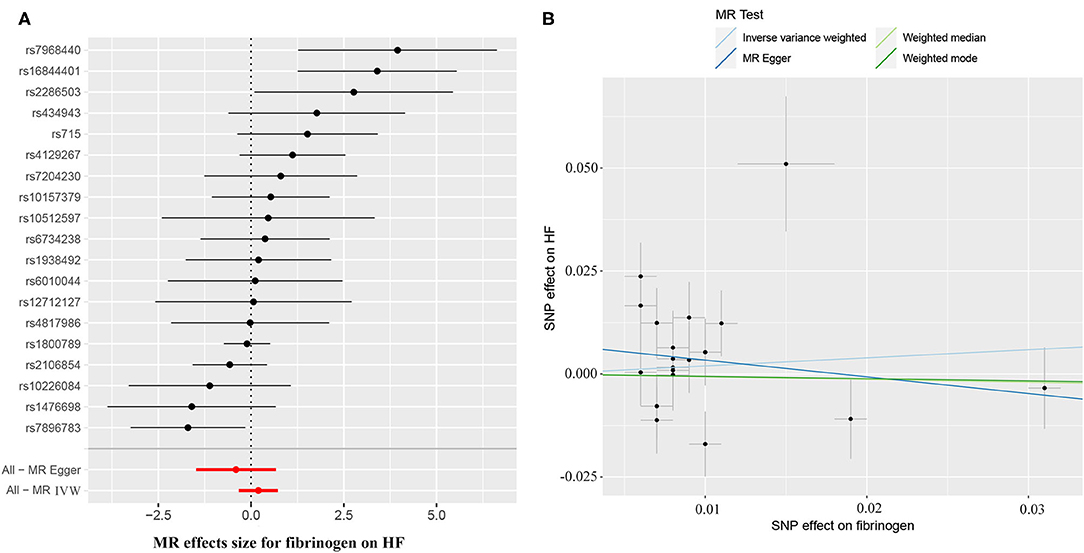
Figure 4. (A) Forest plot and (B) scatter plot of the potential effects of fibrinogen-associated SNPs on HF.
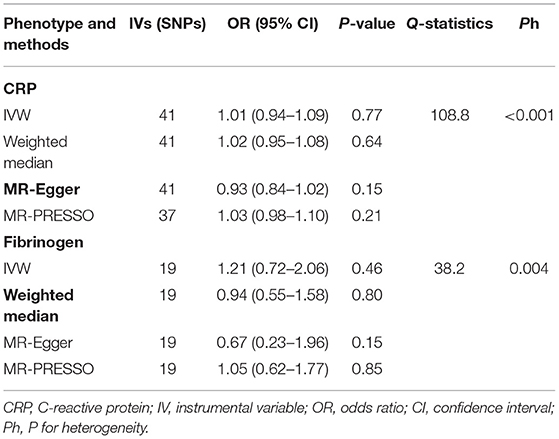
Table 1. Mendelian randomization (MR) estimates of CRP and fibrinogen with heart failure.
The results of the causal association between IL-1 and IL-6 signaling and HF are presented in Table 2. For IL-1 signaling, one SNP (rs6917603) was selected for IL-1β, and two SNPs (rs4251961 and rs6759676) were selected for IL-1ra. For IL-6 signaling, two SNPs were selected for IL-6 (rs4129267, rs643434), and one SNP was selected for sIL-6r (rs2228145). Primary MR analyses revealed an inverse causal effect of IL-6 on HF risk (OR = 0.86, 95% CI 0.81–0.91, p < 0.001), whereas no significant association between genetically determined IL-1β (OR = 1.04, 95% CI 0.86–1.25, p = 0.70), IL-1ra (OR = 0.95, 95% CI 0.83–1.10, p = 0.51), and sIL-6r (OR = 0.96, 95% CI 0.91–1.01, p = 0.14) with HF development was observed (Table 2). However, strong pleiotropic associations were noted between rs4251961 and rs643434 and cardiometabolic traits. After excluding the SNPs with pleiotropy, one SNP remained for IL-1ra and one for IL-6, and no significant causal effect of the IL-1-IL-6 pathway components on HF risk was observed (Table 2).
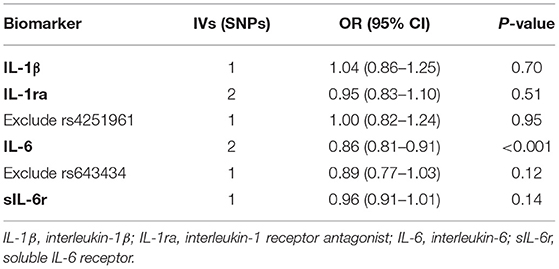
Table 2. Association between IL-1 and IL-6 signaling with heart failure risk estimated in MR analysis.
In our MR analyses of a European population, our findings did not support an important etiological role of CRP and fibrinogen in HF development. Various sensitivity analyses supported our initial findings. In our study investigating IL-1 and IL-6 signaling, there was some evidence of an association between genetically determined IL-6 and HF risk but not for IL-1β, IL-1ra, or sIL-6r. However, this association became non-significant after excluding SNPs with pleiotropy.
Inflammation has been considered to contribute to the pathogenesis and progression of HF through various mechanistic pathways, such as cardiomyocyte apoptosis, cardiac fibrosis, and endothelial dysfunction (14). Activation of systemic inflammation has been widely reported in patients with HF as elevated levels of various markers, such as CRP and fibrinogen (37, 38). Evidence from observational cohort studies has shown that increased CRP and fibrinogen are independent risk factors for HF, suggesting that these inflammation biomarkers may play etiological roles in HF. However, given that the hemodynamic stress of HF itself can induce a state of sterile inflammation (39), whether the elevation of specific biomarkers of inflammation is a reflection of their involvement in disease pathogenesis or an epiphenomenon of HF remains unsolved. Meanwhile, the inherent limitations of conventional observational studies, such as residual confounding and reverse causation, limit the ability to ascertain causal inferences. Random control trials (RCTs) are the most powerful method to demonstrate the etiology hypothesis observed in epidemiological studies (40). However, it is difficult to implement RCTs because of rigorous research designs and expensive costs. In recent years, MR research has been acknowledged as the best alternative to RCTs given that it is a very reliable method that uses genetic variants inherited randomly from parents to infer a causal relationship between risk factors and diseases (40, 41). Previous MR studies have indicated that CRP was unlikely to be a causal factor for ischemic cardiovascular disease (42), and there was only very weak evidence of a causal effect of fibrinogen on coronary heart disease (CHD) (43). In our MR study, we analyzed the correlation between inflammatory biomarkers and HF risk with the aid of large-scale GWAS datasets. Our findings revealed that genetically elevated CRP and fibrinogen were not significantly associated with HF, suggesting that these inflammatory biomarkers may function as bystanders instead of causative factors in HF.
The “upstream” proinflammatory biomarkers IL-1 and IL-6 act as important initiators of inflammation and can trigger a cascade of inflammatory mediators, such as CRP and fibrinogen (44). Although evidence from preclinical and clinical studies suggested that the IL-1-IL-6 signaling pathway led to impaired systolic and diastolic cardiac function (45), clinical trials choosing these pathways as anti-inflammatory therapeutic targets have reported controversial results. Administration of anakinra, an IL-1 receptor antagonist, yielded beneficial effects on aerobic capacity improvement but did not reduce the length of hospital stay or rehospitalization in HF (14, 46). An RCT found a significant reduction in IL-6 and CRP concentrations in 267 patients treated with colchicine compared with those treated with placebo (47). However, there was no improvement in cardiac function or measures of LV remodeling in the colchicine group (47). Recent results from the Canakinumab Anti-Inflammatory Thrombosis Outcomes Study (CANTOS) provided evidence that targeting IL-1β with canakinumab significantly reduced major cardiovascular event rates, including HF hospitalizations (48), and that the magnitude of risk reduction was correlated with the magnitude of IL-6 reduction (44). Thus, canakinumab exhibited minimal improvement in patients who did not achieve substantial reductions in IL-6. Our MR analyses initially found a significant causal effect for IL-6 on HF but not for IL-1β, IL-1ra, or sIL-6r. The inverse association between genetically predicted levels of circulating IL-6 and HF was consistent with previous MR studies that focused on CHD (49, 50), indicating that IL-6 inhibition may be associated with lower risk of HF. However, after excluding an SNP with pleiotropy (rs643434), the association became non-significant. Nevertheless, the MR analysis likely reflects lifelong exposure to risk factors, which may explain the different effects between endogenous inhibition of IL-6 and exogenous blockade of IL-6 signaling. Therefore, it is possible that the administration of an IL-6 inhibitor under a specific condition may reduce HF risk.
A major strength of this study was the design of MR analysis based on the largest GWAS meta-analysis on HF, which can prevent reverse causation and the influence of potential confounders. We also conducted additional analyses excluding SNPs with potential pleiotropy, which can minimize the bias in causal effect estimates. In addition, we performed a comprehensive evaluation of causal inferences of inflammatory biomarkers in the IL-1-IL-6-CRP pathway, which may provide a better understanding of the role of this pathway in the pathogenesis of HF.
Our study had several limitations. First, we restricted the study population to European ancestry to reduce bias from population stratification. This restriction reduced the transferability to individuals with other genetic backgrounds. Second, because of the unavailability of individual data, we could not conduct analyses stratified by subtypes and severity of HF. Since observational studies suggested that a stronger association of inflammatory markers may exist in the context of HF with preserved EF (HFpEF), the etiological role of inflammation in specific subphenotypes requires future research. Third, we only investigated the causal relationship between certain inflammatory biomarkers and HF from a genetic point of view. Therefore, our results should be treated with caution, since the causal effect of SNP exposure on SNP outcome can be modified by compensatory processes during development, and the etiological roles of other inflammatory factors in HF need further exploration.
Our MR analysis did not identify convincing evidence to support the causal relationship between inflammatory biomarkers, such as CRP and fibrinogen, or their upstream IL-1-IL-6 pathway with HF. Additional human and animal studies are needed to confirm our MR results further.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
XL and SP designed this study and conducted the main analysis. BG, SC, GZ, YW, CG, JX, XL, XZ, and SL reviewed and edited the article. All authors contributed to the article and approved the submitted version.
This work was supported by the National Natural Science Foundation of China (Grant No. 81970273).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The authors thank the HERMES Consortium, CIWG Consortium, DIAGRAM Consortium, GLGC Consortium, GIANT Consortium, TAG Consortium and other GWAS involved in our analysis for providing a publicly available GWAS dataset. The authors are grateful to Dr. Lanlan Chen of the First Hospital of Jilin University for the instructions on Mendelian randomization analysis. The authors are also grateful to Pro. Gary Tse of Second Hospital of Tianjin Medical University and Dr. Flores, Ashley Christina of the Pennsylvania State University for English language polishing of this paper.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2021.734400/full#supplementary-material
1. Crespo-Leiro MG, Metra M, Lund LH, Milicic D, Costanzo MR, Filippatos G, et al. Advanced heart failure: a position statement of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail. (2018) 20:1505–35. doi: 10.1002/ejhf.1236
2. Ziaeian B, Fonarow GC. Epidemiology and aetiology of heart failure. Nat Rev Cardiol. (2016) 13:368–78. doi: 10.1038/nrcardio.2016.25
3. Zhang Y, Yuan M, Gong M, Li G, Liu T, Tse G. Associations between prefrailty or frailty components and clinical outcomes in heart failure: a follow-up meta-analysis. J Am Med Dir Assoc. (2019) 20:509–10. doi: 10.1016/j.jamda.2018.10.029
4. de Boer RA, Nayor M, deFilippi CR, Enserro D, Bhambhani V, Kizer JR, et al. Association of cardiovascular biomarkers with incident heart failure with preserved and reduced ejection fraction. JAMA Cardiol. (2018) 3:215–24. doi: 10.1001/jamacardio.2017.4987
5. Kang S, Fan LY, Chen M, Li J, Liu ZM. Relationship of high-sensitivity C-reactive protein concentrations and systolic heart failure. Curr Vasc Pharmacol. (2017) 15:390–6. doi: 10.2174/1570161115666170404121619
6. Benz AP, Aeschbacher S, Krisai P, Moschovitis G, Blum S, Meyre P, et al. Biomarkers of inflammation and risk of hospitalization for heart failure in patients with atrial fibrillation. J Am Heart Assoc. (2021) 10:e019168. doi: 10.1161/JAHA.120.019168
7. Araújo JP, Lourenço P, Azevedo A, Friões F, Rocha-Gonçalves F, Ferreira A, et al. Prognostic value of high-sensitivity C-reactive protein in heart failure: a systematic review. J Card Fail. (2009) 15:256–66. doi: 10.1016/j.cardfail.2008.10.030
8. Eisen A, Benderly M, Behar S, Goldbourt U, Haim M. Inflammation and future risk of symptomatic heart failure in patients with stable coronary artery disease. Am Heart J. (2014) 167:707–14. doi: 10.1016/j.ahj.2014.01.008
9. Suthahar N, Lau ES, Blaha MJ, Paniagua SM, Larson MG, Psaty BM, et al. Sex-specific associations of cardiovascular risk factors and biomarkers with incident heart failure. J Am Coll Cardiol. (2020) 76:1455–65. doi: 10.1016/j.jacc.2020.07.044
10. van der Laan SW, Fall T, Soumaré A, Teumer A, Sedaghat S, Baumert J, et al. Cystatin C and cardiovascular disease: a mendelian randomization study. J Am Coll Cardiol. (2016) 68:934–45. doi: 10.1016/j.jacc.2016.05.092
11. Ridker PM. Targeting inflammatory pathways for the treatment of cardiovascular disease. Eur Heart J. (2014) 35:540–3. doi: 10.1093/eurheartj/eht398
12. Ridker PM, Lüscher TF. Anti-inflammatory therapies for cardiovascular disease. Eur Heart J. (2014) 35:1782–91. doi: 10.1093/eurheartj/ehu203
13. Hanna A, Frangogiannis NG. Inflammatory cytokines and chemokines as therapeutic targets in heart failure. Cardiovasc Drugs Ther. (2020) 34:849–63. doi: 10.1007/s10557-020-07071-0
14. Murphy SP, Kakkar R, McCarthy CP, Januzzi JL. Inflammation in heart failure: JACC state-of-the-art review. J Am Coll Cardiol. (2020) 75:1324–40. doi: 10.1016/j.jacc.2020.01.014
15. Adamo L, Rocha-Resende C, Prabhu SD, Mann DL. Reappraising the role of inflammation in heart failure. Nat Rev Cardiol. (2020) 17:269–85. doi: 10.1038/s41569-019-0315-x
16. Lin J, Wang Y, Wang Y, Pan Y. Inflammatory biomarkers and risk of ischemic stroke and subtypes: a 2-sample Mendelian randomization study. Neurol Res. (2020) 42:118–25. doi: 10.1080/01616412.2019.1710404
17. Robinson T, Martin RM, Yarmolinsky J. Mendelian randomisation analysis of circulating adipokines and C-reactive protein on breast cancer risk. Int J Cancer. (2020) 147:1597–603. doi: 10.1002/ijc.32947
18. Nazarzadeh M, Pinho-Gomes AC, Bidel Z, Dehghan A, Canoy D, Hassaine A, et al. Plasma lipids and risk of aortic valve stenosis: a Mendelian randomization study. Eur Heart J. (2020) 41:3913–20. doi: 10.1093/eurheartj/ehaa070
19. van Oort S, Beulens JWJ, van Ballegooijen AJ, Handoko ML, Larsson SC. Modifiable lifestyle factors and heart failure: a Mendelian randomization study. Am Heart J. (2020) 227:64–73. doi: 10.1016/j.ahj.2020.06.007
20. Burgess S, Scott RA, Timpson NJ, Davey Smith G, Thompson SG. Using published data in Mendelian randomization: a blueprint for efficient identification of causal risk factors. Eur J Epidemiol. (2015) 30:543–52. doi: 10.1007/s10654-015-0011-z
21. Ligthart S, Vaez A, Võsa U, Stathopoulou MG, de Vries PS, Prins BP, et al. Genome analyses of >200,000 individuals identify 58 loci for chronic inflammation and highlight pathways that link inflammation and complex disorders. Am J Hum Genet. (2018) 103:691–706. doi: 10.1016/j.ajhg.2018.09.009
22. Sabater-Lleal M, Huang J, Chasman D, Naitza S, Dehghan A, Johnson AD, et al. Multiethnic meta-analysis of genome-wide association studies in >100 000 subjects identifies 23 fibrinogen-associated Loci but no strong evidence of a causal association between circulating fibrinogen and cardiovascular disease. Circulation. (2013) 128:1310–24. doi: 10.1161/CIRCULATIONAHA.113.002251
23. Sliz E, Kalaoja M, Ahola-Olli A, Raitakari O, Perola M, Salomaa V, et al. Genome-wide association study identifies seven novel loci associating with circulating cytokines and cell adhesion molecules in Finns. J Med Genet. (2019) 56:607–16. doi: 10.1136/jmedgenet-2018-105965
24. Herder C, Nuotio ML, Shah S, Blankenberg S, Brunner EJ, Carstensen M, et al. Genetic determinants of circulating interleukin-1 receptor antagonist levels and their association with glycemic traits. Diabetes. (2014) 63:4343–59. doi: 10.2337/db14-0731
25. Naitza S, Porcu E, Steri M, Taub DD, Mulas A, Xiao X, et al. A genome-wide association scan on the levels of markers of inflammation in Sardinians reveals associations that underpin its complex regulation. PLoS Genet. (2012) 8:e1002480. doi: 10.1371/journal.pgen.1002480
26. Sarwar N, Butterworth AS, Freitag DF, Gregson J, Willeit P, Gorman DN, et al. Interleukin-6 receptor pathways in coronary heart disease: a collaborative meta-analysis of 82 studies. Lancet. (2012) 379:1205–13. doi: 10.1016/S0140-6736(11)61931-4
27. Shah S, Henry A, Roselli C, Lin H, Sveinbjörnsson G, Fatemifar G, et al. Genome-wide association and Mendelian randomisation analysis provide insights into the pathogenesis of heart failure. Nat Commun. (2020) 11:163. doi: 10.1038/s41467-019-13690-5
28. Levin MG, Judy R, Gill D, Vujkovic M, Verma SS, Bradford Y, et al. Genetics of height and risk of atrial fibrillation: A Mendelian randomization study. PLoS Med. (2020) 17:e1003288. doi: 10.1371/journal.pmed.1003288
29. Hartwig FP, Davies NM, Hemani G, Davey Smith G. Two-sample Mendelian randomization: avoiding the downsides of a powerful, widely applicable but potentially fallible technique. Int J Epidemiol. (2016) 45:1717–26. doi: 10.1093/ije/dyx028
30. Jiang Q, Qin D, Yang L, Lin Y, Zhai L, Zhang Y, et al. Causal effects of plasma lipids on the risk of atrial fibrillation: a multivariable mendelian randomization study. Nutr Metab Cardiovasc Dis. (2021) 31:1569–78. doi: 10.1016/j.numecd.2021.02.011
31. Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. (2015) 518:197–206. doi: 10.1038/nature14177
32. Mahajan A, Go MJ, Zhang W, Below JE, Gaulton KJ, Ferreira T, et al. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet. (2014) 46:234–44. doi: 10.1038/ng.2897
33. Evangelou E, Warren HR, Mosen-Ansorena D, Mifsud B, Pazoki R, Gao H, et al. Genetic analysis of over 1 million people identifies 535 new loci associated with blood pressure traits. Nat Genet. (2018) 50:1412–25. doi: 10.1038/s41588-018-0205-x
34. Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S, et al. Discovery and refinement of loci associated with lipid levels. Nat Genet. (2013) 45:1274–83. doi: 10.1038/ng.2797
35. Tobacco and Genetics Consortium. Genome-wide meta-analyses identify multiple loci associated with smoking behavior. Nat Genet. (2010) 42:441–7. doi: 10.1038/ng.571
36. Wang B, Zhang X, Liu D, Zhang J, Cao M, Tian X, et al. The role of C-reactive protein and fibrinogen in the development of intracerebral hemorrhage: a mendelian randomization study in European population. Front Genet. (2021) 12:608714. doi: 10.3389/fgene.2021.608714
37. Redfield MM, Chen HH, Borlaug BA, Semigran MJ, Lee KL, Lewis G, et al. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA. (2013) 309:1268–77. doi: 10.1001/jama.2013.2024
38. Pfisterer M, Buser P, Rickli H, Gutmann M, Erne P, Rickenbacher P, et al. BNP-guided vs symptom-guided heart failure therapy: the Trial of Intensified vs Standard Medical Therapy in Elderly Patients With Congestive Heart Failure (TIME-CHF) randomized trial. JAMA. (2009) 301:383–92. doi: 10.1001/jama.2009.2
39. Nakayama H, Otsu K. Translation of hemodynamic stress to sterile inflammation in the heart. Trends Endocrinol Metab. (2013) 24:546–53. doi: 10.1016/j.tem.2013.06.004
40. Miao L, Deng GX, Yin RX, Nie RJ, Yang S, Wang Y, et al. No causal effects of plasma homocysteine levels on the risk of coronary heart disease or acute myocardial infarction: a Mendelian randomization study. Eur J Prev Cardiol. (2021) 28:227–34. doi: 10.1177/2047487319894679
41. Nitsch D, Molokhia M, Smeeth L, DeStavola BL, Whittaker JC, Leon DA. Limits to causal inference based on Mendelian randomization: a comparison with randomized controlled trials. Am J Epidemiol. (2006) 163:397–403. doi: 10.1093/aje/kwj062
42. Zacho J, Tybjaerg-Hansen A, Jensen JS, Grande P, Sillesen H, Nordestgaard BG. Genetically elevated C-reactive protein and ischemic vascular disease. N Engl J Med. (2008) 359:1897–908. doi: 10.1056/NEJMoa0707402
43. Ward-Caviness CK, de Vries PS, Wiggins KL, Huffman JE, Yanek LR, Bielak LF, et al. Mendelian randomization evaluation of causal effects of fibrinogen on incident coronary heart disease. PLoS ONE. (2019) 14:e0216222. doi: 10.1371/journal.pone.0216222
44. Ridker PM, Rane M. Interleukin-6 signaling and anti-interleukin-6 therapeutics in cardiovascular disease. Circ Res. (2021) 128:1728–46. doi: 10.1161/CIRCRESAHA.121.319077
45. Chia YC, Kieneker LM, van Hassel G, Binnenmars SH, Nolte IM, van Zanden JJ, et al. Interleukin 6 and development of heart failure with preserved ejection fraction in the general population. J Am Heart Assoc. (2021) 10:e018549. doi: 10.1161/JAHA.120.018549
46. Van Tassell BW, Abouzaki NA, Oddi Erdle C, Carbone S, Trankle CR, Melchior RD, et al. Interleukin-1 blockade in acute decompensated heart failure: a randomized, double-blinded, placebo-controlled pilot study. J Cardiovasc Pharmacol. (2016) 67:544–51. doi: 10.1097/FJC.0000000000000378
47. Deftereos S, Giannopoulos G, Panagopoulou V, Bouras G, Raisakis K, Kossyvakis C, et al. Anti-inflammatory treatment with colchicine in stable chronic heart failure: a prospective, randomized study. JACC Heart Fail. (2014) 2:131–7. doi: 10.1016/j.jchf.2013.11.006
48. Everett BM, Cornel JH, Lainscak M, Anker SD, Abbate A, Thuren T, et al. Anti-inflammatory therapy with canakinumab for the prevention of hospitalization for heart failure. Circulation. (2019) 139:1289–99. doi: 10.1161/CIRCULATIONAHA.118.038010
49. Swerdlow DI, Holmes MV, Kuchenbaecker KB, Engmann JE, Shah T, Sofat R, et al. The interleukin-6 receptor as a target for prevention of coronary heart disease: a mendelian randomisation analysis. Lancet. (2012) 379:1214–24. doi: 10.1016/S0140-6736(12)60110-X
Keywords: heart failure, Mendelian randomization, C-reactive protein, fibrinogen, interleukin
Citation: Li X, Peng S, Guan B, Chen S, Zhou G, Wei Y, Gong C, Xu J, Lu X, Zhang X and Liu S (2021) Genetically Determined Inflammatory Biomarkers and the Risk of Heart Failure: A Mendelian Randomization Study. Front. Cardiovasc. Med. 8:734400. doi: 10.3389/fcvm.2021.734400
Received: 01 July 2021; Accepted: 20 October 2021;
Published: 22 November 2021.
Edited by:
Chen Liu, The First Affiliated Hospital of Sun Yat-sen University, ChinaReviewed by:
YuQin Shen, Tongji Hospital Affiliated to Tongji University, ChinaCopyright © 2021 Li, Peng, Guan, Chen, Zhou, Wei, Gong, Xu, Lu, Zhang and Liu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoyu Zhang, aHlkenh5QDEyNi5jb20=; Shaowen Liu, c2hhb3dlbi5saXVAaG90bWFpbC5jb20=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.