 Hengzhi Du
Hengzhi Du Yanru Zhao
Yanru Zhao Dao Wen Wang
Dao Wen Wang Chen Chen
Chen Chen- Division of Cardiology, Hubei Key Laboratory of Genetics and Molecular Mechanisms of Cardiological Disorders, Tongji Medical College, Tongji Hospital, Huazhong University of Science and Technology, Wuhan, China
MicroRNAs (miRNAs) are small non-coding RNAs that participate in heart development and pathological processes mainly by silencing gene expression. Overwhelming evidence has suggested that miRNAs were involved in various cardiovascular pathological processes, including arrhythmias, ischemia-reperfusion injuries, dysregulation of angiogenesis, mitochondrial abnormalities, fibrosis, and maladaptive remodeling. Various miRNAs could regulate myocardial contractility, vascular proliferation, and mitochondrial function. Meanwhile, it was reported that miRNAs could manipulate nutrition metabolism, especially glucose and lipid metabolism, by regulating insulin signaling pathways, energy substrate transport/metabolism. Recently, increasing studies suggested that the abnormal glucose and lipid metabolism were closely associated with a broad spectrum of cardiovascular diseases (CVDs). Therefore, maintaining glucose and lipid metabolism homeostasis in the heart might be beneficial to CVD patients. In this review, we summarized the present knowledge of the functions of miRNAs in regulating cardiac glucose and lipid metabolism, as well as highlighted the miRNA-based therapies targeting cardiac glucose and lipid metabolism.
Introduction
Under normal physiological conditions, in order to fulfill a continuous demand for ATP, the heart can metabolize a range of substrates via mitochondrial oxidative phosphorylation and substrate level phosphorylation, such as fatty acids, glucose, lactate, and amino acids (1). Before feeding into glycolysis or pentose phosphate pathway, glucose in cardiomyocytes is phosphorylated to glucose-6-phosphate (G6P). Activated by acyl CoA synthetase (ACS), cytosolic free fatty acids can form fatty acyl-CoAs, then could enter mitochondria for oxidation or form ceramides, diacylglycerol (DAG) and triacylglycerol (TAG). Although, the adult hearts mainly use fatty acids for ATP production, hearts demonstrate increased reliance on other substrates such as glucose under pathological conditions (2). The glucose and lipid metabolism in the normal and diseased heart have attracted increasing attentions. Under normal circumstances, except that the sources are lactate, ketone bodies, and amino acids, more than 95% of all substrates are derived from fatty acids and glucose to use for ATP generation for maintaining the function of the heart (2). Importantly, the glucose and lipid metabolism could be changed under pathological injury condition in the heart. It is well accepted, e.g., that hypertrophic heart undergoes a reprogramming process in metabolism, characterized by the increased reliance on glucose metabolism and decreased fatty acids oxidation, which is associated with an increase in glycolysis in the hypertrophied heart (3–5). Cardiac ischemia would lead to poor oxygen supply, inadequate washout of metabolic wastes, and increased glycolytic flux, because the amount of oxygen and metabolic substrates that delivered to the myocardium is insufficient to meet the myocardial energy requirements (6). Abnormal glucose metabolism has also been noted in patients with diabetes mellitus and has been associated with cardiac dysfunction (7). Cardiac glucose uptake in diabetic cardiomyopathy is reduced despite hyperglycemia, which could also contribute to the impaired myocardial glucose utilization in diabetes due to decreased protein level of cardiac GLUT-4 (8). MicroRNAs (miRNAs) are small conserved non-coding RNAs which typically inhibits target mRNA translation or promoting target mRNA degradation in physiological or pathological processes (9, 10). Increasing evidence also suggests nuclear or mitochondrial miRNAs could enhance target gene expression through non-canonical mechanisms (11–13). During several phases of cardiac development, many miRNAs have been detected as important regulators to maintain the formation of normal functional heart tissue (14). miR-17-92 cluster, e.g., was suggested as a critical regulator of cardiomyocyte proliferation and might be a therapeutic target for cardiac repairing and heart regeneration (15). Cardiac miRNAs, such as miR-1 (16), miR133a (17), miR-208a/b (18), and miR-499 (19) could enhance regenerative properties and contribute to the reprogramming of mature non-cardiac cells to cardiomyocytes (20). During the progression of various cardiovascular diseases (CVDs), such as hypertrophy, diabetic cardiomyopathy, and myocardial ischemia, many studies also reported that a variety of miRNAs exerted important functions (21–23). Overexpression of miR-297 was found to accelerate the progression of cardiac hypertrophy by increasing the protein expression of ATF4, Xbps1, chaperon Grp78, and calreticulin, the endoplasmic reticulum stress markers (24). Overexpression of miR-200b was reported to prevent diabetes-induced cardiac functional and structural changes by inhibiting endothelial-to-mesenchymal transition (25). Delivery of antisense microribonucleic acid (anti-miR) against miR-21 improved cardiac function, as well as reduced cardiac fibrosis and hypertrophy in a pig model of myocardial ischemia/reperfusion injury (26).
Importantly, it has been reported that miRNAs possess crucial roles in regulating the glucose and lipid metabolism in a variety of organs. miR-146a, e.g., has been found to improve lipid accumulation as well as glucose and insulin tolerance via promoting the oxidative metabolism of fatty acids in the liver (27). In the kidney, through blocking the TLR4/NF-κB pathway, miR-140-5p protected renal tubular epithelial cells against high glucose-induced injury (28). Furthermore, in our recent study, we found miR-320a significantly aggravated diet-induced hyperlipidemia and hepatic steatosis (29). Importantly, the roles of miRNAs in the pathological and physiological regulation of glucose and lipid metabolism in the heart have also been gradually discovered. We previously found that nuclear miR-320a caused lipotoxicity in the diabetic heart and induced cardiac dysfunction by activating transcription of fatty acid metabolic genes (13).
In this review, we focused on the current knowledge to briefly summarize and discuss the regulation of miRNAs in glucose and lipid metabolism during the pathological processes of the heart, and highlighting the potential therapeutic strategies for diseases associated with abnormal cardiac glucose and lipid metabolism.
Roles of miRNAs in Glucose Metabolism in the Heart
In the heart, miRNAs are critical, which participate in cardiac developmental and pathological processes (30). Blocking the expression of all miRNAs in the cardiovascular system has been reported to lead to death in early pregnancy due to severe heart and vascular development defects (31). To support both electrical and mechanical activities, the heart needs a continuous energy supply which are mainly produced by mitochondrial oxidative phosphorylation under normal circumstances (31). A growing number of studies have also shown that miRNAs played crucial roles in the diseased heart by regulating glucose metabolism.
Roles of miRNAs in Glucose Transport in Heart
Glucose has been proven to be transported into cardiomyocytes by the glucose transporters, glucose transporter 4 (GLUT-4) or GLUT-1, in the sarcolemma (32). In response to various stresses, such as insulin stimulation, increased energy demand, or ischemia, GLUT-4 and GLUT-1 are transported from intracellular vesicles to the sarcomembrane to increase the rate of glucose uptake and glucose transport (33, 34). Moreover, previous study has shown that the expression of GLUT4 in cardiomyocytes could be regulated by miRNAs. Lu et al. found that overexpression of miR-223 increased glucose uptake via increasing the GLUT4 protein expression (35). It is worth mentioning that whether miR-223 regulate glucose uptake in cardiomyocytes only by targeting GLUT4 is not clear and remains to be further studied.
Cardiomyocyte hypertrophy, which is characterized by increased size of cardiomyocytes, is one of the compensatory mechanisms of various CVDs (36). Changes in cardiac energy metabolism and substrate utilization are hallmarks of a hypertrophied heart, including increased dependence on glucose, reduction in fatty acid oxidation rate, and decreased high-energy-phosphate content (37). The shift of substrate preference from fatty acid to glucose is therefore considered beneficial in the hypertrophied heart because glucose has a higher oxygen efficiency for ATP production (38). Moreover, many studies suggested that promoting glucose utilization in the hypertrophied heart could be beneficial (39, 40). Studies were performed to explore whether miRNAs affect glucose transport in the hypertrophic cardiomyocytes. Takahiro et al. found that miR-133 decreased the protein level of KLF15 and the level of its downstream target GLUT4, which was involved in metabolic control in the hypertrophic cardiomyocytes (41). Interestingly, Trotta et al. also found the melanocortin 5 receptor agonism reduced the ratio of GLUT1/GLUT4 glucose transporters on the cell membranes and increased the intracellular PI3K activity in the hypertrophic H9c2 cells by decreasing of the levels of miR-133a (42). The important roles of miR-133 in cardiomyocyte glucose transport were confirmed in different models of cardiac hypertrophy. Moreover, Yang et al. found that miR-200a-5p could disturb glucose metabolism by inhibiting selenoprotein n (Seln), selenoprotein t (Selt), selenoprotein 15 (Sep15), and selenoprotein p1 (Sepp1) expression to alter glucose transport, which eventually induce cardiomyocyte hypertrophy (43).
Diabetic cardiomyopathy is a metabolism-related heart disease, which is characterized by clinical heart failure and diastolic relaxation abnormalities in the early stage in the absence of dyslipidemia, hypertension, and coronary artery disease in the advanced stage (43). Due to decreased protein level of cardiac GLUT-4, cardiac glucose uptake is reduced despite hyperglycemia which could also contribute to the impaired myocardial glucose utilization in diabetes (8). Li et al. (44) revealed that the level of let-7 was increased in the myocardium of diabetic rats compared with non-diabetic rats, whereas improved glucose uptake by inhibiting of the let-7 family miRNAs through GLUT4 pathways. Similarly, Ju et al. (45) found miR-150 reduced the glucose utilization by decreasing the translocation and expression of GLUT-4 in the insulin-resistant cardiomyocytes.
In conclusion, miRNAs play important roles in the glucose transport in cardiomyocytes under both pathological and physiological processes (Figure 1A).
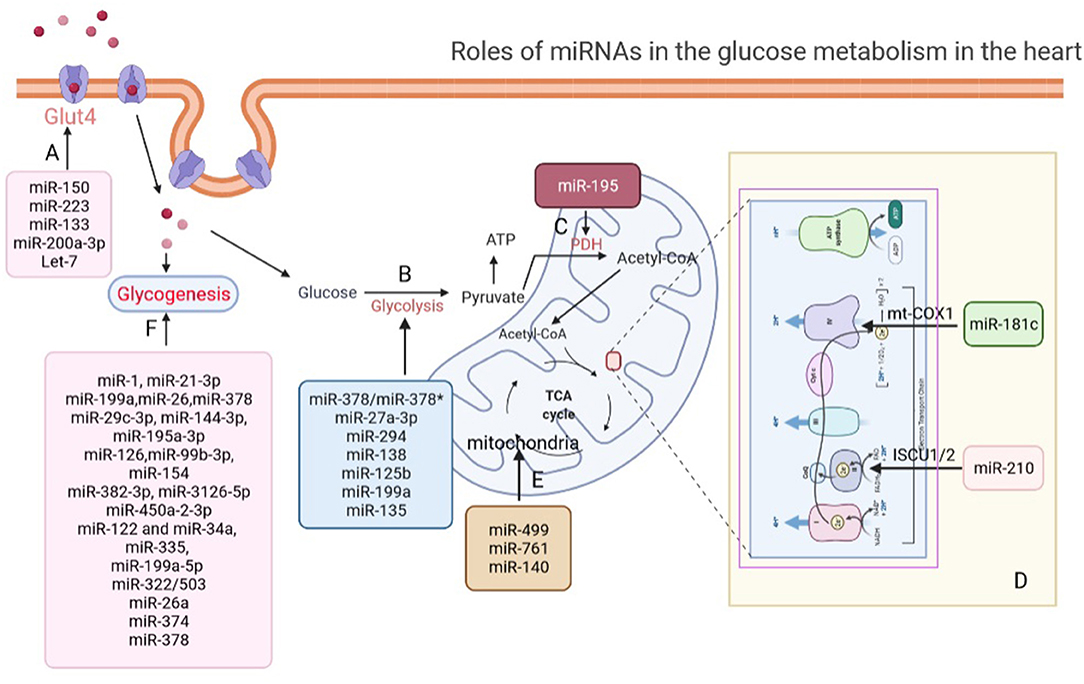
Figure 1. Roles of miRNAs in glucose metabolism in the heart. (A) miRNAs regulate glucose transport via modulating the expression of GLUT4 in the heart. (B) miRNAs participate in glycolysis regulation in the heart. (C) miR-195 increased acetylation of PDH to promote pyruvate and NAD+ convert into acetyl-CoA. (D) miR-181c and miR-210 involve in electron chain complex remodeling in cardiomyocytes by targeting and suppressing mt-COX1 and ISCU1/2. (E) miR-499, miR-761, and miR-140 regulate aerobic glucose oxidation by directly affecting mitochondrial function in the heart. (F) miRNAs regulate glycogenesis in the heart. GLUT4, glucose transporter type 4; PDH, pyruvate dehydrogenase complex; mt-COX1, cytochrome c oxidase subunit 1; ISCU1/2, iron-sulfur cluster assembly proteins.
Roles of miRNAs in Glycolysis in the Heart
After glucose transport into cardiomyocytes, the first step of glucose catabolism is glycolysis, which produces ATP (46). Although, cardiomyocytes use ATP produced by glucose through the process of glycolysis is limited under normal physiological conditions, glycolysis is thought to facilitate some glucose molecules to be diverted into macromolecular precursors required for lipid, amino acid, and nucleotide biosynthesis and the pentose phosphate pathway (47). Importantly, Mallet et al. (48) suggested miRNAs play important roles in glycolysis of normal cardiomyocytes. They found miR-378 inhibited LDHA expression whereas miR-378* indirectly activated its expression to balance between oxidative phosphorylation and glycolysis in cardiomyocytes. However, the detail mechanism that miR-378 and miR-378* regulate the glycolysis pathway of cardiomyocytes under physiological conditions remains to be further studied.
Most cancer cells rely on aerobic glycolysis, a phenomenon known as the Warburg effect, which differs from the fact that normally differentiated cells rely primarily on mitochondrial oxidative phosphorylation to generate energy for cellular process (47). Similarly, under pathological conditions of the heart, the level of glycolysis would change in contrast to normal physiological state in the heart (49).
During the early stage of myocardial ischemia, glycolysis produces ATP and maintains ionic homeostasis, providing a beneficial effect (50). However, under severe ischemia, glycolysis becomes more harmful than beneficial (51). Importantly, multiple studies have shown that miRNAs played two sides function in glycolysis to regulate cardiac function after myocardial ischemia. On one hand, by performing loss- and gain-of-function experiments and glycolysis stress test, Lei et al. (52) detected that miR-27a-3p restoration enhanced cell viability, depleted cell apoptosis, and promoted glycolysis by targeting TNFR-associated factor 5 (TRAF5) in hypoxia-induced AC16 cells. Borden et al. used AAV delivery system to deliver miR-294 in mice and measured oxygen consumption rates (OCR) and extracellular acidification rates (ECAR). They found that miR-294 could significantly promote proliferation of cardiomyocytes and enhance oxidative phosphorylation and glycolysis that lead to improved cardiac function by targeting Wee1/CyclinB-CDK1 complex after myocardial infarction (53). Bartman et al. (54) performed loss- and gain-of-function experiments and measured ECAR, which revealed that the upregulation of miR-21 facilitated glycolysis and cardioprotection through Per2-dependent mechanisms in myocardial ischemia. Pyruvate dehydrogenase kinase 1 (PDK1), a phosphorylate kinase, phosphorylates pyruvate dehydrogenase leading to elevated anaerobic glycolysis. Zhu et al. (55) observed that miR-138 promoted mitochondrial respiration and inhibited glycolysis through directly targeting PDK1 by measuring lactate product, ECAR, and glycolysis key enzyme, which protected against cardiac cell dysfunction during ischemia. On the other hand, many studies have also shown that miRNAs play a key role in glycolysis to deteriorate cardiac function after myocardial ischemia. Fan et al. showed that miR-125b abolished the beneficial effects of lncRNA-XIST in activating glucose metabolism and cardiomyocyte protection under hypoxia by directly targeting hexokinase 2 (HK2), the key enzyme of glycolysis (56). Similarly, Zhang et al. (57) found that miR-34a inhibited the restoration of glycolysis in dysfunctional cardiomyocytes during ischemia reperfusion (I/R) injury. Moreover, Rane et al. (58) detected that miR-199a was rapidly downregulated in cardiomyocytes and the expression of HK2 and pyruvate kinase-M2 (Pkm2) were enhanced during I/R injury.
It is well-known that there is a prominent metabolic shift from fatty acid oxidation to glucose utilization during cardiac hypertrophy and pathological remodeling, which is associated with an increase in glycolysis in the hypertrophied heart (59, 60). Moreover, it was suggested that the elevation of glycolysis during cardiac hypertrophy and pathological remodeling was through the activation of fructose 2,6-BP and phosphofructokinase-1 (PFK1) in response to cardiac pressure overload (59, 60). It is worth mentioning that miR-135 was found to target PFK1 and inhibit aerobic glycolysis in pancreatic cancer cell, which indicated the possible functions of miRNAs in cardiomyocyte glycolysis (61). However, the role of miRNAs in glycolysis during cardiomyocyte hypertrophy has not been fully revealed (Figure 1B).
Roles of miRNAs in Aerobic Oxidation of Glucose in the Heart
Glucose can be converted to pyruvate by glycolysis pathway. As the end-product of glycolysis, pyruvate is ultimately transported into mitochondria and is critical for mitochondrial ATP generation. In mitochondria, pyruvate is the main fuel input to drive several major biosynthetic pathways across the citrate cycle and enhance the carbon flux of the citrate cycle (62).
In humans, the mitochondrial pyruvate carrier (MPC), formed by two paralogous subunits, MPC1 and MPC2, is required to deliver pyruvate from the mitochondrial intermembrane space to the mitochondrial matrix (63). In response to cold and heat stress of common carp by performing high-throughput sequencing, Sun et al. found that miRNAs might regulate the expression of MPC in the liver of fish (64). However, the effects of miRNAs on MPC expression and function in the heart remained to be further explored.
After passing through MPC, pyruvate will be oxidized into carbon dioxide by oxidative phosphorylation to ultimately support the generation of ATP (65). Subsequently, pyruvate and NAD+ are irreversibly converted into acetyl-CoA, NADH, and carbon via the pyruvate dehydrogenase complex (PDH), which serves for bridging glycolytic metabolism in cytoplasm with oxidative phosphorylation and citric acid cycle (66). Importantly, Zhang et al. (67) detected that the expression of miR-195 was increased in failing myocardium, which downregulated the expression of SIRT3 by enhancing global protein acetylation, including PDH complex and ATP synthase directly targeting 3′-untranslated regions that were essential for cardiac energy metabolism (Figure 1C).
In the diseased heart, it was proven that the activity levels of oxidative respiratory chain complex would also change (68). Das et al. indicated that the expression of miR-181c was activated under hypoxic conditions of HF and suppressed cytochrome c oxidase subunit 1 (mt-COX1) to involve in electron chain complex IV remodeling in cardiomyocytes, which in turn increased the production of ROS in the heart (69). Similarly, miR-210 suppressed iron-sulfur cluster assembly protein ISCU1/2 expression, which is a chaperone to assemble iron-sulfur clusters and transport these clusters within the functional position in the cell, in hypoxic conditions of heart (70) (Figure 1D).
The tricarboxylic acid (TCA) cycle, a central route for oxidative phosphorylation in cells, depends on the oxidative respiratory chain to fulfill bioenergetic, biosynthetic, and redox balance requirements (71). The oxidative respiratory chain, containing four complexes, establishes an electrochemical gradient over the inner membrane to connect the transport of electrons to oxygen for ATP synthesis (72). In a healthy heart, the various complexes of the oxidative respiratory chain perform their respective functions to maintain the oxidative phosphorylation of glucose supporting cardiomyocytes. Moreover, it has been suggested that many miRNAs play a critical role in regulating mitochondrial function in the heart (Figure 1E). The downregulation of miR-140, as well as the overexpression of miR-499 or miR-761, e.g., could prevent apoptosis and mitochondrial fission in cardiomyocytes via regulating mitochondrial fusion/fission-related proteins which led to cardiomyocyte apoptosis, mitochondrial fragmentation, and myocardial infarction (73, 74).
Roles of miRNAs in Glycogenesis in the Heart
In addition to the glucose consumed by normal metabolism, the excessive glucose can be converted to glycogen for storage through the glycogen synthesis pathway in the heart (75). Cardiac glycogen is an important source of glucose to support high-energy demands of a normal heart (76). Several studies have revealed that miRNAs played an important role in maintaining the balance of glycogen synthesis in the heart. Wei et al. (77), e.g., suggested that deletion of miR-1s led to a large portion in upregulated genes which associated with the cardiac fetal gene programing including glycolysis, cell proliferation, fetal sarcomere-associated genes, and glycogenesis by massively parallel sequencing. Moreover, they found that cardiac-specific overexpression of Errβ, the primary target of miR-1, could induce glycogen storage, cardiac dilation, and sudden cardiac death.
Several key enzymes such as glycogen synthase kinase-3α (GSK3α) and glycogen synthase kinase-3β (GSK3β) are involved in glycogen synthesis (78). It was reported that miRNAs could target GSK3β in some cardiac pathological processes, such as myocardial I/R injury, cardiac hypertrophy, and cardiac fibrosis, which suggested that miRNAs might be involved in glycogen synthesis. Our previous study, e.g., showed that miR-21-3p suppressed HDAC8 expression and decreased phospho-Akt and phospho-Gsk3β expression to attenuate cardiac hypertrophy (79). Moreover, miR-199a (80), miR-26 (81), miR-378 (82), miR-29c-3p, miR-144-3p, miR-195a-3p (83), and miR-126 (84) were reported to target GSK3β in direct or indirect manners during the occurrence and development of pathological cardiac hypertrophy, respectively. Meanwhile, miR-99b-3p (85), miR-154 (86), miR-382-3p, miR-3126-5p, and miR-450a-2-3p (87) were also found to target GSK3β in the pathological process of myocardial fibrosis. miR-122, miR-34a (88), miR-335 (89), miR-199a-5p (90, 91), miR-322/503 (92), miR-26a (93–95), miR-374 (96), and miR-378 (97) were found to target GSK3β in I/R injury model (Figure 1F). However, these studies did not explicitly indicate that these miRNAs were participated in cardiomyocyte glycogen synthesis during these cardiac pathological processes (Table 1).

Table 1. Roles of microRNAs in the glucose and lipid metabolism in the heart.
Roles of miRNAs in Lipid Metabolism in the Heart
The heart uses ketone bodies, lactate, glucose, fatty acids, and amino acids as energy-providing substrates, among which more than 70% of all substrates are derived from fatty acids to generate ATP in adult heart (98). Moreover, many studies indicate that miRNAs are essential for lipid metabolism in the heart.
Roles of miRNAs in Fatty Acids Transport in the Heart
Fatty acids (FAs) from albumin or lipoprotein triacylglycerol enter cardiomyocytes through passive diffusion or by protein carrier including fatty acid translocase (FAT)/CD36, fatty acid transport protein (FATP), and plasma membrane isoform of fatty acid-binding protein (FABPpm) (99, 100). Importantly, CD36 could translocate FAs across the membrane of cardiac myocytes. Many studies suggested that 50–60% of FA uptake and oxidation in heart was facilitated by FAT/CD36-mediated transport (101, 102). Unlike FATP or FABPpm, in the regulatory control of FA uptake, CD36 can translocate among the intracellular endosome, the sarcolemmal membrane, and the membrane to promote FA uptake (101, 102).
Many studies have reported that different miRNAs targeted CD36 mRNA and regulated its expression at the posttranscriptional level in a tissue-specific manner (103, 104). For example, in the process of bone marrow cell differentiation to the monocytic-macrophage line, Zhou et al. (105) reported that CD36 was increased and its expression level was associated with seven miRNAs, including miR-134, miR-130a, miR-199a, miR-141, miR-152, miR-363, and miR-342-3p. During the erythropoiesis, miR-26a, miR-22, miR-16, and miR-223 were detected to correlate with the level and appearance of CD36 as an erythroid surface antigen by performing the expression profiling of miRNAs (106). It should be noticed that the role of miRNAs targeting CD36 to regulate FA transport in the normal heart remains to be further investigated (Figure 2A).
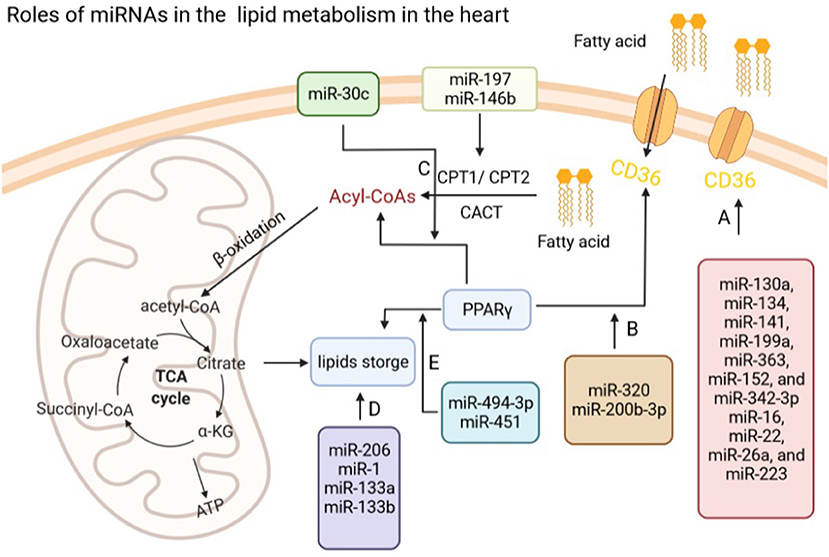
Figure 2. Roles of miRNAs in lipid metabolism in the heart. (A) miRNAs regulate fatty acids transport via modulating the expression of CD36 in the heart. (B) miR-320 and miR-200b-3p regulate fatty acid transport via PPARγ. (C) miR-197 and miR-146b modulate Acyl-CoAs by CPT1/2, while miR-30c regulates the production of Acyl-CoAs through targeting PPARγ. (D) miR-206, miR-1, and miR-133a/b influence lipids storage in the heart. (E) miR-494-3p and miR-451 regulate lipids storage via modulating the expression of PPARγ in the heart. CD36, fatty acid translocase (FAT)/CD36; CPT1/2, carnitine palmitoyltransferase 1/2; PPARγ, peroxisome proliferator-activated receptors (PPARγ).
In the cardiac pathological processes, the FA transport would be changed. During diabetic cardiomyopathy, e.g., energy source will be shifted from glucose to FAs and the FA transport is enhanced to meet the increasing demand of ATP (107). However, lipid accumulation in cardiomyocytes might eventually lead to lipid toxicity that promote contractile abnormalities and cell death (108, 109). Therefore, exploring whether miRNAs are involved in FA transport in diabetic hearts might be helpful in discovering potential therapeutic strategies for diabetes-induced cardiac dysfunction (Figure 2B). Our previous research showed that miR-320 could target the CD36 promoter directly resulting in increased transportation of FAs into diabetic cardiomyocytes via enhancing CD36 transcription by forming a complex with Ago2 (13). In addition, Xu et al. (110) also found miR-200b-3p expression was significantly reduced in diabetic cardiomyopathy tissues and cells, which could target CD36 directly to reduce cardiomyocytes apoptosis in diabetic cardiomyopathy.
Roles of miRNAs in Fatty Acid Oxidation in the Heart
FAs are the main energy source in adult heart. Acyl-CoA synthetases (ACS) activate cytoplasmic FAs to acyl-CoA esters and then imported into mitochondrion by two acyltransferases, carnitine acyl-carnitine translocase (CACT) and carnitine palmitoyl transferases 1 and 2 (CPT1 and CPT2). Acyl-CoAs are degraded via β-oxidation, finally producing acetyl-CoA to fuel the tricarboxylic acid (TCA) cycle inside the mitochondrion (111). Impaired fatty acid oxidation (FAO) led to the decrease of the capacity for ATP production and accumulation of toxic lipid intermediates in the heart, while enhanced FAO was associated with increased oxidative stress (2, 112).
Ekaterina et al. found that miR-146b and miR-197 were upregulation in the failing right ventricular of pulmonary arterial hypertension patients and suppressed genes that drive FAO (CPT1b and FABP4) in primary cardiomyocytes (113). Peroxisome proliferator-activated receptors (PPARs), a class of ligand-activated nuclear receptors, control FAO enzymes expression, while PPARγ coactivator-1β (PGC-1β) is an important coactivator of PPARs (114–116). Our previous work showed that exogenous miR-30c delivery improved lipid and glucose utilization, reduced excessive ROS production and thereby attenuated cardiac dysfunction via PGC-1β/PPARα signals in a mouse model of diabetic cardiomyopathy (117) (Figure 2C).
Roles of miRNAs in Lipid Storage in the Heart
Cardiomyocytes could reserve multiple energy substrates, among which accumulation of non-polar and polar lipids could activate intracellular signaling pathways (98). The FAs are stored as triacylglycerol (TAG) in lipid droplets. The accumulation of excess lipids is prevented by the physiological balance of lipid uptake and oxidation (98). However, various processes that affect this balance might lead to hypoxia, obesity, diabetes mellitus, sepsis, cardiac dysfunction, and even heart failure.
Many studies suggested that miRNAs play an important role in the lipid storage (Figure 2D). For example, Pegoraro et al. (118) suggested that miR-133a, miR-133b, miR-1, and miR-206, might be useful biomarkers for neutral lipid storage disease with myopathy. Ferland-McCollough et al. (119) demonstrated that miR-483-3p modulated the capacity of adipocytes to store lipids and differentiation by manipulating growth/differentiation factor-3 expression. Moreover, overexpression of miR-107 attenuated differentiation and lipid accumulation in pre- and mature human adipocytes of Simpson-Golabi-Behmel syndrome via regulating CDK6 and Notch signaling (120).
Considering the heart, metabolic cardiomyopathy is the main cause of heart failure in obese patients characterized by lipotoxic damage and intramyocardial triglyceride (TG) accumulation (121). JunD could enable transcription of genes involved in TG synthesis, uptake, hydrolysis, and storage by directly binding to PPARγ promoter. Costantino et al. found that miR-494-3p prevented TG synthesis, uptake, hydrolysis, and storage in the heart from diet-induced obese mice by suppressing JunD/PPARγ signaling, which was also associated with myocardial left ventricular (LV) dysfunction and TG accumulation (122). Besides, increased miR-451 were reported in the mouse heart with high-fat diet (HFD), while loss of miR-451 alleviated palmitate-induced lipotoxicity in cardiomyocytes via inhibiting calcium-binding protein 39 (Cab39), which is an AMP-activated protein kinase (AMPK) upstream kinase (123) (Figure 2E).
Potential miRNA-Based Therapy in CVDs
Therapeutic strategies targeting miRNAs for CVDs have been highlighted in many studies (124). For instance, miR-15 family was found to be consistently upregulated during postnatal development of the heart and CVDs, knockdown of the miR-15 family by anti-miRNAs could increase the number of mitotic CMs and reduce the infarct size after ischemia-reperfusion injury in neonatal mice (125–127). As miRNAs can affect different genes simultaneously to alter glucose and lipid metabolism in the pathological processes of diseased heart, they attracted increasing attentions for potential therapeutic targets and treatments (128, 129).
Various strategies were developed for the delivery miRNAs into cardiomyocytes. A novel technique called ultrasound-mediated sonoporation, which carry genetic material to target sites, using albumin-shelled microbubbles, has been considered for miRNA delivery in the myocardium (130). Importantly, Su et al. (131) has used this approach to prevent coronary microembolization-induced cardiac dysfunction by delivering hsa-miR-21-5p in pig myocardium by ultrasound-targeted microbubble. In addition, local injection is a nicely method to overcome the systemic effects on other organs and obtain better transfection efficiency. Many trials have attempted to inject miRNAs via intramyocardial or intracoronary directly during heart surgeries (132). Moreover, with the development of new techniques such as positron emission tomography and electromechanical mapping, clinicians can achieve high efficiency around the site of injection by better targeting site of myocardial ischemia (7, 133).
However, there are limitations of miRNA-based therapy, which should be solved before clinical use. Compared with the physiological miRNA expression levels, gain- and loss-of function assays using synthesized oligonucleotides often induce very high abundance of miRNA into the cells, which may lead to irreproducible and misguided interpretation of the results. Most of miRNA studies have been focused on site-specific phenotypic effects in vivo, which might ignore the signaling pathways responsible for their effects on other organs and the whole genome targets. Moreover, the off-target effects cannot be ignored. Thus, studies are needed to use both site-specific deliveries and systemic approach to focus on the in vivo miRNA effects.
Conclusion
An increasing number of studies have provided important clues of miRNAs and their potential roles in the glucose and lipid metabolism in CVDs. Current studies revealed the biological and pathological process that miRNAs involved, which might broaden the treatment strategies for CVD patients with or without metabolism disorders. In this review, we systematically described the effects of miRNAs on the glucose and lipid metabolism in cardiomyocytes and compared the advantages and limitations in miRNA-based therapy in CVDs. In addition, we provided a summary table to better illustrate the various miRNAs that participate in glucose and lipid metabolism in the heart. However, considering the multiple targets of one certain miRNA, there are still uncertainties that remain regarding the systemic effects of miRNAs on other organs and biological processes. In summary, miRNAs play critical roles in the regulation of glucose and lipid metabolism in CVDs. MiRNAs and miRNA-based therapies are one of the most promising innovative applications in CVD treatment in the future.
Author Contributions
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
Funding
This work was supported by grants from the National Natural Science Foundation of China [81822002 and 31771264 to CC; 31800973 to HL; and 82000387 to YZ]; the Fundamental Research Funds for the Central Universities [2019kfyXMBZ035 to CC]. No funding bodies had any role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank our colleagues from the Division of Cardiology, Tongji Hospital for stimulating discussions during this investigation.
References
1. Taegtmeyer H, Golfman L, Sharma S, Razeghi P, van Arsdall M. Linking gene expression to function: metabolic flexibility in the normal and diseased heart. Ann N Y Acad Sci. (2004) 1015:202–13. doi: 10.1196/annals.1302.017
2. Ritterhoff J, Tian R. Metabolism in cardiomyopathy: every substrate matters. Cardiovasc Res. (2017) 113:411–21. doi: 10.1093/cvr/cvx017
3. Akki A, Smith K, Seymour AM. Compensated cardiac hypertrophy is characterised by a decline in palmitate oxidation. Mol Cell Biochem. (2008) 311:215–24. doi: 10.1007/s11010-008-9711-y
4. Allard MF, Schonekess BO, Henning SL, English DR, Lopaschuk GD. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am J Physiol. (1994) 267:H742–50. doi: 10.1152/ajpheart.1994.267.2.H742
5. Barger PM, Kelly DP. Fatty acid utilization in the hypertrophied and failing heart: molecular regulatory mechanisms. Am J Med Sci. (1999) 318:36–42. doi: 10.1016/S0002-9629(15)40570-1
6. Opie LH. Myocardial ischemia–metabolic pathways and implications of increased glycolysis. Cardiovasc Drugs Ther. (1990) 4 (Suppl. 4):777–90. doi: 10.1007/BF00051275
7. Scheuermann-Freestone M, Madsen PL, Manners D, Blamire AM, Buckingham RE, Styles P, et al. Abnormal cardiac and skeletal muscle energy metabolism in patients with type 2 diabetes. Circulation. (2003) 107:3040–6. doi: 10.1161/01.CIR.0000072789.89096.10
8. Armoni M, Harel C, Bar-Yoseph F, Milo S, Karnieli E. Free fatty acids repress the GLUT4 gene expression in cardiac muscle via novel response elements. J Biol Chem. (2005) 280:34786–95. doi: 10.1074/jbc.M502740200
9. Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. (2004) 116:281–97. doi: 10.1016/S0092-8674(04)00045-5
10. Huntzinger E, Izaurralde E. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat Rev Genet. (2011) 12:99–110. doi: 10.1038/nrg2936
11. Zhang X, Zuo X, Yang B, Li Z, Xue Y, Zhou Y, et al. MicroRNA directly enhances mitochondrial translation during muscle differentiation. Cell. (2014) 158:607–19. doi: 10.1016/j.cell.2014.05.047
12. Li H, Zhang X, Wang F, Zhou L, Yin Z, Fan J, et al. MicroRNA-21 lowers blood pressure in spontaneous hypertensive rats by upregulating mitochondrial translation. Circulation. (2016) 134:734–51. doi: 10.1161/CIRCULATIONAHA.116.023926
13. Li H, Fan J, Zhao Y, Zhang X, Dai B, Zhan J, et al. Nuclear miR-320 mediates diabetes-induced cardiac dysfunction by activating transcription of fatty acid metabolic genes to cause lipotoxicity in the heart. Circ Res. (2019) 125:1106–20. doi: 10.1161/CIRCRESAHA.119.314898
14. Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, et al. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell. (2007) 129:303–17. doi: 10.1016/j.cell.2007.03.030
15. Chen J, Huang ZP, Seok HY, Ding J, Kataoka M, Zhang Z, et al. mir-17-92 cluster is required for and sufficient to induce cardiomyocyte proliferation in postnatal and adult hearts. Circ Res. (2013) 112:1557–66. doi: 10.1161/CIRCRESAHA.112.300658
16. Jayawardena TM, Egemnazarov B, Finch EA, Zhang L, Payne JA, Pandya K, et al. MicroRNA-mediated in vitro and in vivo direct reprogramming of cardiac fibroblasts to cardiomyocytes. Circ Res. (2012) 110:1465–73. doi: 10.1161/CIRCRESAHA.112.269035
17. Nam YJ, Song K, Luo X, Daniel E, Lambeth K, West K, et al. Reprogramming of human fibroblasts toward a cardiac fate. Proc Natl Acad Sci U S A. (2013) 110:5588–93. doi: 10.1073/pnas.1301019110
18. Babiarz JE, Ravon M, Sridhar S, Ravindran P, Swanson B, Bitter H, et al. Determination of the human cardiomyocyte mRNA and miRNA differentiation network by fine-scale profiling. Stem Cells Dev. (2012) 21:1956–65. doi: 10.1089/scd.2011.0357
19. Sluijter JP, van Mil A, van Vliet P, Metz CH, Liu J, Doevendans PA, et al. MicroRNA-1 and−499 regulate differentiation and proliferation in human-derived cardiomyocyte progenitor cells. Arterioscler Thromb Vasc Biol. (2010) 30:859–68. doi: 10.1161/ATVBAHA.109.197434
20. Chistiakov DA, Orekhov AN, Bobryshev YV. Cardiac-specific miRNA in cardiogenesis, heart function, and cardiac pathology (with focus on myocardial infarction). J Mol Cell Cardiol. (2016) 94:107–21. doi: 10.1016/j.yjmcc.2016.03.015
21. Wang K, Long B, Liu F, Wang JX, Liu CY, Zhao B, et al. A circular RNA protects the heart from pathological hypertrophy and heart failure by targeting miR-223. Eur Heart J. (2016) 37:2602–11. doi: 10.1093/eurheartj/ehv713
22. Ucar A, Gupta SK, Fiedler J, Erikci E, Kardasinski M, Batkai S, et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat Commun. (2012) 3:1078. doi: 10.1038/ncomms2090
23. Tijsen AJ, Pinto YM, Creemers EE. Non-cardiomyocyte microRNAs in heart failure. Cardiovasc Res. (2012) 93:573–82. doi: 10.1093/cvr/cvr344
24. Bao Q, Zhao M, Chen L, Wang Y, Wu S, Wu W, et al. MicroRNA-297 promotes cardiomyocyte hypertrophy via targeting sigma-1 receptor. Life Sci. (2017) 175:1–10. doi: 10.1016/j.lfs.2017.03.006
25. Feng B, Cao Y, Chen S, Chu X, Chu Y, Chakrabarti S. miR-200b mediates endothelial-to-mesenchymal transition in diabetic cardiomyopathy. Diabetes. (2016) 65:768–79. doi: 10.2337/db15-1033
26. Hinkel R, Ramanujam D, Kaczmarek V, Howe A, Klett K, Beck C, et al. AntimiR-21 prevents myocardial dysfunction in a pig model of ischemia/reperfusion injury. J Am Coll Cardiol. (2020) 75:1788–800. doi: 10.1016/j.jacc.2020.02.041
27. Li K, Zhao B, Wei D, Wang W, Cui Y, Qian L, et al. miR146a improves hepatic lipid and glucose metabolism by targeting MED1. Int J Mol Med. (2020) 45:543–55. doi: 10.3892/ijmm.2019.4443
28. Su J, Ren J, Chen H, Liu B. MicroRNA-140-5p ameliorates the high glucose-induced apoptosis and inflammation through suppressing TLR4/NF-kappaB signaling pathway in human renal tubular epithelial cells. Biosci Rep. (2020) 40:BSR20192384. doi: 10.1042/BSR20192384
29. Yin Z, Zhao Y, Du H, Nie X, Li H, Fan J, et al. A key GWAS-identified genetic variant contributes to hyperlipidemia by upregulating miR-320a. iScience. (2020) 23:101788. doi: 10.1016/j.isci.2020.101788
30. Wojciechowska A, Braniewska A, Kozar-Kaminska K. MicroRNA in cardiovascular biology and disease. Adv Clin Exp Med. (2017) 26:865–74. doi: 10.17219/acem/62915
31. Hata A. Functions of microRNAs in cardiovascular biology and disease. Annu Rev Physiol. (2013) 75:69–93. doi: 10.1146/annurev-physiol-030212-183737
32. Stanley WC, Lopaschuk GD, Hall JL, McCormack JG. Regulation of myocardial carbohydrate metabolism under normal and ischaemic conditions. Potential for pharmacological interventions. Cardiovasc Res. (1997) 33:243–57. doi: 10.1016/S0008-6363(96)00245-3
33. Young LH, Coven DL, Russell RR III. Cellular and molecular regulation of cardiac glucose transport. J Nucl Cardiol. (2000) 7:267–76. doi: 10.1016/S1071-3581(00)70016-X
34. Young LH, Renfu Y, Russell R, Hu X, Caplan M, Ren J, et al. Low-flow ischemia leads to translocation of canine heart GLUT-4 and GLUT-1 glucose transporters to the sarcolemma in vivo. Circulation. (1997) 95:415–22. doi: 10.1161/01.CIR.95.2.415
35. Lu H, Buchan RJ, Cook SA. MicroRNA-223 regulates Glut4 expression and cardiomyocyte glucose metabolism. Cardiovasc Res. (2010) 86:410–20. doi: 10.1093/cvr/cvq010
36. Nakamura M, Sadoshima J. Mechanisms of physiological and pathological cardiac hypertrophy. Nat Rev Cardiol. (2018) 15:387–407. doi: 10.1038/s41569-018-0007-y
38. Korvald C, Elvenes OP, Myrmel T. Myocardial substrate metabolism influences left ventricular energetics in vivo. Am J Physiol Heart Circ Physiol. (2000) 278:H1345–51. doi: 10.1152/ajpheart.2000.278.4.H1345
39. Liao R, Jain M, Cui L, D'Agostino J, Aiello F, Luptak I, et al. Cardiac-specific overexpression of GLUT1 prevents the development of heart failure attributable to pressure overload in mice. Circulation. (2002) 106:2125–31. doi: 10.1161/01.CIR.0000034049.61181.F3
40. Luptak I, Yan J, Cui L, Jain M, Liao R, Tian R. Long-term effects of increased glucose entry on mouse hearts during normal aging and ischemic stress. Circulation. (2007) 116:901–9. doi: 10.1161/CIRCULATIONAHA.107.691253
41. Horie T, Ono K, Nishi H, Iwanaga Y, Nagao K, Kinoshita M, et al. MicroRNA-133 regulates the expression of GLUT4 by targeting KLF15 and is involved in metabolic control in cardiac myocytes. Biochem Biophys Res Commun. (2009) 389:315–20. doi: 10.1016/j.bbrc.2009.08.136
42. Trotta MC, Maisto R, Alessio N, Hermenean A, D'Amico M, Di Filippo C. The melanocortin MC5R as a new target for treatment of high glucose-induced hypertrophy of the cardiac H9c2 cells. Front Physiol. (2018) 9:1475. doi: 10.3389/fphys.2018.01475
43. Yang T, Liu T, Cao C, Xu S. miR-200a-5p augments cardiomyocyte hypertrophy induced by glucose metabolism disorder via the regulation of selenoproteins. J Cell Physiol. (2019) 234:4095–103. doi: 10.1002/jcp.27206
44. Li J, Ren Y, Shi E, Tan Z, Xiong J, Yan L, Jiang X. Inhibition of the let-7 family microRNAS induces cardioprotection against ischemia-reperfusion injury in diabetic rats. Ann Thorac Surg. (2016) 102:829–35. doi: 10.1016/j.athoracsur.2016.02.016
45. Ju J, Xiao D, Shen N, Zhou T, Che H, Li X, et al. miR-150 regulates glucose utilization through targeting GLUT4 in insulin-resistant cardiomyocytes. Acta Biochim Biophys Sin (Shanghai). (2020) 52:1111–9. doi: 10.1093/abbs/gmaa094
46. Neely JR, Rovetto MJ, Oram JF. Myocardial utilization of carbohydrate and lipids. Prog Cardiovasc Dis. (1972) 15:289–329. doi: 10.1016/0033-0620(72)90029-1
47. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. (2009) 324:1029–33. doi: 10.1126/science.1160809
48. Mallat Y, Tritsch E, Ladouce R, Winter DL, Friguet B, Li Z, et al. Proteome modulation in H9c2 cardiac cells by microRNAs miR-378 and miR-378. Mol Cell Proteomics. (2014) 13:18–29. doi: 10.1074/mcp.M113.030569
49. Savabi F, Kirsch A. Altered functional activity and anoxic tolerance in diabetic rat isolated atria. Arch Biochem Biophys. (1990) 279:183–7. doi: 10.1016/0003-9861(90)90479-I
50. Jaswal JS, Keung W, Wang W, Ussher JR, Lopaschuk GD. Targeting fatty acid and carbohydrate oxidation–a novel therapeutic intervention in the ischemic and failing heart. Biochim Biophys Acta. (2011) 1813:1333–50. doi: 10.1016/j.bbamcr.2011.01.015
51. Liu Q, Docherty JC, Rendell JC, Clanachan AS, Lopaschuk GD. High levels of fatty acids delay the recovery of intracellular pH and cardiac efficiency in post-ischemic hearts by inhibiting glucose oxidation. J Am Coll Cardiol. (2002) 39:718–25. doi: 10.1016/S0735-1097(01)01803-4
52. Lei D, Wang Y, Zhang L, Wang Z. Circ_0010729 regulates hypoxia-induced cardiomyocyte injuries by activating TRAF5 via sponging miR-27a-3p. Life Sci. (2020) 262:118511. doi: 10.1016/j.lfs.2020.118511
53. Borden A, Kurian J, Nickoloff E, Yang Y, Troupes CD, Ibetti J, et al. Transient introduction of miR-294 in the heart promotes cardiomyocyte cell cycle reentry after injury. Circ Res. (2019) 125:14–25. doi: 10.1161/CIRCRESAHA.118.314223
54. Bartman CM, Oyama Y, Brodsky K, Khailova L, Walker L, Koeppen M, et al. Intense light-elicited upregulation of miR-21 facilitates glycolysis and cardioprotection through Per2-dependent mechanisms. PLoS ONE. (2017) 12:e0176243. doi: 10.1371/journal.pone.0176243
55. Zhu H, Xue H, Jin QH, Guo J, Chen YD. MiR-138 protects cardiac cells against hypoxia through modulation of glucose metabolism by targetting pyruvate dehydrogenase kinase 1. Biosci Rep. (2017) 37:BSR20170296. doi: 10.1042/BSR20170296
56. Fan JL, Zhu TT, Xue ZY, Ren WQ, Guo JQ, Zhao HY, et al. lncRNA-XIST protects the hypoxia-induced cardiomyocyte injury through regulating the miR-125b-hexokianse 2 axis. In Vitro Cell Dev Biol Anim. (2020) 56:349–57. doi: 10.1007/s11626-020-00459-0
57. Zhang Y, Liu G, Gao X. Attenuation of miR-34a protects cardiomyocytes against hypoxic stress through maintenance of glycolysis. Biosci Rep. (2017) 37:BSR20170925. doi: 10.1042/BSR20170925
58. Rane S, He M, Sayed D, Vashistha H, Malhotra A, Sadoshima J, et al. Downregulation of miR-199a derepresses hypoxia-inducible factor-1alpha and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ Res. (2009) 104:879–86. doi: 10.1161/CIRCRESAHA.108.193102
59. Donthi RV, Ye G, Wu C, McClain DA, Lange AJ, Epstein PN. Cardiac expression of kinase-deficient 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase inhibits glycolysis, promotes hypertrophy, impairs myocyte function, and reduces insulin sensitivity. J Biol Chem. (2004) 279:48085–90. doi: 10.1074/jbc.M405510200
60. Nascimben L, Ingwall JS, Lorell BH, Pinz I, Schultz V, Tornheim K, et al. Mechanisms for increased glycolysis in the hypertrophied rat heart. Hypertension. (2004) 44:662–7. doi: 10.1161/01.HYP.0000144292.69599.0c
61. Yang Y, Ishak Gabra MB, Hanse EA, Lowman XH, Tran TQ, Li H, et al. MiR-135 suppresses glycolysis and promotes pancreatic cancer cell adaptation to metabolic stress by targeting phosphofructokinase-1. Nat Commun. (2019) 10:809. doi: 10.1038/s41467-019-08759-0
62. Gray LR, Tompkins SC, Taylor EB. Regulation of pyruvate metabolism and human disease. Cell Mol Life Sci. (2014) 71:2577–604. doi: 10.1007/s00018-013-1539-2
63. Bricker DK, Taylor EB, Schell JC, Orsak T, Boutron A, Chen YC, et al. A mitochondrial pyruvate carrier required for pyruvate uptake in yeast, Drosophila, and humans. Science. (2012) 337:96–100. doi: 10.1126/science.1218099
64. Sun J, Liu Q, Zhao L, Cui C, Wu H, Liao L, et al. Potential regulation by miRNAs on glucose metabolism in liver of common carp (Cyprinus carpio) at different temperatures. Comp Biochem Physiol Part D Genomics Proteomics. (2019) 32:100628. doi: 10.1016/j.cbd.2019.100628
65. Harris RA, Bowker-Kinley MM, Huang B, Wu P. Regulation of the activity of the pyruvate dehydrogenase complex. Adv Enzyme Regul. (2002) 42:249–59. doi: 10.1016/S0065-2571(01)00061-9
66. Patel MS, Korotchkina LG. Regulation of the pyruvate dehydrogenase complex. Biochem Soc Trans. (2006) 34:217–22. doi: 10.1042/BST0340217
67. Zhang X, Ji R, Liao X, Castillero E, Kennel PJ, Brunjes DL, et al. MicroRNA-195 regulates metabolism in failing myocardium via alterations in sirtuin 3 expression and mitochondrial protein acetylation. Circulation. (2018) 137:2052–67. doi: 10.1161/CIRCULATIONAHA.117.030486
68. Buchwald A, Till H, Unterberg C, Oberschmidt R, Figulla HR, Wiegand V. Alterations of the mitochondrial respiratory chain in human dilated cardiomyopathy. Eur Heart J. (1990) 11:509–16. doi: 10.1093/oxfordjournals.eurheartj.a059743
69. Das S, Ferlito M, Kent OA, Fox-Talbot K, Wang R, Liu D, et al. Nuclear miRNA regulates the mitochondrial genome in the heart. Circ Res. (2012) 110:1596–603. doi: 10.1161/CIRCRESAHA.112.267732
70. He M, Lu Y, Xu S, Mao L, Zhang L, Duan W, et al. MiRNA-210 modulates a nickel-induced cellular energy metabolism shift by repressing the iron-sulfur cluster assembly proteins ISCU1/2 in Neuro-2a cells. Cell Death Dis. (2014) 5:e1090. doi: 10.1038/cddis.2014.60
71. Anderson NM, Mucka P, Kern JG, Feng H. The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell. (2018) 9:216–37. doi: 10.1007/s13238-017-0451-1
72. Ndi M, Marin-Buera L, Salvatori R, Singh AP, Ott M. Biogenesis of the bc1 complex of the mitochondrial respiratory chain. J Mol Biol. (2018) 430:3892–905. doi: 10.1016/j.jmb.2018.04.036
73. Long B, Wang K, Li N, Murtaza I, Xiao JY, Fan YY, et al. miR-761 regulates the mitochondrial network by targeting mitochondrial fission factor. Free Radic Biol Med. (2013) 65:371–9. doi: 10.1016/j.freeradbiomed.2013.07.009
74. Li J, Donath S, Li Y, Qin D, Prabhakar BS, Li P. miR-30 regulates mitochondrial fission through targeting p53 and the dynamin-related protein-1 pathway. PLoS Genet. (2010) 6:e1000795. doi: 10.1371/journal.pgen.1000795
75. Henning SL, Wambolt RB, Schonekess BO, Lopaschuk GD, Allard MF. Contribution of glycogen to aerobic myocardial glucose utilization. Circulation. (1996) 93:1549–55. doi: 10.1161/01.CIR.93.8.1549
76. Allard MF, Henning SL, Wambolt RB, Granleese SR, English DR, Lopaschuk GD. Glycogen metabolism in the aerobic hypertrophied rat heart. Circulation. (1997) 96:676–82. doi: 10.1161/01.CIR.96.2.676
77. Wei Y, Peng S, Wu M, Sachidanandam R, Tu Z, Zhang S, et al. Multifaceted roles of miR-1s in repressing the fetal gene program in the heart. Cell Res. (2014) 24:278–92. doi: 10.1038/cr.2014.12
78. Cohen P. The Croonian Lecture 1998 identification of a protein kinase cascade of major importance in insulin signal transduction. Philos Trans R Soc Lond B Biol Sci. (1999) 354:485–95. doi: 10.1098/rstb.1999.0399
79. Yan M, Chen C, Gong W, Yin Z, Zhou L, Chaugai S, et al. miR-21-3p regulates cardiac hypertrophic response by targeting histone deacetylase-8. Cardiovasc Res. (2015) 105:340–52. doi: 10.1093/cvr/cvu254
80. Li Z, Song Y, Liu L, Hou N, An X, Zhan D, et al. miR-199a impairs autophagy and induces cardiac hypertrophy through mTOR activation. Cell Death Differ. (2017) 24:1205–13. doi: 10.1038/cdd.2015.95
81. Zhang ZH, Li J, Liu BR, Luo CF, Dong Q, Zhao LN, et al. MicroRNA-26 was decreased in rat cardiac hypertrophy model and may be a promising therapeutic target. J Cardiovasc Pharmacol. (2013) 62:312–9. doi: 10.1097/FJC.0b013e31829b82e6
82. Nagalingam RS, Sundaresan NR, Gupta MP, Geenen DL, Solaro RJ, Gupta M. A cardiac-enriched microRNA, miR-378, blocks cardiac hypertrophy by targeting Ras signaling. J Biol Chem. (2013) 288:11216–32. doi: 10.1074/jbc.M112.442384
83. Guedes EC, Franca GS, Lino CA, Koyama FC, Moreira Ldo N, Alexandre JG, et al. MicroRNA expression signature is altered in the cardiac remodeling induced by high fat diets. J Cell Physiol. (2016) 231:1771–83. doi: 10.1002/jcp.25280
84. Ye X, Hemida MG, Qiu Y, Hanson PJ, Zhang HM, Yang D. MiR-126 promotes coxsackievirus replication by mediating cross-talk of ERK1/2 and Wnt/beta-catenin signal pathways. Cell Mol Life Sci. (2013) 70:4631–44. doi: 10.1007/s00018-013-1411-4
85. Yu YH, Zhang YH, Ding YQ, Bi XY, Yuan J, Zhou H, et al. MicroRNA-99b-3p promotes angiotensin II-induced cardiac fibrosis in mice by targeting GSK-3beta. Acta Pharmacol Sin. (2021) 42:715–25. doi: 10.1038/s41401-020-0498-z
86. Dong P, Liu WJ, Wang ZH. MiR-154 promotes myocardial fibrosis through beta-catenin signaling pathway. Eur Rev Med Pharmacol Sci. (2018) 22:2052–60. doi: 10.26355/eurrev_201804_14735
87. Liu L, Chen Y, Shu J, Tang CE, Jiang Y, Luo F. Identification of microRNAs enriched in exosomes in human pericardial fluid of patients with atrial fibrillation based on bioinformatic analysis. J Thorac Dis. (2020) 12:5617–27. doi: 10.21037/jtd-20-2066
88. Akbari G. Role of zinc supplementation on ischemia/reperfusion injury in various organs. Biol Trace Elem Res. (2020) 196:1–9. doi: 10.1007/s12011-019-01892-3
89. Wu N, Zhang X, Bao Y, Yu H, Jia D, Ma C. Down-regulation of GAS5 ameliorates myocardial ischaemia/reperfusion injury via the miR-335/ROCK1/AKT/GSK-3beta axis. J Cell Mol Med. (2019) 23:8420–31. doi: 10.1111/jcmm.14724
90. Liu DW, Zhang YN, Hu HJ, Zhang PQ, Cui W. Downregulation of microRNA199a5p attenuates hypoxia/reoxygenationinduced cytotoxicity in cardiomyocytes by targeting the HIF1alphaGSK3betamPTP axis. Mol Med Rep. (2019) 19:5335–44. doi: 10.3892/mmr.2019.10197
91. Zuo Y, Wang Y, Hu H, Cui W. Atorvastatin protects myocardium against ischemia-reperfusion injury through inhibiting miR-199a-5p. Cell Physiol Biochem. (2016) 39:1021–30. doi: 10.1159/000447809
92. Dong W, Xie F, Chen XY, Huang WL, Zhang YZ, Luo WB, et al. Inhibition of Smurf2 translation by miR-322/503 protects from ischemia-reperfusion injury by modulating EZH2/Akt/GSK3beta signaling. Am J Physiol Cell Physiol. (2019) 317:C253–61. doi: 10.1152/ajpcell.00375.2018
93. Lu S, Lu Y. MiR-26a inhibits myocardial cell apoptosis in rats with acute myocardial infarction through GSK-3beta pathway. Eur Rev Med Pharmacol Sci. (2020) 24:2659–66. doi: 10.26355/eurrev_202003_20535
94. Gong DD, Yu J, Yu JC, Jiang XD. Effect of miR-26a targeting GSK-3beta/beta-catenin signaling pathway on myocardial apoptosis in rats with myocardial ischemia-reperfusion. Eur Rev Med Pharmacol Sci. (2019) 23:7073–82. doi: 10.26355/eurrev_201908_18751
95. Park H, Park H, Mun D, Kang J, Kim H, Kim M, et al. Extracellular vesicles derived from hypoxic human mesenchymal stem cells attenuate GSK3beta expression via miRNA-26a in an Ischemia-reperfusion injury model. Yonsei Med J. (2018) 59:736–45. doi: 10.3349/ymj.2018.59.6.736
96. Zhang SB, Liu TJ, Pu GH, Li BY, Gao XZ, Han XL. MicroRNA-374 exerts protective effects by inhibiting SP1 through activating the PI3K/Akt pathway in rat models of myocardial ischemia-reperfusion after sevoflurane preconditioning. Cell Physiol Biochem. (2018) 46:1455–70. doi: 10.1159/000489186
97. Kim SW, Kim HW, Huang W, Okada M, Welge JA, Wang Y, et al. Cardiac stem cells with electrical stimulation improve ischaemic heart function through regulation of connective tissue growth factor and miR-378. Cardiovasc Res. (2013) 100:241–51. doi: 10.1093/cvr/cvt192
98. Schulze PC, Drosatos K, Goldberg IJ. Lipid use and misuse by the heart. Circ Res. (2016) 118:1736–51. doi: 10.1161/CIRCRESAHA.116.306842
99. Schwenk RW, Luiken JJ, Bonen A, Glatz JF. Regulation of sarcolemmal glucose and fatty acid transporters in cardiac disease. Cardiovasc Res. (2008) 79:249–58. doi: 10.1093/cvr/cvn116
100. van der Vusse GJ, van Bilsen M, Glatz JF. Cardiac fatty acid uptake and transport in health and disease. Cardiovasc Res. (2000) 45:279–93. doi: 10.1016/S0008-6363(99)00263-1
101. Hafstad AD, Solevag GH, Severson DL, Larsen TS, Aasum E. Perfused hearts from Type 2 diabetic (db/db) mice show metabolic responsiveness to insulin. Am J Physiol Heart Circ Physiol. (2006) 290:H1763–9. doi: 10.1152/ajpheart.01063.2005
102. Luiken JJ, Coort SL, Koonen DP, van der Horst DJ, Bonen A, Zorzano A, et al. Regulation of cardiac long-chain fatty acid and glucose uptake by translocation of substrate transporters. Pflugers Arch. (2004) 448:1–15. doi: 10.1007/s00424-003-1199-4
103. Lin HY, Wang FS, Yang YL, Huang YH. MicroRNA-29a suppresses CD36 to ameliorate high fat diet-induced steatohepatitis and liver fibrosis in mice. Cells. (2019) 8:1298. doi: 10.3390/cells8101298
104. Guo J, Fang W, Sun L, Lu Y, Dou L, Huang X, et al. Ultraconserved element uc.372 drives hepatic lipid accumulation by suppressing miR-195/miR4668 maturation. Nat Commun. (2018) 9:612. doi: 10.1038/s41467-018-03072-8
105. Zhou H, Zhang J, Eyers F, Xiang Y, Herbert C, Tay HL, et al. Identification of the microRNA networks contributing to macrophage differentiation and function. Oncotarget. (2016) 7:28806–20. doi: 10.18632/oncotarget.8933
106. Choong ML, Yang HH, McNiece I. MicroRNA expression profiling during human cord blood-derived CD34 cell erythropoiesis. Exp Hematol. (2007) 35:551–64. doi: 10.1016/j.exphem.2006.12.002
107. Zlobine I, Gopal K, Ussher JR. Lipotoxicity in obesity and diabetes-related cardiac dysfunction. Biochim Biophys Acta. (2016) 1861:1555–68. doi: 10.1016/j.bbalip.2016.02.011
108. Drosatos K, Pollak NM, Pol CJ, Ntziachristos P, Willecke F, Valenti MC, et al. Cardiac myocyte KLF5 regulates Ppara expression and cardiac function. Circ Res. (2016) 118:241–53. doi: 10.1161/CIRCRESAHA.115.306383
109. Wang L, Cai Y, Jian L, Cheung CW, Zhang L, Xia Z. Impact of peroxisome proliferator-activated receptor-alpha on diabetic cardiomyopathy. Cardiovasc Diabetol. (2021) 20:2. doi: 10.1186/s12933-020-01188-0
110. Xu L, Chen W, Ma M, Chen A, Tang C, Zhang C, et al. Microarray profiling analysis identifies the mechanism of miR-200b-3p/mRNA-CD36 affecting diabetic cardiomyopathy via peroxisome proliferator activated receptor-gamma signaling pathway. J Cell Biochem. (2019) 120:5193–206. doi: 10.1002/jcb.27795
111. Houten SM, Violante S, Ventura FV Wanders RJ. The biochemistry and physiology of mitochondrial fatty acid beta-oxidation and its genetic disorders. Annu Rev Physiol. (2016) 78:23–44. doi: 10.1146/annurev-physiol-021115-105045
112. Pillutla P, Hwang YC, Augustus A, Yokoyama M, Yagyu H, Johnston TP, et al. Perfusion of hearts with triglyceride-rich particles reproduces the metabolic abnormalities in lipotoxic cardiomyopathy. Am J Physiol Endocrinol Metab. (2005) 288:E1229–35. doi: 10.1152/ajpendo.00273.2004
113. Legchenko E, Chouvarine P, Borchert P, Fernandez-Gonzalez A, Snay E, Meier M, et al. PPARgamma agonist pioglitazone reverses pulmonary hypertension and prevents right heart failure via fatty acid oxidation. Sci Transl Med. (2018) 10:eaao0303. doi: 10.1126/scitranslmed.aao0303
114. Lefebvre P, Chinetti G, Fruchart JC Staels B. Sorting out the roles of PPAR alpha in energy metabolism and vascular homeostasis. J Clin Invest. (2006) 116:571–80. doi: 10.1172/JCI27989
115. Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. (2000) 106:847–56. doi: 10.1172/JCI10268
116. Rowe GC, Jiang A, Arany Z. PGC-1 coactivators in cardiac development and disease. Circ Res. (2010) 107:825–38. doi: 10.1161/CIRCRESAHA.110.223818
117. Yin Z, Zhao Y, He M, Li H, Fan J, Nie X, et al. MiR-30c/PGC-1beta protects against diabetic cardiomyopathy via PPARalpha. Cardiovasc Diabetol. (2019) 18:7. doi: 10.1186/s12933-019-0811-7
118. Pegoraro V, Missaglia S, Marozzo R, Tavian D, Angelini C. MiRNAs as biomarkers of phenotype in neutral lipid storage disease with myopathy. Muscle Nerve. (2020) 61:253–7. doi: 10.1002/mus.26761
119. Ferland-McCollough D, Fernandez-Twinn DS, Cannell IG, David H, Warner M, Vaag AA, et al. Programming of adipose tissue miR-483-3p and GDF-3 expression by maternal diet in type 2 diabetes. Cell Death Differ. (2012) 19:1003–12. doi: 10.1038/cdd.2011.183
120. Ahonen MA, Haridas PAN, Mysore R, Wabitsch M, Fischer-Posovszky P, Olkkonen VM. miR-107 inhibits CDK6 expression, differentiation, and lipid storage in human adipocytes. Mol Cell Endocrinol. (2019) 479:110–6. doi: 10.1016/j.mce.2018.09.007
121. Nishida K, Otsu K. Inflammation and metabolic cardiomyopathy. Cardiovasc Res. (2017) 113:389–98. doi: 10.1093/cvr/cvx012
122. Costantino S, Akhmedov A, Melina G, Mohammed SA, Othman A, Ambrosini S, et al. Obesity-induced activation of JunD promotes myocardial lipid accumulation and metabolic cardiomyopathy. Eur Heart J. (2019) 40:997–1008. doi: 10.1093/eurheartj/ehy903
123. Kuwabara Y, Horie T, Baba O, Watanabe S, Nishiga M, Usami S, et al. MicroRNA-451 exacerbates lipotoxicity in cardiac myocytes and high-fat diet-induced cardiac hypertrophy in mice through suppression of the LKB1/AMPK pathway. Circ Res. (2015) 116:279–88. doi: 10.1161/CIRCRESAHA.116.304707
124. Kwekkeboom RF, Lei Z, Doevendans PA, Musters RJ Sluijter JP. Targeted delivery of miRNA therapeutics for cardiovascular diseases: opportunities and challenges. Clin Sci. (2014) 127:351–65. doi: 10.1042/CS20140005
125. Boon RA, Dimmeler S. MicroRNAs in myocardial infarction. Nat Rev Cardiol. (2015) 12:135–42. doi: 10.1038/nrcardio.2014.207
126. Hullinger TG, Montgomery RL, Seto AG, Dickinson BA, Semus HM, Lynch JM, et al. Inhibition of miR-15 protects against cardiac ischemic injury. Circ Res. (2012) 110:71–81. doi: 10.1161/CIRCRESAHA.111.244442
127. Porrello ER, Mahmoud AI, Simpson E, Johnson BA, Grinsfelder D, Canseco D, et al. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc Natl Acad Sci U S A. (2013) 110:187–92. doi: 10.1073/pnas.1208863110
128. Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov. (2017) 16:203–22. doi: 10.1038/nrd.2016.246
129. Matsui M, Corey DR. Non-coding RNAs as drug targets. Nat Rev Drug Discov. (2017) 16:167–79. doi: 10.1038/nrd.2016.117
130. Kwekkeboom RF, Sluijter JP, van Middelaar BJ, Metz CH, Brans MA, Kamp O, et al. Increased local delivery of antagomir therapeutics to the rodent myocardium using ultrasound and microbubbles. J Control Release. (2016) 222:18–31. doi: 10.1016/j.jconrel.2015.11.020
131. Su Q, Li L, Liu Y, Zhou Y, Wang J, Wen W. Ultrasound-targeted microbubble destruction-mediated microRNA-21 transfection regulated PDCD4/NF-kappaB/TNF-alpha pathway to prevent coronary microembolization-induced cardiac dysfunction. Gene Ther. (2015) 22:1000–6. doi: 10.1038/gt.2015.59
132. Poggioli T, Sarathchandra P, Rosenthal N, Santini MP. Intramyocardial cell delivery: observations in murine hearts. J Vis Exp. (2014) 83:e51064. doi: 10.3791/51064
Keywords: microRNAs, glucose, lipid, heart, metabolism
Citation: Du H, Zhao Y, Li H, Wang DW and Chen C (2021) Roles of MicroRNAs in Glucose and Lipid Metabolism in the Heart. Front. Cardiovasc. Med. 8:716213. doi: 10.3389/fcvm.2021.716213
Received: 28 May 2021; Accepted: 21 June 2021;
Published: 22 July 2021.
Edited by:
Zhanpeng Huang, The First Affiliated Hospital of Sun Yat-sen University, ChinaReviewed by:
Jinghai Chen, Zhejiang University, ChinaZhi Xin Shan, Guangdong Provincial People's Hospital, China
Copyright © 2021 Du, Zhao, Li, Wang and Chen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Chen Chen, Y2hlbmNoZW5AdGpoLnRqbXUuZWR1LmNu