 Yan Huang1,2,3
Yan Huang1,2,3 Hua-Sheng Ding1,2,3
Hua-Sheng Ding1,2,3 Tao Song1,2,3Yu-Ting Chen2Teng Wang2Yan-Hong Tang1,2,3Hector Barajas-Martinez4,5Cong-Xin Huang1,2,3*
Tao Song1,2,3Yu-Ting Chen2Teng Wang2Yan-Hong Tang1,2,3Hector Barajas-Martinez4,5Cong-Xin Huang1,2,3* Dan Hu1,2,3*
Dan Hu1,2,3*- 1Department of Cardiology, Renmin Hospital of Wuhan University, Wuhan, China
- 2Cardiovascular Research Institute, Wuhan University, Wuhan, China
- 3Hubei Key Laboratory of Cardiology, Wuhan, China
- 4Lankenau Institute for Medical Research, Lankenau Heart Institute, Wynnwood, PA, United States
- 5Jefferson Medical College, Philadelphia, PA, United States
Introduction: Myocardial infarction (MI) triggers structural and electrical remodeling. CC chemokine receptor 9 (CCR9) mediates chemotaxis of inflammatory cells in MI. In our previous study, CCR9 knockout has been found to improve structural remodeling after MI. Here, we further investigate the potential influence of CCR9 on electrical remodeling following MI in order to explore potential new measures to improve the prognosis of MI.
Methods and Results: Mice was used and divided into four groups: CCR9+/+/Sham, CCR9−/−/Sham, CCR9+/+/MI, CCR9−/−/MI. Animals were used at 1 week after MI surgery. Cardiomyocytes in the infracted border zone were acutely dissociated and the whole-cell patch clamp was used to record action potential duration (APD), L-type calcium current (ICa,L) and transient outward potassium current (Ito). Calcium transient and sarcoplasmic reticulum (SR) calcium content under stimulation of Caffeine were measured in isolated cardiomyocytes by confocal microscopy. Multielectrode array (MEA) was used to measure the conduction of the left ventricle. The western-blot was performed for the expression level of connexin 43. We observed prolonged APD90, increased ICa,L and decreased Ito following MI, while CCR9 knockout attenuated these changes (APD90: 50.57 ± 6.51 ms in CCR9−/−/MI vs. 76.53 ± 5.98 ms in CCR9+/+/MI, p < 0.05; ICa,L: −13.15 ± 0.86 pA/pF in CCR9−/−/MI group vs. −17.05 ± 1.11 pA/pF in CCR9+/+/MI, p < 0.05; Ito: 4.01 ± 0.17 pA/pF in CCR9−/−/MI group vs. 2.71 ± 0.16 pA/pF in CCR9+/+/MI, p < 0.05). The confocal microscopy results revealed CCR9 knockout reversed the calcium transient and calcium content reduction in sarcoplasmic reticulum following MI. MEA measurements showed improved conduction velocity in CCR9−/−/MI mice (290.1 ± 34.47 cm/s in CCR9−/−/MI group vs. 113.2 ± 14.4 cm/s in CCR9+/+/MI group, p < 0.05). Western-blot results suggested connexin 43 expression was lowered after MI while CCR9 knockout improved its expression.
Conclusion: This study shows CCR9 knockout prevents the electrical remodeling by normalizing ion currents, the calcium homeostasis, and the gap junction to maintain APD and the conduction function. It suggests CCR9 is a promising therapeutic target for MI-induced arrhythmia, which warrants further investigation.
Introduction
Myocardial infarction (MI) is a disease of continuous ischemia and necrosis of cardiomyocytes caused by thrombus formation due to coronary atherosclerosis and rupture of vascular plaques, resulting in acute vascular occlusion (1). It brings a huge burden to the national economy and seriously affects the quality of life of patients. It is estimated that globally, ischemic heart disease affects around 126 million individuals (1,655 per 100,000), which is approximately 1.72% of the world's population. Nine million deaths were caused by IHD globally. By the year 2030, the prevalence rate is expected to exceed 1,845 per 100,000 population. The cost of MI is expected to account for 1–1.5% of the gross domestic product in countries like the United States (2). Cardiac remodeling including structural and electrical remodeling occurs after MI, which lead to hypertrophy, heart failure, arrhythmias as well as sudden cardiac death (SCD) (3). And ventricular arrhythmia is the leading cause responsible for SCD after MI (4). Electrical remodeling, including changes in ion channel currents and action potentials (APs) by myocardial conduction disorder, is pathological basis of arrhythmias following MI. The changes of repolarization currents, such as reduction in potassium current, including the transient outward current (Ito), the inward rectifier potassium current (IK1), as well as the slow delayed rectifier potassium current (IKs), could lead to the prolongation of ventricular action potential duration (APD), and increased dispersion of ventricular repolarization, which are potential mechanisms for ventricular arrhythmias and SCD (5). As far, targeted drugs and various interventional techniques are the main clinical treatments to improve patient outcomes following MI. However, potential adverse reactions and poor prognosis cannot be totally resolved (6–8). Therefore, noninvasive, nonpharmacological, and clinically applicable technical approaches are being explored, such as stem and progenitor cell therapy or genetic therapy, since these promising strategies are specifically targeted and without drug toxicity compared with traditional means (5, 9, 10).
Chemokine receptors are mainly expressed on inflammatory cells like neutrophils, monocytes and macrophages. They also exist on other cells including endothelial cells and tumor cells (11). Chemokines as well as chemokine receptors play an important role in cardiovascular diseases: chemokine receptor 2 (CCR2)/chemokine ligand 2 (CCL2) participate in atherosclerosis and pathophysiology in MI. Inhibition of CCL2/CCR2 by using inhibitors or genetic technology can reduce inflammation and inhibit detrimental ventricular remodeling (12). Chemokine ligand 21 (CCL21)/chemokine receptor 7 (CCR7) play an important role in MI, neutralization antibody of CCL21 can improve ventricular remodeling by reducing infarct size and suppressing collagen content in myocardium after acute MI (13). Development of atherosclerosis needs the chemokine receptor 1 (CCR1)/chemokine receptor 5 (CCR5) to recruit monocytes (14). Thymus-expressed chemokine (CCL25) is the only known ligand for CC chemokine receptor 9 (CCR9). Like other chemokine receptors, CCR9 is also detected on lymphocytes, monocytes, macrophages and dendritic cells (DCs) (15). CCR9 was found to be involved in inflammatory diseases like inflammatory bowel disease, rheumatoid arthritis, and cancer (16–20). In cardiovascular system, only 2 literatures reported the relationship between CCR9 and heart diseases (21, 22). In the previous study, we found CCR9 abrogation could improve ventricular structural remodeling by reducing infract size, inhibiting inflammation and fibrosis after MI, thus first reported CCR9 plays a crucial role in MI (21). In the other study, CCR9 was found to be related to pathological myocardial hypertrophy (22). Our present study is aimed to use CCR9 gene knockout mice to explore the effect of CCR9 in ventricular electrical remodeling following MI, and to discover novel methods that can prevent deteriorating outcomes after MI.
Materials and Methods
Experimental Mice
Global CCR9 knockout mice (CCR9-KO, C57BL/6J background) were purchased from the European Mouse Mutant Archive (EM:02293. B6;129-Ccr9tm1Dgen/H). The mice were bred and kept in our animal house with specific pathogen-free environment in Renmin hospital of Wuhan university. Male mice with an age of 6–8 weeks (body weight 24–27g) were used in our study. All the protocols in our experiment were authorized by the Animal Care and Use Committee of Renmin Hospital of Wuhan University. The pain and suffering of animals were reduced to minimize during the experiments.
Left Coronary Artery Ligation
The surgery was conducted as described before (21). In short, mice were anesthetized with sodium pentobarbital (intraperitoneal injection, 50 mg/kg). After getting anesthesia, small animal ventilator was used to maintain normal breathing. For subjects in the MI groups, the chest was opened, the heart was fully exposed, and the left anterior descending coronary was ligated. The mice in sham groups underwent the pericardium opening as well but without ligation. The mice were divided into four group: wild type sham group (CCR9+/+/Sham), CCR9 knockout sham group (CCR9−/−/Sham), wild type MI group (CCR9+/+/MI), and CCR9 knockout MI group (CCR9−/−/MI). The animals were used for experiments at 1 week following MI surgery.
Isolation of Single Ventricular Cardiomyocytes
After the mice were anesthetized by sodium pentobarbital, the heart was quickly removed as soon as the chest was opened, then soaked in the iced Ca2+-free Tyrode's solution and appendages were pruned. The ascending aorta cannulation was quickly performed and perfused the heart with Ca2+-free Tyrode's solution for around 5 mins, followed by enzymatic solution for 6–7 mins until the full digestion was achieved. The heart was then transferred to Ca2+-free Tyrode's solution containing BSA (1 mg/ml) to stop digestion. Then, the left ventricle was cut out and the infarct border zone was reserved and dispersed into single cell with a polished Pasteur pipette. The cells were kept in the BSA solution at room temperature after the cell suspension was filtered.
Patch Clamp Recording
Since APD change in the perfused heart was observed in both CCR9+/+/MI mice and CCR9−/−/MI mice in our previous study, we wondered whether there were cardiac ionic currents alternation in single cardiomyocytes. Patch-clamp analyses were performed using acutely isolated ventricular myocytes from the infarcted border zone. Action potential duration (APD), transient outward potassium current (Ito) and L-type calcium current (ICa,L) were recorded by whole-cell patch-clamp. The glass microelectrodes were pulled and the microelectrode resistance ranges from 1 to 3 MΩ. A gentle negative pressure was given to rupture the cell membrane. The pipette solution for AP recording was (mmol/L): NaCl 5; KCl 15; K-glutamate 130; MgCl2 21; MgATP 5; CaCl2 1; HEPES 10; EGTA 5, and Tyrode's solution was used as the extracellular solution. ICa,L was measured with bath solution (mmol/L: CsCl 20; TEA-Cl 135; MgCl2 0.5; HEPES 5; CaCl2 1.8) and pipette solution (mmol/L: CsOH 110; CsCl 20; TEACl 10; Aspartate 90; HEPES 10; EGTA 10; MgATP 5; creatine phosphate sodium 5; GTP 0.4; leupeptin 0.1). For ICa,L recording, a slow voltage ramp with holding potential ranging from −70 to −45 mV was applied to inactivate sodium and T-type calcium current, and then voltage was increased to +65 mV step by step in 10 mV increments. The bath solution for Ito was (mmol/L): NaCl 138; MgCl2 1.0; KCl 5.4; CaCl2 1.8; HEPES 10; Glucose 10; nifedipine 0.02 and pipette solution (mmol/L) was: KOH 130; KCl 15; MgATP 5; CaCl2 1; L-glutamic acid 130; MgCl2 1; NaCl 5; EGTA 5; HEPES 10. Ito was measured with a holding potential −80 mV and using a depolarizing voltage step to +70 mV from a 40 ms pre-pulse of −40 mV to inactivate the sodium channel.
Calcium Image
Isolated ventricular myocytes from infarct border zone were centrifuged (1,200 rpm, 3 min). The supernatant was removed, and the precipitation was resuspended in Tyrode's solution in a 1.5 ml centrifuge tube. Then 10 μM Fluo 4-AM was used to incubate the cells without light for 1 h at 37°C. After incubation, the cells were washed by using Tyrode's solution for three times. Then, cells were kept on the ice and calcium transients were measured using a dual-beam excitation fluorescence photometry setup (Leica, Wetzlar, Germany). The emission wavelength and receiving wavelength for recording was 460–480 nm and 500–550 nm, respectively. A home-made cell groove with two platinum wires at one end was used to pass the electrical stimulation to the cells. The cells were given a field stimulation, of which the stimulus frequency was 0.5 and 1 Hz. The stimulation was repeated six times. After recording Ca2+ transient in both stimulation frequency, we switched the perfusion to normal Tyrode's solution with 10 mM caffeine to empty Ca2+ storage in sarcoplasmic reticulum (SR) to record the SR calcium content.
Multielectrode Array (MEA)
MEA measurements were made in vitro mouse heart 1 week after MI. The preparation of the isolated perfused heart was as above. The isolated heart was perfused with 37°C Tyrode's solution. After recovery of the spontaneous rhythms, the MEA dish containing 32 monopolar electrodes (32 Map) with interelectrode distance of 500 μm to cover a 3 × 3 mm square was placed on left ventricle. Electrical stimulation with the stimulating voltage ranging from 1V to 7V and a duration of 1 ms was applied. Cardio2D+ software (Multi Channel Systems) was used to acquire the data sampled at 10 kHz. Activation maps were produced and the conduction velocity (CV) was calculated by analyzing the whole data.
Western Blot
Structural remodeling occurs after MI, which can lead to changes in gap junctions among cardiac myocytes. In the previous study, we observed CCR9 knockout could improve these serious structural remodeling, so we wondered whether CCR9 knockout could affect these changes in gap junction. Here, western blot was applied to detect the expression level of connexin 43 (Cx43) in ventricular tissue from infracted border zone. The mice were sacrificed to dissect the LV infract border zone. The dissected tissues were grinned to extracted proteins and quantified protein concentrations by BCA assay kit. Forty microgram protein sample was loaded and proteins were separated by 10% sodium dodecylsulphate (SDS)-polyacrylamide gel electrophoresis (PAGE), then transferred to a polyvinylidene difluoride (PVDF) membrane. After the transferring, the PVDF membrane was removed and transferred into sealing solution for mild vibration at 4°C overnight with anti-Cx43 primary antibody (1:1,000, abcam, ab235585) followed by rabbit IgG (1:2,000, abcam, ab6721) for 1 h at room temperature. Development and photographic recording were performed with Bio-RDA gel imaging system.
Statistical Analysis
All data were presented as the means ± SEM. Unpaired student's t-test or one-way analysis of variance (ANOVA) was used to make comparisons between two groups by using graphPad Prism version 5. Chi-Square Test with Fisher's exact test was used for the categorical variables. P ≤ 0.05 was considered significant statistically.
Results
CCR9 Knock Down Inhibits APD Prolongation Following MI
AP was recorded in isolated left ventricular cardiomyocytes around the infraction area using the whole cell patch clamp. MI markly prolonged the APD90 (76.53 ± 5.98 ms in CCR9+/+/MI, n = 6 cells from 3 hearts, vs. 36.91 ± 4.64 ms in CCR9+/+/Sham, n = 11 cells from 3 hearts, p < 0.05). Loss of CCR9 inhibited the plongation of APD induced by MI (50.57 ± 6.51 ms in CCR9−/−/MI, n = 6 cells from 3 hearts vs. 76.53 ± 5.98 ms in CCR9+/+/MI, n = 6 cells from 3 hearts, p < 0.05; Figure 1).

Figure 1. CCR9 knock down inhibits APD prolongation following MI. (A) Representive action potential recordings in mouse ventricular myocytes from each group. (B) Quantified analysis of APD90 in CCR9+/+ and CCR9−/− mice with sham operation or MI surgery at 1 week (*p < 0.05 vs. CCR9+/+/Sham, #p < 0.05 vs. CCR9+/+/MI).
CCR9 Knockout Attenuates MI-Induced Calcium Current Increase and Reduction of Transient Outward Potassium Current After MI
The original ICa,L traces for isolated ventricular myocytes from the infarcted border zone among different groups were shown in Figure 2A. MI markedly increased the amplitude of ICa,L, which can be reduced by CCR9 knockout. The current density-voltage (I-V) correlations for the ICa,L of each group were shown in Figure 2B, which showed that MI significantly increased the peak current when compared with the WT groups (−17.05 ± 1.11 pA/pF in CCR9+/+/MI group, n = 8 cells from 4 hearts vs. −10.02 ± 0.83 pA/pF in CCR9+/+/Sham, n = 9 cells from 3 hearts, p < 0.01; −13.15 ± 0.86 pA/pF in CCR9−/−/MI group, n = 10 cells from 4 hearts vs. −8.76 ± 0.49 pA/pF in CCR9−/−/Sham, n = 9 cells from 3 hearts, p < 0.01), while CCR9 knockout reduced the current density compared with CCR9+/+/MI (−13.15 ± 0.86 pA/pF in CCR9−/−/MI group, n = 10 cells from 4 hearts vs. −17.05 ± 1.11 pA/pF in CCR9+/+/MI, n = 8 cells from 4 hearts, p < 0.05). Besides, we also recorded Ito, the results showed the peak Ito was significantly reduced in MI mice (+60 mV: 2.71 ± 0.16 pA/pF in CCR9+/+/MI group, n = 8 cells from 4 hearts vs. 7.95 ± 0.31 pA/pF in CCR9+/+/Sham, n = 8 cells from 3 hearts, p < 0.05; 4.01 ± 0.17 pA/pF in CCR9−/−/MI group, n = 8 cells from 4 hearts vs. 8.03 ± 0.35 pA/pF in CCR9−/−/Sham, n = 6 cells from 3 hearts, p < 0.05), while CCR9 deficiency notably attenuated MI-induced Ito reduction (4.01 ± 0.17 pA/pF in CCR9−/−/MI group, n = 8 cells from 4 hearts vs. 2.71 ± 0.16 pA/pF in CCR9+/+/MI, n = 8 cells from 4 hearts, p < 0.05, Figures 2C,D). These data showed the MI induced ICa,L increasing and Ito decreasing, while absence of CCR9 can inhibit these ionic currents changing, therefore prevented APD prolongation following MI.
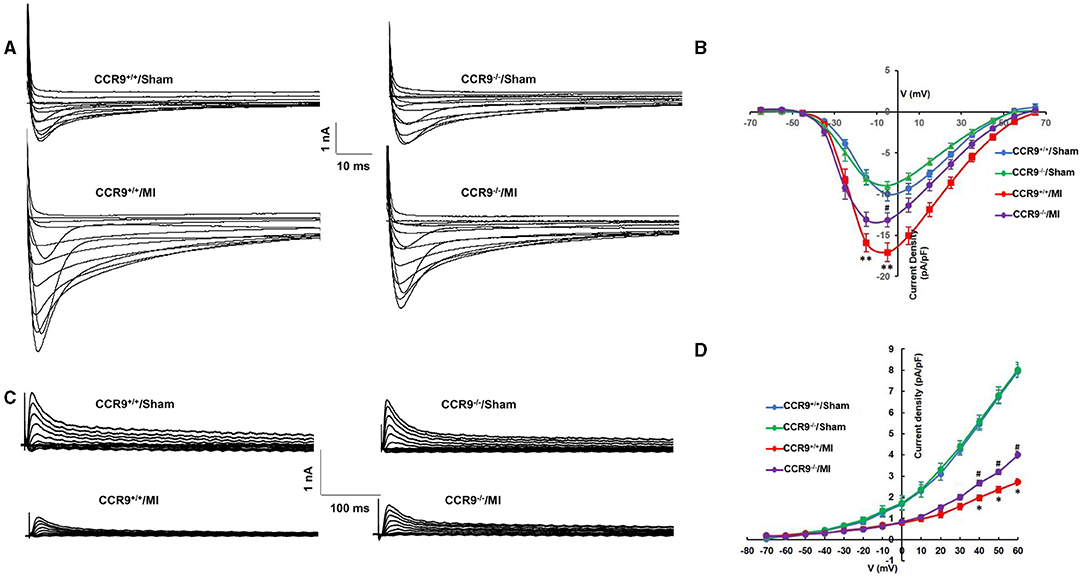
Figure 2. CCR9 knockoutattenuates MI-induced calcium current increasing and reduction of transient outward potassium current after MI. (A) Representive raw traces of ICa,L in mouse ventricular myocytes from each group. (B) Normalized current–voltage relationship for ICa,L (**p < 0.01 vs. CCR9+/+/Sham, #p < 0.05 vs. CCR9+/+/MI). (C) Representive raw traces of Ito in ventricular myocytes from each group. (D) Normalized current–voltage relationship for Ito (*p < 0.05 vs. CCR9+/+/Sham, #p < 0.05 vs. CCR9+/+/MI).
CCR9 Deficiency Restores Calcium Handling After MI
Arrhythmia following MI is associated with calcium homeostasis disorder. We further carried out the calcium image experiment to observe calcium handling in the ventricular cardiomycytes following MI. As shown in Figures 3A,B, MI induced the Ca2+ transient amplitudes decreased in both 0.5 and 1 Hz, but was only statistically significant in 1 Hz (0.53 ± 0.09 in CCR9+/+/MI group, n = 7 cells from 4 hearts, vs. 0.95 ± 0.05 in CCR9+/+/Sham group, n = 9 cells from 3 hearts, p < 0.05; 0.86 ± 0.08 in CCR9−/−/MI group, n = 13 cells from 4 hearts vs. 1.14 ± 0.11 in CCR9−/−/Sham group, n = 12 cells from 3 hearts, p > 0.05). Loss of CCR9 could restore calcium transient amplitude in 1 Hz (0.86 ± 0.08 in CCR9−/−/MI group, n = 13 cells from 4 hearts vs. 0.53 ± 0.09 in CCR9+/+/MI group, n = 7 cells from 4 hearts, p < 0.05). No difference was observed in Ca2+ transient amplitudes between 0.5 and 1 Hz. The SR calcium content experiment revealed decreased SR Ca2+content after MI in CCR9+/+/MI mice but not the CCR9−/−/MI mice (0.90 ± 0.11 in CCR9+/+/MI group, n = 8 cells from 4 hearts vs. 1.23 ± 0.10 in CCR9+/+/Sham group, n = 13 cells from 3 hearts, p = 0.05; 1.24 ± 0.09 in CCR9−/−/MI group, n = 11 cells from 4 hearts vs. 1.37 ± 0.12 in CCR9−/−/Sham group, n = 11 cells from 3 hearts, p > 0.05). CCR9 deficiency restored SR calcium content (1.24 ± 0.09 in CCR9−/−/MI group, n = 11 cells from 4 hearts vs. 0.90 ± 0.11 in CCR9+/+/MI group, n = 8 cells from 4 hearts, p < 0.05, Figure 3C). On the whole, these results indicate that CCR9 deficiency restores abnormal SR Ca2+ storage and Ca2+ release caused by MI.
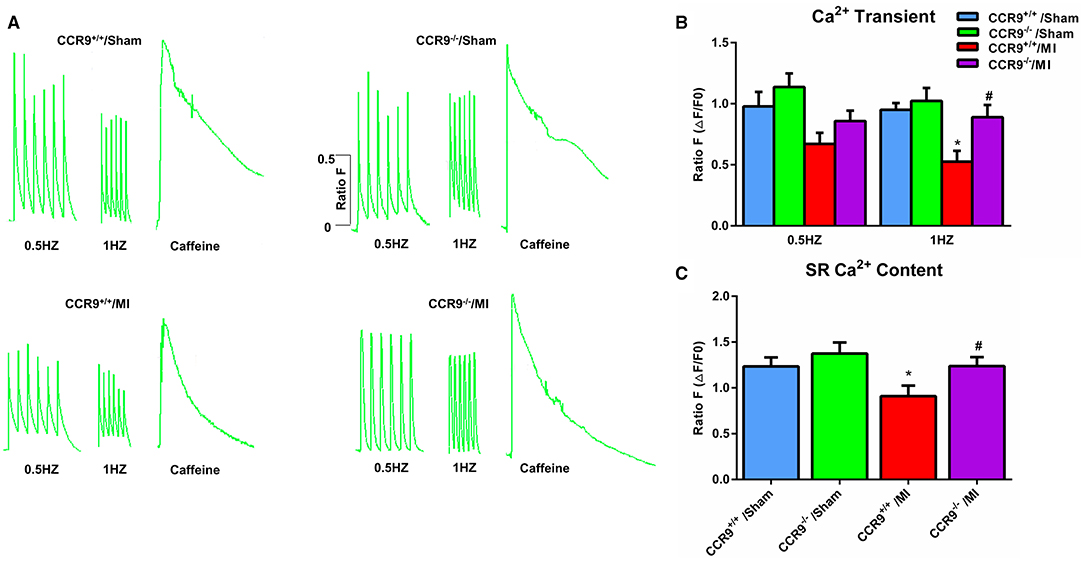
Figure 3. CCR9 deficiency restores calcium handling after MI. (A) Representive Ca2+transients and SR Ca2+content recordings at 0.5 and 1-Hz field stimulation in mouse myocytes from each group. (B) Statistical analysis of Ca2+transient amplitude (ΔF/F0) in each groups (*p < 0.05 vs. CCR9+/+/Sham, #p < 0.05 vs. CCR9+/+/MI). (C) Quantified analysis of SR Ca2+content following 10 mM caffeine perfusion in different groups (*p < 0.05 vs. CCR9+/+/Sham, #p < 0.05 vs. CCR9+/+/MI).
CCR9 Knockout Improves the Conduction Function After MI
Conduction activation maps (Figure 4A) as well as CV maps (Figure 4B) on anterior wall of left ventricle surface diaplayed more crowded isochrones and a slower CV in the MI group than that in the sham group (113.2 ± 14.4 cm/s in CCR9+/+/MI group, n = 5 hearts vs. 636.2 ± 112.1 cm/s in CCR9+/+/sham group, n = 5 hearts, p < 0.01; 290.1 ± 34.47 cm/s in CCR9−/−/MI group, n = 6 hearts vs. 568.6 ± 56.4 cm/s in CCR9−/−/Sham group, n = 4 hearts, p < 0.01). What's more, left ventricle CV was faster and exhibited better homogeneity in the CCR9−/−/MI group (290.1 ± 34.47 cm/s in CCR9−/−/MI group, n=6 hearts vs. 113.2 ± 14.4 cm/s in CCR9+/+/MI group, n = 5 hearts, p < 0.05; Figure 4C).
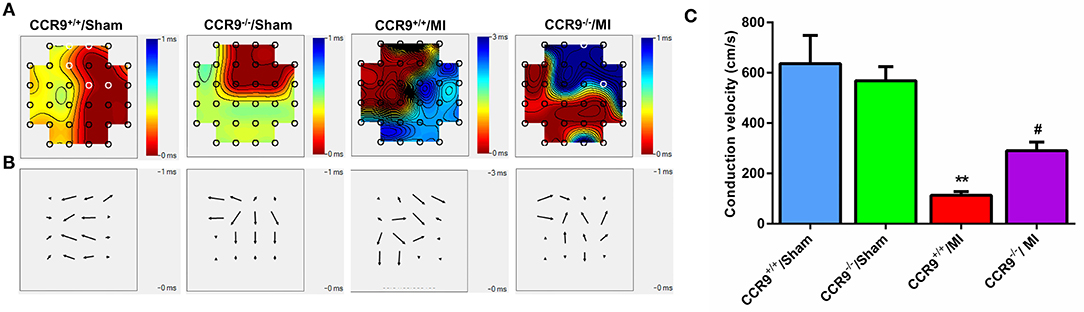
Figure 4. CCR9 knockout improves the conduction function after MI. (A) The conduction maps of excitation in the left ventricle recorded by MEA. MI mice presented crowding isochronal, of which the crowding degree was less in the CCR9−/− MI group. (B) Heterogeneity of conduction in mice left ventricle. There was more heterogeneous in CCR9+/+/MI mice than CCR9−/−/MI mice. (C) Statistical analysis of CV in each group. CCR9 knockout increased the ventricular CV following MI. (**p < 0.01 vs. CCR9+/+/Sham, #p < 0.05 vs. CCR9+/+/MI).
CCR9 Knock Out Preserves the Expression of Cx43
The westernblot result revealed that the expression level of Cx43 was decreased following MI (Protein/GAPDH, 0.16 ± 0.02 in CCR9+/+/MI group, n = 4 hearts vs. 0.74 ± 0.02 in CCR9+/+/sham group, n = 4 hearts, p < 0.01; 0.42 ± 0.04 in CCR9−/−/MI group, n = 4 hearts vs. 0.72 ± 0.03 in CCR9−/−/Sham group, n = 4 hearts, p < 0.01), while loss of CCR9 could preserve the expression of Cx43 (Protein/GAPDH, 0.42 ± 0.04 in CCR9−/−/MI group, n = 4 hearts vs. 0.16 ± 0.02 in CCR9+/+/MI group, n = 4 hearts, p < 0.01, Figures 5A,B).
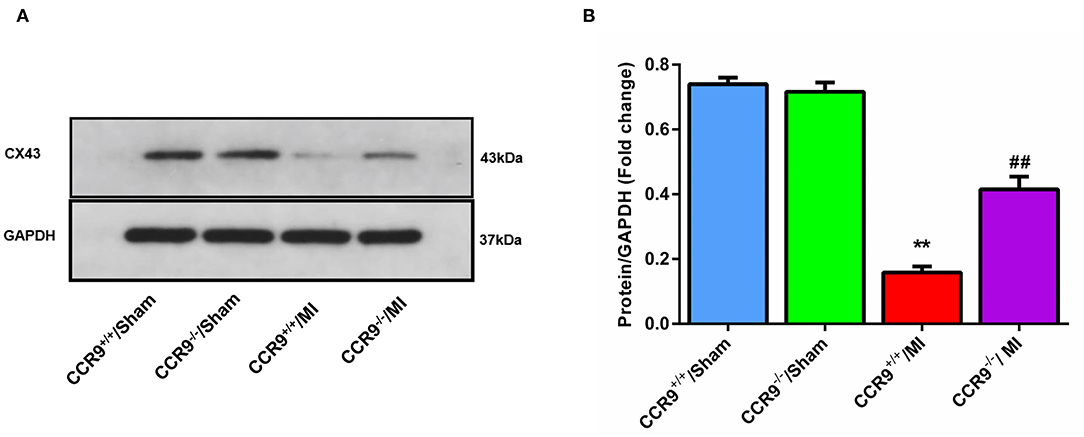
Figure 5. CCR9 knock out preserves the expression of Cx43. (A) Western blot band of Cx43 in sham or MI mice heart tissues. (B) Cx43 expression level in each group (**p < 0.01 vs. CCR9+/+/Sham, ##p < 0.01 vs. CCR9+/+/MI).
Discussion
The major findings in this study include: (1) The APD was prolonged, ICa,L was increased and Ito was decreased following MI in mice, while CCR9 knockout attenuated these changes. (2) CCR9 knockout reversed the calcium transient and calcium content reduction in sarcoplasmic reticulum following MI. (3) The conduction function was preserved in CCR9−/−/MI mice due to the preserved Cx43 expression.
SCD is the main cause of death in patients with acute MI. Arrhythmia usually occurs in the early phase of acute MI, and ventricular arrhythmia is the most common cause of death in MI patients (4). The main factors of arrhythmia after MI include: first, the heterogeneity of scar repair is the risk matrix for arrhythmia after MI, and the more severe myocardial fibrosis, the higher the incidence of arrhythmia (23–25). After MI, collagen fibers were deposited in the surrounding area of infarction, and the surviving myocardial cells and collagen fibers interacted with each other. Due to the electrical heterogeneity between myocardial cells and fibroblasts, reentry produced and electrical conductivity became abnormal, which induce arrhythmia after MI (26–28). Studies have shown that when myocardial cells were co-cultured with fibroblasts, the higher the proportion of fibroblasts, the slower the conduction speed, and the higher incidence of abnormal electrical activity (29). Through in vivo study, Michael and colleagues reported that Cx43 expression at myofibroblast-cardiomyocyte junctions was much less than that in remote region, which may result in low electrical conductivity in myofibroblast-cardiomyocyte junction area (30). Secondly, inflammatory response after MI can promote the occurrence of arrhythmia. Inflammatory response can cause both structural and electrical remodeling. A negative inflammatory factor can activate fibroblasts to differentiate into fibroblasts, increase collagen synthesis and fibrosis, and thus induce arrhythmia. On the other hand, cytokines and other inflammatory factors can directly affect the function of ion channels, affect proteins that regulate intracellular calcium homeostasis and intercellular gap junction (31–34).
Our previous investigation found that severe inflammatory response and myocardial fibrosis occurred after myocardial infarction, and CCR9 gene knockout can alleviate inflammation and myocardial fibrosis. Therefore, we attempted to further explore whether CCR9 gene knockout also has an impact on electrical remodeling after MI. In this part of the study, CCR9 gene knockout mice were studied to establish an acute MI model, and the cell electrophysiological properties were explored. We found that after MI, the APD90 of the ventricular myocytes in the remote area was prolonged, which can be explained by the increased ICa,L and the decreased Ito. These results were accordant to other studies on electrophysiological consequences following MI (35). However, for CCR9−/−/MI mice, the APD90 was shortened compared with wild type mice after MI. Consistent with APD90 alternation, the ICa,L was decreased and Ito was also increased in CCR9−/−/MI mice when compared with wild type MI mice. This finding is novel. CCR9 knockout improves cardiac inflammation and fibrosis by inhibiting CCR9/CCL25 activation as well as inflammatory cytokines chemotaxis, which maintains electrical activity of cardiomyocytes and reduces the APD prolongation as well as the ion current disorder (Figure 6).
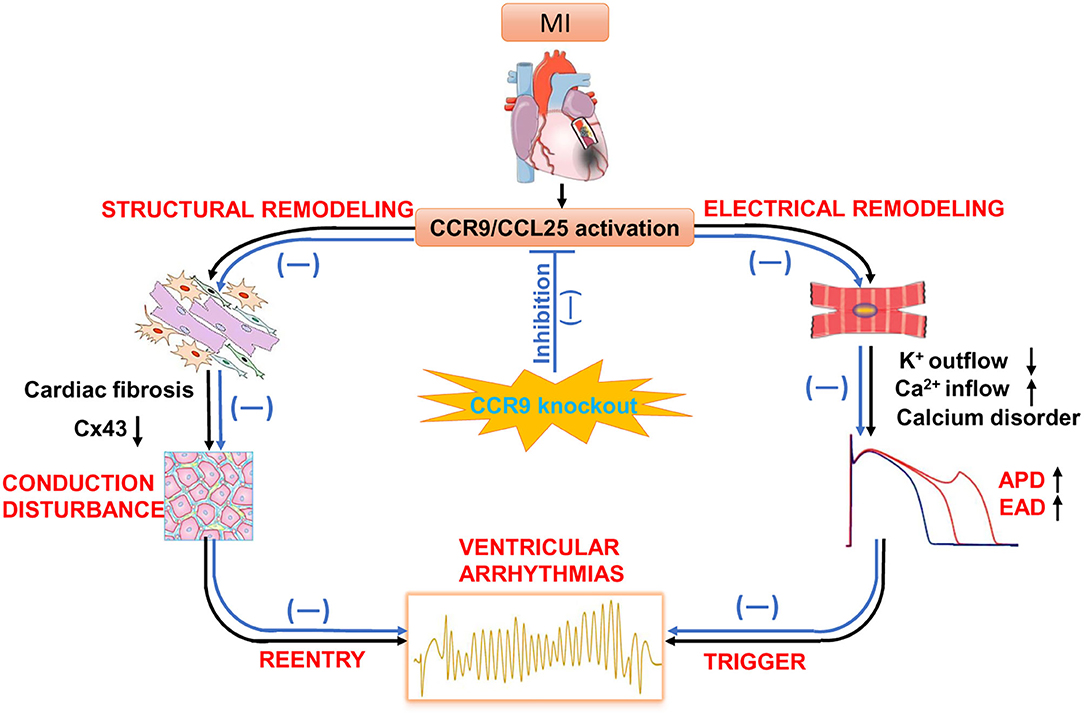
Figure 6. The potential role of CCR9/CCL25 in cardiac remodeling after MI. Following MI, CCR9/CCL25 is activated. Inflammatory cells tend to distribute toward the infarct area and secrete inflammatory factors. Cytokines accelerate structural and electrical remodeling by activating cardiac fibrosis, impairing gap junction via changes in connexin 43 expression, disrupting calcium homeostasis. These pathological changes increase ectopic activity, decrease ventricular conduction and promote reentry, which are substrate for ventricular arrhythmias. Whereas, CCR9 knockout improves cardiac inflammation and fibrosis by inhibiting CCR9/CCL25 activation, as well as inflammatory cytokines chemotaxis, which maintains expression of connexin43 and electrical activity of cardiomyocytes, and preventing ventricular arrhythmias.
Also, we showed that the electrical conductivity was slowed and gap junction protein expression of Cx43 was decreased obviously after MI, which was consistent with the results reported in literatures (36, 37), which have shown that Cx43 knockout mice under myocardial ischemia stress is prone to occur ventricular tachycardia. Cardiac conduction block and anisotropy happened in conditional Cx43 knockout mice, and ventricular arrhythmia susceptibility increase (38–40). Cx43 is the main protein that constitutes the gap junction between myocardial cells. Gap junction mediate movement of ions from cell to cell and is vital for impulse conduction through the Purkinje fibers and ventricular myocardium (41). After MI, the expression of Cx43 is reduced, and the uncoupling between myocardial cells can cause the electrical conduction block between myocardial cells, and subsequently induce malignant arrhythmia. Thus, inflammation, fibrosis, abnormal ion channel function as well as decreased Cx43 expression after MI triggering conduction abnormality, contribute to the reentry. Meanwhile, calcium handling disorder accelerates the occurrence of triggered activity. Triggering and reentry are the pathological basis of arrhythmia and sudden cardiac death (Figure 6).
Our further study previously showed that CCR9 gene knockout can reduce inflammation and improve the structural remodeling after MI. The results of our current study indicated that CCR9 gene knockout can shorten the prolonged APD after MI through influencing the calcium current and potassium current, as well as the calcium handling, which indicated that inhibiting CCR9 can improve the electrical remodeling and reduce the occurrence of malignant arrhythmia after MI. Moreover, CCR9 gene knockout reserves the conduction function and the expression of Cx43 in myocardial tissue after MI, suggesting that CCR9 knockout diminishes the inflammatory response and fibrosis, which can consequently save some Cx43 expression from MI induced degradation, and thus alleviated the electrical uncoupling and conduction abnormalities among myocardial cells. At the same time, CCR9 gene knockout reduces inflammatory response and fibrosis, and these structural remodeling also play an important role in improving electrical remodeling. Therefore, early inhibition of CCR9 expression or chemotaxis of CCR9 positive cells in acute MI is expected to prevent malignant arrhythmia after MI.
Limitations
There are limitations to be addressed for the present study. First, this study is based on mice, which has different pathophysiological procedures from human beings. So, further exploration on larger animals as well as patients is warranted. Secondly, only CCR9 knockout mice are used here. It would be optimal if transgenic CCR9 mice could also be applied as another study group. Additionally, we need to perform more study to explore the underlying mechanisms and pathway in the future.
Conclusion
To address arrhythmias following MI, drugs and implantable cardioverter defibrillators (ICDs) as well as radiofrequency ablation were applied, while the mortality is still high and there are side effects for drugs and life quality is reduced due to devices implanted. Therefore, novel treatment strategies including gene therapy are in urgent need. In this study, CCR9 abrogation was suggested to ameliorate MI-induced electrical remodeling by affecting ion current, AP, calcium homeostasis and cardiac conduction as well as gap junction, suggesting it may be a novel pharmaceutical target for the treatment of MI-induced arrhythmia.
Data Availability Statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics Statement
The animal study was reviewed and approved by the Animal Care and Use Committee of Renmin Hospital of Wuhan University.
Author Contributions
YH, DH, C-XH, and H-SD, conceived and designed the study and drafted the manuscript. YH, DH, H-SD, TS, TW, Y-HT, and Y-TC performed the experiments. YH, DH, H-SD, Y-TC, and HB-M analyzed the experiment data. YH, C-XH, and DH wrote the manuscript. All co-authors participate in editing of the manuscript.
Funding
This research was supported by the National Natural Science Foundation Project of China (Grant No. 81670304 and 8210020476) and the Fundamental Research Funds for the Central Universities of China (No. 2042019kf0058).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Sandoval Y, Jaffe AS. Type 2 myocardial infarction: JACC review topic of the week. J Am Coll Cardiol. (2019) 73:1846–60. doi: 10.1016/j.jacc.2019.02.018
2. Khan MA, Hashim MJ, Mustafa H, Baniyas MY, Al Suwaidi S, AlKatheeri R, et al. Global epidemiology of ischemic heart disease: results from the global burden of disease study. Cureus. (2020) 12:e9349. doi: 10.7759/cureus.9349
3. Huang S, Frangogiannis NG. Anti-inflammatory therapies in myocardial infarction: failures, hopes and challenges. Br J Pharmacol. (2018) 175:1377–400. doi: 10.1111/bph.14155
4. Erne P, Iglesias JF, Urban P, Eberli FR, Rickli H, Simon R, et al. Left bundle-branch block in patients with acute myocardial infarction: presentation, treatment, and trends in outcome from 1997 to 2016 in routine clinical practice. Am Heart J. (2017) 184:106–13. doi: 10.1016/j.ahj.2016.11.003
5. Liu M, Liu H, Parthiban P, Kang GJ, Shi G, Feng F, et al. Inhibition of the unfolded protein response reduces arrhythmic risk after myocardial infarction. J Clin Invest. (2021) 131:e147836. doi: 10.1172/JCI147836
6. Alex Chau YL, Won Yoo J, Chuen Yuen H, Bin Waleed K, Chang D, Liu T, et al. The impact of post-operative atrial fibrillation on outcomes in coronary artery bypass graft and combined procedures. J Geriatr Cardiol. (2021) 18:319–26. doi: 10.11909/j.issn.1671-5411.2021.05.005
7. Lin CF, Chang YH, Su CH, Liu HY, Chien LN. Risk of new-onset atrial fibrillation after drug-eluting stent implantation in patients with stable coronary artery disease. Int J Cardiol. (2019) 291:63–8. doi: 10.1016/j.ijcard.2019.05.053
8. Lee S, Zhou J, Guo CL, Wong WT, Liu T, Wong ICK, et al. Predictive scores for identifying patients with type 2 diabetes mellitus at risk of acute myocardial infarction and sudden cardiac death. Endocrinol Diabetes Metab. (2021) 4:e00240. doi: 10.1002/edm2.240
9. Gao X, Zhang W, Yang F, Ma W, Cai B. Photobiomodulation regulation as one promising therapeutic approach for myocardial infarction. Oxid Med Cell Longev. (2021) 2021:9962922. doi: 10.1155/2021/9962922
10. Keller Iv TCS, Lim L, Shewale SV, McDaid K, Marti-Pamies I, Tang AT, et al. Genetic blockade of lymphangiogenesis does not impair cardiac function after myocardial infarction. J Clin Invest. (2021) 17:147070. doi: 10.1172/JCI147070
11. Schulz O, Hammerschmidt SI, Moschovakis GL, Forster R. Chemokines and chemokine receptors in lymphoid tissue dynamics. Annu Rev Immunol. (2016) 34:203–42. doi: 10.1146/annurev-immunol-041015-055649
12. Wang J, Seo MJ, Deci MB, Weil BR, Canty JM, Nguyen J. Effect of CCR2 inhibitor-loaded lipid micelles on inflammatory cell migration and cardiac function after myocardial infarction. Int J Nanomedicine. (2018) 13:6441–51. doi: 10.2147/IJN.S178650
13. Jiang Y, Bai J, Tang L, Zhang P, Pu J. Anti-CCL21 antibody attenuates infarct size and improves cardiac remodeling after myocardial infarction. Cell Physiol Biochem. (2015) 37:979–90. doi: 10.1159/000430224
14. Huang J, Zhang Z, Guo J, Ni A, Deb A, Zhang L, et al. Genetic modification of mesenchymal stem cells overexpressing CCR1 increases cell viability, migration, engraftment, and capillary density in the injured myocardium. Circ Res. (2010) 106:1753–62. doi: 10.1161/CIRCRESAHA.109.196030
15. Vicari AP, Figueroa DJ, Hedrick JA, Foster JS, Singh KP, Menon S, et al. TECK: a novel CC chemokine specifically expressed by thymic dendritic cells and potentially involved in T cell development. Immunity. (1997) 7:291–301. doi: 10.1016/S1074-7613(00)80531-2
16. Papadakis KA, Prehn J, Moreno ST, Cheng L, Kouroumalis EA, Deem R, et al. CCR9-positive lymphocytes and thymus-expressed chemokine distinguish small bowel from colonic Crohn's disease. Gastroenterology. (2001) 121:246–54. doi: 10.1053/gast.2001.27154
17. Nakamoto N, Ebinuma H, Kanai T, Chu PS, Ono Y, Mikami Y, et al. CCR9+ macrophages are required for acute liver inflammation in mouse models of hepatitis. Gastroenterology. (2012) 142:366–76. doi: 10.1053/j.gastro.2011.10.039
18. Schmutz C, Cartwright A, Williams H, Haworth O, Williams JH, Filer A, et al. Monocytes/macrophages express chemokine receptor CCR9 in rheumatoid arthritis and CCL25 stimulates their differentiation. Arthritis Res Ther. (2010) 12:R161. doi: 10.1186/ar3120
19. Li J, Xiong T, Xiao R, Xiong A, Chen J, Altaf E, et al. Anti-CCL25 antibody prolongs skin allograft survival by blocking CCR9 expression and impairing splenic T-cell function. Arch Immunol Ther Exp (Warsz). (2013) 61:237–44. doi: 10.1007/s00005-013-0223-4
20. Tu Z, Xiao R, Xiong J, Tembo KM, Deng X, Xiong M, et al. CCR9 in cancer: oncogenic role and therapeutic targeting. J Hematol Oncol. (2016) 9:10. doi: 10.1186/s13045-016-0236-7
21. Huang Y, Wang D, Wang X, Zhang Y, Liu T, Chen Y, et al. Abrogation of CC chemokine receptor 9 ameliorates ventricular remodeling in mice after myocardial infarction. Sci Rep. (2016) 6:32660. doi: 10.1038/srep32660
22. Xu Z, Mei F, Liu H, Sun C, Zheng Z. C-C motif chemokine receptor 9 exacerbates pressure overload-induced cardiac hypertrophy and dysfunction. J Am Heart Assoc. (2016) 5:e003342. doi: 10.1161/JAHA.116.003342
23. Zheng ZJ, Croft JB, Giles WH, Mensah GA. Sudden cardiac death in the United States, 1989 to 1998. Circulation. (2001) 104:2158–63. doi: 10.1161/hc4301.098254
24. Schmidt A, Azevedo CF, Cheng A, Gupta SN, Bluemke DA, Foo TK, et al. Infarct tissue heterogeneity by magnetic resonance imaging identifies enhanced cardiac arrhythmia susceptibility in patients with left ventricular dysfunction. Circulation. (2007) 115:2006–14. doi: 10.1161/CIRCULATIONAHA.106.653568
25. Yan AT, Shayne AJ, Brown KA, Gupta SN, Chan CW, Luu TM, et al. Characterization of the peri-infarct zone by contrast-enhanced cardiac magnetic resonance imaging is a powerful predictor of post-myocardial infarction mortality. Circulation. (2006) 114:32–9. doi: 10.1161/CIRCULATIONAHA.106.613414
26. de Bakker JM, van Capelle FJ, Janse MJ, Tasseron S, Vermeulen JT, de Jonge N, et al. Slow conduction in the infarcted human heart. ‘Zigzag' course of activation. Circulation. (1993) 88:915–26. doi: 10.1161/01.CIR.88.3.915
27. de Bakker JM, van Capelle FJ, Janse MJ, Wilde AA, Coronel R, Becker AE, et al. Reentry as a cause of ventricular tachycardia in patients with chronic ischemic heart disease: electrophysiologic and anatomic correlation. Circulation. (1988) 77:589–606. doi: 10.1161/01.CIR.77.3.589
28. Peters NS, Coromilas J, Severs NJ, Wit AL. Disturbed connexin43 gap junction distribution correlates with the location of reentrant circuits in the epicardial border zone of healing canine infarcts that cause ventricular tachycardia. Circulation. (1997) 95:988–96. doi: 10.1161/01.CIR.95.4.988
29. Askar SF, Ramkisoensing AA, Schalij MJ, Bingen BO, Swildens J, van der Laarse A, et al. Antiproliferative treatment of myofibroblasts prevents arrhythmias in vitro by limiting myofibroblast-induced depolarization. Cardiovasc Res. (2011) 90:295–304. doi: 10.1093/cvr/cvr011
30. Rubart M, Tao W, Lu XL, Conway SJ, Reuter SP, Lin SF, et al. Electrical coupling between ventricular myocytes and myofibroblasts in the infarcted mouse heart. Cardiovasc Res. (2018) 114:389–400. doi: 10.1093/cvr/cvx163
31. Abramochkin DV, Kuzmin VS, Mitrochin VM, Kalugin L, Dvorzhak A, Makarenko EY, et al. TNF-alpha provokes electrical abnormalities in rat atrial myocardium via a NO-dependent mechanism. Pflugers Arch. (2013) 465:1741–52. doi: 10.1007/s00424-013-1320-2
32. Adlan AM, Panoulas VF, Smith JP, Fisher JP, Kitas GD. Association between corrected QT interval and inflammatory cytokines in rheumatoid arthritis. J Rheumatol. (2015) 42:421–8. doi: 10.3899/jrheum.140861
33. Lazzerini PE, Capecchi PL, Laghi-Pasini F. Long QT syndrome: an emerging role for inflammation and immunity. Front Cardiovasc Med. (2015) 2:26. doi: 10.3389/fcvm.2015.00026
34. Medenwald D, Kors JA, Loppnow H, Thiery J, Kluttig A, Nuding S, et al. Inflammation and prolonged QT time: results from the Cardiovascular Disease, Living and Ageing in Halle (CARLA) study. PLoS ONE. (2014) 9:e95994. doi: 10.1371/journal.pone.0095994
35. Tao B, Liu Z, Wei F, Fan S, Cui S, Xia H, et al. Over-expression of Kv4.3 gene reverses cardiac remodeling and transient-outward K(+) current (Ito) reduction via CaMKII inhibition in myocardial infarction. Biomed Pharmacother. (2020) 132:110896. doi: 10.1016/j.biopha.2020.110896
36. Rucker-Martin C, Milliez P, Tan S, Decrouy X, Recouvreur M, Vranckx R, et al. Chronic hemodynamic overload of the atria is an important factor for gap junction remodeling in human and rat hearts. Cardiovasc Res. (2006) 72:69–79. doi: 10.1016/j.cardiores.2006.06.016
37. Wang D, Zhu H, Yang Q, Sun Y. Effects of relaxin on cardiac fibrosis, apoptosis, and tachyarrhythmia in rats with myocardial infarction. Biomed Pharmacother. (2016) 84:348–55. doi: 10.1016/j.biopha.2016.09.054
38. Gutstein DE, Morley GE, Tamaddon H, Vaidya D, Schneider MD, Chen J, et al. Conduction slowing and sudden arrhythmic death in mice with cardiac-restricted inactivation of connexin43. Circ Res. (2001) 88:333–9. doi: 10.1161/01.RES.88.3.333
39. Lerner DL, Yamada KA, Schuessler RB, Saffitz JE. Accelerated onset and increased incidence of ventricular arrhythmias induced by ischemia in Cx43-deficient mice. Circulation. (2000) 101:547–52. doi: 10.1161/01.CIR.101.5.547
40. van Rijen HV, Eckardt D, Degen J, Theis M, Ott T, Willecke K, et al. Slow conduction and enhanced anisotropy increase the propensity for ventricular tachyarrhythmias in adult mice with induced deletion of connexin43. Circulation. (2004) 109:1048–55. doi: 10.1161/01.CIR.0000117402.70689.75
Keywords: myocardial infarction, chemokine receptor 9, action potential, ion channel, calcium transient, cardiac conduction, connexin 43
Citation: Huang Y, Ding H-S, Song T, Chen Y-T, Wang T, Tang Y-H, Barajas-Martinez H, Huang C-X and Hu D (2021) Abrogation of CC Chemokine Receptor 9 Ameliorates Ventricular Electrical Remodeling in Mice After Myocardial Infarction. Front. Cardiovasc. Med. 8:716219. doi: 10.3389/fcvm.2021.716219
Received: 28 May 2021; Accepted: 13 September 2021;
Published: 12 October 2021.
Edited by:
Tachapong Ngarmukos, Mahidol University, ThailandReviewed by:
Sunil Jit Ramamoorthy Jeewanlal Logantha, University of Liverpool, United KingdomSharen Lee, The Chinese University of Hong Kong, Hong Kong, SAR China
Jichao Zhao, The University of Auckland, New Zealand
Tao Yang, Vanderbilt University Medical Center, United States
Copyright © 2021 Huang, Ding, Song, Chen, Wang, Tang, Barajas-Martinez, Huang and Hu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Dan Hu, aHVkYW4wNzE2QGhvdG1haWwuY29t; cm0wMDI2NDZAd2h1LmVkdS5jbg==; Cong-Xin Huang, aHVhbmdjb25neGluQHZpcC4xNjMuY29t