
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Cardiovasc. Med. , 27 October 2021
Sec. Atherosclerosis and Vascular Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.708946
 Adem Aksoy1*†
Adem Aksoy1*† Muntadher Al Zaidi1†
Muntadher Al Zaidi1† Elena Repges1
Elena Repges1 Marc Ulrich Becher1Cornelius Müller1
Marc Ulrich Becher1Cornelius Müller1 Johannes Oldenburg2Sebastian Zimmer1Georg Nickenig1Vedat Tiyerili1
Johannes Oldenburg2Sebastian Zimmer1Georg Nickenig1Vedat Tiyerili1Background: Vitamin K antagonists (VKA) are known to promote adverse cardiovascular remodeling. Contrarily, vitamin K supplementation has been discussed to decelerate cardiovascular disease. The recently described VKOR-isoenzyme Vitamin K epoxide reductase complex subunit 1-like 1 (VKORC1L1) is involved in vitamin K maintenance and exerts antioxidant properties. In this study, we sought to investigate the role of VKORC1L1 in neointima formation and on vascular smooth muscle cell (VSMC) function.
Methods and Results: Treatment of wild-type mice with Warfarin, a well-known VKA, increased maladaptive neointima formation after carotid artery injury. This was accompanied by reduced vascular mRNA expression of VKORC1L1. In vitro, Warfarin was found to reduce VKORC1L1 mRNA expression in VSMC. VKORC1L1-downregulation by siRNA promoted viability, migration and formation of reactive oxygen species. VKORC1L1 knockdown further increased expression of key markers of vascular inflammation (NFκB, IL-6). Additionally, downregulation of the endoplasmic reticulum (ER) membrane resident VKORC1L1 increased expression of the main ER Stress moderator, glucose-regulated protein 78 kDa (GRP78). Moreover, treatment with the ER Stress inducer tunicamycin promoted VKORC1L1, but not VKORC1 expression. Finally, we sought to investigate, if treatment with vitamin K can exert protective properties on VSMC. Thus, we examined effects of menaquinone-7 (MK7) on VSMC phenotype switch. MK7 treatment dose-dependently alleviated PDGF-induced proliferation and migration. In addition, we detected a reduction in expression of inflammatory and ER Stress markers.
Conclusion: VKA treatment promotes neointima formation after carotid wire injury. In addition, VKA treatment reduces aortal VKORC1L1 mRNA expression. VKORC1L1 inhibition contributes to an adverse VSMC phenotype, while MK7 restores VSMC function. Thus, MK7 supplementation might be a feasible therapeutic option to modulate vitamin K- and VKORC1L1-mediated vasculoprotection.
Oxidative stress and inflammation contribute to adverse cardiovascular remodeling, eventually resulting in atherosclerosis and vascular dysfunction (1). Vascular smooth muscle cells (VSMC) participate in atherosclerotic plaque growth in different ways. In response to atherosclerotic stimuli, VSMC may turn hyper-proliferative and promote neointima formation. Neointima formation is the main driver of in-stent restenosis and is further enhanced by pro-inflammatory stimuli in the vasculature (2, 3). The ability of VSMC to limit vascular inflammation and to preserve their physiological phenotype is essential to slow the progression of atherosclerosis and neointima formation in particular.
Vitamin K describes a group of fat-soluble vitamins that are required as co-factors for γ-carboxylation of proteins (4). Vitamin K1 and the K2 vitamins menaquione-4 (MK4) and menaquione-7 (MK7) are the best-known members of the vitamin K group. Among these, MK7 is the most potent K vitamin in human physiology (5). In the vitamin K cycle, vitamin K is recycled by reactions catalysed by vitamin K epoxide reductase complex subunit 1 (VKORC1) and its recently described isoenzyme, vitamin K epoxide reductase complex subunit 1-like 1 (VKORC1L1). These two enzymes serve as targets of vitamin K antagonists (VKA), such as Warfarin, that are used in therapeutic anticoagulation regimen (6, 7). Evidence has emerged that VKA also contribute to cardiovascular damage (8–10). In addition, dietary supplementation with vitamin K was shown to be a safe and feasible option to decelerate vascular disease (11–13). Furthermore, vitamin K provides anti-oxidative properties and acts as a potent free-radical scavenger (14, 15). Hence, elucidating the mechanism of VKA-induced vascular dysfunction and vitamin K-dependent vasculoprotection is of relevance. Compared to VKORC1, VKORC1L1 is expressed at lower levels in the liver, the place where coagulation factors are synthesised (16), and thus has a lower ability to γ-carboxylate the coagulation factors (7, 17). Upon oxidative stress, VKORC1L1 is upregulated, while VKORC1 is downregulated (18). Furthermore, VKORC1L1-knockout cells are far more sensitive to oxidative stress as compared to VKORC1-knockout cells (19). Thus far, there is no data regarding the significance of VKORC1L1 in cardiovascular diseases, although a recent mRNA expression analysis described VKORC1L1 as a putative oxidative-stress-related gene in coronary artery disease (20).
Both VKOR proteins are localized in the membrane of the endoplasmic reticulum (ER) (18). In the ER, proteins are folded under sensitive environmental conditions and at a precisely regulated redox state. Disturbances of these conditions constitute ER stress, a major contributor to vascular inflammation (21, 22). The VKOR system is known to interact with protein folding (23), but the role of VKOR proteins in ER stress remains unclear.
Therefore, the aim of this study was to analyse the role of VKORC1L1 in neointima formation and on VSMC inflammation and proliferation.
Animal experiments were performed in accordance with the animal protection law stated in the German civil code and the National Office for Nature, Environment and Consumer Protection in Recklinghausen, North Rhine-Westphalia (Landesamt für Natur, Umwelt und Verbraucherschutz; LANUV). We used six-week-old C57BL/6 wild-type mice (Charles River, Sulzfeld, Germany). Animals were maintained in a 22°C room with a 12 h light/dark cycle and received food and drinking water ad libitum. The surgical intervention was performed under a dissecting microscope (MZ6; Leica). For the carotid-injury procedure, mice were anesthetized with intraperitoneal injections of 150 mg/kg body weight ketamine hydrochloride (Ketanest, Riemser, Greifswald, Germany) and 0.1 mg/kg body weight xylazine hydrochloride (Ceva, Duesseldorf, Germany). Access to the carotid artery was obtained by performing a midline skin incision from directly below the mandible toward the sternum. Careful preparation of the left common carotid artery and carotid bifurcation was performed. Two filaments were placed in the proximal and distal segments of the external carotid artery, and the distal ligature was then pinched with clamps. The internal and common carotid arteries were temporarily occluded to perform a transverse arteriotomy between the ligatures of the external carotid artery and to insert a flexible wire (0.13 mm in diameter), that is slightly curved (30°) at the tip and completely fills out the vessel. For endothelial denudation, a pullback of the wire was performed in a rotating manner for five times per animal and then removed. Then the external carotid artery was closed below the site of puncture with a ligature, and the blood flow of common and internal carotid artery was released. The skin was then sutured. The mice were postoperatively allowed to recover individually. During recovery period, animals were held in a warm environment by use of water-circulating heat pads and were closely observed. Food and water intake were monitored. The recovery period was four h long. Full recovery was confirmed by return of righting reflex and stable respirations. Thereafter, the animals were returned to their littermates and randomized to three different diet groups. Group A received vehicle, group B received a vitamin K1 (1.5 mg/g food)-enriched diet, and group C received a vitamin K1 (1.5 mg/g food) and warfarin (2 mg/g food)-enriched diet. The composition of the food regimes was adapted according to Schurgers et al. (9) After 14 days, the mice were sacrificed. All mice were anesthetized using 2% isoflurane and an intraperitoneal injection of fentanyl (0.05 mg/kg) and midazolam (5 mg/kg), then euthanized by cervical dislocation. All tissue and blood samples were collected and processed immediately after the mice were sacrificed.
For factor x activity measurement, blood samples (100 μl) were collected, immediately added onto citrate-containing tubes in a ratio of 10:1 (blood/citrate) and mixed. After centrifugation, the resulting plasma was diluted in factor x deficient plasma. This step is necessary to achieve that the factor x activity is the limiting factor in the following thromboplastin time measurement. Thromboplastin time corresponding to factor x acticity was then measured and normalized as percentage to a previously determined normal plasma. Factor X activity was measured at the Institute of Experimental Haematology and Transfusion Medicine at the University Hospital of Bonn, Germany.
Vitamin K1 and warfarin were purchased from Sigma-Aldrich (Cat# 47773 and A2250) and provided to Ssniff-Spezialdiäten, Soest, Germany. Ssniff-Spezialdiäten then mixed vitamin K1 or vitamin K1 and warfarin into the regular mice diet.
Primary human coronary artery smooth muscle cells (HCASMC) were purchased (PromoCell, Germany; Cat# C-12511) and cultured in their respective cell medium (PromoCell, Cat# C-22062), using standard conditions (37°C, 5% CO2, 100% relative humidity). Once the cells reached 70–90% confluence, the medium was removed, and cells were washed once before passaging by trypsinisation. Cells from passages 6–8 were used for experiments.
For siRNA transfection, the cells were transfected with HiPerFect Transfection Reagent (Qiagen, Netherlands; Cat# 301705), using the Reverse Transfection Protocol. Briefly, HiPerFect plus an siRNA directed against VKORC1L1 (Qiagen, # SI04138407) or a scrambled siRNA (Qiagen, # 1027280), not directed toward any mRNA, were diluted in serum-free media and mixed by vortexing. The transfection mixtures were incubated at room temperature for ten min and then transferred into empty cell-culture wells. In the meantime, cells were trypsinised and then seeded on top of the transfection complexes. The final siRNA concentration was 10 nM and 0.5% V/V for HiPerFect Transfection Reagent. Following transfection, the cells were incubated under the above-mentioned conditions for 46 h before a readout was taken.
alamarBlueTM HS Cell Viability Reagent (Thermo Fisher Scientific, USA; Cat# A50100) was added onto pre-treated cells. The viability reagent and cell mixtures were incubated for 4 h under standard conditions (37°C, 5% CO2, 100% relative humidity) and protected from light. Then the absorbance was measured using an Infinite M200 Microplate Reader (Tecan, Switzerland).
Cells were lysed with RIPA Buffer (Sigma-Aldrich, Cat# R0278) containing 1:25 Protease Inhibitor Cocktail (Roche, Cat# 4693132001). The lysates were centrifuged at 13,000 x g for 10 min at 4°C. Protein concentration in the supernatant was quantified by a Qubit Protein Assay (Thermo Fisher Scientific, Cat# Q33211) in a Qubit-4 Fluorometer (Thermo Fisher Scientific). 25 μg of the resulting protein were loaded onto an SDS-PAGE gel and electrophoresis was initiated on a Mini Protean system (Bio-Rad). Proteins were then transferred to a nitrocellulose membrane (Carl Roth, HP40.1) for western blotting. For blocking of the membrane, we used 5% BSA for one h at room temperature. Afterwards the membrane was incubated with the respective primary antibody overnight at 4°C.
The next morning, the membrane was washed three times with 0.1% tris-buffered saline containing 0.1% Tween 20, before addition of the secondary antibody. After an one-h long incubation at room temperature, the membrane was washed again three times before detection was performed with an ECL Western Blot Detection Reagent (Sigma-Aldrich, Cat# RPN2232) on a ChemoCam HR16-3200 Imager (Intas).
The following antibodies were used: VKORC1L1 (final concentration: 0.2μg/ml, Sigma-Aldrich, Cat# HPA053954), ß-actin (dilution 1:2000, Sigma-Aldrich, Cat# A1978), anti-mouse-IgG secondary antibody (dilution 1:10,000, Sigma-Aldrich, Cat# A9044) and anti-rabbit-IgG secondary antibody (dilution 1:10,000, Sigma-Aldrich Cat# A0545).
The formation of ROS was measured by using L-012 chemiluminescence and 2',7'-dichlorofluorescein diacetate (DCFDA, Sigma-Aldrich, USA; Cat# D6883) chemiluminescent assays. L-012, formed by derivation of luminol, has a high sensitivity for superoxide radicals and does not exert redox cycling itself. Chemiluminescence was determined over 15 min in a scintillation counter (Lumat LB 9501, Berthold) at one-min intervals.
For DCFDA assay, cells were seeded one day prior to the experiment unto a dark-bottomed 96-well microplate protected from light and were incubated at standard conditions. After 24 h, the cells were stimulated with 75 μM hydrogen peroxide (H2O2) for one h. Since H2O2 is a potent inducer of free radical formation (24), cells were stimulated with H2O2 to measure ROS formation in response to an oxidative stressor. Then, a dilution of 50 μM DCFDA was added for 45 min. Finally, the DCFDA solution was removed and the cells were washed before the addition of PBS. Fluorescence was detected immediately using a microplate reader at maximum excitation and emission of 492 nm and 527 nm, respectively as previously described (25, 26).
To specifically measure the source of ROS, we utilized an AmplexTM Red Hydrogen Peroxide Assay Kit (Thermo Fisher Scientific, Cat# A22188). In presence of HRP, AmplexTM Red reacts with H2O2 to a fluorescent product. Cells were seeded onto 12-well-plates and transfected with siRNA against VKORC1L1 or scrambled siRNA. After 46 h, 50 μl of the medium was collected and incubated for one h in the dark at 37°C with 50 μl of a mixture of HRP (0.1 U/ml) and AmplexTM Red (50 μM). Thereafter fluorescency was measured in an Infinite M200 Microplate Reader (Tecan, Switzerland) microplate reader (excitation 560 nm, emission 590 nm).
Cell proliferation was analysed using a Click-iT™ EdU Proliferation Assay for Microplates (Thermo Fisher Scientific, Cat# C10499). Briefly, the nucleoside analog EdU (5-ethynyl-2'-deoxyuridine) was added to live cells for 4 h. Then the cells were fixed and incubated with horseradish peroxidase (HRP), which becomes ligated to the DNA-incorporated EdU moiety by using click chemistry. Thereafter, AmplexTM UltraRed, a reagent that is converted to a fluorescent product in presence of HRP, was added and the fluorescence was measured on a microplate reader at an excitation of 568 nm and an emission of 585 nm.
Cultured cells were reverse transfected in 12-well plates as described above. After 48 h, cells were at confluence and a vertical scratch was created using a sterile pipette-tip (200 μl). Cells were washed and then cultured in reduced-serum medium (2.5%) to minimize proliferation without inducing extensive apoptosis. A specific position on each well was marked and images using an Axiovert 200M microscope (Carl Zeiss, Germany) were taken after 0, 4, 8, 12, and 24 h. Th area of cell migration into the scratched region was measured and calculated as a percentage of the initially scratched area.
Total RNA from the cells was isolated by using TRIzolTM (Thermo Fisher Scientific, Cat# 15596018) and chloroform phase separation. The amount of RNA collected was quantified using a Nanodrop spectrophotometer (Nanodrop Technologies, USA). 0.5–2 μg of RNA were reverse transcribed by using an Omniscript RT Kit (Qiagen, Cat# 205113). Finally, quantitative real-time PCR was performed on a 7500 HT Real-Time PCR machine (Applied Biosystems, USA) using TaqMan gene expression assays (Thermo Fisher Scientific) and Gene Expression Master Mix (Thermo Fisher Scientific, Cat# 4369542). CT values up to 40 were used for analysis and all samples were run in triplicate. The values were analysed using the ΔΔCT method, by normalising to 18S ribosomal RNA.
The following TaqMan Assays were used: VKORC1L1 (Hs04989728_s1), VKORC1 (Hs00829655_s1), interleukin 6 (IL-6, Hs00174131_m1), nuclear factor ‘kappa-light-chain-enhancer' of activated B-cells (NF-κB, Hs00765730_m1), glucose-regulated protein 78 kDa(GRP78, Hs00607129_gH), C/EBP homologous protein (CHOP, Hs00358796_g1) and 18S ribosomal RNA (18S, Hs99999901_s1).
Cells were seeded into cell-culture wells and allowed to adhere. After 48 h, MK7 (Santa Cruz Biotechnology, USA; Cat# sc-218691) dissolved in DMSO (0.5 mg/ml) was mixed with fresh cell medium and added onto the cells. The incubation time was 24 h and the final MK7 concentrations in the medium ranged from 1 to 10μM. In some cases, cells were co-incubated with oxLDL (Oxidized low-density lipoprotein, Thermo Fisher Scientific, Cat# L34357), tunicamycin (Sigma-Aldrich, Cat# T7765) or platelet-derived growth factor (PDGF, Thermo Fisher Scientific; Cat# PHG0044).
Statistical analyses were performed using the software GraphPad Prism 9. Means of two groups were compared with an unpaired t-test. Means of more than two groups were compared by a one-way ANOVA followed by Bonferroni's multiple comparison test. The number of independent experiments as well as the applied tests for statistical significance are reported in the figure legends. All reported p-values are two- sided.
In order to understand the possible role of VKORC1L1 in vascular remodeling, we utilised a murine vascular-injury model. C57/Bl6 mice were randomized to receive vehicle, a vitamin K1 (1.5 mg/g food)-enriched diet, or a vitamin K1 (1.5 mg/g) and warfarin (2 mg/g)-enriched diet after injury of the carotid artery (Figure 1A). Vitamin K1 was co-administrated with Warfarin in order to prevent the internal bleeding that is normally caused by warfarin and to assess warfarin's extrahepatic effect in isolation (27).
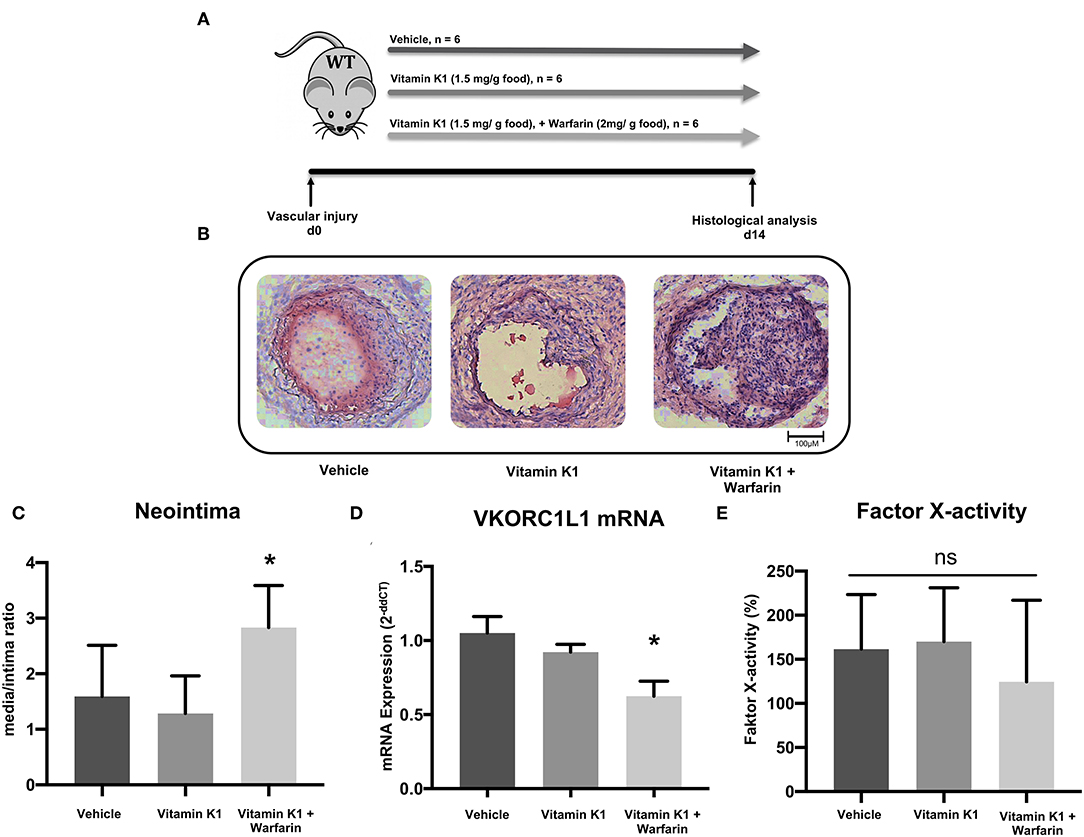
Figure 1. Vitamin K antagonism promotes vascular remodeling. (A) Schematic diagram of the design of the carotid artery injury experiment. After the procedure on 6–8 week-old C57Bl/6J wild-type mice, they were randomized in three groups that received either vehicle (group A), vitamin K1 (1.5 mg/g food)-enriched diet (group B), or vitamin K1 (1.5 mg/g food) and warfarin (2 mg/g food)-enriched diet (group C), n = 6 per group. (B) Images of neointima formation 14 days after carotid artery injury. Vessels were subjected to histological analysis by H.E. staining. (C) Quantitative assessment of neointima formation expressed as the media/intima ratio. (D) VKORC1L1 mRNA expression in the abdominal aorta expressed as 2−ddCT relative to control. (E) Factor X activity measured in citrate plasma samples after 14 days of treatment. Data are presented as the mean ± SEM; *p < 0.05; one-way ANOVA + Bonferroni's multiple comparison test for (C-E).
Mice fed a warfarin-enriched diet had increased neointima formation (media/intima ratio: 1.6 ± 0.9 vs. 1.3 ± 0.7 vs. 2.8 ± 0.7, p = 0.007, Figures 1B,C), and a significantly reduced VKORC1L1-mRNA expression in the abdominal aortic tissue (1.0 ± 0.1 vs. 0.9 ± 0.05 vs. 0.6 ± 0.1, p < 0.0001, Figure 1D). There was no significant difference between the three diet groups for coagulation activity, as assessed by a factor X activity assay (161.5 ± 61.9 % vs. 170.1 ± 61.0 % vs. 124.5 ± 92.5 %, p = 0.52, Figure 1E, control plasma: 100%).
Neointima formation is mainly caused by hyperproliferation of vascular smooth muscle cells (2). Therefore, HCASMC were stimulated with warfarin (1 μM for 24 h). Stimulation with warfarin resulted in significantly higher viability in HCASMC compared to the control group (145.9 ± 21.1% vs. control, p = 0.003, Figure 2A).
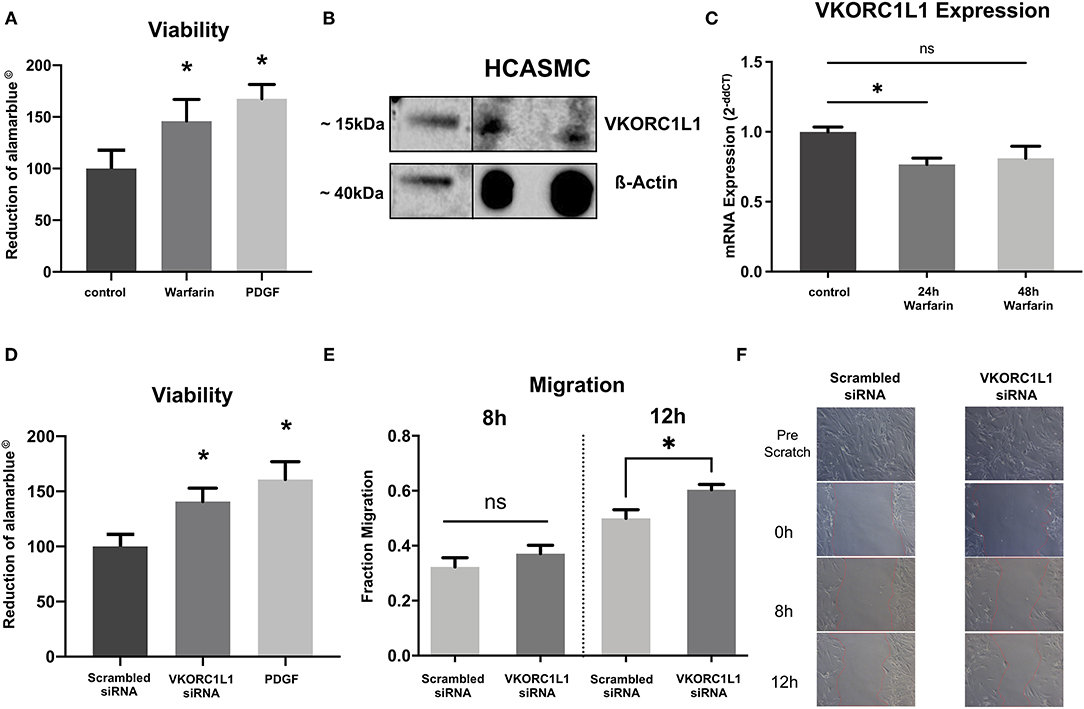
Figure 2. VKORC1L1-downregulation enhances HCASMC viability and migration. (A) Viability of HCASMC measured by alamarblue® assay after treatment with 1 μmol warfarin for 24 h. (B) VKORC1L1 protein expression in untreated HCASMC visualized by western blot. ß-Actin was used as housekeeping protein. (C) mRNA expression of VKORC1L1 in HCASMC after treatment with 1 μmol warfarin for 24 or 48 h. mRNA expression was measured by qPCR and quantified by the 2−ddCT method. n = 5. (D) Viability of HCASMC measured by alamarblue® after transfection of siRNAs directed against VKORC1L1 or a scrambled control. (E) Migration of siRNA-transfected HCASMC following a wound–scratch assay, measured by microscopic visualization after 8 and 12 hours. n = 7. (F) Representative pictures of migration. *p < 0.05. Data are presented as the mean ± SEM; *p < 0.05; one-way ANOVA + Bonferroni's multiple comparison test for (A, C, D).
Since this is the first study investigating the role of VKORC1L1 in vascular cells, we first performed western blot analyses to confirm expression of VKORC1L1. And indeed, VKORC1L1 is expressed significantly in HCASMC (Figure 2B).
Consistent to the in vivo experiments, we found warfarin treatment to reduce VKORC1L1 mRNA expression in vitro (1 μM, 24 h: 0.76-fold, p = 0.03 vs. control; 48 h: 0.81-fold, p = 0.07 vs. control; Figure 2C). To test possible specific effects of VKORC1L1, HCASMC were transfected with VKORC1L1-siRNA, which resulted in a largely reduced expression level of VKORC1L1 mRNA (Supplementary Figure 1). VKORC1L1 downregulated cells showed significantly higher viability compared to the control group (140.8 ± 12.1% vs. control, p = 0.0034, Figure 2D).
Migration of SMC is another major contributor to neointima formation. Therefore, we investigated the effect of VKORC1L1 inhibition on HCASMC migration in vitro. To address this point, wound–scratch assays were performed with transfected HCASMC, and the migration of cells was measured by microscopic visualization after 8 and 12 h. The migration of cells was not different between the VKORC1L1-siRNA transfected cells and the control group after 8 h (0.32 ± 0.09 vs. 0.37 ± 0.09 migration into the wound scratch, p = 0.3), whereas migration was enhanced significantly in knockdown cells compared to control cells after 12 h (0.49 ± 0.08 vs. 0.60 ± 0.05 migration into the wound scratch, p = 0.02, Figures 2E,F). After 24 h, migration into the scratch was fully achieved for both VKORC1L1-siRNA transfected cells and the control cell. Taken together, these data suggest, that VKORC1L1-downregulation enhances HCASMC viability and migration, both of which are crucial steps for neointima formation.
VKORC1L1 was found to exhibit anti-oxidative effects in human embryonic kidney cells. In those cells, VKORC1L1 deficiency was found to increase ROS formation (18, 19). Thus, we sought to investigate, if VKORC1L1 exhibits a similar role in HCASMC.
The release of superoxide radicals in VKORC1L1-downregulated HCASMC was measured by L-012 chemiluminescence and DCFDA assays. Figures 3A,B illustrate that vascular superoxide release was significantly increased in HCASMC after transfection with VKORC1L1-siRNA (L-012: 186.3 ± 22.62 % vs. scrambled siRNA p = 0.009, Figure 3A; DCFDA: 128.7 ± 10.82 % vs. scrambled siRNA, p = 0.04, Figure 3B). In order to investigate, if the generation of a specific oxidant is increased upon VKORC1L1 knockdown, we measured H2O2 generation by an AmplexTM Red Assay. Knockdown of VKORC1L1 by siRNA resulted in slightly, but significantly increased H2O2 generation in HCASMC (Scrambled siRNA: 100 ± 3.68 % vs. VKORC1L1-siRNA: 105 % ± 3.89 p = 0.046, Figure 3C). Thus, VKORC1L1- induced anti-oxidative effects are at least partly mediated by controlling H2O2 generation.
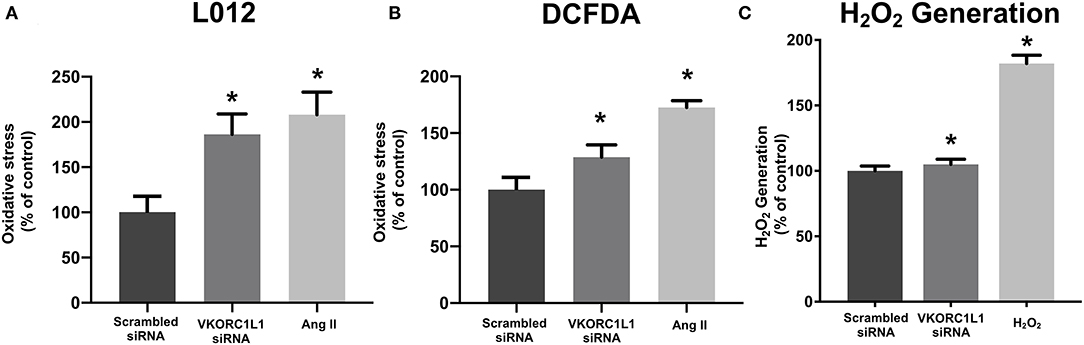
Figure 3. VKORC1L1-inhibition promotes reactive oxygen species formation. (A, B) ROS formation in HCASMC after transfection of siRNAs directed against VKORC1L1 or a scrambled control. Measured by using L-012 (A) and DCFDA assays (B). Angiotensin II (Ang II) was used as a positive control. n = 6–8. *p < 0.05 (C) H2O2 Generation in HCASMC was measured by an AmplexTM Red assay after transfection with siRNA against VKORC1L1 or a scrambled control. Treatment with H2O2 was used as a positive control. n = 6. *p < 0.05. Data are presented as the mean ± SEM; *p < 0.05; one-way ANOVA + Bonferroni's multiple comparison test for (A-C).
ROS formation and inflammation are closely connected with each other in the pathogenesis of cardiovascular diseases, including atherosclerosis. A pathological increase in ROS induces pro-inflammatory pathways including NF-κB signalling (28, 29). Consequently, we next investigated, if VKORC1L1-knockdown also results in increased inflammatory signalling.
To study the effect of VKOR1L1 on vascular inflammation, we measured the mRNA expression of the pro-inflammatory markers NF-κB and IL-6 by RT-PCR after transfection of siRNAs directed against VKORC1L1 or a scrambled control sequence. VKORC1L1 downregulation increased the expression of NF-κB mRNA (1.27-fold ± 0.32 vs. control, p = 0.04) and IL-6 (1.18-fold ± 0.12 vs. control, p = 0.02, Figure 4A).
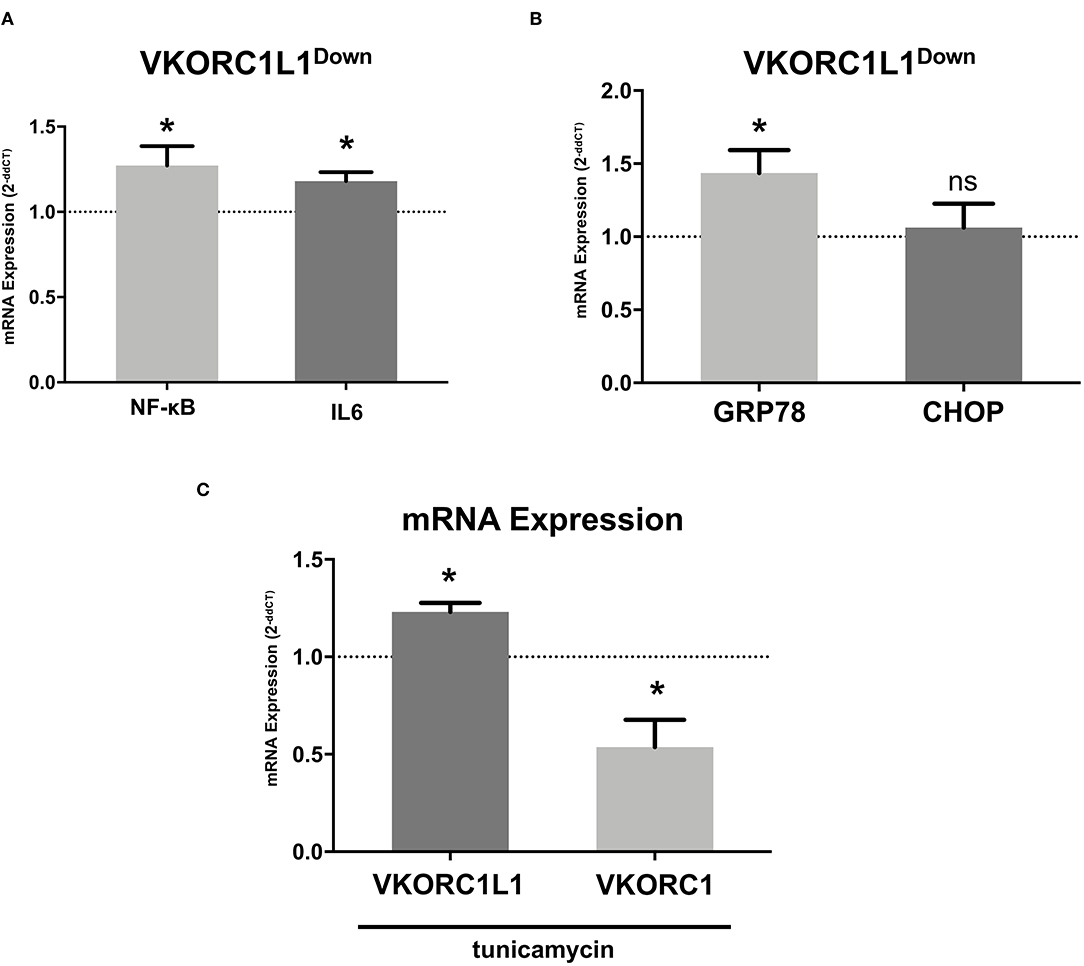
Figure 4. VKORC1L1 regulates vascular inflammation. (A) mRNA expression of the pro-inflammatory markers NF-κB and IL-6 in HCASMC after transfection with siRNAs directed against VKORC1L1 or a scrambled control. Control mRNA expression (=1) is shown as a dotted line. mRNA expression was measured by qPCR and quantified by the 2−ddCT method. n = 3–8. (B) mRNA expression of ER stress markers GRP78 and CHOP in HCASMC after transfection with siRNAs directed against VKORC1L1 or a scrambled control. Control expression (=1) is shown as a dotted line. mRNA expression was measured by qPCR and quantified by the 2−ddCT method. n = 4–8. (C) mRNA expression of VKORC1L1 and VKORC1 in HCASMC after treatment with the ER stress inductor, tunicamycin (6 h, 5 μg/ml). Control expression (DMSO treatment, =1) is shown as a dotted line. n = 4. Data are presented as the mean ± SEM; *p < 0.05. Unpaired t-test for (A-C).
Furthermore, VKORC1L1-downregulated HCASMC showed increased mRNA expression levels of GRP78, the main ER stress regulator (1.44- fold ± 0.44 vs. control, p = 0.027, Figure 4B). However, mRNA expression of CHOP, another ER Stress protein, was unaltered (1.06- fold ± 0.43 vs. control, p = 0.71, Figure 4B). Additionally, stimulation with the ER stress inducer tunicamycin (6 h, 5 μg/ml) promoted mRNA expression of VKORC1L1 while decreasing mRNA expression of VKORC1 (1.23- fold ± 0.09 vs. 0.54- fold ± 0.24, p = 0.029, Figure 4C). Thus, VKORC1L1 appears to play a critical role in the unfolded protein response (UPR) and ER stress.
MK7 is the most potent K-vitamin in humans and is discussed to exert protective properties on progression of cardiovascular disease (5, 30). Therefore, we sought to investigate its role on VSMC remodeling, inflammation and ER Stress in vitro.
HCASMC were treated with DMSO (control), treated with MK7 (1 μM, 24 h), PDGF (30 ng/ml, 24 h) or with PDGF (30 ng/ml, 24 h) and MK7 (1μM or 10 μM, 24 h). Proliferation was measured by EdU-Fluorescency after 24 h. As expected, PDGF treatment increased HCASMC proliferation. Intriguingly, co-Treatment with MK7 attenuated PDGF-induced proliferation (Fluorescence signal: PDGF: 22495 vs. PDGF+MK7 1 μM: 8897, p = 0.0008; PDGF: 22495 vs. PDGF+MK7 1 μM: 10074, p = 0.0018, Figure 5A).
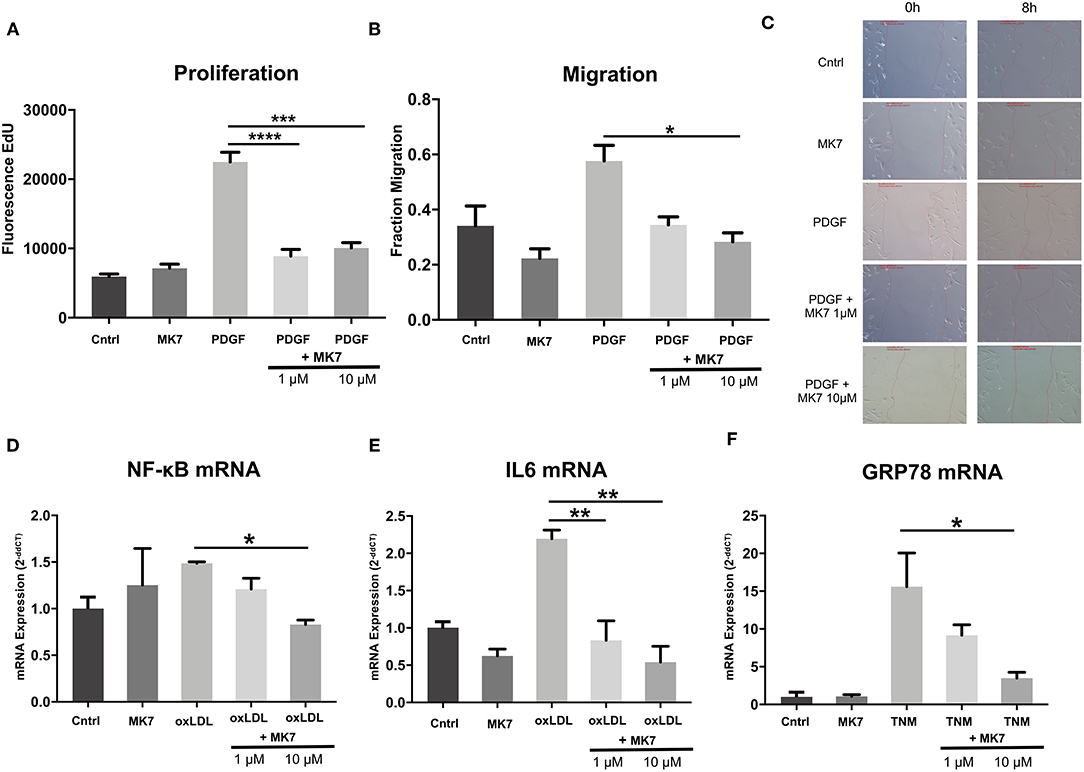
Figure 5. Menaquinone-7 (MK7) alleviates HCASMC remodeling, inflammation and ER stress. (A) Proliferation of HCASMC untreated, treated with MK7 (1 μM, 24 h), PDGF (30 ng/ml, 24 h) or PDGF + MK7 (1 μM or 10 μM, 24 h). Measured by an EdU fluorescence assay. n = 5. (B) Migration of HCASMC after a wound–scratch assay, measured by microscopic visualization after 12 h. Cells treated with MK7 (1 μM, 24 h), PDGF (30 ng/ml, 24 h) or PDGF + MK7 (1 μM or 10 μM, 24 h). n = 4. (C) Representative pictures of cell migration 0 and 8 h after scratch assay. (D, E) mRNA expression of the pro-inflammatory markers NF-κB and IL-6 in HCASMC. Cells treated with MK7 (1 μM, 24 h), oxLDL (25 ng/ml, 24 h) or oxLDL + MK7 (1 μM or 10 μM, 24 h). RNA expression was measured by using qPCR and quantified by the 2−ddCT method. n = 3–4. (F) mRNA expression of the ER stress marker GRP78 in HCASMC. Cells were treated with MK7 (1 μM, 24 h), tunicamycin (TNM, 5 μg/ml, 6 h) or TNM + MK7 (1 μM or 10 μM, 24 h). RNA expression was measured by using qPCR and quantified by the 2−ddCT method. n = 3. Data are presented as the mean ± SEM; *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. One-way ANOVA + Bonferroni's multiple comparison test for (A-E).
Next, HCASMC were treated accordingly and a wound scratch assay was performed. Migration of HCASMC visualized after 12 h showed that PDGF, as expected, induced migration of HCASMC and co-treatment with higher concentrated MK7 reduced PDGF-induced migration (Fraction Migration: PDGF: 0.57 vs. PDGF+MK7 10 μM: 0.28, p = 0.04, Figures 5B,C).
Next, we tested the effect of MK7 on mRNA levels of vascular inflammation markers (Figures 5D,E). Therefore, HCASMC were treated with DMSO (control), treated with MK7 (1μM, 24 h), oxLDL (25 ng/ml, 24 h) or oxLDL and MK7 (1μM or 10 μM, 24 h).
As expected, oxLDL treatment increased expression of NF-κB (1.49-fold ± 0.03) and IL-6 (2.2-fold ± 0.17). When co-treating with MK7, IL-6 expression was reduced at both of the used MK7 concentrations (oxLDL + MK7 1 μM: 0.83-fold, p = 0.004 vs. oxLDL only; oxLDL + MK7 10 μM: 0.54-fold ± 0.36, p = 0.001 vs. oxLDL only) while higher concentration of MK7 abrogated NF-κB expression (oxLDL + MK7 10 μM: 0.82-fold, p = 0.019 vs. oxLDL only).
Finally, we analysed the effect of MK7 on ER Stress induction (Figure 5F). HCASMC were untreated, treated with MK7 (1μM, 24 h), tunicamycin (5 μg/ml, 6 h) or tunicamycin and MK7 (1μM or 10 μM, 24 h). Tunicamycin treatment led to highly increased mRNA expression of GRP78 (15.6-fold ± 7.7). Co-treatment with 10 μM MK7 reduced tunicamycin-induced GRP78 expression (3.5-fold ± 1.3, p = 0.015 vs. tunicamycin only).
In this study, we investigated for the first time the role of the VKOR isoenzyme VKORC1L1 in vascular biology. We demonstrated that VKA-promoted adverse remodeling is accompanied by a downregulation of VKORC1L1 in vivo. In vitro, we showed that VKORC1L1 downregulation promotes maladaptive remodeling and inflammation in vascular smooth muscle cells. Moreover, we describe a novel connection between the vitamin K cycle and oxidative protein folding. VKORC1L1 downregulation promotes ER stress, a major driver of vascular inflammation. Finally, we present data showing that the vitamin K derivative menaquinone-7 is able to alleviate remodeling, inflammation and ER stress in VSMC.
Therapeutic anticoagulation with VKA has been described to be associated with progressive vascular calcification and vulnerable atherosclerotic plaques in both mice and humans (10, 31, 32). While the role of VKA in calcification by way of the matrix Gla-protein pathway has been extensively studied, further mechanisms of VKA-induced cardiovascular disease remain to be elucidated. Although warfarin intake is known to exert a complex influence on the immune system (33), the role of warfarin in vascular inflammation and anti-oxidation has not yet been resolved.
By utilising a widely used carotid-artery injury model, we could confirm that VKA promotes maladaptive vascular remodeling in mice. Recent studies suggested that the VKOR isoenzyme VKORC1L1 has anti-oxidative properties and is less sensitive to VKA than VKORC1 (18, 19). In our injury model, two-week long VKA treatment lead to reduced aortic VKORC1L1 mRNA. In vitro experiments confirmed warfarin treatment to reduce VKORC1L1 expression in VSMC. Our in vitro results hint at an involvement of VKORC1L1-downregulation in warfarin-induced vascular damage. Therefore, the inhibition of VKORC1L1 appears to be a plausible driver of neointima formation and restenosis.
Westhofen et al. demonstrated an increase in ROS formation of HEK-293 cells after VKORC1L1-downregulation via siRNAs (18). We confirmed these findings in HCASMC, suggesting that VKORC1L1 regulates mechanisms of anti-oxidation in the vasculature.
NF-κB is a transcription factor regarded as one of the key regulators of inflammatory processes, including cardiovascular inflammation (34). We showed that siRNA-mediated VKORC1L1-downregulation increases NF-κB expression in HCASMC. Long-term oral intake of warfarin was previously described to be associated with increased IL-6 production in rats (35). In our study, we found that VKORC1L1-downregulation increased the mRNA expression of IL-6. Warfarin-induced vascular inflammation may therefore be at least partly mediated by VKORC1L1 and the NF-κB/IL-6 pathway.
The cellular localization of the VKOR proteins and the effect that VKORC1L1 has on anti-oxidation and inflammation strongly suggest that there is a connection to the UPR. Multiple studies have described that VKOR proteins interact with protein disulfide-isomerases (23, 36). Herein, we present data linking VKORC1L1 to the UPR and ER stress pathways. After VKORC1L1-downregulation, GRP78, a marker for general UPR activation, was found to be increased while CHOP expression remained unchanged. CHOP is mainly activated by the PERK/eIF2α/ATF4 pathway, while downstream activation of GRP78 is induced by the ATF6 pathway (37, 38). Hence, specific UPR pathways are activated upon VKORC1L1 inhibition. Our results extend and support recent data of Furmanik et al., who showed that warfarin induces GRP78 expression and subsequent vascular calcification (39).
Finally, after describing putative mechanisms for VKA-induced vascular dysfunction, we found also that MK7 treatment can dose-dependently reduce vascular remodeling, inflammation and ER stress in vitro. K2 vitamins have previously been attributed anti-inflammatory effects (40, 41). However, we show here for the first time that vitamin K can alleviate inflammation in vascular cells. As far as we are aware, this is also the first evidence for vitamin K-promoted repression of ER stress in cells of any type. A possible mechanism underlying the anti-oxidative effects of vitamin K might be due to a vitamin K-induced increasement in VKORC1L1 activity. The increasement in VKORC1L1 activity could then dampen oxidative stress and subsequent inflammation. Further studies are warranted to elucidate these mechanisms.
In conclusion, we found that VKA promote neointima formation and that this is at least partly mediated by VKORC1L1 inhibition. Contrarily, MK7 supplementation reduces aberrant proliferation and inflammation in VSMC. Due to its favourable risk profile, MK7 may represent a feasible therapeutic target to reduce neointima formation. Main limitation of our study are the limited in vivo read-outs. In this study, we mainly focussed on the in vitro functions of VKORC1L1 on VSMC. Future studies utilising VKORC1L1−/− and VKORC1L1/ApoE−/− mice are currently ongoing and will further elucidate the role of VKORC1L1 during vascular injury and its connection to ER Stress. Additionally we did not yet investigate the presumed in vivo effects of MK7 on inflammation and ER Stress. The association of MK7 and VKORC1L1-deficiency will also be subject of upcoming projects. These results will improve our understanding of the manifold effects of vitamin K on the cardiovascular system.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
The animal study was reviewed and approved by National Office for Nature, Environment, and Consumer Protection in Recklinghausen, North Rhine-Westphalia (Landesamt für Natur, Umwelt und Verbraucherschutz; LANUV.
AA, MA, CM, JO, GN, and VT done conceptualization AA, MA, and VT worked on the methodology. AA, MA, ER, MB, and SZ done formal analysis and investigation. AA, MA, and VT wrote and prepared the original draft. All authors reviewed and edited the manuscript. AA, GN, and VT done funding acquisition. GN and VT provided the resources and supervised the work.
This work was supported by the medical faculty of the University of Bonn (BONFOR Grant No. 2018-1A-02 to AA). ER was supported by the German Heart Foundation (Kaltenbach Scholarship).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We thank Meghan Lucas, Ph.D. for her critical reading.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcvm.2021.708946/full#supplementary-material
CHOP, C/EBP homologous protein; DCFDA, 2', 7'-dichlorofluorescein diacetate; ER, Endoplasmic reticulum; GRP78, Glucose-regulated protein 78kDa; HCASMC, Human coronary artery smooth muscle cells; IL-6, Interleukin 6; MK7, Menaquinone-7; NFκB, Nuclear factor ‘kappa-light-chain-enhancer’ of activated B-cells; oxLDL, Oxidized low-density lipoprotein; PDGF, Platelet-derived growth factor; ROS, Reactive oxygen species; siRNA, Small interfering RNA; VKA, Vitamin K antagonists; VKORC1, Vitamin K epoxide reductase complex subunit 1; VKORC1L1, Vitamin K epoxide reductase complex subunit 1-like 1; VSMC, Vascular smooth muscle cells.
1. Steven S, Frenis K, Oelze M, Kalinovic S, Kuntic M, Jimenez MTB, et al. Vascular inflammation and oxidative stress: Major triggers for cardiovascular disease. Oxid Med Cell Longevity. (2019) doi: 10.1155/2019/7092151
2. Giordano A, Romano A. Inhibition of human in-stent restenosis: a molecular view. Curr Opin Pharmacol. (2011) 11:372–7. doi: 10.1016/j.coph.2011.03.006
3. Dzau VJ, Braun-Dullaeus RC, Sedding DG. Vascular proliferation and atherosclerosis: New perspectives and therapeutic strategies. Nat Med. (2002) 8:1249–56. doi: 10.1038/nm1102-1249
5. Marles RJ, Roe AL, Oketch-Rabah HA. US Pharmacopeial Convention safety evaluation of menaquinone-7, a form of vitamin K. Nutr Rev. (2017) 75:553–78. doi: 10.1093/nutrit/nux022
6. Oldenburg J, Marinova M, Müller-Reible C, Watzka M. The vitamin K cycle. Vitam Horm. (2008) 78:35–62. doi: 10.1016/S0083-6729(07)00003-9
7. Lacombe J, Rishavy MA, Berkner KL, Ferron M. VKOR paralog VKORC1L1 supports vitamin K-dependent protein carboxylation in vivo. JCI insight. (2018) 3:1–11. doi: 10.1172/jci.insight.96501
8. Weijs B, Blaauw Y, Rennenberg RJMW, Schurgers LJ, Timmermans CCMM, Pison L, et al. Patients using vitamin K antagonists show increased levels of coronary calcification: an observational study in low-risk atrial fibrillation patients. Eur Heart J. (2011) 32:2555–62. doi: 10.1093/eurheartj/ehr226
9. Schurgers LJ, Joosen IA, Laufer EM, Chatrou MLL, Herfs M, Winkens MHM, et al. Vitamin K-antagonists accelerate atherosclerotic calcification and induce a vulnerable plaque phenotype. PLoS ONE. (2012) 7:e43229. doi: 10.1371/journal.pone.0043229
10. Krüger T, Oelenberg S, Kaesler N, Schurgers LJ, Van De Sandt, Boor P, et al. Warfarin induces cardiovascular damage in mice. Arterioscler Thromb Vasc Biol. (2013) 33:2618–24. doi: 10.1161/ATVBAHA.113.302244
11. Geleijnse JM, Vermeer C, Grobbee DE, Schurgers LJ, Knapen MHJ, van der Meer, et al. Dietary intake of menaquinone is associated with a reduced risk of coronary heart disease: the rotterdam study. J Nutr. (2004) 134:3100–5. doi: 10.1093/jn/134.11.3100
12. Brandenburg VM, Reinartz S, Kaesler N, Krüger T, Dirrichs T, Kramann R, et al. Slower progress of aortic valve calcification with Vitamin K supplementation: Results from a prospective interventional proof-of-concept study. Circulation. (2017) 135:2081–3. doi: 10.1161/CIRCULATIONAHA.116.027011
13. Shea MK, O'Donnell CJ, Hoffmann U, Dallal GE, Dawson-Hughes B, Ordovas JM, et al. Vitamin K supplementation and progression of coronary artery calcium in older men and women. Am J Clin Nutr. (2009) 89:1799–807. doi: 10.3945/ajcn.2008.27338
14. Vervoort LMT, Ronden JE, Thijssen HHW. The potent antioxidant activity of the vitamin K cycle in microsomal lipid peroxidation. Biochem Pharmacol. (1997) 54:871–6. doi: 10.1016/S0006-2952(97)00254-2
15. Li J, Lin JC, Wang H, Peterson JW, Furie BC, Furie B, et al. Novel role of vitamin K in preventing oxidative injury to developing oligodendrocytes and neurons. J Neurosci. (2003) 23:5816–26. doi: 10.1523/JNEUROSCI.23-13-05816.2003
16. Caspers M, Czogalla KJ, Liphardt K, Müller J, Westhofen P, Watzka M, et al. Two enzymes catalyze vitamin K 2,3-epoxide reductase activity in mouse: VKORC1 is highly expressed in exocrine tissues while VKORC1L1 is highly expressed in brain. Thromb Res. (2015) 135:977–83. doi: 10.1016/j.thromres.2015.01.025
17. Spohn G, Kleinridders A, Wunderlich FT, Watzka M, Zaucke F, Blumbach K, et al. VKORC1 deficiency in mice causes early postnatal lethality due to severe bleeding. Thromb Haemost. (2009) 101:1044–50. doi: 10.1160/TH09-03-0204
18. Westhofen P, Watzka M, Marinova M, Hass M, Kirfel G, Muller J, et al. Human vitamin K 2,3-epoxide reductase complex subunit 1-like 1 (VKORC1L1) mediates vitamin K-dependent intracellular antioxidant function. J Biol Chem. (2011) 286:15085–94. doi: 10.1074/jbc.M110.210971
19. Czogalla KJ, Liphardt K, Höning K, Hornung V, Biswas A, Watzka M, et al. VKORC1 and VKORC1L1 have distinctly different oral anticoagulant dose-response characteristics and binding sites. Blood Advances. (2018) 2:691–702. doi: 10.1182/bloodadvances.2017006775
20. Hueso M, Mallén A, Casas Á, Guiteras J, Sbraga F, Blasco-Lucas A, et al. Integrated miRNA/mRNA counter-expression analysis highlights oxidative stress-related genes CCR7 and FOXO1 as blood markers of coronary arterial disease. Int J Mol Sci. (2020) 21:1–18. doi: 10.3390/ijms21061943
21. Zhang Z, Zhang L, Zhou L, Lei Y, Zhang Y, Huang C. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox Biol. (2019) 25:101047. doi: 10.1016/j.redox.2018.11.005
22. Zhang C, Syed TW, Liu R, Yu J. Role of endoplasmic reticulum stress, autophagy, and inflammation in cardiovascular disease. Front Cardiovasc Med. (2017) 4:1–8. doi: 10.3389/fcvm.2017.00029
23. Wajih N, Hutson SM, Wallin R. Disulfide-dependent protein folding is linked to operation of the vitamin K cycle in the endoplasmic reticulum: a protein disulfide isomerase-VKORC1 redox enzyme complex appears to be responsible for vitamin K1 2,3-epoxide reduction. J Biol Chem. (2007) 282:2626–35. doi: 10.1074/jbc.M608954200
24. Kalyanaraman B, Darley-Usmar V, Davies KJA, Dennery PA, Forman HJ, Grisham MB, et al. Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radical Biology Med. (2012) 52:1–6. doi: 10.1016/j.freeradbiomed.2011.09.030
25. Rosenkranz AR, Schmaldienst S, Stuhlmeier KM, Chen W, Knapp W, Zlabinger GJ, et al. microplate assay for the detection of oxidative products using 2′,7′-dichlorofluorescin-diacetate. J Immunol Methods. (1992) 156:39–45. doi: 10.1016/0022-1759(92)90008-H
26. Wang H, Joseph JA. Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radical Biology Med. (1999) 27:612–6. doi: 10.1016/S0891-5849(99)00107-0
27. Price PA, Faus SA, Williamson MK. Warfarin causes rapid calcification of the elastic lamellae in rat arteries and heart valves. Arterioscler Thromb Vasc Biol. (1998) 18:1400–7. doi: 10.1161/01.ATV.18.9.1400
28. Forrester SJ, Kikuchi DS, Hernandes MS, Xu Q, Griendling KK. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res. (2018) 122:877–902. doi: 10.1161/CIRCRESAHA.117.311401
29. Zhang J, Wang X, Vikash V, Ye Q, Wu D, Liu Y, et al. ROS and ROS-mediated cellular signaling. Oxidative Med Cell Longevity. (2016). doi: 10.1155/2016/4350965
30. Vossen LM, Schurgers LJ, van Varik BJ, Kietselaer BLJH, Vermeer C, Meeder JG, et al. Menaquinone-7 supplementation to reduce vascular calcification in patients with coronary artery disease: Rationale and study protocol (VitaK-CAC Trial). Nutrients. (2015) 7:8905–15. doi: 10.3390/nu7115443
31. Alappan HR, Kaur G, Manzoor S, Navarrete J, O'Neill WC. Warfarin accelerates medial arterial calcification in humans. Arterioscler Thromb Vasc Biol. (2020) 40:1413–9. doi: 10.1161/ATVBAHA.119.313879
32. Andrews J, Psaltis PJ, Bayturan O, Shao M, Stegman B, Elshazly M, et al. Warfarin use is associated with progressive coronary arterial calcification: insights from serial intravascular ultrasound. JACC: Cardiovascular Imaging. (2018) 11:1315–23. doi: 10.1016/j.jcmg.2017.04.010
33. Verma D, Kumar R, Pereira RS, Karantanou C, Zanetti C, Minciacchi VR, et al. Vitamin K antagonism impairs the bone marrow microenvironment and hematopoiesis. Blood. (2019) 134:227–38. doi: 10.1182/blood.2018874214
34. Fiordelisi A, Iaccarino G, Morisco C, Coscioni E, Sorriento D. NfkappaB is a key player in the crosstalk between inflammation and cardiovascular diseases. Int J Mol Sci. (2019) 20:1599. doi: 10.3390/ijms20071599
35. Popov A, Belij S, Subota V, Zolotarevski L, Mirkov I, Kataranovski D, et al. Oral warfarin affects peripheral blood leukocyte IL-6 and TNFα production in rats. J Immunotoxicol. (2013) 10:17–24. doi: 10.3109/1547691X.2012.684159
36. Rutkevich LA, Williams DB. Vitamin K epoxide reductase contributes to protein disulfide formation and redox homeostasis within the endoplasmic reticulum. Mol Biol Cell. (2012) 23:2017–27. doi: 10.1091/mbc.e12-02-0102
37. Bergmann TJ, Molinari M. Three branches to rule them all? UPR signalling in response to chemically versus misfolded proteins-induced ER stress. Biol Cell. (2018) 110:197–204. doi: 10.1111/boc.201800029
38. Hetz C. The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Molec Cell Biol. (2012) 13:89–102. doi: 10.1038/nrm3270
39. Furmanik M, Gorp R, Van Whitehead M, Ahmad S, Bordoloi J, Kapustin A, et al. Endoplasmic reticulum stress mediates vascular smooth muscle cell calcification via increased release of Grp78-loaded extracellular vesicles. Arterioscler Thromb Vasc Biol. (2021) 41:898–914. doi: 10.1161/ATVBAHA.120.315506
40. Ohsaki Y, Shirakawa H, Miura A, Giriwono PE, Sato S, Ohashi A, et al. Vitamin K suppresses the lipopolysaccharide-induced expression of inflammatory cytokines in cultured macrophage-like cells via the inhibition of the activation of nuclear factor αB through the repression of IKKα/β phosphorylation. J Nutr Biochem. (2010) 21:1120–6. doi: 10.1016/j.jnutbio.2009.09.011
Keywords: vascular inflammation, vitamin K, oxidative stress, ER stress, vascular remodeling
Citation: Aksoy A, Al Zaidi M, Repges E, Becher MU, Müller C, Oldenburg J, Zimmer S, Nickenig G and Tiyerili V (2021) Vitamin K Epoxide Reductase Complex Subunit 1-Like 1 (VKORC1L1) Inhibition Induces a Proliferative and Pro-inflammatory Vascular Smooth Muscle Cell Phenotype. Front. Cardiovasc. Med. 8:708946. doi: 10.3389/fcvm.2021.708946
Received: 12 May 2021; Accepted: 05 October 2021;
Published: 27 October 2021.
Edited by:
Leon J. Schurgers, Maastricht University, NetherlandsReviewed by:
Sudhahar Varadarajan, Augusta University, United StatesCopyright © 2021 Aksoy, Al Zaidi, Repges, Becher, Müller, Oldenburg, Zimmer, Nickenig and Tiyerili. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Adem Aksoy, YWRlbS5ha3NveUB1a2Jvbm4uZGU=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.