 William J. Richardson
William J. Richardson Jesse D. Rogers
Jesse D. Rogers Francis G. Spinale2
Francis G. Spinale2- 1Department of Bioengineering, Clemson University, Clemson, SC, United States
- 2Cardiovascular Translational Research Center, University of South Carolina School of Medicine and Columbia Veterans Affairs Health Care System, Columbia, SC, United States
There is a critical need for interventions to control the development and remodeling of scar tissue after myocardial infarction. A significant hurdle to fibrosis-related therapy is presented by the complex spatial needs of the infarcted ventricle, namely that collagenous buildup is beneficial in the ischemic zone but detrimental in the border and remote zones. As a new, alternative approach, we present a case to develop self-adapting, mechano-sensitive drug targets in order to leverage local, microenvironmental mechanics to modulate a therapy's pharmacologic effect. Such approaches could provide self-tuning control to either promote fibrosis or reduce fibrosis only when and where it is beneficial to do so.
Introduction
Roughly 800,000 myocardial infarctions (MIs) occur in the U.S. each year, and while 85% of patients survive the initial ischemic event, these survivors are left with reduced cardiac function and a shortened lifespan (1). Some emerging regenerative therapies may offer potential to restore myocardium in the collagenous infarct scar tissue (2–5), but a critical hurdle remains: how can we control the build-up of collagenous scar tissue in infarct, border zone, and remote myocardial regions post-MI? Reducing collagen content in animal infarct models has shown an ability to enhance the performance of regeneration therapies, enabling increased cell engraftment, capillary density, fractional shortening and ejection fraction (6, 7). Reducing fibrosis in remote viable myocardium post-MI also offers significant therapeutic benefit as excessive remote fibrosis is associated with mechanical and electrical cardiac dysfunction (8–10). However, reducing myocardial fibrosis must be tightly coupled to local structural integrity in order to prevent infarct scar expansion or rupture. This need is highlighted by past clinical trials of steroid treatments that decreased collagen content to disastrous effects, resulting in infarct rupture and multiple patient deaths (11). In this perspective paper, we present a case for self-adapting, mechano-sensitive therapies - i.e., therapies whose effect on fibrosis depends on the localized mechanical microenvironment and thereby reduces matrix content only when and where it is safe to do.
Pros and Cons of Post-Infarct Fibrosis: Essential Mechanics and the Need for Adaptive Control
Myocardial fibrosis is characterized by the accumulation of extracellular matrix (ECM), which is determined by the relative balance of ECM synthesis, assembly, degradation by proteases such as matrix metalloproteinases (MMPs), and protease inhibition by enzymes such as tissue inhibitors of metalloproteinases (TIMPs). These processes are typically balanced in healthy myocardium, where a resident population of quiescent cardiac fibroblasts maintain a network of ECM within the interstitial space between cardiomyocytes providing a scaffold for cell adhesion and regulating ventricle stiffness during diastolic filling (12–14).
MI is caused by a coronary artery occlusion producing immediate ischemia and necrosis of the downstream myocardium. The dynamic wound healing process starts with inflammatory cells infiltrating the infarct zone over the first few days post-MI, releasing a variety of proteolytic enzymes to clear necrotic tissue, and signaling for the recruitment of cardiac fibroblasts via secreted cytokines (15). In the following weeks, fibroblasts migrate, proliferate, and transition to an activated phenotype with heightened contractile forces and heightened synthesis of ECM-related proteins including collagens, fibronectin, laminin, periostin, osteopontin, tenascins, thrombospondins, TIMPs, and many others (16–19). Recent work has found these activated fibroblasts can stem from not only the local resident population but also a range of different lineage sources including epithelial cells, endothelial cells, perivascular cells, fibrocytes, and bone marrow-derived progrenitors (20–22).
From a mechanical perspective, fibrosis in the infarct zone is essential for maintaining structural integrity of the scar, which becomes passive, non-contractile tissue subjected to biaxial tensile stresses from ventricular cavity pressure and remote cardiomyocyte systolic contraction. Decreased levels of collagen in the infarct scar result in reduced material stiffness and reduced thickness, which in turn drive higher wall stresses and exacerbate infarct expansion. This expansion is characterized by wall thinning in the radial direction with dilation in the circumferential-longitudinal plane, and it has long been association with reduced systolic function, increased outward bulging (dyskinesis), increased cavity dilation, increased heart failure risk, and increased wall rupture (17, 23–31). It is important to note that a “small” infarct when defined as the volume of the acutely infarcted zone relative to the uninjured myocardium is strongly correlated with much better post-MI function and clinical outcomes (32–37), so it is tempting to think that less scar tissue should be a top therapeutic objective. But numerous studies urge caution against this overly simplistic goal as drugs to reduce edema and inflammation early post-MI have repeatedly reduced collagen density in the infarct zone without affecting the infarct zone size, thereby exacerbating scar thinning and expansion as a result (38–43).
While fibrosis in the infarct zone is beneficial, excessive fibrosis in the border zone and remote zone can be detrimental to cardiac function (8–10, 44, 45). Most notably, increased ECM levels in the uninjured myocardium are associated with progressive tissue stiffening, which impairs diastolic filling (44–46). Systolic contractile function is also reduced from fibrosis-related electrical dysfunction, possibly diminished Frank-Starling effect, possibly diminished thickening from microenvironment crowding, and impaired radial thickening ability from physical coupling between the borderzone and the infarct scar (17, 28).
Collectively, data suggest that therapeutically increasing infarct stiffness and decreasing remote stiffness may help limit ventricular dilation. Further complicating the pros and cons of post-infarct fibrosis, ECM alignment, preferred orientation, and heterogeneity have demonstrated significant effects on post-MI performance (24, 26, 27, 46–49). The trade-off between pros and cons from post-infarct fibrosis should urge much caution against overly simplistic reports and conclusions in the literature. In other words, it is too simplistic to say we want to “reduce fibrosis” or “limit scar tissue” after a heart attack. We must be clearer to specify our objectives for matrix control - in what location, in what dimension, over what time period, assessed by volume vs. density, etc.? There is a critical need for more nuanced approaches for fibrotic control.
Fibrotic Regulation: Biochemical and Mechanical Signals
Fibroblast expression of matrix-related proteins is regulated by a wide variety of biochemical agonists. Inflammatory cytokines such as tumor necrosis factor-α, interleukin 1, and interleukin 6 are upregulated immediately after injury as a stress response (50, 51) and act to suppress fibroblast activation and upregulate MMP secretion for the removal of necrotic tissue (52–55). Growth factors such as transforming growth factor β and platelet-derived growth factor are later secreted by neutrophils and macrophages as part of both an anti-inflammatory response as well as a pro-fibrotic response via upregulation of proliferation and matrix synthesis (44, 56–59). Hormonal agonists such as angiotensin II, norepinephrine, natriuretic peptides, and endothelin-1 also modulate fibroblast behavior including ECM-related gene expression (60–65).
In addition to biochemical regulation, cardiac fibroblast expression of matrix-related proteins is highly sensitive to mechanical regulation. While beating myocardium contracts with each heartbeat, infarct scar is subjected to tensile stretch, sometimes extending 5–10% in the circumferential-longitudinal dimensions (46, 47, 66). Correspondingly, cardiac fibroblasts in the infarct zone show elevated mechanotransduction signaling activity (18, 19, 67). In vitro mechanobiology studies have subjected fibroblasts to a variety of mechanical deformation environments including uniaxial tension, biaxial tension, and shear (68–91). Across a range of stretch magnitudes, frequencies, and durations, mechanical deformation generally produces a pro-fibrotic effect with ~2-fold increases in collagen production on average (68). In addition to substrate deformation, previous studies have shown that substrate stiffness can similarly induce mechanotransduction pathways and matrix production in cells cultured on acrylamide hydrogels, silicone-based substrates, collagen scaffolds, and other culture environments of tunable stiffness (81, 92–95). These studies have found that increased stiffnesses can induce various cell types to upregulate synthesis of matrix proteins by inducing cytoskeletal contractility and driving tension-dependent signaling pathways via inside-out integrin activation. For example, Herum et al. recently found that cardiac fibroblasts alter a host of ECM-related genes with culture on increasingly stiff substrates, including genes for collagens I and III, tenascin-c, periostin, osteopontin, thrombospondin 1, and secreted protein acidic and rich in cysteine (SPARC) (94).
Not only can mechanical force regulate fibrotic turnover via cellular mechanotransudction, force can also modulate matrix regulation in the extracellular space. A range of studies have confirmed that altered deformation of matrix proteins like collagen fibers induce altered degradation rates by proteases, presumably due to conformational changes in the matrix-protease binding pocket (68, 96–100). Other studies have shown that tension (either from cell-driven contractility or from externally applied tissue loads) can release active growth factors like TGFβ from their matrix-bound latent complexes (101, 102).
Mechano-Adaptive Therapy: A New Strategy
Many preclinical animal studies have sought to develop effective fibrotic-related therapies for the infarcted ventricle, but there remains a striking paucity in adequate therapies. Some interventions have successfully altered matrix levels post-MI at particular time-points or spatial locations, but do so at the cost of exacerbating dysfunction at other time-points or locations. The collection of failed trials reflects the difficulty in developing therapies for the infarcted ventricle context where different spatial regions and directions require different fibrotic responses. For example, Ikeuchi et al. investigated an anti-TGFβ gene therapy in mice following left coronary ligation and found that anti-TGFβ treated mice showed lower collagen volume fraction levels and myocyte hypertrophy as well as improved clinical outcomes such as non-infarct wall thickness, left ventricular end systolic and diastolic diameters at 28 days compared to sham mice (103). However, the same population also experienced a greater degree of wall thinning 9 h post-MI with anti-TGFβ treatment, which was accompanied by heightened neutrophil invasion, inflammatory cytokine expression, and rate of mortality at 24 h, presumably caused by suppression of early reparative scar formation. This finding was later supported by Frantz et al. (104) and demonstrates the temporal nature of fibrosis, in which early scar formation can prevent further dysfunction but late fibrosis can reduce contractility, as well as the spatial nature of fibrosis, in which the infarct zone requires a robust scar formation for continued function while excess scar deposition in remote zones can lead to dysfunction.
Additional studies altering both signaling pathways and matrix-related proteins in a global manner have revealed similar challenges in temporal and spatial control. The Lindsey group has conducted several studies inhibiting specific MMPs in post-MI mouse models, finding that pharmacological inhibition of MMP9 and MMP12 reduced ejection fractions compared to saline-treated controls, with anti-MMP9 treatment increasing leukocyte infiltration and both treatments reducing apoptosis at 1 week post-MI (105, 106). Interestingly, a knockout mouse model of TIMP3 also increased complications related to early ventricular wall thinning, with TIMP3−/− mice demonstrating lower hydroxyproline content, procollagen synthesis, and TGFβ expression 2 days post-MI as well as a 4-fold increase in cardiac rupture compared to wild-type mice (107). The increases in early complications after MI for seemingly opposite modes of treatment suggest that global inhibition of one single pathway or mechanism may produce counterintuitive effects in overall tissue remodeling.
It is increasingly clear that improving post-MI fibrotic control will require therapeutic interventions producing spatially distinct responses in order to reduce matrix buildup in remote and regenerating contractile zones while maintaining or increasing matrix buildup in passive scar zones. As a new, alternative fibrotic intervention strategy, we propose that local mechanics can provide self-adapting feedback to modulate collagen signaling and thereby enable self-adjusting specificity for anti-fibrotic therapy (Figure 1). Localized deformation and stiffness depend on local tissue structure: stiff, collagenous infarct scar stretches while functioning remote muscle contracts, with intermediate stretch levels in the infarct border zone (24, 108–111). The sensitivity of fibroblasts to local mechanical stretch presents an opportunity to design therapies that reduce fibrosis in areas that don't need it (i.e., remote or regenerating myocardium that undergoes repeated contraction) while maintaining or increasing collagen in areas that do need it (i.e., stiffening scar tissue that undergoes persistent stretch). Such a therapy could provide spatial, temporal, and even patient-specific adaptability by tailoring its effect to the localized mechanical need (112).
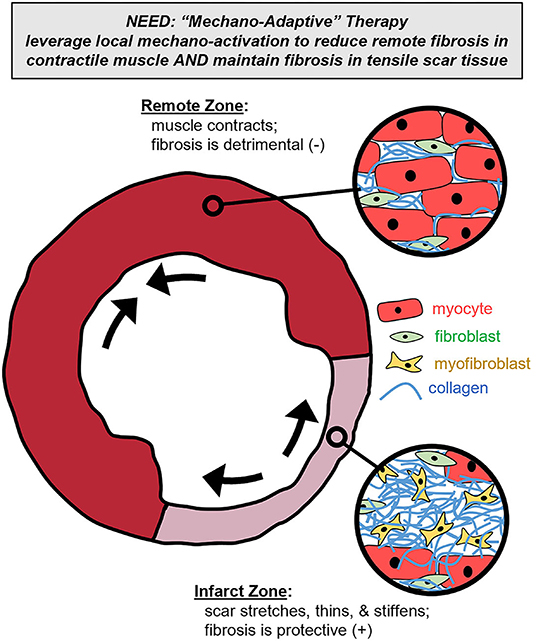
Figure 1. Mechano-adaptive therapy. To meet the spatially complex needs of the left ventricle cross-section post-infarction, we propose that localized mechanical cues could be leveraged to modulate a therapy's pharmacologic effect. Specifically, such a therapy could reduce collagen content in contractile muscle subregions (where fibrosis is detrimental) while simultaneously maintaining collagen content in the stretching and stiffening scar subregions (where fibrosis is beneficial).
The premise of mechano-adaptive therapy is supported by the extensive connection between intracellular mechanotransduction signaling pathways and chemotransduction signaling pathways. Studies investigating biochemical and mechanical signals have identified evidence of crosstalk between modes of signaling, both through common intermediate pathways as well as through secondary activation of one or more additional pathways. Examples of common intermediates include angiotensin II type 1 receptors, which have been shown to mediate cardiac fibroblast gene expression in response to both increased stiffness and interstitial fluid flow (113, 114), and phosphoinositide 3-kinase/Akt signaling, which in addition to regulating TGFβ and TNFα signal transduction (115–117) has been shown to function downstream of β1-integrin signaling to regulate fibroblast apoptosis (118). Several groups have observed the secondary activation of biochemical signaling mechanisms initiated by biomechanical signaling and vice versa. The Hinz group has investigated the activation of TGFβ by fibroblasts in response to mechanical stimuli for example finding that fibroblast-generated forces can release latent TGFβ from the surrounding ECM via β-integrins, thereby inducing a positive feedback loop for further activation (102, 119). This feedback could be amplified by other cellular sources of TGFβ such as macrophages, which are concentrated in the infarcted zone during the inflammatory response to ischemia and thereby influence the local fibrotic niche (120–122). Conversely, canonical transient receptor potential (TRPC) channels have been appreciated for their role as stretch-activated ion channels in promoting cardiac fibroblast activation (67), but Davis et al. observed that the TRPC6 channel was required for TGFβ-induced activation of fibroblasts (123). TRPC6−/− cells displayed attenuated αSMA fiber incorporation and gel contraction in dermal fibroblasts with TGFβ treatment as well as significantly smaller scar size and higher rates of ventricular rupture in a post-MI mouse model, demonstrating an additional mechanism-of-action beyond canonical signaling via Smad signaling.
Many studies have now shown that mechano-chemo-signaling pathway interactions ultimately give rise to mechano-sensitive gene expression responses of cells stimulated by chemical agonists (70, 79, 84, 90, 124–126). In other words, the presence of mechanical stimulation can amplify, dampen, or even reverse the effect of biochemical stimulation. Spatial differences in mechanical cues such as cyclic tension and tissue stiffness observed in the post-MI environment act as local mediators of fibroblast activation compared to remote myocardium, and so it is expected that global pharmacological treatments would exert differential responses on infarct- and remote-localized fibroblasts. Indeed, Ramirez and colleagues observed that in a post-MI mouse model, pharmacological treatments with valsartan (an angiotensin receptor blocker) and aliskiren (a renin inhibitor) primarily affected cardiac gene expression of ECM-related proteins in remote regions with minimal changes in infarcted myocardium (127). We suspect that, in similar fashion, mechano-sensitive therapies could be identified to remove detrimental fibrosis in the remote zone while simultaneously enhancing beneficial fibrosis in the infarct zone. It is also possible, though currently unknown, that cardiac fibroblasts arising from different lineages across the infarcted vs. remote zones could demonstrate region-specific sensitivities to mechano-chemo-stimuli and thereby amplify region-specific therapy responses.
Given the potential capabilities of mechano-adaptive therapies, a pressing question remains: how do we prospectively design such perturbations to leverage local mechanics for spatially and temporally adaptive benefit? One possible approach is the continued advance of higher-throughput tissue culture platforms that provide in vitro screening tools for drug discovery within increasingly physiologic environments. Culture arrays that can modulate the local mechanical environment can help identify pharmacologic perturbations whose effects of matrix turnover are mechano-adaptive (84, 128–132).
Yet even with high-throughput experimental approaches, the complex fibrotic regulatory network presents hundreds of potential pharmacologic targets, and millions of potential target combinations whose combined effects may not be intuitive. This challenge has motivated the advance of computational modeling approaches to predict influential mechanisms-of-action and cellular behavior while accounting for complex conditions and non-linear network dynamics (133–135). Computational models of molecular and cellular systems using mechanistic models, statistical models, and artificial intelligence approaches have been valuable in drug discovery and regulatory approval, both from academic and industry perspectives (136). In recent work, we and others have developed large-scale network models of mechanotransduction signaling capable of computationally predicting how various pathways are activated or inhibited by mechanical stimuli, how these pathways interact with other biochemical stimuli, and how these pathways alter downstream gene expression related to fibrotic turnover (137–141). Zeigler and colleagues, for example, demonstrated sizable cross-talk between TGFβ and biomechanical signaling in a model of cardiac fibroblast signaling, as both model predictions and experimental validation found that the TGFβ1 receptor is necessary for mechanically-induced αSMA expression and contraction of collagen gels (137). The same model was also used to simulate cellular responses to time courses of post-MI biochemical stimuli mimicking the inflammatory, reparative, and maturation phases of wound healing (142). The authors found that different intracellular signaling species mediate collagen I and III expression during early, intermediate, and late phases of wound healing, thus informing potential time courses for treating MI patients with anti-fibrotic therapeutics. In other work, Tan and colleagues demonstrated the potential for network models to enable comprehensive drug screens in silico by predicting cell responses to knockdowns of individual or combinations of signaling species (138). They used a model of cardiomyocyte mechanotransduction to identify several perturbations in combination with angiotensin receptor blockers that could provide therapeutic benefit above the use of angiotensin receptor blockers alone, such as those inhibiting ET-1 receptors or integrins. Applications of this computational approach to the spatially varying post-MI microenvironments can provide a basis for further experimental studies and ultimately improve the efficiency of drug discovery by accounting for the myriad of interconnected signaling pathways between mechano- and chemo-transduction.
Conclusion
Does the heart want what it wants? That is to say, given the complex infarcted ventricle where fibrosis can be both beneficial and detrimental depending on the location, timing, and direction, can we design post-MI therapy that lets the heart's own local mechanical environment dictate the therapeutic effects according to the heart's own personalized, spatial, and temporal needs. While chemo-mechano-interactions complicate the regulatory network driving fibrotic turnover, we have proposed that localized mechanics also offer an exciting opportunity for tailoring fibroblast behavior to localized mechanical cues. Mechano-adaptive therapies could thereby provide self-adapting responses and modulate matrix only when and where it is beneficial to do so.
Data Availability Statement
The original contributions presented in the study are included in the article/supplementary material, further inquiries can be directed to the corresponding author/s.
Author Contributions
All authors contributed substantively to this work including idea generation, review of past studies, text writing, and text editing.
Funding
The authors gratefully acknowledge funding from the National Institutes of Health (HL144927 and GM121342) and the American Heart Association (17SDG33410658).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher's Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
1. Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, et al. Heart disease and stroke statistics−2013 update: a report from the American Heart Association. Circulation. (2013) 127:e6–e245. doi: 10.1161/CIR.0b013e31828124ad
2. Doppler SA, Deutsch M, Lange R, Krane M. Cardiac regeneration : current therapies — future concepts. J Thorac Dis. (2013) 5:683–97. doi: 10.3978/j.issn.2072-1439.2013.08.71
3. Karantalis V, Balkan W, Schulman IH, Hatzistergos KE, Hare JM. Cell-based therapy for prevention and reversal of myocardial remodeling. Am J Physiol Heart Circ Physiol. (2012) 303:H256–70. doi: 10.1152/ajpheart.00221.2012
4. Templin C, Lüscher TF, Landmesser U. Cell-based cardiovascular repair and regeneration in acute myocardial infarction and chronic ischemic cardiomyopathy-current status and future developments. Int J Dev Biol. (2011) 55:407–17. doi: 10.1387/ijdb.103219ct
5. Sanganalmath SK, Bolli R. Cell therapy for heart failure: a comprehensive overview of experimental and clinical studies, current challenges, and future directions. Circ Res. (2013) 113:810–34. doi: 10.1161/CIRCRESAHA.113.300219
6. Dai B, Huang W, Xu M, Millard RW, Gao MH, Hammond HK, et al. Reduced collagen deposition in infarcted myocardium facilitates induced pluripotent stem cell engraftment and angiomyogenesis for improvement of left ventricular function. J Am Coll Cardiol. (2011) 58:2118–27. doi: 10.1016/j.jacc.2011.06.062
7. Huang W, Dai B, Wen Z, Millard RW, Yu X-Y, Luther K, Xu M, et al. Molecular strategy to reduce in vivo collagen barrier promotes entry of NCX1 positive inducible pluripotent stem cells (iPSC(NCX1+)) into ischemic (or injured) myocardium. PLoS ONE. (2013) 8:e70023. doi: 10.1371/journal.pone.0070023
8. Brower GL, Gardner JD, Forman MF, Murray DB, Voloshenyuk T, Levick SP, et al. The relationship between myocardial extracellular matrix remodeling and ventricular function. Eur J Cardiothorac Surg. (2006) 30:604–10. doi: 10.1016/j.ejcts.2006.07.006
9. Nguyen DT, Ding C, Wilson E, Marcus GM, Olgin JE. Pirfenidone mitigates left ventricular fibrosis and dysfunction after myocardial infarction and reduces arrhythmias. Heart Rhythm. (2010) 7:1438–45. doi: 10.1016/j.hrthm.2010.04.030
10. Rutherford SL, Trew ML, Sands GB, Legrice IJ, Smaill BH. High-resolution 3-dimensional reconstruction of the infarct border zone: impact of structural remodeling on electrical activation. Circ Res. (2012) 111:301–11. doi: 10.1161/CIRCRESAHA.111.260943
11. Roberts R, DeMello V, Sobel BE. Deleterious effects of methylprednisolone in patients with myocardial infarction. Circulation. (1976) 53:I204–6.
12. Weber KT. Cardiac interstitium in health and disease: The fibrillar collagen network. J Am Coll Cardiol. (1989) 13:1637–52. doi: 10.1016/0735-1097(89)90360-4
13. Kong P, Christia P, Frangogiannis NG. The pathogenesis of cardiac fibrosis. Cell Mol Life Sci. (2014) 71:549–74. doi: 10.1007/s00018-013-1349-6
14. Fomovsky GM, Thomopoulos S, Holmes JW. Contribution of extracellular matrix to the mechanical properties of the heart. J Mol Cell Cardiol. (2010) 48:490–6. doi: 10.1016/j.yjmcc.2009.08.003
15. Daseke MJ, Valerio FM, Kalusche WJ, Ma Y, DeLeon-Pennell KY, Lindsey ML. Neutrophil proteome shifts over the myocardial infarction time continuum. Basic Res Cardiol. (2019) 114:37. doi: 10.1007/s00395-019-0746-x
16. Chen W, Frangogiannis NG. Fibroblasts in post-infarction inflammation and cardiac repair. Biochim Biophys Acta - Mol Cell Res. (2013) 1833:945–53. doi: 10.1016/j.bbamcr.2012.08.023
17. Richardson WJ, Clarke SA, Quinn TA, Holmes JW. Physiological Implications of Myocardial Scar Structure. Compr Physiol. (2015) 5:1877–909. doi: 10.1002/cphy.c140067
18. Mouton AJ, Ma Y, Rivera Gonzalez OJ, Daseke MJ, Flynn ER, Freeman TC, et al. Fibroblast polarization over the myocardial infarction time continuum shifts roles from inflammation to angiogenesis. Basic Res Cardiol. (2019) 114:6. doi: 10.1007/s00395-019-0715-4
19. Deleon-pennell KY, Iyer RP, Ma Y, Yabluchanskiy A, Zamilpa R, Chiao YA, et al. The mouse heart attack research tool 1.0 database. Am J Physiol Heart Circ Physiol. (2018) 315:H522–30. doi: 10.1152/ajpheart.00172.2018
20. Krenning G, Zeisberg EM, Kalluri R. The Origin of Fibroblasts and Mechanism of Cardiac Fibrosis. J Cell Physiol. (2010) 225:631. doi: 10.1002/jcp.22322
21. Tallquist MD, Molkentin JD. Redefining the identity of cardiac fibroblasts. Nat Rev Cardiol. (2017) 14:484. doi: 10.1038/nrcardio.2017.57
22. Moore-Morris T, Cattaneo P, Puceat M, Evans SM. Origins of cardiac fibroblasts. J Mol Cell Cardiol. (2016) 91:1. doi: 10.1016/j.yjmcc.2015.12.031
23. Weber KT, Sun Y, Bhattacharya SK. a Ahokas R, Gerling IC. Myofibroblast-mediated mechanisms of pathological remodelling of the heart. Nat Rev Cardiol. (2013) 10:15–26. doi: 10.1038/nrcardio.2012.158
24. Fomovsky GM, Holmes JW. Evolution of scar structure, mechanics, and ventricular function after myocardial infarction in the rat. Am J Physiol Heart Circ Physiol. (2010) 298:H221–8. doi: 10.1152/ajpheart.00495.2009
25. Richardson WJ, Holmes JW. Why is infarct expansion such an elusive therapeutic target? J Cardiovasc Transl Res. (2015) 8:421–30. doi: 10.1007/s12265-015-9652-2
26. Fomovsky GM, Macadangdang JR, Ailawadi G, Holmes JW. Model-based design of mechanical therapies for myocardial infarction. J Cardiovasc Transl Res. (2011) 4:82–91. doi: 10.1007/s12265-010-9241-3
27. Korenczuk CE, Barocas VH, Richardson WJ. Effects of collagen heterogeneity on myocardial infarct mechanics in a multiscale fiber network model. J Biomech Eng. (2019) 141:0910151–9. doi: 10.1115/1.4043865
28. Clarke SA, Richardson WJ, Holmes JW. Modifying the mechanics of healing infarcts: Is better the enemy of good? J Mol Cell Cardiol. (2016) 93:115–24. doi: 10.1016/j.yjmcc.2015.11.028
29. Eaton LW, Weiss JL, Bulkley BH, Garrison JB, Weisfeldt ML. Regional cardiac dilatation after acute myocardial infarction: recognition by two-dimensional echocardiography. N Engl J Med. (1979) 300:57–62. doi: 10.1056/NEJM197901113000202
30. Hutchins GM, Bulkley BH. Infarct expansion versus extension: two different complications of acute myocardial infarction. Am J Cardiol. (1978) 41:1127–32. doi: 10.1016/0002-9149(78)90869-X
31. Schuster EH, Bulkley BH. Expansion of transmural myocardial infarction: a pathophysiologic factor in cardiac rupture. Circulation. (1979) 60:1532–8. doi: 10.1161/01.CIR.60.7.1532
32. Sunagawa K, Maughan WL, Sagawa K. Effect of regional ischemia on the left ventricular end-systolic pressure-volume relationship of isolated canine hearts. Circ Res. (1983) 52:170–8. doi: 10.1161/01.RES.52.2.170
33. Savage RM, Guth B, White FC, Hagan AD, Bloor CM. Correlation of regional myocardial blood flow and function with myocardial infarct size during acute myocardial ischemia in the conscious pig. Circulation. (1981) 64:699–707. doi: 10.1161/01.CIR.64.4.699
34. Pfeffer M, Pfeffer J, Fishbein M, Fletcher P, Spadaro J, Kloner R, et al. Myocardial infarct size and ventricular function in rats. Circ Res. (1979) 44:503–12. doi: 10.1161/01.RES.44.4.503
35. Page DL, Caulfield JB, Kastor JA, DeSanctis RW, Sanders CA. Myocardial changes associated with cardiogenic shock. N Engl J Med. (1971) 285:133–7. doi: 10.1056/NEJM197107152850301
36. Mathey D, Biefield W, Hanrath P, Effert S. Attempt to quantitate relation between cardiac function and infarct size in acute myocardial infarction. Br Heart J. (1974) 36:271–9. doi: 10.1136/hrt.36.3.271
37. Fletcher PJ, Pfeffer JM, Pfeffer MA, Braunwald E. Left ventricular diastolic pressure-volume relations in rats with healed myocardial infarction. Effects on systolic function. Circ Res. (1981) 49:618–26. doi: 10.1161/01.RES.49.3.618
38. Hammerman H, Schoen FJ, Braunwald E, Kloner RA. Drug-induced expansion of infarct: Morphologic and functional correlations. Circulation. (1984) 69:611–7. doi: 10.1161/01.CIR.69.3.611
39. Hammerman H, Kloner RA, Hale S, Schoen FJ, Braunwald E. Dose-dependent effects of short-term methylprednisolone on myocardial infarct extent, scar formation, and ventricular function. Circulation. (1983) 68:446–52. doi: 10.1161/01.CIR.68.2.446
40. Brown EJ, Kloner RA, Schoen FJ, Hammerman H, Hale S, Braunwald E. Scar thinning due to ibuprofen administration after experimental myocardial infarction. Am J Cardiol. (1983) 51:877–83. doi: 10.1016/S0002-9149(83)80148-9
41. Hammerman H, Kloner RA, Schoen FJ, Brown EJ, Hale S, Braunwald E. Indomethacin-induced scar thinning after experimental myocardial infarction. Circulation. (1983) 67:1290–5. doi: 10.1161/01.CIR.67.6.1290
42. Mannisi JA, Weisman HF, Bush DE, Dudeck P, Healy B. Steroid administration after myocardial infarction promotes early infarct expansion. A study in the rat. J Clin Invest. (1987) 79:1431–9. doi: 10.1172/JCI112971
43. Ben-Mordechai T, Holbova R, Landa-Rouben N, Harel-Adar T, Feinberg MS, Abd Elrahman I, et al. Macrophage subpopulations are essential for infarct repair with and without stem cell therapy. J Am Coll Cardiol. (2013) 62:1890–901. doi: 10.1016/j.jacc.2013.07.057
44. Sun Y, Weber KT. Infarct scar: a dynamic tissue. Cardiovasc Res. (2000) 46:250–6. doi: 10.1016/S0008-6363(00)00032-8
45. de Boer RA, De Keulenaer G, Bauersachs J, Brutsaert D, Cleland JG, Diez J, et al. Toward better definition, quantification and treatment of fibrosis in heart failure. A scientific roadmap by the Committee of Translational Research of the Heart Failure Association (HFA) of the Eurpoean Society of Cardiology. Eur J Heart Fail. (2019) 21:272–85. doi: 10.1002/ejhf.1406
46. Torres WM, Jacobs J, Doviak H, Barlow SC, Zile MR, Shazly T, et al. Regional and temporal changes in left ventricular strain and stiffness in a porcine model of myocardial infarction. Am J Physiol Circ Physiol. (2018) 315:H958–67. doi: 10.1152/ajpheart.00279.2018
47. Fomovsky GM, Rouillard AD, Holmes JW. Regional mechanics determine collagen fiber structure in healing myocardial infarcts. J Mol Cell Cardiol. (2012) 52:1083–90. doi: 10.1016/j.yjmcc.2012.02.012
48. Zimmerman SD, Karlon WJ, Holmes JW, Omens JH, Covell JW. Structural and mechanical factors influencing infarct scar collagen organization. Am J Physiol Heart Circ Physiol. (2000) 278:H194–200. doi: 10.1152/ajpheart.2000.278.1.H194
49. Richardson WJ, Holmes JW. Emergence of Collagen Orientation Heterogeneity in Healing Infarcts and an Agent-Based Model. Biophys J. (2016) 110:2266–77. doi: 10.1016/j.bpj.2016.04.014
50. Nian M, Lee P, Khaper N, Liu P. Inflammatory cytokines and postmyocardial infarction remodeling. Circ Res. (2004) 94:1543–53. doi: 10.1161/01.RES.0000130526.20854.fa
51. Huang S, Frangogiannis NG. Anti-inflammatory therapies in myocardial infarction: failures, hopes and challenges. Br J Pharmacol. (2018) 175:1377–400. doi: 10.1111/bph.14155
52. Mahler GJ. Butcher JT. Inflammatory Regulation of Valvular Remodeling: The Good(?), the Bad, and the Ugly. Int J Inflam. (2011) 2011:1–13. doi: 10.4061/2011/721419
53. Siwik DA, Chang DLF, Colucci WS. Interleukin-1 β and tumor necrosis factor-α decrease collagen synthesis and increase matrix metalloproteinase activity in cardiac fibroblasts in vitro. Circ Res. (2000) 86:1259–65. doi: 10.1161/01.RES.86.12.1259
54. Awad AE, Kandalam V, Chakrabarti S, Wang X, Penninger JM, Davidge ST, et al. Tumor necrosis factor induces matrix metalloproteinases in cardiomyocytes and cardiofibroblasts differentially via superoxide production in a PI3Kγ-dependent manner. Am J Physiol Physiol. (2010) 298:C679–92. doi: 10.1152/ajpcell.00351.2009
55. Porter KE, Turner NA, O'Regan DJ, Ball SG. Tumor necrosis factor α induces human atrial myofibroblast proliferation, invasion and MMP-9 secretion: Inhibition by simvastatin. Cardiovasc Res. (2004) 64:507–15. doi: 10.1016/j.cardiores.2004.07.020
56. Zymek P, Bujak M, Chatila K, Cieslak A, Thakker G, Entman ML, et al. The role of platelet-derived growth factor signaling in healing myocardial infarcts. J Am Coll Cardiol. (2006) 48:2315–23. doi: 10.1016/j.jacc.2006.07.060
57. Bujak M, Frangogiannis N. The role of TGF-β signaling in myocardial infarction and cardiac remodeling. Cardiovasc Res. (2007) 74:184–95. doi: 10.1016/j.cardiores.2006.10.002
58. Stavropoulou A, Philippou A, Halapas A, Sourla A, Pissimissis N, Koutsilieris M. uPA, uPAR and TGFβ1 expression during early and late post myocardial infarction period in rat myocardium. In Vivo. (2010) 24:647–52.
59. Zhao W, Zhao T, Huang V, Chen Y, Ahokas RA, Sun Y. Platelet-derived growth factor involvement in myocardial remodeling following infarction. J Mol Cell Cardiol. (2011) 51:830–8. doi: 10.1016/j.yjmcc.2011.06.023
60. Burke RM, Lighthouse JK, Mickelsen DM, Small EM. Sacubitril/valsartan decreases cardiac fibrosis in left ventricle pressure overload by restoring PKG signaling in cardiac fibroblasts. Circ Hear Fail. (2019) 12:1–15. doi: 10.1161/CIRCHEARTFAILURE.118.005565
61. Bhambi B, Eghbali M. Effect of norepinephrine on myocardial collagen gene expression and response of cardiac fibroblasts after norepinephrine treatment. Am J Pathol. (1991) 139:1131–42.
62. Nishida M, Onohara N, Sato Y, Suda R, Ogushi M, Tanabe S, et al. Gα12/13-mediated up-regulation of TRPC6 negatively regulates endothelin-1-induced cardiac myofibroblast formation and collagen synthesis through nuclear factor of activated T cells activation. J Biol Chem. (2007) 282:23117–28. doi: 10.1074/jbc.M611780200
63. Loennechen JP, Støylen A, Beisvag V, Wisløff U, Ellingsen Ø. Regional expression of endothelin-1, ANP, IGF-1, and LV wall stress in the infarcted rat heart. Am J Physiol Hear Circ Physiol. (2001) 280:H2902–10. doi: 10.1152/ajpheart.2001.280.6.H2902
64. Omura T, Yoshiyama M, Takeuchi K, Hanatani A, Kim S, Yoshida K, et al. Differences in time course of myocardial mRNA expression in non-infarcted myocardium after myocardial infarction. Basic Res Cardiol. (2000) 995:316–23. doi: 10.1007/s003950070051
65. Flores-Monroy J, Lezama-Martínez D, Fonseca-Coronado S, Martínez-Aguilar L. Differences in the expression of the renin angiotensin system and the kallikrein-kinin system during the course of myocardial infarction in male and female Wistar rats. J Renin Angiotensin Aldosterone Syst. (2020) 21:147032031990003. doi: 10.1177/1470320319900038
66. Pilla JJ, Koomalsingh KJ, McGarvey JR, Witschey WRT, Dougherty L, Gorman JH, et al. Regional Myocardial Three-Dimensional Principal Strains During Postinfarction Remodeling. Ann Thorac Surg. (2015) 99:770–8. doi: 10.1016/j.athoracsur.2014.10.067
67. Herum KM, Lunde IG, Mcculloch AD, Christensen G. The Soft- and Hard-Heartedness of Cardiac Fibroblasts : Mechanotransduction Signaling Pathways in Fibrosis of the Heart. J Clin Med. (2017) 6:53. doi: 10.3390/jcm6050053
68. Rogers JD, Yeganegi A, Richardson WJ. Mechano-regulation of fibrillar collagen turnover by fibroblasts. In: Nagatomi J, Ebong EE, editors. Mechanobiology Handbook. 2nd ed. Boca Raton : Taylor & Francis, CRC Press (2018). p. 511–30.
69. Galie PA, Westfall MV, Stegemann JP. Reduced serum content and increased matrix stiffness promote the cardiac myofibroblast transition in 3D collagen matrices. Cardiovasc Pathol. (2011) 20:325–33. doi: 10.1016/j.carpath.2010.10.001
70. Atance J, Yost MJ, Carver W. Influence of the extracellular matrix on the regulation of cardiac fibroblast behavior by mechanical stretch. J Cell Physiol. (2004) 200:377–86. doi: 10.1002/jcp.20034
71. Carver W, Nagpal ML, Nachtigal M, Borg TK, Terracio L. Collagen expression in mechanically stimulated cardiac fibroblasts. Circ Res. (1991) 69:116–22. doi: 10.1161/01.RES.69.1.116
72. Huang C, Miyazaki K, Akaishi S, Watanabe A, Hyakusoku H, Ogawa R. Biological effects of cellular stretch on human dermal fibroblasts. J Plast Reconstr Aesthetic Surg. (2013) 66:e351–61. doi: 10.1016/j.bjps.2013.08.002
73. Blaauboer ME, Smit TH, Hanemaaijer R, Stoop R, Everts V. Cyclic mechanical stretch reduces myofibroblast differentiation of primary lung fibroblasts. Biochem Biophys Res Commun. (2011) 404:23–7. doi: 10.1016/j.bbrc.2010.11.033
74. Miyake Y, Furumatsu T, Kubota S, Kawata K, Ozaki T, Takigawa M. Mechanical stretch increases CCN2/CTGF expression in anterior cruciate ligament-derived cells. Biochem Biophys Res Commun. (2011) 409:247–52. doi: 10.1016/j.bbrc.2011.04.138
75. Tetsunaga T, Furumatsu T, Abe N, Nishida K, Naruse K, Ozaki T. Mechanical stretch stimulates integrin αVβ3-mediated collagen expression in human anterior cruciate ligament cells. J Biomech. (2009) 42:2097–103. doi: 10.1016/j.jbiomech.2009.06.016
76. Kim S-G, Akaike T, Sasagawa T, Atomi Y, Kurosawa H. Gene expression of type I and type III collagen by mechanical stretch in anterior cruciate ligament cells. Cell Struct Funct. (2002) 27:139–44. doi: 10.1247/csf.27.139
77. Kanazawa Y, Nomura J, Yoshimoto S, Suzuki T, Kita K, Suzuki N, et al. Cyclical cell stretching of skin-derived fibroblasts downregulates connective tissue growth factor (CTGF) production. Connect Tissue Res. (2009) 50:323–9. doi: 10.1080/03008200902836081
78. Husse B, Briest W, Homagk L, Isenberg G, Gekle M. Cyclical mechanical stretch modulates expression of collagen I and collagen III by PKC and tyrosine kinase in cardiac fibroblasts. Am J Physiol Regul Integr Comp Physiol. (2007) 293:R1898–907. doi: 10.1152/ajpregu.00804.2006
79. Lee AA, Delhaas T, McCulloch AD, Villarreal FJ. Differential responses of adult cardiac fibroblasts to in vitro biaxial strain patterns. J Mol Cell Cardiol. (1999) 31:1833–43. doi: 10.1006/jmcc.1999.1017
80. Loesberg WA, Walboomers XF, van Loon JJWA, Jansen JA, Van Loon JJWA, Jansen JA. The effect of combined cyclic mechanical stretching and microgrooved surface topography on the behavior of fibroblasts. J Biomed Mater Res A. (2005) 75:723–32. doi: 10.1002/jbm.a.30480
81. Manuyakorn W, Smart DE, Noto A, Bucchieri F, Haitchi HM, Holgate ST, et al. Mechanical strain causes adaptive change in bronchial fibroblasts enhancing profibrotic and inflammatory responses. PLoS ONE. (2016) 11:e0153926. doi: 10.1371/journal.pone.0153926
82. Shalaw FG, Slimani S, Kolopp-Sarda MN, Marchand M, Faure G, Stoltz J-F, et al. Effect of cyclic stretching and foetal bovine serum (FBS) on proliferation and extra cellular matrix synthesis of fibroblast. Biomed Mater Eng. (2006) 16:S137–44.
83. He Y, Macarak EJ, Korostoff JM, Howard PS. Compression and tension: differential effects on matrix accumulation by periodontal ligament fibroblasts in vitro. Connect Tissue Res. (2004) 45:28–39. doi: 10.1080/03008200490278124
84. Breen EC. Mechanical strain increases type I collagen expression in pulmonary fibroblasts in vitro. J Appl Physiol. (2000) 88:203–9. doi: 10.1152/jappl.2000.88.1.203
85. Yang G, Crawford RC, Wang JHC. Proliferation and collagen production of human patellar tendon fibroblasts in response to cyclic uniaxial stretching in serum-free conditions. J Biomech. (2004) 37:1543–50. doi: 10.1016/j.jbiomech.2004.01.005
86. Watson CJ, Phelan D, Collier P, Horgan S, Glezeva N, Cooke G, et al. Extracellular matrix sub-types and mechanical stretch impact human cardiac fibroblast responses to transforming growth factor beta. Connect Tissue Res. (2014) 55:248–56. doi: 10.3109/03008207.2014.904856
87. Parsons M, Kessler E, Laurent G, Brown R, Bishop JE. Mechanical load enhances procollagen processing in dermal fibroblasts by regulating levels of procollagen C-proteinase. Exp Cell Res. (1999) 252:319–31. doi: 10.1006/excr.1999.4618
88. Petersen A, Joly P, Bergmann C, Korus G, Duda GN. The impact of substrate stiffness and mechanical loading on fibroblast-induced scaffold remodeling. Tissue Eng Part A. (2012) 18:1804–17. doi: 10.1089/ten.tea.2011.0514
89. Liu J, Yu W, Liu Y, Chen S, Huang Y, Li X, et al. Mechanical stretching stimulates collagen synthesis via down-regulating SO2/AAT1 pathway. Sci Rep. (2016) 6:21112. doi: 10.1038/srep21112
90. Jiang M, Qiu J, Zhang L, Lu D, Long M, Luo X. Changes in tension regulates proliferation and migration of fibroblasts by remodeling expression of ECM proteins. Exp Ther Med. (2016) 12:1542–50. doi: 10.3892/etm.2016.3497
91. Rogers JD, Holmes JW, Saucerman JJ, Richardson WJ. Mechano-chemo signaling interactions modulate matrix production by cardiac fibroblasts. Matrix Biol Plus. (2021) 10:100055. doi: 10.1016/j.mbplus.2020.100055
92. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, et al. Role of YAP/TAZ in mechanotransduction. Nature. (2011) 474:179–83. doi: 10.1038/nature10137
93. Qiu Y, Bayomy AF, Gomez MV, Bauer M, Du P, Yang Y, et al. A role for matrix stiffness in the regulation of cardiac side population cell function. Am J Physiol - Hear Circ Physiol. (2015) 308:H990–7. doi: 10.1152/ajpheart.00935.2014
94. Herum KM, Choppe J, Kumar A, Engler AJ, Mcculloch AD, Garcia AJ. Mechanical regulation of cardiac fibroblast profibrotic phenotypes. Mol Biol Cell. (2017) 28:1871–82. doi: 10.1091/mbc.e17-01-0014
95. Xie J, Zhang Q, Zhu T, Zhang Y, Liu B, Xu J, et al. Substrate stiffness-regulated matrix metalloproteinase output in myocardial cells and cardiac fibroblasts: implications for myocardial fibrosis. Acta Biomater. (2014) 10:2463–72. doi: 10.1016/j.actbio.2014.01.031
96. Huang C, Yannas IV. Mechanochemical studies of enzymatic degradation of insoluble collagen fibers. J Biomed Mater Res. (1977) 11:137–54. doi: 10.1002/jbm.820110113
97. Ruberti JW, Hallab NJ. Strain-controlled enzymatic cleavage of collagen in loaded matrix. Biochem Biophys Res Commun. (2005) 336:483–9. doi: 10.1016/j.bbrc.2005.08.128
98. Flynn BP, Tilburey GE, Ruberti JW. Highly sensitive single-fibril erosion assay demonstrates mechanochemical switch in native collagen fibrils. Biomech Model Mechanobiol. (2013) 12:291–300. doi: 10.1007/s10237-012-0399-2
99. Camp RJ, Liles M, Beale J, Saeidi N, Flynn BP, Moore E, et al. Molecular mechanochemistry: low force switch slows enzymatic cleavage of human type I collagen monomer. J Am Chem Soc. (2011) 133:4073–8. doi: 10.1021/ja110098b
100. Ghazanfari S, Driessen-Mol A, Bouten CVC, Baaijens FPT. Modulation of collagen fiber orientation by strain-controlled enzymatic degradation. Acta Biomater. (2016) 35:118–26. doi: 10.1016/j.actbio.2016.02.033
101. Wipff PJ, Rifkin DB, Meister JJ, Hinz B. Myofibroblast contraction activates latent TGF-beta1 from the extracellular matrix. J Cell Biol. (2007) 179:1311–23. doi: 10.1083/jcb.200704042
102. Sarrazy V, Koehler A, Chow ML, Zimina E, Li CX, Kato H, et al. Integrins αvβ5 and αvβ3 promote latent TGF-β1 activation by human cardiac fibroblast contraction. Cardiovasc Res. (2014) 102:407–17. doi: 10.1093/cvr/cvu053
103. Ikeuchi M, Tsutsui H, Shiomi T, Matsusaka H, Matsushima S, Wen J, et al. Inhibition of TGF-B signaling exacerbates early cardiac dysfunction but prevents late remodeling after infarction. Cardiovasc Res. (2004) 64:526–35. doi: 10.1016/j.cardiores.2004.07.017
104. Frantz S, Hu K, Adamek A, Wolf J, Sallam A, Maier SKG, et al. Transforming growth factor beta inhibition increases mortality and left ventricular dilatation after myocardial infarction. Basic Res Cardiol. (2008) 103:485–92. doi: 10.1007/s00395-008-0739-7
105. Iyer RP, de Castro Brás LE, Patterson NL, Bhowmick M, Flynn ER, Asher M, et al. Early matrix metalloproteinase-9 inhibition post-myocardial infarction worsens cardiac dysfunction by delaying inflammation resolution. J Mol Cell Cardiol. (2016) 100:109–17. doi: 10.1016/j.yjmcc.2016.10.005
106. Iyer RP, Patterson NL, Zouein FA, Ma Y, Dive V, de Castro Brás LE, et al. Early matrix metalloproteinase-12 inhibition worsens post-myocardial infarction cardiac dysfunction by delaying inflammation resolution. Int J Cardiol. (2015) 185:198–208. doi: 10.1016/j.ijcard.2015.03.054
107. Hammoud L, Lu X, Lei M, Feng Q. Deficiency in TIMP-3 increases cardiac rupture and mortality post-myocardial infarction via EGFR signaling: beneficial effects of cetuximab. Basic Res Cardiol. (2011) 106:459–71. doi: 10.1007/s00395-010-0147-7
108. Ashikaga H, Mickelsen SR, Ennis DB, Rodriguez I, Kellman P, Wen H, Mcveigh ER, Elec ERM. Electromechanical analysis of infarct border zone in chronic myocardial infarction. Am J Physiol Heart Circ Physiol. (2005) 1061:1099–105. doi: 10.1152/ajpheart.00423.2005
109. Lew WY, Chen ZY, Guth B, Covell JW. Mechanisms of augmented segment shortening in nonischemic areas during acute ischemia of the canine left ventricle. Circ Res. (1985) 56:351–8. doi: 10.1161/01.RES.56.3.351
110. Kass DA, Maughan WL, Ciuffo A, Graves W, Healy B, Weisfeldt ML. Disproportionate epicardial dilation after transmural infarction of the canine left ventricle: acute and chronic differences. J Am Coll Cardiol. (1988) 11:177–85. doi: 10.1016/0735-1097(88)90186-6
111. Theroux P, Ross J, Franklin D, Covell JW, Bloor CM, Sasayama S. Regional myocardial function and dimensions early and late after myocardial infarction in the unanesthetized dog. Circ Res. (1977) 40:158–65. doi: 10.1161/01.RES.40.2.158
112. Schork NJ. Personalized medicine: time for one-person trials. Nature. (2015) 520:609–11. doi: 10.1038/520609a
113. Galie AP, Russell MW, Westfall VM, Stegemann JP. Interstitial fluid flow and cyclic strain differentially regulate cardiac fibroblast activation via AT1R and TGF-β1. Exp Cell Res. (2012) 318:75–84. doi: 10.1016/j.yexcr.2011.10.008
114. Yong KW, Li Y, Liu F, Gao B, Lu TJ, Wan Abas WAB, Wan Safwani WKZ, et al. Paracrine effects of adipose-derived stem cells on matrix stiffness-induced cardiac myofibroblast differentiation via angiotensin II type 1 receptor and smad7. Sci Rep. (2016) 6:1–13. doi: 10.1038/srep33067
115. Voloshenyuk TG, Landesman ES, Khoutorova E, Hart AD, Gardner JD. Induction of cardiac fibroblast lysyl oxidase by TGF-β1 requires PI3K/Akt, Smad3, MAPK signaling. Cytokine. (2011) 55:90–7. doi: 10.1016/j.cyto.2011.03.024
116. Voloshenyuk TG, Hart AD, Khoutorova E, Gardner JD. TNF-alpha increases cardiac fibroblast lysyl oxidase expression through TGF-beta and PI3Kinase signaling pathways. Biochem Biophys Res Commun. (2011) 413:370–5. doi: 10.1016/j.bbrc.2011.08.109
117. Turner NA, Aley PK, Hall KT, Warburton P, Galloway S, Midgley L, et al. Simvastatin inhibits TNFα-induced invasion of human cardiac myofibroblasts via both MMP-9-dependent and -independent mechanisms. J Mol Cell Cardiol. (2007) 43:168–76. doi: 10.1016/j.yjmcc.2007.05.006
118. Xia H, Nho RS, Kahm J, Kleidon J, Henke CA. Focal adhesion kinase is upstream of phosphatidylinositol 3-kinase/Akt in regulating fibroblast survival in response to contraction of type I collagen matrices via a beta 1 integrin viability signaling pathway. J Biol Chem. (2004) 279:33024–34. doi: 10.1074/jbc.M313265200
119. Hinz B. The extracellular matrix and transforming growth factor-β1: tale of a strained relationship. Matrix Biol. (2015) 47:54–65. doi: 10.1016/j.matbio.2015.05.006
120. Ong S, Hernández-Reséndiz S, Crespo-Avilan G, Mukhametshina R, Kwek X, Cabrera-Fuentes H, et al. Inflammation following acute myocardial infarction: multiple players, dynamic roles, and novel therapeutic opportunities. Pharmacol Ther. (2018) 186:73–87. doi: 10.1016/j.pharmthera.2018.01.001
121. Andreadou I, Cabrera-Fuentes HA, Devaux Y, Frangogiannis NG, Frantz S, Guzik T, et al. Immune cells as targets for cardioprotection: new players and novel therapeutic opportunities. Cardiovasc Res. (2019) 115:1117–30. doi: 10.1093/cvr/cvz050
122. Frangogiannis NG. Targeting the inflammatory response in healing myocardial infarcts. Curr Med Chem. (2006) 13:1877–93. doi: 10.2174/092986706777585086
123. Davis J, Burr AR, Davis GF, Birnbaumer L, Molkentin JD. A TRPC6-dependent pathway for myofibroblast transdifferentiation and wound healing in vivo. Dev Cell. (2012) 23:705–15. doi: 10.1016/j.devcel.2012.08.017
124. Butt RP, Bishop JE. Mechanical load enhances the stimulatory effect of serum growth factors on cardiac fibroblast procollagen synthesis. J Mol Cell Cardiol. (1997) 29:1141–51. doi: 10.1006/jmcc.1996.0347
125. Ceccato TL, Starbuck RB, Hall JK, Walker CJ, Brown TE, Killgore JP, et al. Defining the cardiac fibroblast secretome in a fibrotic microenvironment. J Am Heart Assoc. (2020) 9:e017025. doi: 10.1161/JAHA.120.017025
126. Merryman WD, Lukoff HD, Long RA, Engelmayr GC, Hopkins RA, Sacks MS. Synergistic effects of cyclic tension and transforming growth factor-β1 on the aortic valve myofibroblast. Cardiovasc Pathol. (2007) 16:268–76. doi: 10.1016/j.carpath.2007.03.006
127. Ramirez TA, Iyer RP, Ghasemi O, Lopez EF, Levin DB, Zhang J, et al. Aliskiren and valsartan mediate left ventricular remodeling post-myocardial infarction in mice through MMP-9 effects. J Mol. Cell Cardiol. (2014) 72:326–35. doi: 10.1016/j.yjmcc.2014.04.007
128. Matera DL, Wang WY, Baker BM. New directions and dimensions for bioengineered models of fibrosis. Nat Rev Mater. (2021) 6:192–5. doi: 10.1038/s41578-021-00288-x
129. Walker M, Godin M, Pelling PE. A vacuum-actuated microtissue stretcher for long-term exposure to oscillatory strain within a 3D matrix. Biomed Microdevices. (2018) 20:43. doi: 10.1007/s10544-018-0286-4
130. Xu F, Zhao R, Liu AS, Metz T, Shi Y, Bose P, et al. A microfabricated magnetic actuation device for mechanical conditioning of arrays of 3D microtissues. Lab Chip. (2015) 15:2496–503. doi: 10.1039/C4LC01395F
131. Ramade A, Legant WR, Picart C, Chen CS, Boudou T. Microfabrication of a platform to measure and manipulate the mechanics of engineered microtissues. Methods Cell Biol. (2014) 121:191–211. doi: 10.1016/B978-0-12-800281-0.00013-0
132. Mih JD, Sharif AS, Liu F, Marinkovic A, Symer MM, Tschumperlin DJ. A multiwell platform for studying stiffness-dependent cell biology. PLoS ONE. (2011) 6:e19929. doi: 10.1371/journal.pone.0019929
133. Zeigler AC, Richardson WJ, Holmes JW, Saucerman JJ. Computational modeling of cardiac fibroblasts and fibrosis. J Mol Cell Cardiol. (2016) 93:73–83. doi: 10.1016/j.yjmcc.2015.11.020
134. Shim JV, Chun B, van Hasselt JGC, Birtwistle MR, Saucerman JJ, Sobie EA. Mechanistic systems modeling to improve understanding and prediction of cardiotoxicity caused by targeted cancer therapeutics. Front Physiol. (2017) 8:651. doi: 10.3389/fphys.2017.00651
135. Mayourian J, Sobie EA, Costa KD. An Introduction to Computational Modeling of Cardiac Electrophysiology and Arrhythmogenicity. Methods Mol Biol. (2018) 1816:17–35. doi: 10.1007/978-1-4939-8597-5_2
136. Bradshaw EL, Spilker ME, Zang R, Bansal L, He H, Jones RDO, et al. Applications of quantitative systems pharmacology in model-informed drug discovery: perspective on impact and opportunities. CPT Pharmacometrics Syst Pharmacol. (2019) 8:777–91. doi: 10.1002/psp4.12463
137. Zeigler AC, Richardson WJ, Holmes JW, Saucerman JJ. A computational model of cardiac fibroblast signaling predicts context-dependent drivers of myofibroblast differentiation. J Mol Cell Cardiol. (2016) 94:72–81. doi: 10.1016/j.yjmcc.2016.03.008
138. Tan PM, Buchholz KS, Omens JH, McCulloch AD, Saucerman JJ. Predictive model identifies key network regulators of cardiomyocyte mechano-signaling. PLoS Comput Biol. (2017) 13:1–17. doi: 10.1371/journal.pcbi.1005854
139. Saucerman JJ, Tan PM, Buchholz KS, McCulloch AD, Omens JH. Mechanical regulation of gene expression in cardiac myocytes and fibroblasts. Nat Rev Cardiol. (2019) 16:361–78. doi: 10.1038/s41569-019-0155-8
140. Irons L, Humphrey JD. Cell signaling model for arterial mechanobiology. PLoS Comput Biol. (2020) 16:e1008161. doi: 10.1371/journal.pcbi.1008161
141. Wang A, Cao S, Aboelkassem Y, Valdez-Jasso D. Quantification of uncertainty in a new network model of pulmonary arterial adventitial fibroblast pro-fibrotic signalling. Philos Trans R Soc A Math Phys Eng Sci. (2020) 378:20190338. doi: 10.1098/rsta.2019.0338
Keywords: mechanobiology, heterogeneity, cardiac fibroblast, fibrosis, mechanotransduction, myocardial infarction
Citation: Richardson WJ, Rogers JD and Spinale FG (2021) Does the Heart Want What It Wants? A Case for Self-Adapting, Mechano-Sensitive Therapies After Infarction. Front. Cardiovasc. Med. 8:705100. doi: 10.3389/fcvm.2021.705100
Received: 04 May 2021; Accepted: 16 August 2021;
Published: 10 September 2021.
Edited by:
Jingyan Han, Boston University, United StatesReviewed by:
Ivan Luptak, Boston University, United StatesDiego Claudio Humeres, Albert Einstein College of Medicine, United States
Copyright © 2021 Richardson, Rogers and Spinale. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: William J. Richardson, d3JpY2hhNCYjeDAwMDQwO2NsZW1zb24uZWR1