 Jiyoung Bae
Jiyoung Bae Wyatt G. Paltzer
Wyatt G. Paltzer Ahmed I. Mahmoud
Ahmed I. Mahmoud
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med. , 16 July 2021
Sec. Cardiovascular Biologics and Regenerative Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.702920
This article is part of the Research Topic Novel Strategies to Repair the Infarcted Heart View all 11 articles
Heart failure is the leading cause of death worldwide. The inability of the adult mammalian heart to regenerate following injury results in the development of systolic heart failure. Thus, identifying novel approaches toward regenerating the adult heart has enormous therapeutic potential for adult heart failure. Mitochondrial metabolism is an essential homeostatic process for maintaining growth and survival. The emerging role of mitochondrial metabolism in controlling cell fate and function is beginning to be appreciated. Recent evidence suggests that metabolism controls biological processes including cell proliferation and differentiation, which has profound implications during development and regeneration. The regenerative potential of the mammalian heart is lost by the first week of postnatal development when cardiomyocytes exit the cell cycle and become terminally differentiated. This inability to regenerate following injury is correlated with the metabolic shift from glycolysis to fatty acid oxidation that occurs during heart maturation in the postnatal heart. Thus, understanding the mechanisms that regulate cardiac metabolism is key to unlocking metabolic interventions during development, disease, and regeneration. In this review, we will focus on the emerging role of metabolism in cardiac development and regeneration and discuss the potential of targeting metabolism for treatment of heart failure.
Heart failure is the leading cause of morbidity and mortality worldwide. In the United States alone, there are over 6,000,000 people with heart failure (1). This is largely due to the inability of the adult mammalian heart to replenish the lost myocardial tissue following injury, which results in the progressive weakening of the heart muscle and the development of heart failure (2). Current therapies are focused on preventing further remodeling of the remaining myocardial tissue. Heart transplantations are the only treatment in patients with severe heart failure (3). Due to the complexity and complications associated with heart transplants they are not always a suitable treatment; therefore, identifying novel therapeutic approaches to promote adult heart regeneration provides immense opportunities to advance heart failure therapy. Endogenous heart regeneration following injury has been demonstrated in some non-mammalian vertebrates (4, 5). Interestingly, neonatal mice are also capable of regenerating their heart tissue following injury, however this regenerative ability is lost within a few days following birth (6, 7). These models of endogenous regeneration provide us with a platform to elucidate the mechanisms that guide heart regeneration to reactivate these processes to promote adult heart regeneration.
Embryonic and neonatal cardiomyocytes produce energy primarily via glycolysis, where postnatal maturation is accompanied with a metabolic switch to fatty acid oxidation to meet the energy demands of adult cardiomyocytes (8) (Figure 1). This metabolic switch contributes to the postnatal cardiomyocyte cell cycle exit and loss of the regenerative potential of the mammalian heart. This underscores the potential role of cardiac metabolism as a target to promote adult heart regeneration.
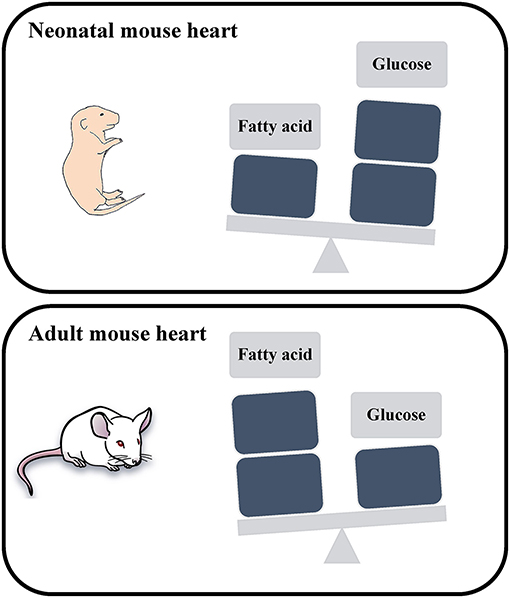
Figure 1. Schematic representation of the energy utilization in neonatal and adult mouse hearts. The neonatal mouse heart generates energy through glucose metabolism, while the adult mouse heart generates energy through fatty acid oxidation.
In this review, we highlight major studies of cardiac metabolism including fatty acid oxidation, glucose, and amino acid metabolism (Figure 2). We also discuss key metabolic targets that may play an important role during cardiomyocyte development and regeneration and their potential as a therapeutic target for adult heart disease.
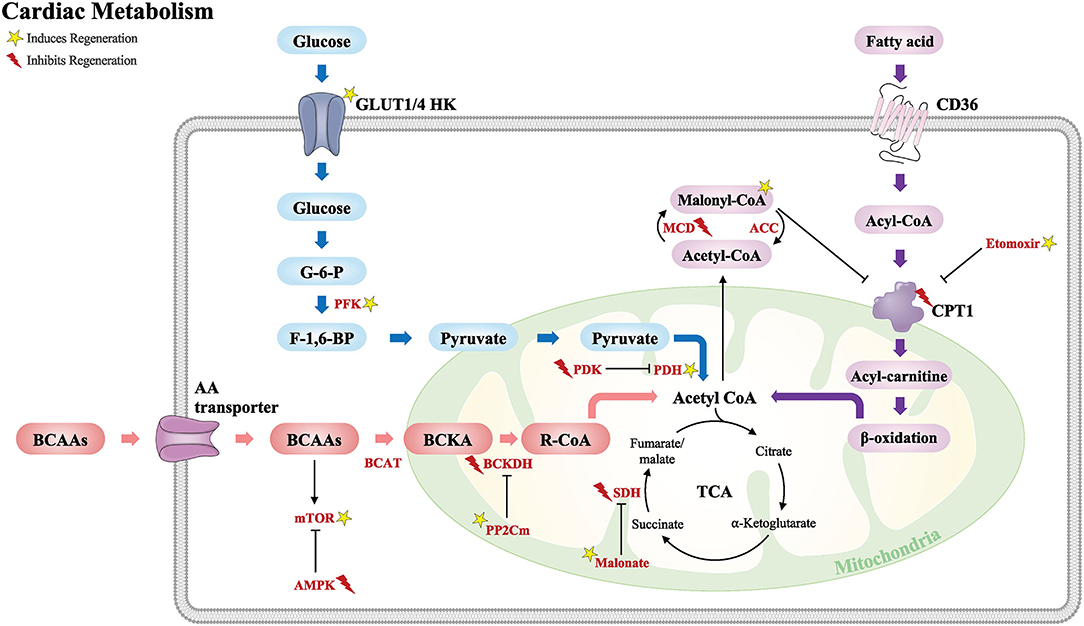
Figure 2. Schematic of the major metabolic pathways that modulate the cardiac regenerative response following injury. Glucose metabolism (blue), fatty acid metabolism (purple), and BCAA metabolism (red). Acetyl CoA from these major metabolic pathways is required for the TCA cycle. GLUT, glucose transporter type; HK, hexokinase; G-6-P, glucose-6-phosphate; F-1,6-BP, fructose-1,6-biphosphate; PFK, phosphofructokinase; PDK, pyruvate dehydrogenase kinase; PDH, pyruvate dehydrogenase; CD36, cluster of differentiation; CPT1, carnitine palmitoyltransferase; MCD, malonyl CoA dehydrogenase; ACC, acetyl CoA carboxylase; BCAAs, branched-chain amino acids; AA, amino acid; BCAT, branched-chain amino-transferase; BCKA, branched-chain alpha keto acids; BCKDH, branched-chain alpha-keto acid dehydrogenase; PP2Cm, protein phosphatase 2Cm; mTOR, the mechanistic target of rapamycin; AMPK, 5′ adenosine monophosphate-activated protein kinase; TCA, tricarboxylic acid cycle; SDH, succinate dehydrogenase. Yellow star induces regeneration and red lightning bolt inhibits regeneration.
The heart is the most energy-consuming tissue (per gram) in the human body (9), and energy production takes place in the mitochondria. The main function of the mitochondria is generating energy as adenosine triphosphate (ATP); thus, mitochondria play an essential role during development, cellular proliferation, and tissue regeneration, all of which are energy demanding processes (10–12).
Heart regenerative capacity differs by model organisms from vertebrates to human. Zebrafish and newts have a remarkable capability to regenerate their hearts after injury. Zebrafish heart regeneration is primarily derived from the proliferation of the pre-existing cardiomyocytes (13, 14). Following injury, cardiomyocyte dedifferentiation and proliferation are required to regenerate the heart following injury. Interestingly, single-cell transcriptional analysis of regenerating zebrafish hearts demonstrate that proliferating border zone cardiomyocytes undergo metabolic reprogramming to glycolysis from oxidative phosphorylation following cryoinjury (15). In contrast, glycolysis inhibitors including 2-deoxyglucose and lonidamine impair cardiomyocyte proliferation and heart regeneration (15). These results suggest that the glycolytic metabolic state mediates cardiomyocyte proliferation and regeneration following injury in zebrafish.
Like zebrafish, embryonic and neonatal mice can regenerate their heart after injury. Both zebrafish and neonatal mouse hearts have lower mitochondrial DNA copy numbers compared to postnatal and adult mice (16). This increase in cardiomyocyte mitochondrial number in the adult heart is due to the switch from glycolytic metabolism in neonatal cardiomyocytes to oxygen-dependent mitochondrial oxidative phosphorylation in adult cardiomyocytes (17). This metabolic switch and increase mitochondrial DNA results in a significant rise in reactive oxygen species (ROS) production from mitochondria which plays an essential role in regulating heart development and regeneration (16). This increase in ROS production contributes to postnatal cardiomyocyte cell cycle arrest. Interestingly, the increased sarcomere contraction in the postnatal heart promotes mitochondrial metabolism, which results in ROS production and DNA damage response activation via p53. As a result, inhibition of sarcomeres in cardiac troponin T knockout cardiomyocytes prevents cell cycle arrest and polyploidy resulting in increased cardiomyocyte proliferation (18). Furthermore, ROS scavengers such as N-acetyl cysteine (NAC) prolongs the postnatal window of cardiomyocyte proliferation and regeneration following ischemia reperfusion (I/R) injury (16).
Significant metabolic shifts occur in response to abnormal heart conditions. A healthy adult heart generates energy through fatty acid oxidation, however conditions such as pressure overload, hypertrophy, and ischemia results in a metabolic transition toward anaerobic glycolytic metabolism to be protect against damage (19). A recent study elegantly demonstrates the different metabolite utilization in human hearts by using arterio-venous metabolomics, which is a powerful tool to measure metabolite utilization in humans by measuring the metabolite intake and release in the blood from human hearts. Similar to mouse studies, healthy human hearts mostly uptake fatty acids as a fuel source while they only uptake limited amounts of glucose. Interestingly, the healthy heart releases amino acids, specifically essential amino acids. In contrast, the failing heart utilizes more ketones and lactate, but less fatty acids (20). These results are consistent with previous animal studies demonstrating that ketones and β-hydroxybutyrate are protective in the failing heart (21, 22). Collectively, these studies demonstrate that cardiac metabolism is dynamic and can switch to different states during development, disease, and regeneration.
The heart requires high amounts of energy to maintain adult cardiac physiology (9). The adult human heart generates ATP via fatty acid oxidation (23–25). Fatty acids are oxidized through the tricarboxylic acid (TCA) cycle in the mitochondria, and the intermediate electrons from the TCA cycle flow through the electron transport chain (ETC) and produce a proton gradient to generate energy through ATP synthesis (26).
The first step for transporting long chain fatty acids from the cytosol into the mitochondria for initiating mitochondrial fatty acid oxidation occurs by carnitine palmitoyltransferase I (CPT1) in the outer mitochondrial membrane. CoA in acyl-CoA, which is derived from fatty acids, is converted to carnitine through CPT1. Thus, CPT1 is a key enzyme in regulating fatty acid oxidation. There are three tissue-specific isoforms of CPT1 that exist in mammalian tissues: CPT1A is expressed in the liver, lung, spleen, pancreas, and kidney; CPT1B is expressed in the heart, skeletal muscle, and adipose tissue; and CPT1C is expressed in the brain (27). Mitochondrial CPT1 activity is very low in the neonatal rat heart. Interestingly, CPT1 level is significantly increased in 7-day-old juvenile mice, which is the timepoint when the majority of mammalian cardiomyocytes have already exited the cell cycle (28). CPT1 expression is increased in adolescent (6 months) sheep hearts compared to fetus (105 days) hearts (29). Thus, CPT1 could be a key regulator of cardiomyocyte proliferation.
CPT1 inhibition reduces fatty acid oxidation due to the blockade of fatty acid transfer into the mitochondria. Inhibition of CPT1 by etomoxir promotes neonatal mouse cardiomyocyte proliferation (30). However, inhibition or activation of CPT1 does not induce cardiomyocyte proliferation in the adult mouse heart (31). Ventricular cardiomyocytes isolated from neonatal mice injected with the CPT1 inhibitor etomoxir show a reduction in fatty acid oxidation genes (30). These results demonstrate that disruption of fatty acid oxidation by inhibition of CPT1 extends neonatal cardiomyocyte proliferation and heart regeneration but is not sufficient to promote adult heart regeneration.
Another metabolite that regulates fatty acid oxidation via CPT1 inhibition is malonyl-CoA (32). Inhibition of malonyl-CoA decarboxylase (MCD), which is responsible for malonyl-CoA decarboxylation, results in increased malonyl-CoA levels which reduces fatty acid oxidation and increases glucose oxidation (33). As a consequence, short-term pharmacological inhibition of MCD increases malonyl-CoA levels in ischemic conditions resulting in improving cardiac function during ischemia/reperfusion (I/R) injury in the swine heart (34) and following myocardial infarction (MI) in the rat heart (35). Genetically MCD deficient mouse hearts show increased glucose oxidation and improved cardiac function following I/R injury (36). These results demonstrate that malonyl-CoA improves cardiac function following injury through CPT1 inhibition.
CPT1 is also regulated by peroxisome proliferator-activated receptors (PPARs), which are lipid receptors that play a critical role in regulating energy metabolism. There are three subtypes of PPAR: PPARα, PPARβ/δ, and PPARγ (37). PPARα, β/δ, γ gene expression levels are lower in the developing mouse heart compared to 14- and 28-day-old mouse hearts (38, 39). The levels of PPARs change during aging, as cardiac PPARα is significantly reduced in aged mice (40). PPARs play multiple roles in cardiac function in several disease states. It has been shown that expression of PPARα and CPT1 is notably reduced in adult mouse hearts following transverse aortic constriction (TAC) injury (41) as well as following I/R injury (42). However, activation of PPARα using the PPARα agonist GW7647 increased CPT1 gene expression which increased fatty acid oxidation and enhanced oxygen consumption rate in the presence of the fatty acid palmitate in isolated mouse cardiomyocytes (30).
However, the role of PPAR in cardiomyocyte proliferation and regeneration remains unclear. The PPARα agonist GW7647 does not promote cardiomyocyte proliferation and cardiac function following MI in adult mouse hearts (31). Furthermore, PPARα activation by agonist WY-14643 reduced cardiac function following I/R injury (42). Moreover, larger infarct size is observed in PPARα knockout mouse heart following I/R injury (43). In contrast, another study showed that PPARα transgenic mouse hearts showed improved cardiac function and reduced left ventricular dilation following TAC injury (41).
Another PPAR family receptor, PPARδ, has been shown to play a role during cardiac injury. The PPARδ ligand, GW501516, has been shown to inhibit cardiac fibroblast proliferation and transdifferentiation to myofibroblasts (44). Furthermore, inhibition of PPARδ reduced cardiomyocyte proliferation following injury in zebrafish hearts, whereas cardiomyocyte-specific PPARδ overexpression induced proliferation and reduced scar size following MI in mouse hearts (45).
Despite the important role of PPAR receptors in a variety of heart disease models, the exact role of these receptors in regulating cardiomyocyte proliferation and heart regeneration remains to be fully defined.
Although the adult mammalian heart utilizes fatty acids as a main source of energy in the heart, glucose plays an important role as an energy source (46–49). Under healthy conditions the heart mostly uses fatty acids to produce energy, however, it will switch to glucose as an energy source during heart failure (50–52). Glucose metabolism is initiated by glucose uptake. In the heart, glucose enters cardiomyocytes via glucose transporters (GLUTs) which are expressed by various cell types. Among 14 members of the GLUT family (53), the most abundant GLUTs in the human heart are the insulin-sensitive glucose transporter GLUT4 (54, 55), and the insulin-independent glucose transporter GLUT1 (54, 56).
Under physiological conditions, GLUT1 is the main glucose transporter in embryonic and neonatal hearts, while GLUT4 is the primary glucose transporter in adult hearts (57, 58). In heart failure, GLUT4 expression is reduced while the levels of GLUT1 increase (59). This results in an increase in GLUT1-mediated glycolysis in heart failure, suggesting that GLUT1 plays an important role in cardiac protection during heart failure. GLUT1 expression is also increased in the heart under hypoxic conditions (60), which is mediated via hypoxia-inducible factor-1α (HIF-1α) (61). Cardiac-specific overexpression of GLUT1 results in increased glucose uptake and glycolysis in the mouse heart (62, 63), whereas cardiac-specific GLUT1 deletion reduces glucose uptake and glycolysis in isolated mouse cardiomyocytes following TAC injury (59). Interestingly, GLUT1 overexpression enhanced the regenerative response of neonatal mice following cryoinjury by increasing the levels of glucose metabolites (64). These results provide new evidence that increased GLUT1 expression promotes cardiomyocyte proliferation and heart regeneration through increased glucose metabolism.
Once glucose enters cardiomyocytes through GLUTs, glucose is phosphorylated and metabolized by key glycolytic enzymes such as hexokinase (HK) and phosphofructokinase (PFK) to form two pyruvate molecules (65). Pyruvate is then oxidized to acetyl CoA by pyruvate dehydrogenase (PDH), a key regulator in pyruvate metabolism (66), to enter the TCA cycle in the mitochondria. These glycolytic enzymes have been demonstrated to play a role in cardiac repair and regeneration following injury. In adult zebrafish, increased glycolysis has been shown to promote cardiomyocyte proliferation through increased cell cycle gene expression following injury (67). In addition, inhibition of glycolysis by 2-deoxyglucose reduced cardiomyocyte proliferation in the injured zebrafish heart (15). Thus, key components of glycolysis play an important role during cardiomyocyte proliferation and heart regeneration.
Hexokinase (HK) is the first enzyme of glycolysis that phosphorylates glucose to glucose-6-phosphate. Among the four distinct HK isozymes (HK 1, 2, 3, and 4) (68), HK-1 and -2 are expressed in the heart and regulate cardiac glucose metabolism (69, 70). Cardiac-specific HK-2 overexpression decreased cardiac hypertrophy in isoproterenol-induced mouse hearts and reduced cardiomyocyte size in neonatal rat ventricular cardiomyocytes (71). In addition, HK-2 overexpression reduced ROS accumulation which is upregulated during cardiac hypertrophy (71). In contrast, reduced HK-2 expression in HK-2+/− mice results in increased cardiac dysfunction due to increase in cell death and fibrosis and reduction of angiogenesis following I//R injury (72). Whether HK plays a role during heart regeneration remains to be determined.
Another important enzyme that regulates glycolysis is phosphofructokinase (PFK) which has two isoforms: PFK-1 and PFK-2. PFK-2 regulates PFK-1 activity since PFK-2 regulates the synthesis of fructose-2,6-biphosphate, which activates PFK1 that promotes glycolysis. Thus, PFK-2 is a crucial enzyme that regulates glycolysis (65). PFK-2 is activated upon insulin stimulation which promotes glycolysis, where PFK-2 is reduced in the insulin-deficient streptozotocin-induced diabetic mice and high-fat diet-induced obese mice (73). Glycolysis and insulin sensitivity are decreased in cardiac-specific kinase-deficient PFK-2 mutant mouse hearts (74, 75). As a result, glycolysis is not increased in cardiac-specific kinase-deficient PFK-2 mice in contrast to wild type mice following TAC surgery (75). On the other hand, overexpression of kinase-active PFK-2 enhances contractility in hypoxic mouse cardiomyocytes (76). Thus, PFK-2 regulates glycolysis and may play a role in cardiac protection following injury.
A key glycolysis enzyme is pyruvate dehydrogenase kinase (PDK). There are four PDK isoforms (PDK 1, 2, 3, and 4). PDKs expression is significantly increased during heart development and is further increased in the adult heart (58). PDKs expression is also increased in the infarct zone following cardiac cryoinjury in zebrafish (67). Among the PDK isoforms, cardiac PDK4 is the most significantly upregulated enzyme in 7-day-old mice, where the majority of mammalian cardiomyocytes exit the cell cycle (58). PDKs play in a role in glycolysis via inhibition of pyruvate dehydrogenase (PDH), which is a limiting step in glucose oxidation. PDK inhibition by dichloroacetate induces PDH activation which promotes cardiac function following KCl-induced cardiac arrest (77). A recent study demonstrated that cardiac-specific deletion of PDK4 promotes adult cardiomyocyte proliferation and heart regeneration following adult MI (78). In summary, PDK plays an important role in glycolysis via inhibition of PDH activity, suggesting that PDKs may be an important therapeutic target to increase glycolysis and promote cardiac repair and regeneration.
Pyruvate kinase muscle isoenzyme 2 (PKM2), a rate-limiting enzyme in the final step of glycolysis, is expressed in embryonic and neonatal mouse hearts; however, it is significantly reduced beyond postnatal day 7 when cardiomyocytes exit the cell cycle (79). Interestingly, overexpression of PKM2 in cardiomyocytes promotes cell cycle and glucose-6-phosphate dehydrogenase expression (79). Cardiomyocyte-specific PKM2 expression by modified RNA (modRNA) promotes adult cardiomyocyte proliferation and cardiac regeneration following adult MI (79). Conversely, loss of PKM2 reduces cardiomyocyte proliferation following injury in zebrafish hearts (67). Moreover, cardiomyocyte-specific deletion of PKM2 impairs heart development as they exhibit smaller heart size and low levels of cardiomyocyte proliferation (79).
Taken together, these studies demonstrate that glycolysis plays an important role in regulating cardiomyocyte proliferation and heart regeneration following injury. Thus, targeting glucose metabolism is a promising approach to promote adult heart regeneration.
Amino acids are key molecules for cell growth and survival. Amino acids are used as the building blocks for protein synthesis as well as inhibiting proteolysis (80). In addition, amino acids serve as precursors to key metabolites (81). Remarkably, amino acids can act as a signaling molecule, such as leucine, which stimulates muscle protein synthesis via the mechanistic target of rapamycin (mTOR) signaling pathway (82–84). The levels of cellular amino acids fluctuate throughout development, increasing in postnatal stages until reaching peak levels around P9 and then decreasing into adult stages suggesting a dynamic role for amino acids during development and maturation (85).
A recent study demonstrated that circulating arterial amino acid levels are reduced in patients with heart failure in comparison to healthy patients (86). Decreasing levels of arterial amino acids correlated with reduced heart function, demonstrating the potential use for arterial amino acid levels as a biomarker of heart failure (86). To understand if this reduction of circulating arterial amino acids was the heart reducing its energy consumption of amino acids a recent study aimed to quantify fuel consumption of the failing and non-failing human heart (20). This study demonstrated that energy consumption of amino acids was unchanged between the non-failing and failing hearts (20), suggesting that the role amino acids play in heart failure is not tied to their function as an energy source.
To further understand the role of amino acid metabolism in heart failure, a main focus was placed on a subset of amino acids, the branched chain amino acids (BCAAs), which are utilized differently than the other amino acids. BCAAs consist of leucine, isoleucine, and valine (87). BCAAs account for nearly 5% of total carbon used within the heart, and they also act as regulatory components for other metabolic processes (20, 88). BCAA catabolism has been shown to play a role in heart failure. This is seen in both humans and rodents where all components in BCAA catabolism have altered expression levels in heart failure (87). A study using a mouse model deficient in protein phosphatase 2Cm (PP2Cm), which is a critical component in the conversion of branch chain ketone acids to acyl-CoA derivatives via the branched-chain alpha-keto acid dehydrogenase complex (BCKDH), demonstrated that the knockout mice have a higher susceptibility to heart failure in response to pressure overload stress (87). This was due to the higher levels of BCAAs in the PP2Cm deficient mice, which reduced glucose breakdown via direct inhibition of pyruvate dehydrogenase (89).
The mechanistic target of rapamycin (mTOR) signaling pathway has been demonstrated to play an important role during heart development and growth (90, 91). Interestingly, BCAAs stimulate mTOR activation which promotes metabolic reprogramming to glycolysis from fatty acid oxidation through HIF-1α (92). In contrast, inhibition of mTOR promotes human iPSC-derived cardiomyocyte maturation and impairs zebrafish heart regeneration following injury (93, 94). mTOR is also inhibited by 5′ adenosine monophosphate-activated protein kinase (AMPK) through tuberous sclerosis complex 2 (TSC2) (95). Pharmacological activation of AMPK by metformin inhibits mTOR pathway activation following TAC injury (96). In addition, AMPK activation by AICAR promotes human iPSC-derived cardiomyocyte maturation (97). Thus, downstream pathways of BCAAs including mTOR and AMPK can regulate cardiomyocyte proliferation and regeneration.
Conversely, stimulating BCAA catabolism can be protective against heart injury and failure. BCAA catabolism can be activated by inhibition of the branched chain ketoacid dehydrogenase kinase (BCKDK), which results in BCKDH activation (87). BCKDK inhibition increased BCAA catabolism, which increased cardiac function following TAC compared to controls (98). In addition, adenoviral overexpression of PP2Cm in infarcted diabetic mice resulted in a significantly smaller scar size compared to controls (99). These studies demonstrate that enhanced BCAA catabolism can be protective against cardiac injury.
This relationship between BCAA catabolism and heart failure demonstrate that amino acid metabolism plays a role in heart disease and repair. Future studies to dissect the role of amino acids in the heart will establish their role as an important therapeutic target in cardiovascular disease.
The metabolic switch from glycolysis in neonatal mice to fatty acid oxidation in adult cardiomyocytes is accompanied by a significant increase in mitochondrial number and high levels of ROS production (16). This increase in ROS levels in the postnatal heart induces cardiomyocyte DNA damage, which contributes to cardiomyocyte cell cycle exit in the adult mammalian heart (16). Thus, elucidating the role of mitochondrial metabolites in regulating this metabolic switch is critical to identify metabolic targets to promote adult heart regeneration.
Succinate dehydrogenase (SDH), also known as mitochondrial complex II, is an important enzyme in regulating cell cycle and metabolic reprogramming in cancer because SDH plays a role in both the TCA cycle and the electron transport chain (100). Metabolic reprogramming has been recognized as a hallmark of various cancers due to the unique metabolic signature of cancer (101). In the presence of oxygen, pyruvate is converted to acetyl-CoA which enters the mitochondrial TCA cycle. However, in the absence of oxygen very limited oxidative phosphorylation takes place, instead lactate production increases aerobic glycolysis (101). Interestingly, pyruvate is mostly converted to lactate in cancer cells regardless of the oxygen levels. This metabolic switch promotes cancer cell survival and cell proliferation (100–102).
Recent studies demonstrated that reverse activity of SDH during ischemia results in succinate accumulation (103, 104). The accumulated succinate is then rapidly oxidized following reperfusion and results in a burst of ROS production via reverse activity of the mitochondrial complex I (105). These studies suggest that ROS production due to reverse activity of SDH and succinate accumulation is a hallmark of I/R injury (105). Interestingly, SDH inhibition reduces infarct size during ischemia in Langendorff-perfused mouse hearts (106). Furthermore, the SDH competitive inhibitor malonate reduces infarct size during I/R injury in pig hearts (107). These results demonstrate that SDH inhibition during I/R injury blocks the SDH-mediated succinate accumulation, thus protecting the heart against the redox insult during I/R injury. Interestingly, a recent study demonstrated that succinate accumulation in ischemia/reperfusion is not due to the reverse activity of SDH, but rather due to canonical TCA cycle activity (108). Thus, although succinate accumulation during ischemia is conserved across vertebrates, the proposed mechanism of succinate accumulation remains to be further understood.
SDH knockdown induces cell proliferation and migration in human hepatocellular carcinoma cell lines and leads to a metabolic shift to glycolysis as demonstrated by increased level of glycolytic enzymes (109). Interestingly, a recent study demonstrated that metabolic reprogramming to glycolysis promotes cardiomyocyte proliferation and heart regeneration following injury in zebrafish (15). Remarkably, SDH inhibition by malonate promotes adult cardiomyocyte proliferation, revascularization, and heart regeneration following adult myocardial infarction (110). In contrast to the cardioprotective role of malonate during I/R injury in mouse and pig hearts (105, 107); malonate did not protect against infarction post-MI but rather promoted regeneration following infarction (110). Furthermore, SDH inhibition by malonate following adult MI was accompanied by increased succinate levels as a consequence of TCA cycle inhibition, which is distinct from the cardioprotective role of malonate that prevents succinate accumulation during I/R injury (105, 110). Interestingly, metabolic profiling of the adult heart demonstrated an increase in glucose metabolism and a decrease in TCA cycle metabolism following SDH inhibition by malonate, consistent with a metabolic reprogramming from oxidative phosphorylation to glycolysis in the adult heart. These results demonstrate that SDH inhibition by malonate promotes adult heart regeneration via metabolic reprogramming (110).
Collectively, these studies demonstrate an important role for mitochondrial metabolites in regulating the cardiac metabolic state, and targeting metabolism has an important therapeutic potential to promote adult heart regeneration.
The role of the complex metabolic interactions in the heart and their potential to promote cardiac repair and regeneration are beginning to be appreciated. The shift in metabolism from glycolysis to fatty acid oxidation after birth coincides with the loss of regenerative potential in the neonatal mouse heart. The studies that are highlighted throughout this review demonstrate that manipulation of metabolic pathways is an area of immense potential for identifying new therapeutics to treat heart diseases (Table 1). Targeting these metabolic pathways can promote or inhibit regeneration depending upon the specific component that is modulated (Figure 2).
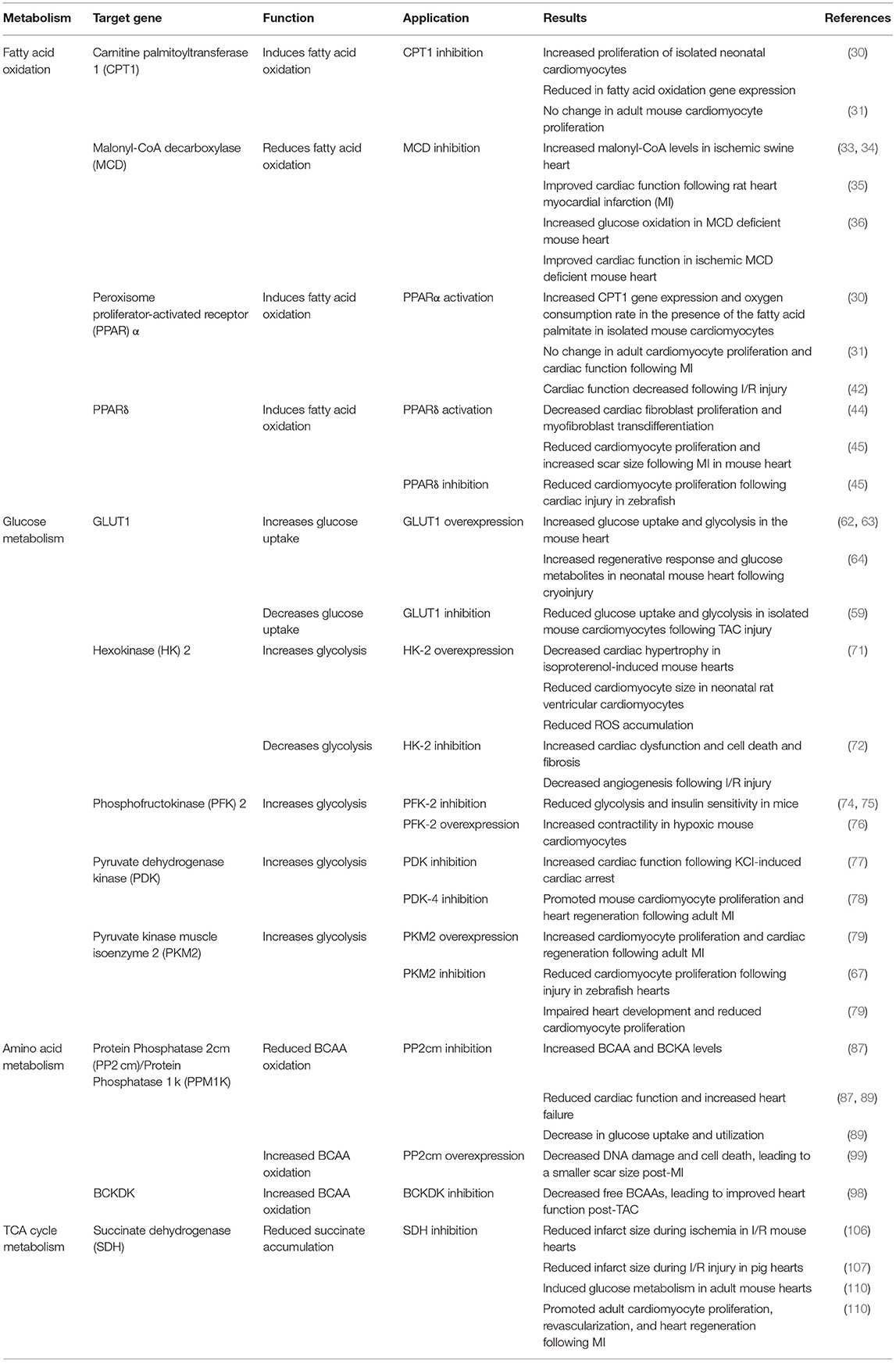
Table 1. Summary of recent studies demonstrating a central role for metabolism in heart failure and regeneration.
Manipulating metabolic components in ways that can stimulate glucose metabolism has been implicated in promoting regeneration, as this shifts the heart's metabolic landscape closer to the metabolic state of the regenerative neonatal heart. This was demonstrated with deletion of PDK4, overexpression of PP2cm, as well as SDH inhibition via malonate, which promoted regeneration by inducing glucose metabolism via modulating their respective metabolic pathways (78, 99, 110).
In contrast, increased fatty acid oxidation has been demonstrated to reduce the cardiac regenerative response following injury. Inducing fatty acid oxidation via treatment with the PPARα agonist WY-14643 results in reduced cardiac function after injury (42). Similarly, inhibition of glycolysis exacerbates cardiac injury, as demonstrated by reduced HK-2 expression (72) and PP2cm deletion (87).
The dynamic role of glycolysis and fatty acid oxidation following injury demonstrates a central role for cardiac metabolism during regeneration. Although multiple key components have already been identified that can be targeted therapeutically, these metabolic pathways play an important role in cardiac homeostasis. Thus, elucidating the mechanisms of these pathways during homeostasis, disease, and regeneration is an essential step prior to targeting these pathways for therapeutic development. For example, targeting succinate dehydrogenase post-MI promoted adult heart regeneration, yet the mechanisms by which succinate dehydrogenase inhibition promotes regeneration needs to be fully understood prior to clinical use (110). Furthermore, harnessing the potential of known pharmacological agents that have been demonstrated to target these metabolic pathways needs to be explored as candidates to induce adult heart regeneration.
Elucidating the role of cardiac metabolism in health and disease will provide us with novel avenues with significant therapeutic potential that could aid in promoting heart repair and regeneration. Advancements in this area of research will provide a better understanding of heart disease and regeneration.
JB and AM contributed to conception and design of the manuscript. JB, WP, and AM wrote the manuscript. All authors contributed to manuscript revision, read, and approved the submitted manuscript.
Funding for this project was provided by the UW School of Medicine and Public Health from the Wisconsin Partnership Program (AM), an American Heart Association Career Development Award 19CDA34660169 (AM), NIH/NCATS through CTSA award UL1TR002373 to the UW Institute for Clinical and Translational Research, a post-doctoral training award from the Stem Cell and Regenerative Medicine Center at UW-Madison (JB) and NIH/NHLBI under Ruth L. Kirschstein NRSA T32 HL007936 to the UW Cardiovascular Research Center (JB).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We apologize to any authors of studies that we might have not discussed.
1. Virani SS, Alonso A, Aparicio HJ, Benjamin EJ, Bittencourt MS, Callaway CW, et al. Heart disease and stroke statistics-2021 update: a report from the American Heart Association. Circulation. (2021) 143:e254–743. doi: 10.1161/CIR.0000000000000950
3. Hashimoto H, Olson EN, Bassel-Duby R. Therapeutic approaches for cardiac regeneration and repair. Nat Rev Cardiol. (2018) 15:585–600. doi: 10.1038/s41569-018-0036-6
4. Becker RO, Chapin S, Sherry R. Regeneration of the ventricular myocardium in amphibians. Nature. (1974) 248:145–7. doi: 10.1038/248145a0
5. Poss KD, Wilson LG, Keating MT. Heart regeneration in zebrafish. Science. (2002) 298:2188–90. doi: 10.1126/science.1077857
6. Mahmoud AI, Porrello ER, Kimura W, Olson EN, Sadek HA. Surgical models for cardiac regeneration in neonatal mice. Nat Protocols. (2014) 9:305–11. doi: 10.1038/nprot.2014.021
7. Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, et al. Transient regenerative potential of the neonatal mouse heart. Science. (2011) 331:1078–80. doi: 10.1126/science.1200708
8. Lopaschuk GD, Collins-Nakai RL, Itoi T. Developmental changes in energy substrate use by the heart. Cardiovasc Res. (1992) 26:1172–80. doi: 10.1093/cvr/26.12.1172
9. Wang Z, Ying Z, Bosy-Westphal A, Zhang J, Schautz B, Later W, et al. Specific metabolic rates of major organs and tissues across adulthood: evaluation by mechanistic model of resting energy expenditure. Am J Clin Nutr. (2010) 92:1369–77. doi: 10.3945/ajcn.2010.29885
10. Fathollahipour S, Patil PS, Leipzig ND. Oxygen regulation in development: lessons from embryogenesis towards tissue engineering. Cells Tissues Organs. (2018) 205:350–71. doi: 10.1159/000493162
11. Son G, Han J. Roles of mitochondria in neuronal development. BMB Rep. (2018) 51:549–56. doi: 10.5483/BMBRep.2018.51.11.226
12. Antico Arciuch VG, Elguero ME, Poderoso JJ, Carreras MC. Mitochondrial regulation of cell cycle and proliferation. Antioxid Redox Signal. (2012) 16:1150–80. doi: 10.1089/ars.2011.4085
13. Lepilina A, Coon AN, Kikuchi K, Holdway JE, Roberts RW, Burns CG, et al. A dynamic epicardial injury response supports progenitor cell activity during zebrafish heart regeneration. Cell. (2006) 127:607–19. doi: 10.1016/j.cell.2006.08.052
14. Jopling C, Sleep E, Raya M, Martí M, Raya A, Izpisúa Belmonte JC. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature. (2010) 464:606–9. doi: 10.1038/nature08899
15. Honkoop H, de Bakker DE, Aharonov A, Kruse F, Shakked A, Nguyen PD, et al. Single-cell analysis uncovers that metabolic reprogramming by ErbB2 signaling is essential for cardiomyocyte proliferation in the regenerating heart. Elife. (2019) 8:e50163. doi: 10.7554/eLife.50163.sa2
16. Puente BN, Kimura W, Muralidhar SA, Moon J, Amatruda JF, Phelps KL, et al. The oxygen-rich postnatal environment induces cardiomyocyte cell-cycle arrest through DNA damage response. Cell. (2014) 157:565–79. doi: 10.1016/j.cell.2014.03.032
17. Ritterhoff J, Tian R. Metabolism in cardiomyopathy: every substrate matters. Cardiovasc Res. (2017) 113:411–21. doi: 10.1093/cvr/cvx017
18. Pettinato AM, Yoo D, VanOudenhove J, Chen YS, Cohn R, Ladha FA, et al. Sarcomere function activates a p53-dependent DNA damage response that promotes polyploidization and limits in vivo cell engraftment. Cell Rep. (2021) 35:109088. doi: 10.1016/j.celrep.2021.109088
19. Tuomainen T, Tavi P. The role of cardiac energy metabolism in cardiac hypertrophy and failure. Exp Cell Res. (2017) 360:12–8. doi: 10.1016/j.yexcr.2017.03.052
20. Murashige D, Jang C, Neinast M, Edwards JJ, Cowan A, Hyman MC, et al. Comprehensive quantification of fuel use by the failing and nonfailing human heart. Science. (2020) 370:364–8. doi: 10.1126/science.abc8861
21. Deng Y, Xie M, Li Q, Xu X, Ou W, Zhang Y, et al. Targeting mitochondria-inflammation circuit by β-hydroxybutyrate mitigates HFpEF. Circ Res. (2021) 128:232–45. doi: 10.1161/CIRCRESAHA.120.317933
22. Horton JL, Davidson MT, Kurishima C, Vega RB, Powers JC, Matsuura TR, et al. The failing heart utilizes 3-hydroxybutyrate as a metabolic stress defense. JCI Insight. (2019) 4:e124079. doi: 10.1172/jci.insight.124079
23. Stanley WC, Recchia FA, Lopaschuk GD. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev. (2005) 85:1093–129. doi: 10.1152/physrev.00006.2004
24. Stanley WC, Chandler MP. Energy metabolism in the normal and failing heart: potential for therapeutic interventions. Heart Fail Rev. (2002) 7:115–30. doi: 10.1023/A:1015320423577
25. Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. (2010) 90:207–58. doi: 10.1152/physrev.00015.2009
26. Azzu V, Brand MD. The on-off switches of the mitochondrial uncoupling proteins. Trends Biochem Sci. (2010) 35:298–307. doi: 10.1016/j.tibs.2009.11.001
27. Bonnefont JP, Djouadi F, Prip-Buus C, Gobin S, Munnich A, Bastin J. Carnitine palmitoyltransferases 1 and 2: biochemical, molecular and medical aspects. Mol Aspects Med. (2004) 25:495–520. doi: 10.1016/j.mam.2004.06.004
28. Brown NF, Weis BC, Husti JE, Foster DW, McGarry JD. Mitochondrial carnitine palmitoyltransferase I isoform switching in the developing rat heart. J Biol Chem. (1995) 270:8952–7. doi: 10.1074/jbc.270.15.8952
29. Lock MC, Darby JRT, Soo JY, Brooks DA, Perumal SR, Selvanayagam JB, et al. Differential response to injury in fetal and adolescent sheep hearts in the immediate post-myocardial infarction period. Front Physiol. (2019) 10:208. doi: 10.3389/fphys.2019.00208
30. Cao T, Liccardo D, LaCanna R, Zhang X, Lu R, Finck BN, et al. Fatty acid oxidation promotes cardiomyocyte proliferation rate but does not change cardiomyocyte number in infant mice. Front Cell Dev Biol. (2019) 7:42. doi: 10.3389/fcell.2019.00042
31. Roy R, Leigh T, Gao E, Zhang X, Tian Y. Activation or inhibition of PPARα-mediated fatty acid β-oxidation does not active cardiomyocyte proliferation in normal or infarcted adult mice. bioRxiv. (2019) 667964. doi: 10.1101/667964
32. Foster DW. Malonyl-CoA: the regulator of fatty acid synthesis and oxidation. J Clin Invest. (2012) 122:1958–9. doi: 10.1172/JCI63967
33. Cuthbert KD, Dyck JR. Malonyl-CoA decarboxylase is a major regulator of myocardial fatty acid oxidation. Curr Hypertens Rep. (2005) 7:407–11. doi: 10.1007/s11906-005-0034-z
34. Dyck JR, Cheng JF, Stanley WC, Barr R, Chandler MP, Brown S, et al. Malonyl coenzyme a decarboxylase inhibition protects the ischemic heart by inhibiting fatty acid oxidation and stimulating glucose oxidation. Circ Res. (2004) 94:e78–84. doi: 10.1161/01.RES.0000129255.19569.8f
35. Wang W, Zhang L, Battiprolu PK, Fukushima A, Nguyen K, Milner K, et al. Malonyl CoA decarboxylase inhibition improves cardiac function post-myocardial infarction. JACC Basic Transl Sci. (2019) 4:385–400. doi: 10.1016/j.jacbts.2019.02.003
36. Dyck JR, Hopkins TA, Bonnet S, Michelakis ED, Young ME, Watanabe M, et al. Absence of malonyl coenzyme A decarboxylase in mice increases cardiac glucose oxidation and protects the heart from ischemic injury. Circulation. (2006) 114:1721–8. doi: 10.1161/CIRCULATIONAHA.106.642009
37. Tyagi S, Gupta P, Saini AS, Kaushal C, Sharma S. The peroxisome proliferator-activated receptor: a family of nuclear receptors role in various diseases. J Adv Pharm Technol Res. (2011) 2:236–40. doi: 10.4103/2231-4040.90879
38. Kliewer SA, Forman BM, Blumberg B, Ong ES, Borgmeyer U, Mangelsdorf DJ, et al. Differential expression and activation of a family of murine peroxisome proliferator-activated receptors. Proc Natl Acad Sci USA. (1994) 91:7355–9. doi: 10.1073/pnas.91.15.7355
39. Murphy SA, Miyamoto M, Kervadec A, Kannan S, Tampakakis E, Kambhampati S, et al. PGC1/PPAR drive cardiomyocyte maturation at single cell level via YAP1 and SF3B2. Nat Commun. (2021) 12:1648. doi: 10.1038/s41467-021-21957-z
40. Iemitsu M, Miyauchi T, Maeda S, Tanabe T, Takanashi M, Irukayama-Tomobe Y, et al. Aging-induced decrease in the PPAR-alpha level in hearts is improved by exercise training. Am J Physiol Heart Circ Physiol. (2002) 283:H1750–60. doi: 10.1152/ajpheart.01051.2001
41. Kaimoto S, Hoshino A, Ariyoshi M, Okawa Y, Tateishi S, Ono K, et al. Activation of PPAR-α in the early stage of heart failure maintained myocardial function and energetics in pressure-overload heart failure. Am J Physiol Heart Circ Physiol. (2017) 312:H305–13. doi: 10.1152/ajpheart.00553.2016
42. Dewald O, Sharma S, Adrogue J, Salazar R, Duerr GD, Crapo JD, et al. Downregulation of peroxisome proliferator-activated receptor-alpha gene expression in a mouse model of ischemic cardiomyopathy is dependent on reactive oxygen species and prevents lipotoxicity. Circulation. (2005) 112:407–15. doi: 10.1161/CIRCULATIONAHA.105.536318
43. Yue TL, Bao W, Jucker BM, Gu JL, Romanic AM, Brown PJ, et al. Activation of peroxisome proliferator-activated receptor-alpha protects the heart from ischemia/reperfusion injury. Circulation. (2003) 108:2393–9. doi: 10.1161/01.CIR.0000093187.42015.6C
44. Teunissen BE, Smeets PJ, Willemsen PH, De Windt LJ, Van der Vusse GJ, Van Bilsen M. Activation of PPARdelta inhibits cardiac fibroblast proliferation and the transdifferentiation into myofibroblasts. Cardiovasc Res. (2007) 75:519–29. doi: 10.1016/j.cardiores.2007.04.026
45. Magadum A, Ding Y, He L, Kim T, Vasudevarao MD, Long Q, et al. Live cell screening platform identifies PPARδ as a regulator of cardiomyocyte proliferation and cardiac repair. Cell Res. (2017) 27:1002–19. doi: 10.1038/cr.2017.84
46. Schulze PC, Drosatos K, Goldberg IJ. Lipid use and misuse by the heart. Circ Res. (2016) 118:1736–51. doi: 10.1161/CIRCRESAHA.116.306842
47. Neely JR, Rovetto MJ, Oram JF. Myocardial utilization of carbohydrate and lipids. Prog Cardiovasc Dis. (1972) 15:289–329. doi: 10.1016/0033-0620(72)90029-1
48. Zierler K. Whole body glucose metabolism. Am J Physiol. (1999) 276:E409–26. doi: 10.1152/ajpendo.1999.276.3.E409
49. Young LH, Russell RR, Yin R, Caplan MJ, Ren J, Bergeron R, et al. Regulation of myocardial glucose uptake and transport during ischemia and energetic stress. Am J Cardiol. (1999) 83:25H−30H. doi: 10.1016/S0002-9149(99)00253-2
50. Barger PM, Kelly DP. Fatty acid utilization in the hypertrophied and failing heart: molecular regulatory mechanisms. Am J Med Sci. (1999) 318:36–42. doi: 10.1097/00000441-199907000-00006
51. Sack MN, Kelly DP. The energy substrate switch during development of heart failure: gene regulatory mechanisms (Review). Int J Mol Med. (1998) 1:17–24. doi: 10.3892/ijmm.1.1.17
52. Dávila-Román VG, Vedala G, Herrero P, de las Fuentes L, Rogers JG, Kelly DP, et al. Altered myocardial fatty acid and glucose metabolism in idiopathic dilated cardiomyopathy. J Am Coll Cardiol. (2002) 40:271–7. doi: 10.1016/S0735-1097(02)01967-8
53. Thorens B, Mueckler M. Glucose transporters in the 21st century. Am J Physiol Endocrinol Metab. (2010) 298:E141–5. doi: 10.1152/ajpendo.00712.2009
55. Szablewski L. Glucose transporters in healthy heart and in cardiac disease. Int J Cardiol. (2017) 230:70–5. doi: 10.1016/j.ijcard.2016.12.083
56. Montessuit C, Lerch R. Regulation and dysregulation of glucose transport in cardiomyocytes. Biochim Biophys Acta. (2013) 1833:848–56. doi: 10.1016/j.bbamcr.2012.08.009
57. Shao D, Tian R. Glucose transporters in cardiac metabolism and hypertrophy. Compr Physiol. (2015) 6:331–51. doi: 10.1002/cphy.c150016
58. Sugden MC, Langdown ML, Harris RA, Holness MJ. Expression and regulation of pyruvate dehydrogenase kinase isoforms in the developing rat heart and in adulthood: role of thyroid hormone status and lipid supply. Biochem J. (2000) 352:731–8. doi: 10.1042/bj3520731
59. Pereira RO, Wende AR, Olsen C, Soto J, Rawlings T, Zhu Y, et al. GLUT1 deficiency in cardiomyocytes does not accelerate the transition from compensated hypertrophy to heart failure. J Mol Cell Cardiol. (2014) 72:95–103. doi: 10.1016/j.yjmcc.2014.02.011
60. Sivitz WI, Lund DD, Yorek B, Grover-McKay M, Schmid PG. Pretranslational regulation of two cardiac glucose transporters in rats exposed to hypobaric hypoxia. Am J Physiol. (1992) 263:E562–9. doi: 10.1152/ajpendo.1992.263.3.E562
61. Huang Y, Lei L, Liu D, Jovin I, Russell R, Johnson RS, et al. Normal glucose uptake in the brain and heart requires an endothelial cell-specific HIF-1α-dependent function. Proc Natl Acad Sci USA. (2012) 109:17478–83. doi: 10.1073/pnas.1209281109
62. Liao R, Jain M, Cui L, D'Agostino J, Aiello F, Luptak I, et al. Cardiac-specific overexpression of GLUT1 prevents the development of heart failure attributable to pressure overload in mice. Circulation. (2002) 106:2125–31. doi: 10.1161/01.CIR.0000034049.61181.F3
63. Pereira RO, Wende AR, Olsen C, Soto J, Rawlings T, Zhu Y, et al. Inducible overexpression of GLUT1 prevents mitochondrial dysfunction and attenuates structural remodeling in pressure overload but does not prevent left ventricular dysfunction. J Am Heart Assoc. (2013) 2:e000301. doi: 10.1161/JAHA.113.000301
64. Fajardo VM, Feng I, Chen BY, Perez-Ramirez CA, Shi B, Clark P, et al. GLUT1 overexpression enhances glucose metabolism and promotes neonatal heart regeneration. Sci Rep. (2021) 11:8669. doi: 10.1038/s41598-021-88159-x
65. Tran DH, Wang ZV. Glucose metabolism in cardiac hypertrophy and heart failure. J Am Heart Assoc. (2019) 8:e012673. doi: 10.1161/JAHA.119.012673
66. Zhang S, Hulver MW, McMillan RP, Cline MA, Gilbert ER. The pivotal role of pyruvate dehydrogenase kinases in metabolic flexibility. Nutr Metab (Lond). (2014) 11:10. doi: 10.1186/1743-7075-11-10
67. Fukuda R, Marín-Juez R, El-Sammak H, Beisaw A, Ramadass R, Kuenne C, et al. Stimulation of glycolysis promotes cardiomyocyte proliferation after injury in adult zebrafish. EMBO Rep. (2020) 21:e49752. doi: 10.15252/embr.201949752
68. Wilson JE. Isozymes of mammalian hexokinase: structure, subcellular localization and metabolic function. J Exp Biol. (2003) 206:2049–57. doi: 10.1242/jeb.00241
69. Southworth R, Davey KA, Warley A, Garlick PB. A reevaluation of the roles of hexokinase I and II in the heart. Am J Physiol Heart Circ Physiol. (2007) 292:H378–86. doi: 10.1152/ajpheart.00664.2006
70. Smeele KM, ter Horst LH, Koeman A, Heikkinen S, Laakso M, Weber NC, et al. The effect of standard chow and reduced hexokinase II on growth, cardiac and skeletal muscle hexokinase and low-flow cardiac ischaemia-reperfusion injury. Lab Anim. (2011) 45:160–6. doi: 10.1258/la.2011.010096
71. McCommis KS, Douglas DL, Krenz M, Baines CP. Cardiac-specific hexokinase 2 overexpression attenuates hypertrophy by increasing pentose phosphate pathway flux. J Am Heart Assoc. (2013) 2:e000355. doi: 10.1161/JAHA.113.000355
72. Wu R, Smeele KM, Wyatt E, Ichikawa Y, Eerbeek O, Sun L, et al. Reduction in hexokinase II levels results in decreased cardiac function and altered remodeling after ischemia/reperfusion injury. Circ Res. (2011) 108:60–9. doi: 10.1161/CIRCRESAHA.110.223115
73. Bockus LB, Matsuzaki S, Vadvalkar SS, Young ZT, Giorgione JR, Newhardt MF, et al. Cardiac insulin signaling regulates glycolysis through phosphofructokinase 2 content and activity. J Am Heart Assoc. (2017) 6:e007159. doi: 10.1161/JAHA.117.007159
74. Donthi RV, Ye G, Wu C, McClain DA, Lange AJ, Epstein PN. Cardiac expression of kinase-deficient 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase inhibits glycolysis, promotes hypertrophy, impairs myocyte function, and reduces insulin sensitivity. J Biol Chem. (2004) 279:48085–90. doi: 10.1074/jbc.M405510200
75. Wang J, Xu J, Wang Q, Brainard RE, Watson LJ, Jones SP, et al. Reduced cardiac fructose 2,6 bisphosphate increases hypertrophy and decreases glycolysis following aortic constriction. PLoS ONE. (2013) 8:e53951. doi: 10.1371/journal.pone.0053951
76. Wang Q, Donthi RV, Wang J, Lange AJ, Watson LJ, Jones SP, et al. Cardiac phosphatase-deficient 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase increases glycolysis, hypertrophy, and myocyte resistance to hypoxia. Am J Physiol Heart Circ Physiol. (2008) 294:H2889–97. doi: 10.1152/ajpheart.91501.2007
77. Piao L, Fang YH, Kubler MM, Donnino MW, Sharp WW. Enhanced pyruvate dehydrogenase activity improves cardiac outcomes in a murine model of cardiac arrest. PLoS ONE. (2017) 12:e0185046. doi: 10.1371/journal.pone.0185046
78. Cardoso AC, Lam NT, Savla JJ, Nakada Y, Pereira AHM, Elnwasany A, et al. Mitochondrial substrate utilization regulates cardiomyocyte cell cycle progression. Nat Metab. (2020) 2:167–78. doi: 10.1038/s42255-020-0169-x
79. Magadum A, Singh N, Kurian AA, Munir I, Mehmood T, Brown K, et al. Pkm2 regulates cardiomyocyte cell cycle and promotes cardiac regeneration. Circulation. (2020) 141:1249–65. doi: 10.1161/CIRCULATIONAHA.119.043067
80. Kadowaki M, Kanazawa T. Amino acids as regulators of proteolysis. J Nutr. (2003) 133(Suppl. 1):2052S-6S. doi: 10.1093/jn/133.6.2052S
81. Wu G. Amino acids: metabolism, functions, and nutrition. Amino Acids. (2009) 37:1–17. doi: 10.1007/s00726-009-0269-0
82. Kimball SR, Shantz LM, Horetsky RL, Jefferson LS. Leucine regulates translation of specific mRNAs in L6 myoblasts through mTOR-mediated changes in availability of eIF4E and phosphorylation of ribosomal protein S6. J Biol Chem. (1999) 274:11647–52. doi: 10.1074/jbc.274.17.11647
83. Norton LE, Layman DK. Leucine regulates translation initiation of protein synthesis in skeletal muscle after exercise. J Nutr. (2006) 136:533S-7S. doi: 10.1093/jn/136.2.533S
84. Sanchez Canedo C, Demeulder B, Ginion A, Bayascas JR, Balligand JL, Alessi DR, et al. Activation of the cardiac mTOR/p70(S6K) pathway by leucine requires PDK1 and correlates with PRAS40 phosphorylation. Am J Physiol Endocrinol Metab. (2010) 298:E761–9. doi: 10.1152/ajpendo.00421.2009
85. Talman V, Teppo J, Poho P, Movahedi P, Vaikkinen A, Karhu ST, et al. Molecular atlas of postnatal mouse heart development. J Am Heart Assoc. (2018) 7:e010378. doi: 10.1161/JAHA.118.010378
86. Aquilani R, La Rovere MT, Corbellini D, Pasini E, Verri M, Barbieri A, et al. Plasma amino acid abnormalities in chronic heart failure. mechanisms, potential risks and targets in human myocardium metabolism. Nutrients. (2017) 9:1251. doi: 10.3390/nu9111251
87. Sun H, Olson KC, Gao C, Prosdocimo DA, Zhou M, Wang Z, et al. Catabolic defect of branched-chain amino acids promotes heart failure. Circulation. (2016) 133:2038–49. doi: 10.1161/CIRCULATIONAHA.115.020226
88. Nie C, He T, Zhang W, Zhang G, Ma X. Branched chain amino acids: beyond nutrition metabolism. Int J Mol Sci. (2018) 19:954. doi: 10.3390/ijms19040954
89. Li T, Zhang Z, Kolwicz SC Jr, Abell L, Roe ND, Kim M, et al. Defective branched-chain amino acid catabolism disrupts glucose metabolism and sensitizes the heart to ischemia-reperfusion injury. Cell Metab. (2017) 25:374–85. doi: 10.1016/j.cmet.2016.11.005
90. Mazelin L, Panthu B, Nicot AS, Belotti E, Tintignac L, Teixeira G, et al. mTOR inactivation in myocardium from infant mice rapidly leads to dilated cardiomyopathy due to translation defects and p53/JNK-mediated apoptosis. J Mol Cell Cardiol. (2016) 97:213–25. doi: 10.1016/j.yjmcc.2016.04.011
91. Zhang P, Shan T, Liang X, Deng C, Kuang S. Mammalian target of rapamycin is essential for cardiomyocyte survival and heart development in mice. Biochem Biophys Res Commun. (2014) 452:53–9. doi: 10.1016/j.bbrc.2014.08.046
92. Hu J, Nie Y, Chen S, Xie C, Fan Q, Wang Z, et al. Leucine reduces reactive oxygen species levels via an energy metabolism switch by activation of the mTOR-HIF-1α pathway in porcine intestinal epithelial cells. Int J Biochem Cell Biol. (2017) 89:42–56. doi: 10.1016/j.biocel.2017.05.026
93. Chávez MN, Morales RA, López-Crisosto C, Roa JC, Allende ML, Lavandero S. Autophagy activation in zebrafish heart regeneration. Sci Rep. (2020) 10:2191. doi: 10.1038/s41598-020-59106-z
94. Garbern JC, Helman A, Sereda R, Sarikhani M, Ahmed A, Escalante GO, et al. Inhibition of mTOR signaling enhances maturation of cardiomyocytes derived from human-induced pluripotent stem cells via p53-induced quiescence. Circulation. (2020) 141:285–300. doi: 10.1161/CIRCULATIONAHA.119.044205
95. Agarwal S, Bell CM, Rothbart SB, Moran RG. AMP-activated protein kinase (AMPK) control of mTORC1 Is p53- and TSC2-independent in pemetrexed-treated carcinoma cells. J Biol Chem. (2015) 290:27473–86. doi: 10.1074/jbc.M115.665133
96. Xu X, Lu Z, Fassett J, Zhang P, Hu X, Liu X, et al. Metformin protects against systolic overload-induced heart failure independent of AMP-activated protein kinase α2. Hypertension. (2014) 63:723–8. doi: 10.1161/HYPERTENSIONAHA.113.02619
97. Ye L, Zhang X, Zhou Q, Tan B, Xu H, Yi Q, et al. Activation of AMPK promotes maturation of cardiomyocytes derived from human induced pluripotent stem cells. Front Cell Dev Biol. (2021) 9:644667. doi: 10.3389/fcell.2021.644667
98. Uddin GM, Zhang L, Shah S, Fukushima A, Wagg CS, Gopal K, et al. Impaired branched chain amino acid oxidation contributes to cardiac insulin resistance in heart failure. Cardiovasc Diabetol. (2019) 18:86. doi: 10.1186/s12933-019-0892-3
99. Lian K, Guo X, Wang Q, Liu Y, Wang RT, Gao C, et al. PP2Cm overexpression alleviates MI/R injury mediated by a BCAA catabolism defect and oxidative stress in diabetic mice. Eur J Pharmacol. (2020) 866:172796. doi: 10.1016/j.ejphar.2019.172796
100. Martínez-Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun. (2020) 11:102. doi: 10.1038/s41467-019-13668-3
101. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. (2009) 324:1029–33. doi: 10.1126/science.1160809
102. Faubert B, Solmonson A, DeBerardinis RJ. Metabolic reprogramming and cancer progression. Science. (2020) 368:eaaw5473. doi: 10.1126/science.aaw5473
103. Scialò F, Fernández-Ayala DJ, Sanz A. Role of mitochondrial reverse electron transport in ROS signaling: potential roles in health and disease. Front Physiol. (2017) 8:428. doi: 10.3389/fphys.2017.00428
104. Scialò F, Sriram A, Fernández-Ayala D, Gubina N, Lõhmus M, Nelson G, et al. Mitochondrial ROS produced via reverse electron transport extend animal lifespan. Cell Metab. (2016) 23:725–34. doi: 10.1016/j.cmet.2016.03.009
105. Chouchani ET, Pell VR, Gaude E, Aksentijevic D, Sundier SY, Robb EL, et al. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature. (2014) 515:431–5. doi: 10.1038/nature13909
106. Valls-Lacalle L, Barba I, Miro-Casas E, Alburquerque-Bejar JJ, Ruiz-Meana M, Fuertes-Agudo M, et al. Succinate dehydrogenase inhibition with malonate during reperfusion reduces infarct size by preventing mitochondrial permeability transition. Cardiovasc Res. (2016) 109:374–84. doi: 10.1093/cvr/cvv279
107. Valls-Lacalle L, Barba I, Miro-Casas E, Ruiz-Meana M, Rodriguez-Sinovas A, Garcia-Dorado D. Selective inhibition of succinate dehydrogenase in reperfused myocardium with intracoronary malonate reduces infarct size. Sci Rep. (2018) 8:2442. doi: 10.1038/s41598-018-20866-4
108. Milliken AS, Kulkarni CA, Brookes PS. Acid enhancement of ROS generation by complex-I reverse electron transport is balanced by acid inhibition of complex-II: relevance for tissue reperfusion injury. Redox Biol. (2020) 37:101733. doi: 10.1016/j.redox.2020.101733
109. Tseng PL, Wu WH, Hu TH, Chen CW, Cheng HC, Li CF, et al. Decreased succinate dehydrogenase B in human hepatocellular carcinoma accelerates tumor malignancy by inducing the Warburg effect. Sci Rep. (2018) 8:3081. doi: 10.1038/s41598-018-21361-6
Keywords: heart regeneration, development, metabolism, heart failure, cell cycle
Citation: Bae J, Paltzer WG and Mahmoud AI (2021) The Role of Metabolism in Heart Failure and Regeneration. Front. Cardiovasc. Med. 8:702920. doi: 10.3389/fcvm.2021.702920
Received: 30 April 2021; Accepted: 23 June 2021;
Published: 16 July 2021.
Edited by:
Monika Gladka, Academic Medical Center, NetherlandsReviewed by:
Yuka Morikawa, Texas Heart Institute, United StatesCopyright © 2021 Bae, Paltzer and Mahmoud. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ahmed I. Mahmoud, YWltYWhtb3VkQHdpc2MuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.