
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med. , 01 September 2021
Sec. Cardiovascular Biologics and Regenerative Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.724846
 Bowen Li1,2†
Bowen Li1,2† Kun Chen3†
Kun Chen3† Fangfang Liu4†Juan Zhang5
Fangfang Liu4†Juan Zhang5 Xihui Chen1,2Tangdong Chen1,2Qi Chen1,2Yan Yao6Weihong Hu6Li Wang1,2,7*Yuanming Wu1,2*
Xihui Chen1,2Tangdong Chen1,2Qi Chen1,2Yan Yao6Weihong Hu6Li Wang1,2,7*Yuanming Wu1,2*Background: Mitochondrial aminoacyl-tRNA synthetases (mtARSs) catalyze the binding of specific amino acids to their cognate tRNAs and play an essential role in the synthesis of proteins encoded by mitochondrial DNA. Defects in mtARSs have been linked to human diseases, but their tissue-specific pathophysiology remains elusive. Here we examined the role of mitochondrial phenylalanyl-tRNA synthetase (FARS2) in developmental angiogenesis and its potential contribution to the pathogenesis of cardiovascular disease.
Methods: Morpholinos were injected into fertilized zebrafish ova to establish an in vivo fars2 knock-down model. A visualization of the vasculature was achieved by using Tg (fli1: EGFP) y1 transgenic zebrafish. In addition, small interference RNAs (siRNAs) were transferred into human umbilical vein endothelial cells (HUVECs) to establish an in vitro FARS2 knock-down model. Cell motility, proliferation, and tubulogenesis were determined using scratch-wound CCK8, transwell-based migration, and tube formation assays. In addition, mitochondria- and non-mitochondria-related respiration were evaluated using a Seahorse XF24 analyzer and flow cytometry assays. Analyses of the expression levels of transcripts and proteins were performed using qRT-PCR and western blotting, respectively.
Results: The knock-down of fars2 hampered the embryonic development in zebrafish and delayed the formation of the vasculature in Tg (fli1: EGFP) y1 transgenic zebrafish. In addition, the siRNA-mediated knock-down of FARS2 impaired angiogenesis in HUVECs as indicated by decreased cell motility and tube formation capacity. The knock-down of FARS2 also produced variable decreases in mitochondrial- and non-mitochondrial respiration in HUVECs and disrupted the regulatory pathways of angiogenesis in both HUVECs and zebrafish.
Conclusion: Our current work offers novel insights into angiogenesis defects and cardiovascular diseases induced by FARS2 deficiency.
Aminoacyl-tRNA synthetases pair amino acids with their cognate tRNAs and are therefore crucial for protein synthesis (1). Human mitochondrial DNA (mtDNA) encodes 13 indispensable subunits of the oxidative phosphorylation system (OXPHOs), 22 tRNAs, and two rRNAs (2). Mitochondrial aminoacyl-tRNA synthetases (mtARSs) play an important role in the translation of mtDNA coding genes. Although deficiencies in mtARSs are naturally assumed to impact mitochondrial protein synthesis, they have also been associated with various tissue-specific phenotypes (3–5). In particular, mutations in several mtARSs result in encephalopathies (RARS2, NARS2, CARS2, IARS2, FARS2, PARS2, TARS2, and VARS2), leukodystrophies (AARS2, DARS2, EARS2, and MARS2), or Perrault syndrome (HARS2 and LARS2). In addition, mutations in three other aminoacyl-tRNA synthetases cause cardiomyopathies (AARS2, GARS, and KARS), while mutations in YARS2 and SARS2 cause MLASA syndrome and HUPRA syndrome, respectively. Notably, different clinical features have been reported in patients with mutations in the same mtARSs gene (5, 6).
Mitochondrial phenylalanyl-tRNA synthetase, encoded by the nuclear gene FARS2, catalyzes the recognition and binding of Phe and mt-tRNAPhe in the mitochondria (5). Mutations in the FARS2 gene are associated with central nervous system (CNS) diseases, such as autosomal recessive spastic paraplegia (7), epileptic encephalopathy (8–10), and infantile mitochondrial Alpers encephalopathy (11–13). In addition, our group reported that a missense homozygous mutation [c.424 G > T (p.D142Y)] in the FARS2 gene was the underlying cause of hereditary spastic paraplegia in a Chinese family (7). Because CNS disorders are recognized as the major manifestations of FARS2 gene mutations, previous research into the potential molecular mechanisms involved in the pathogenicity of these mutations has focused on the CNS (7, 8, 10, 12–16), and little is known about their effects on the cardiovascular system.
Cardiovascular diseases (CVDs), including stroke, heart failure, coronary artery disease, cardiomyopathy, and hypertensive heart disease, are some of the leading causes of death worldwide (17–19). Nonetheless, the etiology of CVDs has not been well investigated on account of their multi-factorial causes, covering inherited and environmental factors (20). Endothelial cells (ECs) play an indispensable role in angiogenesis and vascular remodeling, and endothelial dysfunction occurs in the early stages of CVDs such as coronary artery disease (21, 22). Angiogenesis, a process in which new blood vessels are formed from pre-existing vessels, is crucial for embryogenesis, tissue healing, and placental vascularization (23). In response to angiogenic stimuli, ECs differentiate into two distinct subtypes that perform characteristic functions: the tip cells extend the filopodia of the vascular branch frontlines, and the stalk cells extend the vascular branches behind the tip cells. Following the formation of the vascular network and blood perfusion, ECs are trans-differentiated into quiescent phalanx cells that line the new vessels (24–26). This complex process of EC specialization is regulated by multifarious signaling molecules, including paracrine and autocrine factors, as well as by oxidative respiratory metabolism. The mitochondria play an essential role in cellular oxidative respiration; however, although angiogenesis is an energy-intensive process, the respiratory metabolism in ECs is highly glycolytic and relies little on the mitochondria (27–31). Nonetheless, the mitochondria not only play a major role in aerobic oxidation but are also key intracellular structures that regulate several EC functions (32–34). While mitochondria-related metabolism resulting from angiogenic stimuli has been studied extensively (34, 35), the functions of mitochondrial protein synthesis in angiogenesis are only partially understood.
Angiogenesis is regulated by a complex network of molecules. As one of the indispensable pathways regulating embryonic development, the Wnt signaling pathway regulates a variety of complex biological processes (36, 37). The high expression levels of Wnt signaling genes in ECs during vasculature development support the pivotal role of this pathway in angiogenesis (38, 39). The Notch pathway, another evolutionarily conserved signaling system, is required for normal embryonic development, tissue homeostasis, and adult stem cell maintenance (40) and controls the specification of ECs in multiple vertebrates, such as chicken, zebrafish, and mice. Although the intracellular signaling pathways regulated by angiogenic stimulation have been investigated widely, the relationship between FARS2 and signaling transduction in angiogenesis is unknown. Here, to determine whether the FARS2 gene plays an essential role in developmental angiogenesis, we established two FARS2 deficiency models. In the in vivo model, Tg (fli1: EGFP) y1 transgenic zebrafish were treated with fars2-specific morpholinos (41). In the in vitro model, HUVECs were transfected with FARS2-specific small interference RNAs (siRNAs). By combining imaging, post-transcriptional manipulations of FARS2, and gene expression detection techniques, we found that FARS2 might participate in the pathological process of CVD by affecting the mitochondrial protein synthesis in ECs. Our data demonstrate a previously unanticipated role of FARS2 in coordinating the angiogenic process.
Adult wild-type AB strain zebrafish were maintained at 28.5 °C on a 14-h light/10-h dark cycle. Five to six pairs of zebrafish were set up for natural mating every time. On average, 200–300 embryos were generated. The embryos were maintained at 28.5 °C in fish water (0.2% Instant Ocean Salt in deionized water). The embryos were washed and staged according to (41). The establishment and characterization of fli1a-EGFP transgenic lines have been described elsewhere (42). The zebrafish facility at Shanghai Model Organisms Center is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International.
Gene Tools, LLC (http://www.gene-tools.com/) designed the morpholinos (MOs). Antisense MOs (GeneTools) were microinjected into fertilized one-cell-stage embryos according to standard protocols (43). The sequences of the fars2 translation-blocking and splice-blocking morpholinos were 5′-CATAGTAGCTGGTCCATAAGCCTCT-3′ (ATG-MO) and 5′-GAACATGGCAGGATTCCTACCTTCC-3′ (E3I3-MO), respectively. The sequence for the standard control morpholino was 5′-CCTCTTACCTCAGTTACAATTTATA-3′ (Gene Tools). The amount of the MOs used for injection was as follows: control-MO and ATG-MO, 4 ng per embryo; and E3I3-MO, 8 ng per embryo. Primers spanning fars2 exon 2 (forward primer: 5′-CACTATCCCGTCTTCCATCAG-3′) and exon 4 (reverse primer: 5′-TGAAAGAACACCTCCATCTCG-3′) were used for RT-PCR analysis for confirmation of the efficacy of E3I3-MO. The primer ef1α sequences used as the internal control were 5′-GGAAATTCGAGACCAGCAAATAC-3′ (forward) and 5′-GATACCAGCCTCAAACTCACC-3′ (reverse).
For zebrafish, total RNA was extracted from 30 to 50 embryos per group in Trizol (Roche) according to the instructions of the manufacturer. The RNA was reverse-transcribed using the PrimeScript RT reagent Kit with gDNA Eraser (Takara). The quantification of gene expression was performed in triplicates using Bio-rad iQ SYBR Green Supermix (Bio-rad) with detection on the Realplex system (Eppendorf). The relative gene expression quantification was based on the comparative threshold cycle method (2−ΔΔCt) using ef1α as endogenous control gene. The primer sequences are given in Supplementary Table 1.
For HUVECs, total RNA was extracted from cells by using Axypre™ Multisource Total RNA Miniprep Kit (Axygen, cat. #365). The total RNA was reverse-transcribed with PrimeScript™ RT Master Mix (Takara, cat. #RR036A). Real-time fluorescent quantitative PCR was implemented by SYBR® Premix Ex Taq™ II (Takara, #RR820A) using 7500 system (Applied Biosystems). The procedures of the qRT-PCR were as follows: 95°C for 30 s for the first step and then for the ensuing 40 cycles–95 °C for 5 s and 60 °C for 30 s. Relative gene expression quantification was based on the comparative threshold cycle method (2−ΔΔCt) using GAPDH as the endogenous control gene (44). The primer sequences are given in Supplementary Table 1. All experiments were performed in triplicate and repeated three times independently.
To evaluate blood vessel formation in zebrafish, fertilized one-cell fli1a-EGFP transgenic line embryos were injected with fars2-MO and control-MO. At 48 hpf, the embryos were dechorionated and anesthetized with 0.016% MS-222 (tricaine methanesulfonate, Sigma-Aldrich, St. Louis, MO). The zebrafish were then oriented on the lateral side (anterior, left; posterior, right; dorsal, top) and mounted with 3% methylcellulose in a depression slide for observation by fluorescence microscopy. The phenotypes of complete intersegmental vessels (ISVs) [i.e., the number of ISVs that connect the dorsal anastomotic vessels to the dorsal longitudinal anastomotic vessels (DLAVs)], caudal vein plexus (CVP), DLAVs, and parachordal vessels (PAVs) were quantitatively analyzed. A total of 10 animals from at least three independent MO injections in each group were used in this experiment.
Human umbilical vein endothelial cells (HUVECs, Sciencell cat. # 8000) were used from passages 3–9 and cultured in endothelial cell medium (ECM, Sciencell cat. # 1001) containing 500 ml of basal medium, 5% fetal bovine serum (FBS, Sciencell cat. #0025), 1% endothelial cell growth supplement (Sciencell cat. #1052), and 1% antibiotic solution (P/S, Sciencell cat. #0503) in 5% CO2 at 37°C. Then, 2 × 105 cells, 105 cells, and 104 cells per well were seeded in six-well, 12-well, and 96-well plates for siRNA transfection. The cells were transfected with the following siRNAs: a FAM-labeled non-relevant control (50 nM), a non-relevant control (siCtrl, 50 nM), and FARS2 siRNA (si-FARS2, 50 nM) from Ribobio™ (Guangzhou, China). The specific target sequences of these siRNAs are listed in Supplementary Table 2. X-tremeGENE siRNA Transfection Reagent (Roche) was used to build cells in the transfection process. In brief, X-tremeGENE siRNA Transfection Reagent and siRNA were separately diluted in Opti-MEM (Gibco cat. # 31985070) and mixed for 15 min at room temperature. Then, the mixture was added into the plates. The evaluation of transfection efficiency and functional assays on HUVECs was performed at 48 h after transfection. The transfection efficiency was monitored by calculating the percentage of FAM-positive cells under a fluorescence microscope.
We studied western blotting as described previously (44). The RIPA buffer (Biotime Biotechnology, cat. #P0013B, China), which included a protease inhibitor and a phosphatase inhibitor (Roche), was used for cell lysates. Then, the protein concentration was quantified by using a bicinchoninic acid (BCA) protein assay kit (Biovision, cat. #K813-2500). After mixing with 6 × loading buffer (Tiangen, cat#RT201), the protein samples were boiled for 10 mins in a metal bath for sufficient denaturation. Then, 10 μl (2 μg/μl) protein samples were measured in this study. After separating the different-molecular-weight proteins by 10% SDS-PAGE, all the proteins were transferred to the polyvinylidene difluoride membrane (Millipore, Germany, 0.45 μm) and blocked with 5% skim milk for 1 h at room temperature. The primary antibodies used included anti-FARS2 (1:1,000, Invitrogen), NOTCH1 (1:1,000, Abcam cat. #ab52627), β-catenin (1:1,000, Abcam cat. #ab16051), and GAPDH (1:20,000, Proteintech cat. #60004-1-Ig), followed by corresponding secondary antibodies (anti-mouse, 1:8,000 and anti-rabbit, 1:8,000, coupled to horseradish peroxidase). Proteins were revealed by chemiluminescence using the ECL kit (Millipore) (44). All experiments were performed in triplicate and repeated three times independently.
The HUVEC cell proliferation assay in vitro was evaluated by CCK8 assay (44). The HUVECs were seeded in a 96-well plate with 100 μl ECM per well at 24 h before transfecting with siRNAs. Then, 10 μl CCK8 (HanBio, cat. # HB-CCK8-500T, Shanghai, China) reagent was added to each well for 1 h at 48 h after transfection with siRNAs. We measured the absorbance at 450 nm to detect proliferation of cells. All experiments were performed in triplicate and repeated three times independently.
The HUVEC migration assay was as described previously (45). In brief, for one well of a 24-well plate, the HUVECs transfected for 48 h were re-seeded in the upside of the transwell chamber (Corning) with 500 μl basal medium; 700 μl ECM (containing 5% FBS) was added in the bottom of the well. After cultivating for 24 h, the chamber was wiped with a cotton swab. The cells were fixed with 4% paraformaldehyde, stained with crystal violet solution, and counted under a microscope (× 20 objective). At least three different fields were averaged, and the experiment was repeated three times independently.
The HUVEC scratch-wound migration assay was evaluated by wound-healing assay (46). Briefly, the cells were transfected with siRNAs for 48 h (cultured upon reaching 90–95% confluence) in a six-well plate with 2 ml ECM; the HUVECs were scratched with the head of a 200-μl tip. The motility of the cells into the wound was imaged under a microscope (× 10 objective) at 0 and 6 h after wounding. The blank area in the wound was detected using Fiji Image J (NIH, Bethesda, MD, United States). All experiments were performed in triplicate and repeated three times independently.
The method of tube network formation was studied as described previously (47). After transfection for 48 h, 300 μl of HUVEC suspension (4 × 105 cells/ml) was re-seeded in a 24-well plate pre-coated with 289 μl Matrigel (10mg/ml, Corning cat. #354248) per well, which was polymerized by incubating in 37 °C for 30 min. Then, an Olympus microscope, with × 10 objectives, was used to take brightfield images of the 24-well plate. Fiji Image J (NIH, Bethesda, MD, United States) was employed to count the number of intersections in each field, and the total length of the structures was measured (48). At least three different fields were averaged, and the experiment was repeated three times independently.
We studied mitochondrial stress testing as described previously (49). Seahorse Bioscience XFp extracellular flux analyzer (Agilent) was used to measure the mitochondrial stress test of HUVECs. This device works by creating a sealed chamber to measure oxygen consumption by the mitochondria in real time in the microplates under various stimuli. Mitochondrial reagents (Seahorse Bioscience Cell Mito Stress Test Kit, Agilent cat. #103010-100) were optimized at 2 μg/ml oligomycin (complex V inhibitor), 5 μM FCCP (a respiratory uncoupler), and 2 μM rotenone/antimycin A (inhibitors of complex I and complex III). A total of 30,000 HUVECs transfected with siRNAs for 48 h were seeded into the seahorse cell culture plate per well with 500 μl ECM and cultured at 37 °C in 5% CO2 humid atmosphere overnight. The sensor cartridge was incubated at 37 °C in a non-CO2 incubator for 24 h before detection. The cell culture plate and sensor cartridge were placed on XFp extracellular flux analyzer for Mito Stress Test. After detection, all the data were normalized to the BCA quantification of each well. This synthetic bioenergy spectrum provides detailed information on the various components of the respiratory chain. In brief, six essential parameters of mitochondrial respiration function were calculated from the results: basal respiration, ATP production, proton leakage, maximum respiration, spare respiration capacity, and non-mitochondrial respiration.
The intracellular reactive oxygen species (ROS) was analyzed by Reactive Oxygen Species Assay Kit (Beyotime cat. # S0033S, China). HUVECs (2 × 105 per well of six-well plates) were seeded and transfected with siRNAs for 48 h. Then, the cells were washed with PBS once; 1 ml of DCFH-DA (1:1,000 dilution) was added to each collecting tube in the dark and incubated at 37 °C for 30 min. The labeled cells were collected and analyzed by flow cytometry at 488 nm. All experiments were repeated three times independently.
ATP dissolved in cells was detected by enhanced ATP assay kits (Beyotime cat. #S0027, China). According to the recommendations of the manufacturer, the standard curve was established and the concentration was detected by an enzyme reader (TECAN cat. #30086376, Switzerland). Finally, the ATP concentration was normalized by the BCA protein concentration method to eliminate the error caused by the difference of protein content. The quantification of the total ATP levels in HUVECs was conducted 48 h after transfection with siRNAs. All experiments were repeated three times independently.
For zebrafish, embryos and larvae were analyzed with a Nikon SMZ 18 fluorescence microscope and subsequently photographed with digital cameras. A subset of images was adjusted for level, brightness, contrast, hue, and saturation with Adobe Photoshop 7.0 software (Adobe, San Jose, California) to optimally visualize the expression patterns. Quantitative image analyses was processed using image-based morphometric analysis (NIS-Elements D4.6, Japan) and Fiji Image J (NIH, Bethesda, MD, United States). Ten animals for each treatment were quantified, and the total signal per animal was averaged.
For HUVECs, all the experiment images were taken with an Olympus IX73 fluorescence microscope. Quantitative image analyses were processed using image Fiji Image J (NIH, Bethesda, MD, United States).
All data were presented as mean ± SEM. Statistical analysis and graphical representation of the data were performed using GraphPad Prism 8.3 (GraphPad Software, San Diego, CA). Statistical significance was performed using Student's t-test or ANOVA as appropriate. Statistical significance is indicated by an asterisk where P < 0.05; two asterisks, where P < 0.01; three asterisks, where P < 0.001; and four asterisks, where P < 0.0001.
Zebrafish (Danio rerio) is used extensively in angiogenesis studies because it undergoes rapid growth. The development of the vasculature in zebrafish can be divided into five major stages (50, 51). Compared with humans, the fars2 gene was highly homologous, and the sequence similarity of Fars2 protein in zebrafish reached 71.39%. To explore its role in angiogenesis, we investigated the expression of fars2 during the embryonic development of zebrafish. The qRT-PCR analyses of total embryos revealed that fars2 transcription increased between 6 and 24 h post-fertilization (hpf) and then again between 72 and 96 hpf, which are the critical stages of vascular formation in zebrafish (Figure 1A). At 20 hpf, primary sprouts start to emerge bilaterally from the dorsal aorta at each vertical myoseptal boundary and then elongate dorsally, ramify, and interconnect along the dorsolateral roof of the neural tube to form paired dorsal longitudinal anastomotic vessels. The primary sprouts grow in a saltatory pattern, with numerous filopodia actively extending and retracting in all directions around the stretchy vessels (50, 52). The 3–6 days post-fertilization stage is the key period for the establishment of the systemic circulation in zebrafish embryos (50).
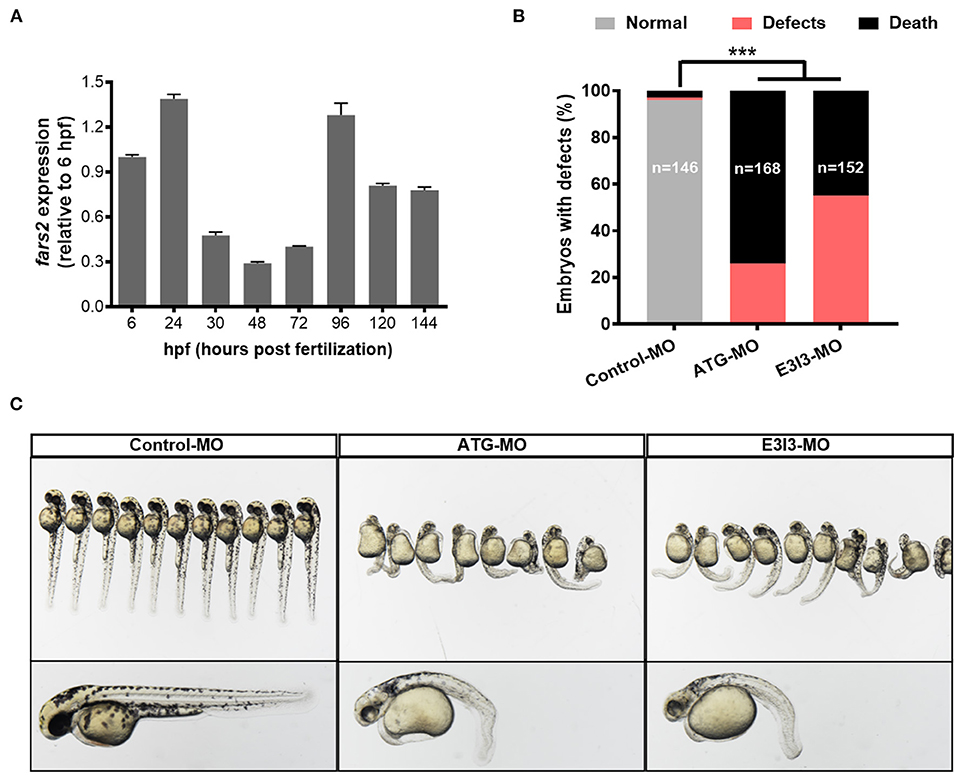
Figure 1. The expression of fars2 is essential in the early stage of zebrafish embryo development. (A) The expression patterns of fars2 during zebrafish embryonic development. The qRT-PCR analyses were performed at eight embryo development stages (6, 24, 30, 48, 72, 96, 120, and 144 hpf). (B) The percentages of embryos with developmental defects in zebrafish injected with a non-specific control or fars2-specific morpholinos (MOs). (C) Representative images of zebrafish at 50 hpf, following an injection with a non-specific control or fars2-specific MOs. ***P < 0.001.
To investigate the role of fars2 in zebrafish embryo development further, two specific MOs (ATG-MO and E3I3-MO) were designed to reduce its expression in vivo (Supplementary Figure 1A). Quantitative analyses performed after injecting one-cell fertilized ova with a non-specific control MO or the fars2-specific MOs confirmed the successful knock-down of fars2 by the latter (Supplementary Figures 1B,C). Approximately 26.2% of fars2 ATG morphants and 55.3% of E3I3 morphants presented an enlarged yolk sac, with the embryos displaying delayed growth and curved trunks (Figures 1B,C). The remaining embryos injected with the fars2 MOs all died (Figure 1B).
Overall, these findings demonstrate that fars2 is expressed at high levels during the critical period of angiogenesis in zebrafish and that the loss of fars2 impairs embryonic development.
To examine its role in zebrafish developmental angiogenesis, fars2 was knocked down in Tg (fli1:EGFP) y1 transgenic zebrafish, which display a steady expression of EGFP within vascular ECs, allowing easy visualization of the vascular structures (41). The labeled ISVs and DLAVs showed regular development in the embryos injected with the control MO. By contrast, embryos injected with fars2-specific MOs displayed lower numbers of ISVs and ectopic sprouts (Figure 2A). The PAVs, the precursors to the lymphatic system, formed normally in control embryos, whereas fars2 morphants displayed deficient PAV formation (Figure 2A). In addition, the number of complete ISVs (Figure 2B) and the mean length of ISVs (Figure 2C) were significantly lower in the fars2 morphants than in the controls.
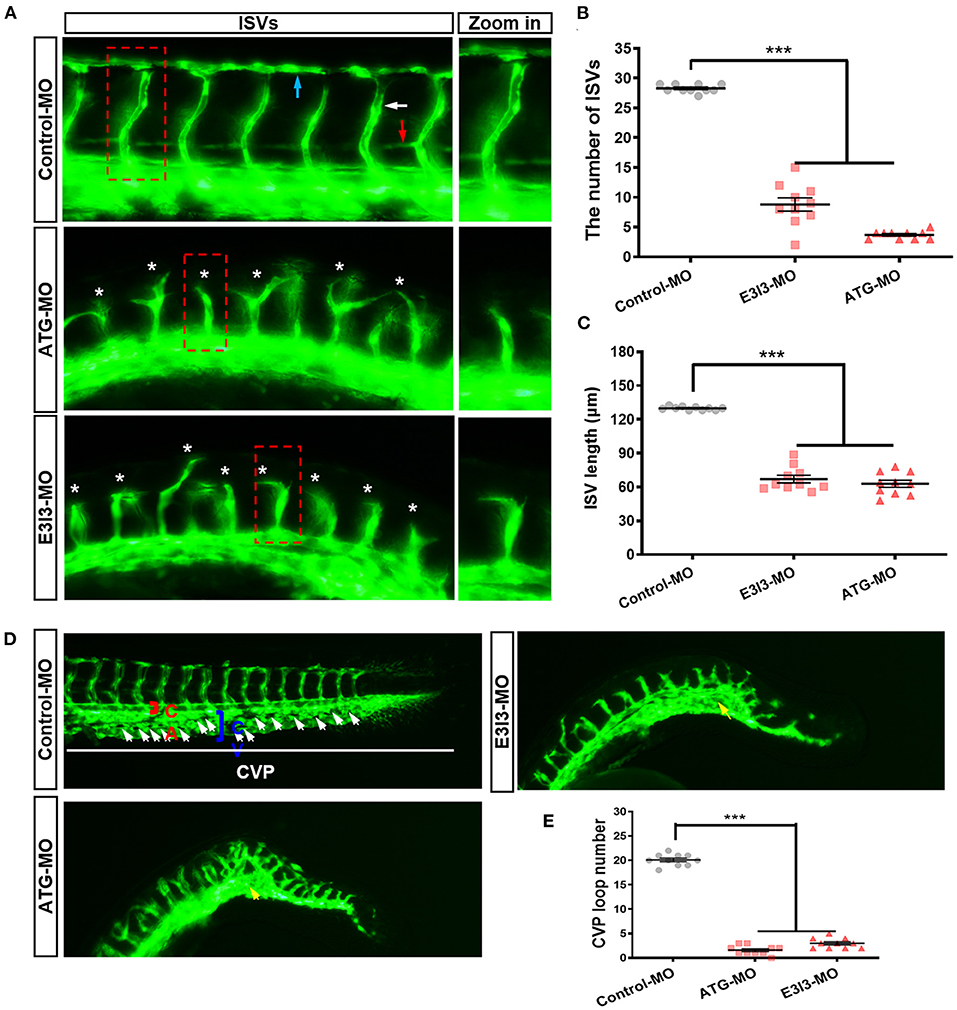
Figure 2. The morpholino-mediated knock-down of fars2 delays vascular formation in zebrafish. (A) Representative images of the trunk regions of Tg (fli1: EGFP) y1 embryos taken at 50 hpf. The intersegmental vessel (white arrow), dorsal longitudinal anastomotic vessel (blue arrow), parachordal vessel (red arrow), and ectopic sprouts (asterisks) are indicated. (B,C) The number of complete intersegmental vessels (ISVs) and the mean lengths of the ISVs in control and fars2 morphants. The horizontal bars show the mean ± SEM (n = 10 per group). ***P < 0.001 via ANOVA. (D) Representative images of the caudal artery, caudal vein, and caudal vein plexus (CVP; arrows) in Tg (fli1: EGFP) y1 embryos taken at 50 hpf. In the control embryo, the CVP formed a typical honeycomb structure in the tail (white arrows). The knock-down of fars2 resulted in specific defects in CVP formation (yellow arrows). (E) Quantification of the loop number at the CVP. The horizontal bars show the mean ± SEM (n = 10 per group). ***P < 0.001 via ANOVA.
During zebrafish angiogenesis, new vessels that arise from axial veins and dorsal aortas form a primitive circulatory loop (53, 54). At 26–32 hpf, the posterior axial vein stretches ventrally and ultimately forms a “honeycomb-like” network named the CVP at 38 hpf. The shape of the CVP is produced by dorsal veins, ventral veins, and interlacing vessels (55, 56). In embryos injected with the control MO, the CVP formed canonical honeycomb-like structures at the tail at around 50 hpf. By contrast, fars2 knock-down caused specific defects in CVP formation (Figure 2D). Furthermore, the number of loops at the CVP was lower in the fars2 knock-down embryos than in the control embryos (Figure 2E). Overall, these findings demonstrate that MO-mediated knock-down of fars2 disrupted the formation of ISVs, DLAVs, and the CVP during embryonic development in zebrafish.
To gain further insight into the function of FARS2 in angiogenesis, we established an in vitro FARS2 knock-down model using HUVECs and siRNAs. Western blot and qRT-PCR analyses confirmed the efficient knock-down of FARS2 by three different siRNAs (si-FARS2). Compared with those in cells transfected with a control siRNA (siCtrl), the expression levels of the FARS2 gene and protein were reduced by at least 30% following transfection with si-FARS2 (Supplementary Figure 2).
Scratch-wound assays, CCK8-based cell proliferation tests, and transwell-based migration assays revealed that the loss of FARS2 reduced the motility, proliferation, and migration capacity of HUVECs (Figures 3A–E). In addition, tubulogenesis was also reduced in cells transfected with si-FARS2 (Figure 3F). Compared with those in cells transfected with siCtrl, the number of intersections in one field (Figure 3G) and the total length of the tube structures (Figure 3H) were lower following FARS2 silencing. To our knowledge, this is the first report of an in vitro FARS2 knock-down cell model created using siRNAs. Our findings demonstrate that the loss of FARS2 in HUVECs impairs cell motility, proliferation, invasion, and tube formation.
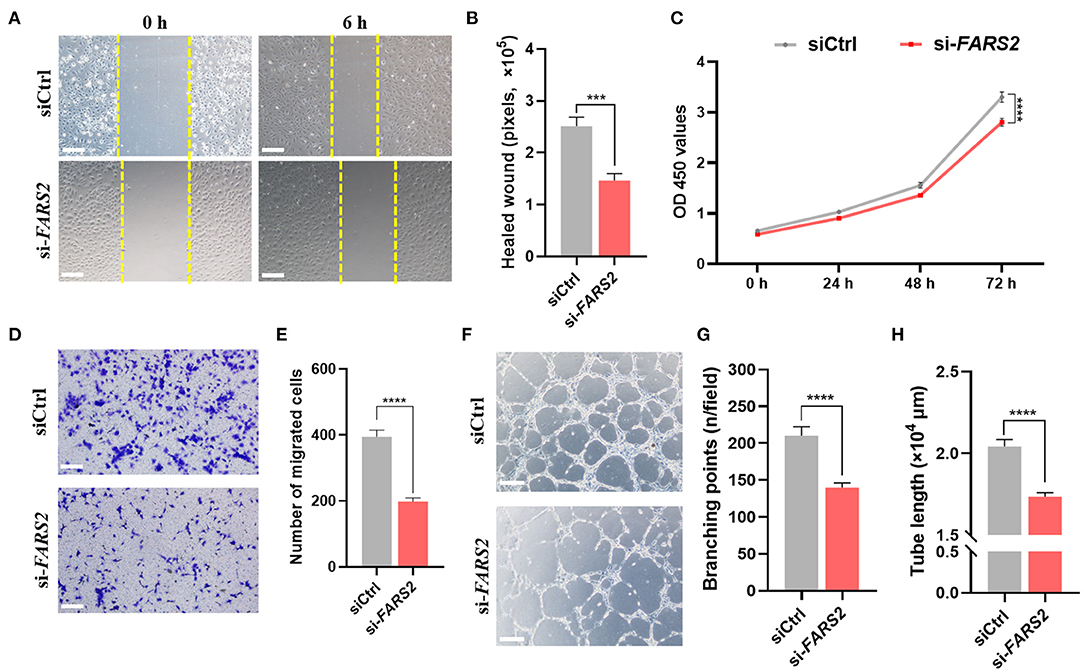
Figure 3. The deficiency of FARS2 impairs cell motility, proliferation, invasion, and tube formation in human umbilical vein endothelial cells (HUVECs). (A) Representative images of scratch-wound assays of HUVECs 0 and 6 h at 48 h after transfection with a control (siCtrl) or FARS2-specific (si-FARS2) siRNA. Scale bar = 200 μm. (B) Quantification of the healed wound area from (A). Data are prepresented as the mean and SEM (n = 10). ***P < 0.001 via ANOVA. (C) A CCK8-based cell proliferation assay of HUVECs at the indicated time-points after transfection with siCtrl or si-FARS2. The measurements were made in triplicate (mean and SEM), and the results are indicative of three independent experiments. ****P < 0.0001. (D) Representative images of transwell-based migration assays of HUVECs 48 h after transfection with siCtrl or si-FARS2. Scale bar = 200 μm. (E) Quantification of the number of migrated cells from (D). ***P < 0.001. (F) Representative images of tube network assays of HUVECs 48 h after transfection with siCtrl or si-FARS2. Scale bar = 200 μm. (G,H) Quantification of the branching points (G) and tube lengths (H) from (F). The measurements were made in triplicate (mean and SEM), and the results are indicative of three independent experiments. ****P < 0.0001.
As the FARS2 gene encodes the mitochondrial phenylalanyl-tRNA synthetase, which is involved in the synthesis of mtDNA-coded OXPHOs subunits, we investigated mitochondrial respiration in HUVECs after FARS2 silencing. To this end, a Seahorse Bioscience XF24 analyzer was used to measure the rates of non-mitochondrial respiration, basal respiration, maximal respiration, proton leak, ATP production, and spare respiratory capacity (57) in HUVECs transfected with siCtrl or si-FARS2 for 48 h. Basal mitochondrial respiration, represented by the oxygen consumption rate (OCR), was lower in HUVECs transfected with si-FARS2 than in non-transfected HUVECs or those transfected with siCtrl (Figures 4A,B). Following the addition of oligomycin, an inhibitor of ATP synthase, ATP production and proton leak were lower in si-FARS2-treated cells than in siCtrl-treated cells (Figure 4B). FARS2 silencing also attenuated the OCR after the cells were treated with FCCP to maximize mitochondrial respiration (Figure 4B). In addition, after treatment with rotenone to uncouple the oxidation respiratory chain, the loss of FARS2 attenuated the OCR. Finally, the spare respiratory capacity, which was calculated based on the basal and maximal respiration values, was also lower in si-FARS2-treated cells than in siCtrl-treated cells (Figure 4B). The mitochondria produce ATP and are a main source of ROS. Reduced ATP production and increased levels of ROS are thought to occur as a result of mitochondrial dysfunction. Compared with the control cells, the FARS2-deficient HUVECs showed lower levels of sector ATP and increased levels of ROS (Figures 4C,D). Overall, these results suggest that silencing of the FARS2 gene impairs mitochondria- and non-mitochondria-related respiration, leading to mitochondrial dysfunction in HUVECs.
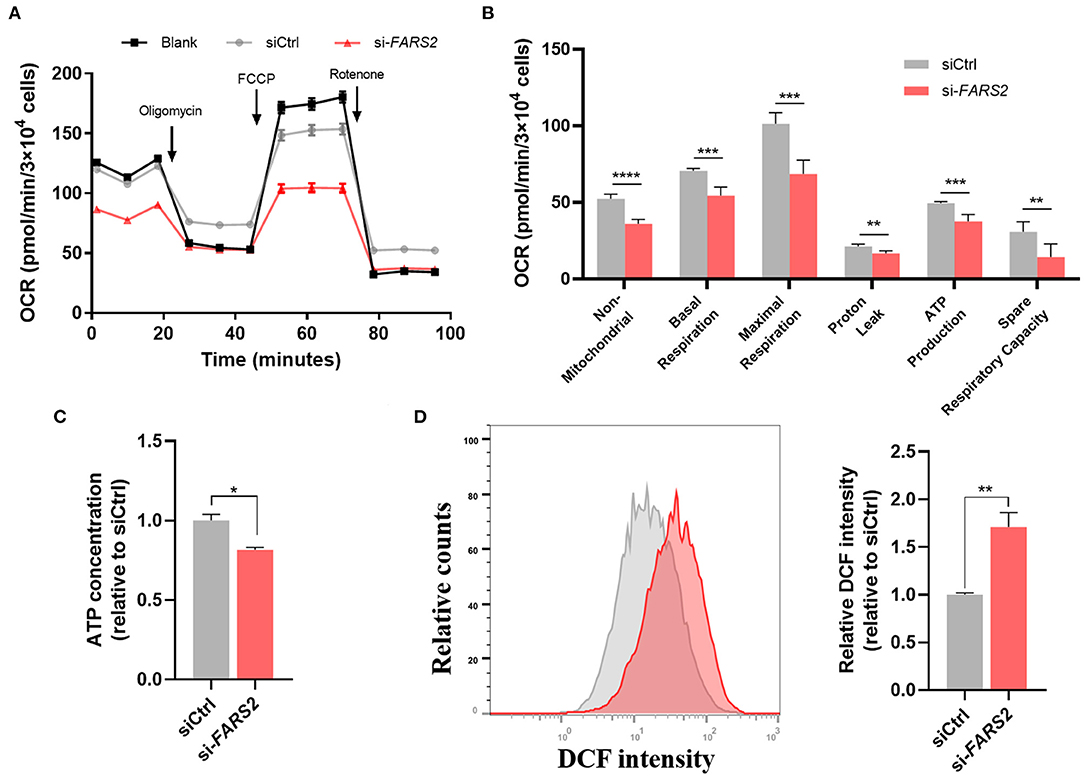
Figure 4. FARS2 silencing causes mitochondrial dysfunction in human umbilical vein endothelial cells (HUVECs). (A) The oxygen consumption rate (OCR) in HUVECs transfected with siCtrl or si-FARS2. The HUVECs were seeded 48 h after transfection with siRNAs and 12 h before analysis using a Seahorse XF24 analyzer. The OCR was measured continuously throughout the experimental period, both at baseline and in the presence of the indicated drugs. (B) Non-mitochondrial respiration, basal respiration, maximal respiration, proton leak, ATP production, and spare respiratory capacity in control and FARS2-deficient HUVECs. The measurements were made in triplicate (mean and SEM). **P < 0.01, ***P < 0.001, ****P < 0.0001. (C) The effects of FARS2 knock-down on intracellular reactive oxygen species production by HUVECs. The measurements were made in triplicate (mean and SEM). *P < 0.05, (D) Quantification of total ATP levels in HUVECs 48 h after transfection with the indicated siRNAs. The measurements were made in triplicate (mean and SEM). **P < 0.01.
To explore the potential molecular mechanisms underlying the suppression of angiogenesis following MO-mediated knock-down of fars2 in zebrafish, the expression levels of key genes in the Notch and Wnt pathways were examined using qRT-PCR. In zebrafish, fars2 deficiency upregulated the notch1b (a Notch receptor) and hey2 (a downstream gene in the Notch pathway) expression levels, indicating the activation of the Notch pathway (Figure 5A). In addition, fars2 deficiency increased the expression level of dkk1b and decreased those of other downstream genes in the Wnt pathway, indicating an inhibition of Wnt signaling (Figure 5A).
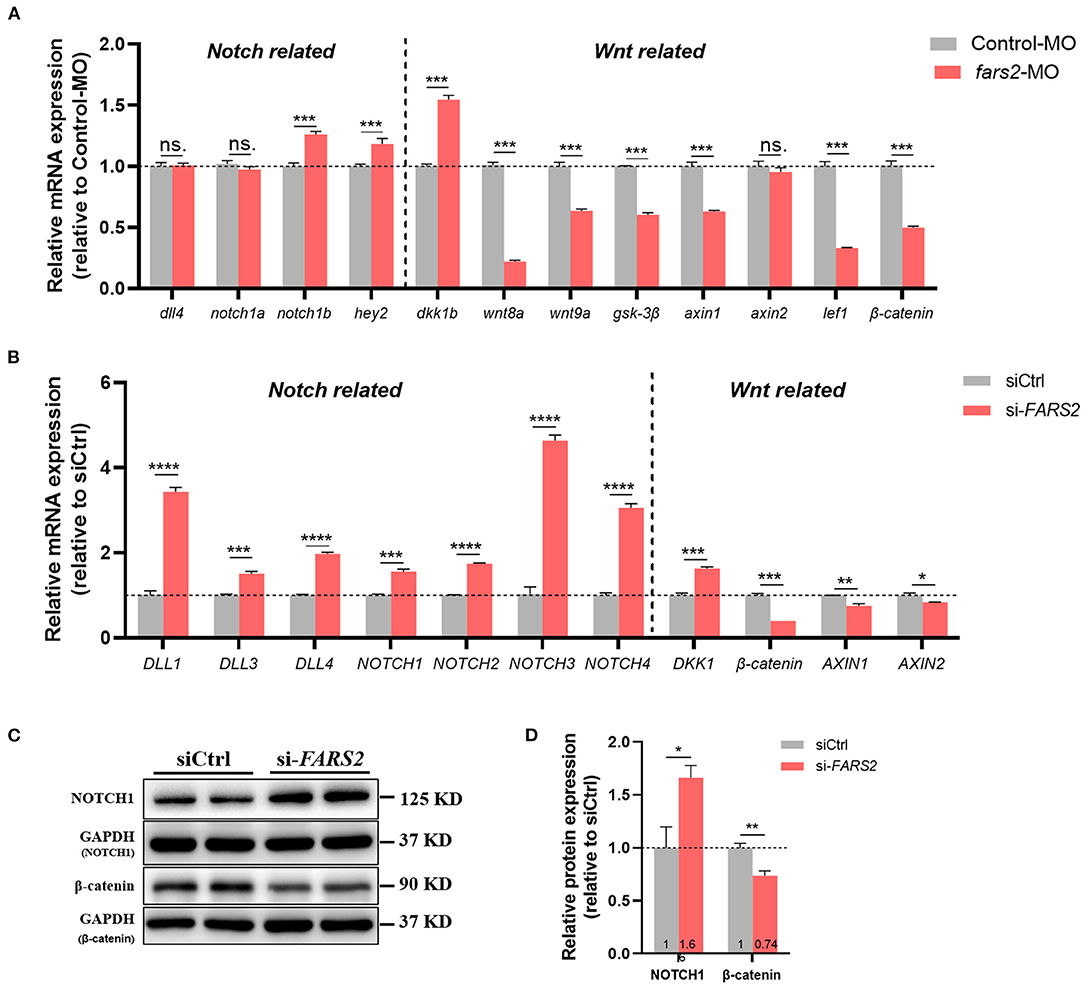
Figure 5. The deficiency of FARS2 impairs angiogenesis by disrupting the Notch and Wnt signaling pathways. (A) The expression levels of genes involved in the Notch/Wnt pathways in control and fars2 zebrafish morphants, as determined by qRT-PCR analyses (n = 6–10 individual embryos). ***P < 0.001, **P < 0.01, *P < 0.05; ns, not significant. (B) The relative mRNA expression levels of Notch/Wnt pathway-related genes. The human umbilical vein endothelial cells (HUVECs) were transfected with the indicated siRNAs for 48 h and then harvested for qRT-PCR analysis. The measurements were made in triplicate (mean and SEM), and the results are indicative of three independent experiments. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. (C) Western blot analyses of NOTCH1 and β-catenin protein levels. The HUVECs were transfected with the indicated siRNAs for 48 h prior to analysis. (D) Quantification of the western blotting data described in (C). The measurements were made in triplicate (mean and SEM). *P < 0.05, **P < 0.01.
As seen in zebrafish, siRNA-mediated knock-down of FARS2 in HUVECs also activated the Notch signaling pathway by upregulating all four mammalian Notch receptors (NOTCH1–4) and three ligands (DLL1, 3, and 4) to varying degrees (Figure 5B). In addition, the Wnt signaling pathway was inhibited after FARS2 silencing, as indicated by the downregulation of Wnt downstream genes (β-catenin, AXIN1, and AXIN2) and upregulation of the Wnt signaling inhibitor gene DKK1 (Figure 5B). Western blot analyses confirmed that the NOTCH1 and β-catenin protein levels were increased and decreased, respectively, following the siRNA-mediated knock-down of FARS2 (Figures 5C,D). Overall, these findings demonstrate that the loss of FARS2 affects angiogenesis by disrupting the Notch and Wnt signaling pathways.
The results presented here show that mitochondrial phenylalanyl-tRNA synthetase plays an essential role in angiogenesis both in vivo and in vitro. Our initial analysis of the expression pattern of fars2 during zebrafish embryonic development suggested that it plays a role in developmental angiogenesis. Subsequently, using MOs, we found that fars2 deficiency caused the delayed development of zebrafish embryos and impaired vascular formation, including those of ISVs, DLAVs, PAVs, and the CVP. Similarly, we found that siRNA-mediated knock-down of FARS2 in HUVECs impaired cell motility, proliferation, migration, and tube formation, confirming the role of FARS2 in angiogenesis. We also found that the loss of FARS2 led to mitochondrial dysfunction in HUVECs. Finally, we explored the possible mechanisms underlying the disruption of angiogenesis and found that FARS2 deficiency may disrupt the Notch and Wnt signaling pathways, both of which are involved in angiogenesis (Figure 6).
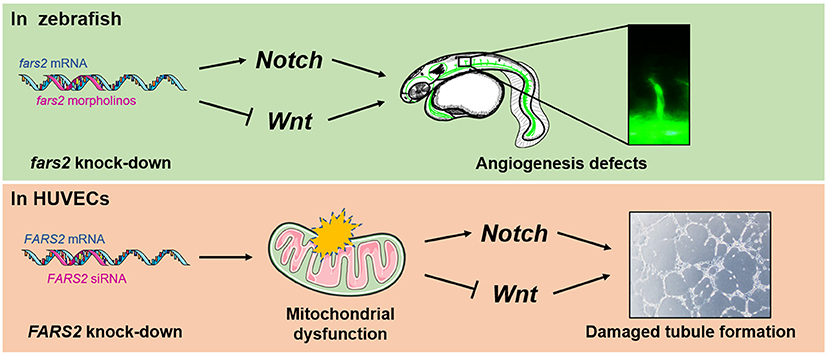
Figure 6. Developmental angiogenesis requires the mitochondrial phenylalanyl-tRNA synthetase. An overview of the mechanisms by which the deficiency of mitochondrial phenylalanyl-tRNA synthetase impairs angiogenesis by disrupting the Notch/Wnt pathways in zebrafish and human umbilical vein endothelial cells.
The lethality of defects in ECs to mammalian embryos confirms the pivotal function of the vasculature in development. During embryonic development, two essential processes, vasculogenesis and angiogenesis, form the vasculature consisting of arterial, venous, and lymphatic vessels. Vasculogenesis is defined as the de novo emergence of vessels through the differentiation of angioblasts. Angiogenesis describes new vascular formation after the proliferation of ECs from pre-existing vessels (58–60). Much effort has been focused on investigating the key stages of vasculature development in mammalian embryos. The first sign of vascular formation occurs in the extraembryonic yolk sac blood island at the gastrulation stage as early as embryonic day 7.5. Subsequently, the blood island fuses to constitute the primary plexus, which leads to the establishment of the complex yolk sac vasculature (61, 62). Next, under the influence of complex transcriptional regulation and critical signaling components of angiogenesis, the newborn vessels of the developing embryo specialize further and differentiate into arteries, veins, and capillaries. Our results presented here not only identify the phenotype of delayed embryonic development in zebrafish caused by fars2 deficiency but also preliminarily suggest that this phenotype may be caused by impaired angiogenesis.
Although angiogenesis is an energy-intensive process, glycolysis is the primary energy-producing mechanism in vascular ECs, a feature that is attributable to their special physiological distribution and high levels of exposure to oxygen (27, 28, 63). Historically, the role of mitochondrial respiration in angiogenesis has been overlooked, that is, until the discovery of the essential role of mitochondrial fatty acid and amino acid oxidation pathways in angiogenesis (34). A growing body of evidence suggests that, by acting as important organelles that sense ambient oxygen concentrations and generate energy, the mitochondria play an integral role in controlling metabolism and in regulating the proliferation and survival of ECs during angiogenesis. The mutation of mitochondrial tRNA and aberrant tRNA metabolism induce mitochondrial dysfunction, leading to apoptosis and impaired angiogenesis in HUVECs (64). The mitochondrial permeability transition pore also plays a role in regulating mitochondrial metabolism in ECs and in the maintenance of vascular integrity (65). In addition, mitochondrial dynamics (44) and mitochondria–endoplasmic reticulum contacts (66, 67) are critical for the regulation of angiogenesis and vascular remodeling. In our current study, we found that the impairment of HUVEC proliferation, migration, and tube formation by FARS2 deficiency was caused by abnormal mitochondrial respiratory function.
The hierarchical organization of ECs into tip cells (leading role) and stalk cells (trailing role) is required by angiogenesis. Tip cells lead the sprouts toward the signaling sources of angiogenesis in tissues, and the tip cells are followed by stalk cells, which elongate the sprout (24, 68). These processes are orchestrated by a complex molecular network, like Notch, Wnt, and VEGF/VEGFR. In tip cells, the activation of VEGFR2 induced the expression of DLL4 in response to VEGF from the signaling source (69). Then, DLL4 activates Notch in stalk cells to restrict branching. Studies in zebrafish and mice reveal that Notch is essential for restricting EC behavior to tip cells, reflected in the excessive sprouting of arteries in the absence of the Notch and the damage of angiogenesis in the activation of the Notch (70, 71). In ECs, Wnt signals could induce a Notch-like phenotype in a reciprocal feedback role, characterized by vascular remodeling and branching defects (39). Studies in mice reveal that Wnt is also required for angiogenesis, reflected in vascular defects after gene-inactivation of the Wnt genes (72). In our study, the activation of Notch and the inhibition of Wnt caused by FARS2 deficiency might damage angiogenesis by breaking the determination of EC fate and disrupting the signaling system in ECs. In addition, we detected that the transcript of dll4 and notch1a had no significant changes in zebrafish, which was inconsistent with the results of HUVECs. However, the regulation of angiogenesis in vivo is an extremely complex process involving various network pathways. In fars2 deficiency in zebrafish, the upregulation of hey2 and notch1b could partially indicate the activation of Notch signaling pathway (73, 74), but no changes in dll4 and notch1a were potentially due to the crosstalk with other signaling pathways, like VEGF/VEGFR (75). Moreover, we are eager to explore the specific molecular mechanisms involved in these processes during future research.
Expanding research into brain science has produced a large amount of evidence showing that angiogenesis plays a neurotrophic role in neurodegenerative disorders such as Alzheimer's disease. The relationship between cerebrovascular abnormalities and cognitive decline is supported by the fact that Alzheimer's disease brains display vascular pathology, with microvasculature changes occurring before cognitive decline and preceding neurodegenerative changes (76–78). In addition, there is sufficient evidence to suggest that vascular endothelial growth factor-based gene or protein therapies could be used to treat amyotrophic lateral sclerosis patients (79). Although mutations in the FARS2 gene have a strong association with neurological diseases, the relationship between neural microvascular networks and disease phenotypes in patients with these mutations has not been characterized. Our study may provide new insights into the progression of neurovascular diseases and the diagnosis and treatment of FARS2 mutation-related genetic diseases.
In summary, using in vivo and in vitro knock-down models, we report that FARS2 is essential for angiogenesis. In this study, we focused on elucidating the phenotypes associated with angiogenic defects caused by FARS2 deficiency. However, the specific molecular mechanisms linking cardiovascular system defects to the impairment of mitochondrial respiratory function due to FARS2 deficiency have not been investigated thoroughly. In addition, the interaction between the pathogenesis of neurodegenerative diseases and impairment of angiogenesis caused by FARS2 defects requires further exploration.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
The animal study was reviewed and approved by the Fourth Military Medical University.
BL contributed to the conceptualization, data curation, investigation, statistical analysis, visualization, and writing of the original draft. KC and FL contributed to the conceptualization, project administration, methodology, software, editing, and writing of the original draft. JZ contributed to the conceptualization, data collection, and writing of the original draft. XC contributed to the conceptualization, methodology, and writing of the original draft. TC, QC, YY, and WH contributed to the methodology, data collection, data validation, formal analysis, and resources. YW and LW contributed to the conceptualization, project administration, and writing (editing). All authors contributed to the article and approved the submitted version.
This study was supported by the key research and development plan in Shaanxi, Grant/Award Number: 2019SF-059 and 2020SF-204; the Key Innovative Project in Shaanxi, Grant/Award Number: 2021ZDLSF02-02; National Natural Science Foundation of China, Grant/Award Number: 81671476 and 31570906.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2021.724846/full#supplementary-material
mtARSs, mitochondrial aminoacyl-tRNA synthetases; FARS2, mitochondrial phenylalanyl-tRNA synthetase; siRNAs, small interference RNAs; HUVECs, human umbilical vein endothelial cells; ARSs, aminoacyl-tRNA synthetases; mtDNA, mitochondrial DNA; OXPHOs, oxidative phosphorylation system; Phe, Phenylalanine; mt-tRNAPhe, mitochondrial phenylalanyl-tRNA; CNS, central nervous system; CVDs, cardiovascular diseases; ECs, endothelial cells; MOs, Morpholinos; ISVs, intersegmental vessels; DLAVs, dorsal longitudinal anastomotic vessels; PAVs, parachordal vessels; CVP, caudal vein plexus; hpf, hours post-fertilization; OCR, oxygen consumption rate; ROS, reactive oxygen species.
1. Ibba M, Soll D. Aminoacyl-tRNA synthesis. Annu Rev Biochem. (2000) 69:617–50. doi: 10.1146/annurev.biochem.69.1.617
2. Andrews RM, Kubacka I, Chinnery PF, Lightowlers RN, Turnbull DM, Howell N. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA. Nat Genet. (1999) 23:147. doi: 10.1038/13779
3. Yao P, Fox PL. Aminoacyl-tRNA synthetases in medicine and disease. EMBO Mol Med. (2013) 5:332–43. doi: 10.1002/emmm.201100626
4. Moulinier L, Ripp R, Castillo G, Poch O, Sissler M. MiSynPat: an integrated knowledge base linking clinical, genetic, and structural data for disease-causing mutations in human mitochondrial aminoacyl-tRNA synthetases. Hum Mutat. (2017) 38:1316–24. doi: 10.1002/humu.23277
5. Sissler M, González-Serrano LE, Westhof E. Recent advances in mitochondrial aminoacyl-tRNA synthetases and disease. Trends Mol Med. (2017) 23:693–708. doi: 10.1016/j.molmed.2017.06.002
6. Ardissone A, Piscosquito G, Legati A, Langella T, Lamantea E, Garavaglia B, et al. A slowly progressive mitochondrial encephalomyopathy widens the spectrum of AIFM1 disorders. Neurology. (2015) 84:2193–5. doi: 10.1212/wnl.0000000000001613
7. Yang Y, Liu W, Fang Z, Shi J, Che F, He C, et al. A newly identified missense mutation in FARS2 causes autosomal-recessive spastic paraplegia. Hum Mutat. (2016) 37:165–9. doi: 10.1002/humu.22930
8. Cho JS, Kim SH, Kim HY, Chung T, Kim D, Jang S, et al. FARS2 mutation and epilepsy: possible link with early-onset epileptic encephalopathy. Epilepsy Res. (2017) 129:118–24. doi: 10.1016/j.eplepsyres.2016.11.022
9. Vantroys E, Larson A, Friederich M, Knight K, Swanson MA, Powell CA, et al. New insights into the phenotype of FARS2 deficiency. Mol Genet Metab. (2017) 122:172–81. doi: 10.1016/j.ymgme.2017.10.004
10. Almannai M, Wang J, Dai H, El-Hattab AW, Faqeih EA, Saleh MA, et al. FARS2 deficiency; new cases, review of clinical, biochemical, and molecular spectra, and variants interpretation based on structural, functional, and evolutionary significance. Mol Genet Metab. (2018) 125:281–91. doi: 10.1016/j.ymgme.2018.07.014
11. Elo JM, Yadavalli SS, Euro L, Isohanni P, Götz A, Carroll CJ, et al. Mitochondrial phenylalanyl-tRNA synthetase mutations underlie fatal infantile Alpers encephalopathy. Hum Mol Genet. (2012) 21:4521–9. doi: 10.1093/hmg/dds294
12. Kartvelishvili E, Tworowski D, Vernon H, Moor N, Wang J, Wong LJ, et al. Kinetic and structural changes in HsmtPheRS, induced by pathogenic mutations in human FARS2. Protein Sci. (2017) 26:1505–16. doi: 10.1002/pro.3176
13. Chakraborty S, Ibba M, Banerjee R. Biophysical characterization Of Alpers encephalopathy associated mutants of human mitochondrial phenylalanyl-tRNA synthetase. IUBMB Life. (2019) 71:1141–9. doi: 10.1002/iub.2114
14. Vernon HJ, McClellan R, Batista DA, Naidu S. Mutations in FARS2 and non-fatal mitochondrial dysfunction in two siblings. Am J Med Genet A. (2015) 167:1147–51. doi: 10.1002/ajmg.a.36993
15. Chen Z, Zhang Y. A patient with juvenile-onset refractory status epilepticus caused by two novel compound heterozygous mutations in FARS2 gene. Int J Neurosci. (2019) 129:1094–7. doi: 10.1080/00207454.2019.1634071
16. Hotait M, Nasreddine W, El-Khoury R, Dirani M, Nawfal O, Beydoun A. FARS2 mutations: more than two phenotypes? A case report. Front Genet. (2020) 11:787. doi: 10.3389/fgene.2020.00787
17. Lopez AD, Mathers CD, Ezzati M, Jamison DT, Murray CJ. Global and regional burden of disease and risk factors, 2001: systematic analysis of population health data. Lancet. (2006) 367:1747–57. doi: 10.1016/s0140-6736(06)68770-9
18. Mathers CD, Loncar D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. (2006) 3:e442. doi: 10.1371/journal.pmed.0030442
19. North BJ, Sinclair DA. The intersection between aging and cardiovascular disease. Circ Res. (2012) 110:1097–108. doi: 10.1161/circresaha.111.246876
20. Sing CF, Stengård JH, Kardia SL. Genes, environment, and cardiovascular disease. Arterioscler Thromb Vasc Biol. (2003) 23:1190–6. doi: 10.1161/01.atv.0000075081.51227.86
21. Lähteenvuo J, Rosenzweig A. Effects of aging on angiogenesis. Circ Res. (2012) 110:1252–64. doi: 10.1161/circresaha.111.246116
22. Donato AJ, Machin DR, Lesniewski LA. Mechanisms of dysfunction in the aging vasculature and role in age-related disease. Circ Res. (2018) 123:825–48. doi: 10.1161/circresaha.118.312563
23. Carmeliet P. Angiogenesis in life, disease and medicine. Nature. (2005) 438:932–6. doi: 10.1038/nature04478
24. Gerhardt H, Golding M, Fruttiger M, Ruhrberg C, Lundkvist A, Abramsson A, et al. VEGF guides angiogenic sprouting utilizing endothelial tip cell filopodia. J Cell Biol. (2003) 161:1163–77. doi: 10.1083/jcb.200302047
25. De Smet F, Segura I, De Bock K, Hohensinner PJ, Carmeliet P. Mechanisms of vessel branching: filopodia on endothelial tip cells lead the way. Arterioscler Thromb Vasc Biol. (2009) 29:639–49. doi: 10.1161/atvbaha.109.185165
26. Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell. (2011) 146:873–87. doi: 10.1016/j.cell.2011.08.039
27. Dobrina A, Rossi F. Metabolic properties of freshly isolated bovine endothelial cells. Biochim Biophys Acta. (1983) 762:295–301. doi: 10.1016/0167-4889(83)90084-8
28. Krützfeldt A, Spahr R, Mertens S, Siegmund B, Piper HM. Metabolism of exogenous substrates by coronary endothelial cells in culture. J Mol Cell Cardiol. (1990) 22:1393–404. doi: 10.1016/0022-2828(90)90984-a
29. Harjes U, Bensaad K, Harris AL. Endothelial cell metabolism and implications for cancer therapy. Br J Cancer. (2012) 107:1207–12. doi: 10.1038/bjc.2012.398
30. Harjes U, Verfaillie C, Carmeliet P. Endothelial barrier and metabolism: new kids on the block regulating bone marrow vascular niches. Dev Cell. (2016) 37:210–2. doi: 10.1016/j.devcel.2016.04.016
31. Zecchin A, Kalucka J, Dubois C, Carmeliet P. How endothelial cells adapt their metabolism to form vessels in tumors. Front Immunol. (2017) 8:1750. doi: 10.3389/fimmu.2017.01750
32. Kluge MA, Fetterman JL, Vita JA. Mitochondria and endothelial function. Circ Res. (2013) 112:1171–88. doi: 10.1161/circresaha.111.300233
33. Pernas L, Scorrano L. Mito-morphosis: mitochondrial fusion, fission, and cristae remodeling as key mediators of cellular function. Annu Rev Physiol. (2016) 78:505–31. doi: 10.1146/annurev-physiol-021115-105011
34. Potente M, Carmeliet P. The link between angiogenesis and endothelial metabolism. Annu Rev Physiol. (2017) 79:43–66. doi: 10.1146/annurev-physiol-021115-105134
35. Reichard A, Asosingh K. The role of mitochondria in angiogenesis. Mol Biol Rep. (2019) 46:1393–400. doi: 10.1007/s11033-018-4488-x
36. van de Schans VA, Smits JF, Blankesteijn WM. The Wnt/frizzled pathway in cardiovascular development and disease: friend or foe? Eur J Pharmacol. (2008) 585:338–45. doi: 10.1016/j.ejphar.2008.02.093
37. Franco CA, Liebner S, Gerhardt H. Vascular morphogenesis: a Wnt for every vessel? Curr Opin Genet Dev. (2009) 19:476–83. doi: 10.1016/j.gde.2009.09.004
38. Goodwin AM, Sullivan KM, D'Amore PA. Cultured endothelial cells display endogenous activation of the canonical Wnt signaling pathway and express multiple ligands, receptors, and secreted modulators of Wnt signaling. Dev Dyn. (2006) 235:3110–20. doi: 10.1002/dvdy.20939
39. Corada M, Nyqvist D, Orsenigo F, Caprini A, Giampietro C, Taketo MM, et al. The Wnt/beta-catenin pathway modulates vascular remodeling and specification by upregulating Dll4/Notch signaling. Dev Cell. (2010) 18:938–49. doi: 10.1016/j.devcel.2010.05.006
40. Artavanis-Tsakonas S, Rand MD, Lake RJ. Notch signaling: cell fate control and signal integration in development. Science. (1999) 284:770–6. doi: 10.1126/science.284.5415.770
41. Lawson ND, Weinstein BM. In vivo imaging of embryonic vascular development using transgenic zebrafish. Dev Biol. (2002) 248:307–18. doi: 10.1006/dbio.2002.0711
42. Kimmel CB, Ballard WW, Kimmel SR, Ullmann B, Schilling TF. Stages of embryonic development of the zebrafish. Dev Dyn. (1995) 203:253–310. doi: 10.1002/aja.1002030302
43. Nasevicius A, Ekker SC. Effective targeted gene “knockdown” in zebrafish. Nat Genet. (2000) 26:216–20. doi: 10.1038/79951
44. Herkenne S, Ek O, Zamberlan M, Pellattiero A, Chergova M, Chivite I, et al. Developmental and tumor angiogenesis requires the mitochondria-shaping protein opa1. Cell Metab. (2020) 31:987–1003.e1008. doi: 10.1016/j.cmet.2020.04.007
45. Tian X, Liu Y, Wang Z, Wu S. miR-144 delivered by nasopharyngeal carcinoma-derived EVs stimulates angiogenesis through the FBXW7/HIF-1α/VEGF-A axis. Mol Ther Nucleic Acids. (2021) 24:1000–11. doi: 10.1016/j.omtn.2021.03.016
46. Rodriguez LG, Wu X, Guan JL. Wound-healing assay. Methods Mol Biol. (2005) 294:23–9. doi: 10.1385/1-59259-860-9:023
47. DeCicco-Skinner KL, Henry GH, Cataisson C, Tabib T, Gwilliam JC, Watson NJ, et al. Endothelial cell tube formation assay for the in vitro study of angiogenesis. J Vis Exp. (2014) (91):e51312. doi: 10.3791/51312
48. Galeano-Otero I, Del Toro R, Khatib AM, Rosado JA, Ordóñez-Fernández A, Smani T. SARAF and orai1 contribute to endothelial cell activation and angiogenesis. Front Cell Dev Biol. (2021) 9:639952. doi: 10.3389/fcell.2021.639952
49. Balogh E, Veale DJ, McGarry T, Orr C, Szekanecz Z, Ng CT, et al. Oxidative stress impairs energy metabolism in primary cells and synovial tissue of patients with rheumatoid arthritis. Arthritis Res Ther. (2018) 20:95. doi: 10.1186/s13075-018-1592-1
50. Isogai S, Lawson ND, Torrealday S, Horiguchi M, Weinstein BM. Angiogenic network formation in the developing vertebrate trunk. Development. (2003) 130:5281–90. doi: 10.1242/dev.00733
51. Brown DR, Samsa LA, Qian L, Liu J. Advances in the study of heart development and disease using zebrafish. J Cardiovasc Dev Dis. (2016) 3:13. doi: 10.3390/jcdd3020013
52. Chico TJ, Ingham PW, Crossman DC. Modeling cardiovascular disease in the zebrafish. Trends Cardiovasc Med. (2008) 18:150–5. doi: 10.1016/j.tcm.2008.04.002
53. Betz C, Lenard A, Belting HG, Affolter M. Cell behaviors and dynamics during angiogenesis. Development. (2016) 143:2249–60. doi: 10.1242/dev.135616
54. Xia Z, Bi X, Lian J, Dai W, He X, Zhao L, et al. Slc39a5-mediated zinc homeostasis plays an essential role in venous angiogenesis in zebrafish. Open Biol. (2020) 10:200281. doi: 10.1098/rsob.200281
55. Wiley DM, Kim JD, Hao J, Hong CC, Bautch VL, Jin SW. Distinct signalling pathways regulate sprouting angiogenesis from the dorsal aorta and the axial vein. Nat Cell Biol. (2011) 13:686–92. doi: 10.1038/ncb2232
56. Schuermann A, Helker CS, Herzog W. Angiogenesis in zebrafish. Semin Cell Dev Biol. (2014) 31:106–14. doi: 10.1016/j.semcdb.2014.04.037
57. Brand MD, Nicholls DG. Assessing mitochondrial dysfunction in cells. Biochem J. (2011) 435:297–312. doi: 10.1042/bj20110162
58. Poole TJ, Coffin JD. Vasculogenesis and angiogenesis: two distinct morphogenetic mechanisms establish embryonic vascular pattern. J Exp Zool. (1989) 251:224–31. doi: 10.1002/jez.1402510210
59. Flamme I, Frölich T, Risau W. Molecular mechanisms of vasculogenesis and embryonic angiogenesis. J Cell Physiol. (1997) 173:206–10. doi: 10.1002/(sici)1097-4652(199711)173:2<206::aid-jcp22>3.0.co;2-c
61. Choi K. The hemangioblast: a common progenitor of hematopoietic and endothelial cells. J Hematother Stem Cell Res. (2002) 11:91–101. doi: 10.1089/152581602753448568
62. Park C, Kim TM, Malik AB. Transcriptional regulation of endothelial cell and vascular development. Circ Res. (2013) 112:1380–400. doi: 10.1161/circresaha.113.301078
63. Cruys B, Wong BW, Kuchnio A, Verdegem D, Cantelmo AR, Conradi LC, et al. Glycolytic regulation of cell rearrangement in angiogenesis. Nat Commun. (2016) 7:12240. doi: 10.1038/ncomms12240
64. Jia Z, Zhang Y, Li Q, Ye Z, Liu Y, Fu C, et al. A coronary artery disease-associated tRNAThr mutation altered mitochondrial function, apoptosis and angiogenesis. Nucleic Acids Res. (2019) 47:2056–74. doi: 10.1093/nar/gky1241
65. Marcu R, Kotha S, Zhi Z, Qin W, Neeley CK, Wang RK, et al. The mitochondrial permeability transition pore regulates endothelial bioenergetics and angiogenesis. Circ Res. (2015) 116:1336–45. doi: 10.1161/circresaha.116.304881
66. Göbel J, Engelhardt E, Pelzer P, Sakthivelu V, Jahn HM, Jevtic M, et al. Mitochondria-endoplasmic reticulum contacts in reactive astrocytes promote vascular remodeling. Cell Metab. (2020) 31:791–808.e798. doi: 10.1016/j.cmet.2020.03.005
67. Wang C, Dai X, Wu S, Xu W, Song P, Huang K. FUNDC1-dependent mitochondria-associated endoplasmic reticulum membranes are involved in angiogenesis and neoangiogenesis. Nat Commun. (2021) 12:2616. doi: 10.1038/s41467-021-22771-3
68. Adams RH, Eichmann A. Axon guidance molecules in vascular patterning. Cold Spring Harb Perspect Biol. (2010) 2:a001875. doi: 10.1101/cshperspect.a001875
69. Carmeliet P, Jain RK. Molecular mechanisms and clinical applications of angiogenesis. Nature. (2011) 473:298–307. doi: 10.1038/nature10144
70. Hellström M, Phng LK, Hofmann JJ, Wallgard E, Coultas L, Lindblom P, et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature. (2007) 445:776–80. doi: 10.1038/nature05571
71. Siekmann AF, Lawson ND. Notch signalling limits angiogenic cell behaviour in developing zebrafish arteries. Nature. (2007) 445:781–4. doi: 10.1038/nature05577
72. Dejana E. The role of wnt signaling in physiological and pathological angiogenesis. Circ Res. (2010) 107:943–52. doi: 10.1161/circresaha.110.223750
73. Bill BR, Balciunas D, McCarra JA, Young ED, Xiong T, Spahn AM, et al. Development and Notch signaling requirements of the zebrafish choroid plexus. PLoS ONE. (2008) 3:e3114. doi: 10.1371/journal.pone.0003114
74. Rochon ER, Wright DS, Schubert MM, Roman BL. Context-specific interactions between Notch and ALK1 cannot explain ALK1-associated arteriovenous malformations. Cardiovasc Res. (2015) 107:143–52. doi: 10.1093/cvr/cvv148
75. Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol. (2007) 8:464–78. doi: 10.1038/nrm2183
76. Farkas E, Luiten PG. Cerebral microvascular pathology in aging and Alzheimer's disease. Prog Neurobiol. (2001) 64:575–611. doi: 10.1016/s0301-0082(00)00068-x
77. Kalaria RN. Neuropathological diagnosis of vascular cognitive impairment and vascular dementia with implications for Alzheimer's disease. Acta Neuropathol. (2016) 131:659–85. doi: 10.1007/s00401-016-1571-z
78. Kalaria RN. The pathology and pathophysiology of vascular dementia. Neuropharmacology. (2018) 134:226–39. doi: 10.1016/j.neuropharm.2017.12.030
Keywords: mtARSs, FARS2, mitochondrial dysfunction, angiogenesis, zebrafish, HUVECs
Citation: Li B, Chen K, Liu F, Zhang J, Chen X, Chen T, Chen Q, Yao Y, Hu W, Wang L and Wu Y (2021) Developmental Angiogenesis Requires the Mitochondrial Phenylalanyl-tRNA Synthetase. Front. Cardiovasc. Med. 8:724846. doi: 10.3389/fcvm.2021.724846
Received: 14 June 2021; Accepted: 04 August 2021;
Published: 01 September 2021.
Edited by:
Gianfranco Pintus, University of Sharjah, United Arab EmiratesReviewed by:
Eric First, Louisiana State University Health Shreveport, United StatesCopyright © 2021 Li, Chen, Liu, Zhang, Chen, Chen, Chen, Yao, Hu, Wang and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yuanming Wu, d3V5bUBmbW11LmVkdS5jbg==; Li Wang, amNiZnpyQGZtbXUuZWR1LmNu
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.