 Junyi Li
Junyi Li Li Zhou2†
Li Zhou2† Xinping Ouyang
Xinping Ouyang Pingping He
Pingping He
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Cardiovasc. Med., 09 September 2021
Sec. Atherosclerosis and Vascular Medicine
Volume 8 - 2021 | https://doi.org/10.3389/fcvm.2021.701279
This article is part of the Research TopicLipids and Inflammation in Health and DiseaseView all 26 articles
Transcription factor-7-like-2 (TCF7L2), a vital member of the T-cell factor/lymphoid enhancer factor (TCF/LEF) family, plays an important role in normal human physiological and pathological processes. TCF7L2 exhibits multiple anti-atherosclerotic effects through the activation of specific molecular mechanisms, including regulation of metabolic homeostasis, macrophage polarization, and neointimal hyperplasia. A single-nucleotide substitution of TCF7L2, rs7903146, is a genetic high-risk factor for type 2 diabetes and indicates susceptibility to cardiovascular disease as a link between metabolic disorders and atherosclerosis. In this review, we summarize the anti-atherosclerosis effect and novel mechanisms underlying the function of TCF7L2 to elucidate its potential as an anti-atherosclerosis biomarker and provide a novel therapeutic target for cardiovascular diseases.
Atherosclerosis precedes and forecasts the pathological process of cardiovascular disease (CVD), the leading cause of mortality worldwide (1, 2). Atherosclerosis is characterized by complex pathological progression involving endothelial damage, local chronic inflammation, and metabolic disorders (3). The onset of atherosclerosis in endothelium stimulates chemokine releases and recruits circulating monocytes, which then differentiate into M1 macrophages in the local inflammatory environment (4). Additionally, subendothelial M1 macrophages not only secrete proinflammatory cytokines but also transform into foam cells through excessive lipid uptake. However, foam cells are prone to apoptosis or necrosis owing to cytotoxicity, and the debris and lipids from foam cells inflate the necrotic core of atherosclerotic plaques. Vascular smooth muscle cell (VSMC) migration is another pathologic feature of atherogenesis, which thickens the vascular walls and narrows the vascular lumen. Even worse, once these atherosclerotic plaques rupture and the necrotic core breaks through into the vascular lumen, patients will suffer from acute CVD events, such as stroke or myocardial infarction. In addition to the local microenvironment, atherogenesis is affected by multiple systemic factors such as obesity, dyslipidemia, and insulin resistance (IR). Recently, metabolic syndrome, the simultaneous presence of various risk factors related to metabolism, has been identified as a prominent risk factor for atherosclerosis (5).
TCF7L2 is well characterized as a member of the T-cell factor/lymphoid enhancer factor (TCF/LEF) family, whose remaining members are LEF1, TCF7, and TCF7L1 in humans. It was also previously called T-cell factor 4 (TCF4), not to be mistaken for transcription factor 4 (6). Korinek et al. (7) discovered TCF7L2 via the hybridization screening of TCF7 cDNA for the first time and described TCF7L2 as an important transcription factor of Wnt signaling. During embryogenesis, TCF7L2 promotes embryonic organ development as a key player in Wnt signaling (7, 8). In adults, TCF7L2 has attracted widespread attention because of its significant genetic correlation with the high risk of type 2 diabetes (T2D), which alters the function and survival of pancreatic β cells (9–13). Currently, TCF7L2 has drawn increasing academic attention because it has been reported to be associated with inflammation, metabolism, and atherosclerosis (14, 15). Studies have shown that silencing TCF7L2 leads to insufficient insulin secretion, impaired adipogenic differentiation, blood-vessel dysplasia, and lipid accumulation (16–18). In contrast, overexpression of TCF7L2 promotes macrophage polarization (M2) and inhibits neointimal hyperplasia (19, 20). This evidence indicates the anti-atherosclerotic effects of TCF7L2. With an in-depth study of genomics, single-nucleotide polymorphisms (SNPs) have been found to be related to the genetic susceptibility to atherosclerosis and the distribution of high-risk populations; of these, TC7FL2 rs7903146 is one of the most notable SNPs (21–23). These findings indicate that TCF7L2 plays a vital role in anti-atherosclerosis and can be considered as a potential biomarker for the treatment of CVD. Therefore, in this review, we summarize the structure, function, and anti-atherosclerosis effects of TCF7L2 in order to provide insight for the development of an alternative treatment strategy for CVD.
In humans, TCF7L2 is located on chromosome 10q25.2-q25.3; it spans over 217,432 bp of DNA and consists of 17 exons. The full-length TCF7L2 protein contains 596 amino-acid residues in a certain subsequence of the N terminal, β catenin-binding domain, Groucho-binding sequence, HMG box-DNA-binding domain (HGM-DBD), cysteine clamp (C clamp), and C terminal (24, 25). With the help of HGM-DBD, TCF7L2 can recognize specific DNA subsequences (5′-xCTTTGATx-3′) in the double-helix dimple and trigger transcription factor activity (17, 26). Until now, the C clamp has been considered to assist the binding of HGM-DBD with certain DNA sequences, although the C clamp contains an alternative DNA-binding domain (5′-xTGCCGCx-3′) without transcription regulatory activity (27). Remarkably, TCF7L2 exerts dual transcription regulatory effects on target genes influenced by the transcriptional co-activator β-catenin or transcriptional co-repressor transducin-like enhancer of split (TLE)/Groucho (28–30). With Wnt signaling stimulation, increased amounts of β-catenin are imported into the nucleus, where they subsequently assemble into the β-catenin/TCF7L2 complex. In addition, β-catenin functions as a scaffold to assist the binding of the β-catenin/TCF7L2 complex to the promoter of target genes and thus enhance promoter activity. In the absence of Wnt/β-catenin signaling, the co-repressor TLEs preferentially occupy TCF7L2 by the glutamine-rich (Q) domain and recruit histone methyltransferases or histone deacetylases to silence downstream genes (31, 32). Taken together, TCF7L2 contains two DNA-binding domains (HGM-DBD and C clamp), but only HGM-DBD can activate transcription. Furthermore, TCF7L2 is subject to dual regulation by the transcriptional co-activator β-catenin or transcriptional co-repressor TLE/Groucho.
Wnt signaling plays a protective role in the development of atherosclerotic CVDs. Many studies have shown that Wnt signaling prevents the development of atherosclerosis via a series of processes, including glucolipid metabolism, macrophage polarization, and neointimal hyperplasia (33–36). Upon activation of canonical Wnt signaling, accumulated β-catenin binds to TCF7L2 in the nucleus and regulates the expression of downstream genes. This signaling pathway is widely present in pancreatic islets and adipocytes, where it balances glucose and lipid metabolism. TCF7L2 is the terminal executor of the Wnt signaling pathway, which directly acts on the promoters of downstream genes. Insulin and glucagon exert opposite effects on blood glucose levels; glucagon-like peptide 1 (GLP-1) promotes insulin secretion, while inhibiting glucagon secretion in a blood glucose concentration-dependent manner. Yi et al. (37) found that the promoters of both GLP-1 and glucagon contain TCF7L2-binding sites. However, TCF7L2 upregulates GLP-1 expression in gut endocrine cells but downregulates glucagon expression in pancreatic α cells, exerting powerful effects on insulin synthesis and balance of blood glucose levels (37). Additionally, macrophages have been proven to be a source and effector of Wnt signaling, and Wnt signaling greatly affects macrophage phenotypes. Wnt signaling can induce myeloblasts to differentiate into monocytes in the bone marrow (38) and can promote macrophage polarization into M2 phenotypes in the peripheral residences, with a pattern of decreasing expression of TNF-α and IL-6 and increasing expression of the M2 marker CD206 (39–41). Taken together, the Wnt signaling pathway has been proven to balance blood glucose levels and alleviate local inflammation, which is defined as an anti-atherosclerosis action. Considering TCF7L2 to be a determinant element of Wnt signaling, we will further explore the role of TCF7L2 in metabolism, inflammation, and atherosclerosis.
With the advances in genome sequencing, the identification of gene polymorphisms in populations has been shown to be important in disease risk prediction (42). SNPs comprise a type of gene polymorphisms and specifically refer to single-nucleotide substitutions of one base for another that have more than 1% incidence in the general population (43). Many gene polymorphisms have been confirmed to be involved in the development of atherosclerosis, such as rs7903146 in TCF7L2.
SNPs can be classified into two categories according to their distribution on DNA: linked SNPs and causative SNPs (44, 45). Linked SNPs do not reside within genes and do not affect protein production or function. Causative SNPs are located within the coding region and regulatory subsequence and can thus influence gene transcription or translation. Among causative SNPs, rs7903146, located in exon 4 of TCF7L2, is well known for conferring a genetic predisposition to T2D (Figure 1) (9, 46). rs7903146 influences the activity of the TCF7L2 promoter and alters the alternative splicing of exon 4 (47–50). The T-allele genotype of TCF7L2 shows a higher degree of open chromatin, and the promoter is accessible to transcription factors. Additionally, rs7903146 is regarded as a cis-regulatory variation wherein the T-allele genotype exhibits robust enhancer activity to promote TCF7L2 transcription (49, 50). Conversely, TCF7L2 protein levels are decreased in T-allele carriers, in contrast to elevated TCF7L2 mRNA levels (11, 51). A possible hypothesis for this inconsistency is that rs7903146 reduces exon 4 cutting from TCF7L2 transcripts (47, 48). Notably, this TCF7L2 transcript containing exon 4 exhibits high mRNA but gets rarely encoded into mature protein, which caused the discrepancy in transcription and translation. In addition, the expression of this TCF7L2 splice variant increases endoplasmic reticulum stress, triggers the ER-associated degradation pathway to degrade, and ultimately induces irreversible apoptosis (48, 52, 53). Thus, rs7903146 in TCF7L2 not only affects the expression of TCF7L2 but also causes cell dysfunction.
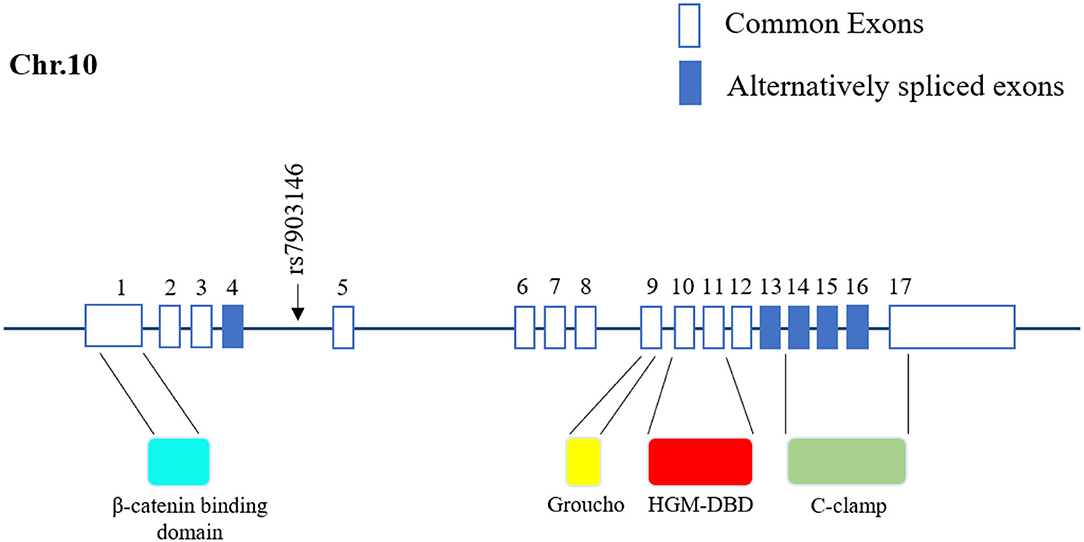
Figure 1. Location of rs7903146 in human TCF7L2. Representative 17 exons and location of rs7903146 on human TCF7L2. Alternatively spliced exons are colored, and the black arrow represents alternative transcription start sites. rs7903146 is located in exon 4 of TCF7L2. TCF7L2 comprises several domains, including the β catenin-binding domain, Groucho-binding sequence, HMG box-DNA-binding domain (HGM-DBD), and cysteine clamp (C-clamp).
Atherosclerosis is well documented to be the primary pathological basis of CVD and results from subendothelial lipid accumulation and local chronic inflammation. rs7903146 induces postprandial dyslipidemia, causing high triglyceride (TG), high low-density lipoprotein (LDL)-C, and low high-density lipoprotein (HDL)-C levels (54, 55). Some evidence implies that Apo-B is overproduced, but ApoA-I is lacking in T-allele carriers with a high-fat diet (54). ApoA-I is well documented to be a beneficial apolipoprotein in the cardiovascular system (56, 57); ApoA-I accepts intracellular cholesterol efflux from the membrane transporter ABCA1, assembles it into HDL, and transports the excess cholesterol to the liver for metabolism. Deficiency of ApoA-I reduces the feedback regulation of the cardiovascular system in response to dyslipidemia and increases subendothelial lipid accumulation (21, 58). Additionally, TCF7L2 rs7903146 aggravates pancreatic β-cell dysfunction and IR and thus presents as a metabolic correlation in CVD and T2D. Islet morphology analysis showed a 20% decrease in β-cell mass and a 30% increase in α-cell mass within islets from T/T genotype carriers (59). As for islet function, T/T-allele homozygotes exhibited 3.2 mg/dl higher baseline fasting glucose levels than C/C allele homozygotes. In addition, TCF7L2 rs7903146 reduces the efficiency of glipizide and metformin in pre-diabetic or newly diagnosed T2D subjects (60, 61). As previously mentioned, TCF7L2 can promote GLP-1 expression and sensitize pancreatic β-cells to blood glucose, but TCF7L2 rs7903146 causes a decrease in protein levels. These underlying mechanisms are consistent with the function and pathological changes in the islets of T/T allele homozygotes.
In summary, TCF7L2 rs7903146 has a great impact on metabolic balance, linking dyslipidemia and hyperglycemia, and results in greater risk of CVD in T-allele carriers, especially in T2D patients (21). Decades ago, Bielinski et al. (62) found that rs7903146 slightly increased the CVD risk in patients with early-onset diabetes, and they attributed this risk to diabetes without further research. However, this explanation was not all-inclusive, because atherosclerosis was regarded as a multi-cause disease. On the one hand, TCF7L2 rs7903146 induces the development of T2D by damaging pancreatic β-cell function and stimulating IR, which causes the vascular endothelium to be more accessible to atherosclerosis (18, 63). On the other hand, rs7903146 can lead to dyslipidemia and arterial intimal hyperplasia, both of which causes irreversible damage to the cardiovascular system (54, 64). Above all, TCF7L2 rs7903146 appears to have an intricate relationship with atherosclerosis. Although T2D is an indispensable factor, dyslipidemia and other factors are also involved in this relationship. Therefore, TCF7L2 rs7903146 can be regarded as a genetic risk factor for CVD, which could be useful to guide clinical treatment.
As mentioned previously, TCF7L2 rs7903146 is a genetic risk factor for CVD, which cause a reduction in TCF7L2 protein. However, TCF7L2 is a functional transcription factor with pleiotropic anti-atherosclerotic effects on glucose and lipid metabolism. Metabolic syndrome comprises a complex group of pathological circumstances associated with metabolic and proinflammatory states, which play an important role in the atherosclerotic process by gathering atherogenic risk factors (Figure 2) (65). It is noteworthy that TCF7L2 is widely distributed in pancreatic islets, adipose tissue, and the liver and regulates their function and metabolism.
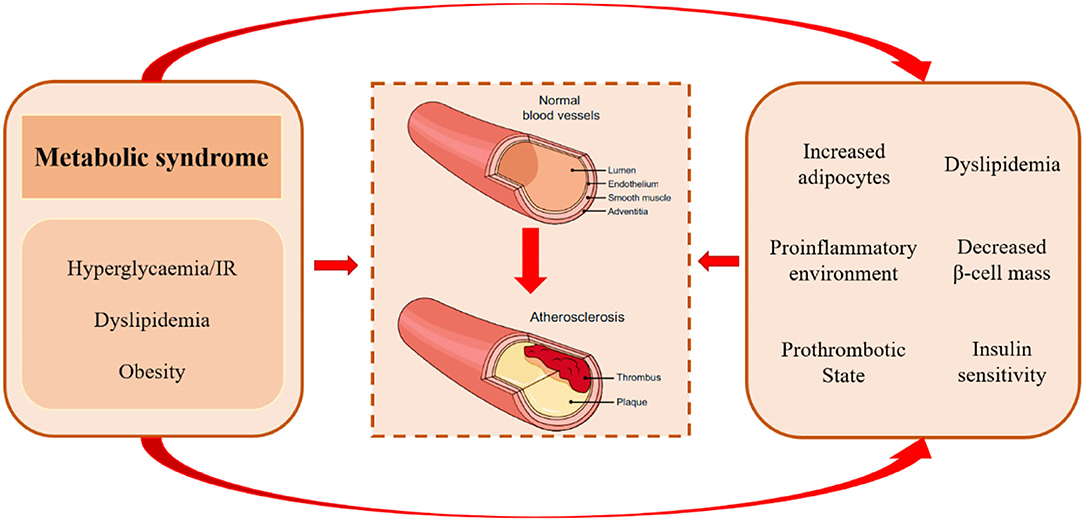
Figure 2. Pathological mechanisms of atherosclerosis in metabolic syndrome. Metabolic syndrome comprises a complex group of metabolic disorders characterized by hyperglycemia/insulin resistance, dyslipidemia, and obesity. It may accelerate atherogenic progression via increased adipocytes, proinflammatory environment, prothrombotic state, excess concentration of free fatty acids, unfavorable lipidomics, and decreased insulin levels and insulin sensitivity.
A large sample investigation involving 584 participants revealed that compared to non-T2D participants, T2D patients suffer from more severe carotid atherosclerotic stenosis, appearing as larger lipid necrosis cores with increasing calcification (66). T2D patients are extremely susceptible to CVD, which has become the most common cause of death in T2D (67). Similar to other metabolic diseases, T2D shares several pathological factors with atherosclerosis. Insulin insufficiency, the chief culprit in T2D, also causes damage to vessel walls and increases inflammasome activity (68–70). All these pathologies propel the progression of atherosclerosis. TCF7L2 mutations or deficiency have been shown to impair both pancreatic β-cell and insulin function in in vivo and in vitro experiments (18, 71, 72).
As the exclusive source of insulin, β cells are vital for maintaining metabolic balance and organ functions. On the one hand, TCF7L2 is positively related to β-cell mass (16, 59). As quiescent cells, β cells have a long-life span with low proliferation ability, but their regeneration can be affected by tissue damage and increasing demands (73–75). In these situations, TCF7L2 is required for β-cell renewal and regeneration through JAK2/STAT3/Ngn3 signaling. In addition, TCF7L2 directly reduces mitochondrial permeability and inhibits β-cell apoptosis via the GSK-3β/p53-dependent pathway (76–78). On the other hand, TCF7L2 affects the most important β-cell function, insulin secretion, from two aspects: proinsulin maturation and Ca2+ voltage-gated channel (CAV) activity. To enhance insulin synthesis, TCF7L2 significantly promotes the expression of the proinsulin genes Ins1 and Ins2. Subsequently, TCF7L2 can upregulate PSCK1 and PSCK2 to cleave proinsulin into mature insulin and the C peptide (79, 80). Insulin secretion relies on islet β-cell exocytosis, which is controlled by the L-type Ca2+ channel CAV1.2. Upon membrane depolarization, Ca2+ influx through CAV1.2 triggers β-cell exocytosis (81–83). Ye et al. (83) found that TCF7L2 silencing reduced the expression of α2δ-1, an auxiliary subunit of CAV1.2, and alleviated the Ca2+ current, causing a voltage-insensitive response to high-glucose/depolarization-evoked stimulation, accompanied by insufficient insulin secretion. In addition, TCF7L2 can stimulate the PI3K/AKT pathway to promote insulin sensitivity and secretion, because TCF7L2 can bind to conserved TCAAAG motifs in the promoter of PIK3R1 to upregulate PI3K (84). Li et al. found that TCF7L2 protein expression in the adipocytes, liver, and skeletal muscle is positively correlated with changes in homeostasis model assessment (HOMA)-IR (85), which indicates that TCF7L2 deficiency may induce IR. A recent study found that selective deletion of TCF7L2 in adipocytes leads to IR (63). In conclusion, accumulating evidence confirms that TCF7L2 plays a key role in metabolism and is responsible for maintaining β-cell function and insulin sensitivity. Under TCF7L2 dysfunction, T2D patients suffer more severe insulin deficiency and glucose metabolism disorders with a high risk of CVD (21, 36, 86).
Adipose tissue is the most abundant fat storehouse and exerts a chronic and profound influence on lipid metabolism and blood lipids. Blood lipid homeostasis is jointly maintained by adipose tissue and the cardiovascular system and is mutually beneficial (87, 88). A heat map of gene expression and gene set enrichment analysis showed alterations in several signaling pathways related to adipogenesis and metabolism in TCF7L2−/− mice (63). Wnt signaling is well-documented to inhibit adipogenesis, in which the formation of the β-catenin/TCF7L2 complex is indispensable (89, 90). Inducible deletion of TCF7L2 promotes adipose differentiation and subcutaneous fat accumulation in TCF7L2F/F mice (63).
Atherogenic dyslipidemia is characterized by high TG, high fatty free acid (FFA), high LDL, and low HDL levels (91–94); particularly, high serum TG levels are very dangerous signals for myocardial infarction (95). TGs, which are small particles, can easily enter the arterial intima and induce subendothelial lipid accumulation and local inflammation (96). However, TCF7L2 expression can decrease serum TG levels with the help of lipoprotein lipase (LPL) and triglyceride hydrolases (TGH), both of which can hydrolyze TGs (22, 63). LPL is mainly anchored in the vascular endothelium and is sensitive to serum TG. Our previous study showed that TCF7L2 can bind to the promoter of LPL and promote LPL expression to decrease serum TG levels (97, 98). Another study showed using RNA seq analysis that the expression of TGH was decreased in TCF7L2F/F mice, but the underlying mechanism remains unknown. Additionally, FFAs play a major role in inducing the endothelial prothrombotic state (99). A recent study showed that serum FFA concentrations increased dramatically after knocking out TCF7L2 in mice (72). In conclusion, TCF7L2 is a crucial regulatory element in metabolic balance. On the one hand, TCF7L2 regulates adipose differentiation via Wnt/β-catenin signaling and prevents adipocyte hypertrophy; on the other hand, TCF7L2 upregulates serum TG and FFA levels (Figure 3).
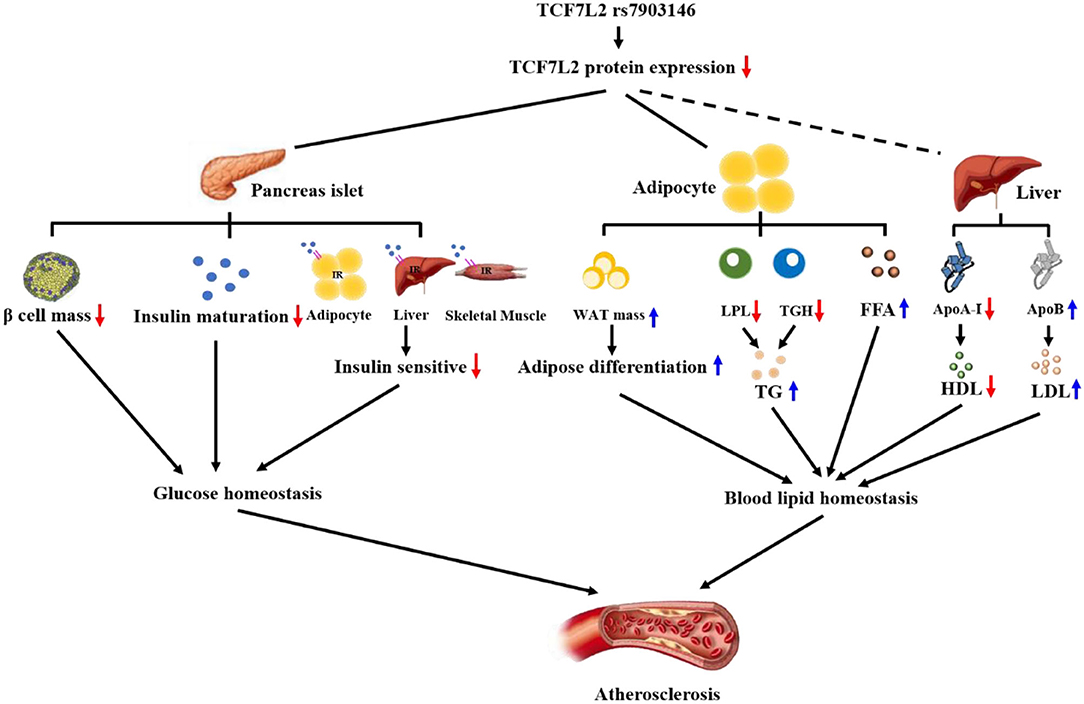
Figure 3. TCF7L2 rs7903146 is a link between metabolism disorders and atherosclerosis. TCF7L2 rs7903146 inhibits the expression of TCF7L2, thereby influencing glucose and blood lipid homeostasis. On the one hand, TCF7L2 rs7903146 induces hyperglycemia and insulin resistance by decreasing β-cell mass, proinsulin gene expression, Ca2+ voltage-gated channel activity, and insulin sensitivity of cells. On the other hand, TCF7L2 rs7903146 disrupts blood lipid homeostasis by increasing white adipocyte tissue (WAT) mass and harmful lipid components and decreasing HDL levels.
In addition to metabolic disorders, inflammatory infiltrates and neointimal hyperplasia aggravate the progression of atherosclerosis. Previously, we have illustrated TCF7L2 as a regulatory element in metabolism, but recent research has revealed other anti-atherosclerotic effects of TCF7L2, such as those against inflammation and neointimal hyperplasia (Figure 4).
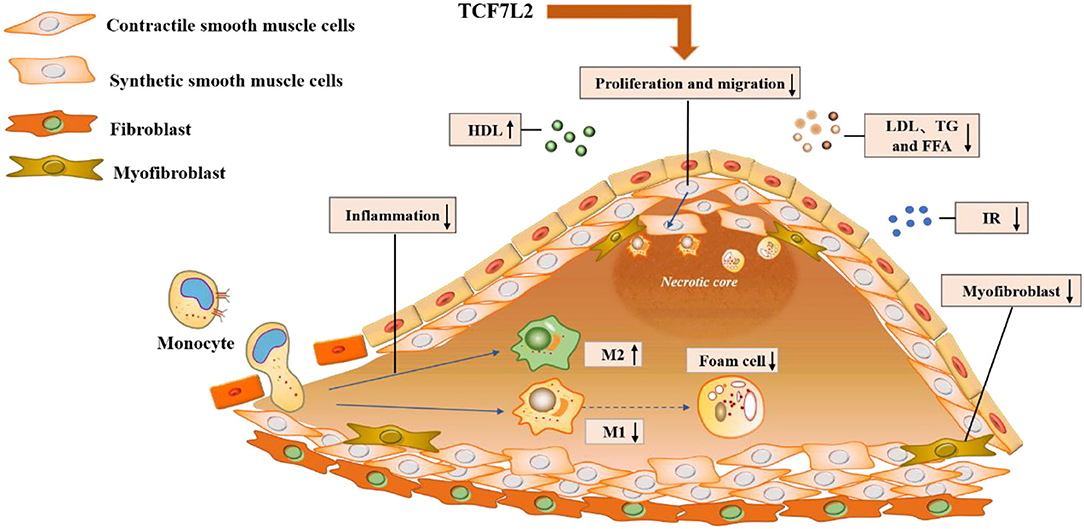
Figure 4. Molecular mechanisms underlying the action of TCF7L2 in anti-atherosclerosis. TCF7L2 exerts a protective role in atherosclerosis through multiple molecular mechanisms, including the inhibition of inflammation, VSMC proliferation and migration, dyslipidemia, insulin resistance, and foam cell and myofibroblast formation.
Macrophages are the major inflammatory effectors in atherosclerotic lesions. After endothelial injury, monocytes are recruited by chemokines and differentiate into macrophages in the vascular wall. Macrophages are divided through the process of macrophage polarization into two primary phenotypes according to their roles in inflammation: pro-inflammatory M1 macrophages and anti-inflammatory M2 macrophages. Under IFN-γ or TNF-α stimulation, macrophages polarize into the M1 phenotype and intensify local inflammation. M1 macrophages secrete numerous pro-inflammatory factors, including ROS, IL-1, and TNF-α, in the atherosclerotic microenvironment where they can destroy the structure or function of blood vessels (100). Additionally, M1 macrophages are more likely to transform into foam cells because of excessive lipid intake. In contrast, M2 macrophages can delay the development of atherosclerosis by relieving local inflammation (101, 102). TCF7L2 partially regulates macrophage polarization (19, 103). In vitro, curcumin can upregulate TCF7L2 to suppress M1 macrophage-derived inflammation (103). Consistently, TCF7L2 can induce macrophage polarization toward the M2 phenotype by downregulating the lncRNA XIST (19). These results indicate that TCF7L2 may be an upstream factor that regulates macrophage polarization and exerts anti-inflammatory effects in the atherosclerotic microenvironment, but the underlying molecular mechanism needs further exploration.
During atherogenesis, VSMCs are characterized by two phenotypes: a contractile phenotype and a synthetic phenotype. VSMCs usually exhibit a contractile phenotype to maintain vascular elasticity. However, VSMCs transform from this contractile phenotype into a synthetic phenotype during atherogenesis and show strong proliferation and migration activities (104, 105). TCF7L2 has been proved to be negatively related to VSMC plasticity (20, 36). Mechanistically, TCF7L2 can directly bind to the promoter of GATA6 and upregulate the expression of SM-MHC, a well-established marker of the contractile VSMC phenotype. In addition, TCF7L2 can downregulate Sp1 expression to suppress the VSMC phenotypic switch (36, 106). Notably, Wnt/TCF7L2 dysfunction enhances Sp1/PDGF/JNK signaling and subsequently leads to extensive VSMC proliferation (107, 108). Taken together, TCF7L2 upregulates GATA6 expression while downregulating Sp1 expression to inhibit VSMC plasticity and neointimal hyperplasia. Understanding the mechanisms underlying the recruitment of VSMCs by TCF7L2 may provide novel insights into the pathogenesis of atherosclerosis.
Myofibroblasts play an active role in neointimal hyperplasia and have been reported in all stages of atherosclerotic CVD from plaque formation and rupture to restenosis after percutaneous coronary intervention (109). Transformation of fibroblasts into neointimal myofibroblasts causes collagen deposition and neointimal expansion under TGF-β stimulation (110–112). Meanwhile, myofibroblasts also secrete pro-inflammatory cytokines to aggravate atherosclerosis (113, 114). Contreras et al. found that TCF7L2 is highly expressed in fibroblasts, while it is barely expressed in myofibroblasts, and it behaves as a key control switch in the differentiation of fibroblasts to myofibroblasts (115, 116). During this process, TGF-β accelerates the degradation of TCF7L2 to induce myofibroblast formation via the ubiquitin-protease system (115).
TCF7L2 exerts multiple anti-atherosclerotic effects, including IR, lipid accumulation, and local inflammation. TCF7L2 is expected to be an alternative therapeutic target for atherosclerotic CVDs. Thus, the promotion of TCF7L2 expression is a more intuitive therapeutic strategy. Numerous studies have indicated that noncoding RNAs, including short noncoding RNAs (miRNAs) and long noncoding RNAs (lncRNAs), are master regulators of gene expression, and many are involved in the development of atherosclerosis (117). Serum miR-217 levels have been reported to be significantly increased in patients or mice with atherosclerosis (118). Mechanistically, miR-217 aggravates the progression of atherosclerosis by promoting endothelial dysfunction and inflammatory responses (119, 120). Yu et al. (121) showed that TCF7L2 is a downstream target of miR-217, as confirmed by dual luciferase reporter assays. Thus, suppression of miR-214 expression may be a possible treatment for atherosclerotic CVDs. Other miRNAs, such as miR-26b-5p, miR-17-5p, and miR-181-5p, show inhibitory effects on TCF7L2 expression similar to those of miR-217 (122, 123). Moreover, miR-26b-5p and miR-17-5p can promote adipogenic differentiation by downregulating TCF7L2 expression (124). MiR-17-5p can specifically bind to the 3′UTR of TCF7L2 mRNA and recruit the RNA-induced silencing complex (RISC) to degrade TCF7L2 mRNA (125). LncRNA also participates in the anti-atherosclerotic action of TCF7L2, wherein TCF7L2 downregulates the lncRNA XIST and promotes M2 polarization in THP-1 macrophages (19). Interestingly, TCF7L2 also participates in the protective effects of estradiol on the cardiovascular system (126). Tian et al. (126) generated a dominant-negative TCF7L2 mouse model via adenovirus transfection and found that overexpression of dominant-negative TCF7L2 significantly increased serum TG and hepatic lipid accumulation in male transgenic mice, but not in females; this finding caused the researchers to be curious about the relationship between TCF7L2 and estradiol. In the subsequent estradiol treatment, male transgenic mice showed an amelioration in serum TG and hepatic lipid accumulation, perhaps owing to the increased expression of TCF7L2 and the nuclear translocation of β-catenin. In general, TCF7L2 exerts a variety of anti-atherosclerotic effects by regulating downstream genes and is itself regulated by multiple controllable factors, making its use in clinical treatment applications potentially advantageous.
TCF7L2, an important element in Wnt signaling, has attracted extensive attention since its discovery in 1998 (7). There are four functional structures in the TCF7L2 protein: the β catenin-binding domain, Groucho-binding sequence, HGM-DBD, and C-clamp (24, 25). HMG-DBD is responsible for identifying and binding to specific nucleotide sequences on the promoters of target genes to regulate their expression (17, 26). However, the activity of TCF7L2 is controlled by two contradictory regulatory elements: the co-activator β-catenin and co-repressor TLE/Groucho (28–30). TCF7L2 is mainly distributed in the nucleus and is usually occupied by TLE/Groucho, which suppresses the regulatory activity of TCF7L2 on target genes. However, activating the Wnt signaling pathway promotes the nuclear import of β-catenin, which competitively binds with TCF7L2 and exerts positive effects on TCF7L2. With the assistance of β-catenin, TCF7L2 participates in insulin secretion, adipogenesis, and maintenance of the blood lipid balance. During insulin secretion, TCF7L2 can increase proinsulin and PSCK1/PSCK2 to provide abundant insulin (79, 80). In addition, TCF7L2 upregulates the L-type Ca2+ channel CAV1.2, which promotes insulin efflux through exocytosis (81–83). TCF7L2-binding sites are present on the promoters of LPL and TGH genes, both of which are related to TG hydrolysis and can reduce serum TG levels (91–94).
In addition to metabolism, TCF7L2 can influence cell fate to alleviate atherosclerosis in the vascular endothelium. It is well established that local inflammation is an essential and forceful factor in the development of atherosclerosis, and macrophages are major participants in the inflammatory response (100–102). With different stimulations, macrophages can polarize into the pro-inflammatory M1 phenotype or anti-inflammatory M2 phenotype. TCF7L2 is involved in polarization of macrophages into the M2 phenotype, alleviating the local inflammatory response and atherogenesis (19, 103). Neointimal hyperplasia mainly occurs in the middle or late stages of atherosclerosis, especially after PCI (20, 36, 115, 116). During neointimal hyperplasia, VSMCs or fibroblasts proliferate and migrate to the vascular intima, where they transform into synthetic VSMCs or myofibroblasts, respectively, which synthesize and secrete large amounts of extracellular matrix. These changes eventually cause a decline in blood-vessel elasticity and lumen area, causing CVD patients to have more obvious clinical manifestations.
In terms of CVD prevention and treatment, we should pay attention to TCF7L2 rs7903146, which has been proven to be a causative SNP close to exon 4 (9, 46). Previous studies have shown that rs7903146 is closely associated with genetic susceptibility to T2D, and accumulating evidence indicates that rs7903146 increases CVD risk in the population, especially in subjects with diabetes (21, 127). Causally, rs7903146 alters the sequence of TCF7L2 mRNA, inserting an extra exon 4, and results in the reduction of TCF7L2 protein (47, 48). Owing to insufficient TCF7L2 protein, rs7903146 T-allele carriers are more likely to suffer from islet atrophy, dyslipidemia, and atherosclerosis, as shown in Figure 3. Thus, rs7903146 is a genetic biomarker to identify a population with a high risk of CVDs. Furthermore, TCF7L2 is a promising therapeutic target; we have provided a detailed description of its role in atherosclerosis from metabolism and inflammation to neointimal hyperplasia, which makes it suitable for clinical applications. However, a comprehensive understanding of TCF7L2 remains necessary to ensure that any future TCF7L2-based clinical treatment for CVD is safe and effective. Therefore, the following issues are worthy of further exploration: (i) the mechanism of TCF7L2 in independent Wnt signaling; (ii) factors affecting the conversion of TCF7L2 between co-repressors and co-activators; (iii) potential effects of TCF7L2 on FFA uptake in the intestine; (iv) mechanism underlying the induction of M2 polarization by TCF7L2; (v) potential inhibition of foam cells by TCF7L2; and (vi) potential regressive effect of TCF7L2 on vascular intimal hyperplasia. A better understanding of these issues will further reveal the molecular mechanism underlying the action of TCF7L2 in atherosclerosis, which is necessary for the development of new anti-atherosclerotic drugs in the future.
JYL, LZ, and PPH designed the writing framework. JYL and LZ wrote the original draft and drew figures. LZ, PPH, and XPOY modified and polished the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by the Natural Science Foundation of China (No. 82170485), Natural Science Foundation of Hunan Province, China (No. 2019JJ40249), Outstanding Young Aid Program for Education Department of Human Province (No. 18B274), the Major Project of social science achievement review committee in Hunan Province (XSP20ZDI013), Key Project of Hunan Provincial Department of Education (20A427), and Graduate Research innovation Project of School of Nursing, University of South China (2021CX22).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
1. Herrington W, Lacey B, Sherliker P, Armitage J, Lewington S. Epidemiology of atherosclerosis and the potential to reduce the global burden of atherothrombotic disease. Circ Res. (2016) 118:535–46. doi: 10.1161/CIRCRESAHA.115.307611
2. Townsend N, Nichols M, Scarborough P, Rayner M. Cardiovascular disease in Europe–epidemiological update 2015. Eur Heart J. (2015) 36:2696–705. doi: 10.1093/eurheartj/ehv428
3. Falk E. Pathogenesis of atherosclerosis. J Am Coll Cardiol. (2006) 47:C7–12. doi: 10.1016/j.jacc.2005.09.068
4. Mudau M, Genis A, Lochner A, Strijdom H. Endothelial dysfunction: the early predictor of atherosclerosis. Cardiovasc J Afr. (2012) 23:222–31. doi: 10.5830/CVJA-2011-068
5. Aboonabi A, Meyer RR, Singh I. The association between metabolic syndrome components and the development of atherosclerosis. J Hum Hypertens. (2019) 33:844–55. doi: 10.1038/s41371-019-0273-0
6. Weedon MN. The importance of TCF7L2. Diabet Med. (2007) 24:10. doi: 10.1111/j.1464-5491.2007.02258.x
7. Korinek V, Barker N, Willert K, Molenaar M, Roose J, Wagenaar G, et al. Two members of the Tcf family implicated in Wnt/beta-catenin signaling during embryogenesis in the mouse. Mol Cell Biol. (1998) 18:1248–56. doi: 10.1128/mcb.18.3.1248
8. Ye B, Li L, Xu H, Chen Y, Li F. Opposing roles of TCF7/LEF1 and TCF7L2 in cyclin D2 and Bmp4 expression and cardiomyocyte cell cycle control during late heart development. Lab Invest. (2019) 99:807–18. doi: 10.1038/s41374-019-0204-2
9. Grant SF, Thorleifsson G, Reynisdottir I, Benediktsson R, Manolescu A, Sainz J, et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet. (2006) 38:320–3. doi: 10.1038/ng1732
10. Hattersley AT. Prime suspect: the TCF7L2 gene and type 2 diabetes risk. J Clin Invest. (2007) 117:2077–9. doi: 10.1172/JCI33077
11. Lyssenko V, Lupi R, Marchetti P, Del Guerra S, Orho-Melander M, Almgren P, et al. Mechanisms by which common variants in the TCF7L2 gene increase risk of type 2 diabetes. J Clin Invest. (2007) 117:2155–63. doi: 10.1172/JCI30706
12. Schafer SA, Tschritter O, Machicao F, Thamer C, Stefan N, Gallwitz B, et al. Impaired glucagon-like peptide-1-induced insulin secretion in carriers of transcription factor 7-like 2 (TCF7L2) gene polymorphisms. Diabetologia. (2007) 50:2443–50. doi: 10.1007/s00125-007-0753-6
13. Shu L, Sauter NS, Schulthess FT, Matveyenko AV, Oberholzer J, Maedler K. Transcription factor 7-like 2 regulates beta-cell survival and function in human pancreatic islets. Diabetes. (2008) 57:645–53. doi: 10.2337/db07-0847
14. Sousa AG, Marquezine GF, Lemos PA, Martinez E, Lopes N, Hueb WA, et al. TCF7L2 polymorphism rs7903146 is associated with coronary artery disease severity and mortality. PLoS ONE. (2009) 4:e7697. doi: 10.1371/journal.pone.0007697
15. Corella D, Carrasco P, Sorli JV, Estruch R, Rico-Sanz J, Martinez-Gonzalez MA, et al. Mediterranean diet reduces the adverse effect of the TCF7L2-rs7903146 polymorphism on cardiovascular risk factors and stroke incidence: a randomized controlled trial in a high-cardiovascular-risk population. Diabetes Care. (2013) 36:3803–11. doi: 10.2337/dc13-0955
16. Mitchell RK, Mondragon A, Chen L, McGinty JA, French PM, Ferrer J, et al. Selective disruption of Tcf7l2 in the pancreatic beta cell impairs secretory function and lowers beta cell mass. Hum Mol Genet. (2015) 24:1390–9. doi: 10.1093/hmg/ddu553
17. Chen X, Ayala I, Shannon C, Fourcaudot M, Acharya NK, Jenkinson CP, et al. the diabetes gene and Wnt pathway effector TCF7L2 regulates adipocyte development and function. Diabetes. (2018) 67:554–68. doi: 10.2337/db17-0318
18. Facchinello N, Tarifeno-Saldivia E, Grisan E, Schiavone M, Peron M, Mongera A, et al. Tcf7l2 plays pleiotropic roles in the control of glucose homeostasis, pancreas morphology, vascularization and regeneration. Sci Rep. (2017) 7:9605. doi: 10.1038/s41598-017-09867-x
19. Sun Y, Xu J. TCF-4 regulated lncRNA-XIST promotes M2 polarization of macrophages and is associated with lung cancer. Onco Targets Ther. (2019) 12:8055–62. doi: 10.2147/OTT.S210952
20. Srivastava R, Rolyan H, Xie Y, Li N, Bhat N, Hong L, et al. TCF7L2 (Transcription Factor 7-Like 2) regulation of GATA6 (GATA-binding protein 6)-dependent and -independent vascular smooth muscle cell plasticity and intimal hyperplasia. Arterioscler Thromb Vasc Biol. (2019) 39:250–62. doi: 10.1161/ATVBAHA.118.311830
21. Muendlein A, Saely CH, Geller-Rhomberg S, Sonderegger G, Rein P, Winder T, et al. Single nucleotide polymorphisms of TCF7L2 are linked to diabetic coronary atherosclerosis. PLoS One. (2011) 6:e17978. doi: 10.1371/journal.pone.0017978
22. Huertas-Vazquez A, Plaisier C, Weissglas-Volkov D, Sinsheimer J, Canizales-Quinteros S, Cruz-Bautista I, et al. TCF7L2 is associated with high serum triacylglycerol and differentially expressed in adipose tissue in families with familial combined hyperlipidaemia. Diabetologia. (2008) 51:62–9. doi: 10.1007/s00125-007-0850-6
23. Palizban A, Rezaei M, Khanahmad H, Fazilati M. Transcription factor 7-like 2 polymorphism and context-specific risk of metabolic syndrome, type 2 diabetes, and dyslipidemia. J Res Med Sci. (2017) 22:40. doi: 10.4103/1735-1995.202141
24. Zhang Y, Liu C, Duan X, Ren F, Li S, Jin Z, et al. CREPT/RPRD1B, a recently identified novel protein highly expressed in tumors, enhances the beta-catenin.TCF4 transcriptional activity in response to Wnt signaling. J Biol Chem. (2014) 289:22589–99. doi: 10.1074/jbc.M114.560979
25. Yang S, Liu Y, Li MY, Ng CSH, Yang SL, Wang S, et al. FOXP3 promotes tumor growth and metastasis by activating Wnt/beta-catenin signaling pathway and EMT in non-small cell lung cancer. Mol Cancer. (2017) 16:124. doi: 10.1186/s12943-017-0700-1
26. Norton L, Fourcaudot M, Abdul-Ghani MA, Winnier D, Mehta FF, Jenkinson CP, et al. Chromatin occupancy of transcription factor 7-like 2 (TCF7L2) and its role in hepatic glucose metabolism. Diabetologia. (2011) 54:3132–42. doi: 10.1007/s00125-011-2289-z
27. Atcha FA, Syed A, Wu B, Hoverter NP, Yokoyama NN, Ting JH, et al. A unique DNA binding domain converts T-cell factors into strong Wnt effectors. Mol Cell Biol. (2007) 27:8352–63. doi: 10.1128/MCB.02132-06
28. Poy F, Lepourcelet M, Shivdasani RA, Eck MJ. Structure of a human Tcf4-beta-catenin complex. Nat Struct Biol. (2001) 8:1053–7. doi: 10.1038/nsb720
29. Lu FI, Sun YH, Wei CY, Thisse C, Thisse B. Tissue-specific derepression of TCF/LEF controls the activity of the Wnt/beta-catenin pathway. Nat Commun. (2014) 5:5368. doi: 10.1038/ncomms6368
30. Chodaparambil JV, Pate KT, Hepler MR, Tsai BP, Muthurajan UM, Luger K, et al. Molecular functions of the TLE tetramerization domain in Wnt target gene repression. EMBO J. (2014) 33:719–31. doi: 10.1002/embj.201387188
31. Daniels DL, Weis WI. Beta-catenin directly displaces Groucho/TLE repressors from Tcf/Lef in Wnt-mediated transcription activation. Nat Struct Mol Biol. (2005) 12:364–71. doi: 10.1038/nsmb912
32. Hanson AJ, Wallace HA, Freeman TJ, Beauchamp RD, Lee LA, Lee E. XIAP monoubiquitylates Groucho/TLE to promote canonical Wnt signaling. Mol Cell. (2012) 45:619–28. doi: 10.1016/j.molcel.2011.12.032
33. Hasegawa H, Bitoh S, Obashi J, Hiraga S, Higuchi M. [Lateral approach to the anterior foramen magnum tumor: report of two cases]. No Shinkei Geka. (1988) 16:1517–20.
34. Behari J, Yeh TH, Krauland L, Otruba W, Cieply B, Hauth B, et al. Liver-specific beta-catenin knockout mice exhibit defective bile acid and cholesterol homeostasis and increased susceptibility to diet-induced steatohepatitis. Am J Pathol. (2010) 176:744–53. doi: 10.2353/ajpath.2010.090667
35. Abaricia JO, Shah AH, Chaubal M, Hotchkiss KM, Olivares-Navarrete R. Wnt signaling modulates macrophage polarization and is regulated by biomaterial surface properties. Biomaterials. (2020) 243:119920. doi: 10.1016/j.biomaterials.2020.119920
36. Srivastava R, Zhang J, Go GW, Narayanan A, Nottoli TP, Mani A. Impaired LRP6-TCF7L2 activity enhances smooth muscle cell plasticity and causes coronary artery disease. Cell Rep. (2015) 13:746–59. doi: 10.1016/j.celrep.2015.09.028
37. Yi F, Brubaker PL, Jin T. TCF-4 mediates cell type-specific regulation of proglucagon gene expression by beta-catenin and glycogen synthase kinase-3beta. J Biol Chem. (2005) 280:1457–64. doi: 10.1074/jbc.M411487200
38. Brown AL, Peters M, D'Andrea RJ, Gonda TJ. Constitutive mutants of the GM-CSF receptor reveal multiple pathways leading to myeloid cell survival, proliferation, and granulocyte-macrophage differentiation. Blood. (2004) 103:507–16. doi: 10.1182/blood-2003-05-1435
39. Duan Y, Liao AP, Kuppireddi S, Ye Z, Ciancio MJ, Sun J. beta-Catenin activity negatively regulates bacteria-induced inflammation. Lab Invest. (2007) 87:613–24. doi: 10.1038/labinvest.3700545
40. Lee H, Bae S, Choi BW, Yoon Y. WNT/beta-catenin pathway is modulated in asthma patients and LPS-stimulated RAW264.7 macrophage cell line. Immunopharmacol Immunotoxicol. (2012) 34:56–65. doi: 10.3109/08923973.2011.574704
41. Yochum GS, McWeeney S, Rajaraman V, Cleland R, Peters S, Goodman RH. Serial analysis of chromatin occupancy identifies beta-catenin target genes in colorectal carcinoma cells. Proc Natl Acad Sci USA. (2007) 104:3324–9. doi: 10.1073/pnas.0611576104
42. Konuma T, Okada Y. Statistical genetics and polygenic risk score for precision medicine. Inflamm Regen. (2021) 41:18. doi: 10.1186/s41232-021-00172-9
43. Shastry BS. SNPs: impact on gene function and phenotype. Methods Mol Biol. (2009) 578:3–22. doi: 10.1007/978-1-60327-411-1_1
44. Xu Z, Taylor JA. SNPinfo: integrating GWAS and candidate gene information into functional SNP selection for genetic association studies. Nucleic Acids Res. (2009) 37:W600–5. doi: 10.1093/nar/gkp290
45. Brodie A, Azaria JR, Ofran Y. How far from the SNP may the causative genes be? Nucleic Acids Res. (2016) 44:6046–54. doi: 10.1093/nar/gkw500
46. Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, et al. A genome-wide association study identifies novel risk loci for type 2 diabetes. Nature. (2007) 445:881–5. doi: 10.1038/nature05616
47. Le Bacquer O, Shu L, Marchand M, Neve B, Paroni F, Kerr Conte J, et al. TCF7L2 splice variants have distinct effects on beta-cell turnover and function. Hum Mol Genet. (2011) 20:1906–15. doi: 10.1093/hmg/ddr072
48. Pradas-Juni M, Nicod N, Fernandez-Rebollo E, Gomis R. Differential transcriptional and posttranslational transcription factor 7-like regulation among nondiabetic individuals and type 2 diabetic patients. Mol Endocrinol. (2014) 28:1558–70. doi: 10.1210/me.2014-1065
49. Ruiz-Narvaez EA. Redundant enhancers and causal variants in the TCF7L2 gene. Eur J Hum Genet. (2014) 22:1243–6. doi: 10.1038/ejhg.2014.17
50. Xia Q, Deliard S, Yuan CX, Johnson ME, Grant SF. Characterization of the transcriptional machinery bound across the widely presumed type 2 diabetes causal variant, rs7903146, within TCF7L2. Eur J Hum Genet. (2015) 23:103–9. doi: 10.1038/ejhg.2014.48
51. Shu L, Matveyenko AV, Kerr-Conte J, Cho JH, McIntosh CH, Maedler K. Decreased TCF7L2 protein levels in type 2 diabetes mellitus correlate with downregulation of GIP- and GLP-1 receptors and impaired beta-cell function. Hum Mol Genet. (2015) 24:3004. doi: 10.1093/hmg/ddv075
52. Back SH, Kaufman RJ. Endoplasmic reticulum stress and type 2 diabetes. Annu Rev Biochem. (2012) 81:767–93. doi: 10.1146/annurev-biochem-072909-095555
53. Ghosh R, Colon-Negron K, Papa FR. Endoplasmic reticulum stress, degeneration of pancreatic islet beta-cells, and therapeutic modulation of the unfolded protein response in diabetes. Mol Metab. (2019) 27S:S60–S8. doi: 10.1016/j.molmet.2019.06.012
54. Perez-Martinez P, Perez-Caballero AI, Garcia-Rios A, Yubero-Serrano EM, Camargo A, Gomez-Luna MJ, et al. Effects of rs7903146 variation in the Tcf7l2 gene in the lipid metabolism of three different populations. PLoS ONE. (2012) 7:e43390. doi: 10.1371/journal.pone.0043390
55. Delgado-Lista J, Perez-Martinez P, Garcia-Rios A, Phillips CM, Williams CM, Gulseth HL, et al. Pleiotropic effects of TCF7L2 gene variants and its modulation in the metabolic syndrome: from the LIPGENE study. Atherosclerosis. (2011) 214:110–6. doi: 10.1016/j.atherosclerosis.2010.10.027
56. Zhang M, Zhao GJ, Yin K, Xia XD, Gong D, Zhao ZW, et al. Apolipoprotein A-1 binding protein inhibits inflammatory signaling pathways by binding to Apolipoprotein A-1 in THP-1 macrophages. Circ J. (2018) 82:1396–404. doi: 10.1253/circj.CJ-17-0877
57. Zhao GJ, Yin K, Fu YC, Tang CK. The interaction of ApoA-I and ABCA1 triggers signal transduction pathways to mediate efflux of cellular lipids. Mol Med. (2012) 18:149–58. doi: 10.2119/molmed.2011.00183
58. Cropano C, Santoro N, Groop L, Dalla Man C, Cobelli C, Galderisi A, et al. The rs7903146 variant in the TCF7L2 gene increases the risk of Prediabetes/Type 2 diabetes in obese adolescents by impairing beta-cell function and hepatic insulin sensitivity. Diabetes Care. (2017) 40:1082–9. doi: 10.2337/dc17-0290
59. Le Bacquer O, Kerr-Conte J, Gargani S, Delalleau N, Huyvaert M, Gmyr V, et al. TCF7L2 rs7903146 impairs islet function and morphology in non-diabetic individuals. Diabetologia. (2012) 55:2677–81. doi: 10.1007/s00125-012-2660-8
60. Srinivasan S, Kaur V, Chamarthi B, Littleton KR, Chen L, Manning AK, et al. TCF7L2 genetic variation augments incretin resistance and influences response to a sulfonylurea and metformin: the study to understand the genetics of the acute response to metformin and glipizide in humans (SUGAR-MGH). Diabetes Care. (2018) 41:554–61. doi: 10.2337/dc17-1386
61. Dujic T, Bego T, Malenica M, Velija-Asimi Z, Ahlqvist E, Groop L, et al. Effects of TCF7L2 rs7903146 variant on metformin response in patients with type 2 diabetes. Bosn J Basic Med Sci. (2019) 19:368–74. doi: 10.17305/bjbms.2019.4181
62. Bielinski SJ, Pankow JS, Folsom AR, North KE, Boerwinkle E. TCF7L2 single nucleotide polymorphisms, cardiovascular disease and all-cause mortality: the Atherosclerosis Risk in Communities (ARIC) study. Diabetologia. (2008) 51:968–70. doi: 10.1007/s00125-008-1004-1
63. Geoghegan G, Simcox J, Seldin MM, Parnell TJ, Stubben C, Just S, et al. Targeted deletion of Tcf7l2 in adipocytes promotes adipocyte hypertrophy and impaired glucose metabolism. Mol Metab. (2019) 24:44–63. doi: 10.1016/j.molmet.2019.03.003
64. Yan Y, Klein R, Heiss G, Girman CJ, Lange EM, Klein BE, et al. The transcription factor 7-like 2 (TCF7L2) polymorphism may be associated with focal arteriolar narrowing in Caucasians with hypertension or without diabetes: the ARIC Study. BMC Endocr Disord. (2010) 10:9. doi: 10.1186/1472-6823-10-9
65. Zhao Y, Evans MA, Allison MA, Bertoni AG, Budoff MJ, Criqui MH, et al. Multisite atherosclerosis in subjects with metabolic syndrome and diabetes and relation to cardiovascular events: the multi-ethnic study of atherosclerosis. Atherosclerosis. (2019) 282:202–9. doi: 10.1016/j.atherosclerosis.2018.12.005
66. Gao X, Song J, Watase H, Hippe DS, Zhao X, Canton G, et al. Differences in carotid plaques between symptomatic patients with and without diabetes mellitus. Arterioscler Thromb Vasc Biol. (2019) 39:1234–9. doi: 10.1161/ATVBAHA.118.312092
67. Liu M, Zhuang X, Chen X, Zhang S, Yang D, Zhong X, et al. Antiplatelet strategy in primary and secondary prevention of cardiovascular disease in patients with type 2 diabetes mellitus: a perspective from the guideline appraisal. J Diabetes Investig. (2021) 12:99–108. doi: 10.1111/jdi.13324
68. DeFronzo RA. Insulin resistance, lipotoxicity, type 2 diabetes and atherosclerosis: the missing links. The claude bernard lecture 2009. Diabetologia. (2010) 53:1270–87. doi: 10.1007/s00125-010-1684-1
69. Poznyak A, Grechko AV, Poggio P, Myasoedova VA, Alfieri V, Orekhov AN. The diabetes mellitus-atherosclerosis connection: the role of lipid and glucose metabolism and chronic inflammation. Int J Mol Sci. (2020) 21:1835. doi: 10.3390/ijms21051835
70. Kaur R, Kaur M, Singh J. Endothelial dysfunction and platelet hyperactivity in type 2 diabetes mellitus: molecular insights and therapeutic strategies. Cardiovasc Diabetol. (2018) 17:121. doi: 10.1186/s12933-018-0763-3
71. Savic D, Ye H, Aneas I, Park SY, Bell GI, Nobrega MA. Alterations in TCF7L2 expression define its role as a key regulator of glucose metabolism. Genome Res. (2011) 21:1417–25. doi: 10.1101/gr.123745.111
72. Nguyen-Tu MS, Martinez-Sanchez A, Leclerc I, Rutter GA, da Silva Xavier G. Adipocyte-specific deletion of Tcf7l2 induces dysregulated lipid metabolism and impairs glucose tolerance in mice. Diabetologia. (2021) 64:129–41. doi: 10.1007/s00125-020-05292-4
73. Shu L, Zien K, Gutjahr G, Oberholzer J, Pattou F, Kerr-Conte J, et al. TCF7L2 promotes beta cell regeneration in human and mouse pancreas. Diabetologia. (2012) 55:3296–307. doi: 10.1007/s00125-012-2693-z
74. Desgraz R, Bonal C, Herrera PL. beta-cell regeneration: the pancreatic intrinsic faculty. Trends Endocrinol Metab. (2011) 22:34–43. doi: 10.1016/j.tem.2010.09.004
75. Tschen SI, Dhawan S, Gurlo T, Bhushan A. Age-dependent decline in beta-cell proliferation restricts the capacity of beta-cell regeneration in mice. Diabetes. (2009) 58:1312–20. doi: 10.2337/db08-1651
76. Zhou Y, Zhang E, Berggreen C, Jing X, Osmark P, Lang S, et al. Survival of pancreatic beta cells is partly controlled by a TCF7L2-p53-p53INP1-dependent pathway. Hum Mol Genet. (2012) 21:196–207. doi: 10.1093/hmg/ddr454
77. Yao DD, Yang L, Wang Y, Liu C, Wei YJ, Jia XB, et al. Geniposide promotes beta-cell regeneration and survival through regulating beta-catenin/TCF7L2 pathway. Cell Death Dis. (2015) 6:e1746. doi: 10.1038/cddis.2015.107
78. Wanet A, Caruso M, Domelevo Entfellner JB, Najar M, Fattaccioli A, Demazy C, et al. The transcription factor 7-Like 2-peroxisome proliferator-activated receptor gamma coactivator-1 alpha axis connects mitochondrial biogenesis and metabolic shift with stem cell commitment to hepatic differentiation. Stem Cells. (2017) 35:2184–97. doi: 10.1002/stem.2688
79. Zhou Y, Park SY, Su J, Bailey K, Ottosson-Laakso E, Shcherbina L, et al. TCF7L2 is a master regulator of insulin production and processing. Hum Mol Genet. (2014) 23:6419–31. doi: 10.1093/hmg/ddu359
80. Shao W, Szeto V, Song Z, Tian L, Feng ZP, Nostro MC, et al. The LIM homeodomain protein ISL1 mediates the function of TCF7L2 in pancreatic beta cells. J Mol Endocrinol. (2018) 61:1–12. doi: 10.1530/JME-17-0181
81. Braun M, Ramracheya R, Rorsman P. Autocrine regulation of insulin secretion. Diabetes Obes Metab. (2012) 14(Suppl. 3):143–51. doi: 10.1111/j.1463-1326.2012.01642.x
82. Mastrolia V, Flucher SM, Obermair GJ, Drach M, Hofer H, Renstrom E, et al. Loss of alpha2delta-1 calcium channel subunit function increases the susceptibility for diabetes. Diabetes. (2017) 66:897–907. doi: 10.2337/db16-0336
83. Ye Y, Barghouth M, Luan C, Kazim A, Zhou Y, Eliasson L, et al. The TCF7L2-dependent high-voltage activated calcium channel subunit alpha2delta-1 controls calcium signaling in rodent pancreatic beta-cells. Mol Cell Endocrinol. (2020) 502:110673. doi: 10.1016/j.mce.2019.110673
84. Wu HH, Li YL, Liu NJ, Yang Z, Tao XM, Du YP, et al. TCF7L2 regulates pancreatic beta-cell function through PI3K/AKT signal pathway. Diabetol Metab Syndr. (2019) 11:55. doi: 10.1186/s13098-019-0449-3
85. Li R, Ou J, Li L, Yang Y, Zhao J, Wu R. The Wnt Signaling Pathway Effector TCF7L2 Mediates Olanzapine-Induced Weight Gain and Insulin Resistance. Front Pharmacol. (2018) 9:379. doi: 10.3389/fphar.2018.00379
86. Ku EJ, Won GW, Lee YH, Lee DH, Jeon HJ, Oh TK. Genetic variation in TCF7L2 rs7903146 correlating with peripheral arterial disease in long-standing type 2 diabetes. Diab Vasc Dis Res. (2020) 17:1479164119888475. doi: 10.1177/1479164119888475
87. Zhang T, Chen J, Tang X, Luo Q, Xu D, Yu B. Interaction between adipocytes and high-density lipoprotein:new insights into the mechanism of obesity-induced dyslipidemia and atherosclerosis. Lipids Health Dis. (2019) 18:223. doi: 10.1186/s12944-019-1170-9
88. Kirton JP, Crofts NJ, George SJ, Brennan K, Canfield AE. Wnt/beta-catenin signaling stimulates chondrogenic and inhibits adipogenic differentiation of pericytes: potential relevance to vascular disease? Circ Res. (2007) 101:581–9. doi: 10.1161/CIRCRESAHA.107.156372
89. Ross SE, Hemati N, Longo KA, Bennett CN, Lucas PC, Erickson RL, et al. Inhibition of adipogenesis by Wnt signaling. Science. (2000) 289:950–3. doi: 10.1126/science.289.5481.950
90. Bennett CN, Ross SE, Longo KA, Bajnok L, Hemati N, Johnson KW, et al. Regulation of Wnt signaling during adipogenesis. J Biol Chem. (2002) 277:30998–1004. doi: 10.1074/jbc.M204527200
91. Okopien B, Buldak L, Boldys A. Fibrates in the management of atherogenic dyslipidemia. Expert Rev Cardiovasc Ther. (2017) 15:913–21. doi: 10.1080/14779072.2017.1408410
93. Tietge UJ. Hyperlipidemia and cardiovascular disease: inflammation, dyslipidemia, and atherosclerosis. Curr Opin Lipidol. (2014) 25:94–5. doi: 10.1097/MOL.0000000000000051
94. Nordestgaard BG. Triglyceride-Rich Lipoproteins and Atherosclerotic Cardiovascular Disease: New Insights From Epidemiology, Genetics, and Biology. Circ Res. (2016) 118:547–63. doi: 10.1161/CIRCRESAHA.115.306249
95. Suzuki K, Oikawa T, Nochioka K, Miura M, Kasahara S, Sato M, et al. Elevated serum non-HDL (High-density lipoprotein) cholesterol and triglyceride levels as residual risks for myocardial infarction recurrence under statin treatment. Arterioscler Thromb Vasc Biol. (2019) 39:934–44. doi: 10.1161/ATVBAHA.119.312336
96. Welty FK. How do elevated triglycerides and low HDL-cholesterol affect inflammation and atherothrombosis? Curr Cardiol Rep. (2013) 15:400. doi: 10.1007/s11886-013-0400-4
97. He LH, Gao JH, Yu XH, Wen FJ, Luo JJ, Qin YS, et al. Artesunate inhibits atherosclerosis by upregulating vascular smooth muscle cells-derived LPL expression via the KLF2/NRF2/TCF7L2 pathway. Eur J Pharmacol. (2020) 884:173408. doi: 10.1016/j.ejphar.2020.173408
98. He PP, Ouyang XP, Tang YY, Liao L, Wang ZB, Lv YC, et al. MicroRNA-590 attenuates lipid accumulation and pro-inflammatory cytokine secretion by targeting lipoprotein lipase gene in human THP-1 macrophages. Biochimie. (2014) 106:81–90. doi: 10.1016/j.biochi.2014.08.003
99. Yisireyili M, Takeshita K, Hayashi M, Wu H, Uchida Y, Yamamoto K, et al. Dipeptidyl peptidase- IV inhibitor alogliptin improves stress-induced insulin resistance and prothrombotic state in a murine model. Psychoneuroendocrinology. (2016) 73:186–95. doi: 10.1016/j.psyneuen.2016.08.004
100. de Couto G, Liu W, Tseliou E, Sun B, Makkar N, Kanazawa H, et al. Macrophages mediate cardioprotective cellular postconditioning in acute myocardial infarction. J Clin Invest. (2015) 125:3147–62. doi: 10.1172/JCI81321
101. Colin S, Chinetti-Gbaguidi G, Staels B. Macrophage phenotypes in atherosclerosis. Immunol Rev. (2014) 262:153–66. doi: 10.1111/imr.12218
102. Momtazi-Borojeni AA, Abdollahi E, Nikfar B, Chaichian S, Ekhlasi-Hundrieser M. Curcumin as a potential modulator of M1 and M2 macrophages: new insights in atherosclerosis therapy. Heart Fail Rev. (2019) 24:399–409. doi: 10.1007/s10741-018-09764-z
103. Khalooghi K, Hashemi S, Mehraban N, Amiri P, Bazzaz JT, Larijani B, et al. In vitro modulation of TCF7L2 gene expression in human pancreatic cells. Mol Biol Rep. (2009) 36:2329–32. doi: 10.1007/s11033-009-9452-3
104. Liu M, Gomez D. Smooth Muscle Cell Phenotypic Diversity. Arterioscler Thromb Vasc Biol. (2019) 39:1715–23. doi: 10.1161/ATVBAHA.119.312131
105. Gomez D, Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res. (2012) 95:156–64. doi: 10.1093/cvr/cvs115
106. Azahri NS, Di Bartolo BA, Khachigian LM, Kavurma MM. Sp1, acetylated histone-3 and p300 regulate TRAIL transcription: mechanisms of PDGF-BB-mediated VSMC proliferation and migration. J Cell Biochem. (2012) 113:2597–606. doi: 10.1002/jcb.24135
107. Wilson E, Mai Q, Sudhir K, Weiss RH, Ives HE. Mechanical strain induces growth of vascular smooth muscle cells via autocrine action of PDGF. J Cell Biol. (1993) 123:741–7. doi: 10.1083/jcb.123.3.741
108. Marmur JD, Poon M, Rossikhina M, Taubman MB. Induction of PDGF-responsive genes in vascular smooth muscle. Implications for the early response to vessel injury. Circulation. (1992) 86:III53–60.
109. Sartore S, Chiavegato A, Faggin E, Franch R, Puato M, Ausoni S, et al. Contribution of adventitial fibroblasts to neointima formation and vascular remodeling: from innocent bystander to active participant. Circ Res. (2001) 89:1111–21. doi: 10.1161/hh2401.100844
110. Shi Y, O'Brien JE Jr., Fard A, Zalewski A. Transforming growth factor-beta 1 expression and myofibroblast formation during arterial repair. Arterioscler Thromb Vasc Biol. (1996) 16:1298–305. doi: 10.1161/01.atv.16.10.1298
111. Hundae A, McCullough PA. Cardiac and renal fibrosis in chronic cardiorenal syndromes. Nephron Clin Pract. (2014) 127:106–12. doi: 10.1159/000363705
112. Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, et al. Fibroblast-specific TGF-beta-Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest. (2017) 127:3770–83. doi: 10.1172/JCI94753
113. Singh S, Torzewski M. Fibroblasts and their pathological functions in the fibrosis of aortic valve sclerosis and atherosclerosis. Biomolecules. (2019) 9:472. doi: 10.3390/biom9090472
114. Siow RC, Mallawaarachchi CM, Weissberg PL. Migration of adventitial myofibroblasts following vascular balloon injury: insights from in vivo gene transfer to rat carotid arteries. Cardiovasc Res. (2003) 59:212–21. doi: 10.1016/s0008-6363(03)00292-x
115. Contreras O, Soliman H, Theret M, Rossi FMV, Brandan E. TGF-beta-driven downregulation of the transcription factor TCF7L2 affects Wnt/beta-catenin signaling in PDGFRalpha(+) fibroblasts. J Cell Sci. (2020) 133:jcs242297 doi: 10.1242/jcs.242297
116. Fu X, Khalil H, Kanisicak O, Boyer JG, Vagnozzi RJ, Maliken BD, et al. Specialized fibroblast differentiated states underlie scar formation in the infarcted mouse heart. J Clin Invest. (2018) 128:2127–43. doi: 10.1172/JCI98215
117. Fasolo F, Di Gregoli K, Maegdefessel L, Johnson JL. Non-coding RNAs in cardiovascular cell biology and atherosclerosis. Cardiovasc Res. (2019) 115:1732–56. doi: 10.1093/cvr/cvz203
118. Zhang L, Chen J, He Q, Chao Z, Li X, Chen M. MicroRNA217 is involved in the progression of atherosclerosis through regulating inflammatory responses by targeting sirtuin 1. Mol Med Rep. (2019) 20:3182–90. doi: 10.3892/mmr.2019.10581
119. de Yebenes VG, Briones AM, Martos-Folgado I, Mur SM, Oller J, Bilal F, et al. Aging-associated miR-217 aggravates atherosclerosis and promotes cardiovascular dysfunction. Arterioscler Thromb Vasc Biol. (2020) 40:2408–24. doi: 10.1161/ATVBAHA.120.314333
120. Zhang B, Zhang YF, Li R, Zhao L, Qin SG, Pan LF, et al. MiR-217 inhibits apoptosis of atherosclerotic endothelial cells via the TLR4/PI3K/Akt/NF-kappaB pathway. Eur Rev Med Pharmacol Sci. (2020) 24:12867–77. doi: 10.26355/eurrev_202012_24190
121. Yu B, Ye X, Du Q, Zhu B, Zhai Q, Li XX. The long non-coding RNA CRNDE Promotes colorectal carcinoma progression by competitively binding miR-217 with TCF7L2 and enhancing the Wnt/beta-catenin signaling pathway. Cell Physiol Biochem. (2017) 41:2489–502. doi: 10.1159/000475941
122. Chen Y, Li S, Zhang Y, Wang M, Li X, Liu S, et al. The lncRNA Malat1 regulates microvascular function after myocardial infarction in mice via miR-26b-5p/Mfn1 axis-mediated mitochondrial dynamics. Redox Biol. (2021) 41:101910. doi: 10.1016/j.redox.2021.101910
123. Du J, Han R, Li Y, Liu X, Liu S, Cai Z, et al. LncRNA HCG11/miR-26b-5p/QKI5 feedback loop reversed high glucose-induced proliferation and angiogenesis inhibition of HUVECs. J Cell Mol Med. (2020) 24:14231–46. doi: 10.1111/jcmm.16040
124. Luo Y, Ji H, Cao Y, Ding X, Li M, Song H, et al. miR-26b-5p/TCF-4 controls the adipogenic differentiation of human adipose-derived mesenchymal stem cells. Cell Transplant. (2020) 29:963689720934418. doi: 10.1177/0963689720934418
125. Tian L, Song Z, Shao W, Du WW, Zhao LR, Zeng K, et al. Curcumin represses mouse 3T3-L1 cell adipogenic differentiation via inhibiting miR-17-5p and stimulating the Wnt signalling pathway effector Tcf7l2. Cell Death Dis. (2017) 8:e2559. doi: 10.1038/cddis.2016.455
126. Tian L, Shao W, Ip W, Song Z, Badakhshi Y, Jin T. The developmental Wnt signaling pathway effector beta-catenin/TCF mediates hepatic functions of the sex hormone estradiol in regulating lipid metabolism. PLoS Biol. (2019) 17:e3000444. doi: 10.1371/journal.pbio.3000444
Keywords: TCF7L2, rs7903146, metabolism, atherosclerosis, CVD
Citation: Li JY, Zhou L, Ouyang XP and He PP (2021) Transcription Factor-7-Like-2 (TCF7L2) in Atherosclerosis: A Potential Biomarker and Therapeutic Target. Front. Cardiovasc. Med. 8:701279. doi: 10.3389/fcvm.2021.701279
Received: 27 April 2021; Accepted: 09 August 2021;
Published: 09 September 2021.
Edited by:
Vasily Sukhorukov, Research Institute of Human Morphology, RussiaReviewed by:
Voahanginirina Randriamboavonjy, Goethe University Frankfurt, GermanyCopyright © 2021 Li, Zhou, Ouyang and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xinping Ouyang, eTE2NTVAMTYzLmNvbQ==; Pingping He, aHBwLTYxMkAxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.