 Natalie N. Khalil
Natalie N. Khalil Megan L. McCain
Megan L. McCain
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med., 14 July 2021
Sec. Atherosclerosis and Vascular Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.709871
This article is part of the Research TopicTechnological Innovations in Cardiovascular Medicine: Imaging, nanotechnology, tissue regeneration, genetic engineering, deep learning and beyondView all 15 articles
Myocardial infarctions are one of the most common forms of cardiac injury and death worldwide. Infarctions cause immediate necrosis in a localized region of the myocardium, which is followed by a repair process with inflammatory, proliferative, and maturation phases. This repair process culminates in the formation of scar tissue, which often leads to heart failure in the months or years after the initial injury. In each reparative phase, the infarct microenvironment is characterized by distinct biochemical, physical, and mechanical features, such as inflammatory cytokine production, localized hypoxia, and tissue stiffening, which likely each contribute to physiological and pathological tissue remodeling by mechanisms that are incompletely understood. Traditionally, simplified two-dimensional cell culture systems or animal models have been implemented to elucidate basic pathophysiological mechanisms or predict drug responses following myocardial infarction. However, these conventional approaches offer limited spatiotemporal control over relevant features of the post-infarct cellular microenvironment. To address these gaps, Organ on a Chip models of post-infarct myocardium have recently emerged as new paradigms for dissecting the highly complex, heterogeneous, and dynamic post-infarct microenvironment. In this review, we describe recent Organ on a Chip models of post-infarct myocardium, including their limitations and future opportunities in disease modeling and drug screening.
Cardiovascular disease is the leading cause of death worldwide, responsible for over 17.9 million deaths annually (1). One of the most common causes of cardiovascular disease is coronary artery disease, in which plaque buildup in the coronary arteries deprives downstream myocardium of vital, oxygenated blood. Complete occlusion of the coronary arteries can ultimately lead to myocardial infarction, commonly referred to as a heart attack. Myocardial infarction is among the top five most expensive conditions treated by US hospitals annually and is a common cause of heart failure (2–4).
When a patient presents with a myocardial infarction, percutaneous coronary intervention is often performed, which is a catheterization procedure to restore flow to the interrupted artery and minimize the initial insult (5, 6). Rapid identification of myocardial infarction is essential for timely reperfusion of the occluded coronary artery, which can, in some cases, lead to reperfusion injury that further increases the size of the initial insult (7). After revascularization procedures, treatment is predominantly focused on pharmaceutical interventions to reduce adverse cardiovascular events, including stroke, recurrent infarctions, and death (8). Common treatments include renin-angiotensin-aldosterone system (RAAS) inhibitors, such as angiotensin-converting enzyme inhibitors, which decrease the load on the heart by lowering blood pressure (9) and may also reduce fibrotic remodeling (10). Another therapeutic target is beta adrenergic receptors, which are activated by epinephrine or norepinephrine to stimulate heart rate, strength of contraction, and cardiac output. Similar to RAAS inhibitors, beta blockers, such as bisoprolol, carvedilol, and metoprolol, can reduce blood pressure following myocardial infarction (8, 10). Thus, existing pharmacological interventions are primarily focused on reducing the load on the damaged heart instead of attempting to repair the initial injury or mitigate the ensuing fibrotic remodeling process.
One challenge facing the development of new therapies for post-infarct myocardium is that conventional preclinical models are limited primarily to non-human animal models or static, uniform monolayers of cultured cells that poorly replicate the clinical setting, especially the complex, dynamic remodeling that occurs post-infarction (3). Thus, developing the next generation of treatments for myocardial infarction can be accelerated by new preclinical model systems that more closely replicate native pathophysiology across multiple spatial scales, including the post-infarct cellular microenvironment.
Organs on Chips are engineered in vitro systems that mimic the fundamental structural and functional units of native tissues and have been shown to mimic responses to drugs at clinical doses in various tissue models with higher fidelity than conventional in vitro methods (11). Organs on Chips can also provide enhanced spatiotemporal control over multiple distinct features of the cellular microenvironment, which is especially relevant to modeling post-infarct myocardium. Thus, Organ on Chip approaches have vast potential to provide new mechanistic insights into post-infarct remodeling and to inform future pharmacological interventions.
In this review, we will first describe the cellular and molecular remodeling that occurs after myocardial infarction in humans, which will provide a framework for the microenvironmental features that are important to model on a chip. Next, we will describe existing strategies for engineering aspects of the cellular microenvironment of post-infarct myocardium on a chip, which have until now focused on hypoxia, fibrosis, and strain in both 2-dimensional (2-D) and 3-dimensional (3-D) tissue constructs. Lastly, we will describe considerations for future work to promote clinical mimicry and translation of Organ on a Chip models of myocardial infarction.
The initial coronary occlusion of a myocardial infarction deprives downstream myocardium of vital oxygen and nutrients, causing cardiac myocyte necrosis within hours (10, 12, 13). Due to tissue necrosis, the infarct zone is vulnerable to deformation and thinning, which can lead to infarct expansion (14, 15). Cardiac myocytes in the surviving infarct border zone myocardium also begin to rapidly remodel. For example, the normal distribution of intercalated disc protein complexes (gap junctions, desmosomes, and adherens junctions) is lost as early as 6 h post-infarction. Because intercalated disks are necessary for the heart to function as an electromechanical syncytium, changes in their expression and localization are thought to contribute to arrhythmogenesis after infarction (16).
As a result of infarct expansion, diastolic and systolic wall stresses increase (17, 18). To normalize the increased load, the myocardium undergoes cardiac hypertrophy in the weeks and months after an infarction by increasing muscle mass and wall thickness (19). These changes are reflected in the morphology of individual cardiac myocytes, which can increase in both length and diameter. Due to the spatial arrangement of cardiac myocytes in the ventricles, increased diameter and lengthening of individual cells ultimately results in changes in chamber geometry (6, 20, 21), which contributes to ventricular enlargement. Increasing chamber volume through ventricular enlargement may be compensatory in order to maintain stroke volume after the initial loss of contractility (14, 22) but is ultimately associated with increased likelihood of mortality (14, 23).
Tissue necrosis and infarct expansion can also increase the likelihood of myocardial rupture (14, 15). Because mammalian cardiac myocytes have limited regenerative capacity, maintaining tissue integrity is dependent on the formation of a scar. Scar formation occurs through three overlapping phases: the inflammatory phase, proliferative phase, and maturation phase (Figure 1). The inflammatory phase occurs in the first few days following a myocardial infarction and is initiated when necrotic cardiac myocytes release their intracellular contents, which activate pro-inflammatory signaling pathways in innate immune cells (24). Next, inflammatory chemokine and cytokine gradients [comprised of tumor necrosis factor-α (TNF-α), interleukin (IL)-1β, and IL-6] promote leukocyte migration into the infarct zone to clear dead cell debris and damaged extracellular matrix. After 1 week, tissue inhibitors of metalloproteinases are upregulated to conclude this degradative phase (24). Ultimately, as neutrophils undergo apoptosis, macrophages are directed toward a resolving phenotype and begin secreting anti-inflammatory signals [such as transforming growth factor-β (TGF-β) and IL-10] that repress the inflammatory response and drive cardiac fibroblast activation in the subsequent proliferative phase (10).
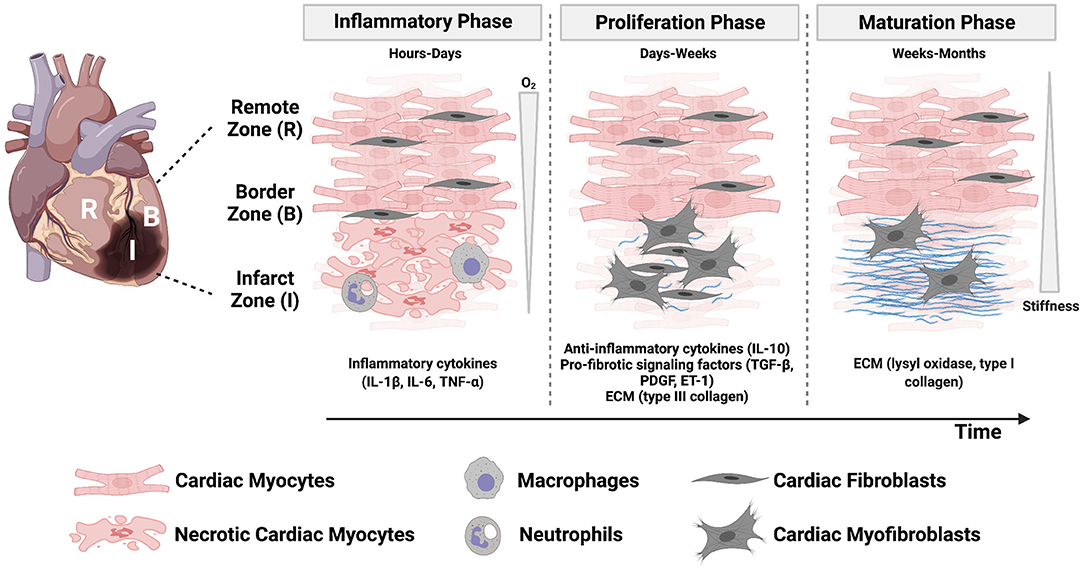
Figure 1. The post-infarct microenvironment is characterized by dynamic physical, mechanical, and biochemical cues that orchestrate the three phases of healing. Regional and temporal changes of oxygen concentration, stiffness, inflammatory cytokines, cell, and matrix composition characterize the myocardium as injured tissue is replaced with a scar. Created with BioRender.com.
The proliferative phase occurs over the next few weeks and is characterized by cardiac fibroblast proliferation, migration into the site of injury, and differentiation into an activated myofibroblast phenotype. Cardiac fibroblasts are the most abundant non-myocyte cell population in the mammalian myocardium (24) and, after expansion in the proliferative phase in combination with cardiac myocyte necrosis, they become the most abundant cell type in the infarcted region (10). Myofibroblasts play an important role in depositing collagen, fibronectin, and other matrix proteins to maintain tissue integrity and prevent myocardial rupture. They also express α-smooth muscle actin (α-SMA) and non-muscle myosin, which provide them with the ability to generate force to migrate and facilitate wound contracture (10).
The final phase is scar maturation, which occurs on a timescale of weeks to months. In this phase, myofibroblasts initiate collagen turnover and begin secreting type I collagen, which has the tensile strength of steel (12), in place of type III collagen. The depletion of growth factors necessary for myofibroblast survival ultimately leads to myofibroblast apoptosis (10, 25). Further enzymatic cross-linking of collagen occurs through lysyl oxidase, which progressively increases the tensile strength of the myocardium for months after myocardial infarction (10). While myofibroblasts play a vital role in maintaining structural integrity after the initial loss of tissue, they may also be driven by many cellular and molecular events toward a pathological fibrotic response. Myofibroblast persistence in the myocardium can occur for months, or even years, after injury and is a common feature of heart failure (12). Fibrotic tissue, which was historically considered an inert tissue, is now known to secrete factors (such as angiotensin II and TGF-β) that can traverse the interstitial space and promote fibrosis in non-infarcted regions (12, 26).
In summary, post-infarct myocardium is characterized by multiple biochemical and biomechanical properties changing in both space and time, which are correlated to complex remodeling of several cell types and the extracellular matrix in parallel (Figure 1). Some or all of these remodeling processes ultimately impact cardiac function and patient outcomes. However, the relationships between these biochemical, biomechanical, cellular, and molecular factors are incompletely understood, hindering the discovery of new therapies to mitigate the effects of the initial injury. Thus, there is a great need for controlled experimental models of post-infarct myocardium that account for remodeling of the cellular microenvironment to uncover mechanisms of pathophysiology.
Due to the high metabolic activity of cardiac myocytes, the myocardium is a highly vascularized tissue, with capillaries separated by approximately 20 μm (27). This translates to about one blood vessel between every two cardiac myocytes (28). As a result, hypoxia is one of the most injurious effects of a myocardial infarction. Conventionally, hypoxia has been induced by culturing cells in environments with uniformly low oxygen. However, phosphorescent oxygen probes have demonstrated that a spatial gradient ranging from 0 to 10% oxygen bridges injured tissue with neighboring viable tissue in post-infarct myocardium (29, 30). In addition, oxygen concentrations change over time as the infarct zone is re-oxygenated during reperfusion. Thus, in vitro systems that can modulate oxygen concentrations in space or time have more recently been developed to mimic the hypoxic landscape of post-infarct myocardium. In this section, we will describe conventional hypoxia models that replicate uniform hypoxia as well as engineered systems that offer spatial or temporal control over oxygen tension.
To recapitulate myocardial hypoxia in vitro (3), one of the most common approaches is to place cardiac cells in incubators or hypoxia chambers and replace oxygen with nitrogen. This enables the stable, long-term induction of hypoxia, with tunable control over global oxygen levels by selecting a gas composition of choice. To enable cell handling, hypoxia workstations have also been developed that contain gloveboxes to allow for the manipulation of cells in a hypoxic enclosure. However, these approaches for physical induction of hypoxia require access to specialized equipment, such as incubators with oxygen regulation, and are limited to uniform gas concentrations.
Hypoxia can also be simulated in cells cultured in ambient oxygen by adding hypoxia mimetic agents, such as cobalt chloride, to cell media. Hypoxia mimetic agents often work by stabilizing hypoxia inducible factors (HIF), a family of transcription factors that facilitates the cellular response to hypoxia by upregulating genes associated with survival in low oxygen. In normoxia, HIF is constantly degraded through hydroxylation of the HIF-α subunit by the enzyme prolyl hydroxylase, which marks it for ubiquitination by Von Hippel Lindau protein and subsequent degradation. Cobalt chloride chemically stabilizes HIF in normoxia by replacing Fe2+ with Co2+ in the prolyl hydroxylase active site, thereby inhibiting its hydroxylation of HIF-α and the ensuing degradation pathway. Other successful hypoxia mimetics include dimethyloxalylglycine and deferoxamine, which similarly work by inhibiting prolyl hydroxylase activity (31). Hypoxia mimetics enable easy access to cells during cell culture, are inexpensive, and can quickly simulate hypoxic conditions in vitro. However, they can have adverse effects on other signaling pathways that are not affected by low oxygen (31, 32), may be cytotoxic (33), and likely do not capture all the effects of true hypoxia.
Conventional in vitro models have shown that hypoxia results in structural and functional changes that may contribute to the development of arrhythmias. Specifically, the gap junction protein connexin 43 (Cx43) is known to be affected, which plays a critical role in conducting electrical impulses in the myocardium. Hypoxia has been shown to drive electrical uncoupling in vitro, with effects including decreased Cx43 signal at gap junctions (34, 35), increased Cx43 internalization (35) and dephosphorylation (36), and decreased conduction velocity (34). Hypoxia is also associated with inactivation of sodium potassium pumps (Na,K ATPase), which regulate cardiac action potentials (37, 38). Hypoxic cardiac myocytes also upregulate fetal, T type calcium channels, which are absent in healthy, adult myocardium, in a mechanism dependent on HIF-1α (39). Finally, hypoxia has also been shown to promote apoptosis in cardiac myocytes (40–43). These remodeling processes are possibly related to the increased incidence of arrhythmias observed in post-infarct myocardium.
Studies have also shown that cardiac fibroblasts and other non-myocyte cell populations are generally more resistant to hypoxia (40–42). In response to hypoxic stimuli in vitro, cardiac fibroblasts undergo differentiation into the activated, myofibroblast phenotype, which is signified by increased α-SMA expression (44–46), collagen type I expression (44–47), and migration capacity (46). However, there have been differing reports of cardiac fibroblast proliferation in response to hypoxia (44–46), which may result from differences in basal levels of fibroblast differentiation, or from differences in cell source, oxygen concentration, and hypoxia duration. In response to prolonged hypoxia exposure in vitro, cardiac fibroblasts ultimately undergo apoptosis (48–50).
Hypoxia also changes the secretion of paracrine factors that are involved in many processes of infarct healing, such as angiogenesis, fibroblast differentiation, and remodeling of the extracellular matrix. Hypoxic cardiac myocytes upregulate vascular endothelial growth factor (VEGF) (51, 52), insulin-like growth factor 2 (52), inflammatory cytokines (TNF-α, IL-1β, IL-6), and TGF-β (53). As TGF-β is known to promote fibroblast differentiation into myofibroblasts, medium conditioned by hypoxic myocytes has been reported to drive cardiac fibroblast migration (54) and facilitate faster wound closure in cultured skin fibroblasts (53). Similar to cardiac myocytes, hypoxic cardiac fibroblasts upregulate TGF-β1 and also its receptor, TGFβ-R1, which can play a role in autocrine signaling pathways to promote fibroblast differentiation (46, 48). Hypoxic fibroblasts also exhibit increased secretion of inflammatory cytokines [TNF-α (55–57) and IL-6 (57)], matrix metalloproteinases [MMP-2 and MMP-9 (56)] and VEGF (56), and conditioned medium from hypoxic fibroblasts has been shown to reduce cardiac myocyte viability (55, 58). Thus, hypoxia likely alters cellular cross-talk between distinct cardiac cell types in post-infarct myocardium.
Although conventional hypoxia systems have revealed valuable insights into oxygen-dependent remodeling of cardiac cell types, they do not replicate the spatial or temporal changes in oxygen that are characteristic of post-infarct myocardium. To mimic spatial oxygen gradients, more complex in vitro systems have been engineered. In one example, a microfluidic device was fabricated with a central channel designated for cell culture embedded between two lateral media channels. By flowing media containing a chemical hypoxia mimetic through one channel and standard media through the other, a chemical hypoxia gradient was established in the central cell-containing channel. Cardiac myoblasts near the hypoxic end of the gradient exhibited altered morphology, including reduced cell area and actin disintegration, which was accompanied by mitochondrial dysfunction and decreased cell viability (59). In addition to chemical methods (60–63), microfluidic devices have also been developed to generate physical oxygen gradients by culturing cells on a gas-permeable membrane above microchannels for gas flow (64–66). However, these methods have not been extensively applied to cardiac cell types.
Ischemic gradients have also been developed by stacking thin layers of hydrogels that are mechanically supported by a paper scaffold, a technique termed “cells-in-gels-in-paper.” To control oxygen and nutrient diffusion into the stack, one end of the construct was placed in a base that is impermeable to gases and liquids. Nutrients become depleted as they diffuse into the stack, which creates an ischemic environment in the lower layers. Cardiac myocytes in the ischemic, lower layers exhibited reduced viability and circular morphology when compared with upper layers. Cell-tracking demonstrated that cardiac fibroblasts embedded in upper layers migrate toward ischemic cardiac myocytes. Fibroblast migration increased when myocytes were exposed to higher levels of ischemia (generated through taller stacks) and was reduced in the absence of cardiac myocytes or with the pharmacological inhibition of TGF-β (27). Other methods have been developed that similarly modify hydrogels to generate oxygen gradients (67) by using oxygen-consuming enzymes during hydrogel cross-linking (68, 69), embedding a hydrogel between gas flow channels (70), or linearly increasing cell density and thus oxygen consumption rates (71), but these methods have not yet been implemented to model post-infarct myocardium.
Lastly, cardiac spheroids have also been implemented to mimic hypoxia gradients. Cardiac spheroids are 3-D aggregates of cardiac cells that recapitulate select aspects of native tissue structure and function (72–76). Because the diffusion limit of oxygen in tissues is around 100–200 microns (77), cardiac spheroids intrinsically generate oxygen gradients, for which oxygen tension is highest at the surface and decreases toward the necrotic core. Though this is conventionally thought of as a limitation of spheroids, recent work has harnessed this property to develop “infarct spheroids” that are exposed to ambient 10% oxygen to generate spatial hypoxia gradients (0–10% oxygen) that mimic the infarct, border, and remote zones after infarction (Figure 2A). These infarct spheroids contained cardiac myocytes, endothelial cells, and stromal cells and were treated with noradrenaline to mimic neurohormonal stimulation after infarction. Infarct spheroids demonstrated similar global gene-expression profiles to human ischemic cardiomyopathy and animal myocardial infarction samples. Furthermore, when compared with control spheroids in ambient oxygen, infarct spheroids exhibited a metabolic shift toward glycolysis, increased stiffness, increased expression of myofibroblast markers, decreased cardiac myocyte contraction amplitude (Figure 2A), and asynchronization of contractions (78).
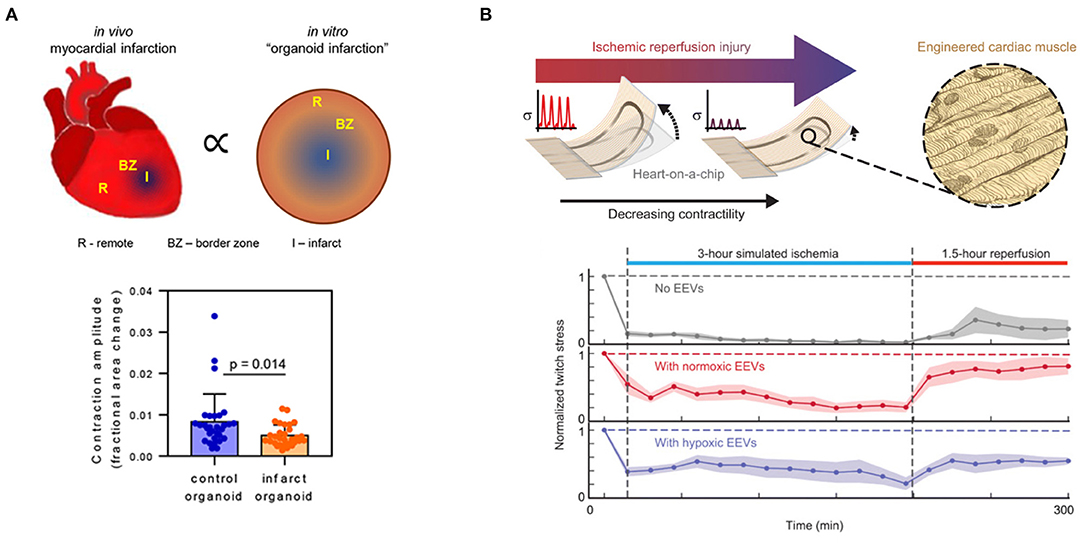
Figure 2. Engineered in vitro hypoxia models that modulate oxygen in (A) space and (B) time. (A) Infarct spheroids generate intrinsic spatial oxygen gradients to model the infarct, border, and remote zones after myocardial infarction. Infarct spheroids exhibit higher levels of myofibroblast marker expression, exhibit asynchronous contraction of myocytes, and have lower contraction amplitudes than control spheroids. Reprinted by permission from Springer Nature: Nat. Biomed. Eng. (78). (B) Muscular thin films with integrated strain gauges can measure changes in contractility in cardiac myocytes pre-treated with endothelial extracellular vesicles in a simulated ischemia-reperfusion injury. Endothelial extracellular vesicles enhanced the contractility of engineered cardiac tissues during both ischemia and reperfusion. From Yadid et al. (79). Reprinted with permission from AAAS.
In vitro models have also been engineered to replicate dynamic changes in oxygen characteristic of ischemia-reperfusion. Microfluidic devices are particularly suitable for this application because they contain chambers of small volumes that can be rapidly filled with hypoxic gas or cell medium. For example, a microfluidic device with integrated bioelectronics was used to measure intracellular action potential and extracellular beat rate and propagation velocity in cardiac myocytes cultured in a microchannel. The microchannel was rapidly filled with hypoxic cell medium followed by recovery medium to mimic ischemia-reperfusion. Hypoxic cardiac myocytes demonstrated substantially reduced depolarization times and beat rates, as well as irregular propagation patterns, which recovered within 30 min after reintroducing normoxic cell media (80). A similar microfluidic device was fabricated to contain small chamber volumes that can be quickly filled with hypoxic gas. Gas from the upper chamber diffused across a thin, gas-permeable membrane to reach cells cultured in lower microfluidic channels. This device demonstrated that hypoxic conditions below 5% oxygen induce changes in cardiac myocyte calcium transients, including a decrease in amplitude that could be mimicked using L-type calcium channel antagonists. After a subsequent 10 min of reperfusion with normoxic gas, cardiac myocytes recovered with normal calcium transients (81). Together, these studies suggest that hypoxia induces reversible alterations in cardiac myocyte electrophysiology.
In another model of ischemia-reperfusion, cardiac myocytes were cultured as an aligned tissue on silicone cantilever substrates with embedded strain sensors (Figure 2B). The cardiac myocytes were aligned using microcontact printing, which involves the preparation of silicone “stamps” that are used to transfer extracellular matrix proteins to a substrate in desired geometries. With a spatial resolution of approximately 1 μm, microcontact printing can direct tissue orientation (82–84), single-cell shape (85), and even subcellular structures (86). When the aligned cardiac tissues contracted on the cantilever substrates, the cantilevers deflected, leading to a resistance change in the embedded strain sensors proportional to the contractile stress (87, 88). This system was used to provide real-time measurements of contractile stress in a simulated ischemia-reperfusion injury by switching cells from ischemic media at 1% oxygen to standard media at ambient 21% oxygen. Integrated sensor readouts demonstrated that cardiac tissues stopped contracting during ischemia and displayed a minor recovery of twitch stress during reperfusion. In contrast, pre-treatment with endothelial cell-derived extracellular vesicles was cardioprotective and enabled cardiac tissues to continue contracting during simulated ischemia and exhibit higher recovery of twitch stress after reperfusion (Figure 2B) (79).
In summary, several technologies, including microfluidic devices, hydrogels, spheroids, and strain sensors, have been implemented to mimic the spatial and temporal oxygen gradients characteristic of post-infarct myocardium and subsequently quantify changes in cardiac cell phenotypes. Unlike conventional systems, these approaches can be used to explore oxygen-dependent regional and temporal changes in cellular phenotypes at high resolution and uncover cross-talk between cells in distinct oxygen environments to identify new mechanisms of infarct remodeling.
Healthy myocardium is a moderately stiff tissue, with an elastic modulus of around 10 kPa. After myocardial infarction, local elastic modulus in the infarcted region increases to 20–100 kPa due to scar formation and fibrosis (89–92). Rat models with coronary artery ligation demonstrate myocardial stiffening over time, with elastic modulus increasing from 18 to 55 kPa (90), and increased stiffness has been observed in the infarct zone as early as 1 day post-infarction (93). In addition to temporal changes, regional stiffness varies between the infarct zone, border zone, and remote zone by 3 days post-infarction (94). Elastic modulus progressively decreases in the border zone at a rate of 8.5 kPa/mm toward remote tissue (90). Because investigating the effects of tissue stiffness with in vivo models is confounded by many other concurrent changes, including matrix composition (93), in vitro models have been implemented to elucidate the effects of stiffness and other aspects of fibrosis on cardiac cell phenotypes. In this section, we will describe 2-D and 3-D in vitro models of cardiac fibrosis that focus primarily on recapitulating uniform or spatiotemporal changes in stiffness.
Because standard polystyrene dishes used for cell culture are nearly five orders of magnitude more stiff than the native myocardium (95), mechanically tunable biomaterials have been developed to mimic the rigidity of healthy or fibrotic myocardium (96). For example, hydrogels are cross-linked, hydrophilic polymers with high water content that are commonly used as cell culture substrates because they can be tuned to resemble the elasticity of soft tissue and allow for efficient mass transfer. Hydrogels can incorporate natural polymer chains, such as mammalian matrix proteins, or synthetic polymer chains, such as polyacrylamide or polyethylene glycol (97, 98). Other biomaterials commonly used to recapitulate physiological or pathological stiffness in vitro include elastomers like polydimethylsiloxane (PDMS), which is biocompatible, mechanically tunable, and transparent (99–102).
When cultured on rigid hydrogel or elastomer substrates, cardiac myocytes exhibit disorganized sarcomeres, reduced sarcoplasmic calcium stores (103), lower amplitude calcium currents (103, 104), decreased cell shortening during contraction (103), and a progressive decrease in beat frequency over time (105) when compared to substrates that mimic the elasticity of healthy myocardium. Using traction force microscopy, in which fluorescent beads embedded in substrates are displaced during cell contraction, several studies have established non-monotonic relationships between force generation and substrate rigidity. In these studies, cardiac myocytes generally generate maximum forces on physiological stiffness, which decrease on substrates that are either more soft or stiff in both isotropic (103, 106) and aligned microtissues (107). However, some studies have reported linearly increasing force generation with increased substrate stiffness (101, 108, 109), which may be attributed to differences in experimental methods, such as cell source, biomaterial substrate, or analysis techniques. Similar results are observed in cocultures of cardiac myocytes and fibroblasts on polyacrylamide substrates, in which increased stiffness results in reduced troponin I staining, increased fibroblast density, and poor electrical excitability (106).
Micropatterning has also been used in combination with tunable hydrogel or elastomer substrates to modulate both cellular architecture and substrate stiffness because both of these features remodel concurrently in post-infarct myocardium. Substrate stiffness and cellular architecture has been shown to modulate metabolic activity (99, 100, 107) and mitochondrial structure in cardiac myocytes (102). Microcontact printed hydrogels have also been used to characterize the contractility of single (85) or coupled (110) cardiac myocytes as a function of both cellular architecture and substrate stiffness. At the single cell level, cardiac myocytes with low cell aspect ratios that mimic concentric hypertrophy do more work on stiff substrates that resemble fibrotic myocardium, demonstrating a functional advantage of cell shape remodeling in response to mechanical overload (85). In coupled myocytes, stiff substrates caused increased focal adhesion formation at the cell-cell interface, possibly contributing to cellular uncoupling in post-infarct myocardium (110).
To model both the cellular and biomechanical aspects of fibrosis, tissues have also been engineered with cardiac myocytes and fibroblasts on substrates with tunable stiffness. For example, microcontact printing has been implemented to engineer aligned microtissues on polyacrylamide hydrogels with both cardiac myocytes and fibroblasts. Microtissues generated less work on rigid substrates, irrespective of cell adhesion ligand or presence of fibroblasts, revealing the dominant role of substrate elasticity in regulating contractile output (111). To engineer an artificial infarct boundary, cardiac myocytes and fibroblasts have been cocultured on separate halves of cell culture substrates with rigidities that range from healthy, 1-week post-infarct, and 2- to 6-weeks post-infarct myocardium. The presence of cardiac fibroblasts in this coculture setting attenuated mechanical signal propagation across the infarct boundary in a stiffness-dependent manner (112).
In addition to affecting cardiac myocytes, rigid substrates that mimic fibrotic myocardium also promote fibroblast activation to myofibroblasts. On stiff substrates, cardiac fibroblasts notably activate into a myofibroblast phenotype, exhibiting increased α-SMA coverage (113–116), increased contractile force generation measured through traction force microscopy (117), and increased nuclear localization of the mechanosensitive transcription factors yes-associated protein (YAP) and transcriptional co-activator with PDZ-binding motif (TAZ) (114). Knockdown of YAP and TAZ reversed or attenuated stiffness-dependent changes in cell morphology and function, suggesting YAP and TAZ coordinate fibroblast mechanoactivation (114). In other work, limiting focal adhesion size through microcontact printing was also sufficient to interrupt the recruitment of α-SMA to stress fibers on stiff substrates, indicating that focal adhesion size may control α-SMA localization (113). Studies that establish mechanisms behind fibroblast mechanoactivation may reveal new targets for anti-fibrotic strategies to mitigate adverse remodeling following myocardial infarction.
In vitro models have also identified stiffness-dependent secretion of paracrine factors, which may regulate several processes involved in infarct remodeling. For example, cardiac myocytes on stiff substrates secrete more VEGF. Consistent with this finding, media conditioned by myocytes on stiff substrates promotes angiogenesis, including increased migration capacity and tube length of microvascular endothelial cells (118). Fibroblasts cultured on stiff hydrogels and treated with TGF-β have also been shown to upregulate several cytokines, including osteopontin, a known regulator of collagen cross-linking via lysyl oxidase, and insulin-like growth factor 1, which regulates cardiac myocyte hypertrophy. As a result, conditioned media from TGF-β-treated fibroblasts cultured on stiff hydrogels has been shown to induce cardiac myocyte hypertrophy, indicated by increased cell volume, when compared with medium from cardiac fibroblasts without TGF-β, regardless of substrate stiffness (119). This echoes previous work that demonstrates TGF-β may be a more potent regulator of the myofibroblast phenotype than substrate rigidity (116). Together, 2-D models that resemble the elasticity of fibrotic myocardium recapitulate many cellular and molecular events following myocardial infarction.
2-D models with uniform stiffness do not encompass regional changes in stiffness between the infarct, border, or remote zones following myocardial infarction. Spatial stiffness gradients have been fabricated through graded material cross-linking (120), including gradient-patterned (121–124) or sliding (125, 126) photomasks, layering of hydrogels of different elasticities (127), applying a temperature gradient to PDMS during curing (128), or microfluidic-mixing of prepolymer solutions with different cross-linker concentrations (129). However, most of these have not been implemented with cardiac cell types to model post-infarct myocardium. In one example, a polyethylene glycol hydrogel was patterned with soft and stiff concentric circles using a photomask. Cardiac fibroblasts cultured on stiff regions of the substrate expressed increased α-SMA and collagen when compared with soft regions. Live imaging demonstrated a directional cellular migration toward the inner stiff region. Treatment with a ROCK inhibitor reduced the population of myofibroblasts, demonstrating that the model can be used as an antifibrotic drug screening platform (130). To investigate the effects of pathological matrix stiffening in lung fibroblasts, a stiffness gradient was made from polyacrylamide gels polymerized through gradient photomasks. Human lung fibroblasts cultured on the stiffness gradient show a progressive increase in fibroblast activation, indicated by increased proliferation and matrix synthesis, toward the stiff end of the gradient. Addition of prostaglandin E2, an inhibitor of fibrogenesis, inhibited fibroblast activation (131). Similar phenotypes may also be observed in cardiac fibroblasts over stiffness gradients, though this has not yet been tested.
Mechanical properties can also be controlled in situ to model changes in stiffness over time, which is characteristic of infarct scar maturation. This can be achieved with materials that polymerize in response to light exposure (132, 133) or by varying the molecular weight of the cross-linking agent in real-time (134). Engineered models to capture dynamic stiffening have been used to model development, wound healing, and disease (135), but few have been implemented in the context of myocardial infarction. In one study, hyaluronic acid hydrogels seeded with cardiac fibroblasts were modified to dynamically increase in stiffness in response to UV exposure. Dynamic stiffening to model scar maturation resulted in increased cell spreading, α-SMA formation, and collagen I expression (Figure 3A) (115). Although fibroblast activation correlates with increased stiffness in 2-D spatiotemporal models, cardiac myocyte phenotype and function has been relatively unexplored in these settings.
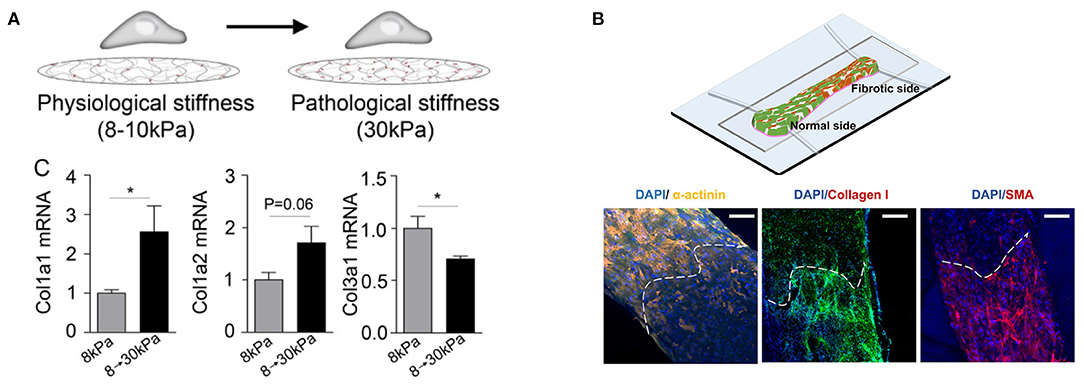
Figure 3. Engineered in vitro fibrosis models that modulate stiffness in (A) time and (B) space. (A) A hyaluronic acid hydrogel is polymerized in situ upon exposure to UV light to mimic scar maturation after myocardial infarction. Stiffening from 8 to 30 kPa results in collagen turnover (*p < 0.05). Republished with permission of the American Society for Cell Biology, from Herum et al. (115); permission conveyed through Copyright Clearance Center, Inc. (B) A heteropolar biowire integrates normal (7.6 kPa) and fibrotic (61.1 kPa) cardiac tissue. The white dashed line indicates the interface between normal and fibrotic regions. Normal regions exhibit increased α-actinin, a component of sarcomeres in cardiac myocytes, while fibrotic regions have increased collagen and α-smooth muscle actin, indicating increased presence of myofibroblasts. Images reproduced with permission of the American Chemical Society, from https://pubs.acs.org/doi/full/10.1021/acscentsci.9b00052 (136). Further permissions related to the material excerpted should be directed to the American Chemical Society.
Modulating stiffness in 2-D only exposes one side of cells to the fibrotic microenvironments experienced in vivo. To more closely mimic cell-cell and cell-matrix interactions that occur in native myocardium, cardiac cells have been mixed into hydrogels to form 3-D tissues. Similar to findings in 2-D, cardiac myocytes encapsulated in rigid polyethylene glycol hydrogels demonstrate reduced cell shortening and increased relaxation time when compared with soft hydrogels, which was also accompanied by increased intracellular localization of the mechanosensitive transcription factor YAP (133). In 3-D, matrix stiffness promotes fibroblast differentiation into myofibroblasts, demonstrated by increased stellate morphology, α-SMA and collagen type III levels, and gel compaction (137), consistent with findings in 2-D. The simple aggregation of cardiac fibroblasts in 3-D using low-attachment plates has also been shown to induce gene expression changes associated with adverse cardiac remodeling and the extracellular matrix. Conditioned media from 3-D fibroblast aggregates also causes cardiac myocyte hypertrophy relative to media from fibroblasts cultured in 2-D (138), indicating that phenotypes in 2-D do not always translate to 3-D.
Using microfabricated templates, 3-D cardiac tissues have also been engineered with control over cell composition, matrix stiffness, and tissue architecture. In one model, cardiac myocytes and fibroblasts were embedded in collagen hydrogels of varying fibroblast cell densities or collagen concentrations and suspended between uniaxial PDMS microposts. Microposts serve as tissue constraints that promote alignment. Increasing fibroblast density decreased tissue contraction force and hampered beating frequency, as measured by displacement of the microposts (139). In a similar paper, the system was modified to contain biaxial PDMS microposts to generate isotropic cardiac matrices, designed to mimic “diseased” architecture. 3-D microtissues of cardiac myocytes and fibroblasts in isotropic matrices display more stellate morphology, characteristic of myofibroblasts, and more heterogeneous force distribution when compared with “healthy” aligned matrices. Furthermore, increasing the proportion of fibroblasts in the tissues reduces the overall tissue beating frequency, suggesting that both matrix organization and cellular composition regulate cardiac function (140).
Although hydrogels are mechanically tunable, they fail to recapitulate the fibrous architecture of native cardiac extracellular matrix. A 3-D fibrous network functionalized with fibronectin, which anchors cardiac cells in vivo, was fabricated through electrospinning. Spin speed was adjusted to tune fiber alignment while photo-initiated cross-linking was used to tune fiber stiffness to mimic physiologic (9–14 kPa) or pathophysiologic (>20 kPa) tissues. Cardiac myocytes in stiff, fibrous networks exhibit slower calcium flux, indicated by increased decay time and increased peak-to-peak irregularity (141). In another example, fibrous scaffolds with varying fiber stiffness were fabricated through two-photon polymerization and seeded with cardiac myocytes that lack expression of cardiac myosin binding protein C, which is thought to play a role in sarcomere sliding during contraction. Mutations in this protein are also associated with hypertrophic and dilated cardiomyopathy. While control cells were able to adapt to the increased load with increasing contraction force, cells with the mutation displayed impaired contraction on stiffer fibers. This work demonstrates the combined effects of mechanical stress and genetic factors on contraction deficits (142).
Interestingly, fibroblasts in 3-D fibrous matrices depart from the conventional relationships established between stiffness and fibroblast activation in 2-D cell culture or 3-D hydrogels (143). In human lung fibroblasts seeded in the same fibrous matrices, increasing fiber stiffness actually reduced proliferation and myofibroblast activation (α-SMA) when compared with cells on soft and deformable fibrous matrices. This is correlated with reduced cell spreading and focal adhesion formation that was also observed with increasing stiffness (144). Fiber density, on the other hand, has been shown to promote differentiation in lung fibroblasts, signified by increased fibronectin synthesis, nuclear localization of YAP, proliferation, and cytokine secretion (145). Similar relationships may also exist for cardiac fibroblasts but have yet to be investigated.
Engineered 3-D cardiac tissues have also been fabricated with increasing spatiotemporal control over stiffness. A 3-D fibrosis model was developed using the biowire platform, in which cardiac cells are encapsulated in a fibrin-based hydrogel and suspended between a pair of polymer wires that function to promote microtissue alignment. Tissue contractile stress is measured based on the deflection of the intrinsically fluorescent polymer wires. To model healthy (7.6 kPa) or fibrotic (61.1 kPa) myocardium, cardiac myocytes were cocultured with 25 or 75% cardiac fibroblasts, respectively. Fibrotic tissues underwent more rapid compaction and had higher collagen content, disrupted myofibril structures, altered Cx43 distribution, prolonged time to peak, and lower contractile force generation when compared with healthy tissues. To next create a spatially heterogeneous stiffness model, which can mimic the interface between the infarct zone and viable tissue, fibrotic and healthy tissue were integrated at opposing sides of a single biowire platform (Figure 3B). The fibrotic side of the microtissue underwent more rapid compaction, contained increased collagen content and myofibroblast activation, and had slower calcium transients with a lower amplitude compared to the healthy side. In addition, propagation velocity at the healthy side was diminished when compared with uniform healthy biowire tissues. Arrhythmic waves were also observed, especially in the interface region. This platform was also used to screen antifibrotic drugs (136), demonstrating the potential impact of these approaches in drug development.
To alter substrate rigidity over time, one approach is to encapsulate cells into hydrogels that either degrade or cross-link in response to specific wavelengths of light. In one example, a 3-D photodegradable hydrogel was used to demonstrate that valvular myofibroblasts can be redirected into a quiescent phenotype by decreasing stiffness. This work demonstrates fibroblast phenotypic plasticity and the potential role of the mechanical environment in de-differentiating fibroblasts, which has therapeutic applications in resolving fibrotic disease (146). Lastly, one study demonstrated that cardiac myocytes encapsulated in photopolymerizable polyethylene glycol hydrogels do not exhibit differences in cell viability after UV exposure (133), though the impact of progressive stiffening in 3-D on cardiac cell phenotypes has not been further established.
Cardiac cells are constantly under cyclic stretch in the healthy, beating heart. Myocardial infarction results in an initial loss of contractility in the infarct zone followed by arrhythmogenesis, which alters strain rates experienced by surviving cardiac cells, as quantified through echocardiographic imaging (147). To stretch myocytes and non-myocytes in vitro, experimental platforms include microchips with stretchable silicone membranes, custom-built bioreactors, or commercially available cell straining units in which strain can be applied to cell culture plates with integrated loading posts.
Chronic cyclic stretch over several days to mimic the diastolic and systolic movement of cardiac muscle has been shown to promote the maturation of “engineered heart tissues,” which are generally defined as cardiac myocytes embedded in hydrogels and cast around uniaxial tissue constraints or circular molds. Stretched heart tissues exhibit increased cell alignment (148–151), sarcomere organization (151, 152), Cx43 expression (150, 151), and contractile force generation (148, 150, 153). In some studies, morphological changes were also observed that indicate cardiac myocyte hypertrophy through increased cell size (148, 152) and mitochondrial density (148). In 2-D aligned cardiac tissues fabricated through microcontact printing, chronic cyclic stretch has also been shown to induce pathological changes in cell aspect ratio and sarcomere alignment, promote gene expression profiles associated with pathological remodeling, and diminish calcium transients and force generation (Figure 4A) (154). Thus, chronic cyclic stretch can be beneficial or detrimental to cardiac myocytes, depending on the specific parameters.
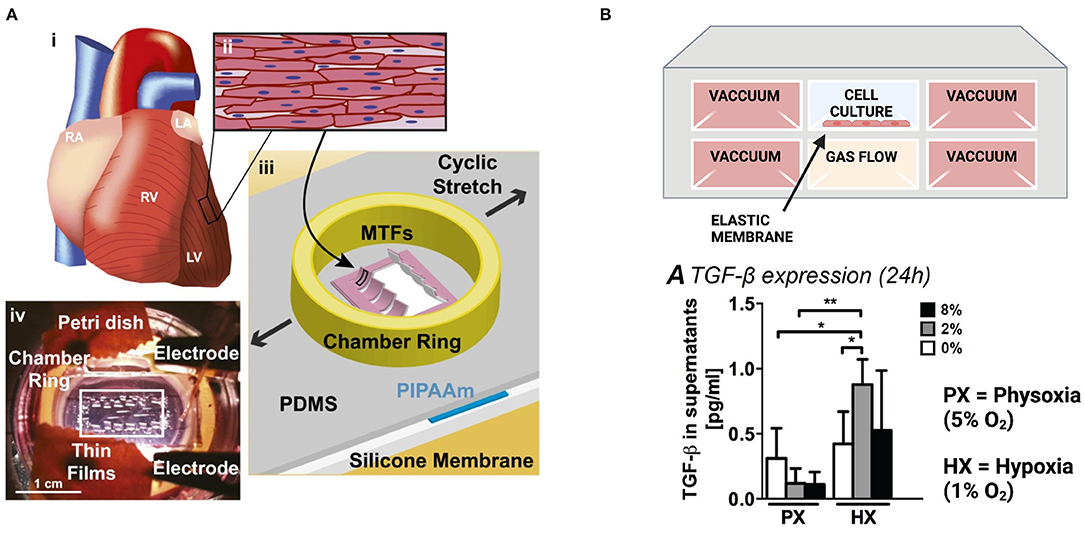
Figure 4. Engineered in vitro models to mimic pathological strain post-infarction. (A) Muscular thin films on a stretchable silicone membrane provide real-time measurements of contractile stress during stretch. Images reproduced from McCain et al. (154). (B) A microfluidic device to evaluate the combined effects of hypoxia and strain on cardiac fibroblasts. Created with BioRender.com. Both hypoxia (1% O2) and reduced contractility (2% strain) are required for cardiac fibroblasts to upregulate TGF-β and IL-1β (*p < 0.05, **p < 0.01). Image reproduced from Ugolini et al. (155) (https://creativecommons.org/licenses/by/4.0/).
Cardiac fibroblast responses to strain have also been relatively inconsistent. In some cases, stretching activates many hallmarks of cardiac fibrosis, including increased fibroblast proliferation, hydrogel stiffening (156), increased gel compaction and strength (157, 158), extracellular matrix deposition (156–158), and enhanced secretion of TNF-α (159). However, responses are dependent on baseline levels of fibroblast activation, which is highly sensitive to culture conditions. Cardiac fibroblasts that are cultured for 1 day on rigid substrates and have lower initial levels of α-SMA respond to static tensile forces with increased α-SMA, while cells cultured for 3 days with higher basal levels of α-SMA respond to the same force with decreased α-SMA production (160). Consistent with this, fibroblasts grown on soft hydrogels with minimal basal α-SMA expression and exposed to static stretch show elevated α-SMA mRNA levels and expression of various extracellular matrix proteins, including collagen and fibronectin (115). Fibroblasts also show differing proliferative behavior in response to mechanical strain, which may be dependent on baseline α-SMA levels, strain rate (161), ECM composition (162, 163), serum concentration (164), or substrate stiffness (115), which highlights a need for more standardized cell culture methods (165).
To assess the combined effects of strain and hypoxia, cardiac fibroblasts have been cultured in a microfluidic device containing a stretchable, gas-permeable membrane situated above a microchannel for gas flow and between lateral actuation channels (Figure 4B). Uniform hypoxia (1% oxygen) or reduced contractility to mimic post-infarct myocardium (2% strain) are alone sufficient to induce proliferation and collagen type 1 production, although the combined effects of hypoxia and reduced strain are required to trigger fibroblast secretion of IL-1β or TGF-β (155).
Paracrine signals secreted by stretched cardiac cells may also regulate critical aspects of infarct healing. Recent work has characterized the transcriptomic profile of stretched cardiac myocytes, which show differentially expressed genes and regulatory networks that may lead to hypertrophic growth of cardiac myocytes (166). Consistent with this, stretched cardiac myocytes upregulate miR208, a mediator of cardiac hypertrophy, hypertrophic proteins, such as β-myosin heavy chain, and secretion of TGF-β (167). Neonatal rat cardiac myocytes on stretched silicone membranes have also been reported to undergo apoptosis, accompanied by mitochondrial dysfunction (168). One study explored factors secreted by stretched cardiac myocytes by fabricating a coculture device that enables paracrine signaling between cardiac myocytes and fibroblasts while exposing cardiac myocytes to strains that mimic the border zone after infarction in vivo. In this device, coculture with stretched cardiac myocytes increases cardiac fibroblast proliferation. A media screen indicated the presence of cytokines such as colony stimulating factor 1 and platelet derived growth factor B, which were sufficient to increase proliferation in fibroblast monocultures (115).
As described above, post-infarct myocardium is characterized by distinct biochemical and biomechanical changes in the cellular microenvironment that vary in both space and time and are thought to contribute to excessive fibrosis, hypertrophy, and arrhythmias. Unlike conventional in vitro and in vivo models, Organs on Chips are able to dissect the impact of these complex and dynamic changes to the post-infarct microenvironment by offering a unique combination of multi-modal microenvironmental control and accessibility to physiological readouts. For example, the gradient systems described above showed that cardiac fibroblasts migrate toward both ischemic cardiac myocytes (27) and stiffer environments (130), two hallmark features of post-infarct myocardium. These studies also demonstrated that myofibroblast phenotypes can be reduced by inhibiting TGF-β (27) or ROCK (130), suggesting that these molecules or pathways could be exploited as anti-fibrotic therapies. As another example, microfluidic devices that offer precise control over oxygen concentration showed that the electrophysiology of cardiac myocytes becomes irregular in response to hypoxia but can recover after 10–30 min of reperfusion (80, 81). Another Organ on a Chip system showed that pre-treatment with endothelial cell-derived vesicles reduces ischemia-reperfusion injury in engineered cardiac tissues (79). Collectively, these and the other examples highlighted above demonstrate how Organ on a Chip models of post-infarct myocardium are powerful for determining how disease evolution is regulated by spatiotemporal heterogeneity while also serving as platforms for therapeutic development.
Despite the advantages of Organs on Chips, there are still many challenges that limit their widescale adoption for disease modeling and drug discovery. First, some findings in response to hypoxia, stiffness, or strain have produced conflicting results. For example, studies have reported increased (45, 155), decreased (46), or unchanged (44) proliferation of cardiac fibroblasts in response to hypoxia. Similarly, fibroblast proliferation has been shown to increase (155, 161) or decrease (162) in response to strain. This may be due to the inherent heterogeneity of the biological responses or may highlight a need for more standardized experimental methods. There is also a need for more characterization of injured myocardium in vivo and ex vivo through techniques such as atomic force microscopy, fluorescent oxygen probes, and high-resolution imaging to ensure that Organs on Chips are accurately modeling relevant features of post-infarct myocardium and to establish more universal design parameters.
Another limitation of existing Organ on a Chip models of post-infarct myocardium is their over-simplified architecture. As described above, current in vitro models have been predominantly 2-D monocultures that can be micropatterned to control tissue architecture or 3-D cocultured tissues or spheroids with relatively random tissue architecture. Thus, future work should focus on engineering cardiac tissues with distinct control over the positioning of multiple cell types and matrix components, leading to more granular models of post-infarct myocardium. Emerging methods to pattern multiple cell types in 2-D (169) and 3-D (170, 171) can improve the architectural relevance and reproducibility of engineered tissues. 3-D bioprinting has also advanced considerably in recent years to provide increasing structural complexity (172, 173), including spatial gradients in porosity (174) and material and cell composition (175), and can be implemented to make more precise tissue models. Model systems should also strive to incorporate relevant cell types beyond cardiac myocytes and fibroblasts, such as neurons (176) and immune cells (177). In addition, there are other types of spatial and temporal gradients beyond those described above, such as cytokine and chemokine gradients that orchestrate the inflammatory cascade after myocardial infarction. Recent approaches to control gradients of soluble factors using microfluidics (178, 179) or 3-D hydrogels (180) will enable more complex modeling of small molecule gradients in the context of myocardial infarction.
Another limitation of many of the studies described above is their reliance on primary cardiac cells from other species (usually neonatal rats), which have historically been the most accessible cardiac cell source. Cells from neonatal rats exhibit species-specific differences and are relatively resistant to hypoxia, a key feature of the infarct microenvironment (3). Thus, model systems will be improved as the field continues to adopt human induced pluripotent stem cell (hiPSC)-derived cardiac myocytes. In addition to providing human relevance, hiPSC-derived cardiac myocytes can also help identify genetic contributions to post-infarct remodeling (181) and contribute to patient-specific models and treatment regimens. However, a major concern is that these cells demonstrate fetal-like maturity, which is especially problematic for modeling myocardial infarctions, a condition that affects almost exclusively adults. Recent approaches to mature hiPSC-derived cardiac myocytes with electromechanical or biochemical stimuli may help mitigate this concern (182, 183), but achieving adult-like maturity in hiPSC-derived cardiac myocytes remains a major hurdle for the field.
Lastly, Organ on a Chip systems need to be more high-throughput and scalable to be integrated into the drug discovery pipeline. Thus, the field also needs more scalable fabrication methods, such as rapid, multimaterial bioprinting of cardiac biowire scaffolds into 96-well plate formats (184) or the development of substrates with integrated electrodes to streamline electrical stimulation (185). Throughput can also be improved by integrating sensors for real-time readouts of parameters such as tissue contractility (87, 88), action potentials (186), the consumption or secretion of biomolecules (187, 188), or physical aspects of the microenvironment, such as oxygen concentration and temperature (189). These types of multi-sensor systems will provide more continuous and detailed insight into cellular phenotypes in response to drug treatments while also requiring less manual handling, thereby increasing throughput and reproducibility.
In summary, engineered Organ on a Chip models of post-infarct myocardium have exciting potential to address many of the gaps presented by oversimplified 2-D cell culture models and animal models that lack human relevance. As technologies continue to develop, next-generation multi-dimensional models could provide simultaneous control over spatial and temporal changes in the physical, biochemical, and mechanical microenvironment that correspond to the phases of infarct healing. When further advanced with patient-derived cells, scalable fabrication techniques, and integrated sensors, these models have potential to emerge as new standards for disease modeling and drug screening and lead to new breakthrough therapies for mitigating post-infarction remodeling.
Both authors were involved in the conceptualization and writing of this manuscript.
This work was supported by NIH R01 HL153286 (MM), NSF CAREER 1944734 (MM), and the Provost Fellowship at the USC Viterbi School of Engineering (NK).
MM is an inventor on US patent US9857356B2 filed by Harvard University and licensed to Emulate, Inc.
The remaining author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
1. Cardiovascular Diseases [Online]. World Health Organization (2020). Available online at: https://www.who.int/health-topics/cardiovascular-diseases/#tab=tab_1 (accessed May 14, 2021).
2. Torio CM, Moore BJ. National Inpatient Hospital Costs: The Most Expensive Conditions by Payer [Online]. Healthcare Cost and Utilization Project (HCUP): Agency for Healthcare Research and Quality (2016). Available online at: https://www.hcup-us.ahrq.gov/reports/statbriefs/sb204-Most-Expensive-Hospital-Conditions.jsp
3. Lindsey ML, Bolli R, Canty JM, Du XJ, Frangogiannis NG, Frantz S, et al. Guidelines for experimental models of myocardial ischemia and infarction. Am J Physiol Heart Circ Physiol. (2018) 314:H812–38. doi: 10.1152/ajpheart.00335.2017
4. Heart Disease Facts [Online]. Centers for Disease Control and Prevention (2020). Available online at: https://www.cdc.gov/heartdisease/facts.htm (accessed May 14, 2021).
5. Montalescot G, Andersen HR, Antoniucci D, Betriu A, de Boer MJ, Grip L, et al. Recommendations on percutaneous coronary intervention for the reperfusion of acute ST elevation myocardial infarction. Heart. (2004) 90:e37. doi: 10.1136/hrt.2003.016014
6. Bagai A, Dangas GD, Stone GW, Granger CB. Reperfusion strategies in acute coronary syndromes. Circ Res. (2014) 114:1918–28. doi: 10.1161/CIRCRESAHA.114.302744
7. Hausenloy DJ, Botker HE, Engstrom T, Erlinge D, Heusch G, Ibanez B, et al. Targeting reperfusion injury in patients with ST-segment elevation myocardial infarction: trials and tribulations. Eur Heart J. (2017) 38:935–41. doi: 10.1093/eurheartj/ehw145
8. Mercado MG, Smith DK, McConnon ML. Myocardial infarction: management of the subacute period. Am Fam Physician. (2013) 88:581–8.
9. Iravanian S, Dudley SC. The renin-angiotensin-aldosterone system (RAAS) and cardiac arrhythmias. Heart Rhythm. (2008) 5:S12–17. doi: 10.1016/j.hrthm.2008.02.025
10. Talman V, Ruskoaho H. Cardiac fibrosis in myocardial infarction-from repair and remodeling to regeneration. Cell Tissue Res. (2016) 365:563–81. doi: 10.1007/s00441-016-2431-9
11. Ingber DE. Is it time for reviewer 3 to request human organ chip experiments instead of animal validation studies? Adv Sci. (2020) 7:2002030. doi: 10.1002/advs.202002030
12. Weber KT, Sun Y, Bhattacharya SK, Ahokas RA, Gerling IC. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat Rev Cardiol. (2013) 10:15–26. doi: 10.1038/nrcardio.2012.158
13. Prabhu SD, Frangogiannis NG. The biological basis for cardiac repair after myocardial infarction: from inflammation to fibrosis. Circ Res. (2016) 119:91–112. doi: 10.1161/CIRCRESAHA.116.303577
14. Pfeffer MA, Braunwald E. Ventricular remodeling after myocardial infarction. Experimental observations and clinical implications. Circulation. (1990) 81:1161–72. doi: 10.1161/01.CIR.81.4.1161
15. Azevedo PS, Polegato BF, Minicucci MF, Paiva SA, Zornoff LA. Cardiac remodeling: concepts, clinical impact, pathophysiological mechanisms and pharmacologic treatment. Arq Bras Cardiol. (2016) 106:62–9. doi: 10.5935/abc.20160005
16. Matsushita T, Oyamada M, Fujimoto K, Yasuda Y, Masuda S, Wada Y, et al. Remodeling of cell-cell and cell-extracellular matrix interactions at the border zone of rat myocardial infarcts. Circ Res. (1999) 85:1046–55. doi: 10.1161/01.RES.85.11.1046
17. Cohn JN, Ferrari R, Sharpe N. Cardiac remodeling–concepts and clinical implications: a consensus paper from an international forum on cardiac remodeling. Behalf of an international forum on cardiac remodeling. J Am Coll Cardiol. (2000) 35:569–82. doi: 10.1016/S0735-1097(99)00630-0
18. Sutton MG, Sharpe N. Left ventricular remodeling after myocardial infarction: pathophysiology and therapy. Circulation. (2000) 101:2981–88. doi: 10.1161/01.CIR.101.25.2981
19. Lorell BH, Carabello BA. Left ventricular hypertrophy: pathogenesis, detection, and prognosis. Circulation. (2000) 102:470–9. doi: 10.1161/01.CIR.102.4.470
20. Konstam MA, Kramer DG, Patel AR, Maron MS, Udelson JE. Left ventricular remodeling in heart failure: current concepts in clinical significance and assessment. JACC Cardiovasc Imaging. (2011) 4:98–108. doi: 10.1016/j.jcmg.2010.10.008
21. Savinova OV, Gerdes AM. Myocyte changes in heart failure. Heart Fail Clin. (2012) 8:1–6. doi: 10.1016/j.hfc.2011.08.004
22. Korup E, Dalsgaard D, Nyvad O, Jensen TM, Toft E, Berning J. Comparison of degrees of left ventricular dilation within three hours and up to six days after onset of first acute myocardial infarction. Am J Cardiol. (1997) 80:449–53. doi: 10.1016/S0002-9149(97)00393-7
23. Armstrong PW. Left ventricular dysfunction: causes, natural history, and hopes for reversal. Heart. (2000) 84(Suppl 1):i15–7; discussion: i50. doi: 10.1136/heart.84.suppl_1.i15
24. Frangogiannis NG. The inflammatory response in myocardial injury, repair, and remodelling. Nat Rev Cardiol. (2014) 11:255–65. doi: 10.1038/nrcardio.2014.28
25. Shinde AV, Frangogiannis NG. Fibroblasts in myocardial infarction: a role in inflammation and repair. J Mol Cell Cardiol. (2014) 70:74–82. doi: 10.1016/j.yjmcc.2013.11.015
26. Rog-Zielinska EA, Norris RA, Kohl P, Markwald R. The living scar–cardiac fibroblasts and the injured heart. Trends Mol Med. (2016) 22:99–114. doi: 10.1016/j.molmed.2015.12.006
27. Mosadegh B, Dabiri BE, Lockett MR, Derda R, Campbell P, Parker KK, et al. Three-dimensional paper-based model for cardiac ischemia. Adv Healthc Mater. (2014) 3:1036–43. doi: 10.1002/adhm.201300575
28. Chiu LL, Radisic M, Vunjak-Novakovic G. Bioactive scaffolds for engineering vascularized cardiac tissues. Macromol Biosci. (2010) 10:1286–301. doi: 10.1002/mabi.201000202
29. Rumsey WL, Pawlowski M, Lejavardi N, Wilson DF. Oxygen pressure distribution in the heart in vivo and evaluation of the ischemic “border zone”. Am J Physiol. (1994) 266(4 Pt 2):H1676–80. doi: 10.1152/ajpheart.1994.266.4.H1676
30. Sen CK, Khanna S, Roy S. Perceived hyperoxia: oxygen-induced remodeling of the reoxygenated heart. Cardiovasc Res. (2006) 71:280–8. doi: 10.1016/j.cardiores.2006.01.003
31. Muñoz-Sánchez J, Chánez-Cárdenas ME. The use of cobalt chloride as a chemical hypoxia model. J Appl Toxicol. (2019) 39:556–70. doi: 10.1002/jat.3749
32. Zhao RZ, Jiang S, Ru NY, Jiao B, Yu ZB. Comparison of hypoxic effects induced by chemical and physical hypoxia on cardiomyocytes. Can J Physiol Pharmacol. (2019) 97:980–8. doi: 10.1139/cjpp-2019-0092
33. Pavlacky J, Polak J. Technical feasibility and physiological relevance of hypoxic cell culture models. Front Endocrinol. (2020) 11:57. doi: 10.3389/fendo.2020.00057
34. Zeevi-Levin N, Barac YD, Reisner Y, Reiter I, Yaniv G, Meiry G, et al. Gap junctional remodeling by hypoxia in cultured neonatal rat ventricular myocytes. Cardiovasc Res. (2005) 66:64–73. doi: 10.1016/j.cardiores.2005.01.014
35. Danon A, Zeevi-Levin N, Pinkovich DY, Michaeli T, Berkovich A, Flugelman M, et al. Hypoxia causes connexin 43 internalization in neonatal rat ventricular myocytes. Gen Physiol Biophys. (2010) 29:222–33. doi: 10.4149/gpb_2010_03_222
36. Turner MS, Haywood GA, Andreka P, You L, Martin PE, Evans WH, et al. Reversible connexin 43 dephosphorylation during hypoxia and reoxygenation is linked to cellular ATP levels. Circ Res. (2004) 95:726–33. doi: 10.1161/01.RES.0000144805.11519.1e
37. Xu KY, Kuppusamy SP, Wang JQ, Li H, Cui H, Dawson TM, et al. Nitric oxide protects cardiac sarcolemmal membrane enzyme function and ion active transport against ischemia-induced inactivation. J Biol Chem. (2003) 278:41798–803. doi: 10.1074/jbc.M306865200
38. Yakushev S, Band M, Tissot van Patot MC, Gassmann M, Avivi A, Bogdanova A. Cross talk between S-nitrosylation and S-glutathionylation in control of the Na,K-ATPase regulation in hypoxic heart. Am J Physiol Heart Circ Physiol. (2012) 303:H1332–43. doi: 10.1152/ajpheart.00145.2012
39. González-Rodríguez P, Falcón D, Castro MJ, Ureña J, López-Barneo J, Castellano A. Hypoxic induction of T-type Ca(2+) channels in rat cardiac myocytes: role of HIF-1α and RhoA/ROCK signalling. J Physiol. (2015) 593:4729–45. doi: 10.1113/JP271053
40. Tanaka M, Ito H, Adachi S, Akimoto H, Nishikawa T, Kasajima T, et al. Hypoxia induces apoptosis with enhanced expression of Fas antigen messenger RNA in cultured neonatal rat cardiomyocytes. Circ Res. (1994) 75:426–33. doi: 10.1161/01.RES.75.3.426
41. Long X, Boluyt MO, Hipolito ML, Lundberg MS, Zheng JS, O'Neill L, et al. p53 and the hypoxia-induced apoptosis of cultured neonatal rat cardiac myocytes. J Clin Invest. (1997) 99:2635–43. doi: 10.1172/JCI119452
42. Chen SJ, Bradley ME, Lee TC. Chemical hypoxia triggers apoptosis of cultured neonatal rat cardiac myocytes: modulation by calcium-regulated proteases and protein kinases. Mol Cell Biochem. (1998) 178:141–9. doi: 10.1023/A:1006893528428
43. Kang PM, Haunstetter A, Aoki H, Usheva A, Izumo S. Morphological and molecular characterization of adult cardiomyocyte apoptosis during hypoxia and reoxygenation. Circ Res. (2000) 87:118–25. doi: 10.1161/01.RES.87.2.118
44. Gao Y, Chu M, Hong J, Shang J, Xu D. Hypoxia induces cardiac fibroblast proliferation and phenotypic switch: a role for caveolae and caveolin-1/PTEN mediated pathway. J Thorac Dis. (2014) 6:1458–68. doi: 10.3978/j.issn.2072-1439.2014.08.31
45. Watson CJ, Collier P, Tea I, Neary R, Watson JA, Robinson C, et al. Hypoxia-induced epigenetic modifications are associated with cardiac tissue fibrosis and the development of a myofibroblast-like phenotype. Hum Mol Genet. (2014) 23:2176–88. doi: 10.1093/hmg/ddt614
46. Zhao X, Wang K, Liao Y, Zeng Q, Li Y, Hu F, et al. MicroRNA-101a inhibits cardiac fibrosis induced by hypoxia via targeting TGFβRI on cardiac fibroblasts. Cell Physiol Biochem. (2015) 35:213–26. doi: 10.1159/000369689
47. Tamamori M, Ito H, Hiroe M, Marumo F, Hata RI. Stimulation of collagen synthesis in rat cardiac fibroblasts by exposure to hypoxic culture conditions and suppression of the effect by natriuretic peptides. Cell Biol Int. (1997) 21:175–80. doi: 10.1006/cbir.1997.0130
48. Chu W, Li X, Li C, Wan L, Shi H, Song X, et al. TGFBR3, a potential negative regulator of TGF-β signaling, protects cardiac fibroblasts from hypoxia-induced apoptosis. J Cell Physiol. (2011) 226:2586–94. doi: 10.1002/jcp.22604
49. Yang B, He K, Zheng F, Wan L, Yu X, Wang X, et al. Over-expression of hypoxia-inducible factor-1 alpha in vitro protects the cardiac fibroblasts from hypoxia-induced apoptosis. J Cardiovasc Med. (2014) 15:579–86. doi: 10.2459/JCM.0b013e3283629c52
50. Zhao X, Wang K, Hu F, Qian C, Guan H, Feng K, et al. MicroRNA-101 protects cardiac fibroblasts from hypoxia-induced apoptosis via inhibition of the TGF-β signaling pathway. Int J Biochem Cell Biol. (2015) 65:155–64. doi: 10.1016/j.biocel.2015.06.005
51. Ladoux A, Frelin C. Hypoxia is a strong inducer of vascular endothelial growth factor mRNA expression in the heart. Biochem Biophys Res Commun. (1993) 195:1005–10. doi: 10.1006/bbrc.1993.2144
52. Hwang JM, Weng YJ, Lin JA, Bau DT, Ko FY, Tsai FJ, et al. Hypoxia-induced compensatory effect as related to Shh and HIF-1alpha in ischemia embryo rat heart. Mol Cell Biochem. (2008) 311:179–87. doi: 10.1007/s11010-008-9708-6
53. Ontoria-Oviedo I, Dorronsoro A, Sánchez R, Ciria M, Gómez-Ferrer M, Buigues M, et al. Extracellular vesicles secreted by hypoxic AC10 cardiomyocytes modulate fibroblast cell motility. Front Cardiovasc Med. (2018) 5:152. doi: 10.3389/fcvm.2018.00152
54. Shi H, Zhang X, He Z, Wu Z, Rao L, Li Y. Metabolites of hypoxic cardiomyocytes induce the migration of cardiac fibroblasts. Cell Physiol Biochem. (2017) 41:413–21. doi: 10.1159/000456531
55. Shivakumar K, Sollott SJ, Sangeetha M, Sapna S, Ziman B, Wang S, et al. Paracrine effects of hypoxic fibroblast-derived factors on the MPT-ROS threshold and viability of adult rat cardiac myocytes. Am J Physiol Heart Circ Physiol. (2008) 294:H2653–8. doi: 10.1152/ajpheart.91443.2007
56. Gao Q, Guo M, Zeng W, Wang Y, Yang L, Pang X, et al. Matrix metalloproteinase 9 secreted by hypoxia cardiac fibroblasts triggers cardiac stem cell migration in vitro. Stem Cells Int. (2015) 2015:836390. doi: 10.1155/2015/836390
57. Wang JH, Zhao L, Pan X, Chen NN, Chen J, Gong QL, et al. Hypoxia-stimulated cardiac fibroblast production of IL-6 promotes myocardial fibrosis via the TGF-β1 signaling pathway. Lab Invest. (2016) 96:1035. doi: 10.1038/labinvest.2016.84
58. Cosme J, Guo H, Hadipour-Lakmehsari S, Emili A, Gramolini AO. Hypoxia-induced changes in the fibroblast secretome, exosome, and whole-cell proteome using cultured, cardiac-derived cells isolated from neonatal mice. J Proteome Res. (2017) 16:2836–47. doi: 10.1021/acs.jproteome.7b00144
59. Ren L, Liu W, Wang Y, Wang JC, Tu Q, Xu J, et al. Investigation of hypoxia-induced myocardial injury dynamics in a tissue interface mimicking microfluidic device. Anal Chem. (2013) 85:235–44. doi: 10.1021/ac3025812
60. Wang L, Liu W, Wang Y, Wang JC, Tu Q, Liu R, et al. Construction of oxygen and chemical concentration gradients in a single microfluidic device for studying tumor cell-drug interactions in a dynamic hypoxia microenvironment. Lab Chip. (2013) 13:695–705. doi: 10.1039/C2LC40661F
61. Chang CW, Cheng YJ, Tu M, Chen YH, Peng CC, Liao WH, et al. A polydimethylsiloxane-polycarbonate hybrid microfluidic device capable of generating perpendicular chemical and oxygen gradients for cell culture studies. Lab Chip. (2014) 14:3762–72. doi: 10.1039/C4LC00732H
62. Shih HC, Lee TA, Wu HM, Ko PL, Liao WH, Tung YC. Microfluidic collective cell migration assay for study of endothelial cell proliferation and migration under combinations of oxygen gradients, tensions, and drug treatments. Sci Rep. (2019) 9:8234. doi: 10.1038/s41598-019-44594-5
63. Kang YBA, Eo J, Bulutoglu B, Yarmush ML, Usta OB. Progressive hypoxia-on-a-chip: an in vitro oxygen gradient model for capturing the effects of hypoxia on primary hepatocytes in health and disease. Biotechnol Bioeng. (2020) 117:763–75. doi: 10.1002/bit.27225
64. Polinkovsky M, Gutierrez E, Levchenko A, Groisman A. Fine temporal control of the medium gas content and acidity and on-chip generation of series of oxygen concentrations for cell cultures. Lab Chip. (2009) 9:1073–84. doi: 10.1039/b816191g
65. Lo JF, Sinkala E, Eddington DT. Oxygen gradients for open well cellular cultures via microfluidic substrates. Lab Chip. (2010) 10:2394–401. doi: 10.1039/c004660d
66. Rexius-Hall ML, Rehman J, Eddington DT. A microfluidic oxygen gradient demonstrates differential activation of the hypoxia-regulated transcription factors HIF-1α and HIF-2α. Integr Biol. (2017) 9:742–50. doi: 10.1039/C7IB00099E
67. Lavrentieva A. Advances in biochemical engineering/biotechnology. In: Lavrentieva A, Pepelanova I, Seliktar D, editors. Tunable Hydrogels. Cham: Springer. (2020). p. 1–25. doi: 10.1007/978-3-030-76769-3
68. Park KM, Gerecht S. Hypoxia-inducible hydrogels. Nat Commun. (2014) 5:4075. doi: 10.1038/ncomms5075
69. Blatchley M, Park KM, Gerecht S. Designer hydrogels for precision control of oxygen tension and mechanical properties. J Mater Chem B. (2015) 3:7939–49. doi: 10.1039/C5TB01038A
70. Boyce MW, Simke WC, Kenney RM, Lockett MR. Generating linear oxygen gradients across 3D cell cultures with block-layered oxygen controlled chips (BLOCCs). Anal Methods. (2020) 12:18–24. doi: 10.1039/C9AY01690B
71. Pedron S, Becka E, Harley BA. Spatially gradated hydrogel platform as a 3D engineered tumor microenvironment. Adv Mater. (2015) 27:1567–72. doi: 10.1002/adma.201404896
72. Khademhosseini A, Eng G, Yeh J, Kucharczyk PA, Langer R, Vunjak-Novakovic G, et al. Microfluidic patterning for fabrication of contractile cardiac organoids. Biomed Microdev. (2007) 9:149–57. doi: 10.1007/s10544-006-9013-7
73. Iyer RK, Chui J, Radisic M. Spatiotemporal tracking of cells in tissue-engineered cardiac organoids. J Tissue Eng Regen Med. (2009) 3:196–207. doi: 10.1002/term.153
74. Chiu LL, Iyer RK, King JP, Radisic M. Biphasic electrical field stimulation aids in tissue engineering of multicell-type cardiac organoids. Tissue Eng A. (2011) 17:1465–77. doi: 10.1089/ten.tea.2007.0244
75. Iyer RK, Odedra D, Chiu LL, Vunjak-Novakovic G, Radisic M. Vascular endothelial growth factor secretion by nonmyocytes modulates Connexin-43 levels in cardiac organoids. Tissue Eng A. (2012) 18:1771–83. doi: 10.1089/ten.tea.2011.0468
76. Mills RJ, Titmarsh DM, Koenig X, Parker BL, Ryall JG, Quaife-Ryan GA, et al. Functional screening in human cardiac organoids reveals a metabolic mechanism for cardiomyocyte cell cycle arrest. Proc Natl Acad Sci USA. (2017) 114:E8372–81. doi: 10.1073/pnas.1707316114
77. Cai Y, Zhang J, Wu J, Li ZY. Oxygen transport in a three-dimensional microvascular network incorporated with early tumour growth and preexisting vessel cooption: numerical simulation study. Biomed Res Int. (2015) 2015:476964. doi: 10.1155/2015/476964
78. Richards DJ, Li Y, Kerr CM, Yao J, Beeson GC, Coyle RC, et al. Human cardiac organoids for the modelling of myocardial infarction and drug cardiotoxicity. Nat Biomed Eng. (2020) 4:446–62. doi: 10.1038/s41551-020-0539-4
79. Yadid M, Lind JU, Ardoña HAM, Sheehy SP, Dickinson LE, Eweje F, et al. Endothelial extracellular vesicles contain protective proteins and rescue ischemia-reperfusion injury in a human heart-on-chip. Sci Transl Med. (2020) 12:eaax8005. doi: 10.1126/scitranslmed.aax8005
80. Liu H, Bolonduro OA, Hu N, Ju J, Rao AA, Duffy BM, et al. Heart-on-a-chip model with integrated extra- and intracellular bioelectronics for monitoring cardiac electrophysiology under acute hypoxia. Nano Lett. (2020) 20:2585–93. doi: 10.1021/acs.nanolett.0c00076
81. Martewicz S, Michielin F, Serena E, Zambon A, Mongillo M, Elvassore N. Reversible alteration of calcium dynamics in cardiomyocytes during acute hypoxia transient in a microfluidic platform. Integr Biol (Camb). (2012) 4:153–64. doi: 10.1039/C1IB00087J
82. Bursac N, Parker KK, Iravanian S, Tung L. Cardiomyocyte cultures with controlled macroscopic anisotropy: a model for functional electrophysiological studies of cardiac muscle. Circ Res. (2002) 91:e45–54. doi: 10.1161/01.RES.0000047530.88338.EB
83. Feinberg AW, Alford PW, Jin H, Ripplinger CM, Werdich AA, Sheehy SP, et al. Controlling the contractile strength of engineered cardiac muscle by hierarchal tissue architecture. Biomaterials. (2012) 33:5732–41. doi: 10.1016/j.biomaterials.2012.04.043
84. Petersen AP, Lyra-Leite DM, Ariyasinghe NR, Cho N, Goodwin CM, Kim JY, et al. Microenvironmental modulation of calcium wave propagation velocity in engineered cardiac tissues. Cell Mol Bioeng. (2018) 11:337–52. doi: 10.1007/s12195-018-0522-2
85. McCain ML, Yuan H, Pasqualini FS, Campbell PH, Parker KK. Matrix elasticity regulates the optimal cardiac myocyte shape for contractility. Am J Physiol Heart Circ Physiol. (2014) 306:H1525–39. doi: 10.1152/ajpheart.00799.2013
86. Watson JL, Aich S, Oller-Salvia B, Drabek AA, Blacklow SC, Chin J, et al. High-efficacy subcellular micropatterning of proteins using fibrinogen anchors. J Cell Biol. (2021) 220:e202009063. doi: 10.1083/jcb.202009063
87. Lind JU, Busbee TA, Valentine AD, Pasqualini FS, Yuan H, Yadid M, et al. Instrumented cardiac microphysiological devices via multimaterial three-dimensional printing. Nat Mater. (2017) 16:303–8. doi: 10.1038/nmat4782
88. Lind JU, Yadid M, Perkins I, O'Connor BB, Eweje F, Chantre CO, et al. Cardiac microphysiological devices with flexible thin-film sensors for higher-throughput drug screening. Lab Chip. (2017) 17:3692–703. doi: 10.1039/C7LC00740J
89. Litwin SE, Litwin CM, Raya TE, Warner AL, Goldman S. Contractility and stiffness of noninfarcted myocardium after coronary ligation in rats. Effects of chronic angiotensin converting enzyme inhibition. Circulation. (1991) 83:1028–37. doi: 10.1161/01.CIR.83.3.1028
90. Berry MF, Engler AJ, Woo YJ, Pirolli TJ, Bish LT, Jayasankar V, et al. Mesenchymal stem cell injection after myocardial infarction improves myocardial compliance. Am J Physiol Heart Circ Physiol. (2006) 290:H2196–203. doi: 10.1152/ajpheart.01017.2005
91. van Putten S, Shafieyan Y, Hinz B. Mechanical control of cardiac myofibroblasts. J Mol Cell Cardiol. (2016) 93:133–42. doi: 10.1016/j.yjmcc.2015.11.025
92. Herum KM, Lunde IG, McCulloch AD, Christensen G. The soft- and hard-heartedness of cardiac fibroblasts: mechanotransduction signaling pathways in fibrosis of the heart. J Clin Med. (2017) 6:53. doi: 10.3390/jcm6050053
93. Rusu M, Hilse K, Schuh A, Martin L, Slabu I, Stoppe C, et al. Biomechanical assessment of remote and postinfarction scar remodeling following myocardial infarction. Sci Rep. (2019) 9:16744. doi: 10.1038/s41598-019-53351-7
94. Torres WM, Jacobs J, Doviak H, Barlow SC, Zile MR, Shazly T, et al. Regional and temporal changes in left ventricular strain and stiffness in a porcine model of myocardial infarction. Am J Physiol Heart Circ Physiol. (2018) 315:H958–67. doi: 10.1152/ajpheart.00279.2018
95. Landry NM, Rattan SG, Dixon IMC. An improved method of maintaining primary murine cardiac fibroblasts in two-dimensional cell culture. Sci Rep. (2019) 9:12889. doi: 10.1038/s41598-019-49285-9
96. Ariyasinghe NR, Lyra-Leite DM, McCain ML. Engineering cardiac microphysiological systems to model pathological extracellular matrix remodeling. Am J Physiol Heart Circ Physiol. (2018) 315:H771–89. doi: 10.1152/ajpheart.00110.2018
97. Zhu J, Marchant RE. Design properties of hydrogel tissue-engineering scaffolds. Expert Rev Med Devices. (2011) 8:607–26. doi: 10.1586/erd.11.27
98. Hu W, Wang Z, Xiao Y, Zhang S, Wang J. Advances in crosslinking strategies of biomedical hydrogels. Biomater Sci. (2019) 7:843–55. doi: 10.1039/C8BM01246F
99. Lyra-Leite DM, Andres AM, Petersen AP, Ariyasinghe NR, Cho N, Lee JA, et al. Mitochondrial function in engineered cardiac tissues is regulated by extracellular matrix elasticity and tissue alignment. Am J Physiol Heart Circ Physiol. (2017) 313:H757–67. doi: 10.1152/ajpheart.00290.2017
100. Lyra-Leite DM, Andres AM, Cho N, Petersen AP, Ariyasinghe NR, Kim SS, et al. Matrix-guided control of mitochondrial function in cardiac myocytes. Acta Biomater. (2019) 97:281–95. doi: 10.1016/j.actbio.2019.08.007
101. Guo J, Simmons DW, Ramahdita G, Munsell MK, Oguntuyo K, Kandalaft B, et al. Elastomer-grafted iPSC-derived micro heart muscles to investigate effects of mechanical loading on physiology. ACS Biomater Sci Eng. (2020). doi: 10.1021/acsbiomaterials.0c00318
102. Lyra-Leite DM, Petersen AP, Ariyasinghe NR, Cho N, McCain ML. Mitochondrial architecture in cardiac myocytes depends on cell shape and matrix rigidity. J Mol Cell Cardiol. (2021) 150:32–43. doi: 10.1016/j.yjmcc.2020.10.004
103. Jacot JG, McCulloch AD, Omens JH. Substrate stiffness affects the functional maturation of neonatal rat ventricular myocytes. Biophys J. (2008) 95:3479–87. doi: 10.1529/biophysj.107.124545
104. Boothe SD, Myers JD, Pok S, Sun J, Xi Y, Nieto RM, et al. The effect of substrate stiffness on cardiomyocyte action potentials. Cell Biochem Biophys. (2016) 74:527–35. doi: 10.1007/s12013-016-0758-1
105. Engler AJ, Carag-Krieger C, Johnson CP, Raab M, Tang HY, Speicher DW, et al. Embryonic cardiomyocytes beat best on a matrix with heart-like elasticity: scar-like rigidity inhibits beating. J Cell Sci. (2008) 121(Pt 22):3794–802. doi: 10.1242/jcs.029678
106. Bhana B, Iyer RK, Chen WL, Zhao R, Sider KL, Likhitpanichkul M, et al. Influence of substrate stiffness on the phenotype of heart cells. Biotechnol Bioeng. (2010) 105:1148–60. doi: 10.1002/bit.22647
107. Pasqualini FS, Agarwal A, O'Connor BB, Liu Q, Sheehy SP, Parker KK. Traction force microscopy of engineered cardiac tissues. PLoS ONE. (2018) 13:e0194706. doi: 10.1371/journal.pone.0194706
108. Bajaj P, Tang X, Saif TA, Bashir R. Stiffness of the substrate influences the phenotype of embryonic chicken cardiac myocytes. J Biomed Mater Res A. (2010) 95:1261–9. doi: 10.1002/jbm.a.32951
109. Hersch N, Wolters B, Dreissen G, Springer R, Kirchgeßner N, Merkel R, et al. The constant beat: cardiomyocytes adapt their forces by equal contraction upon environmental stiffening. Biol Open. (2013) 2:351–61. doi: 10.1242/bio.20133830
110. McCain ML, Lee H, Aratyn-Schaus Y, Kléber AG, Parker KK. Cooperative coupling of cell-matrix and cell-cell adhesions in cardiac muscle. Proc Natl Acad Sci USA. (2012) 109:9881–6. doi: 10.1073/pnas.1203007109
111. Ariyasinghe NR, Reck CH, Viscio AA, Petersen AP, Lyra-Leite DM, Cho N, et al. Engineering micromyocardium to delineate cellular and extracellular regulation of myocardial tissue contractility. Integr Biol. (2017) 9:730–41. doi: 10.1039/C7IB00081B
112. Nguyen DT, Nagarajan N, Zorlutuna P. Effect of substrate stiffness on mechanical coupling and force propagation at the infarct boundary. Biophys J. (2018) 115:1966–80. doi: 10.1016/j.bpj.2018.08.050
113. Goffin JM, Pittet P, Csucs G, Lussi JW, Meister JJ, Hinz B. Focal adhesion size controls tension-dependent recruitment of alpha-smooth muscle actin to stress fibers. J Cell Biol. (2006) 172:259–68. doi: 10.1083/jcb.200506179
114. Liu F, Lagares D, Choi KM, Stopfer L, Marinković A, Vrbanac V, et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol. (2015) 308:L344–57. doi: 10.1152/ajplung.00300.2014
115. Herum KM, Choppe J, Kumar A, Engler AJ, McCulloch AD. Mechanical regulation of cardiac fibroblast profibrotic phenotypes. Mol Biol Cell. (2017) 28:1871–82. doi: 10.1091/mbc.e17-01-0014
116. Cho N, Razipour SE, McCain ML. Featured Article: TGF-β1 dominates extracellular matrix rigidity for inducing differentiation of human cardiac fibroblasts to myofibroblasts. Exp Biol Med. (2018) 243:601–12. doi: 10.1177/1535370218761628
117. Marinković A, Mih JD, Park JA, Liu F, Tschumperlin DJ. Improved throughput traction microscopy reveals pivotal role for matrix stiffness in fibroblast contractility and TGF-β responsiveness. Am J Physiol Lung Cell Mol Physiol. (2012) 303:L169–80. doi: 10.1152/ajplung.00108.2012
118. Shen J, Xie Y, Liu Z, Zhang S, Wang Y, Jia L, et al. Increased myocardial stiffness activates cardiac microvascular endothelial cell via VEGF paracrine signaling in cardiac hypertrophy. J Mol Cell Cardiol. (2018) 122:140–51. doi: 10.1016/j.yjmcc.2018.08.014
119. Ceccato TL, Starbuck RB, Hall JK, Walker CJ, Brown TE, Killgore JP, et al. Defining the cardiac fibroblast secretome in a fibrotic microenvironment. J Am Heart Assoc. (2020) 9:e017025. doi: 10.1161/JAHA.120.017025
120. Li C, Ouyang L, Armstrong JPK, Stevens MM. Advances in the fabrication of biomaterials for gradient tissue engineering. Trends Biotechnol. (2021) 39:150–64. doi: 10.1016/j.tibtech.2020.06.005
121. Marklein RA, Burdick JA. Spatially controlled hydrogel mechanics to modulate stem cell interactions. Soft Matter. (2010) 6:136–43. doi: 10.1039/B916933D
122. Tse JR, Engler AJ. Stiffness gradients mimicking in vivo tissue variation regulate mesenchymal stem cell fate. PLoS ONE. (2011) 6:e15978. doi: 10.1371/journal.pone.0015978
123. Major LG, Holle AW, Young JL, Hepburn MS, Jeong K, Chin IL, et al. Volume adaptation controls stem cell mechanotransduction. ACS Appl Mater Interfaces. (2019) 11:45520–30. doi: 10.1021/acsami.9b19770
124. Kim C, Young JL, Holle AW, Jeong K, Major LG, Jeong JH, et al. Stem cell mechanosensation on gelatin methacryloyl (GelMA) stiffness gradient hydrogels. Ann Biomed Eng. (2020) 48:893–902. doi: 10.1007/s10439-019-02428-5
125. Sunyer R, Jin AJ, Nossal R, Sackett DL. Fabrication of hydrogels with steep stiffness gradients for studying cell mechanical response. PLoS ONE. (2012) 7:e46107. doi: 10.1371/journal.pone.0046107
126. Dou J, Mao S, Li H, Lin JM. Combination stiffness gradient with chemical stimulation directs glioma cell migration on a microfluidic chip. Anal Chem. (2020) 92:892–8. doi: 10.1021/acs.analchem.9b03681
127. Hadden WJ, Young JL, Holle AW, McFetridge ML, Kim DY, Wijesinghe P, et al. Stem cell migration and mechanotransduction on linear stiffness gradient hydrogels. Proc Natl Acad Sci USA. (2017) 114:5647–52. doi: 10.1073/pnas.1618239114
128. Wang PY, Tsai WB, Voelcker NH. Screening of rat mesenchymal stem cell behaviour on polydimethylsiloxane stiffness gradients. Acta Biomater. (2012) 8:519–30. doi: 10.1016/j.actbio.2011.09.030
129. Lavrentieva A, Fleischhammer T, Enders A, Pirmahboub H, Bahnemann J, Pepelanova I. Fabrication of stiffness gradients of GelMA hydrogels using a 3D printed micromixer. Macromol Biosci. (2020) 20:e2000107. doi: 10.1002/mabi.202000107
130. Zhao H, Li X, Zhao S, Zeng Y, Zhao L, Ding H, et al. Microengineered in vitro model of cardiac fibrosis through modulating myofibroblast mechanotransduction. Biofabrication. (2014) 6:045009. doi: 10.1088/1758-5082/6/4/045009
131. Liu F, Mih JD, Shea BS, Kho AT, Sharif AS, Tager AM, et al. Feedback amplification of fibrosis through matrix stiffening and COX-2 suppression. J Cell Biol. (2010) 190:693–706. doi: 10.1083/jcb.201004082
132. Kloxin AM, Kasko AM, Salinas CN, Anseth KS. Photodegradable hydrogels for dynamic tuning of physical and chemical properties. Science. (2009) 324:59–63. doi: 10.1126/science.1169494
133. Crocini C, Walker CJ, Anseth KS, Leinwand LA. Three-dimensional encapsulation of adult mouse cardiomyocytes in hydrogels with tunable stiffness. Prog Biophys Mol Biol. (2020) 154:71–9. doi: 10.1016/j.pbiomolbio.2019.04.008
134. Young JL, Engler AJ. Hydrogels with time-dependent material properties enhance cardiomyocyte differentiation in vitro. Biomaterials. (2011) 32:1002–9. doi: 10.1016/j.biomaterials.2010.10.020
135. Burdick JA, Murphy WL. Moving from static to dynamic complexity in hydrogel design. Nat Commun. (2012) 3:1269. doi: 10.1038/ncomms2271
136. Wang EY, Rafatian N, Zhao Y, Lee A, Lai BFL, Lu RX, et al. Biowire model of interstitial and focal cardiac fibrosis. ACS Cent Sci. (2019) 5:1146–58. doi: 10.1021/acscentsci.9b00052
137. Galie PA, Westfall MV, Stegemann JP. Reduced serum content and increased matrix stiffness promote the cardiac myofibroblast transition in 3D collagen matrices. Cardiovasc Pathol. (2011) 20:325–33. doi: 10.1016/j.carpath.2010.10.001
138. Yu J, Seldin MM, Fu K, Li S, Lam L, Wang P, et al. Topological arrangement of cardiac fibroblasts regulates cellular plasticity. Circ Res. (2018) 123:73–85. doi: 10.1161/CIRCRESAHA.118.312589
139. van Spreeuwel ACC, Bax NAM, van Nierop BJ, Aartsma-Rus A, Goumans MTH, Bouten CVC. Mimicking cardiac fibrosis in a dish: fibroblast density rather than collagen density weakens cardiomyocyte function. J Cardiovasc Transl Res. (2017) 10:116–27. doi: 10.1007/s12265-017-9737-1
140. van Spreeuwel AC, Bax NA, Bastiaens AJ, Foolen J, Loerakker S, Borochin M, et al. The influence of matrix (an)isotropy on cardiomyocyte contraction in engineered cardiac microtissues. Integr Biol. (2014) 6:422–9. doi: 10.1039/C3IB40219C
141. DePalma SJ, Davidson CD, Stis AE, Helms AS, Baker BM. Microenvironmental determinants of organized iPSC-cardiomyocyte tissues on synthetic fibrous matrices. Biomater Sci. (2021) 9:93–107. doi: 10.1039/D0BM01247E
142. Ma Z, Huebsch N, Koo S, Mandegar MA, Siemons B, Boggess S, et al. Contractile deficits in engineered cardiac microtissues as a result of MYBPC3 deficiency and mechanical overload. Nat Biomed Eng. (2018) 2:955–67. doi: 10.1038/s41551-018-0280-4
143. Baker BM, Trappmann B, Wang WY, Sakar MS, Kim IL, Shenoy VB, et al. Cell-mediated fibre recruitment drives extracellular matrix mechanosensing in engineered fibrillar microenvironments. Nat Mater. (2015) 14:1262–8. doi: 10.1038/nmat4444
144. Davidson CD, Jayco DKP, Matera DL, DePalma SJ, Hiraki HL, Wang WY, et al. Myofibroblast activation in synthetic fibrous matrices composed of dextran vinyl sulfone. Acta Biomater. (2020) 105:78–86. doi: 10.1016/j.actbio.2020.01.009
145. Matera DL, DiLillo KM, Smith MR, Davidson CD, Parikh R, Said M, et al. Microengineered 3D pulmonary interstitial mimetics highlight a critical role for matrix degradation in myofibroblast differentiation. Sci Adv. (2020) 6:eabb5069. doi: 10.1126/sciadv.abb5069
146. Wang H, Haeger SM, Kloxin AM, Leinwand LA, Anseth KS. Redirecting valvular myofibroblasts into dormant fibroblasts through light-mediated reduction in substrate modulus. PLoS ONE. (2012) 7:e39969. doi: 10.1371/journal.pone.0039969
147. Gorcsan J, Tanaka H. Echocardiographic assessment of myocardial strain. J Am Coll Cardiol. (2011) 58:1401–13. doi: 10.1016/j.jacc.2011.06.038
148. Fink C, Ergün S, Kralisch D, Remmers U, Weil J, Eschenhagen T. Chronic stretch of engineered heart tissue induces hypertrophy and functional improvement. FASEB J. (2000) 14:669–79. doi: 10.1096/fasebj.14.5.669
149. Zimmermann WH, Schneiderbanger K, Schubert P, Didié M, Münzel F, Heubach JF, et al. Tissue engineering of a differentiated cardiac muscle construct. Circ Res. (2002) 90:223–30. doi: 10.1161/hh0202.103644
150. Salazar BH, Cashion AT, Dennis RG, Birla RK. Development of a cyclic strain bioreactor for mechanical enhancement and assessment of bioengineered myocardial constructs. Cardiovasc Eng Technol. (2015) 6:533–45. doi: 10.1007/s13239-015-0236-8
151. Massai D, Pisani G, Isu G, Rodriguez Ruiz A, Cerino G, Galluzzi R, et al. Bioreactor platform for biomimetic culture and in situ monitoring of the mechanical response of in vitro engineered models of cardiac tissue. Front Bioeng Biotechnol. (2020) 8:733. doi: 10.3389/fbioe.2020.00733
152. Tulloch NL, Muskheli V, Razumova MV, Korte FS, Regnier M, Hauch KD, et al. Growth of engineered human myocardium with mechanical loading and vascular coculture. Circ Res. (2011) 109:47–59. doi: 10.1161/CIRCRESAHA.110.237206
153. Naito H, Melnychenko I, Didié M, Schneiderbanger K, Schubert P, Rosenkranz S, et al. Optimizing engineered heart tissue for therapeutic applications as surrogate heart muscle. Circulation. (2006) 114:I72–8. doi: 10.1161/CIRCULATIONAHA.105.001560
154. McCain ML, Sheehy SP, Grosberg A, Goss JA, Parker KK. Recapitulating maladaptive, multiscale remodeling of failing myocardium on a chip. Proc Natl Acad Sci USA. (2013) 110:9770–5. doi: 10.1073/pnas.1304913110
155. Ugolini GS, Pavesi A, Rasponi M, Fiore GB, Kamm R, Soncini M. Human cardiac fibroblasts adaptive responses to controlled combined mechanical strain and oxygen changes in vitro. eLife. (2017) 6:e22847. doi: 10.7554/eLife.22847.020
156. Occhetta P, Isu G, Lemme M, Conficconi C, Oertle P, Räz C, et al. A three-dimensional in vitro dynamic micro-tissue model of cardiac scar formation. Integr Biol. (2018) 10:174–83. doi: 10.1039/C7IB00199A
157. Balestrini JL, Billiar KL. Equibiaxial cyclic stretch stimulates fibroblasts to rapidly remodel fibrin. J Biomech. (2006) 39:2983–90. doi: 10.1016/j.jbiomech.2005.10.025
158. Balestrini JL, Billiar KL. Magnitude and duration of stretch modulate fibroblast remodeling. J Biomech Eng. (2009) 131:051005. doi: 10.1115/1.3049527
159. Yokoyama T, Sekiguchi K, Tanaka T, Tomaru K, Arai M, Suzuki T, et al. Angiotensin II and mechanical stretch induce production of tumor necrosis factor in cardiac fibroblasts. Am J Physiol. (1999) 276:H1968–76. doi: 10.1152/ajpheart.1999.276.6.H1968
160. Wang J, Chen H, Seth A, McCulloch CA. Mechanical force regulation of myofibroblast differentiation in cardiac fibroblasts. Am J Physiol Heart Circ Physiol. (2003) 285:H1871–81. doi: 10.1152/ajpheart.00387.2003
161. Ugolini GS, Rasponi M, Pavesi A, Santoro R, Kamm R, Fiore GB, et al. On-chip assessment of human primary cardiac fibroblasts proliferative responses to uniaxial cyclic mechanical strain. Biotechnol Bioeng. (2016) 113:859–69. doi: 10.1002/bit.25847
162. Atance J, Yost MJ, Carver W. Influence of the extracellular matrix on the regulation of cardiac fibroblast behavior by mechanical stretch. J Cell Physiol. (2004) 200:377–86. doi: 10.1002/jcp.20034
163. Watson CJ, Phelan D, Collier P, Horgan S, Glezeva N, Cooke G, et al. Extracellular matrix sub-types and mechanical stretch impact human cardiac fibroblast responses to transforming growth factor beta. Connect Tissue Res. (2014) 55:248–56. doi: 10.3109/03008207.2014.904856
164. Butt RP, Bishop JE. Mechanical load enhances the stimulatory effect of serum growth factors on cardiac fibroblast procollagen synthesis. J Mol Cell Cardiol. (1997) 29:1141–51. doi: 10.1006/jmcc.1996.0347
165. Li X, Garcia-Elias A, Benito B, Nattel S. The effects of cardiac stretch on atrial fibroblasts: analysis of the evidence and potential role in atrial fibrillation. Cardiovasc Res. (2021). doi: 10.1093/cvr/cvab035. [Epub ahead of print].
166. Rysä J, Tokola H, Ruskoaho H. Mechanical stretch induced transcriptomic profiles in cardiac myocytes. Sci Rep. (2018) 8:4733. doi: 10.1038/s41598-018-23042-w
167. Wang BW, Wu GJ, Cheng WP, Shyu KG. Mechanical stretch via transforming growth factor-β1 activates microRNA-208a to regulate hypertrophy in cultured rat cardiac myocytes. J Formos Med Assoc. (2013) 112:635–43. doi: 10.1016/j.jfma.2013.01.002
168. Liao XD, Wang XH, Jin HJ, Chen LY, Chen Q. Mechanical stretch induces mitochondria-dependent apoptosis in neonatal rat cardiomyocytes and G2/M accumulation in cardiac fibroblasts. Cell Res. (2004) 14:16–26. doi: 10.1038/sj.cr.7290198
169. Stevens KR, Ungrin MD, Schwartz RE, Ng S, Carvalho B, Christine KS, et al. InVERT molding for scalable control of tissue microarchitecture. Nat Commun. (2013) 4:1847. doi: 10.1038/ncomms2853
170. Hoang P, Wang J, Conklin BR, Healy KE, Ma Z. Generation of spatial-patterned early-developing cardiac organoids using human pluripotent stem cells. Nat Protoc. (2018) 13:723–37. doi: 10.1038/nprot.2018.006
171. Nikolaev M, Mitrofanova O, Broguiere N, Geraldo S, Dutta D, Tabata Y, et al. Homeostatic mini-intestines through scaffold-guided organoid morphogenesis. Nature. (2020) 585:574–8. doi: 10.1038/s41586-020-2724-8
172. Skylar-Scott MA, Uzel SGM, Nam LL, Ahrens JH, Truby RL, Damaraju S, et al. Biomanufacturing of organ-specific tissues with high cellular density and embedded vascular channels. Sci Adv. (2019) 5:eaaw. doi: 10.1126/sciadv.aaw2459
173. Mirdamadi E, Tashman JW, Shiwarski DJ, Palchesko RN, Feinberg AW. FRESH 3D bioprinting a full-size model of the human heart. ACS Biomater Sci Eng. (2020) 6:6453–9. doi: 10.1021/acsbiomaterials.0c01133
174. Bittner SM, Smith BT, Diaz-Gomez L, Hudgins CD, Melchiorri AJ, Scott DW, et al. Fabrication and mechanical characterization of 3D printed vertical uniform and gradient scaffolds for bone and osteochondral tissue engineering. Acta Biomater. (2019) 90:37–48. doi: 10.1016/j.actbio.2019.03.041
175. Liu W, Zhang YS, Heinrich MA, De Ferrari F, Jang HL, Bakht SM, et al. Rapid continuous multimaterial extrusion bioprinting. Adv Mater. (2017) 29:1604630. doi: 10.1002/adma.201604630
176. Hausenloy DJ, Bøtker HE, Ferdinandy P, Heusch G, Ng GA, Redington A, et al. Cardiac innervation in acute myocardial ischaemia/reperfusion injury and cardioprotection. Cardiovasc Res. (2019) 115:1167–77. doi: 10.1093/cvr/cvz053
177. Gentek R, Hoeffel G. The innate immune response in myocardial infarction, repair, and regeneration. Adv Exp Med Biol. (2017) 1003:251–72. doi: 10.1007/978-3-319-57613-8_12
178. Frick C, Dettinger P, Renkawitz J, Jauch A, Berger CT, Recher M, et al. Nano-scale microfluidics to study 3D chemotaxis at the single cell level. PLoS ONE. (2018) 13:e0198330. doi: 10.1371/journal.pone.0198330
179. Li Y, Xuan J, Hu R, Zhang P, Lou X, Yang Y. Microfluidic triple-gradient generator for efficient screening of chemical space. Talanta. (2019) 204:569–75. doi: 10.1016/j.talanta.2019.06.018
180. Gandavarapu NR, Azagarsamy MA, Anseth KS. Photo-click living strategy for controlled, reversible exchange of biochemical ligands. Adv Mater. (2014) 26:2521–6. doi: 10.1002/adma.201304847
181. Dai X, Wiernek S, Evans JP, Runge MS. Genetics of coronary artery disease and myocardial infarction. World J Cardiol. (2016) 8:1–23. doi: 10.4330/wjc.v8.i1.1
182. Yang X, Pabon L, Murry CE. Engineering adolescence: maturation of human pluripotent stem cell-derived cardiomyocytes. Circ Res. (2014) 114:511–23. doi: 10.1161/CIRCRESAHA.114.300558
183. Ronaldson-Bouchard K, Ma SP, Yeager K, Chen T, Song L, Sirabella D, et al. Advanced maturation of human cardiac tissue grown from pluripotent stem cells. Nature. (2018) 556:239–43. doi: 10.1038/s41586-018-0016-3
184. Zhao Y, Wang EY, Davenport LH, Liao Y, Yeager K, Vunjak-Novakovic G, et al. A multimaterial microphysiological platform enabled by rapid casting of elastic microwires. Adv Healthc Mater. (2019) 8:e1801187. doi: 10.1002/adhm.201801187
185. Yip JK, Sarkar D, Petersen AP, Gipson JN, Tao J, Kale S, et al. Contact photolithography-free integration of patterned and semi-transparent indium tin oxide stimulation electrodes into polydimethylsiloxane-based heart-on-a-chip devices for streamlining physiological recordings. Lab Chip. (2021) 21:674–87. doi: 10.1039/D0LC00948B
186. Dipalo M, Rastogi SK, Matino L, Garg R, Bliley J, Iachetta G, et al. Intracellular action potential recordings from cardiomyocytes by ultrafast pulsed laser irradiation of fuzzy graphene microelectrodes. Sci Adv. (2021) 7:eabd5175. doi: 10.1126/sciadv.abd5175
187. Marasco CC, Enders JR, Seale KT, McLean JA, Wikswo JP. Real-time cellular exometabolome analysis with a microfluidic-mass spectrometry platform. PLoS ONE. (2015) 10:e0117685. doi: 10.1371/journal.pone.0117685
188. McKenzie JR, Cognata AC, Davis AN, Wikswo JP, Cliffel DE. Real-time monitoring of cellular bioenergetics with a multianalyte screen-printed electrode. Anal Chem. (2015) 87:7857–64. doi: 10.1021/acs.analchem.5b01533
Keywords: tissue engineering, myocardial infarction, organ on a chip, hypoxia, stiffness, strain, cardiac myocytes, cardiac fibroblasts
Citation: Khalil NN and McCain ML (2021) Engineering the Cellular Microenvironment of Post-infarct Myocardium on a Chip. Front. Cardiovasc. Med. 8:709871. doi: 10.3389/fcvm.2021.709871
Received: 14 May 2021; Accepted: 14 June 2021;
Published: 14 July 2021.
Edited by:
David Wu, University of Chicago, United StatesReviewed by:
Walter Emerson Cromer, Texas A&M Health Science Center, United StatesCopyright © 2021 Khalil and McCain. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Megan L. McCain, bWxtY2NhaW5AdXNjLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.