 Ahmed Elsanhoury
Ahmed Elsanhoury Vivian Nelki3
Vivian Nelki3 Sophie Van Linthout
Sophie Van Linthout Carsten Tschöpe
Carsten Tschöpe
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 17 September 2021
Sec. Cardiovascular Metabolism
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.720690
Heart failure with preserved ejection fraction (HFpEF) is a heterogeneous syndrome with diverse etiologies and pathophysiological factors. Obesity and type 2 diabetes mellitus (T2DM), conditions that coexist frequently, induce a cluster of metabolic and non-metabolic signaling derangements which are in favor to induce inflammation, fibrosis, myocyte stiffness, all hallmarks of HFpEF. In contrast to other HFpEF risk factors, obesity and T2DM are often associated with the generation of enlarged epicardial adipose tissue (EAT). EAT acts as an endocrine tissue that may exacerbate myocardial inflammation and fibrosis via various paracrine and vasocrine signals. In addition, an abnormally large EAT poses mechanical stress on the heart via pericardial restrain. HFpEF patients with enlarged EAT may belong to a unique phenotype that can benefit from specific EAT-targeted interventions, including life-style modifications and pharmacologically via statins and fat modifying anti-diabetics drugs; like metformin, sodium-glucose cotransporter 2 inhibitors, or glucagon-like peptide-1 receptor agonists, respectively.
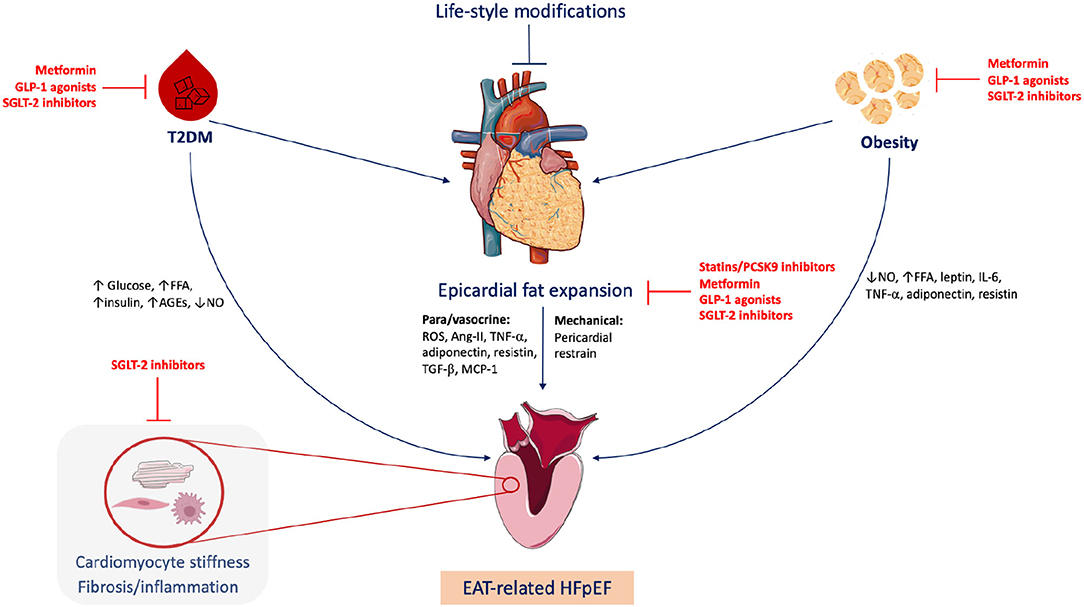
Graphical Abstract. Epicardial adipose tissue (EAT)-related heart failure with preserved ejection fraction (HFpEF). Obesity and type 2 diabetes mellitus (T2DM) are common triggers of HFpEF, frequently associated with EAT expansion. EAT plays metabolic and mechanical roles in HFpEF development via para/vasocrine factors and pericardial restrain, respectively. Life-style modifications including healthy diet and regular exercise can quash the EAT expansion. Statins, proprotein convertase subtilisin/kexin type 9 (PCSK9) inhibitors and fat-modulating antidiabetics including metformin, sodium-glucose cotransporter 2 (SGLT2) inhibitors and glucagon-like peptide-1 (GLP-1) agonists can target EAT. FFA, free fatty acids; AGEs, advanced glycation end-products; NO, nitric oxide; ROS, reactive oxygen species; Ang-II, angiotensin II; TGF-β, Transforming growth factor beta; MCP-1, monocyte chemoattractant protein 1; IL-6, interleukin 6; TNF-α, tumor necrosis factor alpha. Figure created via Servier Medical Art and BioRender tools.
Nearly one-half of heart failure (HF) patients have a preserved ejection fraction (HFpEF), with rising prevalence in the United States of America and Western populations (1, 2). Common hemodynamic features of HFpEF include diastolic dysfunction and reduced ventricular compliance (3). The pathophysiology of HFpEF is complex, exacerbated by a variety of comorbidities including age, hypertension, renal dysfunction, diabetes mellitus (DM), and obesity (4, 5), and may reflect also different phenotypes and differences in pathology (6). This could be especially important for the obese and DM subgroup of HFpEF. In dispersion through large outcome studies and registries, around 80% of HFpEF patients are obese and 20–45% have type 2 DM (T2DM) (7–10). Thirty-percent of HFpEF patients have both obesity and T2DM (11). As such, HFpEF is perceived as an inflammatory cardiometabolic disease, which includes all major mechanisms discussed in HFpEF (12, 13). However, even in this subgroup of HFpEF some patients, differ with respect to the existences of epicardial adipose tissue (EAT). EAT, which is the visceral fat depot of the heart, may play an important extra role in the development of HFpEF (10, 14). Compared to non-obese HFpEF patients, HFpEF patients with obese phenotype show 20–50% higher EAT thickness despite similar body mass index (BMI) (15). The volume of EAT is directly proportional with the presence of atrial fibrillation and T2DM and with myocardial injury biomarkers (16). It has been recognized as a metabolically active depot that affects the myocardium via production of cytokines and adipokines (17).
There is growing evidence that HFpEF with enlarged EAT is a clinically relevant HF phenotype that may require specific treatments (15, 17). We hypothesize that EAT quantification would allow the differentiation of HFpEF patients with enlarged EAT. This specific subgroup of patients might benefit from EAT-modifying therapies.
HFpEF is a systemic disorder involving multiple organ systems where circulating proinflammatory mediators originating from multiple comorbidities trigger abnormalities in both the heart and the skeletal muscles (18). An expansion in body fat mass causes hemodynamic, metabolic, inflammatory, and hormonal disruption, which affect the vascular endothelium and the heart (19, 20). Obesity is a principal HFpEF component via triggering a systemic proinflammatory environment and inducing endothelial production of reactive oxygen species and reduces nitric oxide (NO) bioavailability, which especially affect the coronary microvasculature and the neighboring cardiomyocytes (21–23). Moreover, cardiomyocytes have no lipid storage capacity, their exposure to excess blood lipids, typically occurring in obese patients can lead to cardiomyocyte steatosis and reduction of function (14, 24).
Accordingly, HFpEF patients can be further classified into non-obese and obese phenotypes. The latter is characterized by high body mass index >30 kg/m2, lower natriuretic peptide levels and higher left ventricular (LV) mass to volume ratio compared to the non-obese phenotype which is more common in the elderly (15). Moreover, obese HFpEF patients have abnormal cardiac and skeletal muscle composition with infiltration of adipose tissue (25). Furthermore, in comparison with non-obese HFpEF patients, obese HFpEF patients have unique pathophysiological features including larger volume overload, abnormal right ventricular-pulmonary arterial coupling, worse exercise capacity, subtle hemodynamic perturbations, increased epicardial fat mass and higher LV filling pressure and exaggerated biventricular remodeling (15, 26, 27). Increased biventricular pressure can be attributed to greater dependence on plasma volume expansion, and enhanced ventricular interaction, and is further amplified as pulmonary pressure load increases (15, 26). This increase in the pulmonary pressure is a consequence of impaired pulmonary vasodilatation that could be related to the existence of adipokines which reduce NO bioavailability (28, 29). However, obese patients can also develop a significant enlargement in EAT, which may represent an additional factor in the HFpEF hemodynamic features due to an increased pericardial restraint.
Like obesity, T2DM plays a fundamental role in the pathophysiology of HFpEF via diabetic specific mechanisms which culminate in matrix changes, vascular endothelial dysfunction, and myocardial stiffness, respectively (30, 31). In HFpEF patients, T2DM is associated with poor prognosis manifested in an increased risk of hospitalization for worsening HF and HF-related death (32, 33). In obese T2DM patients, metabolic disturbances including hyperglycemia, lipotoxicity, abundance of advanced glycation end-products (AGEs), and hyperinsulinemia provoke coronary microvascular dysfunction, and the development of HFpEF (21, 30). Hyperglycemia impairs endothelial NO generation and reduces cyclic guanosine monophosphate (cGMP) production which in turn reduce protein kinase G (PKG) activity in cardiomyocytes and consequently titin protein function and diastolic distensibility (21, 34). Similarly, AGEs impair endothelial NO production and predisposes to concentric LV remodeling and myocardial stiffness as observed in diabetic cardiomyopathy patients with HFpEF (35–38). In addition, in T2DM there is an increase in glucose-auto-oxidation and free-fatty acid concentrations which creates oxidative stress in the myocardium and subsequently concentric ventricular remodeling (39). Lam (30) recognized T2DM-related HFpEF as a unique diabetic cardiomyopathy phenotype, which can be defined by the presence of left ventricular diastolic dysfunction in diabetic patients without coronary artery disease, hypertension, or other potential etiologies (40). This phenotype is in contrast with the well-established diabetic cardiomyopathy definitions where LV dilatation, reduced EF and systolic dysfunction are the main characteristics (30). The features of both phenotypes have been compared in detail elsewhere (21). Common HFpEF mechanisms associated with obesity and T2DM are summarized in Table 1. Like obesity, T2DM patients may develop EAT expansion, which contributed to HFpEF pathogenesis via several mechanisms.

Table 1. Common obesity and type 2 diabetes mellitus related HFpEF mechanisms.
EAT is the visceral fat depot of the heart. In the adult, EAT is physiologically found in the atrioventricular and interventricular grooves of the heart. HFpEF patients have 20–50% higher EAT mass compared to both patients with non-obese HFpEF and control subjects with similar body mass index (14–16). EAT expansion includes the intensification of perivascular fat which causes coronary inflammation and accelerated atherosclerosis, and ultimately leads to myocardial stiffness and fibrosis (41, 42). The EAT interacts directly with the heart, metabolically and mechanically (Figure 1) (15, 17). Since there is no muscle fascia between the EAT and the myocardium, the two tissues depend on the same microvasculature and could interact directly via paracrine and vasocrine secretions (43, 44). In obese patients, the EAT secretes several pro-inflammatory chemokines and cytokines, collectively called adipokines such as tumor necrosis factor alpha (TNF-α), monocyte chemoattractant protein-1 (MCP-1), interleukin-6 (IL-6), IL-1β, plasminogen activator inhibitor-1 (PAI-1), resistin, S100A9, and many others (16). All together create a proinflammatory state in the myocardium associated with cardiomyocyte stiffness, coronary endothelial dysfunction, and fibrosis, which are all implicated in the development of HFpEF (14, 15, 45). In line, Karastergiou et al. (46) have reported a strong presence of activated macrophages in the EAT obtained from obese patients with coronary artery disease (CAD) (46). Moreover, the EAT produces high levels of reactive oxygen species (ROS) products, which drives oxidative stress in the myocardium and the coronary vasculature (47). Furthermore, the EAT is a source of angiotensin II, which provokes coronary vasoconstriction enhancing ischemia, especially in patients with DM due to coexisting vasculopathies (48). Also, the EAT transcriptome contributes to the development of CAD via higher levels of renitol-binding protein 4 (RBP4) and lower levels of glucose transporter-4 (GLUT4), leading to adverse lipid and glucose metabolic profile augmented by the proinflammatory secretome (49, 50). Metabolically, EAT expansion is associated with intramyocardial accumulation of triglycerides causing cardiac steatosis (51). It has been shown that myocardial triglyceride content is independently associated with reduced pump function (52), and impaired ventricular strain parameters (53). Cardiac steatosis induces fetal gene transcription that favors myocardial glucose utilization instead of free fatty acids under physiological conditions, which further aggravates lipid accumulation (54, 55).
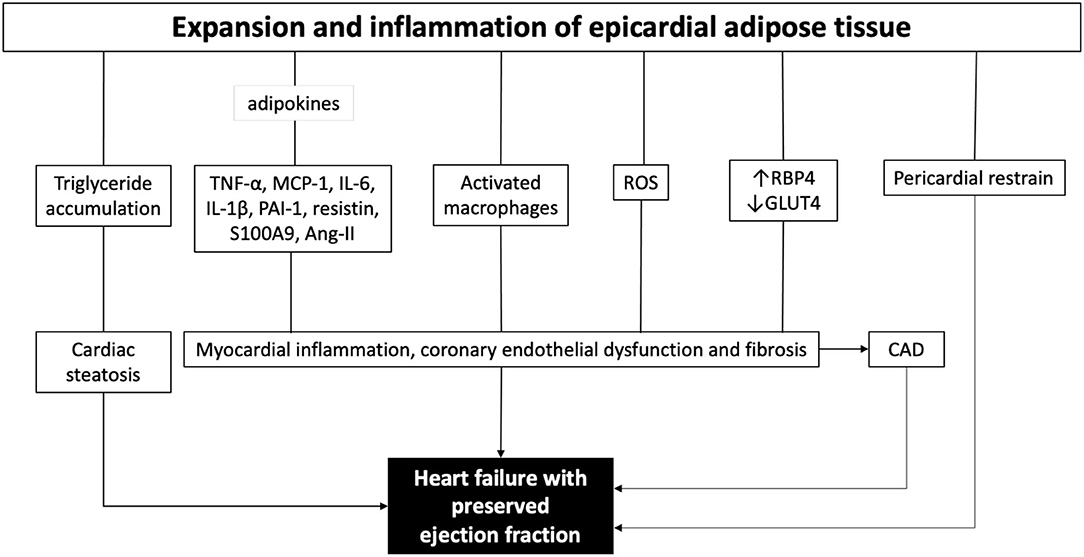
Figure 1. The role of epicardial adipose tissue in the pathogenesis of heart failure with preserved ejection fraction. ROS, reactive oxygen species; TNF-α, tumor necrosis factor alpha; MCP-1, monocyte chemoattractant protein 1; IL-6, interleukin 6; IL-1b, interleukin 1b; PAI-1, plasminogen activator inhibitor-1; Ang-II, angiotensin II; ROS, reactive oxygen species; RBP4, renitol-binding protein 4; GLUT4, glucose transporter-4; CAD, coronary artery disease.
Mechanically, the EAT occupies space in the cardiac fossa. In obese patients, large EAT mass causes an increase in intracardiac pressures, particularly during exercise (15, 56). In HFpEF patients, an expansion in the EAT volume is commonly observed jointly with biventricular hypertrophy, however, the pericardium does not expand at the same proportion (15, 56). Hence, the pericardium exerts a compressive contact force on the heart and consequently increases pericardial restrain and enhances ventricular interaction (57). The abnormal mechanical interaction between the heart and the pericardium causes an increase in pericardial pressure and LV end-diastolic pressure, and a decrease in LV transmural pressure and LV end-diastolic diameter which together elevate the pulmonary capillary hydrostatic pressures promoting dyspnea (58). Further studies have substantiated the deleterious effect of EAT expansion on the cardiac muscle function including strain abnormalities (59). In a 2-dimensional speckle tracking echocardiography study, Maimaituxun et al. (60) have demonstrated that EAT volume is a determinant of global longitudinal strain (GLS) in HFpEF patients. Similarly, DM was a sole determinant of GLS (60).
Visceral fat accumulation is a fundamental element of T2DM (43, 61). HFpEF patients with T2DM have higher EAT mass compared to those without T2DM at similar BMI (16, 61). Being a marker of visceral adiposity, EAT is a risk factor for T2DM, cardiovascular complications and metabolic syndrome (14, 44, 62, 63). In diabetics, the EAT transcriptome is rich in proinflammatory and innate immune genes like Pentraxin 3 (PTX3) and endothelial lipase G (LIPG) compared with the subcutaneous fat transcriptome from the same patients (64). Van Woerden et al. (16) have observed that in HF patients creatine kinase-MB, troponin T, and glycated hemoglobin positively correlate with EAT volume. It appears that EAT expansion is associated with DM, however, whether there is a causal association between the two disorders is not yet clear.
The diagnosis of EAT enlargement is imperative for the identification of EAT-related HFpEF phenotypes. Transthoracic echocardiography can estimate the EAT thickness, measured as the echo-lucent area between the epicardial surface and parietal pericardium. However, echocardiography cannot be used to estimate EAT volume and has relatively poor inter-observer and intra-observer variability among other limitations (65).
Ideally, EAT volume can be evaluated via cardiac magnetic resonance (CMR) (66, 67) (Figure 2). In line, the European society of cardiology (ESC) consensus recommends the use of a stepwise score-based algorithm to diagnose HFpEF (68). The algorithm suggests the use of more sophisticated tools including CMR to identify specific etiologies in patients with confirmed HFpEF. CMR-based stratification of HFpEF patients based on EAT volume might support the rational to use of EAT-modulating therapies. In addition, CMR opens the possibility to a combined evaluation of potential underlying myocardial ischemia, storage diseases like amyloidosis, diffuse myocardial fibrosis (extracellular volume), and epicardial fat. Recently, a growing number of publications also demonstrate the easiness of epicardial fat quantification using artificial intelligence (AI) algorithms (69). Beside the heart, the same approach can also be applied to other organ regions in patients with HFpEF like the abdomen (70). Since CMR allows imaging also in severely obese patients, functional evaluation of the systolic and diastolic function including myocardial work is feasible (71). Recent guidelines of the ESC recommend the use of the imaging modality which is locally available with the best expertise and confidence in results. Cardiac computed tomography (CT) is widely available and easy to use and has demonstrated its ability to quantify epicardial fat. However, the necessary use of radiation limits its application for serial measurements needed to control treatment effects. However, since newest generation CT-scanner can be used with very low radiation doses, this might be an interesting field for future research. In addition, quantification of EAT by cardiac CT can be used to predict outcome even in inflammatory diseases like Covid-19 (72).
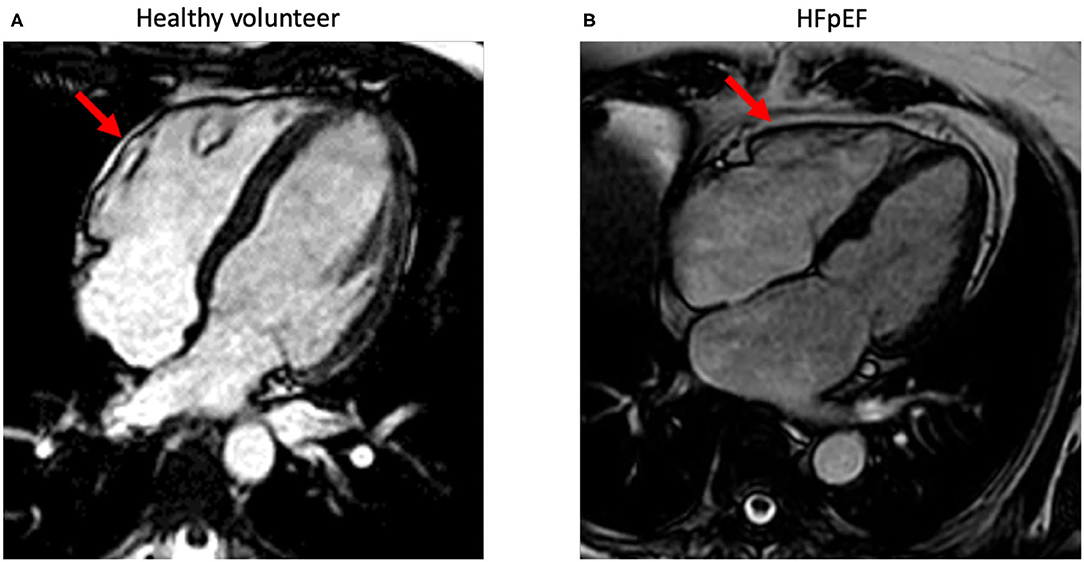
Figure 2. Standard-4-chamber orientations acquired using standard steady-state free precession-cardiac magnetic imaging sequences. The images demonstrate epicardial fat (red arrow) with a minimal (normal) amount in a healthy volunteer (A) and in a patient with HFpEF (B) with increased epicardial fat surrounding the whole heart.
Unlike HF with reduced EF (HFrEF), patients with HFpEF phenotype do not benefit from most standard HF therapies, but rather from lifestyle modifications (73, 74). Similarly, diabetic cardiomyopathy patients with an HFpEF phenotype benefit more from lifestyle modifications compared to standard heart failure therapies (21). Implementation of a healthy lifestyle including smoking cessation, weight reduction, exercise, and healthy diet is the mainstay of HFpEF management (75). Looking at obese-HFpEF patients, weight reduction, and exercise might regress the EAT volume, restore its physiological role (14), and mitigate excessive pericardial restraint (15).
In addition to lifestyle modifications, the ESC 2016 guidelines have recommended the symptomatic use of diuretics in HFpEF (3). Owing to the strong dependency between filling pressures and blood volume, obese-HFpEF patients might benefit from diuretics (15, 76). By reducing blood volume, diuretics reduce right ventricular volume and ventricular interaction, which in turn improves LV end-diastolic volume (LVEDV) and stroke volume (SV), and relives pericardial restrain (15, 77). However, the diuretic dose should be carefully adjusted to avoid hypovolemia and severe preload reduction (3).
Statins are lead prescribed medications that can achieve substantial serum cholesterol lowering via inhibiting 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, the rate-limiting enzyme of cholesterol synthesis (78). Members of this class are known pleiotropic agents having the potential to affect different body tissues independent of lipid lowering (79, 80). Statins restore endothelial redox balance and NO bioavailability (81). Although the use of statins in HFpEF has been a topic of debate, several studies support the beneficial effect of statins in HFpEF (82, 83). Various metanalysis studies advocate the potential benefit of statins on HFpEF-mortality (84–86). In cohort studies statins have demonstrated the ability to reduce EAT volume up to 15% with atorvastatin, independent of low-density lipoprotein cholesterol-lowering (79, 87). Prominently, a multi-center study comparing 87 aortic stenosis patients on statins to 106 not on statins, concluded that statin treatment was significantly associated with reduced EAT thickness and proinflammatory cytokines secretome (88). The expression of LDL and VLDL receptors by the EAT, advocates the role of statins in modulating the metabolism of this fat depot (89). Besides, it has been suggested that statins reduce the EAT metabolic activity (79). Ultimately, the exact mechanism by which statins affect the EAT volume is so far unknown.
Ezetimibe is another antihypercholesterolemic agent that inhibits cholesterol absorption from the gut. In combination with simvastatin, ezetimibe could not achieve better reduction in EAT thickness compared to atorvastatin monotherapy (87).
Proprotein convertase subtilisin/kexin type 9 inhibitors (PCSK9i) are novel cholesterol lowering agents which act via inhibiting the PCSK9 enzyme which is responsible the downregulation of low-density lipoprotein (LDL) receptors on the surface of hepatocytes (90). A study by Galvez et al. concluded that 6 months of treatment with PSCK9i could achieve a 20% reduction in EAT thickness (91).
Metformin is the first-line treatment for T2DM, particularly in obese patients. Alongside its glucose-lowering effect, metformin use is associated with weight loss independent of diabetes, primary due to reduction of VAT (92, 93). A recent meta-analysis by Halabi et al. concluded that metformin treatment is associated with a reduction in mortality in patients with HFpEF (94). In addition, two recent studies have demonstrated that metformin monotherapy reduces EAT thickness (95, 96). A standard metformin monotherapy for 3 months could reduce the EAT thickness by 10% (95). The exact mechanism by which metformin affects EAT is unclear but most reasonably attributed to the well-established metabolic effects of metformin, specifically shifting metabolism into fat oxidation and upregulation of thermogenesis (92, 95).
Semaglutide, liraglutide, and dulaglutide are glucagon-like peptide-1 (GLP-1) receptor agonists, indicated for the treatment T2DM. They act via enhancing glucose-dependent insulin secretion, reducing glucagon secretion, and delaying gastric emptying, resulting in adequate T2DM control and weight loss (97). Several studies have demonstrated that the use of GLP-1 associates with reduced cardiovascular risk (98, 99). In a randomized placebo-controlled trial on patients with T2DM, liraglutide exhibited favorable cardiovascular outcomes including LV filling pressure reduction and diastolic function improvement. Both parameters are relevant for diabetic cardiomyopathy and HFpEF (100). In addition, a systematic review comparing the effect of GLP-1 agonists to different antidiabetics on LV diastolic function has concluded that liraglutide monotherapy offers a considerably beneficial outcome (101). Interestingly, the EAT was found to express GLP-1 receptors in contrast to subcutaneous fat in the same patient (102). In a cohort of patients with T2DM and obesity, Lacobellis and his colleagues demonstrated that weekly administration of either semaglutide or dulaglutide causes rapid, substantial, and dose dependent reduction in EAT thickness, attaining 20% reduction in 12 weeks (103). Similarly, liraglutide treatment on top of metformin has resulted in 29 and 36% reduction in EAT after 3 and 6 months, respectively, together with a reduction in BMI and glycated hemoglobin (104). GLP-1 agonists are suggested to regulate EAT adipocyte formation and metabolism as an outcome of promoting preadipocyte differentiation, increasing sensitivity to insulin and stimulating thermogenesis and adipocyte browning via a complex signaling system (105–107).
Endogenous GLP-1 is susceptible to cleavage by dipeptidyl peptidase-4 (DPP-4). Sitagliptin, a DPP-4 inhibitor, has recently been shown to substantially reduce EAT in subjects with obesity and T2DM via prolonging GLP-1 half-life (108). DDP-4 inhibitors are recommended in T2DM patients without cardiovascular risk. Whether DPP-4 inhibitors would offer long-term benefit for HFpEF patients or not is debatable. Several studies have shown that DPP-4 inhibitors exhibit cardioprotective anti-inflammatory properties that may have beneficial effects on EAT (109–111). Those effects were described to be mediated via different mechanisms including downregulation of the receptor for AGE (RAGE) (112), activation of cAMP/PKA signaling and IL-6 production (113), reduction of ROS generation and ICAM-1 expression (114), and diminishing DPP-4-activated phosphatidylinositol 3-kinase signaling which favors adipocyte maturation (115). In contrast, other studies have concluded that DPP-4 inhibitors might increase the inflammatory products of the EAT and adversely affect the myocardium especially in diabetic patients via potentiating the actions of endogenous proinflammatory chemokines like CXCL12 and mineralocorticoid receptor signaling (116–118).
Sodium-glucose cotransporter 2 inhibitors (SGLT2i), for example dapagliflozin and empagliflozin are relatively new medications indicated for T2DM patients, but they are also sufficient in HFrEF patients without DM (119, 120). Agents of this class lower plasma glucose concentration via increasing urinary glucose excretion (121). In addition, they cause significant weight reduction, comparable to GLP-1 agonists, via caloric loss, osmotic diuresis, and stimulation of visceral fat burn (122, 123). Like diuretics, SGLT2i can reduce plasma volume and consequently ventricular filling pressures, offering benefit to HFpEF patients (15). With respect to HFpEF animal data, showing that SGLT2 inhibition exhibited beneficial cardiovascular effects in several non-diabetic HFpEF-animal models (124, 125). Cardiomyocytes from empagliflozin-treated HFpEF patients showed improved NO–sGC–cGMP–cascade and PKG-activity, suggesting favorable cardiovascular outcomes (126). In patients with T2DM and recent worsening HF, sotagliflozin treatment reduced the total number of deaths from cardiovascular causes and hospitalizations and urgent visits for HF, particularly in patients with preserved EF (127). Press release about the EMPEROR-Preserved study, which has been recently completed on HFpEF patients, stated that the study met its primary endpoint showing that empagliflozin has reduced mortality and hospitalization in the treated cohort [NCT03057951]. Moreover, optimistic results are expected from the DELIVER trial, which investigates the benefit of dapagliflozin in HFpEF patients (128), all showing that SGLTi inhibition can exert cardio-beneficial effects in HFpEF. In addition, in T2DM patients dapagliflozin could reduce the EAT volume and the occurrence cardiovascular events (94). Similarly, dapagliflozin was recently shown to cause significant EAT thickness reduction reaching 20% after 24 weeks of treatment in obese patients with T2DM, independent of weight loss (129). Weight loss independent mechanisms include improvement of EAT cells-insulin sensitivity and reduction of local proinflammatory chemokines secretion (e.g., CCL2) (130). Overall, SGLT2i showed favorable results in HFpEF patients including EAT reduction. Whether this effect is mediated via weight loss, or a direct metabolic mechanism warrants further investigation.
Anterior pericardiotomy through minimally invasive percutaneous procedure is a potentially novel last-option treatment for HFpEF patients with severe LV restriction, since it eliminates the external restraint of the pericardium (57, 131, 132). In canine HFpEF animal models, resection of the pericardium improved LV compliance and filling pressure (132). This technique is also applicable in humans, however further studies are warranted to evaluate its long-term benefit and safety (131).
Inflammation is an important driver of HF, by which its role in the pathogenesis of HFrEF and HFpEF differs (133, 134). Although it plays a pathological role in the EAT of obese patients (46), pharmacological treatment with anti-inflammatory agents like steroids (135) cannot be recommended in HF patients with DM and/or metabolic syndrome (136). Theoretically, several biological agents like interleukin (IL)-1 and IL-6 inhibitors can target EAT-induced myocardial inflammation (136). The small D-HARD study has demonstrated the beneficial effects of the competitive IL-1 receptor antagonist anakinra in HFpEF patients (137). However, the subsequent phase II (D-HART2) study has failed to corroborate favorable outcomes (138). Eventually, the study was underpowered and most of the study participants suffered from obesity which independently affects the co-primary endpoints peak oxygen uptake (VO2) and ventilatory efficiency (VE/VCO2) (138). Whether the D-HART2 study results be different in HFpEF patients with enlarged EAT warrants further investigation. Canakinumab is another monoclonal antibody that binds and neutralizes IL-1β. Results from the CANTOS study demonstrated that canakinumab significantly reduces the recurrence of new cardiovascular events (139). These findings form the rationale to consider investigating whether canakinumab would influence EAT-inflammation. Ultimately, clinical studies testing whether targeting EAT-related proinflammatory cytokines can benefit HFpEF patients with enlarged EAT or not are necessary.
HFpEF is a diverse disease resulting from wide range of comorbidities. Obesity and DM are principal drivers of HFpEF. Stratifying HFpEF patients based on phenotypic data results in novel classifications including obese and diabetic HFpEF phenotypes. There is a close association between EAT volume and HFpEF. HFpEF patients can be further classified according to EAT volume using advanced imaging techniques including CMR and CT. EAT functions as endocrine tissue that contributes to myocardial inflammation. In addition, EAT expansion acts as space-occupying lesion that causes pericardial restrain, increase in ventricular filling pressures, and enhanced ventricular interaction. HFpEF patients with enlarged EAT may benefit from lifestyle modifications and symptomatic treatment with diuretics. Besides, statins, PCSK9i and fat-modulating anti-diabetic agents like metformin, SGLT2i or GLP-1 agonists can be especially effective in this subgroup of patients, being able to induce EAT regression. In addition, direct effects of SGLT2i and GLP-1 agonists on HFpEF are currently under clinical investigation.
AE wrote most of the manuscript and prepared the figures. VN wrote short parts of the manuscript. SV revised the manuscript and applied changes to the text and figures. CT provided the main ideas of the manuscript and guided the other authors, revised the manuscript, and added some parts. SK provided Figure 2 and wrote short parts of the manuscript. All authors contributed to the article and approved the submitted version.
CT has received speaker fees and/or contributions to congresses from Abbott, Abiomed, Astra Zeneca, Bayer, Böhringer-Ingelheim, Novartis Pfizer, and Servier; all outside the submitted work.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Prenner SB, Mather PJ. Obesity and heart failure with preserved ejection fraction: a growing problem. Trends Cardiovasc Med. (2018) 28:322–7. doi: 10.1016/j.tcm.2017.12.003
2. Sundaram V, Deo S, Rana M, Selvaganesan P, Rubelowsky J, Elgudin Y, et al. Rising prevalence of obese heart failure with preserved ejection fraction in the United States. J Am Coll Cardiol. (2021) 77:680. doi: 10.1016/S0735-1097(21)02039-8
3. Ponikowski P, Voors AA, Anker SD, Bueno H, Cleland JG, Coats AJ, et al. 2016 ESC Guidelines for the diagnosis treatment of acute chronic heart failure: The Task Force for the diagnosis treatment of acute chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur J Heart Fail. (2016) 18:891–975. doi: 10.1093/eurheartj/ehw128
4. Reddy YN, Borlaug BA. Heart failure with preserved ejection fraction. Curr Probl Cardiol. (2016) 41:145–88. doi: 10.1016/j.cpcardiol.2015.12.002
5. Shah SJ, Kitzman DW, Borlaug BA Heerebeek, van L, Zile MR, Kass DA, et al. Phenotype-specific treatment of heart failure with preserved ejection fraction: a multiorgan roadmap. Circulation. (2016) 134:73–90. doi: 10.1161/CIRCULATIONAHA.116.021884
6. Cohen JB, Schrauben SJ, Zhao L, Basso MD, Cvijic ME, Li Z, et al. Clinical phenogroups in heart failure with preserved ejection fraction: detailed phenotypes, prognosis, and response to spironolactone. JACC Heart Fail. (2020) 8:172–84. doi: 10.1016/j.jchf.2019.09.009
7. Obokata M, Borlaug BA. Response by obokata and borlaug to letters regarding article, “evidence supporting the existence of a distinct obese phenotype of heart failure with preserved ejection fraction. Circulation. (2018) 137:416–7. doi: 10.1161/CIRCULATIONAHA.117.031394
8. Dunlay SM, Roger VL, Redfield MM. Epidemiology of heart failure with preserved ejection fraction. Nat Rev Cardiol. (2017) 14:591–602. doi: 10.1038/nrcardio.2017.65
9. Haass M, Kitzman DW, Anand IS, Miller A, Zile MR, Massie BM, et al. Body mass index and adverse cardiovascular outcomes in heart failure patients with preserved ejection fraction: results from the Irbesartan in Heart Failure with Preserved Ejection Fraction (I-PRESERVE) trial. Circ Heart Fail. (2011) 4:324–31. doi: 10.1161/CIRCHEARTFAILURE.110.959890
10. Lam CSP, Chandramouli C. Fat, female, fatigued: features of the obese HFpEF phenotype. JACC Heart Fail. (2018) 6:710–3. doi: 10.1016/j.jchf.2018.06.006
11. Lam CS, Donal E, Kraigher-Krainer E, Vasan RS. Epidemiology and clinical course of heart failure with preserved ejection fraction. Eur J Heart Fail. (2011) 13:18–28. doi: 10.1093/eurjhf/hfq121
12. Paulus WJ, Tschope C. A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol. (2013) 62:263–71. doi: 10.1016/j.jacc.2013.02.092
13. Tschope C, Van Linthout S. New insights in (inter)cellular mechanisms by heart failure with preserved ejection fraction. Curr Heart Fail Rep. (2014) 11:436–44. doi: 10.1007/s11897-014-0219-3
14. Iacobellis G, Bianco AC. Epicardial adipose tissue: emerging physiological, pathophysiological and clinical features. Trends Endocrinol Metab. (2011) 22:450–7. doi: 10.1016/j.tem.2011.07.003
15. Obokata M, Reddy YNV, Pislaru SV, Melenovsky V, Borlaug BA. Evidence supporting the existence of a distinct obese phenotype of heart failure with preserved ejection fraction. Circulation. (2017) 136:6–19. doi: 10.1161/CIRCULATIONAHA.116.026807
16. van Woerden G, Gorter TM, Westenbrink BD, Willems TP, van Veldhuisen DJ, Rienstra M. Epicardial fat in heart failure patients with mid-range and preserved ejection fraction. Eur J Heart Fail. (2018) 20:1559–66. doi: 10.1002/ejhf.1283
17. Iacobellis G, Corradi D, Sharma AM. Epicardial adipose tissue: anatomic, biomolecular and clinical relationships with the heart. Nat Clin Pract Cardiovasc Med. (2005) 2:536–43. doi: 10.1038/ncpcardio0319
18. Ather S, Chan W, Bozkurt B, Aguilar D, Ramasubbu K, Zachariah AA, et al. Impact of noncardiac comorbidities on morbidity and mortality in a predominantly male population with heart failure and preserved versus reduced ejection fraction. J Am Coll Cardiol. (2012) 59:998–1005. doi: 10.1016/j.jacc.2011.11.040
19. Abel ED, Litwin SE, Sweeney G. Cardiac remodeling in obesity. Physiol Rev. (2008) 88:389–419. doi: 10.1152/physrev.00017.2007
20. Kitzman DW, Shah SJ. The HFpEF obesity phenotype: the elephant in the room. J Am Coll Cardiol. (2016) 68:200–3. doi: 10.1016/j.jacc.2016.05.019
21. Seferovic PM, Paulus WJ. Clinical diabetic cardiomyopathy: a two-faced disease with restrictive and dilated phenotypes. Eur Heart J. (2015) 36:1718–27. doi: 10.1093/eurheartj/ehv134
22. Van Linthout S, Rimoldi O, Tschope C, Camici PG. Coronary microvascular dysfunction in heart failure with preserved ejection fraction - adding new pieces to the jigsaw puzzle. Eur J Heart Fail. (2020) 22:442–4. doi: 10.1002/ejhf.1720
23. Camici PG, Tschope C, Di Carli MF, Rimoldi O, Van Linthout S. Coronary microvascular dysfunction in hypertrophy and heart failure. Cardiovasc Res. (2020) 116:806–16. doi: 10.1093/cvr/cvaa023
24. Spillmann F, Trimpert C, Peng J, Eckerle LG, Staudt A, Warstat K, et al. High-density lipoproteins reduce palmitate-induced cardiomyocyte apoptosis in an AMPK-dependent manner. Biochem Biophys Res Commun. (2015) 466:272–7. doi: 10.1016/j.bbrc.2015.09.034
25. Berezin AE, Berezin AA, Lichtenauer M. Emerging role of adipocyte dysfunction in inducing heart failure among obese patients with prediabetes and known diabetes mellitus. Front Cardiovasc Med. (2020) 7:583175. doi: 10.3389/fcvm.2020.583175
26. Lavie CJ, Alpert MA, Arena R, Mehra MR, Milani RV, Ventura HO. Impact of obesity and the obesity paradox on prevalence and prognosis in heart failure. JACC Heart Fail. (2013) 1:93–102. doi: 10.1016/j.jchf.2013.01.006
27. Lam CS, Roger VL, Rodeheffer RJ, Bursi F, Borlaug BA, Ommen SR, et al. Cardiac structure and ventricular-vascular function in persons with heart failure and preserved ejection fraction from Olmsted County, Minnesota. Circulation. (2007) 115:1982–90. doi: 10.1161/CIRCULATIONAHA.106.659763
28. Gorter TM, van Veldhuisen DJ, Bauersachs J, Borlaug BA, Celutkiene J, Coats AJS, et al. Right heart dysfunction and failure in heart failure with preserved ejection fraction: mechanisms and management. Position statement on behalf of the Heart Failure Association of the European Society of Cardiology. Eur J Heart Fail. (2018) 20:16–37. doi: 10.1002/ejhf.1029
29. Yudkin JS, Eringa E, Stehouwer CD. Vasocrine” signalling from perivascular fat: a mechanism linking insulin resistance to vascular disease. Lancet. (2005) 365:1817–20. doi: 10.1016/S0140-6736(05)66585-3
30. Lam CS. Diabetic cardiomyopathy: an expression of stage B heart failure with preserved ejection fraction. Diab Vasc Dis Res. (2015) 12:234–8. doi: 10.1177/1479164115579006
31. Meagher P, Adam M, Civitarese R, Bugyei-Twum A, Connelly KA. Heart failure with preserved ejection fraction in diabetes: mechanisms and management. Can J Cardiol. (2018) 34:632–43. doi: 10.1016/j.cjca.2018.02.026
32. MacDonald MR, Petrie MC, Varyani F, Ostergren J, Michelson EL, Young JB, et al. Impact of diabetes on outcomes in patients with low and preserved ejection fraction heart failure: an analysis of the Candesartan in Heart failure: Assessment of Reduction in Mortality and morbidity (CHARM) programme. Eur Heart J. (2008) 29:1377–85. doi: 10.1093/eurheartj/ehn153
33. Tribouilloy C, Rusinaru D, Mahjoub H, Tartiere JM, Kesri-Tartiere L, Godard S, et al. Prognostic impact of diabetes mellitus in patients with heart failure and preserved ejection fraction: a prospective five-year study. Heart. (2008) 94:1450–5. doi: 10.1136/hrt.2007.128769
34. Zhazykbayeva S, Pabel S, Mugge A, Sossalla S, Hamdani N. The molecular mechanisms associated with the physiological responses to inflammation and oxidative stress in cardiovascular diseases. Biophys Rev. (2020) 12:947–68. doi: 10.1007/s12551-020-00742-0
35. van Heerebeek L, Hamdani N, Handoko ML, Falcao-Pires I, Musters RJ, Kupreishvili K, et al. Diastolic stiffness of the failing diabetic heart: importance of fibrosis, advanced glycation end products, and myocyte resting tension. Circulation. (2008) 117:43–51. doi: 10.1161/CIRCULATIONAHA.107.728550
36. Falcao-Pires I, Hamdani N, Borbely A, Gavina C, Schalkwijk CG, van der Velden J, et al. Diabetes mellitus worsens diastolic left ventricular dysfunction in aortic stenosis through altered myocardial structure and cardiomyocyte stiffness. Circulation. (2011) 124:1151–9. doi: 10.1161/CIRCULATIONAHA.111.025270
37. Miteva K, Pappritz K, El-Shafeey M, Dong F, Ringe J, Tschope C, et al. Mesenchymal stromal cells modulate monocytes trafficking in coxsackievirus B3-induced myocarditis. Stem Cells Transl Med. (2017) 6:1249–61. doi: 10.1002/sctm.16-0353
38. Van Linthout S, Hamdani N, Miteva K, Koschel A, Muller I, Pinzur L, et al. Placenta-derived adherent stromal cells improve diabetes mellitus-associated left ventricular diastolic performance. Stem Cells Transl Med. (2017) 6:2135–45. doi: 10.1002/sctm.17-0130
39. Jay D, Hitomi H, Griendling KK. Oxidative stress and diabetic cardiovascular complications. Free Radic Biol Med. (2006) 40:183–92. doi: 10.1016/j.freeradbiomed.2005.06.018
40. Lukic L, Lalic NM, Rajkovic N, Jotic A, Lalic K, Milicic T, et al. Hypertension in obese type 2 diabetes patients is associated with increases in insulin resistance and IL-6 cytokine levels: potential targets for an efficient preventive intervention. Int J Environ Res Public Health. (2014) 11:3586–98. doi: 10.3390/ijerph110403586
41. Packer M. The epicardial adipose inflammatory triad: coronary atherosclerosis, atrial fibrillation, and heart failure with a preserved ejection fraction. Eur J Heart Fail. (2018) 20:1567–9. doi: 10.1002/ejhf.1294
42. Packer M. Epicardial adipose tissue may mediate deleterious effects of obesity and inflammation on the myocardium. J Am Coll Cardiol. (2018) 71:2360–72. doi: 10.1016/j.jacc.2018.03.509
43. Corradi D, Maestri R, Callegari S, Pastori P, Goldoni M, Luong TV, et al. The ventricular epicardial fat is related to the myocardial mass in normal, ischemic and hypertrophic hearts. Cardiovasc Pathol. (2004) 13:313–6. doi: 10.1016/j.carpath.2004.08.005
44. Sacks HS, Fain JN. Human epicardial adipose tissue: a review. Am Heart J. (2007) 153:907–17. doi: 10.1016/j.ahj.2007.03.019
45. Iacobellis G. Epicardial adipose tissue in endocrine and metabolic diseases. Endocrine. (2014) 46:8–15. doi: 10.1007/s12020-013-0099-4
46. Karastergiou K, Evans I, Ogston N, Miheisi N, Nair D, Kaski JC, et al. Epicardial adipokines in obesity and coronary artery disease induce atherogenic changes in monocytes and endothelial cells. Arterioscler Thromb Vasc Biol. (2010) 30:1340–6. doi: 10.1161/ATVBAHA.110.204719
47. Salgado-Somoza A, Teijeira-Fernandez E, Fernandez AL, Gonzalez-Juanatey JR, Eiras S. Proteomic analysis of epicardial and subcutaneous adipose tissue reveals differences in proteins involved in oxidative stress. Am J Physiol Heart Circ Physiol. (2010) 299:H202–9. doi: 10.1152/ajpheart.00120.2010
48. Baker AR, Silva NF, Quinn DW, Harte AL, Pagano D, Bonser RS, et al. Human epicardial adipose tissue expresses a pathogenic profile of adipocytokines in patients with cardiovascular disease. Cardiovasc Diabetol. (2006) 5:1. doi: 10.1186/1475-2840-5-1
49. Mazurek T, Zhang L, Zalewski A, Mannion JD, Diehl JT, Arafat H, et al. Human epicardial adipose tissue is a source of inflammatory mediators. Circulation. (2003) 108:2460–6. doi: 10.1161/01.CIR.0000099542.57313.C5
50. Salgado-Somoza A, Teijeira-Fernandez E, Rubio J, Couso E, Gonzalez-Juanatey JR, Eiras S. Coronary artery disease is associated with higher epicardial retinol-binding protein 4 (RBP4) and lower glucose transporter (GLUT) 4 levels in epicardial and subcutaneous adipose tissue. Clin Endocrinol. (2012) 76:51–8. doi: 10.1111/j.1365-2265.2011.04140.x
51. Nyman K, Graner M, Pentikainen MO, Lundbom J, Hakkarainen A, Siren R, et al. Cardiac steatosis and left ventricular function in men with metabolic syndrome. J Cardiovasc Magn Reson. (2013) 15:103. doi: 10.1186/1532-429X-15-103
52. Gaborit B, Kober F, Jacquier A, Moro PJ, Cuisset T, Boullu S, et al. Assessment of epicardial fat volume and myocardial triglyceride content in severely obese subjects: relationship to metabolic profile, cardiac function and visceral fat. Int J Obes. (2012) 36:422–30. doi: 10.1038/ijo.2011.117
53. Ng AC, Delgado V, Bertini M, van der Meer RW, Rijzewijk LJ, Hooi Ewe S, et al. Myocardial steatosis and biventricular strain and strain rate imaging in patients with type 2 diabetes mellitus. Circulation. (2010) 122:2538–44. doi: 10.1161/CIRCULATIONAHA.110.955542
54. Rajabi M, Kassiotis C, Razeghi P, Taegtmeyer H. Return to the fetal gene program protects the stressed heart: a strong hypothesis. Heart Fail Rev. (2007) 12:331–43. doi: 10.1007/s10741-007-9034-1
55. Han Y, Ferrari VA. Fatty heart and subclinical left ventricular dysfunction: rediscovery of a pathological metabolic process: with a twist. Circ Cardiovasc Imaging. (2013) 6:614–6. doi: 10.1161/CIRCIMAGING.113.000973
56. Koepp KE, Obokata M, Reddy YNV, Olson TP, Borlaug BA. Hemodynamic and functional impact of epicardial adipose tissue in heart failure with preserved ejection fraction. JACC Heart Fail. (2020) 8:657–66. doi: 10.1016/j.jchf.2020.04.016
57. Borlaug BA, Reddy YNV. The role of the pericardium in heart failure: implications for pathophysiology and treatment. JACC Heart Fail. (2019) 7:574–85. doi: 10.1016/j.jchf.2019.03.021
58. Obokata M, Olson TP, Reddy YNV, Melenovsky V, Kane GC, Borlaug BA. Haemodynamics, dyspnoea, and pulmonary reserve in heart failure with preserved ejection fraction. Eur Heart J. (2018) 39:2810–21. doi: 10.1093/eurheartj/ehy268
59. Cho DH, Joo HJ, Kim MN, Lim DS, Shim WJ, Park SM. Association between epicardial adipose tissue, high-sensitivity C-reactive protein and myocardial dysfunction in middle-aged men with suspected metabolic syndrome. Cardiovasc Diabetol. (2018) 17:95. doi: 10.1186/s12933-018-0735-7
60. Maimaituxun G, Kusunose K, Yamada H, Fukuda D, Yagi S, Torii Y, et al. Deleterious effects of epicardial adipose tissue volume on global longitudinal strain in patients with preserved left ventricular ejection fraction. Front Cardiovasc Med. (2020) 7:607825. doi: 10.3389/fcvm.2020.607825
61. Lebovitz HE, Banerji MA. Point: visceral adiposity is causally related to insulin resistance. Diabetes Care. (2005) 28:2322–5. doi: 10.2337/diacare.28.9.2322
62. Iacobellis G, Ribaudo MC, Assael F, Vecci E, Tiberti C, Zappaterreno A, et al. Echocardiographic epicardial adipose tissue is related to anthropometric and clinical parameters of metabolic syndrome: a new indicator of cardiovascular risk. J Clin Endocrinol Metab. (2003) 88:5163–8. doi: 10.1210/jc.2003-030698
63. Unnikrishnan AG, Babu T. A new marker of the metabolic syndrome is closer to the heart. Natl Med J India. (2004) 17:27–8.
64. Camarena V, Sant D, Mohseni M, Salerno T, Zaleski ML, Wang G, et al. Novel atherogenic pathways from the differential transcriptome analysis of diabetic epicardial adipose tissue. Nutr Metab Cardiovasc Dis. (2017) 27:739–50. doi: 10.1016/j.numecd.2017.05.010
65. Eroglu S. How do we measure epicardial adipose tissue thickness by transthoracic echocardiography? Anatol J Cardiol. (2015) 15:416–9. doi: 10.5152/akd.2015.5991
66. Doesch C, Streitner F, Bellm S, Suselbeck T, Haghi D, Heggemann F, et al. Epicardial adipose tissue assessed by cardiac magnetic resonance imaging in patients with heart failure due to dilated cardiomyopathy. Obesity. (2013) 21:E253–61. doi: 10.1002/oby.20149
67. Homsi R, Meier-Schroers M, Gieseke J, Dabir D, Luetkens JA, Kuetting DL, et al. 3D-Dixon MRI based volumetry of peri- and epicardial fat. Int J Cardiovasc Imaging. (2016) 32:291–9. doi: 10.1007/s10554-015-0778-8
68. Pieske B, Tschope C, de Boer RA, Fraser AG, Anker SD, Donal E, et al. How to diagnose heart failure with preserved ejection fraction: the HFA-PEFF diagnostic algorithm: a consensus recommendation from the Heart Failure Association (HFA) of the European Society of Cardiology (ESC). Eur Heart J. (2019) 40:3297–317. doi: 10.1093/eurheartj/ehz641
69. Bard A, Raisi-Estabragh Z, Ardissino M, Lee AM, Pugliese F, Dey D, et al. Automated quality-controlled cardiovascular magnetic resonance pericardial fat quantification using a convolutional neural network in the UK biobank. Front Cardiovasc Med. (2021) 8:677574. doi: 10.3389/fcvm.2021.677574
70. Ying W, Sharma K, Yanek LR, Vaidya D, Schar M, Markl M, et al. Visceral adiposity, muscle composition, and exercise tolerance in heart failure with preserved ejection fraction. ESC Heart Fail. (2021). 8:2535–45. doi: 10.1002/ehf2.13382
71. Faragli A, Alogna A, Lee CB, Zhu M, Ghorbani N, Lo Muzio FP, et al. Non-invasive CMR-based quantification of myocardial power and efficiency under stress and ischemic conditions in landrace pigs. Front Cardiovasc Med. (2021) 8:689255. doi: 10.3389/fcvm.2021.689255
72. Conte C, Esposito A, De Lorenzo R, Di Filippo L, Palmisano A, Vignale D, et al. Epicardial adipose tissue characteristics, obesity and clinical outcomes in COVID-19: a post-hoc analysis of a prospective cohort study. Nutr Metab Cardiovasc Dis. (2021) 31:2156–64. doi: 10.1016/j.numecd.2021.04.020
73. Jeong EM, Dudley SC Jr. Diastolic dysfunction. Circ J. (2015) 79:470–7. doi: 10.1253/circj.CJ-15-0064
74. Tschope C, Van Linthout S, Kherad B. Heart failure with preserved ejection fraction and future pharmacological strategies: a glance in the crystal ball. Curr Cardiol Rep. (2017) 19:70. doi: 10.1007/s11886-017-0874-6
75. Cilia L, Saeed A, Ganga HV, Wu WC. Heart failure with preserved ejection fraction: prevention and management. Am J Lifestyle Med. (2019) 13:182–9. doi: 10.1177/1559827617695219
76. Howard PA. Treating heart failure with preserved ejection fraction: a challenge for clinicians. Hosp Pharm. (2015) 50:454–9. doi: 10.1310/hpj5006-454
77. Atherton JJ, Moore TD, Lele SS, Thomson HL, Galbraith AJ, Belenkie I, et al. Diastolic ventricular interaction in chronic heart failure. Lancet. (1997) 349:1720–4. doi: 10.1016/S0140-6736(96)05109-4
78. Baigent C, Keech A, Kearney PM, Blackwell L, Buck G, Pollicino C, et al. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet. (2005) 366:1267–78. doi: 10.1016/S0140-6736(05)67394-1
79. Raggi P, Gadiyaram V, Zhang C, Chen Z, Lopaschuk G, Stillman AE. Statins reduce epicardial adipose tissue attenuation independent of lipid lowering: a potential pleiotropic effect. J Am Heart Assoc. (2019) 8:e013104. doi: 10.1161/JAHA.119.013104
80. Van Linthout S, Riad A, Dhayat N, Spillmann F, Du J, Dhayat S, et al. Anti-inflammatory effects of atorvastatin improve left ventricular function in experimental diabetic cardiomyopathy. Diabetologia. (2007) 50:1977–86. doi: 10.1007/s00125-007-0719-8
81. Ramasubbu K, Estep J, White DL, Deswal A, Mann DL. Experimental and clinical basis for the use of statins in patients with ischemic and nonischemic cardiomyopathy. J Am Coll Cardiol. (2008) 51:415–26. doi: 10.1016/j.jacc.2007.10.009
82. Fukuta H, Sane DC, Brucks S, Little WC. Statin therapy may be associated with lower mortality in patients with diastolic heart failure: a preliminary report. Circulation. (2005) 112:357–63. doi: 10.1161/CIRCULATIONAHA.104.519876
83. Alehagen U, Benson L, Edner M, Dahlstrom U, Lund LH. Association between use of statins and mortality in patients with heart failure and ejection fraction of >/=50. Circ Heart Fail. (2015) 8:862–70. doi: 10.1161/CIRCHEARTFAILURE.115.002143
84. Fukuta H, Goto T, Wakami K, Ohte N. The effect of statins on mortality in heart failure with preserved ejection fraction: a meta-analysis of propensity score analyses. Int J Cardiol. (2016) 214:301–6. doi: 10.1016/j.ijcard.2016.03.186
85. Liu G, Zheng XX, Xu YL, Ru J, Hui RT, Huang XH. Meta-analysis of the effect of statins on mortality in patients with preserved ejection fraction. Am J Cardiol. (2014) 113:1198–204. doi: 10.1016/j.amjcard.2013.12.023
86. Bielecka-Dabrowa A, Bytyci I, Von Haehling S, Anker S, Jozwiak J, Rysz J, et al. Association of statin use and clinical outcomes in heart failure patients: a systematic review and meta-analysis. Lipids Health Dis. (2019) 18:188. doi: 10.1186/s12944-019-1135-z
87. Park JH, Park YS, Kim YJ, Lee IS, Kim JH, Lee JH, et al. Effects of statins on the epicardial fat thickness in patients with coronary artery stenosis underwent percutaneous coronary intervention: comparison of atorvastatin with simvastatin/ezetimibe. J Cardiovasc Ultrasound. (2010) 18:121–6. doi: 10.4250/jcu.2010.18.4.121
88. Parisi V, Petraglia L, D'Esposito V, Cabaro S, Rengo G, Caruso A, et al. Statin therapy modulates thickness and inflammatory profile of human epicardial adipose tissue. Int J Cardiol. (2019) 274:326–30. doi: 10.1016/j.ijcard.2018.06.106
89. Nasarre L, Juan-Babot O, Gastelurrutia P, Llucia-Valldeperas A, Badimon L, Bayes-Genis A, et al. Low density lipoprotein receptor-related protein 1 is upregulated in epicardial fat from type 2 diabetes mellitus patients and correlates with glucose and triglyceride plasma levels. Acta Diabetol. (2014) 51:23–30. doi: 10.1007/s00592-012-0436-8
90. Roth EM, Davidson MH. PCSK9 inhibitors: mechanism of action, efficacy, and safety. Rev Cardiovasc Med. (2018) 19:S31−46.
91. Rivas Galvez RE, Morales Portano JD, Trujillo Cortes R, Gomez Alvarez EB, Sanchez Cubias SM, Zelaya SM. Reduction of epicardial adipose tissue thickness with PCSK9 inhibitors. Euro Heart J. (2020) (accessed November 2020), 41. doi: 10.1093/ehjci/ehaa946.3008
92. Tokubuchi I, Tajiri Y, Iwata S, Hara K, Wada N, Hashinaga T, et al. Beneficial effects of metformin on energy metabolism and visceral fat volume through a possible mechanism of fatty acid oxidation in human subjects and rats. PLoS ONE. (2017) 12:e0171293. doi: 10.1371/journal.pone.0171293
93. Koshizaka M, Ishikawa K, Ishibashi R, Takahashi S, Sakamoto K, Yokoh H, et al. Comparison of visceral fat reduction by ipragliflozin and metformin in elderly type 2 diabetes patients: sub-analysis of a randomized-controlled study. Diabetes Ther. (2021) 12:183–96. doi: 10.1007/s13300-020-00949-0
94. Halabi A, Sen J, Huynh Q, Marwick TH. Metformin treatment in heart failure with preserved ejection fraction: a systematic review and meta-regression analysis. Cardiovasc Diabetol. (2020) 19:124. doi: 10.1186/s12933-020-01100-w
95. Ziyrek M, Kahraman S, Ozdemir E, Dogan A. Metformin monotherapy significantly decreases epicardial adipose tissue thickness in newly diagnosed type 2 diabetes patients. Rev Port Cardiol. (2019) 38:419–23. doi: 10.1016/j.repc.2018.08.010
96. Gunes H, Gunes H, Ozmen S, Celik E, Temiz F. Effects of metformin on epicardial adipose tissue and atrial electromechanical delay of obese children with insulin resistance. Cardiol Young. (2020) 30:1429–32. doi: 10.1017/S1047951120002103
97. Gentilella R, Pechtner V, Corcos A, Consoli A. Glucagon-like peptide-1 receptor agonists in type 2 diabetes treatment: are they all the same? Diabetes Metab Res Rev. (2019) 35:e3070. doi: 10.1002/dmrr.3070
98. Del Olmo-Garcia MI, Merino-Torres JF. GLP-1 receptor agonists and cardiovascular disease in patients with type 2 diabetes. J Diabetes Res. (2018) 2018:4020492. doi: 10.1155/2018/4020492
99. Sheahan KH, Wahlberg EA, Gilbert MP. An overview of GLP-1 agonists and recent cardiovascular outcomes trials. Postgrad Med J. (2020) 96:156–61. doi: 10.1136/postgradmedj-2019-137186
100. Bizino MB, Jazet IM, Westenberg JJM, van Eyk HJ, Paiman EHM, Smit JWA, et al. Effect of liraglutide on cardiac function in patients with type 2 diabetes mellitus: randomized placebo-controlled trial. Cardiovasc Diabetol. (2019) 18:55. doi: 10.1186/s12933-019-0905-2
101. Ida S, Kaneko R, Imataka K, Okubo K, Shirakura Y, Azuma K, et al. Effects of oral antidiabetic drugs and glucagon-like peptide-1 receptor agonists on left ventricular diastolic function in patients with type 2 diabetes mellitus: a systematic review and network meta-analysis. Heart Fail Rev. (2020). 26:1151–8. doi: 10.1007/s10741-020-09936-w
102. Iacobellis G, Camarena V, Sant DW, Wang G. Human epicardial fat expresses glucagon-like peptide 1 and 2 receptors genes. Horm Metab Res. (2017) 49:625–30. doi: 10.1055/s-0043-109563
103. Iacobellis G, Villasante Fricke AC. Effects of semaglutide versus dulaglutide on epicardial fat thickness in subjects with type 2 diabetes and obesity. J Endocr Soc. (2020) 4:bvz042. doi: 10.1210/jendso/bvz042
104. Iacobellis G, Mohseni M, Bianco SD, Banga PK. Liraglutide causes large and rapid epicardial fat reduction. Obesity. (2017) 25:311–6. doi: 10.1002/oby.21718
105. Yang J, Ren J, Song J, Liu F, Wu C, Wang X, et al. Glucagon-like peptide 1 regulates adipogenesis in 3T3-L1 preadipocytes. Int J Mol Med. (2013) 31:1429–35. doi: 10.3892/ijmm.2013.1350
106. Beiroa D, Imbernon M, Gallego R, Senra A, Herranz D, Villarroya F, et al. GLP-1 agonism stimulates brown adipose tissue thermogenesis and browning through hypothalamic AMPK. Diabetes. (2014) 63:3346–58. doi: 10.2337/db14-0302
107. Smith NK, Hackett TA, Galli A, Flynn CR. GLP-1: molecular mechanisms and outcomes of a complex signaling system. Neurochem Int. (2019) 128:94–105. doi: 10.1016/j.neuint.2019.04.010
108. Lima-Martinez MM, Paoli M, Rodney M, Balladares N, Contreras M, D'Marco L, et al. Effect of sitagliptin on epicardial fat thickness in subjects with type 2 diabetes and obesity: a pilot study. Endocrine. (2016) 51:448–55. doi: 10.1007/s12020-015-0710-y
109. Aroor A, McKarns S, Nistala R, DeMarco V, Gardner M, Garcia-Touza M, et al. DPP-4 inhibitors as therapeutic modulators of immune cell function and associated cardiovascular and renal insulin resistance in obesity and diabetes. Cardiorenal Med. (2013) 3:48–56. doi: 10.1159/000348756
110. Shah Z, Kampfrath T, Deiuliis JA, Zhong J, Pineda C, Ying Z, et al. Long-term dipeptidyl-peptidase 4 inhibition reduces atherosclerosis and inflammation via effects on monocyte recruitment and chemotaxis. Circulation. (2011) 124:2338–49. doi: 10.1161/CIRCULATIONAHA.111.041418
111. Tomovic K, Lazarevic J, Kocic G, Deljanin-Ilic M, Anderluh M, Smelcerovic A. Mechanisms and pathways of anti-inflammatory activity of DPP-4 inhibitors in cardiovascular and renal protection. Med Res Rev. (2019) 39:404–22. doi: 10.1002/med.21513
112. Sakata K, Hayakawa M, Yano Y, Tamaki N, Yokota N, Eto T, et al. Efficacy of alogliptin, a dipeptidyl peptidase-4 inhibitor, on glucose parameters, the activity of the advanced glycation end product (AGE) - receptor for AGE (RAGE) axis and albuminuria in Japanese type 2 diabetes. Diabetes Metab Res Rev. (2013) 29:624–30. doi: 10.1002/dmrr.2437
113. Sato N, Nakamura Y, Yamadera S, Inagaki M, Kenmotsu S, Saito H, et al. Linagliptin inhibits lipopolysaccharide-induced inflammation concentration-dependently and -independently. J Inflamm Res. (2019) 12:285–91. doi: 10.2147/JIR.S221761
114. Ishibashi Y, Matsui T, Maeda S, Higashimoto Y, Yamagishi S. Advanced glycation end products evoke endothelial cell damage by stimulating soluble dipeptidyl peptidase-4 production and its interaction with mannose 6-phosphate/insulin-like growth factor II receptor. Cardiovasc Diabetol. (2013) 12:125. doi: 10.1186/1475-2840-12-125
115. Dobrian AD, Ma Q, Lindsay JW, Leone KA, Ma K, Coben J, et al. Dipeptidyl peptidase IV inhibitor sitagliptin reduces local inflammation in adipose tissue and in pancreatic islets of obese mice. Am J Physiol Endocrinol Metab. (2011) 300:E410–21. doi: 10.1152/ajpendo.00463.2010
116. McMurray JJV, Ponikowski P, Bolli GB, Lukashevich V, Kozlovski P, Kothny W, et al. Effects of vildagliptin on ventricular function in patients with type 2 diabetes mellitus and heart failure: a randomized placebo-controlled trial. JACC Heart Fail. (2018) 6:8–17. doi: 10.1016/j.jchf.2017.08.004
117. Mulvihill EE, Varin EM, Ussher JR, Campbell JE, Bang KW, Abdullah T, et al. Inhibition of dipeptidyl peptidase-4 impairs ventricular function and promotes cardiac fibrosis in high fat-fed diabetic mice. Diabetes. (2016) 65:742–54. doi: 10.2337/db15-1224
118. Packer M. Have dipeptidyl peptidase-4 inhibitors ameliorated the vascular complications of type 2 diabetes in large-scale trials? The potential confounding effect of stem-cell chemokines. Cardiovasc Diabetol. (2018) 17:9. doi: 10.1186/s12933-017-0648-x
119. Zinman B, Lachin JM, Inzucchi SE. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N Engl J Med. (2016) 374:1094. doi: 10.1056/NEJMc1600827
120. Packer M, Anker SD, Butler J, Filippatos G, Pocock SJ, Carson P, et al. Cardiovascular and renal outcomes with empagliflozin in heart failure. N Engl J Med. (2020) 383:1413–24. doi: 10.1056/NEJMoa2022190
121. Kalra S. Sodium glucose co-transporter-2 (SGLT2) inhibitors: a review of their basic and clinical pharmacology. Diabetes Ther. (2014) 5:355–66. doi: 10.1007/s13300-014-0089-4
122. Bolinder J, Ljunggren O, Kullberg J, Johansson L, Wilding J, Langkilde AM, et al. Effects of dapagliflozin on body weight, total fat mass, and regional adipose tissue distribution in patients with type 2 diabetes mellitus with inadequate glycemic control on metformin. J Clin Endocrinol Metab. (2012) 97:1020–31. doi: 10.1210/jc.2011-2260
123. Cai X, Ji L, Chen Y, Yang W, Zhou L, Han X, et al. Comparisons of weight changes between sodium-glucose cotransporter 2 inhibitors treatment and glucagon-like peptide-1 analogs treatment in type 2 diabetes patients: a meta-analysis. J Diabetes Investig. (2017) 8:510–7. doi: 10.1111/jdi.12625
124. Santos-Gallego CG, Requena-Ibanez JA, San Antonio R, Garcia-Ropero A, Ishikawa K, Watanabe S, et al. Empagliflozin ameliorates diastolic dysfunction and left ventricular fibrosis/stiffness in nondiabetic heart failure: a multimodality study. JACC Cardiovasc Imaging. (2021) 14:393–407. doi: 10.1016/j.jcmg.2020.07.042
125. Pabel S, Wagner S, Bollenberg H, Bengel P, Kovacs A, Schach C, et al. Empagliflozin directly improves diastolic function in human heart failure. Eur J Heart Fail. (2018) 20:1690–700. doi: 10.1002/ejhf.1328
126. Kolijn D, Pabel S, Tian Y, Lodi M, Herwig M, Carrizzo A, et al. Empagliflozin improves endothelial and cardiomyocyte function in human heart failure with preserved ejection fraction via reduced pro-inflammatory-oxidative pathways and protein kinase Galpha oxidation. Cardiovasc Res. (2021) 117:495–507. doi: 10.1093/cvr/cvaa123
127. Bhatt DL, Szarek M, Steg PG, Cannon CP, Leiter LA, McGuire DK, et al. Sotagliflozin in patients with diabetes and recent worsening heart failure. N Engl J Med. (2021) 384:117–28. doi: 10.1056/NEJMoa2030183
128. Solomon SD, de Boer RA, DeMets D, Hernandez AF, Inzucchi SE, Kosiborod MN, et al. Dapagliflozin in heart failure with preserved and mildly reduced ejection fraction: rationale and design of the DELIVER trial. Eur J Heart Fail. (2021) 23:1217–25. doi: 10.1002/ejhf.2249
129. Iacobellis G, Gra-Menendez S. Effects of dapagliflozin on epicardial fat thickness in patients with type 2 diabetes and obesity. Obesity. (2020) 28:1068–74. doi: 10.1002/oby.22798
130. Diaz-Rodriguez E, Agra RM, Fernandez AL, Adrio B, Garcia-Caballero T, Gonzalez-Juanatey JR, et al. Effects of dapagliflozin on human epicardial adipose tissue: modulation of insulin resistance, inflammatory chemokine production, and differentiation ability. Cardiovasc Res. (2018) 114:336–46. doi: 10.1093/cvr/cvx186
131. Borlaug BA, Schaff HV, Pochettino A, Pedrotty DM, Asirvatham SJ, Abel MD, et al. Pericardiotomy enhances left ventricular diastolic reserve with volume loading in humans. Circulation. (2018) 138:2295–7. doi: 10.1161/CIRCULATIONAHA.118.036006
132. Borlaug BA, Carter RE, Melenovsky V, DeSimone CV, Gaba P, Killu A, et al. Percutaneous pericardial resection: a novel potential treatment for heart failure with preserved ejection fraction. Circ Heart Fail. (2017) 10:e003612. doi: 10.1161/CIRCHEARTFAILURE.116.003612
133. Van Linthout S, Tschope C. The quest for antiinflammatory and immunomodulatory strategies in heart failure. Clin Pharmacol Ther. (2019) 106:1198–208. doi: 10.1002/cpt.1637
134. Van Linthout S, Tschope C. Inflammation - cause or consequence of heart failure or both? Curr Heart Fail Rep. (2017) 14:251–65. doi: 10.1007/s11897-017-0337-9
135. Baker AR, Harte AL, Howell N, Pritlove DC, Ranasinghe AM, da Silva NF, et al. Epicardial adipose tissue as a source of nuclear factor-kappaB and c-Jun N-terminal kinase mediated inflammation in patients with coronary artery disease. J Clin Endocrinol Metab. (2009) 94:261–7. doi: 10.1210/jc.2007-2579
136. Elsanhoury A, Tschope C, Van Linthout S. A toolbox of potential immune-related therapies for inflammatory cardiomyopathy. J Cardiovasc Transl Res. (2021) 14:75–87. doi: 10.1007/s12265-020-10025-4
137. Van Tassell BW, Arena R, Biondi-Zoccai G, Canada JM, Oddi C, Abouzaki NA, et al. Effects of interleukin-1 blockade with anakinra on aerobic exercise capacity in patients with heart failure and preserved ejection fraction (from the D-HART pilot study). Am J Cardiol. (2014) 113:321–7. doi: 10.1016/j.amjcard.2013.08.047
138. Van Tassell BW, Trankle CR, Canada JM, Carbone S, Buckley L, Kadariya D, et al. IL-1 blockade in patients with heart failure with preserved ejection fraction. Circ Heart Fail. (2018) 11:e005036. doi: 10.1161/CIRCHEARTFAILURE.118.005036
Keywords: heart failure with a preserved ejection fraction, epicardiac adipose tissue, diabetes, obesity, SGLT2, inhibitor, GLP-1 agonists
Citation: Elsanhoury A, Nelki V, Kelle S, Van Linthout S and Tschöpe C (2021) Epicardial Fat Expansion in Diabetic and Obese Patients With Heart Failure and Preserved Ejection Fraction—A Specific HFpEF Phenotype. Front. Cardiovasc. Med. 8:720690. doi: 10.3389/fcvm.2021.720690
Received: 08 June 2021; Accepted: 09 August 2021;
Published: 17 September 2021.
Edited by:
Kristin Stanford, The Ohio State University, United StatesReviewed by:
Giuseppe Danilo Norata, University of Milan, ItalyCopyright © 2021 Elsanhoury, Nelki, Kelle, Van Linthout and Tschöpe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carsten Tschöpe, Y2Fyc3Rlbi50c2Nob2VwZUBjaGFyaXRlLmRl
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.